

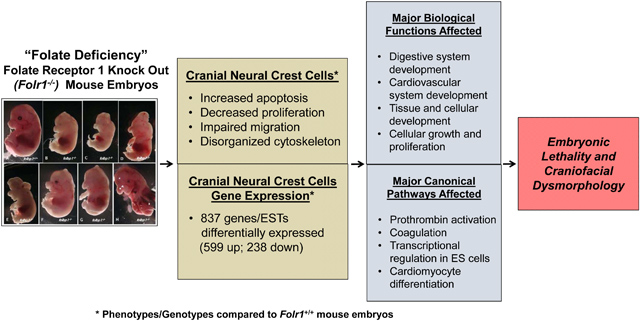
Abstract
Folic acid is a nutrient essential for embryonic development. Folate deficiency can cause embryonic lethality or neural tube defects and orofacial anomalies. Folate receptor 1 (Folr1) is a folate binding protein that facilitates the cellular uptake of dietary folate. To better understand the biological processes affected by folate deficiency, gene expression profiles of gestational day 9.5 (gd9.5) Folr1−/− embryos were compared to those of gd9.5 Folr1+/+ embryos. The expression of 837 genes/ESTs was found to be differentially altered in Folr1−/− embryos, relative to those observed in wild-type embryos. The 837 differentially expressed genes were subjected to Ingenuity Pathway Analysis. Among the major biological functions affected in Folr1−/− mice were those related to ‘digestive system development/function’, ‘cardiovascular system development/function’, ‘tissue development’, ‘cellular development’, and ‘cell growth and differentiation’, while the major canonical pathways affected were those associated with blood coagulation, embryonic stem cell transcription and cardiomyocyte differentiation (via BMP receptors). Cellular proliferation, apoptosis and migration were all significantly affected in the Folr1−/− embryos. Cranial neural crest cells (NCCs) and neural tube explants, grown under folate-deficient conditions, exhibited marked reduction in directed migration that can be attributed, in part, to an altered cytoskeleton caused by perturbations in F-actin formation and/or assembly. The present study revealed that several developmentally relevant biological processes were compromised in Folr1−/− embryos.
Keywords: Folr1, neural crest cells, proliferation, cell migration, F-actin, apoptosis
Graphical Abstract
INTRODUCTION
In the United States, birth defects affect 7 percent of all newborns and are the leading cause of infant mortality. While the etiology of over 60% of birth defects remains unknown, both genetics and environmental factors are known to be contributing factors. Craniofacial anomalies, including neural tube defects (NTDs) and oro/facial clefting constitute one of the most prevalent types of congenital dysmorphologies (Deguen et al., 2016; Juriloff and Harris, 2018). Gestational folate deficiency has been documented as a contributing factor for the etiology of many of these abnormalities (Hernandez-Diaz et al., 2000; Lucock, 2000; Beaudin and Stover; 2007; Wallingford et al., 2013; Chen et al., 2019).
Nearly half of orofacial clefts can be prevented with folic acid supplementation prior to and during early pregnancy (Czeizel, 1993; Shaw et al., 1995; Czeizel and Hirschberg, 1997; Loffredo et al., 2001; Wehby et al., 2013; Figueiredo et al., 2015; López-Gordillo et al., 2019) suggesting an essential role for folate in normal orofacial development. Moreover, pregnant women using folate antagonists as part of a treatment regimen for cancerous and non-cancerous diseases, exhibited increased risk for abnormal pregnancy outcomes (Amitai and Leventhal, 2001; Matok et al., 2009; Martino et al., 2016) including orofacial clefts (Hernandez-Diaz et al., 2000; Hyoun et al., 2012). This latter risk was reduced if the mother supplemented her diet before and during her pregnancy with multivitamins containing folic acid (Hernandez-Diaz et al., 2000; Jahanbin et al., 2018). While conflicting evidence can be found (Shaw et al., 1998; Sözen et al., 2009), a linkage appears to exist between polymorphisms in genes associated with folate metabolism in the maternal genome of some human cohorts and the occurrence of orofacial clefts in their offspring (Bufalino et al., 2010; Blanton et al., 2011; Marini et al., 2016). Collectively, these observations suggest that any alterations in optimal folate levels, or folate utilization, during early pregnancy can adversely impact normal orofacial development.
Animal studies provide further support for the critical role of folate in orofacial development. Orofacial clefts were observed in the offspring of experimental animals on folic acid-deficient diets (Maldonado et al., 2011, 2018) and those treated with folate antimetabolites (Jordan et al., 1977; Natsume et al., 1998; Wahl et al., 2015). Mice lacking Folr1 (the gene encoding folate receptor 1) also demonstrated a variety of orofacial defects (Piedrahita et al., 1999; Finnell et al., 2002; Tang and Finnell, 2003; Spiegelstein et al., 2004). However, maternal periconceptional folate supplementation in animal models with compromised folate transport decreased the rate and severity of orofacial anomalies in the offspring (Gelineau-van Waes et al., 2008b).
Folate, the naturally occurring form of folic acid (vitamin B9), is needed to provide one-carbon groups for methylation reactions and nucleic acid synthesis, and its derivatives are substrates in a number of single-carbon-transfer reactions (Lucock, 2000; Beaudin and Stover, 2007; Salbaum and Kappen, 2012; Guéant et al., 2013). Folate deficiency can thus result in: (1) the reduction of nucleotide precursors leading to decreased rates of DNA synthesis and reduced DNA repair capacity (Blount et al., 1997; Duthie, 2001; Bach et al., 2017); (2) the failure to transmethylate homocysteine, resulting in increased homocysteine levels that can, in turn, induce DNA strand breakage, oxidative stress and apoptosis (Mattson and Shea, 2003; Kasture et al., 2018); (3) a deficiency in the cellular level of SAM (S-adenosyl methionine), thereby contributing to a reduction in DNA and protein methylation reactions (Grillo and Colombatto, 2005; Fenech, 2010); and, (4) global hypomethylation and gene specific methylation changes (Chang et al., 2013; Li et al., 2019).
Eukaryotic animal cells cannot synthesize folate de novo and must obtain folate from dietary sources. Uptake of extracellular folate is achieved through folate receptors (or folate binding proteins; FBPs), and the reduced folate carrier, Slc19a1 (formerly Rfc1) (Spiegelstein et al., 2000). Three other folate transporters, Slc46a1 (or proton-coupled folate transporter (PCFT)), Slc25a32 (or mitochondrial folate transporter (MFT)), and Folh1 (Folate hydrolase 1) have also been suggested to play vital roles in transporting folate from the maternal intestinal lumen to the developing embryo (Findley et al., 2017). In mice, three FBPs - Folr1, Folr2 and Folr3 - are known to exist, whereas humans have four - FR (Folate Receptor) α, β, γ, and δ. FBPs have high affinity for folic acid, whereas Slc19a1 has a high affinity for 5-methyltetrahydrofolate (5-MTHF), the natural, active form of folic acid (Doucette and Stevens, 2001). FBPs are crucial for transplacental maternal-to-fetal folate transport (Henderson et al., 1995) as well as for the embryonic utilization of folate (Piedrahita et al., 1999). Hence, genes encoding these receptors/binding proteins represent excellent candidates for investigating the mechanisms underlying maternal folate deficiency-induced birth defects. Moreover, a high concentration of folate receptors/binding proteins resides on the embryonic neuroepithelium at the time of neural tube closure (Barber et al., 1999; Saitsu et al., 2003). Findings from our laboratory indicate that these FBPs are also expressed by embryonic oro/facial tissue at midgestation during the period of secondary palate development (Mukhopadhyay et al., 2004). These observations provide further support for a possible functional linkage between folate deficiency, neural tube and orofacial defects.
The most compelling evidence suggesting a role for FBPs in normal craniofacial development come from studies in which the gene encoding the high affinity receptor, Folr1, was functionally deleted in mice. The embryonic development of Folr1−/− mice is severely compromised in that they fail to complete cranial neural tube closure and eventually die by approximately gd10 due to cardiovascular defects (Piedrahita et al., 1999). However, daily maternal folate supplementation, before and throughout gestation, significantly reduced embryonic mortality and ameliorated the adverse developmental effects in these animals (Tang and Finnell, 2003; Spiegelstein et al., 2004). Maternal supplementation with lower doses of folate before and throughout gestation resulted in partial “rescue” wherein the Folr1−/− embryos survived past gd10, but presented with a variety of craniofacial abnormalities including, cleft lip and/or cleft palate (Zhu et al., 2007). Murine embryos with functional deletion of the gene encoding the low affinity receptor, Folr2, developed normally, suggesting distinct roles for Folr1 and Folr2 in the embryo (Piedrahita et al., 1999). Subsequently, Slc19a1 was functionally inactivated in mice and Slc19a1−/− embryos died shortly after implantation at gd5 (Gelineau-van Waes et al., 2008b). Maternal supplementation with low doses of folate before and throughout gestation, however, resulted in survival of these embryos to mid gestation (gd10.5). Embryos were nonetheless developmentally delayed and displayed multiple malformations including severe NTDs, and craniofacial and cardiovascular abnormalities (Gelineau-van Waes et al., 2008b). Recently, mutations in Slc46a1 and Slc25a32 were also suggested to be associated with NTDs and orofacial clefting (VanderMeer et al., 2016; Kim et al., 2018).
Collectively, these findings suggest that functional Folr1 and Slc19a1 are indispensable for normal neural tube, cardiovascular and orofacial development, and that folate supplementation can partially compensate for inherent genetic defects in folate uptake in the embryo during development. With the long term view of understanding the role of folate in orofacial development, experiments were conducted in the present study to investigate the effects of gestational folate deficiency on genome-wide gene expression levels in gd9.5 Folr1−/− knockout mice. In addition, the effects of folate deficiency on cellular migration, proliferation and apoptosis, key developmental processes affected in Folr1−/− embryos, have been examined in greater detail.
MATERIALS AND METHODS
Animals
Folr1+/− mice were provided by Dr. Richard H. Finnell (Baylor College of Medicine, Houston, TX. USA) and ICR mice were purchased from Harlan Laboratories (Indianapolis, IN, USA). Animals were housed at the University of Louisville Research Resources Center in a climate-controlled room with a 12-hour alternating light/dark cycle and maintained on standard Purina Mouse Chow #5015 and water ad libitum. Mature male and female mice were mated overnight. The presence of a vaginal plug the following morning was considered evidence of mating and was designated as gd0. Folr1−/− mice were generated by mating female Folr1+/− mice, maintained on the folate supplemented diet (Purina Mouse Chow #5015 supplemented with 40 ppm folate) for 2 weeks, prior to mating with Folr1+/− sires. Pregnant mice were maintained on this diet throughout gestation. Animal protocols utilized were reviewed and approved by the University of Louisville Institutional Animal Care and Use Committee (IACUC).
Embryo harvest
Pregnant female mice were euthanized by CO2 asphyxiation and Folr1−/− and Folr1+/+ embryos collected on gd9.5. Embryos were harvested by caesarian section, placed in ice cold PBS and dissected free from the decidual capsule, chorion, and amnion. Folr1−/− and Folr1+/+ embryos were genotyped by tail PCR. Embryos were examined under a Nikon SMZ1500 stereomicroscope and documented by digital photomicroscopy using a Nikon DXM1200 digital camera (Nikon Corporation, Tokyo, Japan).
RNA extraction and mRNA profiling of Folr1+/+ and Folr1−/− embryos
Total RNA from whole embryos was isolated using the Picopure RNA isolation kit (Arcturus, Mountain View, CA, USA) following the manufacturer’s recommendations. Antisense RNA (aRNA) was produced using the RiboAmp OA Amplification Kit (Arcturus) according to the manufacturer’s protocols. The RNA was subjected to one round of first strand cDNA synthesis, followed by second strand cDNA synthesis and in vitro transcription. This was followed by another round of first and second strand cDNA synthesis. The double stranded cDNA was transcribed in vitro using the High-Yield RNA Transcript Labeling Kit (Enzo, Farmingdale, NY, USA) according to the manufacturer’s instructions using biotinylated CTP and UTP (Affymetrix, Santa Clara, CA, USA). The resultant biotin-labeled cRNA was purified with RNeasy columns (Qiagen, Germantown, MD, USA) and eluted in 40 μL of RNase-free water. The quality and quantity of biotin-labeled cRNA were determined using the Agilent 2100 Bioanalyzer (Agilent Technologies, Santa Clara, CA, USA). Fifteen μg of labeled cRNA was fragmented in 40 μL of 1X fragmentation buffer (40 mM Tris-acetate pH 8.1, 100 mM K-acetate, 30 mM Mg-acetate) for 35 min at 94°C and assessed by agarose gel electrophoresis. Fragmented cRNA was brought to a total volume of 300 μL with 1X hybridization buffer (100 mM MES, 1 M NaCl, 20 mM EDTA, 0.01%Tween 20), 100 μg/mL herring sperm DNA, 500 μg/mL acetylated BSA, 50 pM biotinylated control oligonucleotide B2 and 1X eukaryotic hybridization controls (1.5 pM BioB, 5.0 pM BioC, 25 pM BioD and 100 pM cre; Affymetrix). Each cRNA preparation derived from either Folr1+/+ or Folr1−/− embryos was hybridized to an individual GeneChip® from an identical lot of Affymetrix Mouse Expression Array 430A for 16 hours. GeneChip® arrays were washed and stained using antibody-mediated signal amplification and the Affymetrix Fluidics Station’s standard Eukaryotic GE Wash 2 protocol. Three independent biological samples were prepared from either Folr1+/+ or Folr1−/− embryos, and then each of the three biological replicates was hybridized to a single GeneChip. Although a total of six samples and six GeneChips were used, one chip from Folr1−/− embryos failed quality control tests and did not yield any results.
mRNA microarray data analysis
Individual GeneChip® arrays were scanned with the GeneChip® Scanner 3000 (Affymetrix) and images were processed using Affymetrix Microarray Analysis Suite (MAS) 5.0 software, according to Affymetrix protocols. For analysis of microarray data, the GeneChip® images of the gd9.5 Folr1−/− embryo samples were normalized to the corresponding images of the gd9.5 Folr1+/+ embryo samples across all probe pair sets. The full dataset (difference call, fold change, average difference value, and absolute call data) from the three Folr1+/+ and two Folr1−/− samples was obtained using Affymetrix MAS 5.0 and contained expression levels for all 23,000 genes and expressed sequence tags (ESTs), as well as logarithm (base 2) of the estimated fold changes. The CEL files containing individual raw chip data (probe intensities) were imported to GeneSpring 7.2 (Silicon Genetics, Redwood City, CA, USA) and were pre-processed using Robust Multi-chip Average, with GC-content background correction (GCRMA). These GC-RMA normalized data were then further normalized using the ‘per gene normalization’ step in which all the samples were normalized against the median of the control samples. The raw and processed microarray data have been deposited in NCBI GEO under accession number GSE155428. Principal component analysis (PCA) was performed as a quality control procedure, to examine within-and between-group variability of the tested samples. To define a set of statistically significant, differentially expressed genes, a one-way ANOVA (parametric test, assuming equal variances) was applied with “Benjamini and Hochberg False Discovery Rate” (FDR) as the multiple testing correction (p<0.05). From this list, probes displaying >1.5-fold increase or decrease in expression with FDR-adjusted p-values equal or below 0.05, in knockout vs. wild-type embryos were selected. Hierarchical clustering analysis was performed to create a condition tree representing all of the differentially expressed genes based on their expression profiles.
Verification of mRNA microarray results by TaqMan® Real-Time PCR
Total RNA prepared from gd9.5 wild-type and Folr1−/− whole embryos was first treated with DNase I in the presence of RNaseOUT (Invitrogen Life Technologies, Carlsbad, CA, USA) to remove any contaminating DNA. cDNAs were synthesized as described by Mukhopadhyay et al (2004). mRNA levels were normalized to GAPDH mRNA present in each sample.
Establishment of cranial NCC monolayer cultures
First branchial arch tissues (containing NCCs) from gd10.5 embryos were microdissected in ice-cold Ca2+, Mg2+-free phosphate buffered saline (CMF-PBS), pH 7.2. Gd10.5 was chosen because of the difficulty in procuring enough NCCs from gd9.5 first branchial arches. The tissue was dissociated with 0.05 % trypsin-EDTA (GIBCO, Grand Island, NY, USA) at 37°C for 10 min, followed by pelleting and resuspension of the cells in high glucose DMEM/F12 (GIBCO) media containing 10% fetal bovine serum (FBS; Atlanta Biologicals, GA, USA) and two growth factors - FGF-2 (20 ng/ml; Chemicon, Temecula, CA, USA) and 1% N2 (GIBCO). The resuspended cells were seeded at 105 cells/dish in 60 mm fibronectin-coated (15 μg/mL; Sigma, St. Louis, MO, USA) culture dishes and allowed to grow in a humidified atmosphere at 37°C in 95% air / 5% CO2. After 24 hours, the medium was replaced either with the above medium, termed the ‘folate-replete medium’, or a ‘folate-free medium’ (RPMI media (GIBCO) containing 10% Knockout™ Serum Replacement; GIBCO) and the two growth factors at the concentrations noted above. After 48 hours, the cells were isolated for analysis of various cellular endpoints (see below).
