

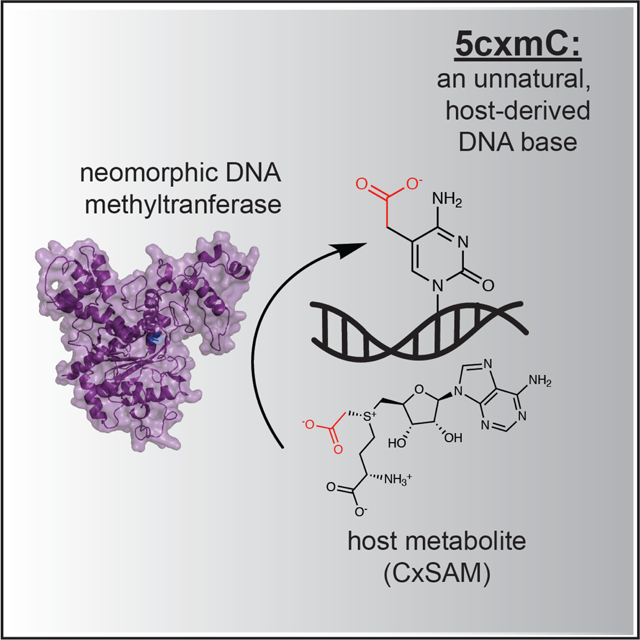
Summary
Despite widespread interest for understanding how modified bases have evolved their contemporary functions, limited experimental evidence exists for measuring how close an organism is to accidentally creating a new, modified base within the framework of its existing genome. Here, we describe the biochemical and structural basis for how a single point mutation in E. coli’s naturally occurring cytosine methyltransferase can surprisingly endow a neomorphic ability to create the unnatural DNA base, 5-carboxymethylcytosine (5cxmC), in vivo. Mass spectrometry, bacterial genetics, and structure-guided biochemistry reveal this base to be exclusively derived from the natural but sparse secondary metabolite carboxy-S-adenosyl-L-methionine (CxSAM). Our discovery of a new, unnatural DNA modification reveals insights into the substrate selectivity of DNA methyltransferase enzymes, offers a promising new biotechnological tool for the characterization of the mammalian epigenome, and provides an unexpected model for how neomorphic bases could arise in nature from repurposed host metabolites.
Keywords: DNA modifications, synthetic biology, epigenetics, enzymology
eTOC blurb
In Wang and Kohli, 5-carboxymethylcytosine is revealed as an unnatural genomic DNA modification that can be generated in vivo from a unique combination of a sparse natural host metabolite and a neomorphic cytosine DNA methyltransferase.
Graphical Abstract
Introduction
Manipulation of genomes to accommodate non-canonical nucleobases creates opportunities for increasing DNA coding capacity and offers insights into the requirements for sustaining life (Krueger and Kool, 2009). Significant progress has been made in the propagation of unnatural nucleobases within the E. coli genome (Malyshev et al., 2014). Similarly, non-native but naturally-occurring nucleobases, such as those derived from bacteriophages, have been introduced into E. coli (Mehta et al., 2016a; Mehta et al., 2016b). Both strategies rely on the manipulation of nucleoside triphosphate (dNTP) pools. Although dNTP manipulation represents one way to imbue the genome with chemical diversity, marks covalently introduced after DNA replication provide an alternative route to encoding additional complexity in DNA, a strategy akin to nature’s existing epigenetic bases. To our knowledge, there have been no previous reports describing an organism with an unnatural DNA modification derived from its own native metabolome, a finding that may show the feasibility for an organism to accidentally accrue a gain-of-function ability to create a new DNA base in vivo.
Within the natural realm, an array of different DNA modifications have been described, but the vast majority of this diversity is confined to bacteriophage genomes and their prokaryotic hosts. Modifications to all canonical nucleobases have been described in phages (Weigele and Raleigh, 2016). In prokaryotes, the predominant modifications are found at the N6 position of adenine and N4 or C5 position of cytosine. Methylation of these bases primarily serves rudimentary functions to distinguish self from non-self in the arms race against bacteriophages (Nabel et al., 2012; Wilson and Murray, 1991), although emerging models suggest that some modifications may impact genome regulation (Sánchez-Romero and Casadesús, 2020).
5-methylcytosine (5mC) is a genomic DNA modification that extends from prokaryotes to higher organisms. Phylogenetic evidence shows that DNA cytosine methyltransferases (MTases), the enzymes that make 5mC, are conserved from prokaryotic restriction-modification systems to eukaryotic gene regulatory machinery (Iyer et al., 2011). In mammals, 5mC generation is largely confined to cytosine-guanine (CpG) dinucleotides (Portela and Esteller, 2010). Adding further complexity to this model, 5mC is a substrate for the Ten-Eleven Translocation (TET) family enzymes, which iteratively oxidize 5mC to create 5-hydroxymethyl-, 5-formyl-, and 5-carboxylcytosine (He et al., 2011; Ito et al., 2011; Tahiliani et al., 2009). While predominantly implicated as intermediates towards 5mC erasure, the potential independent epigenetic identities of each oxidized 5mC base is the subject of numerous provocative hypotheses (Bilyard et al., 2020). Across phylogeny, there is therefore compelling evidence for a functional role for diverse DNA modifications, providing the motivation for understanding how new DNA modifications can arise.
Here, we describe how our efforts to understand the structural basis of 5mC generation by the CpG MTase M.MpeI unexpectedly led to the identification of the previously undescribed DNA modification, 5-carboxymethylcytosine (5cxmC), which originates from the naturally-occurring, trace secondary metabolite carboxy-S-adenosyl-L-methionine (CxSAM). Additional biochemistry and structure-guided rationalization led to the accurate prediction that E. coli’s native MTase, Dcm, only requires a single point mutation to generate 5cxmC as a metabolite-derived, unnatural nucleobase in genomic DNA in vivo, offering a model for how new modifications can arise in nature.
Results
Saturation mutagenesis of a CpG MTase reveals a new DNA modification:
Given our interest in developing tools to understand mammalian modifications in the CpG context, we began by examining the crystal structure of a recently described bacterial CpG methyltransferase, M.MpeI (Wojciechowski et al., 2013). M.MpeI employs a canonical cytosine DNA MTase mechanism to make 5mC from S-adenosyl-L-methionine (SAM) and cytosine (Figure 1A). We focused on Asn374 of M.MpeI to assimilate two competing observations from the literature. The Asn sidechain, which is heavily conserved across MTases, has been proposed to act as part of a network of hydrogen bonds with active site water molecules to promote the final elimination step for 5mC generation (Jurkowski and Jeltsch, 2011; Zhang, X. and Bruice, 2006). Despite this model, however, mutation of this Asn to Ala is tolerated in homologous MTases and permits transfer of bulky SAM analogs in vitro (Lukinavicius et al., 2012). We thus pursued saturation mutagenesis of N374 as an unbiased way to understand its core role in MTase catalysis.
Figure 1: Saturation mutagenesis of M.MpeI N374X shows activity inconsistent with methylation.