Establishment of cranial NCC outgrowth cultures (neural tube explants)
Cranial NCC outgrowth cultures (neural tube explants) were derived from gd8.5 embryos so as to mimic the in vivo emigration of NCCs from the neural folds (neural tubes). Embryos were dissected in ice-cold CMF-PBS, pH 7.2. The region containing cranial NCCs was obtained by dissecting the neural folds extending from the optic vesicles (anteriorly) to the otic placodes (posteriorly) using tungsten needles. The tissue was explanted onto fibronectin-coated (15 μg/mL, bovine fibronectin) 4-well chamber slides and cultured in DMEM/F12 (GIBCO) containing 10% FBS (Atlanta Biologicals), 1% antibiotic-antimycotic solution (GIBCO) and 2 mM L-Glutamine (GIBCO). Cultures were maintained at 37°C in a humidified 95% air / 5% CO2 atmosphere. After 24 hours, the medium was replaced with either ‘folate-replete medium’ (RPMI [GIBCO] + 10% FBS) or ‘folate-free medium’ (RPMI [GIBCO] + 10% Knockout™ Serum Replacement; GIBCO). The NCC outgrowth cultures were maintained under these conditions for at least 24 hours, at the end of which the cultures were processed for various endpoints as noted below.
Determination of cranial NCC proliferation via incorporation of bromodeoxyuridine into DNA
NCCs were cultured for 48 hours until the cells reached 80% confluence. Two hours prior to the end of the experiment, bromodeoxyuridine (BrdU, 10 μM; Calbiochem, La Jolla, CA, USA) was added to the cells. Cultures were then washed and incubated with fresh medium for 30 min, followed by a wash with PBS. Cells were trypsinized with 0.25 % trypsin/EDTA (GIBCO) at 37°C for 5 minutes followed by a PBS wash. Cells were fixed with 10% paraformaldehyde in PBS for 30 min and washed with PBS. NCCs were dehydrated using 70% ethanol for 20 min at room temperature and then treated with 2N HCl for 30 min at 37°C. After neutralization with boric buffer (0.05 M, pH 8.5) for 10 min at 37°C, cells were washed with PBS and permeabilized with 0.3% Triton X-100/PBS. Non-specific staining was blocked with normal mouse serum at a 1/20 dilution in PBS, containing 0.1% BSA and 0.1% Triton X, for 30 min at 37°C. The cells were then labeled with Alexa Fluor® 488 conjugated anti-BrdU antibody (Molecular Probes/Invitrogen, Eugene, OR, USA) for 60 min at 37°C. Propidium iodide (PI; Molecular Probes) was used as a marker for DNA content. After labeling, cells were washed, acquired and analyzed using the BD FACS Calibur flow cytometer and Cell Quest Pro software (BD Bioscience, San Jose, CA, USA). The experiment was conducted on three independent biological samples (i.e. NCC cultures established at three different times from independent litters of embryos) and each was assayed in triplicate.
Determination of cranial NCC migration (transwell cell migration assay)
NCC migration was assessed using a transwell cell migration assay. Dissociated NCCs from the first branchial arches of gd10.5 embryos (105 cells/mL, prepared as detailed above) were seeded into 6.5 mm fibronectin-coated transwell inserts (8 μm pore; Corning; Lowell, MA, USA). The NCCs were plated in either folate-replete or folate-free media as noted previously. After 24 hours, the cells on each side of the transwell membrane were processed separately as detailed below. Cells were rinsed with CMF-PBS, pH 7.2, trypsinized from each side of the membrane using 0.05% Trypsin/EDTA solution (GIBCO) for 10 min at 37°C, pelleted again by centrifugation (10 min at 100 × g), resuspended and washed in CMF-PBS, and then pelleted by centrifugation. To quantify the relative number of non-migrated and migrated cells on either side of the transwell membrane, the CyQuant® Assay (Invitrogen Life Technologies), which determines relative cell numbers based on DNA content, was utilized. Cell pellets derived from each side of the transwell membrane were resuspended in Hank’s balanced salt solution containing a 1/500 dilution of CyQuant® DNA binding dye (Molecular Probes/Invtirogen). Cell samples were incubated at 37°C for 60 min to allow equilibration of dye binding to DNA. Fluorescence was measured on a Victor™ Plate Reader (Perkin Elmer; Waltham, MA, USA) with excitation at 488 nm and emission detection at 515 nm. Fluorescent dye binding/intensity is proportional to cell number. The experiment was conducted on three independent biological samples (i.e., NCC cultures established at three different times from independent litters of embryos) and each sample was assayed in triplicate.
Determination of cranial NCC migration (visualization of F-actin staining)
Tissue explants were cultured in 4-well-chamber slides, as described above, from gd8.5 Folr1+/+ and Folr1−/− embryos. After 48 hours, media was removed and the tissue explants were washed with PBS three times, 5 min each. Tissue explants were fixed with 3.7% formaldehyde (Sigma) for 15 min, washed with PBS and permeabilized with 0.1% Triton X-100/PBS for 5 min at room temperature. The tissue explants were then incubated with AF594-Phalloidin (300 μM; Molecular Probes/Invitrogen) for 30 min at 37°C to localize F-actin and with DAPI (300 μM; Molecular Probes/Invitrogen) for 5 min at room temperature to stain nuclei. Finally, tissue explants were washed with PBS three times for 5 min, air dried, and mounted under Fluoromount-G to preserve fluorescence (Southern Biotech; Birmingham, Ala, USA). Photomicrographs were obtained using a Nikon TE2000 microscope with fluorescence optics. The experiment was repeated using three biological replicates.
Determination of cranial NCC death (TUNEL assay)
Neural tube explant cultures from gd8.5 Folr1−/− or Folr1+/+ embryos were established as described above. Apoptosis in the explant cultures of Folr1−/− and Folr1+/+ embryonic neural crest was evaluated after 48 hours. Formaldehyde-fixed tissue explants were incubated overnight with anti-p75NGFR (rabbit polyclonal anti-nerve growth factor receptor, Advanced Targeting Systems, San Diego, CA, USA) to immuno-label cranial NCCs. Subsequently, a TUNEL assay (terminal deoxynucleotidyl transferase [TdT] dUTP nick labeling kit; Roche, Indianapolis, IN, USA) was conducted on tissue explants utilizing the manufacturer’s recommended protocol. After 2 washes in PBS, incubation with TUNEL mix (terminal deoxynucleotidyl transferase + labeled nucleotide mixture) was performed in association with the secondary anti-rabbit AF-488 antibody (Molecular Probes/Invitrogen) for 1 hour at 37°C in the dark, followed by 2 washes in PBS, to reveal NCCs. Slides were mounted with Fluoromount-G (Southern Biotech). Photomicrographs of explants were obtained using a Nikon TE2000 microscope with fluorescence optics and analysis of apoptosis was executed using Metamorph™ software (Universal Imaging Corporation; a subsidiary of Molecular Devices, San Jose, CA, USA). The experiment was repeated using three biological replicates.
Statistical Analyses
Data were analyzed with the Statistical Package for Social Sciences© (SPSS; Chicago, IL, USA) version 13.0. Statistical analyses for RT-qPCR, BrdU incorporation, transwell cell migration and cell migration in neural tube explants were conducted using a Student’s t-Test. All data are presented as mean ± standard error of mean (S.E.M.), unless otherwise stated. Statistical significance of p < 0.05 was noted. Where applicable, statistical significance was evaluated by One-Way ANOVA.
RESULTS
Effect of gestational folate deficiency on gene expression in developing mouse embryo
The effects of gestational folate deficiency on genome-wide gene expression in murine embryos were examined using gd9.5 Folr1−/− knockout mice and a chronological wild-type control. Folr1−/− mice exhibited the varied developmental defects associated with folate deficiency, such as anencephaly, exencephaly, cleft lip/palate, and retrognathia/agnathia (Supplementary Figure 1). Transcriptional profiling was performed using Affymetrix high-density Mouse Expression Array 430A. Examination of within- and between-group variability of the samples was assessed via principal component analysis (PCA). The three-dimensional PCA plot (Figure 1), colored by sample type, shows that the Folr1−/− and Folr1+/+ samples cluster separately and are quite distinct from each other.
Figure 1: Principal component analysis of gene expression in Folr1−/− knockout and Folr1+/+ wild-type samples.
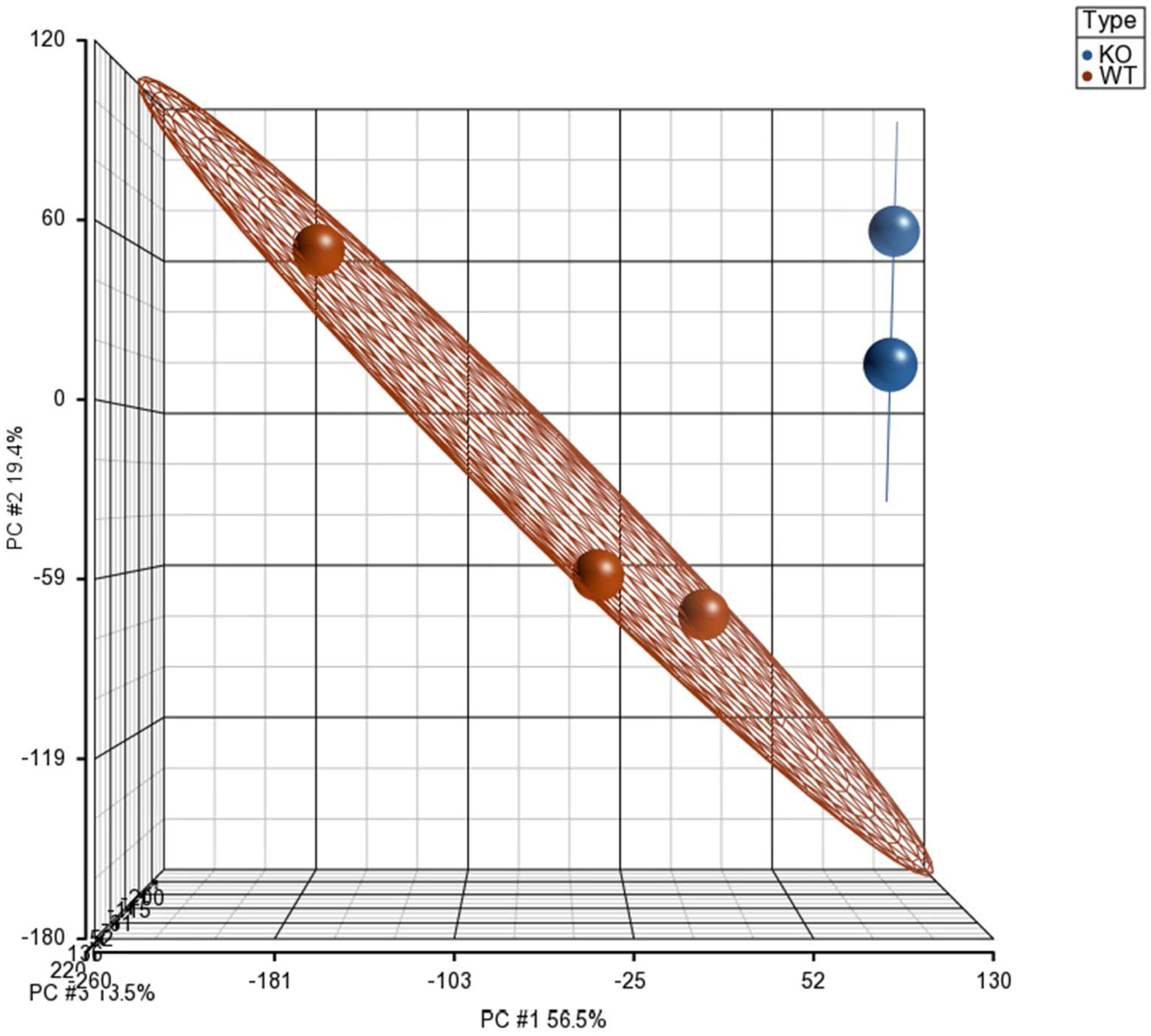
The plot shows two distinct clusters: Folr1−/− knockout samples represented by blue spheres and Folr1+/+ wild-type samples represented by red spheres. The X, Y and Z-axes represent principal components 1, 2 and 3 (PC#1, PC#2, and PC#3), respectively.
Out of ~ 23,000 genes and ESTs examined on the microarray, >50% were expressed in both Folr1+/+ and Folr1−/− mice. A total of 949 probe sets identified 837 genes/ESTs that were differentially expressed as a function of folate deficiency (≥ 1.5-fold change; adjusted p-value ≤ 0.05); of these 599 and 238 displayed increased or decreased expression, respectively, in Folr1−/− embryos, compared to their wild-type counterparts. The complete list of the differentially expressed genes, with their corresponding fold changes and adjusted p-values, is presented in Supplementary Table 1. Hierarchical clustering analysis generated a clustered image map (heat map) comprising all 837 differentially expressed genes demonstrating consistent differences between the two treatments (Figure 2). These genes/ESTs were classified into broad functional categories using GeneSpring Software (Silicon Genetics) and NetAffx Analysis Center (Affymetrix) and include genes encoding: extracellular proteins and those associated with extracellular matrix (ECM) function (Supplementary Table 2); transcription factors (including those associated with nucleic acid metabolism) (Supplementary Table 3); growth and differentiation factors (including those regulating apoptosis and cell migration) (Supplementary Table 4); signaling mediators (Supplementary Table 5); and, those that do not fit into any of these categories (Unclassified) (Supplementary Table 6). Graphical representation of the number of genes in the five distinct categories, whose expression was altered as a function of folate deficiency is provided in Figure 3.
Figure 2: Heat maps (hierarchical clusters) of all genes differentially expressed between gd9.5 Folr1−/− knockout and Folr1+/+ wild-type embryos.
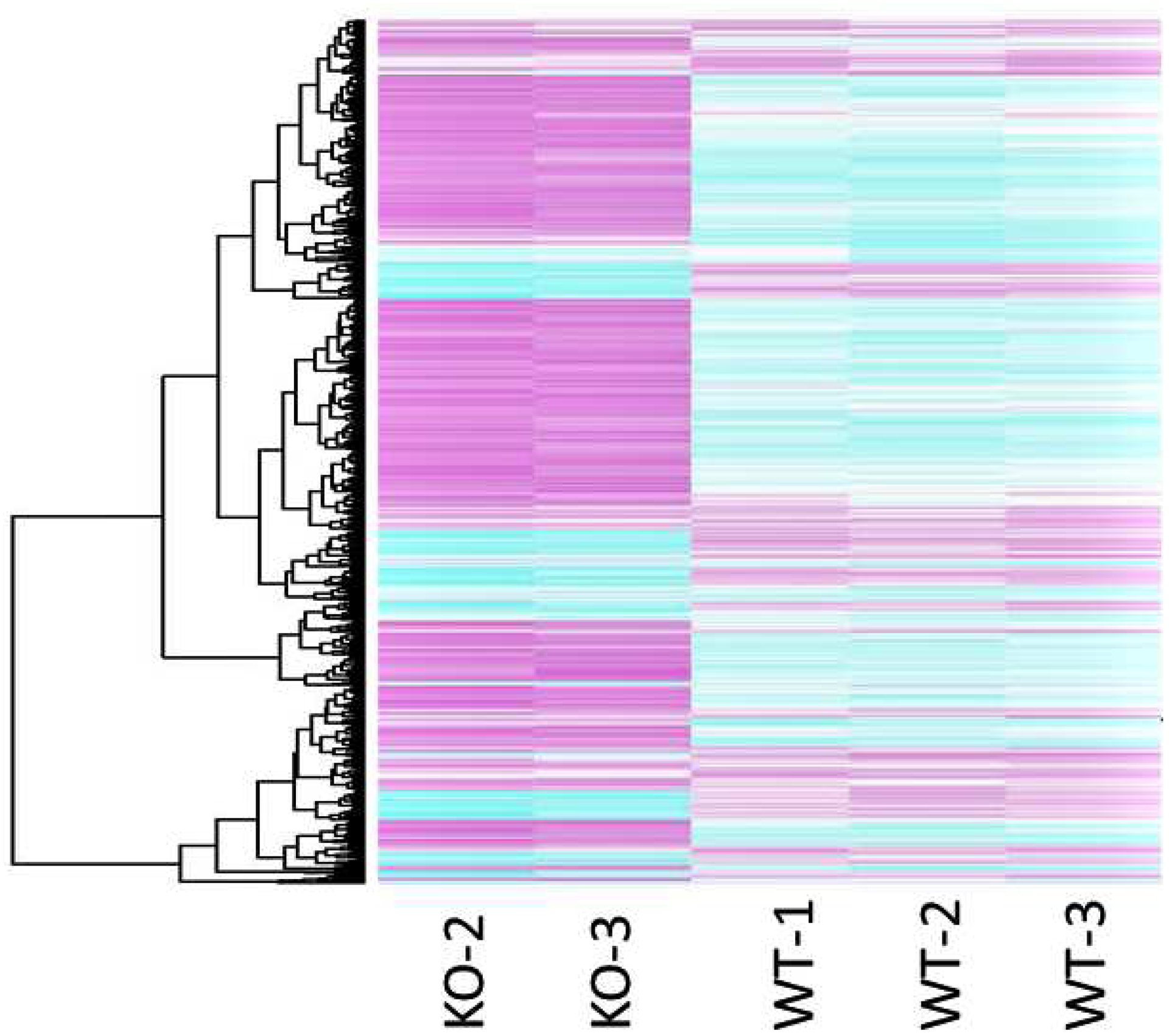
Each row of the clustered image map represents a gene, and each column represents one array for the sample type examined (KO or WT). Genes demonstrating a 1.5-fold or greater difference (adjusted p < 0.05) in expression are included in the map. Pink indicates an increase in gene expression (relative to the other expression measurements in the same row), whereas blue indicates a decrease.