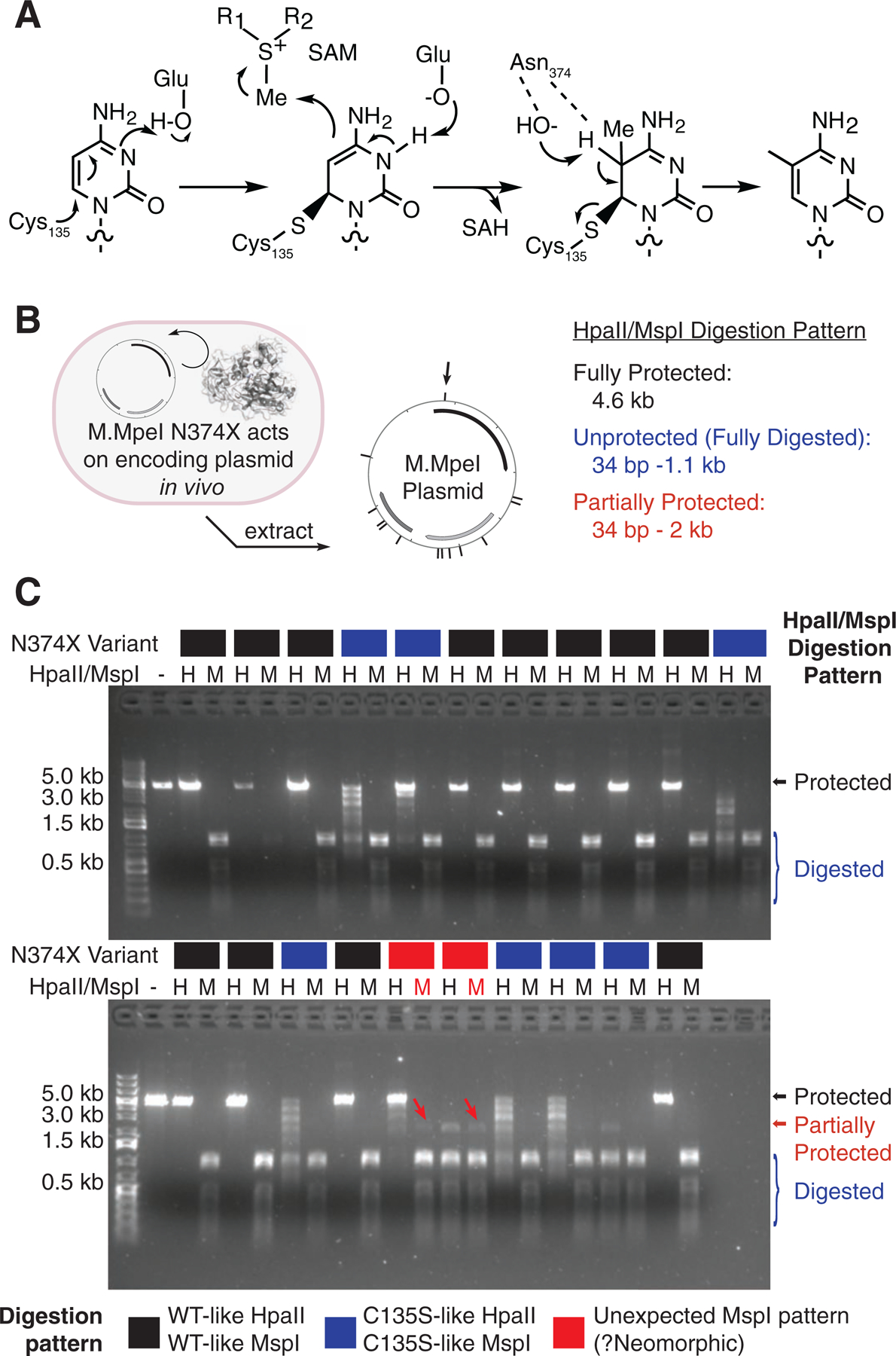
(A) Mechanism of DNA cytosine methyltransferases. (B) Experimental design. Individual M.MpeI N374X constructs were transformed and expressed in E. coli. In vivo modification was detected by restriction digestion of purified plasmids. The CCGG sites recognized by HpaII or MspI are visualized by hashmarks and include CpGs that could be modified. Partial resistance to cleavage at the site marked with a black arrow above the plasmid can result in the generation of a ~2 kb fragment highlighted with the red arrows on the gel. (C) HpaII (“H”, modification-sensitive) and MspI (“M”, methylation-insensitive) digestion. Each N374X mutant digestion pattern is WT-like, C135S catalytic mutant like, or potentially neomorphic. The red arrows highlight MspI digestion bands inconsistent with methylation.
We performed an in vivo activity screen that relies upon the linkage of the M.MpeI mutant genotype with a cytosine-methylating phenotype. We separately transformed each of the twenty N374X variants, along with a C135S catalytic mutant, into E. coli. After inducing expression, the plasmids were recovered and assessed for the ability of each MTase to modify its own encoding plasmid in vivo (Figure 1B). Extracted plasmids were digested with one of two CCGG recognizing restriction enzymes, HpaII and MspI. HpaII is methylation-sensitive and blocked by any covalent modification at the 5-position of the underlined cytosine. Its isoschizomer MspI is methylation-insensitive and was intended to serve as a positive control for methylation (Figure 1C).
In our in vivo screen, both the HpaII and MspI digestion patterns were similar to WT M.MpeI for the majority of our variants, suggesting that conversion to C5mCGG was achieved across all plasmid sites. This finding suggests that, contrary to its postulated mechanistic role, N374 is generally tolerant to variation, although our saturating in vivo overexpression conditions and indirect assay may overestimate tolerance to mutagenesis. Nonetheless, partial protection from HpaII digestion, suggesting impaired catalysis, was observed with hydrophobic β-branched (Ile/Val), constrained (Pro), or bulky aromatic (Phe/Tyr/Trp) mutations at position N374. Most notably and unexpectedly, in both positively-charged variants, N374K and N374R, there emerged a faint ~2 kb band resistant to MspI digestion, inconsistent with cytosine methylation (Figure 1C, red arrows). Upon reexamination of the plasmid map, we considered the possibility that if the CpG dinucleotides in the plasmid were to be randomly and partially modified to something other than 5mC in vivo, a CCGG protection event at a position between two larger fragments normally generated by MspI digestion (black arrow, Figure 1B) could account for this new ~2-kb band (red arrows, Figure 1C).
Identification of 5-carboxymethylcytosine:
While MspI cleaves at C5mCGG, it is blocked by bulkier modifications such as the oxidized 5mCs (Liu et al., 2016). To explore the possibility that we were detecting an unexpected DNA modification, we degraded each plasmid to its individual nucleosides and performed liquid chromatography tandem mass spectrometry (LC-MS/MS), scanning for nucleoside masses larger than 5mC (Figure 2A). In the N374K and N374R mutants but not WT or C135S, we identified a peak with a unique retention time of 2.2 min and m/z 286.1 [M + H]+ → 170.1 [M + H − dR]+ (Figure 2B/C).
Figure 2: M.MpeI mutants create 5-carboxymethylcytosine (5cxmC) in vivo.