Figure 3: Numbers and categories of differentially expressed genes in folate deficient (Folr1−/−) embryos compared to wild-type (Folr1+/+) embryos.
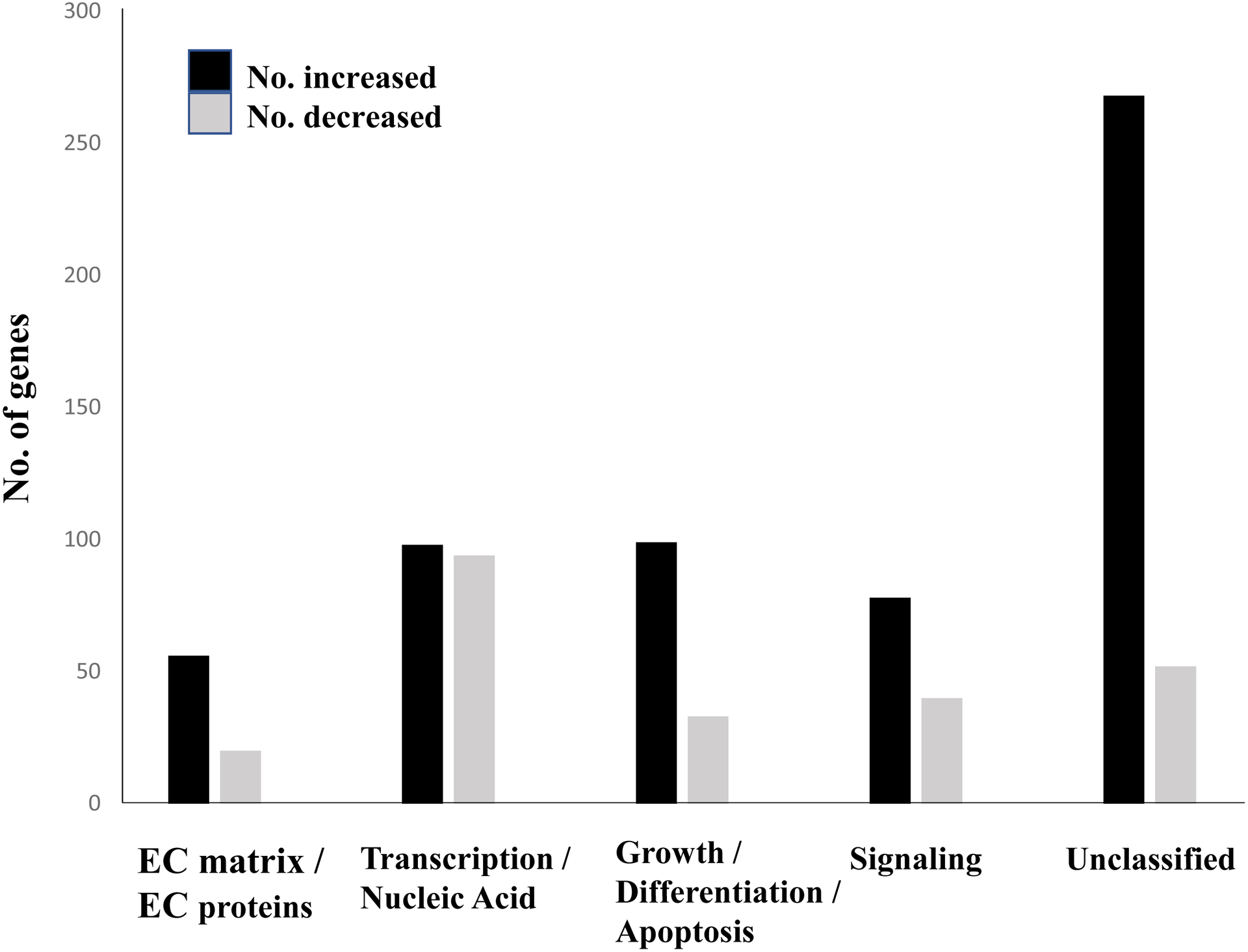
The figure broadly groups five categories of proteins encoded by the 837 differentially expressed genes in Folr1−/− embryos compared to wild-type Folr1+/+ embryos: Extracellular (EC) matrix and EC proteins; Transcription and Nucleic Acid Metabolism; Cell Growth, Differentiation and Apoptosis; Signaling Molecules; and Unclassified (those that did not fit into any of the aforementioned categories). The black and gray bars indicate genes whose expression levels were increased or decreased, respectively, in Folr1−/− embryos compared to those in Folr1+/+ embryos. The Y-axis indicates the number of genes. A complete list of genes in each category, ranked by expression levels, is provided in Supplementary Tables 2–6.
Ingenuity Pathway Analysis
In order to determine signaling pathways and biological functions that were significantly affected in Folr1−/− embryos, the 837 differentially expressed genes in these embryos were subjected to Ingenuity Pathway Analysis (IPA; Ingenuity Systems, Inc., Redwood City, CA, USA). The bar graph in Figure 4 categorizes the major (top ten) ‘Biological Functions’ that were affected as a result of folate deficiency. Digestive system development/function was the most affected category, followed by cardiovascular development/function, tissue and cellular development and cellular growth and proliferation. Figure 5 depicts the major (top ten) ‘Canonical Pathways’ affected in Folr1−/− mice and includes the prothrombin activation pathways, the coagulation system, transcriptional regulation in embryonic stem cells and cardiomyocyte differentiation via BMP receptors. The genes constituting some of the various categories depicted in Figures 4 and 5 are separately listed in Supplementary Tables 7 and 8, respectively. A more extensive list of ‘Biological functions’ and ‘Canonical pathways’ associated with folate deficiency can be found in Supplementary Figures 2 and 3, respectively. Specific categories of ‘Biological Functions’ that are critical for embryonic development and affected by folate metabolism, such as apoptosis, cell migration, cell proliferation, cell differentiation, and embryogenesis were selected for Network/Pathway analysis (Supplementary Figures 4–8, respectively). Specific signaling pathways likely compromised in embryos as a result of folate deficiency (see Discussion) are shown in Supplementary Figures 9A–F and include tight junction signaling, cholesterol biosynthesis, TGFβ-BMP signaling, embryonic stem cell pluripotency, Wnt β-catenin signaling, and mitochondrial dysfunction, respectively.
Figure 4: Major biological functions affected by folate deficiency in Folr1−/− embryos.
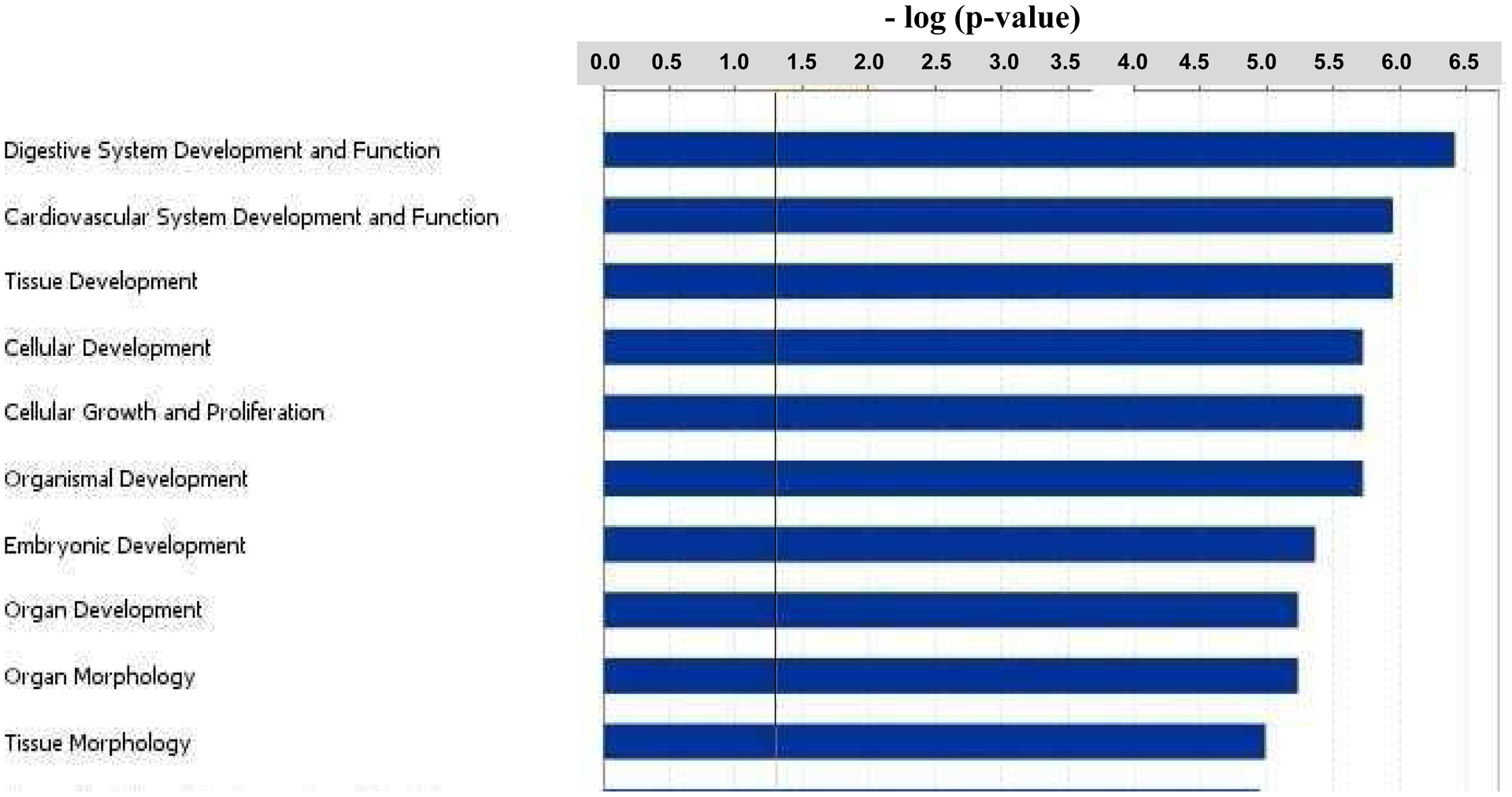
The 837 genes that were differentially expressed in Folr1−/− embryos compared to wild-type Folr1+/+ embryos, were subjected to Ingenuity Pathway Analysis (Ingenuity Systems) to identify key biological functions that were potentially affected as a result of folate deficiency. These functions are ranked, based on the number of affected genes participating in each affected category, from top to bottom in the figure. The biological functions were considered significant (Fisher’s Exact Test; p-value ≤ 0.05) if they were above the threshold line (in black). This figure depicts the top ten biological functions. A more extensive list is provided in Supplementary Figure 2.
Figure 5: Major canonical pathways affected by folate deficiency in Folr1−/− embryos.
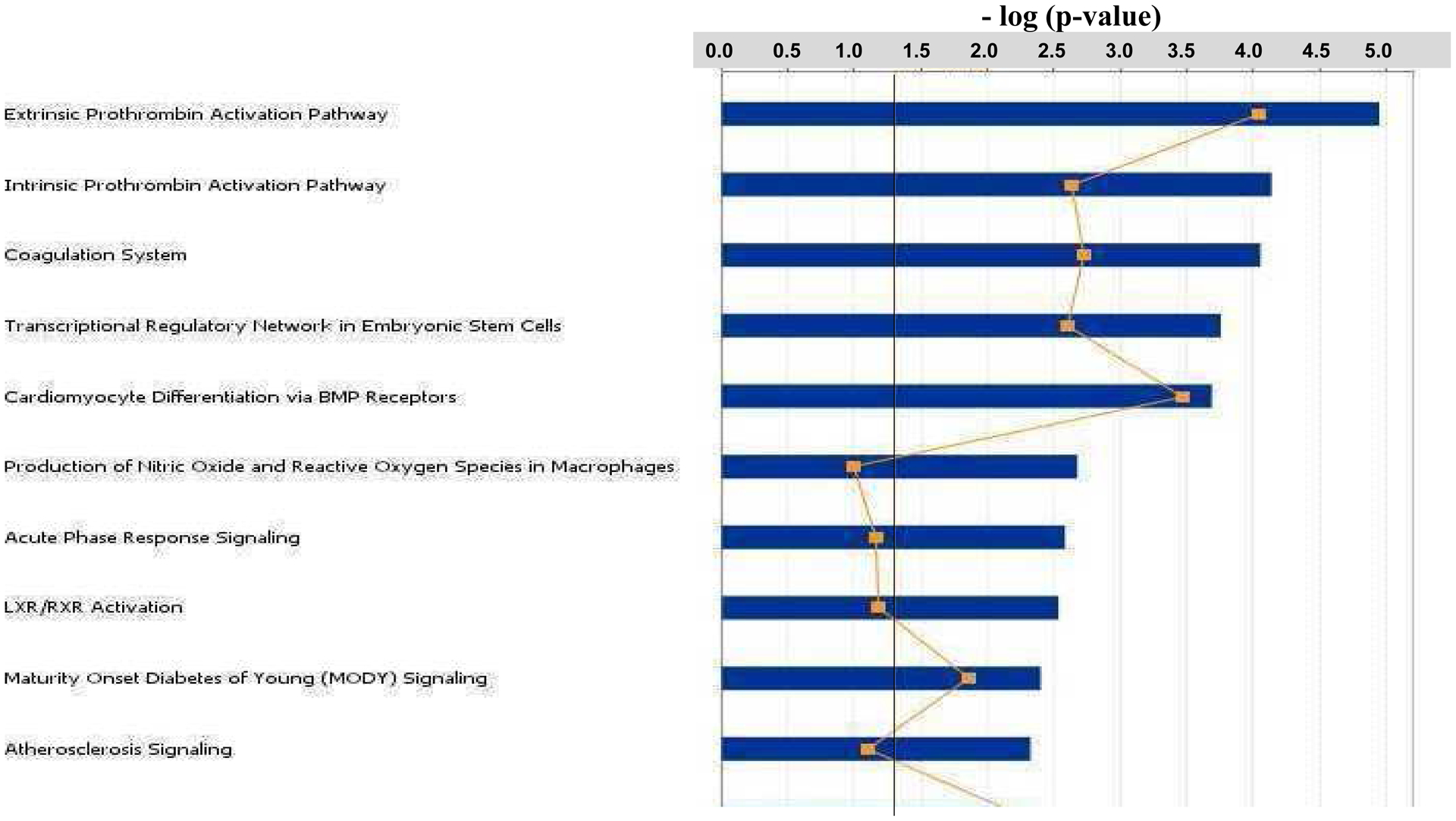
The 837 genes that were differentially expressed in Folr1−/− embryos compared to wild-type Folr1+/+ embryos were subjected to Ingenuity Pathway Analysis (Ingenuity Systems) and key canonical pathways potentially affected as a result of folate deficiency were identified. These functions are ranked, based on the number of affected genes participating in each affected category, from top to bottom in the figure. Pathways were considered significant (Fisher’s Exact Test; p-value ≤ 0.05) if they were above the threshold line (black). The ratio, represented by the line graph (-□-), is calculated by dividing the number of molecules in a given pathway that meet the cutoff criteria (i.e. ≥ 1.5-fold change), by the total number of molecules that constitute the pathway. This figure depicts the top ten canonical pathways. A more extensive list is provided in Supplementary Figure 3.
Quantitative Real Time-PCR validation of microarray gene expression profiling data
A small subset of genes with varying fold changes in expression was selected for verification using TaqMan® RT-qPCR. These target genes were selected based on their well established and central roles in embryonic development, in general, and NCC function, in particular. Comparison of expression levels (fold changes) of these genes by microarray and RT-qPCR analyses is presented in Tables 1 and 2. Selected genes whose expression decreased as a function of folate deficiency in Folr1−/− embryos (Table 1) include: Pdgfra (PDGF receptor α), Tgfbr1 (Transforming growth factor β, receptor 1), Msi1 (Musashi homolog 1), Tfap2α (AP2α; Adaptor protein 2α), Lhx2 (LIM homeobox 2), Hoxa2 (Homeobox A2), Foxd1 (Forkhead box D1) and Sema3a (Semaphorin 3A), whereas, those whose expression increased as a function of folate deficiency (Table 2) include: Apob (Apolipoprotein B), Cdkn1c (Cyclin-dependent kinase inhibitor 1C), Cubn (Cubilin), Tbx2 (T-box 2), Bmps 2 and 4 (Bone morphogenetic proteins 2 and 4), Ccng1 (Cyclin G1), Apoe (Apolipoprotein E) and Dkk1 (Dickkopf 1). Both the directionality and the relative level of change in expression of these genes were confirmed by RT-qPCR, with concordance in results between the two methods (Tables 1 and 2).
Table 1:
Genes whose expression decreased as a function of folate deficiency in Folr1−/− embryos
| Gene Symbol | Gene Name | Microarray Mean Fold Change ± SEM |
Real-Time PCR Mean Fold Change ± SEM |
|---|---|---|---|
| Pdgfra | PDGF receptor α | − 1.79 ± 0.11 | − 1.43 ± 0.08 |
| Tgfbr1 | TGFβ receptor 1 | − 1.89 ± 0.19 | − 1.79 ± 0.15 |
| Msi 1 | Musashi homolog 1 | − 2.01 ± 0.22 | − 2.38 ± 0.34 |
| Tfap2α | Transcription factor AP2α | − 2.02 ± 0.06 | − 1.61 ± 0.09 |
| Lhx2 | LIM homeobox 2 | − 1.52 ± 0.06 | − 3.45 ± 0.35 |
| Hoxa2 | Homeobox A2 | − 2.53 ± 0.22 | − 3.70 ± 0.42 |
| Foxd1 | Forkhead box D1 | − 3.41 ± 0.35 | − 4.17 ± 0.56 |
| Sema3a | Semaphorin 3A | − 1.92 ± 0.23 | −1.79 ± 0.21 |
Note: Expression of individual genes in Folr1−/− embryos was compared to that of Folr1+/+ at the same chronological gestational day (gd9.5). For microarray analysis, 2 Folr1−/− and 3 Folr1+/+ samples were used. For Real-Time PCR experiments, 3 Folr1−/− and 3 Folr1+/+ samples were used and assayed in triplicate.
Table 2:
Genes whose expression increased as a function of folate deficiency in Folr1−/− embryos
| Gene Symbol | Gene Name | Microarray Mean Fold Change ± SEM |
Real-Time PCR Mean Fold Change ± SEM |
|---|---|---|---|
| Apob | Apolipoprotein B | 6.02 ± 0.31 | 7.7 ± 0.48 |
| Cdkn1c | CDK inhibitor 1C | 3.27 ± 0.34 | 4.4 ± 0.26 |
| Cubn | Cubilina | 5.53 ± 0.07 | 11.6 ± 0.33 |
| Tbx2 | T-box 2 | 1.98 ± 0.22 | 3.1 ± 0.28 |
| Bmp2 | Bone morphogenetic protein 2 | 2.01 ± 0.33 | 4.5 ± 0.32 |
| Bmp4 | Bone morphogenetic protein 4 | 2.97 ± 0.38 | 6.4 ± 0.19 |
| Ccng1 | Cyclin G1 | 3.28 ± 0.41 | 3.3 ± 0.14 |
| Apoe | Apolipoprotein E | 6.37 ± 0.29 | 11.1 ± 0.40 |
| Dkk1 | Dickkopf 1 | 2.50 ± 0.22 | 5.2 ± 0.21 |
Note: Expression of individual genes in Folr1−/− embryos was compared to that of Folr1+/+ at the same chronological gestational day (gd9.5). For microarray analysis, 2 Folr1−/− and 3 Folr1+/+ samples were used. For Real-Time PCR experiments, 3 Folr1−/− and 3 Folr1+/+ samples were used and assayed in triplicate.
Averaged from two probes (Supplementary Table 6).
Effect of folate deficiency on embryonic cranial NCC proliferation: Flow cytometric analysis of bromodeoxyuridine incorporation and propidium iodide staining
The impact of folate deficiency on proliferation of embryonic cranial NCC in monolayer culture was evaluated by flow cytometry following BrdU incorporation and propidium iodide (PI) staining. Representative two-parameter flow cytometry histograms depicting BrdU incorporation (Y-axis) and PI staining (X-axis) of embryonic cranial NCCs cultured under folate-replete (Panel A) and folate-free (Panel B) conditions are presented in Figure 6. Embryonic NCCs grown in folate-free conditions incorporated significantly less BrdU into cellular DNA than those grown in folate-replete conditions. Graphical representation of the cumulative flow cytometry data (Panel C) indicates that growth of NCC in the absence of folate resulted in a statistically significant decrease in the percentage of NCCs that were proliferating, relative to growth of NCCs in the presence of folate (n=3; p ≤ 0.05).
Figure 6: Flow cytometric histograms of 5-Bromo-2’-deoxyuridine (BrdU) incorporation in embryonic cranial NCCs cultured in folate-replete or folate-free media.
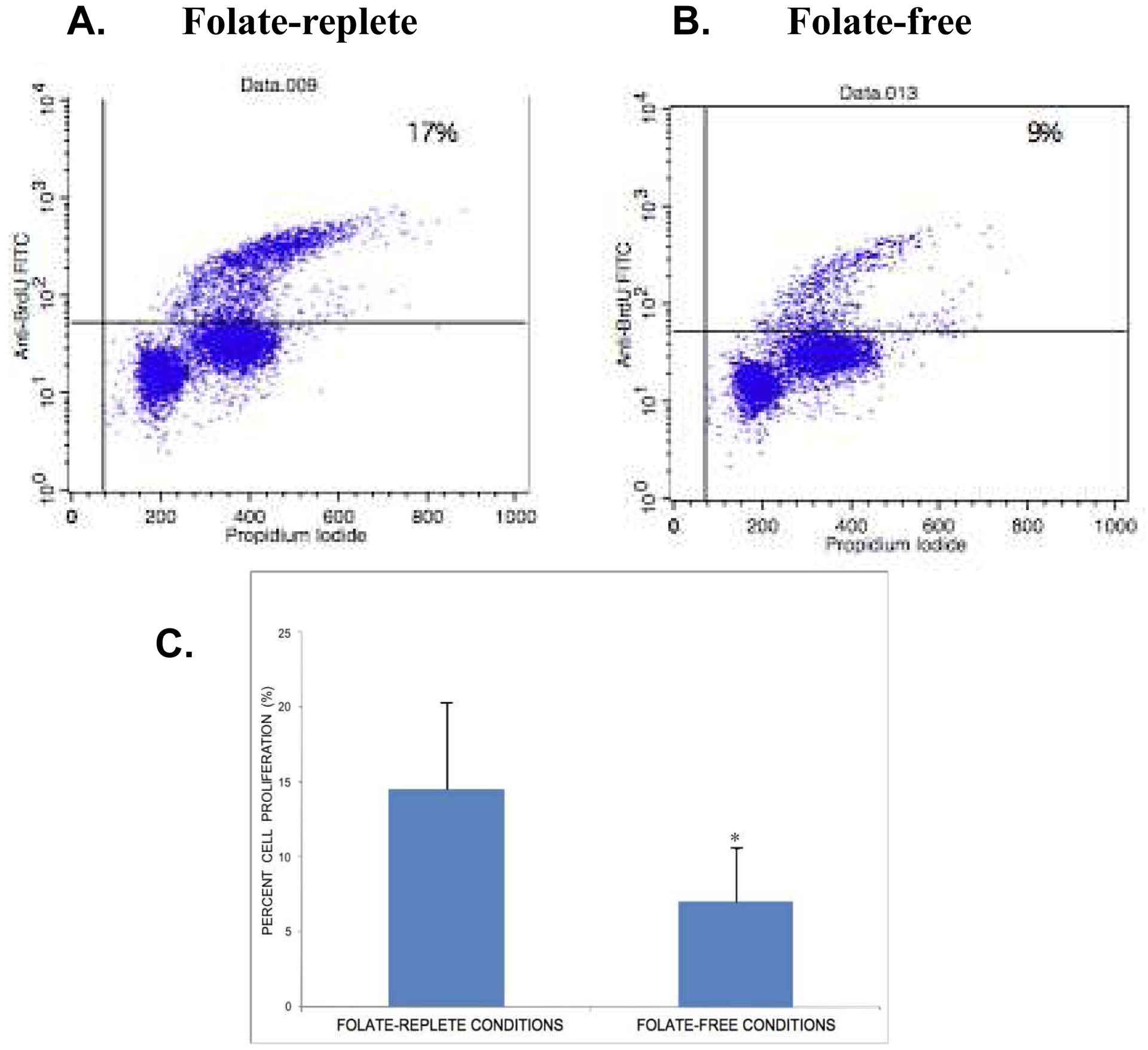
Flow cytometric histograms of BrdU incorporation vs. propidium iodide (PI) staining of embryonic cranial NCCs cultured in folate-replete (Panel A) or folate-free (Panel B) media. Primary cultures of embryonic cranial NCCs were established and grown in folate-replete or folate-free conditions for 48 hours. BrdU was added for an additional 2 hours, and the cells subsequently processed for flow cytometry assessment of cell proliferation. Graphical representation of the cumulative flow cytometry data is presented in Panel C. Percent cell proliferation represents the number of BrdU-positive cells compared to total cell number, and is reported as mean ± SEM. Growth of NCCs in the absence of folate resulted in a statistically significant decrease in the percentage of cells that were proliferating when compared to the growth of NCCs in the presence of folate (n=3; * p < 0.05).
F-actin staining of cranial NCC explant cultures from Folr1+/+ and Folr1−/− embryos
During routine culturing of cranial neural tube explants, a difference in the overall morphology of NCCs emigrating from wild-type explants compared to those emigrating from explants of folate deficient embryos was noted. To pursue this observation further, the basic cellular architecture of NCCs emigrating from the cranial neural tube explants of both Folr1+/+ and Folr1−/− embryos was examined by staining cultures for filamentous F-actin. The polymerization of G-actin to F-actin is required for polarized assembly of the cytoskeleton and cell migration. A notable effect of folate deficiency on NCC cellular phenotype/morphology was that F-actin in NCC ‘outgrowths’ from Folr1−/− explants (Figure 7, Panel B) was distributed in a pericellular pattern indicative of impaired directed migration. F-actin in NCC ‘outgrowths’ from wild-type explants (Figure 7, Panel A) was primarily distributed in a filamentous pattern of radiating stress fibers throughout the cytoplasm, indicative of typical motile cells engaged in directed migration.
Figure 7: F-actin staining in cranial NCC explant cultures from Folr1+/+ and Folr1−/− embryos.
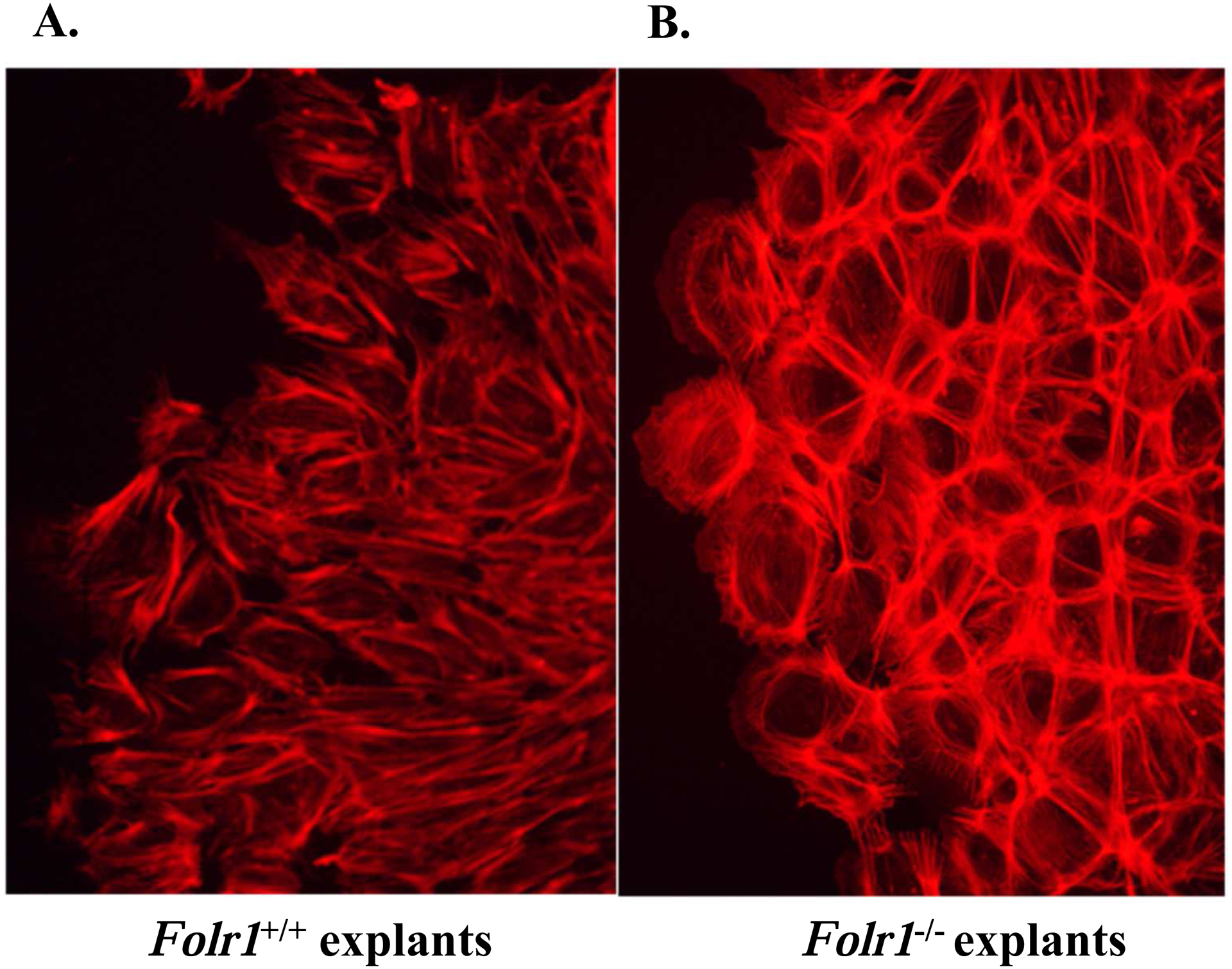
Cranial neural tubes from gd8.5 Folr1+/+ (Panel A) and Folr1−/− (Panel B) embryos were cultured for 48 hours. The explants were then fixed and processed for F-actin staining with AF594-Phalloidin and photographed under epifluorescence optics. Note that F-actin staining in the NCC of a representative Folr1−/− explant (Panel B) is primarily distributed in a pericellular pattern indicative of cells with impaired directed migratory ability, as opposed to those from a representative Folr1+/+ explant (Panel A), which is primarily distributed in a filamentous pattern of radiating stress fibers throughout the cytoplasm indicative of motile cells with directed migration. Magnification of both Figures 7A and 7B: 20X.
Effect of folate deficiency on embryonic cranial NCC migration: Transwell cell migration assay
Cell migration was one of the major developmental processes affected by folate deficiency. An in vitro transwell migration assay was used to quantify the effect of folate on the migratory activity of wild-type NCCs. Percent cell migration, representing the number of cells that have migrated through the transwell membrane as a function of total cell number, is reported as mean ± SEM. As seen in Figure 8, the migration of embryonic cranial NCCs across the transwell membrane in the absence of folate was significantly inhibited in comparison to the percentage of cells that migrated across the membrane in the presence of folate (n=3; * p≤ 0.05, Student’s t-test).
Figure 8: Effect of folate deficiency on embryonic cranial NCC migration: Transwell cell migration assay.
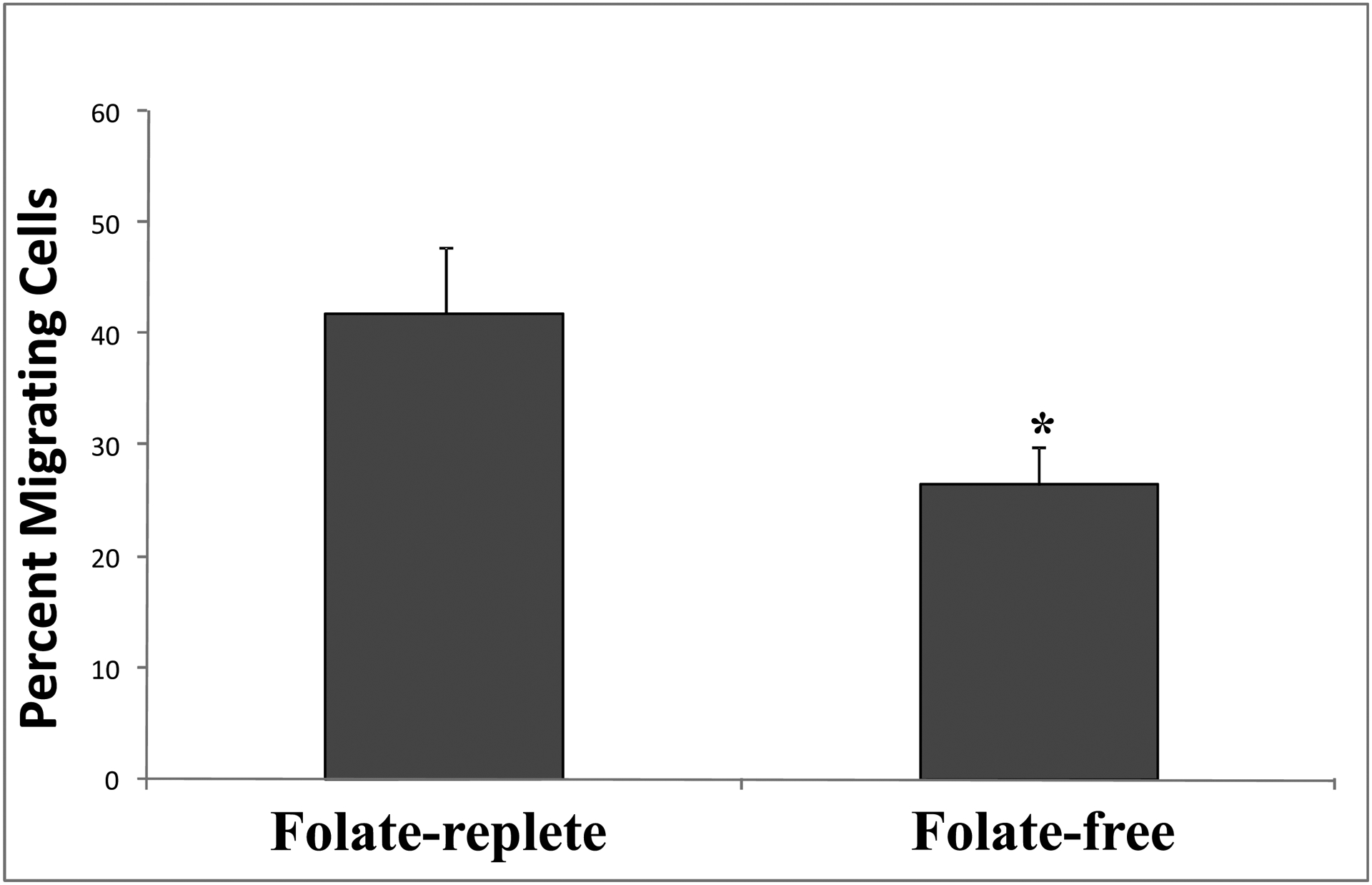
Primary cultures of wild-type embryonic cranial NCCs were grown in transwells in either folate-replete or folate-free conditions for 24 hours, at which time NCCs on either side of the transwell membrane were separately processed for determination of cell number, as described in MATERIALS AND METHODS. Three biologically independent NCC cultures were evaluated (with and without folate), and each assayed in triplicate. Percent migration represents the percentage of total cells that have migrated through the transwell membrane during 24 hours, reported as mean ± SEM. Migration of NCCs (expressed as percentage) across the transwell membrane in the absence of folate was significantly inhibited in comparison to cells that migrated across the membrane in the presence of folate (n=3; Student’s t-Test: * p≤ 0.05).