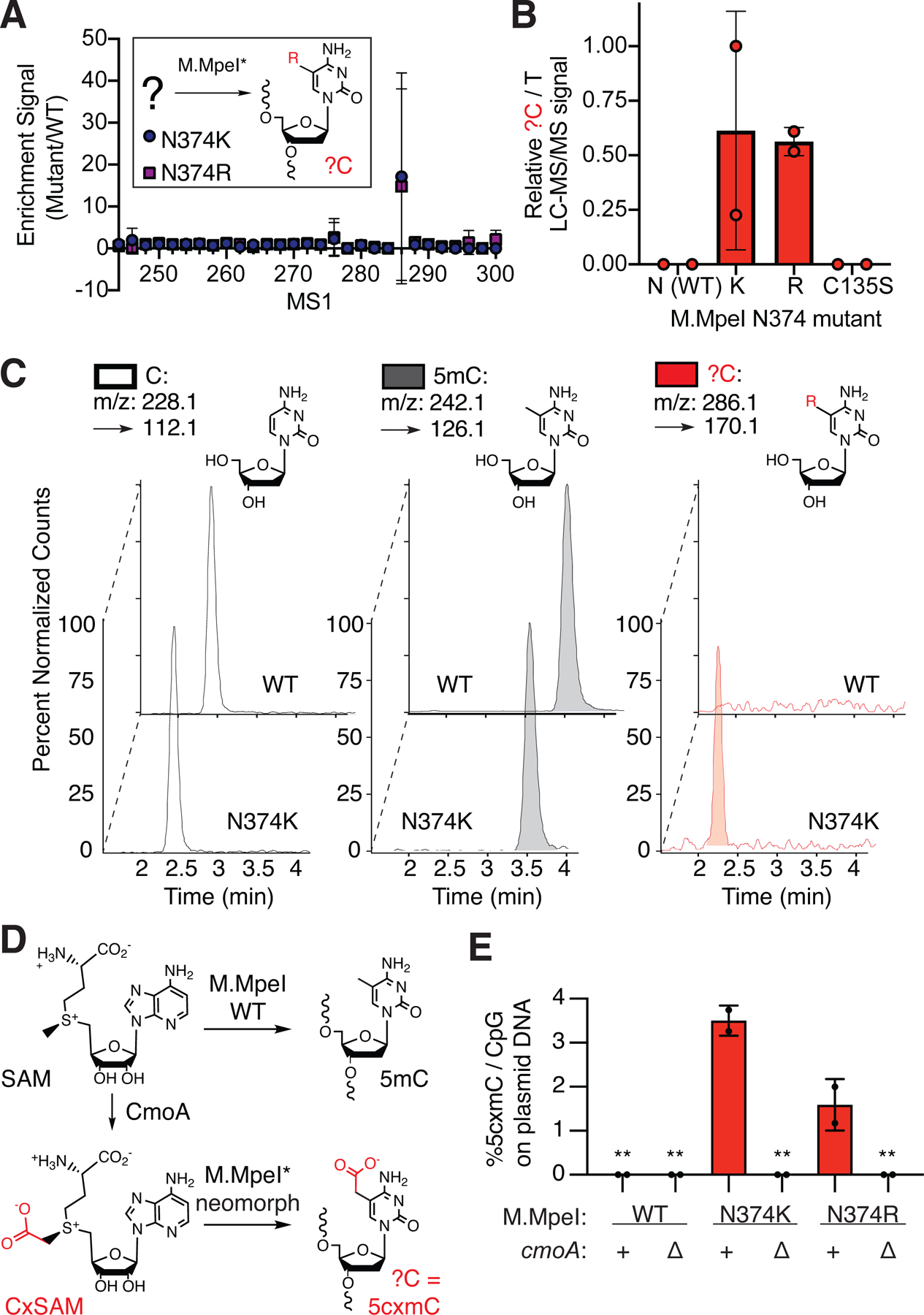
(A-C) Nucleoside LC-MS/MS of plasmid DNA modified in vivo by M.MpeI mutants. (A) Scanning mode identifies a candidate mass. Listed are the parent ion (MS1) m/z [M + H]+ associated with the fragment ion (MS2) [M + H − dR]+ resulting from loss of the deoxyribose sugar. (B) Quantitative detection normalized to input (T), expressed relative to maximum signal observed. (C) New modification shows a distinct retention time and detectable m/z in N374K but not WT M.MpeI plasmids. Peaks are normalized for maximal detection. Panels (A-C) show the same samples (n = 2 biological replicates). Error bars represent s.d. (D) CxSAM synthesized by CmoA in vivo provides a putative substrate for carboxymethylation. (E) 5cxmC is derived exclusively from CxSAM as assessed by cmoA knockout. Shown is the %5cxmC relative to total CpGs. Graphs show mean ± s.d. (n = 2 biological replicates). **below limit of detection.
Upon literature review, we identified carboxy-S-adenosyl-L-methionine (CxSAM) as a candidate MTase substrate worth further investigation. CxSAM is a sparse secondary metabolite in E. coli generated from SAM and prephenate by the non-essential enzyme CxSAM synthase (CmoA) and has recently been shown to be involved in tRNA modifications of uridine in E. coli (Kim et al., 2013). Although CxSAM is 400-fold less prevalent than SAM in vivo (Kim et al., 2015), we noted that the reaction of CxSAM with a target cytosine would yield 5-carboxymethylcytosine (5cxmC) (Figure 2D), a bulky modification that could resist MspI digestion. The nucleoside LC-MS/MS signal could also be consistent with this possibility: it has a more polar retention time relative to cytosine and 5mC, and the parent and fragmentation mass patterns fit with the addition of a carboxymethyl group.
To rigorously assess if CxSAM was in fact the substrate for our mutant MTase in vivo, we generated a cmoA knockout strain (Figure S1A–C). While in vivo plasmid carboxymethylation by M.MpeI N374K and N374R can be detected in the cmoA+ E. coli strain by LC-MS/MS, these signals are lost in the ΔcmoA strain (Figure 2D, Figure S2D/E). We also expressed M.MpeI WT, C135S, N374K and a double mutant C135S N374K and showed that only the combination of M.MpeI N374K in cmoA+ E. coli could create an MspI resistant modification (Figure S1F). Thus, 5cxmC is generated via a similar mechanism to 5mC and is solely derived from the activity of mutant M.MpeI utilizing endogenous CxSAM as a substrate.
Validation of DNA carboxymethyltransferase activity:
Having established the identity and origin of the 5cxmC base, we aimed to reproduce this activity in vitro. We purified both the WT and N374K M.MpeI variants (Figure S2A) and synthesized CxSAM as a diastereomeric mixture (Figures 2D, S1D, and S1). We then incubated enzyme with a pUC19 plasmid DNA substrate and either SAM or CxSAM. The plasmids were subsequently digested with modification-sensitive HpaII (CCGG, 13 sites) (Figure 3A). Consistent with our in vivo analysis, WT M.MpeI is capable of completely protecting the pUC19 plasmid with SAM but not CxSAM. M.MpeI N374K, by contrast, transfers SAM less efficiently than the WT enzyme, but gains the neomorphic ability to transfer CxSAM.
Figure 3: M.MpeI N374K creates 5-carboxymethylcytosine (5cxmC) in vitro.