Effect of folate deficiency on embryonic cranial NCC death: Determination of apoptosis by TUNEL staining
Cranial neural tube explants from gd8.5 mouse embryos provide a rich source of NCCs which proliferate, migrate, differentiate and respond to physiologic signals in a manner similar to their response in vivo. To understand the effects of folate deficiency on NCC apoptosis, we examined NCCs using TUNEL in explanted tissue derived directly from Folr1+/+ and Folr1−/− embryos (Figure 9). As seen in Panel E, cells within explants, as well as those emigrating from the explant, are immunopositive for p75NGFR, a well characterized marker of undifferentiated cranial NCCs (Sauka-Spengler and Bronner-Fraser, 2008). Explants from Folr1−/− embryos processed for TUNEL indicated markedly greater NCC apoptosis in both the core of the explant and in emigrating cells (Panel D), when compared to explants from wild-type embryos (Panel C). Explants processed for TUNEL are shown at higher magnification in Panels E and F. These data demonstrate that p75NGFR -positive cells (i.e., NCCs) in neural tube explants from functionally folate deficient embryos exhibit dramatically more apoptosis than do NCCs in explants from wild-type embryos.
Figure 9: Determination of apoptosis by TUNEL assay in cranial NCC “Outgrowth” (explant) cultures from Folr1+/+ and Folr1−/− embryos.
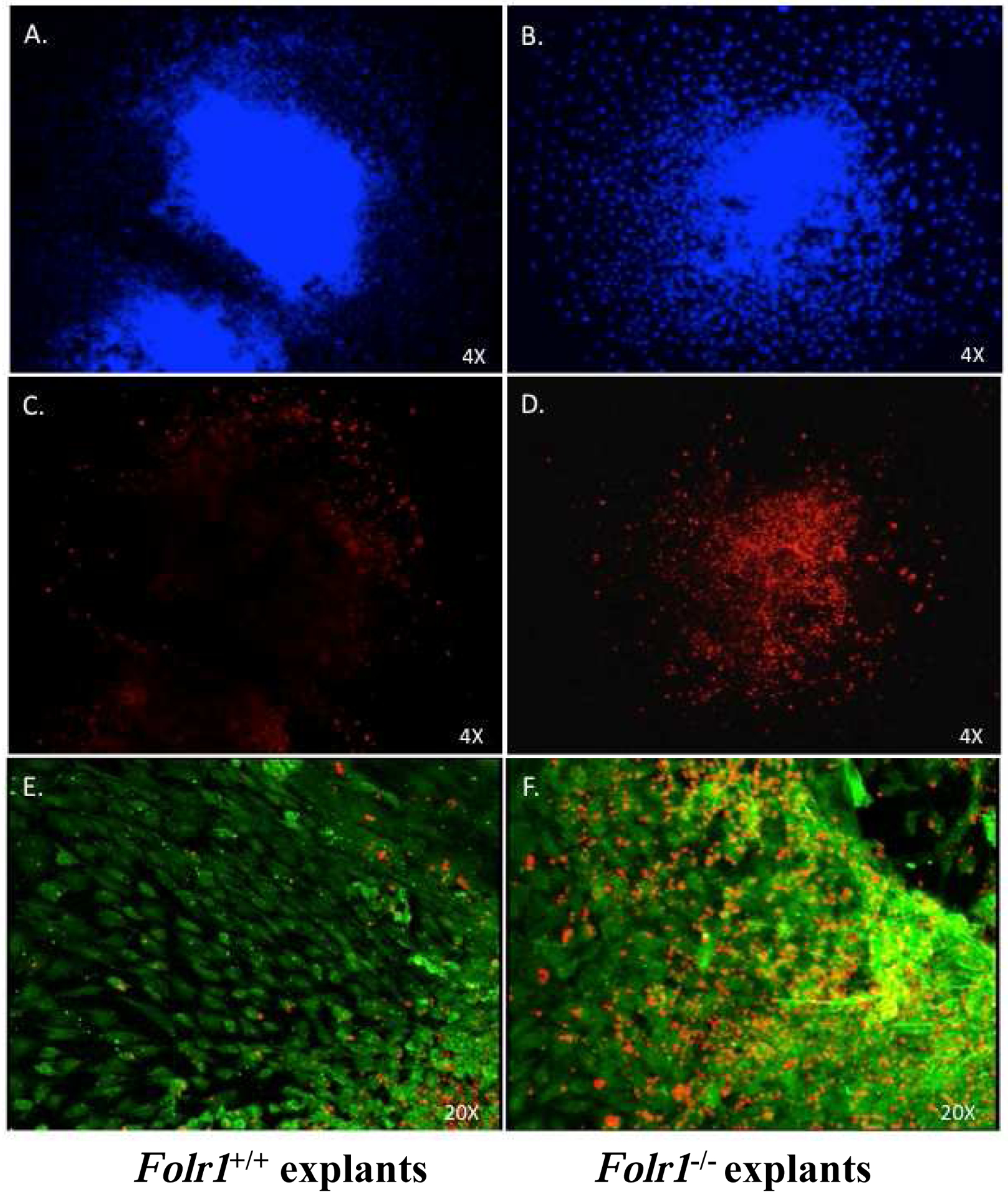
Cranial neural tubes from gd8.5 wild-type (Panels A, C, E) and folate deficient (Panels B, D, F) embryos were cultured for 48 hours at which time the explants were processed for TUNEL evaluation. Apoptosis (TMR-red label), immunolocalization of p75NGFR (Alexafluor-488, green label), and DAPI staining (blue label) are indicated in the figure. Explants were stained with DAPI (Panels A and B) to visualize cell nuclei, processed for TUNEL to visualize apoptotic cells with fragmented DNA (Panels C-F) and immunostained with p75NGFR to verify the identity of cranial NCCs in the explant (Panels E and F). Panels E and F demonstrate co-localization of TUNEL-positive cells with p75NGFR positive NCC. Note the markedly increased TUNEL staining of NCC in the representative “outgrowth” culture from the cranial neural tube of a Folr1−/− embryo (Panel F) compared to that from a wild-type embryo, indicative of increased apoptosis in the folate-deficient NCCs.
DISCUSSION
Inadequate dietary folate and/or genetic alterations in folate metabolism have been associated with various developmental defects (congenital NTDs, congenital cardiovascular anomalies and orofacial clefting), and adult diseases (adult neurodegenerative and cardiovascular diseases, and certain cancers) (Czeizel, 1993; Shaw et al., 1995; Czeizel and Hirschberg, 1997; Piedrahita et al., 1999; Loffredo et al., 2001; Kim, 2003; Tchantchou et al., 2006; Gelineau-van Waes et al., 2008b; Guéant et al., 2013; Wallingford et al., 2013; Figueiredo et al., 2015; López-Gordillo et al., 2019). Increased dietary folate consumption has been shown to attenuate or entirely prevent many of these adverse conditions although the cellular and molecular mechanisms by which folate does so are poorly defined. Studies reported here sought to address this gap in our knowledge by utilizing an animal model to investigate how folate deficiency affects normal developmental processes, including craniofacial ontogenesis.
Transcription profiling using high-density microarrays identified 837 differentially expressed genes - 599 upregulated; 238 downregulated (Supplementary Table 1). Although three microarray chips were used for Folr1+/+ and two for Folr1−/− (one chip failed quality control), PCA (Figure 1) and hierarchical clustering (Figure 2) analyses indicate that samples for each condition tended to cluster together and each cluster remained distinctly separate from the other. Together, these results demonstrate the biological reproducibility for key genes between the replicates and that the samples were well correlated with one another. To identify critical biological functions and pathways significantly affected in Folr1 null embryos, the 837 differentially expressed genes were subjected to Ingenuity Pathway Analysis. Among the top five affected biological functions in Folr1−/− mice were those related to ‘digestive system development/function’, ‘cardiovascular system development/function’, ‘tissue development’, ‘cellular development’, and ‘cell growth and differentiation’ (Figure 4), while the major canonical pathways affected were those associated with blood coagulation (constituting the top three), embryonic stem cell transcription and cardiomyocyte differentiation (via BMP receptors) (Figure 5). That disturbances in the blood coagulation system were among the most affected in Folr1−/− embryos is consistent with the finding that transcripts expressing the embryonic chains of hemoglobin (X, Y and Z) were the most downregulated (Supplementary Table 6), an observation similar to that seen in Slc19a1−/− embryos (Gelineau-van Waes et al., 2008b; see Table 3). Interestingly, studies on subjects with atherosclerosis indicate that folic acid supplementation could reduce the risk of thrombosis and cardiovascular disease (Li et al., 2016). Slc19a1−/− mice exhibit heart defects (Zhao et al., 2001; Gelineuvan Waes et al., 2008a) which are consistent with human studies reporting cardiovascular diseases in those exhibiting hyperhomocysteinemia arising due to inadequate folate supply (Eskes, 2002; Essouma and Noubiap, 2015). The 837 differentially expressed genes in Folr1−/− mice target several processes essential for proper embryonic development, including apoptosis, cell migration, cell proliferation, cell differentiation, and embryogenesis (Supplementary Figures 4–8, respectively) (Tang et al., 2005; Bliek et al., 2008; Han et al., 2009; Zhang et al., 2009). Based on the number of genes differentially expressed within the Folr1 null embryos forming gene interaction networks, apoptosis (90 genes; Supplementary Figure 4) and cell migration (87 genes; Supplementary Figure 5) were the most affected cellular processes, followed by embryogenesis (57 genes; Supplementary Figure 8), cell differentiation (46 genes; Supplementary Figure 7) and cell proliferation (29 genes; Supplementary Figure 6). These observations are consistent with the results of Tang et al. (2005) who observed massive apoptosis in Folr1−/− defective tissues. Increased Bax levels, a hallmark of apoptosis, were observed in these tissues, as well as in Folr1−/− embryos (~1.7- fold; Supplementary Table 4). Of particular relevance to our study is the finding that many of the genes affecting embryogenesis in Folr1−/− embryos were associated with the development of the neural tube, neural crest, branchial arch and craniofacial regions and neural tube closure (Supplementary Figure 8).
Table 3:
Comparison of expression profiles of genes associated with the cubilin-megalin endocytotic receptor complex and hematopoiesis in gd9.5 Slc19a1−/− and Folr1−/− embryos
| Cubilin-Megalin Endocytotic Receptor Complex | |||
|---|---|---|---|
| Gene Symbol | Gene Name | Fold Change | |
| Slc19a1−/− a | Folr1−/− | ||
| Apoa1 | Apolipoprotein A-Ib | 66.0 | 8.4 ± 0.3 |
| Cubn | Cubilinb | 35.5 | 5.5 ± 0.1 |
| Apom | Apolipoprotein Mb | 23.4 | 5.7 ± 1.7 |
| Ttr | Transthyretinb | 18.0 | 7.8 ± 1.7 |
| Rbp4 | Retinol binding protein 4, plasma | 14.4 | 5.2 ± 0.2 |
| Timd2 | T-cell immunoglobulin and mucin domain containing 2 | 12.0 | 4.5 ± 0.4 |
| Trf | Transferrin | 9.4 | 5.9 ± 0.2 |
| Apoe | Apolipoprotein E | 4.7 | 6.4 ± 0.3 |
| Dab2 | Disabled homolog 2b | 3.8 | 3.1 ± 0.0 |
| Myo6 | Myosin VIb | 3.6 | 2.3 ± 0.2 |
| Fabp1 | Fatty acid binding protein 1 | 3.1 | ns |
| Lrpap1 | Low density lipoprotein receptor-related protein associated protein1 | 2.2 | 3.1 ± 0.4b |
| Hematopoiesis | |||
| Hbb-bh1 | Hemoglobin Z, β-like embryonic chain (human hemoglobin, gamma A) | −25.4 | −16.5 ± 1.1b |
| Hba-x | Hemoglobin X, α-like embryonic chain in Hba complex (human hemoglobin, zeta) | −34.6 | −10.6 ± 1.0 |
| Hbb-y | Hemoglobin Y, β-like embryonic chain (human hemoglobin, gamma G) | −53.2 | −26.4 ± 8.4b |
Note:
column data from Gelineau-van Waes et al. (2008b);
Slc19a1 (formerly Rfc1);
fold changes were averaged where multiple probes targeted the same transcript (± SEM); ns, not significant (<1.5-fold change).
Early development is characterized by the rapid proliferation of embryonic cells. Results from gene expression profiling of Folr1−/− embryos indicate that folate deficiency affects numerous genes involved in cell proliferation. In particular, the expression of a number of transcription factors, required for cell cycle regulation, and impinging on cell proliferation, was found to be altered as a function of folate deficiency in Folr1−/− embryos. Included among these was the Fox (Forkhead) group of genes encoding the winged helix transcription factors: a2, c1, c2, d1, d4, h1 and m1. FOX proteins play key roles in embryonic development and facilitate cross-talk between a number of signaling pathways, including TGF-β/SMAD, MAPK, Sonic-Hedgehog, Wnt/β-catenin and insulin/IGF pathways (Golson and Kaestner, 2016). In addition to their role in development, these molecules also regulate cell differentiation, proliferation and apoptosis in adult life (Golson and Kaestner, 2016). Null mutants for several Fox genes, including Foxa2, −c1, −c2, −d1, −h1, and −m1, are developmentally deleterious (Lehmann et al., 2003; Golson and Kaestner, 2016). Aberrant regulation of FOX proteins, due to folate deficiency, can thus affect key signaling systems intrinsic to proper fetal development. In the current study, expression of genes encoding two SRY-box containing transcription factors (Sox 10 and Sox17) was also differentially altered in Folr1−/− embryos. It is intriguing that Sox10 expression was significantly reduced (~ 3.4-fold), whereas Sox17 expression was significantly increased (~ 3.0-fold) by Folr1 deletion (Supplementary Table 3). Expressed in the peripheral nervous system and in certain neural crest-derived cells (Cook et al., 2005), Sox10 plays a critical role in proliferation of NCCs as they contribute to the formation of certain adult structures (Sonnenberg-Riethmacher et al., 2001). It is plausible that Sox10 contributes to regulation of NCC proliferation as they populate the first branchial arch. Indeed, PACS1 mutations which cause anomalous craniofacial features have been attributed to the defective migration of Sox10-positive cranial NCCs (Schuurs-Hoeijmakers et al., 2012). Overexpression of Sox17 led to suppression of cyclin D1 expression and inhibition of cell proliferation in oligodendrocyte progenitor cells (Chew et al., 2011). In the present study, expression of the gene (Ccnd2) encoding cyclin D2 was significantly reduced (~2.0-fold; Supplementary Table 3) in Folr1−/− embryos. Reduction in Sox10 expression and increase in Sox17 expression in NCCs could adversely affect growth of the first branchial arch with attendant craniofacial defects seen in Folr1−/− embryos. Significant upregulation of genes encoding two inhibitors of cell proliferation (e.g. Cdkn1a: 4.0-fold and Cdkn1c: 3.3-fold; Supplementary Table 4) in Folr1−/− embryos, also support this conjecture.
Analysis of our data on the effect of folate deficiency on cranial NCC proliferation revealed a significant reduction in the number of proliferating NCCs. These findings correlate well with the aforementioned speculations and the developmental phenotypes noted in embryos from folate deficient animal models. In models of dietary folate deficiency, or genetic models where Folr1 or Rfc1 have been knocked out, the embryos are smaller in size and developmentally delayed, in addition to exhibiting dysmorphology of folate sensitive structures (Piedrahita et al., 1999; Gelineau-van Waes et al., 2008b).