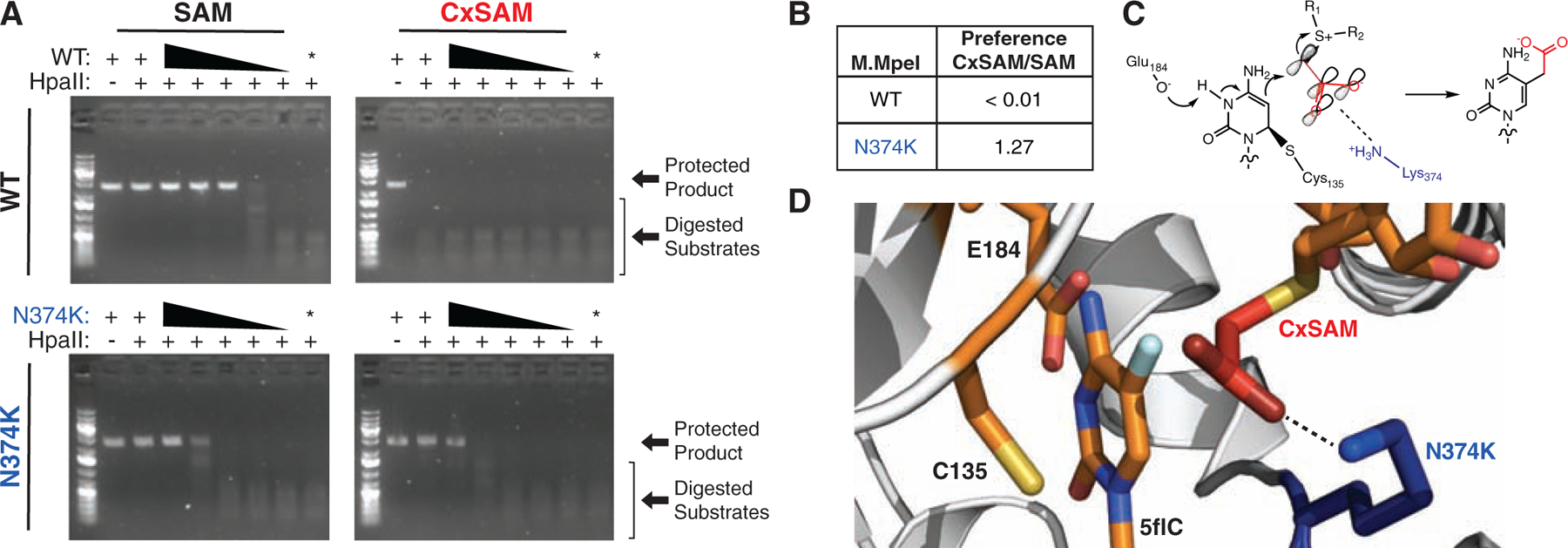
(A) Unmodified pUC19 plasmid DNA was incubated with an excess of SAM or CxSAM and serial dilutions of M.MpeI WT or N374K. The negative control lane (*) contains the highest concentration of enzyme with no SAM or CxSAM substrate. Digestion with HpaII fragments only unmodified DNA. M.MpeI N374K transfers both CxSAM and SAM in vitro while M.MpeI WT only transfers SAM. (B) M.MpeI N374K prefers CxSAM over SAM in a quantitative oligonucleotide assay shown in Figure S3 (n = 3 independent replicates). (C) Mechanism of DNA carboxymethylation visualizing a π-system which is favorable for CxSAM electrophilicity. Adjacent to catalytic residues E184 and C135, N374K (blue) could form a gain-of-function salt bridge (dashed-line) with the carboxylate (red) of CxSAM. (D) Structural model of M.MpeI with same elements as in (C). The model was obtained by the introduction of a N374K mutation and manually overlaying CxSAM from PDB 4QNV in place of SAH in the 5-fluorocytosine (5flC)-bound M.MpeI structure (PDB 4DKJ).
For a more quantitative comparison of in vitro activity, we devised an oligonucleotide-based assay, whereby modification of a CpG on a fluorophore labeled strand can be tracked by monitoring its resistance to HpaII digestion (Figure S3A). Consistent with the prior pUC19-based assay, we found that for WT M.MpeI, only SAM and not CxSAM was a substrate for cytosine modification. For the N374K variant, CxSAM was 1.3-fold preferred over SAM (Figure 3B, Figure S3B/C). While our in vitro studies show that N374K M.MpeI has a modest preference for CxSAM over SAM, our in vivo experiments suggest that the oligonucleotide assay may underestimate the extent of this preference, possibly due to our inability to separate CxSAM diastereomers or other factors that enhance CxSAM selectivity in vivo.
Structural model accurately predicts neomorphic carboxymethylation by a mutant E. coli MTase:
Prior work with synthetic SAM analogs has suggested that transfer can be promoted by the presence of a conjugated π-system at the β-carbon relative to the electrophilic carbon (Figure 3C) (Dalhoff et al., 2006). This mechanism alone, however, cannot explain why our mutant and WT M.MpeI behave differently towards the same CxSAM substrate. We thus turned to the crystal structure of M.MpeI with the S-adenosyl-L-homocysteine (SAH) product bound and manually overlaid CxSAM in place of SAH (Figure 3D). In the model, we observed that a mutant Lys374 is poised to form a potential salt bridge with the carboxylate anion of CxSAM, offering a likely explanation for this enzyme’s ability to accept this substrate.
Given this structural model for cytosine carboxymethylation, we wondered if this neomorphic activity was also accessible for homologous MTases. We specifically chose to focus on E. coli’s naturally occurring DNA Cytosine Methyltransferase (Dcm) because this enzyme allowed us to address the question, “What does it take for an organism to make a new DNA modification, in vivo?”. While M.MpeI is native to Mycoplasma penetrans and generates 5mC in the CpG context, Dcm generates 5mC in CCWGG (W = A/G) contexts. Structural alignment showed that Asn436 of Dcm and Asn374 of M.MpeI are both positioned adjacent to C5 of the target cytosine (Figure 4A/1A), and a simple transversion mutation in dcm could create an N436K mutation (Figure 4B).
Figure 4: E. coli’s native methyltransferase Dcm is a single point mutation away from accruing a gain-of-function ability to carboxymethylate genomic DNA in vivo, redefining the boundary between unnatural and natural nucleobases.
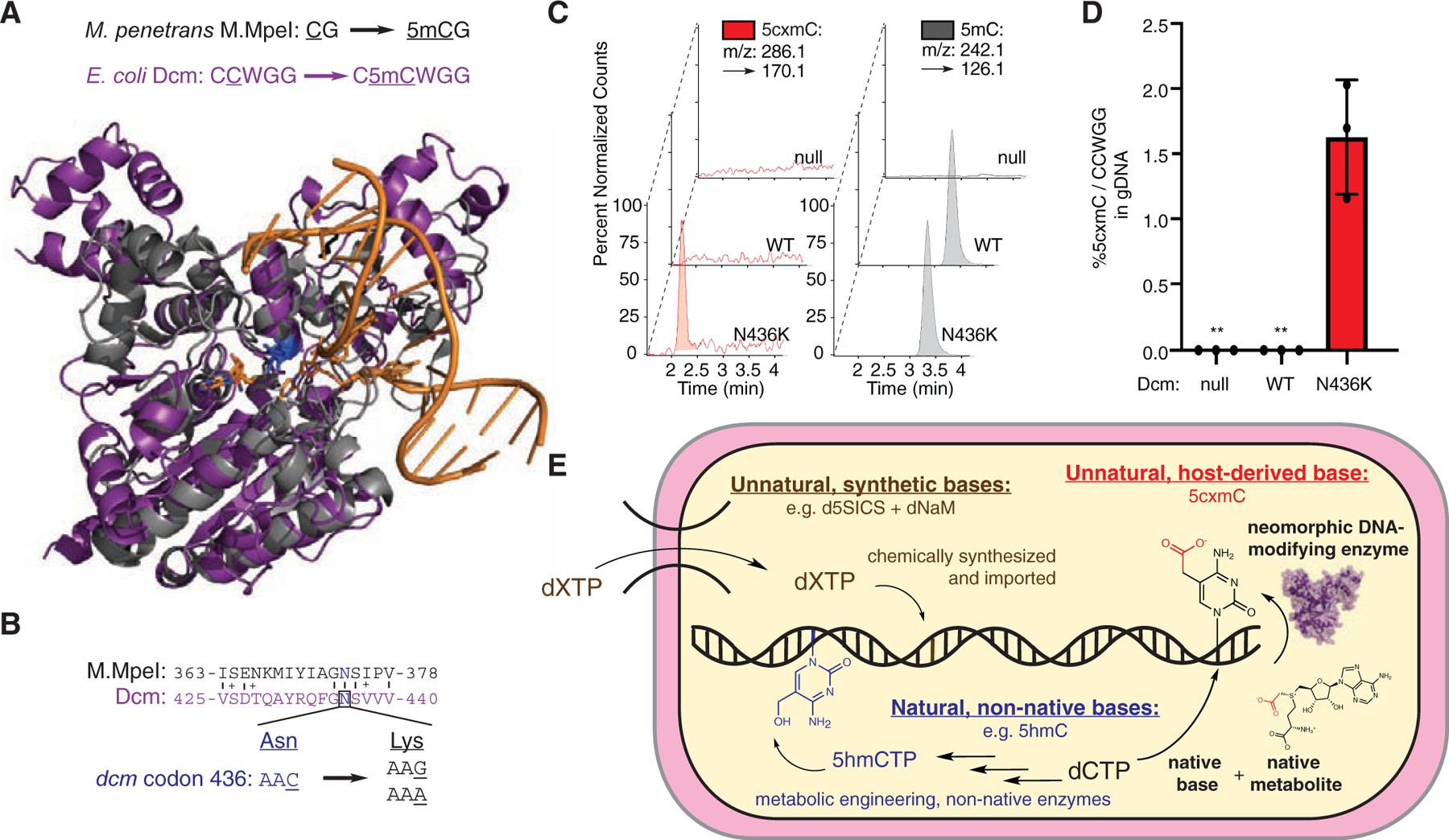
(A) Alignment of Dcm (purple, Swiss-Model: P0AED9) with M.MpeI (gray, PDB 4DKJ). (B) Asn436 codon (C) Qualitative nucleoside LC-MS/MS showing that only Dcm N436K can carboxymethylate E. coli genomic DNA in vivo. Peaks are normalized for maximal detection. (D) Quantitative 5cxmC LC-MS/MS signal in Dcm mutants. Shown is the %5cxmC relative to total CCWGGs (W = A/T) with each Dcm variant (null = no plasmid). Error bars represent mean ± s.d. (n = 3 biological replicates). **below limit of detection. (E). Multiple routes to expanded genomic diversity are shown. Brown: Unnatural dXTPs can be imported and replicated in vivo (Malyshev et al., 2014). Blue: Non-native nucleoside triphosphates (e.g. 5hmCTPs) can be incorporated into E. coli genomes using bacteriophage biosynthetic machinery (Mehta et al., 2016). Red: 5-carboxymethylcytosine (5cxmC) in DNA, synthesized by a neomorphic DNA-modifying enzyme, is a new, unnatural DNA base, derived from the native substrates cytosine and CxSAM, without dNTP manipulation
Encouraged by our biochemical understanding of M.MpeI-mediated DNA carboxymethylation, we moved to dam−/dcm− E. coli and introduced either Dcm WT or N436K on a plasmid. After induction of MTase expression, we extracted the genomic DNA (gDNA) and performed nucleoside LC-MS/MS (Figure 4C). In this setting, both the WT and N436K enzymes could methylate cytosine. However, only the N436K mutant enzyme could create 5cxmC in the native E. coli genome. Quantification of 5cxmC showed that >1.5% of the CCWGG sites were carboxymethylated (Figure 4D). Thus, as with M.MpeI, despite the excess of SAM over CxSAM, a substantial amount of 5cxmC can be detected in vivo (Figure S4A). Additionally, tracking E. coli growth curves for the Dcm WT, N436K, or C177A catalytic mutants suggests that the presence of 5cxmC does not lead to any apparent deficiency in growth under these conditions (Figure S4B).
To further compare to the M.MpeI variants, we next purified the Dcm WT and N436K mutant enzymes and assayed for their ability to modify pUC19 plasmid DNA with either SAM or CxSAM using the modification-sensitive restriction enzyme EcoRII (CCWGG, 7 sites). Consistent with our in vivo observations, WT Dcm can protect the pUC19 plasmid with SAM but not CxSAM. By contrast, Dcm N436K has comparable ability to transfer both CxSAM and SAM (Figure S4C/D). Given the extensive conservation of the active site Asn in homologous MTases (Jurkowski and Jeltsch, 2011), the consistent findings from M.MpeI to Dcm indicate that this residue may have neomorphic potential across the cytosine MTase family. Our results highlight that a single point mutation in the native dcm coding sequence is sufficient to create an unnatural DNA modification in E. coli.
Discussion
Here, we describe the discovery of a new genomic DNA base derived exclusively from the combination of a neomorphic methyltransferase and a native, but sparse, metabolite. Although not our original intent, the realization that our findings occupy a distinct space relative to similar, yet methodologically divergent synthetic biology efforts has afforded us unique insights into both technology development and the chemical determinants of genomic composition and evolution (Figure 4E).
Non-canonical nucleobases can originate from a variety of sources. While prior efforts have shown that synthetic and non-native sources of dNTPs can be used to create new bases in vivo, this study instead identifies the metabolome as an underappreciated source of genomic diversification. Outside of SAM, metabolites have been well documented to potentiate (Blaschke et al., 2013; Chen et al., 2013) or inhibit (Dang et al., 2009; Xu et al., 2016) the production of naturally-occurring modified nucleobases, but only very rarely are they considered as substrates which can directly be used to modify genomic DNA. An interesting exception is provided by ascorbic acid (vitamin C), which was recently shown to be an unexpected co-substrate for generating the natural, modified base 5-glycerylmethylcytosine in the algae Chlamydomonas reinhardtii (Xue et al., 2019). In the case of CxSAM, while no role in DNA modification was previously known, the metabolite has been shown to act as a direct substrate for modified uridines in tRNA (Kim et al., 2013; Kim et al., 2015) and small molecule cofactor modifications (Serebryakova et al., 2016). These precedents helped us to uncover that CxSAM can also be utilized by neomorphic, mutant DNA MTases. Our study thus highlights the possibility that rare metabolites similar to CxSAM, with other biological functions, could be redirected towards the creation of new, modified nucleic acids, in agreement with previous phylogenetic predictions that invoked modified SAM substrates as possible sources for undiscovered genomic diversity (Iyer et al., 2013).