Apoptosis plays an integral role during embryogenesis. Dysregulated apoptosis during development can result in abnormal formation of tissues and subsequent birth defects (Haouzi et al., 2018). Folate deficiency has been shown to induce apoptosis by increasing blood homocysteine levels (hyperhomocysteinemia) (Blom and Smulders, 2011), inducing DNA strand breaks (Fenech, 2010), increasing uracil misincorporation into DNA (Fenech, 2010) and/or impairing DNA repair (Blount et al., 1997; Duthie, 2001; Bach et al., 2017). Gene expression data from the present study reveal that transcript levels for Bax, a proapoptotic protein, were found to be increased (~1.7-fold) in Folr1−/− embryos (Supplementary Table 4). Furthermore, an increase in gelsolin (Gsn) transcript levels (~3.8 fold) was also observed. The gelsolin family of actin-binding proteins has been identified as novel transcriptional co-activators and genetic evidence has linked gelsolin-mediated reactive oxygen species (ROS) production to TNF-induced apoptosis in MCF-7 cells (Li et al., 2009). While it is unclear whether folate deficiency mediates NCC apoptosis via ROS generation, increased gelsolin levels, as a result of folate deficiency, could potentially increase apoptosis in developing embryonic tissue. In Folr1−/− embryos, enhanced expression of Msx1 (~2.1-fold; Supplementary Table 3), reported to promote apoptosis of NCCs, could also mediate increased apoptosis of these cells (Tríbulo et al., 2004). Since BMPs were documented as direct triggers of apoptosis during limb development (Kaltcheva et al., 2016), significant upregulation of Bmp4 (~ 3.0-fold), Bmp2 (~2.0-fold), and Bmp7 (1.7-fold) could also underlie folate-deficiency-induced apoptosis in Folr1−/− embryos (Supplementary Table 4). Enhanced apoptosis within Folr1−/− embryos is in concordance with the results of TUNEL staining of NCCs in explanted tissue derived directly from Folr1+/+ and Folr1−/− embryos (Figure 9). From Figure 9, it is apparent that p75NGFR-positive cells (i.e., NCCs) in neural tube explants from folate deficient embryos display significantly augmented apoptosis when compared to NCCs in explants from wild-type embryos.
The expression of several genes governing cellular migration was found to be altered in Folr1−/− embryos. Cadherins are a family of cell-cell adhesion molecules expressed in a broad range of embryonic tissues including the neural tube and NCCs, and decreased expression of the genes encoding the cadherins is essential for cell motility (Rogers et al., 2018). Results presented here indicate that Folr1−/− embryos have an increased expression of the E-cadherin gene (Cdh1; 3.4-fold; Supplementary Table 3) when compared to their wild-type counterparts, implying that migration of embryonic cells was likely compromised. This observation was confirmed using transwell migration assays and explant cultures (see below). In addition, it has been reported that epithelium-to-mesenchyme transition (EMT), a critical process in cranial NCC formation and initial migration in the embryo, is associated with simultaneous repression of the genes encoding E-cadherin and claudins (Ikenouchi et al., 2003). Indeed, we observe that several members of the claudin family (claudins-2, −6 and −7) have increased gene transcript levels in folate deficient embryos (Supplementary Tables 4 and 6). These findings suggest that folate deficiency may impair EMT associated with the formation of the cranial neural crest, emigration from the neural tube, and initial migration of these cells into the developing oro/facial region. Expression of Hoxa2, a gene involved in cell migration during craniofacial development, was also reduced in Folr1−/− embryos (~ 2.5-fold; Supplementary Table 3). In cranial NCCs, Hoxa2 is expressed throughout the period of NCC migration and its expression is maintained until the NCCs populate branchial arch 2 (Trainor and Krumlauf, 2001). Expression of Pdgfra and Sox10, two key regulators of NCC migration during craniofacial development, was also diminished in Folr1−/− embryos (McCarthy et al., 2016; Alkobtawi et al., 2018). Collectively, our data suggest that folate deficiency alters the expression of an array of critical genes directing cell adhesion, cell migration and EMT in the craniofacial region, including the ones that are important for NCC migration. These data support the hypothesis that craniofacial defects associated with gestational folate deficiency could potentially be the result of impaired formation and migration of cranial NCCs from the neural folds and/or impaired NCC migration into the developing orofacial region.
The most important and characteristic feature of NCCs is their ability to migrate extensively throughout the embryo (Szabó and Mayor, 2018). Developmental folate deficiency perturbs neural fold elevation and orofacial morphogenesis, processes dependent on the migration of cranial NCCs (Maldonado et al., 2018). Therefore, in the current study, the effect of folate availability and/or uptake on cranial NCCs was investigated using transwell analysis and explant cultures to assess their migratory properties. Transwell analysis using wild-type NCCs indicated a significant decrease in the number of migrated cells when grown in a folate-free medium, relative to those grown in folate-replete medium (Figure 8). This may be due, in part, to aberrant cellular architecture of NCCs emigrating from the cranial neural tube in folate deficient embryos. Supportive of this, we demonstrated a pronounced difference in the overall morphology of NCCs emigrating from the Folr1−/− neural tube explants compared to those emigrating from wild-type explants when explant cultures were stained for filamentous F-actin (Figure 7). The polymerization of G-actin to F-actin is necessary for the polarized assembly of the cytoskeleton requisite for cell migration. In Folr1+/+embryos, F-actin was found to be distributed in a filamentous pattern of radiating fibers throughout the cytoplasm, indicating that the NCCs were typically motile exhibiting directed migration; in contrast, F-actin in NCCs of Folr1−/− embryos exhibited a pericellular pattern associated with impaired directional migration. Impaired migration was also evident when NCCs from wild-type explant cultures were grown in folate-free media, compared to growth in folate-replete media. Collectively, results from the transwell migration analyses, digital morphometric analyses, and F-actin staining of cultures of embryonic cranial NCCs suggest that folate deficiency markedly alters the cytoarchitecture of these cells by perturbing filamentous actin formation and/or distribution, and significantly inhibits the directed migration of these cells. This suggestion is supported by examination of the putative ‘tight junction’ signaling pathway generated by IPA analysis of the differentially expressed genes observed in our study (Supplementary Figure 9A). The figure indicates that increased expression of several members of the claudin family in Folr1−/− embryos can affect cytoskeletal organization via actin nucleation and branching, while, increased expression of the F11 receptor (F11R/Jam) can inhibit actin-based cell motility, thereby impeding cell migration. Although folate has been shown to alter the expression of genes associated with adhesion and migration in a variety of cancer cell lines, this is the first demonstration that folate depletion has a disorganizing effect on the cytoskeleton of embryonic cells and perturbs their migration.
Expression of genes encoding several members of the apolipoprotein (Apo) family, namely Apoa1, -a4, -a5, -b, -e and -m, were consistently and, in some cases, markedly increased in Folr1−/− embryos compared to expression in wild-type embryos. Indeed, five of the top ten upregulated genes belong to this family (Supplementary Table 1). Interestingly, ApoA4 has been shown to attenuate oxidant-induced apoptosis in mitotically competent, undifferentiated cells by modulating intracellular glutathione redox balance (Spaulding et al., 2006). Functioning as antioxidants, ApoA1 and ApoA4 have been reported to counter oxidative stress (Carnicer et al., 2007). The marked upregulation of Apoa1 and Apoa4 in Folr1−/− embryos may represent a physiological response of the embryo to perceived cellular stress, and an attempt to counter the increased cell death and/or oxidative stress induced by folate deficiency in the developing embryo. This notion is supported by other studies demonstrating that ApoA4 can physically bind to LDL (low density lipoproteins) and act as a site-specific antioxidant (Gallagher et al., 2004; Carnicer et al., 2007).
ApoE plays a major role in lipoprotein metabolism and cholesterol homeostasis in the brain (Mahley, 2016). It has also been implicated in the decline in cognitive functions and late-onset Alzheimer’s disease and has been demonstrated to directly bind to the β-amyloid-precursor protein (APP), which is central to the pathogenesis of Alzheimer’s disease (Chang et al., 2017). Of relevance to our studies is the finding that both APP and ApoE receptors have functions in neuronal migration during development (Pohlkamp et al., 2017). In addition, other findings have suggested that ApoE has cytostatic functions in the blood vessel wall and may regulate smooth muscle cell migration and proliferation (Ishigami et al., 1998). The substantial increase in the expression of apolipoprotein genes in Folr1−/− embryos (Table 3; Supplementary Table 1) was particularly intriguing, especially in view of the complexity of apolipoproteins found in the embryonic nervous system, and the ability of apolipoproteins to modulate survival and migration of varied cell types. Maintenance of proper levels of proteins encoded by genes associated with cholesterol metabolism is essential for neurogenesis, as any alteration or defect in cholesterol metabolism results in abnormal CNS development (Saher et al., 2011; Cunningham et al., 2015). Changes in Apoe and Apob expression affect cholesterol levels and deletion of Apob can lead to an abnormal neuroepithelium, incomplete neural tube closure, exencephaly and hydrocephaly (Huang et al., 1995; Harris and Juriloff, 1999). Indeed, key enzymes of cholesterol biosynthesis such as squalene epoxidase, 3-hydroxy-3-methylglutaryl-CoA synthase 1 and mevalonate (diphospho) decarboxylase were all found to be downregulated in Folr1−/− embryos in our study, affecting not only the biosynthesis of cholesterol, but also 1,25-dihydroxy vitamin D3 (Supplementary Figure 9B). Interestingly, 1,25-dihydroxy vitamin D3 and the vitamin D receptor are known to increase intestinal PCFT (proton-coupled folate transporter) expression which enhances cellular folate uptake (Eloranta et al., 2009). Whether the loss of Folr1 also compromises PCFT function is not known. It is well established that folate deficiency results in decreased SAM levels (Fenech, 2010; Guéant et al., 2013). Key nutritional deficiencies, including folate and SAM deprivation, can be further exacerbated by decreased ApoE functionality (Serra et al., 2008). Therefore, in the current study, the striking increase in the expression of the Apo family of genes in Folr1−/− embryos, could potentially be an attempt by the developing embryo to compensate for such defects.
The present study also permitted comparison of gene expression changes reported in Slc19a1 (encoding the reduced folate carrier) knockout embryos with those in Folr1−/− embryos. Slc19a1 null embryos do not survive beyond gd9.5 due to congenital malformations of the heart and lung and the absence of erythropoiesis (Zhao et al., 2001; Gelineau-van Waes et al., 2008a). However, these embryos can be rescued by supplementing the pregnant dams with a low dose of folate (25mg/kg/day/S.Q.). The primary gene targets affected in Slc19a1−/− embryos are those encoding ligands and receptors associated with the cubilin-megalin multiligand endocytic receptor complex and those regulating hematopoiesis (Gelineau-van Waes et al., 2008b). The cubilin-megalin multiligand endocytic receptor complex is involved in the maternal-fetal transport of folate, key nutrients, and morphogens (Moestrup and Verroust, 2001; Christensen and Birn, 2002; Moestrup, 2006). Of particular interest was the observation from the current study that the expression of genes encoding ligands and interacting proteins of this receptor complex (Apoa1, Cubn, Apom, Ttr, Rbp4, Timd2, Trf, Apoe, Dab2, Myo6, Fabp1, and Lrpap1, among others) were significantly increased in Folr1−/− embryos (Table 3), emphasizing a key developmental role for this complex in folate utilization and transport. As in Slc19a1−/− embryos, Apoa1 exhibited the highest expression among the interacting members of this receptor complex in Folr1−/− embryos (Table 3) and, as observed by Gelineau-van Waes et al (2008b), the upregulation of genes encoding proteins associated with this complex is likely a compensatory mechanism by the Folr1−/− embryos to offset the defective internalization of the receptor complex. Remarkably, as in Slc19a1−/− embryos, the predominant gene expression changes in Folr1−/− embryos appear to target cardiovascular development and erythropoiesis (as it relates to hemoglobin synthesis; discussed above).
CONCLUSIONS
Collectively, data from the present study document potential adverse impact(s) of the absence of Folr1, the high affinity folate receptor, on a range of growth factor-mediated signaling cascades and critical biological pathways governing key cellular processes central to normal cranial neural crest and orofacial morphogenesis. Such negative impacts potentially underlie the abnormal cellular proliferation, migration, and apoptosis as well as altered cellular growth and differentiation, and energy metabolism, as well as DNA damage that we report. Such alterations likely contribute to the etiology of developmental, particularly craniofacial, anomalies observed within Folr1-deficient embryos.
Supplementary Material
Supplementary Figure 1: Phenotypes of Folr1−/− mouse embryos born to Folr1+/− dams supplemented with folic acid. Representative examples of various phenotypes of Folr1−/− mouse embryos derived from Folr1+/− dams supplemented with 20 ppm folic acid and observed on gd13.5. A: wild-type mouse embryo; B-H: embryos with varied types of neural tube defects and orofacial malformations. These defects include: anencephaly (An) (lack of brain tissue) / exencephaly (Ex) (protruding brain tissue); cleft lip (Cl) / cleft palate (Cp); retrognathia / agnathia (RA) (recessed / missing jaw).
Supplementary Figure 2: Biological functions affected by folate deficiency in Folr1−/− embryos. This Figure provides an extensive list of the biological functions affected by folate deficiency. See legend to Figure 4 for details.
Supplementary Figure 3: Canonical pathways affected by folate deficiency in Folr1−/− embryos. This Figure provides an extensive list of the canonical pathways affected by folate deficiency. See legend to Figure 5 for details.
Supplementary Figure 4: A gene network representing differentially expressed genes associated with apoptosis in Folr1−/− embryos. A network with selected genes encoding mRNAs with increased (red) and decreased (green) expression in Folr1−/− embryos (when compared to that in Folr1+/+ embryos) was constructed using Ingenuity Pathway Analysis software (Ingenuity Systems). Color intensity correlates with level of expression. Solid lines specify direct relationships, while dotted lines indicate indirect interactions. For ‘apoptosis’, 90 genes (out of the 837) formed a network. The cell types/tissues/specific processes potentially affected by changes in gene expression are shown in yellow ovals.
{kind=link}
Supplementary Figure 5: A gene network representing differentially expressed genes associated with cell migration in Folr1−/− embryos. For ‘cell migration’, 87/837 genes formed an interacting network. All other details are the same as in the legend to Supplementary Figure 4.
{kind=link}
Supplementary Figure 6: A gene network representing differentially expressed genes associated with cell proliferation in Folr1−/− embryos. For ‘cell proliferation’, 29/837 genes formed a network. Shh (shown in white) was not identified in our study but was incorporated by the program to generate a more integrated network. All other details are the same as in the legend to Supplementary Figure 4.
{kind=link}
Supplementary Figure 7: A gene network representing differentially expressed genes associated with cell differentiation in Folr1−/− embryos. For ‘cell differentiation’, 46/837 genes formed a network. All other details are the same as in the legend to Supplementary Figure 4.
{kind=link}
Supplementary Figure 8: A gene network representing differentially expressed genes associated with embryogenesis in Folr1−/− embryos. For ‘embryogenesis’, 57/837 genes formed a network. All other details are the same as in the legend to Supplementary Figure 4.
{kind=link}
Supplementary Figure 9A: Major signaling pathways affected in Folr1−/− embryos - Tight junction signaling. Some of the major signaling pathways potentially affected by the differentially expressed genes (increases, in red; decreases, in green) in Folr1−/− embryos, as analyzed by Ingenuity Pathway Analysis (Ingenuity Systems), are indicated in Figs. 9A–9F. Color intensity correlates with level of expression. Solid lines specify direct relationships, while dotted lines indicate indirect interactions.
{kind=link}
Supplementary Figure 9B: Major signaling pathways affected in Folr1−/− embryos - Cholesterol biosynthesis. For details, see legend to Fig. 9A.
{kind=link}
Supplementary Figure 9C: Major signaling pathways affected in Folr1−/− embryos - TGFβ-BMP signaling. For details, see legend to Fig. 9A.
{kind=link}
Supplementary Figure 9D: Major signaling pathways affected in Folr1−/− embryos - Mammalian embryonic stem cell pluripotency. For details, see legend to Fig. 9A.
{kind=link}
Supplementary Figure 9E: Major signaling pathways affected in Folr1−/− embryos - Wnt β-catenin signaling. For details, see legend to Fig. 9A.
{kind=link}
Supplementary Figure 9F: Major signaling pathways affected in Folr1−/− embryos - Mitochondrial dysfunction. For details, see legend to Fig. 9A.
{kind=link}
Highlights.
Gene expression profiling was determined in Folate receptor 1 KO (Folr1−/−) embryos.
837 genes were differentially expressed in Folr1−/−, relative to Folr1+/+, embryos.
Increased apoptosis and decreased proliferation were noted in Folr1−/− cranial NCCs.
Directed migration of cranial NCCs was markedly affected in Folr1−/− embryos.
F-actin mediated cytoskeletal changes may impair NCC migration in Folr1−/− embryos.
Acknowledgements
Folr1+/− mice were kindly provided by Dr. Richard H. Finnell (Baylor College of Medicine, Houston, TX, USA). This work was supported in part through NIH grants HD053509, DE018215, and P20-RR017702 from the COBRE program of the National Center for Research Resources.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Declaration of Interests
None
REFERENCES
- Alkobtawi M, Ray H, Barriga EH, Moreno M, Kerney R, Monsoro-Burq AH, Saint-Jeannet JP, Mayor R, 2018. Characterization of Pax3 and Sox10 transgenic Xenopus laevis embryos as tools to study neural crest development. Dev. Biol 444 Suppl 1, S202–S208. doi: 10.1016/j.ydbio.2018.02.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Amitai Y, Leventhal A, 2001. Folic acid antagonists during pregnancy and risk of birth defects. N Engl. J. Med 344, 933. [PubMed] [Google Scholar]
- Bach M, Savini C, Krufczik M, Cremer C, Rösl F, Hausmann M, 2017. Super-resolution localization microscopy of γ-H2AX and heterochromatin after folate deficiency. Int. J. Mol. Sci 18, 1726. doi: 10.3390/ijms18081726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barber RC, Bennett GD, Greer KA, Finnell RH, 1999. Expression patterns of folate binding proteins one and two in the developing mouse embryo. Mol. Genet. Metab 66, 31–39. doi: 10.1006/mgme.1998.2772. [DOI] [PubMed] [Google Scholar]
- Beaudin AE, Stover PJ, 2007. Folate-mediated one-carbon metabolism and neural tube defects: balancing genome synthesis and gene expression. Birth Defects Res. C Embryo Today 81, 183–203. doi: 10.1002/bdrc.20100. [DOI] [PubMed] [Google Scholar]
- Blanton SH, Henry RR, Yuan Q, Mulliken JB, Stal S, Finnell RH, Hecht JT, 2011. Folate pathway and nonsyndromic cleft lip and palate. Birth Defects Res. A Clin. Mol. Teratol 91, 50–60. doi: 10.1002/bdra.20740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bliek BJ, Steegers-Theunissen RP, Blok LJ, Santegoets LA, Lindemans J, Oostra BA, Steegers EA, de Klein A, 2008. Genome-wide pathway analysis of folate-responsive genes to unravel the pathogenesis of orofacial clefting in man. Birth Defects Res. A Clin. Mol. Teratol 82, 627–635. doi: 10.1002/bdra.20488. [DOI] [PubMed] [Google Scholar]
- Blom HJ, Smulders Y, 2011. Overview of homocysteine and folate metabolism. With special references to cardiovascular disease and neural tube defects. J. Inherit. Metab. Dis 34, 75–81. doi: 10.1007/s10545-010-9177-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blount BC, Mack MM, Wehr CM, MacGregor JT, Hiatt RA, Wang G, Wickramasinghe SN, Everson RB, Ames BN, 1997. Folate deficiency causes uracil misincorporation into human DNA and chromosome breakage: implications for cancer and neuronal damage. Proc. Natl. Acad. Sci. USA 94, 3290–3295. doi: 10.1073/pnas.94.7.3290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bufalino A, Ribeiro Paranaíba LM, Nascimento de Aquino S, Martelli-Júnior H, Oliveira Swerts MS, Coletta RD, 2010. Maternal polymorphisms in folic acid metabolic genes are associated with nonsyndromic cleft lip and/or palate in the Brazilian population. Birth Defects Res. A Clin. Mol. Teratol 88, 980–986. doi: 10.1002/bdra.20732. [DOI] [PubMed] [Google Scholar]
- Carnicer R, Navarro MA, Arbonés-Mainar JM, Acín S, Guzmán MA, Surra JC, Arnal C, de Las Heras M, Blanco-Vaca F, Osada J, 2007. Folic acid supplementation delays atherosclerotic lesion development in apoE-deficient mice. Life Sci. 80, 638–643. doi: 10.1016/j.lfs.2006.10.014. [DOI] [PubMed] [Google Scholar]
- Chang S, Wang L, Guan Y, Shangguan S, Du Q, Wang Y, Zhang T, Zhang Y, 2013. Long interspersed nucleotide element-1 hypomethylation in folate-deficient mouse embryonic stem cells. J. Cell Biochem 114, 1549–1558. doi: 10.1002/jcb.24496. [DOI] [PubMed] [Google Scholar]
- Chang TY, Yamauchi Y, Hasan MT, Chang C, 2017. Cellular cholesterol homeostasis and Alzheimer’s disease. J. Lipid Res 58, 2239–2254. doi: 10.1194/jlr.R075630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen MY, Rose CE, Qi YP, Williams JL, Yeung LF, Berry RJ, Hao L, Cannon MJ, Crider KS, 2019. Defining the plasma folate concentration associated with the red blood cell folate concentration threshold for optimal neural tube defects prevention: a population-based, randomized trial of folic acid supplementation. Am. J. Clin. Nutr 109, 1452–1461. doi: 10.1093/ajcn/nqz027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chew LJ, Shen W, Ming X, Senatorov VV Jr., Chen HL, Cheng Y, Hong E, Knoblach S, Gallo V, 2011. SRY-box containing gene 17 regulates the Wnt/β-catenin signaling pathway in oligodendrocyte progenitor cells. J. Neurosci 31, 13921–13935. doi: 10.1523/JNEUROSCI.3343-11.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Christensen EI, Birn H, 2002. Megalin and cubilin: multifunctional endocytic receptors. Nat. Rev. Mol. Cell Biol 3, 256–266. doi: 10.1038/nrm778. [DOI] [PubMed] [Google Scholar]
- Cook AL, Smith AG, Smit DJ, Leonard JH, Sturm RA, 2005. Co-expression of SOX9 and SOX10 during melanocytic differentiation in vitro. Exp. Cell Res 308, 222–235. doi: 10.1016/j.yexcr.2005.04.019. [DOI] [PubMed] [Google Scholar]
- Cunningham D, DeBarber AE, Bir N, Binkley L, Merkens LS, Steiner RD, Herman GE, 2015. Analysis of hedgehog signaling in cerebellar granule cell precursors in a conditional Nsdhl allele demonstrates an essential role for cholesterol in postnatal CNS development. Hum. Mol. Genet 24, 2808–25. doi: 10.1093/hmg/ddv042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Czeizel A, 1993. Periconceptional multivitamin supplementation and nonneural midline defects. Am. J. Med. Genet 46, 611. doi: 10.1002/ajmg.1320460536. [DOI] [PubMed] [Google Scholar]
- Czeizel AE, Hirschberg J, 1997. Orofacial clefts in Hungary. Epidemiological and genetic data, primary prevention. Folia Phoniatr. Logop 49, 111–116. doi: 10.1159/000266446. [DOI] [PubMed] [Google Scholar]
- Deguen S, Kihal W, Jeanjean M, Padilla C, Zmirou-Navier D, 2016. Neighborhood deprivation and risk of congenital heart defects, neural tube defects and orofacial clefts: A systematic review and meta-analysis. PLoS One. 11, e0159039. doi: 10.1371/journal.pone.0159039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doucette MM, Stevens VL, 2001. Folate receptor function is regulated in response to different cellular growth rates in cultured mammalian cells. J. Nutr 131, 2819–2825. doi: 10.1093/jn/131.11.2819. [DOI] [PubMed] [Google Scholar]
- Duthie SJ, 2001. Folic-acid-mediated inhibition of human colon-cancer cell growth. Nutrition. 17, 736–737. doi: 10.1016/s0899-9007(01)00595-0. [DOI] [PubMed] [Google Scholar]
- Eloranta JJ, Zaïr ZM, Hiller C, Häusler S, Stieger B, Kullak-Ublick GA, 2009. Vitamin D3 and its nuclear receptor increase the expression and activity of the human proton-coupled folate transporter. Mol. Pharmacol 76, 1062–1071. doi: 10.1124/mol.109.055392. [DOI] [PubMed] [Google Scholar]
- Eskes TKAB, 2002. Vascular disease in women. Folate and homocysteine In ‘Folate and human development’. Massaro EJ, Rogers JM (eds). Totowa, NJ; Humana Press Inc. [Google Scholar]
- Essouma M, Noubiap JJ, 2015. Therapeutic potential of folic acid supplementation for cardiovascular disease prevention through homocysteine lowering and blockade in rheumatoid arthritis patients. Biomark. Res 3, 24. doi: 10.1186/s40364-015-0049-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fenech M, 2010. Folate, DNA damage and the aging brain. Mech. Ageing Dev 131, 36–41. doi: 10.1016/j.mad.2010.02.004. [DOI] [PubMed] [Google Scholar]
- Figueiredo RF, Figueiredo N, Feguri A, Bieski I, Mello R, Espinosa M, Damazo AS, 2015. The role of the folic acid to the prevention of orofacial cleft: an epidemiological study. Oral Dis. 21, 240–247. doi: 10.1111/odi.12256. [DOI] [PubMed] [Google Scholar]
- Findley TO, Tenpenny JC, O’Byrne MR, Morrison AC, Hixson JE, Northrup H, Au KS, 2017. Mutations in folate transporter genes and risk for human myelomeningocele. Am. J. Med. Genet. A 173, 2973–2984. doi: 10.1002/ajmg.a.38472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Finnell RH, Waes JG, Eudy JD, Rosenquist TH, 2002. Molecular basis of environmentally induced birth defects. Annu. Rev. Pharmacol. Toxicol 42, 181–208. doi: 10.1146/annurev.pharmtox.42.083001.110955. [DOI] [PubMed] [Google Scholar]
- Gallagher JW, Weinberg RB, Shelness GS, 2004. apoA-IV tagged with the ER retention signal KDEL perturbs the intracellular trafficking and secretion of apoB. J. Lipid Res 45, 1826–1834. doi: 10.1194/jlr.M400188-JLR200. [DOI] [PubMed] [Google Scholar]
- Gelineau-van Waes J, Heller S, Bauer LK, Wilberding J, Maddox JR, Aleman F, Rosenquist TH, Finnell RH, 2008a. Embryonic development in the reduced folate carrier knockout mouse is modulated by maternal folate supplementation. Birth Defects Res. A Clin. Mol. Teratol 82, 494–507. doi: 10.1002/bdra.20453. [DOI] [PubMed] [Google Scholar]
- Gelineau-van Waes J, Maddox JR, Smith LM, van Waes M, Wilberding J, Eudy JD, Bauer LK Finnell RH, 2008b. Microarray analysis of E9.5 reduced folate carrier (RFC1; Slc19a1) knockout embryos reveals altered expression of genes in the cubilin-megalin multiligand endocytic receptor complex. BMC Genomics. 9, 156. doi: 10.1186/1471-2164-9-156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Golson ML, Kaestner KH, 2016. Fox transcription factors: from development to disease. Development. 143, 4558–4570. doi: 10.1242/dev.112672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grillo MA, Colombatto S, 2005. S-adenosylmethionine and protein methylation. Amino Acids. 28, 357–362. doi: 10.1007/s00726-005-0197-6. [DOI] [PubMed] [Google Scholar]
- Guéant JL, Namour F, Guéant-Rodriguez RM, Daval JL, 2013. Folate and fetal programming: a play in epigenomics? Trends Endocrinol. Metab 24, 279–289. doi: 10.1016/j.tem.2013.01.010. [DOI] [PubMed] [Google Scholar]
- Han SP, Pan Y, Peng YZ, Gu XQ, Chen RH, Guo XR, 2009. Folbp1 promotes embryonic myocardial cell proliferation and apoptosis through the WNT signal transduction pathway. Int. J. Mol. Med 23, 321–330. doi: 10.3892/ijmm_00000134. [DOI] [PubMed] [Google Scholar]
- Haouzi D, Boumela I, Chebli K, Hamamah S, 2018. Global, survival, and apoptotic transcriptome during mouse and human early embryonic development. Biomed. Res. Int 2018, 5895628. doi: 10.1155/2018/5895628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harris MJ, Juriloff DM, 1999. Mini-review: toward understanding mechanisms of genetic neural tube defects in mice. Teratology. 60, 292–305. doi: . [DOI] [PubMed] [Google Scholar]
- Henderson GI, Perez T, Schenker S, Mackins J, Antony AC, 1995. Maternal-to-fetal transfer of 5-methyltetrahydrofolate by the perfused human placental cotyledon: evidence for a concentrative role by placental folate receptors in fetal folate delivery. J. Lab. Clin. Med 126, 184–203. [PubMed] [Google Scholar]
- Hernandez-Díaz S, Werler MM, Walker AM, Mitchell AA, 2000. Folic acid antagonists during pregnancy and the risk of birth defects. N. Engl. J. Med 343, 1608–1614. doi: 10.1056/NEJM200011303432204. [DOI] [PubMed] [Google Scholar]
- Huang LS, Voyiaziakis E, Markenson DF, Sokol KA, Hayek T, Breslow JL, 1995. Apo B gene knockout in mice results in embryonic lethality in homozygotes and neural tube defects, male infertility, and reduced HDL cholesterol ester and apo A-I transport rates in heterozygotes. J. Clin. Invest 96, 2152–2161. doi: 10.1172/JCI118269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hyoun SC, Običan SG, Scialli AR, 2012. Teratogen update: methotrexate. Birth Defects Res. A Clin. Mol. Teratol 94, 187–207. doi: 10.1002/bdra.23003. [DOI] [PubMed] [Google Scholar]
- Ikenouchi J, Matsuda M, Furuse M, Tsukita S, 2003. Regulation of tight junctions during the epithelium-mesenchyme transition: direct repression of the gene expression of claudins/occludin by Snail. J. Cell Sci 116, 1959–1967. doi: 10.1242/jcs.00389. [DOI] [PubMed] [Google Scholar]
- Ishigami M, Swertfeger DK, Granholm NA, Hui DY, 1998. Apolipoprotein E inhibits platelet-derived growth factor-induced vascular smooth muscle cell migration and proliferation by suppressing signal transduction and preventing cell entry to G1 phase. J. Biol. Chem 273, 20156–20161. doi: 10.1074/jbc.273.32.20156. [DOI] [PubMed] [Google Scholar]
- Jahanbin A, Shadkam E, Miri HH, Shirazi AS, Abtahi M, 2018. Maternal folic acid supplementation and the risk of oral clefts in offspring. J. Craniofac. Surg 29, e534–e541. doi: 10.1097/SCS.0000000000004488. [DOI] [PubMed] [Google Scholar]
- Jordan RL, Wilson JG, Schumacher HJ, 1977. Embryotoxicity of the folate antagonist methotrexate in rats and rabbits. Teratology. 15, 73–79. doi: 10.1002/tera.1420150110. [DOI] [PubMed] [Google Scholar]
- Juriloff DM, Harris MJ, 2018. Insights into the etiology of mammalian neural tube closure defects from developmental, genetic and evolutionary studies. J. Dev. Biol 6, 22. doi: 10.3390/jdb6030022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaltcheva MM, Anderson MJ, Harfe BD, Lewandoski M, 2016. BMPs are direct triggers of interdigital programmed cell death. Dev. Biol 411, 266–276. doi: 10.1016/j.ydbio.2015.12.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kasture VV, Sundrani DP, Joshi SR, 2018. Maternal one carbon metabolism through increased oxidative stress and disturbed angiogenesis can influence placental apoptosis in preeclampsia. Life Sci. 206, 61–69. doi: 10.1016/j.lfs.2018.05.029. [DOI] [PubMed] [Google Scholar]
- Kim J, Lei Y, Guo J, Kim SE, Wlodarczyk BJ, Cabrera RM, Lin YL, Nilsson TK, Zhang T, Ren A, Wang L, Yuan Z, Zheng YF, Wang HY, Finnell RH, 2018. Formate rescues neural tube defects caused by mutations in Slc25a32. Proc. Natl. Acad. Sci. USA 115, 4690–4695. doi: 10.1073/pnas.1800138115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim YI, 2003. Role of folate in colon cancer development and progression. J. Nutr 133, 3731S–3739S. doi: 10.1093/jn/133.11.3731S. [DOI] [PubMed] [Google Scholar]
- Lehmann OJ, Sowden JC, Carlsson P, Jordan T, Bhattacharya SS, 2003. Fox’s in development and disease. Trends Genet. 19, 339–344. doi: 10.1016/S0168-9525(03)00111-2. [DOI] [PubMed] [Google Scholar]
- Li J, Sausville EA, Klein PJ, Morgenstern D, Leamon CP, Messmann RA, LoRusso P, 2009. Clinical pharmacokinetics and exposure-toxicity relationship of a folate-Vinca alkaloid conjugate EC145 in cancer patients. J. Clin. Pharmacol 49, 1467–1476. doi: 10.1177/0091270009339740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Y, Huang T, Zheng Y, Muka T, Troup J, Hu FB, 2016. Folic acid supplementation and the risk of cardiovascular diseases: a meta-analysis of randomized controlled trials. J. Am. Heart Assoc 5, e003768. doi: 10.1161/JAHA.116.003768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li B, Chang S, Liu C, Zhang M, Zhang L, Liang L, Li R, Wang X, Qin C, Zhang T, Niu B, Wang L, 2019. Low maternal dietary folate alters retrotranspose by methylation regulation in intrauterine growth retardation (IUGR) fetuses in a mouse model. Med. Sci. Monit 25, 3354–3365. doi: 10.12659/MSM.914292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loffredo LC, Souza JM, Freitas JA, Mossey PA, 2001. Oral clefts and vitamin supplementation. Cleft Palate Craniofac. J 38, 76–83. doi: 10.1597/1545-1569_2001_038_0076_ocavs_2.0.co_2. [DOI] [PubMed] [Google Scholar]
- López-Gordillo Y, Maldonado E, Nogales L, Del Río A, Barrio MC, Murillo J, Martínez-Sanz E, Paradas-Lara I, Alonso MI, Partearroyo T, Martínez-Álvarez C, 2019. Maternal folic acid supplementation reduces the severity of cleft palate in Tgf-β(3) null mutant mice. Pediatr. Res 85, 566–573. doi: 10.1038/s41390-018-0267-6. [DOI] [PubMed] [Google Scholar]