Because CxSAM is a derivative of the parent metabolite SAM, substrate competition is a notable aspect of this study. This competition recapitulates a challenge faced in other synthetic biology efforts, outside of nucleic acids. For example, genetic code expansion can be limited by poor unnatural amino acid bioavailability and competition with endogenous elongation factor Tu (EF-Tu) (Chin, 2014). The experiments here establish that competition from a ubiquitous substrate (SAM) can be overcome. For technology development, this is an important observation because the synthetic SAM analog field continues to expand but is currently predominantly limited to in vitro settings. Given this study and others (Wang et al., 2013), it is now more feasible to consider whether SAM analogs with useful chemical handles can covalently modify DNA or proteins in vivo, despite their inevitable competition with native SAM.
As unnatural cytosines derived from SAM analogs have already been applied for in vitro sequencing applications (Kriukienė et al., 2013), the discovery of this DNA-modifying enzyme is additionally notable for its biotechnological potential. Here, we show that our enzyme can completely carboxymethylate a 2.6 kb plasmid substrate (Figure 3A). Given its proficiency, we speculate that our CpG DNA carboxymethyltransferase, which would selectively react with unmodified CpGs, can be leveraged to distinguish unmethylated, methylated, and oxidized CpGs when coupled to either third-generation or enzymatic methods for epigenetic sequencing (Figure S4E) (Schutsky et al., 2018).
Finally, we note that 5cxmC, but not 5mC, showed a gain-of-function ability to resist digestion by the modification-sensitive endonuclease MspI (Figure 1C and S1D/F). Given the growing body of evidence that suggests that restriction-modification systems have the capacity to coevolve, it is feasible that selection focused on 5cxmC could be harnessed to improve the stability and abundance of 5cxmC modifications in vivo (Iyer et al., 2011; Sánchez-Romero and Casadesús, 2020). However, we caution that the levels of 5cxmC remain relatively low (~1.5% of possible sites in gDNA for Dcm N436K, Figure 4), and the neomorphic CxMTases not only can create 5cxmC from CxSAM but also have a diminished ability to create 5mC from SAM. Both properties have unknown and potentially independent effects on physiology that will require future investigation (Figure S4).
When considering the potential advantages of modified DNA bases such as 5cxmC, it is particularly surprising that the answer to the question, “How close is an organism to accidentally procuring a new DNA modification?” may be only a point mutation. Interpreting this finding with knowledge of the extensive structural conservation within the cytosine MTase family and phylogenetic conservation of the enzyme CmoA illustrates how the boundary separating natural and unnatural DNA modifications is significantly blurrier than what might have been previously imagined.
STAR Methods
RESOURCE AVAILABILITY
Lead Contact:
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Rahul Kohli (rkohli@pennmedicine.upenn.edu).
Materials Availability:
Plasmids generated in this study can be obtained with a Materials Transfer Agreement.
Data and Code Availability:
This study did not generate any datasets/code.
EXPERIMENTAL MODELS AND SUBJECT DETAILS
E. Coli Strains:
ER1821 E. coli (New England Biolabs (NEB), F− glnV44 e14-(McrA-) rfbD1? relA1? endA1 spoT1? thi-1 Δ(mcrC-mrr)114::IS10) were used in M.MpeI experiments, including cloning. This strain is void of all methylation-specific restriction factors which recognize CpG methylation as foreign. ER1821 ΔcmoA was created with P1vir phage transduction using the ΔcmoA strain (JW1859) from the KEIO collection and kanamycin selection (Figure S1A–C) (Baba et al., 2006; Miller, 1992). For Dcm experiments, dcm− /dam− E. coli were used (NEB C2925I, ara-14 leuB6 fhuA31 lacY1 tsx78 glnV44 galK2 galT22 mcrA dcm-6 hisG4 rfbD1 R(zgb210::Tn10) TetS endA1 rspL136 (StrR) dam13::Tn9 (CamR) xylA-5 mtl-1 thi-1 mcrB1 hsdR2).
METHOD DETAILS
Cloning:
The WT M.MpeI sequence was obtained from the protein FASTA file from the PDB deposited (4DKJ) crystal structure (Wojciechowski et al., 2013) and ordered as an E. coli codon-optimized GeneBlock from Integrated DNA Technologies (IDT). The gene was PCR amplified with primers containing BsaI-HF and HindIII-HF overhangs using Phusion Polymerase (NEB) and ligated into digested pMG81 plasmid, a medium copy number vector with an anhydrotetracycline-inducible promoter (Kubiak et al., 2017). The WT dcm gene was obtained by directly amplifying from ER1821 gDNA with Phusion Polymerase and primers introducing a C-terminal His tag and appropriate BsaI overhangs. This gene was then assembled using Golden-Gate cloning (Engler et al., 2008) into a compatible pMG81 plasmid. All mutations were obtained by performing Q5 Site Directed Mutagenesis (NEB) and confirmed by Sanger sequencing.
In vivo methyltransferase assays:
Individual pMG81-MMpeI or pMG81-Dcm plasmids were transformed into chemically-competent E. coli and plated separately. Single colonies were grown in cultures (3 mL LB, 100 μg/mL carbenicillin). Cultures were allowed to grow at 37 °C until log phase (OD ~ 0.4–0.7) before induction with 20 ng/mL anhydrotetracycline (ATc) and then grown overnight. Plasmid extractions (Qiagen) or gDNA extractions (Qiagen DNeasy) were then performed, eluted in 10 mM Tris-Cl, pH 8.0, and quantified by nanodrop.
Nucleoside LC-MS/MS:
Plasmid or gDNA was digested with Nucleoside Digestion Mix (NEB) in a 10 μL total volume for 4 hours at 37 °C, and the mixture was diluted 10-fold into 0.1% formic acid with the addition of 770 fmol thymidine-D3 (T-D3) internal standard (ITSD) into a volume of 20 μL. LC-MS/MS was performed as previously described (DeNizio et al., 2019). Briefly, 5 μL was injected onto an Agilent 1200 Series HPLC instrument equipped with a 5 μm, 2.1 mm × 250 mm Supelcosil LC18-S analytical column (Sigma) equilibrated to 45 °C in buffer A (0.1% formic acid). The nucleosides were separated in a gradient of 0 to 10% buffer B (0.1% formic acid and 30% (v/v) acetonitrile) over 8 min at a flow rate of 0.5 mL/min. MS/MS was performed by positive ion mode ESI on an Agilent 6460 triple-quadrupole mass spectrometer, with a gas temperature of 225 °C, a gas flow of 12 L/min, a nebulizer at 35 psi, a sheath gas temperature of 300 °C, a sheath gas flow of 11 L/min, a capillary voltage of 3500 V, a fragmentor voltage of 70 V, and a delta EMV of 1000 V. Energies were 10 V for all bases except for 5cxmC (25V). When scanning in multiple reaction monitoring (MRM) mode, mass transitions were collected in increments of two mass units from 242.1 → 126.1 to 300.1 → 184.1. For targeted MRM, mass transitions were (C: 228.1 → 112.1, T: 243.1 → 127.1, T-D3: 246.1 → 130.1, 5mC: 242.1 → 126.1, 5cxmC: 286.1 → 170.1). The amount of total input DNA injected was first obtained using T and the T-D3 ITSD using the equations below, where A signifies area measured by the MS/MS instrument. This number was then used to calculate a relative quantity of 5cxmC nucleoside in the experiments that lack a chemical standard for 5cxmC. This approach allows for accurate comparisons across conditions and is used in Figure 2A–C.
A standard for 5cxmC was synthesized using a chemoenzymatic approach. Excess M.MpeI N374K was reacted with CxSAM and hemimethylated substrate (see oligonucleotide assay methods). The substrate was verified for complete carboxymethylation by MspI digestion and visualized for fluorescein (FAM) fluorescence (excitation at 488 nm, emission at 520 nm) after separation on a 20% TBE Acrylamide Denaturing PAGE. The fully carboxymethylated standard was purified using an oligonucleotide spin column (Zymo), and quantified using the FAM fluorophore. This purified hemi-carboxymethylated oligonucleotide was then digested with Nucleoside Digestion Mix (New England Biolabs) in a 10 μL total volume for 4 hours at 37 °C and diluted 10-fold into 0.1% formic acid before LC-MS/MS injection. An LC-MS/MS standard curve with serial dilutions of digested nucleoside (Figure S1D–E) was used to convert the integrated area of an experimental sample to fmol 5cxmC detected. The lowest concentration on the LC-MS/MS standard curve was determined to be the limit of detection (0.26 fmol Figures 2 and 4).
With knowledge of the amount of T and 5cxmC injected, it was possible to calculate the total amount of 5cxmC relative to either total CpG sites (M.MpeI) or CCWGG sites (Dcm, W = A/T). For M.MpeI experiments, the amount of T injected was converted to total amount of CpGs injected by dividing by the molar ratio of Ts to CpGs in the pMG81-MMpeI plasmid (5.07). For Dcm samples, the complete genome of K-12 MG1655, the parent strain of the dam−/dcm− E. coli strain (GenBank: U00096.3) was analyzed. The molar ratio (101) comparing total instances of T (2,284,124) to CCWGG (22,716) was used to calculate the total amount of 5cxmC relative to total CCWGG sites.
Protein Purification:
All variants were purified using a C-terminal His tag. Single colonies from transformation of pMG81-MMpeI, pMG81-M.MpeI-N374K, pMG81-Dcm, or pMG81-Dcm-N436K into ER1821 were started in overnight cultures (10 mL LB, 100 μg/mL carbenicillin), inoculated into large scale cultures (1 L LB, 100 μg/mL ampicillin) in the morning, and allowed to grow at 37 °C until log phase (OD ~ 0.4 – 0.7) before switching the temperature to 16 °C. After 20 minutes, 20 ng/mL anhydrotetracycline (ATc) was used to induce protein overexpression and cultures were left at 16 °C overnight. Cells were harvested by centrifugation (8000g, 30 min, 4 °C) before resuspending in 25 mL Buffer A (50 mM Tris Cl, pH 7.5 at 25 °C, 150 mM NaCl, 25 mM Imidazole, 10% Glycerol (v/v)) + 1 EDTA-free Protease Inhibitor Tablet (Sigma) + 10 μL RNase A (Thermo Fisher). Resuspended cells were frozen overnight at −80 °C.
Cells were lysed using a sonicator and centrifuged at 4 °C for 30 min at 27,000g. Soluble lysate was passed through a gravity column containing 4 mL His Cobalt Resin, pre-equilibrated in Buffer A. After loading, the columns were washed with 25 column volumes (CV) of Buffer A for WT M.MpeI and WT Dcm, or 25 CV of Buffer B (50 mM Tris Cl, pH 7.5 at 25°C, 1 M NaCl, 25 mM Imidazole, 10% Glycerol (v/v)) followed by 5 CV of Buffer A for M.MpeI N374K and Dcm N436K. Protein was eluted with sequential fractions of Buffer C (50 mM Tris Cl, pH 7.5 at 25 °C, 150 mM NaCl, 150 mM Imidazole, 10% Glycerol (v/v)). Samples were dialyzed (8,000 MWCO, Thermo Fisher) overnight at 4 °C in 2 L of prechilled Dialysis Buffer (20 mM Tris HCl pH 7.5 at 25 °C, 0.2 mM EDTA, 2 mM DTT, 150 mM NaCl, 10% Glycerol (v/v)). Cold 40% (v/v) glycerol was added to the protein to dilute the dialyzed protein 2-fold before flash freezing with liquid nitrogen and long-term storage at −80 °C. All proteins were quantified by comparison to a BSA standard curve after running SDS-PAGE and visualizing with Coomassie Blue.
CxSAM Synthesis:
Reactions were performed as previously described (Kim et al., 2015). 2-iodoacetic acid (667 mg) was added to a solution of S-adenosyl-L-homocysteine (20 mg) in 3.3 mL of 150 mM aqueous ammonium bicarbonate. The reaction was incubated at 37°C for 24 hrs. 80 mL of methanol was then added and incubated at 4°C overnight. Precipitates were collected by centrifugation at 4°C (2000g, 30 min) and washed twice with cold methanol to yield CxSAM. CxSAM was dissolved in nuclease free water and stored at −20°C. We attempted purification using a Zorbax 300SB-C18 column (9.4 × 250 mm, 5 μm particle size) (Agilent Technologies) with Buffer A = 10 mM Na2HPO4, pH 5.9 and Buffer B = 100% acetonitrile as described previously (Serebryakova et al., 2016). The absence of other products under these conditions suggested that no further purifications were necessary (Figure S2). CxSAM was quantified using absorbance measured at 260 nm: (15,400 L mol−1 cm−1). High resolution mass spectrometry (HRMS) was obtained to 443.1360 (mDa = −0.2, PPM = −0.5, Theoretical Mass: 443.1343).
Restriction Digest Based Assays:
Restriction digestions were performed at 37°C for 1 hr in 1x NEB CutSmart Buffer in the specified volume. EcoRII restriction digests were performed at 37°C for 1 hr in 1x Thermo Scientific Buffer O.
pUC19 assay:
3-fold serial dilutions of M.MpeI (0.78 μM – 3.2 nM) were incubated with 160 μM SAM or CxSAM substrate and pUC19 plasmid DNA (100 ng) for 4 hrs at 37°C in M.MpeI reaction buffer (10 mM Tris Cl, 50 mM NaCl, 1 mM DTT, 1 mM EDTA, pH 7.9 at 25°C) in a 5 μL volume. 2.5 μL of the reaction was then incubated with the appropriate restriction enzyme, and the plasmid DNA was simultaneously linearized with HindIII-HF (NEB) in a final digestion volume of 25 μL. HpaII (NEB) recognizes CCGGs (13 sites). Samples were treated with 1 μL Proteinase K at 37°C for 10 min, separated on 1% TAE Agarose gel, and visualized with SYBR Safe DNA Gel Stain (Thermo-Fisher).
A similar assay was performed with the Dcm proteins except that serial 3-fold dilutions encompassed 0.26 μM – 3.2 nM and activity was instead assessed with EcoRII (Thermo), which recognizes CCWGGs (7 sites). The plasmid was also linearized with NdeI (NEB).