- Lucock M, 2000. Folic acid: nutritional biochemistry, molecular biology, and role in disease processes. Mol. Genet. Metab 71, 121–138. doi: 10.1006/mgme.2000.3027. [DOI] [PubMed] [Google Scholar]
- Mahley RW, 2016. Central nervous system lipoproteins: ApoE and regulation of cholesterol metabolism. Arterioscler. Thromb. Vasc. Biol 36, 1305–1315. doi: 10.1161/ATVBAHA.116.307023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maldonado E, Murillo J, Barrio C, del Río A, Pérez-Miguelsanz J, López-Gordillo Y, Partearroyo T, Paradas I, Maestro C, Martínez-Sanz E, Varela-Moreiras G, Martínez-Álvarez C, 2011. Occurrence of cleft-palate and alteration of Tgf-β(3) expression and the mechanisms leading to palatal fusion in mice following dietary folic-acid deficiency. Cells Tissues Organs. 194, 406–420. doi: 10.1159/000323213. [DOI] [PubMed] [Google Scholar]
- Maldonado E, López Y, Herrera M, Martínez-Sanz E, Martínez-Álvarez C, Pérez-Miguelsanz J, 2018. Craniofacial structure alterations of foetuses from folic acid deficient pregnant mice. Ann. Anat 218, 59–68. doi: 10.1016/j.aanat.2018.02.010. [DOI] [PubMed] [Google Scholar]
- Marini NJ, Yang W, Asrani K, Witte JS, Rine J, Lammer EJ, Shaw GM, 2016. Sequence variation in folate pathway genes and risks of human cleft lip with or without cleft palate. Am. J. Med. Genet. A 170, 2777–2787. doi: 10.1002/ajmg.a.37874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martino A, Towner MNW, Towner JE, 2016. Fetal myelomeningocele after maternal methotrexate administration: a case report. J. Reprod. Med 61, 609–611. [PubMed] [Google Scholar]
- Matok I, Gorodischer R, Koren G, Landau D, Wiznitzer A, Levy A, 2009. Exposure to folic acid antagonists during the first trimester of pregnancy and the risk of major malformations. Br. J. Clin. Pharmacol 68, 956–962. doi: 10.1111/j.1365-2125.2009.03544.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mattson MP, Shea TB, 2003. Folate and homocysteine metabolism in neural plasticity and neurodegenerative disorders. Trends Neurosci. 26, 137–146. doi: 10.1016/S0166-2236(03)00032-8. [DOI] [PubMed] [Google Scholar]
- McCarthy N, Liu JS, Richarte AM, Eskiocak B, Lovely CB, Tallquist MD, Eberhart JK, 2016. Pdgfra and Pdgfrb genetically interact during craniofacial development. Dev. Dyn 245, 641–652. doi: 10.1002/dvdy.24403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moestrup SK, 2006. New insights into carrier binding and epithelial uptake of the erythropoietic nutrients cobalamin and folate. Curr. Opin. Hematol 13, 119–123. doi: 10.1097/01.moh.0000219654.65538.5b. [DOI] [PubMed] [Google Scholar]
- Moestrup SK, Verroust PJ, 2001. Megalin- and cubilin-mediated endocytosis of protein-bound vitamins, lipids, and hormones in polarized epithelia. Annu. Rev. Nutr 21, 407–428. 10.1146/annurev.nutr.21.1.407. [DOI] [PubMed] [Google Scholar]
- Mukhopadhyay P, Greene RM, Zacharias W, Weinrich MC, Singh S, Young WW Jr., Pisano MM (2004). Developmental gene expression profiling of mammalian, fetal orofacial tissue. Birth Defects Res. A Clin. Mol. Teratol 70, 912–926. doi: 10.1002/bdra.20095. [DOI] [PubMed] [Google Scholar]
- Natsume N, Nagatsu Y, Kawai T, 1998. Direct effect of vitamins at the time of palatal fusion. Plast. Reconstr. Surg 102, 2512–2513. doi: 10.1097/00006534-199812000-00050. [DOI] [PubMed] [Google Scholar]
- Piedrahita JA, Oetama B, Bennett GD, van Waes J, Kamen BA, Richardson J, Lacey SW, Anderson RG, Finnell RH, 1999. Mice lacking the folic acid-binding protein Folbp1 are defective in early embryonic development. Nat. Genet 23, 228–232. doi: 10.1038/13861. [DOI] [PubMed] [Google Scholar]
- Pohlkamp T, Wasser CR, Herz J, 2017. Functional roles of the interaction of APP and lipoprotein receptors. Front. Mol. Neurosci 10, 54. doi: 10.3389/fnmol.2017.00054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rogers CD, Sorrells LK, Bronner ME, 2018. A catenin-dependent balance between N-cadherin and E-cadherin controls neuroectodermal cell fate choices. Mech. Dev 152, 44–56. doi: 10.1016/j.mod.2018.07.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saher G, Quintes S, Nave KA, 2011. Cholesterol: a novel regulatory role in myelin formation. Neuroscientist. 17, 79–93. doi: 10.1177/1073858410373835. [DOI] [PubMed] [Google Scholar]
- Saitsu H, Ishibashi M, Nakano H, Shiota K, 2003. Spatial and temporal expression of folate-binding protein 1 (Fbp1) is closely associated with anterior neural tube closure in mice. Dev. Dyn 226, 112–117. doi: 10.1002/dvdy.10203. [DOI] [PubMed] [Google Scholar]
- Sauka-Spengler T, Bronner-Fraser M, 2008. A gene regulatory network orchestrates neural crest formation. Nat. Rev. Mol. Cell Biol 9, 557–68. doi: 10.1038/nrm2428. [DOI] [PubMed] [Google Scholar]
- Salbaum JM, Kappen C, 2012. Genetic and epigenetic footprints of folate. Prog. Mol. Biol. Transl. Sci 108, 129–158. doi: 10.1016/B978-0-12-398397-8.00006-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schuurs-Hoeijmakers JH, Oh EC, Vissers LE, Swinkels ME, Gilissen C, Willemsen MA, Holvoet M, Steehouwer M, Veltman JA, de Vries BB, van Bokhoven H, de Brouwer AP, Katsanis N, Devriendt K, Brunner HG, 2012. Recurrent de novo mutations in PACS1 cause defective cranial-neural-crest migration and define a recognizable intellectual-disability syndrome. Am. J. Hum. Genet 91, 1122–1127. doi: 10.1016/j.ajhg.2012.10.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Serra M, Chan A, Dubey M, Gilman V, Shea TB, 2008. Folate and S-adenosylmethionine modulate synaptic activity in cultured cortical neurons: acute differential impact on normal and apolipoprotein-deficient mice. Phys. Biol 5, 044002. doi: 10.1088/1478-3975/5/4/044002. [DOI] [PubMed] [Google Scholar]
- Shaw GM, Rozen R, Finnell RH, Wasserman CR, Lammer EJ, 1998. Maternal vitamin use, genetic variation of infant methylenetetrahydrofolate reductase, and risk for spina bifida. Am. J. Epidemiol 148, 30–37. doi: 10.1093/oxfordjournals.aje.a009555. [DOI] [PubMed] [Google Scholar]
- Shaw GM, Schaffer D, Velie EM, Morland K, Harris JA, 1995. Periconceptional vitamin use, dietary folate, and the occurrence of neural tube defects. Epidemiology. 6, 219–226. doi: 10.1097/00001648-199505000-00005. [DOI] [PubMed] [Google Scholar]
- Sonnenberg-Riethmacher E, Miehe M, Stolt CC, Goerich DE, Wegner M, Riethmacher D, 2001. Development and degeneration of dorsal root ganglia in the absence of the HMG-domain transcription factor Sox10. Mech. Dev 109, 253–265. doi: 10.1016/s0925-4773(01)00547-0. [DOI] [PubMed] [Google Scholar]
- Sözen MA, Tolarova MM, Spritz RA, 2009. The common MTHFR C677T and A1298C variants are not associated with the risk of non-syndromic cleft lip/palate in northern Venezuela. J. Genet. Genomics 36, 283–288. doi: 10.1016/S1673-8527(08)60116-2. [DOI] [PubMed] [Google Scholar]
- Spaulding HL, Saijo F, Turnage RH, Alexander JS, Aw TY, Kalogeris TJ, 2006. Apolipoprotein A-IV attenuates oxidant-induced apoptosis in mitotic competent, undifferentiated cells by modulating intracellular glutathione redox balance. Am. J. Physiol. Cell Physiol 290, C95–C103. doi: 10.1152/ajpcell.00388.2005. [DOI] [PubMed] [Google Scholar]
- Spiegelstein O, Cabrera RM, Bozinov D, Wlodarczyk B, Finnell RH, 2004. Folate-regulated changes in gene expression in the anterior neural tube of folate binding protein-1 (Folbp1)-deficient murine embryos. Neurochem. Res 29, 1105–1112. doi: 10.1023/b:nere.0000023597.37698.13. [DOI] [PubMed] [Google Scholar]
- Spiegelstein O, Eudy JD, Finnell RH, 2000. Identification of two putative novel folate receptor genes in humans and mouse. Gene. 258, 117–125. doi: 10.1016/s0378-1119(00)00418-2. [DOI] [PubMed] [Google Scholar]
- Szabó A, Mayor R, 2018. Mechanisms of neural crest migration. Annu. Rev. Genet 52, 43–63. doi: 10.1146/annurev-genet-120417-031559. [DOI] [PubMed] [Google Scholar]
- Tang LS, Finnell RH, 2003. Neural and orofacial defects in Folp1 knockout mice. Birth Defects Res. A Clin. Mol. Teratol 67, 209–218. doi: 10.1002/bdra.10045. [Erratum: 67: 473]. [DOI] [PubMed] [Google Scholar]
- Tang LS, Santillano DR, Wlodarczyk BJ, Miranda RC, Finnell RH, 2005. Role of Folbp1 in the regional regulation of apoptosis and cell proliferation in the developing neural tube and craniofacies. Am. J. Med. Genet. C Semin. Med. Genet 135C, 48–58. doi: 10.1002/ajmg.c.30053. [DOI] [PubMed] [Google Scholar]
- Tchantchou F, Graves M, Shea TB, 2006. Expression and activity of methionine cycle genes are altered following folate and vitamin E deficiency under oxidative challenge: modulation by apolipoprotein E-deficiency. Nutr. Neurosci 9, 17–24. doi: 10.1080/10284150500502561. [DOI] [PubMed] [Google Scholar]
- Trainor PA, Krumlauf R, 2001. Hox genes, neural crest cells and branchial arch patterning. Curr. Opin. Cell Biol 13, 698–705. doi: 10.1016/s0955-0674(00)00273-8. [DOI] [PubMed] [Google Scholar]
- Tríbulo C, Aybar MJ, Sánchez SS, Mayor R, 2004. A balance between the anti-apoptotic activity of Slug and the apoptotic activity of msx1 is required for the proper development of the neural crest. Dev. Biol 275, 325–342. doi: 10.1016/j.ydbio.2004.07.041. [DOI] [PubMed] [Google Scholar]
- VanderMeer JE, Carter TC, Pangilinan F, Mitchell A, Kurnat-Thoma E, Kirke PN, Troendle JF, Molloy AM, Munger RG, Feldkamp ML, Mansilla MA, Mills JL, Murray JC, Brody LC, 2016. Evaluation of proton-coupled folate transporter (SLC46A1) polymorphisms as risk factors for neural tube defects and oral clefts. Am. J. Med. Genet. A 170A, 1007–1016. doi: 10.1002/ajmg.a.37539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wahl SE, Kennedy AE, Wyatt BH, Moore AD, Pridgen DE, Cherry AM, Mavila CB, Dickinson AJ, 2015. The role of folate metabolism in orofacial development and clefting. Dev. Biol 405, 108–122. doi: 10.1016/j.ydbio.2015.07.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wallingford JB, Niswander LA, Shaw GM, Finnell RH, 2013. The continuing challenge of understanding, preventing, and treating neural tube defects. Science. 339, 1222002 DOI: 10.1126/science.1222002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wehby GL, Félix TM, Goco N, Richieri-Costa A, Chakraborty H, Souza J, Pereira R, Padovani C, Moretti-Ferreira D, Murray JC, 2013. High dosage folic acid supplementation, oral cleft recurrence and fetal growth. Int. J. Environ. Res. Public Health 10, 590–605. doi: 10.3390/ijerph10020590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang XM, Huang GW, Tian ZH, Ren DL, Wilson JX, 2009. Folate deficiency induces neural stem cell apoptosis by increasing homocysteine in vitro. J. Clin. Biochem. Nutr 45, 14–19. doi: 10.3164/jcbn.08-223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao R, Russell RG, Wang Y, Liu L, Gao F, Kneitz B, Edelmann W, Goldman ID, 2001. Rescue of embryonic lethality in reduced folate carrier-deficient mice by maternal folic acid supplementation reveals early neonatal failure of hematopoietic organs. J. Biol. Chem 276, 10224–10228. doi: 10.1074/jbc.c000905200. [DOI] [PubMed] [Google Scholar]
- Zhu H, Wlodarczyk BJ, Scott M, Yu W, Merriweather M, Gelineau-van Waes J, Schwartz RJ, Finnell RH, 2007. Cardiovascular abnormalities in Folr1 knockout mice and folate rescue. Birth Defects Res. A Clin. Mol. Teratol 79, 257–268. doi: 10.1002/bdra.20347. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Figure 1: Phenotypes of Folr1−/− mouse embryos born to Folr1+/− dams supplemented with folic acid. Representative examples of various phenotypes of Folr1−/− mouse embryos derived from Folr1+/− dams supplemented with 20 ppm folic acid and observed on gd13.5. A: wild-type mouse embryo; B-H: embryos with varied types of neural tube defects and orofacial malformations. These defects include: anencephaly (An) (lack of brain tissue) / exencephaly (Ex) (protruding brain tissue); cleft lip (Cl) / cleft palate (Cp); retrognathia / agnathia (RA) (recessed / missing jaw).
Supplementary Figure 2: Biological functions affected by folate deficiency in Folr1−/− embryos. This Figure provides an extensive list of the biological functions affected by folate deficiency. See legend to Figure 4 for details.
Supplementary Figure 3: Canonical pathways affected by folate deficiency in Folr1−/− embryos. This Figure provides an extensive list of the canonical pathways affected by folate deficiency. See legend to Figure 5 for details.
Supplementary Figure 4: A gene network representing differentially expressed genes associated with apoptosis in Folr1−/− embryos. A network with selected genes encoding mRNAs with increased (red) and decreased (green) expression in Folr1−/− embryos (when compared to that in Folr1+/+ embryos) was constructed using Ingenuity Pathway Analysis software (Ingenuity Systems). Color intensity correlates with level of expression. Solid lines specify direct relationships, while dotted lines indicate indirect interactions. For ‘apoptosis’, 90 genes (out of the 837) formed a network. The cell types/tissues/specific processes potentially affected by changes in gene expression are shown in yellow ovals.
Supplementary Figure 5: A gene network representing differentially expressed genes associated with cell migration in Folr1−/− embryos. For ‘cell migration’, 87/837 genes formed an interacting network. All other details are the same as in the legend to Supplementary Figure 4.
Supplementary Figure 6: A gene network representing differentially expressed genes associated with cell proliferation in Folr1−/− embryos. For ‘cell proliferation’, 29/837 genes formed a network. Shh (shown in white) was not identified in our study but was incorporated by the program to generate a more integrated network. All other details are the same as in the legend to Supplementary Figure 4.
Supplementary Figure 7: A gene network representing differentially expressed genes associated with cell differentiation in Folr1−/− embryos. For ‘cell differentiation’, 46/837 genes formed a network. All other details are the same as in the legend to Supplementary Figure 4.
Supplementary Figure 8: A gene network representing differentially expressed genes associated with embryogenesis in Folr1−/− embryos. For ‘embryogenesis’, 57/837 genes formed a network. All other details are the same as in the legend to Supplementary Figure 4.
Supplementary Figure 9A: Major signaling pathways affected in Folr1−/− embryos - Tight junction signaling. Some of the major signaling pathways potentially affected by the differentially expressed genes (increases, in red; decreases, in green) in Folr1−/− embryos, as analyzed by Ingenuity Pathway Analysis (Ingenuity Systems), are indicated in Figs. 9A–9F. Color intensity correlates with level of expression. Solid lines specify direct relationships, while dotted lines indicate indirect interactions.
Supplementary Figure 9B: Major signaling pathways affected in Folr1−/− embryos - Cholesterol biosynthesis. For details, see legend to Fig. 9A.
Supplementary Figure 9C: Major signaling pathways affected in Folr1−/− embryos - TGFβ-BMP signaling. For details, see legend to Fig. 9A.
Supplementary Figure 9D: Major signaling pathways affected in Folr1−/− embryos - Mammalian embryonic stem cell pluripotency. For details, see legend to Fig. 9A.
Supplementary Figure 9E: Major signaling pathways affected in Folr1−/− embryos - Wnt β-catenin signaling. For details, see legend to Fig. 9A.
Supplementary Figure 9F: Major signaling pathways affected in Folr1−/− embryos - Mitochondrial dysfunction. For details, see legend to Fig. 9A.