Oligonucleotide Assay:
A FAM labelled top strand oligonucleotide with a single unmethylated CCGG and unlabeled complementary bottom strand oligonucleotides with an unmethylated or methylated CCGG were obtained from IDT as above. 200 nM of the duplexed, hemimethylated oligo was reacted with serial dilutions of M.MpeI and 40 μM SAM or CxSAM substrate at 37°C in M.MpeI reaction buffer and a final volume of 5 μL for 30 min before heat inactivation at 95°C for 5 min. 25x excess of unmethylated bottom strand was reannealed before restriction digestion with HpaII in a final volume of 50 μL (Nabel et al., 2017). In vitro carboxymethylation was also confirmed by purifying the reaction mixture before the strand exchange step with an Oligo Clean & Concentrator column (Zymo) and analyzing by electrospray ionization mass spectrometry (ESI-MS, Novatia, Figure S3).
Protein Structures:
The structure of M.MpeI bound to SAH and a 5-fluorocytosine containing DNA substrate was used in modeling (PDB 4DKJ). The mutant N374K residue was manually created in PyMOL. Subsequently, CxSAM (PDB 4QNV) was manually overlaid on top of SAH with no energy minimization calculations to determine bond angles.
Growth Curves:
Dam−/dcm− E. coli were transformed with pMG81-Dcm expression plasmids and plated on amplicillin. Overnight cultures were started from single colonies. The next morning, cultures were quantified by OD600 before dilution (LB, 100 μg/mL carbenicillin). Protein overexpression was induced with or without ATc (20 ng/mL). Cultures were monitored for 20 hours at 37°C with cycled agitation (3-mm orbital shaking, 450 rpm) in a 96-well plate (Mo et al., 2016). Optical density at 595 nm (OD595) measurements were obtained every 5 minutes on a plate reader (Tecan).
QUANTIFICATION AND STATISTICAL ANALYSIS
All replicates are explicitly specified within figure legends.
FAM-labelled gels were quantified on a Typhoon imager and using Fiji software. Curves in Figure S3 were fit using GraphPad Prism 8 using non-linear least squares fit to extract EC50 values and 95% confidence intervals.
Supplementary Material
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Bacterial and Virus Strains | ||
| ER1821 E. coli | NEB | N/A |
| dcm−/dam− E. coli | NEB | C2925I |
| ΔcmoA E. coli | KEIO, Baba et al. 2006 | JW1859 |
| ER1821 ΔcmoA | This study | N/A |
| Chemicals, Peptides, and Recombinant Proteins | ||
| HpaII | NEB | R0171 |
| MspI | NEB | R0106 |
| HindIII-HF | NEB | R3104 |
| NdeI | NEB | R0111S |
| EcoRII | Thermo Fisher | ER1921 |
| Nucleoside Digestion Mix | NEB | M0649 |
| M.MpeI WT His | This study | N/A |
| M.MpeI N374K His | This study | N/A |
| Dcm WT His | This study | N/A |
| Dcm N374K His | This study | N/A |
| Proteinase K | Zymo | D3001 |
| RNAse A | Thermo | EN0531 |
| SAM | NEB | B9003S |
| SAH | Sigma | A9384 |
| Iodoacetic acid | Sigma | I4386 |
| CxSAM | This study | N/A |
| Thymidine-D3 ITSD | Toronto Research Chemicals | T412002 |
| Critical Commercial Assays | ||
| Oligo Clean and Concentrator kit | Zymo | D4060 |
| Plasmid extraction kit | Qiagen | 27104 |
| gDNA extraction kit | Qiagen | 69504 |
| Oligonucleotides | ||
| 27-Top-C-FAM: GTATCTAGTTCAATCCGGTTCATAGCA-FAM |
IDT | N/A |
| 27-Bottom-mC: TGCTATGAAC/5mC/GGATTGAACTAGATAC |
IDT | N/A |
| 27-Bottom-C: TGCTATGAACCGGATTGAACTAGATAC |
IDT | N/A |
| 27-cxmC-standard | This study | N/A |
| Recombinant DNA | ||
| pUC19 | Miniprepped from NEB C2925I | N/A |
| Software and Algorithms | ||
| Prism 8 | Graphpad | www.graphpad.com |
| Fiji | This study | https://fiji.sc/ |
Highlights.
Mutagenesis unexpectedly identifies a neomorphic activity in a DNA methyltransferase
Bacterial genetics uncovers the modification 5-carboxymethylcytosine (5cxmC)
Structure-guided biochemistry reveals enzymatic basis for DNA carboxymethylation
5cxmC provides a model for how gain-of-function genomic bases may arise in nature
Significance.
Efforts to manipulate and create synthetic organisms tolerant of unnatural or non-native nucleobases have collectively shown that life is remarkably accepting of new genomic chemistries. While these efforts have helped establish that exotic chemistries can be accommodated, here we describe and characterize a biochemical route for an unnatural DNA base to accidentally appear in vivo. We show that a single mutation in the active site of cytosine DNA methyltransferases confers neomorphic activity on these enzymes, allowing them to accept a sparse natural metabolite as a substrate and to thereby create 5-carboxymethylcytosine in genomic DNA. In addition to contributing a promising biotechnology for future exploration, our discovery of a host-derived, unnatural DNA modification provides evidence for the metabolome as a potentially rich and underappreciated source for accessing genomic diversity and offers a model for how new genomic DNA bases could arise in nature.
Acknowledgments
The authors thank Michael Cory, Juan Serrano, all members of the Kohli Laboratory, and Walraj Gosal (Cambridge Epigenetix, CEGX) for insightful discussions and feedback. In addition, we thank E. James Petersson for drawing our attention to the CxSAM metabolite, Mark Goulian and Kiran Gajula for assistance with bacteriology, Jamie DeNizio and Clementina Mesaros for providing mass spectrometry training and guidance, Uday Ghanty and Charles Ross III for CxSAM characterization support.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Declaration of Interests
T.W. is supported by a Cambridge Epigenetix (CEGX) Training Fellowship. This work was supported by the NIH (R01-GM118501 and R01-HG010646 to R.M.K.). R.M.K. is supported by an Investigator in the Pathogenesis of Infectious Disease Award (Burroughs Wellcome Fund). Patent filing on the DNA carboxymethyltransferase is pending.
References
- Baba T, Ara T, Hasegawa M, Takai Y, Okumura Y, Baba M, Datsenko KA, Tomita M, Wanner BL, and Mori H (2006). Construction of Escherichia coli K-12 in-frame, single-gene knockout mutants: the Keio collection. Mol. Syst. Biol 2, 2006.0008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bilyard MK, Becker S, and Balasubramanian S (2020). Natural, modified DNA bases. Current Opinion in Chemical Biology 57, 1–7. [DOI] [PubMed] [Google Scholar]
- Blaschke K, Ebata KT, Karimi MM, Zepeda-Martinez JA, Goyal P, Mahapatra S, Tam A, Laird DJ, Hirst M, Rao A, Lorincz MC, and Ramalho-Santos M (2013). Vitamin C induces Tet-dependent DNA demethylation and a blastocyst-like state in ES cells. Nature 500, 222–226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen J, Guo L, Zhang L, Wu H, Yang J, Liu H, Wang X, Hu X, Gu T, Zhou Z, et al. (2013). Vitamin C modulates TET1 function during somatic cell reprogramming. Nat. Genet 45, 1504–1509. [DOI] [PubMed] [Google Scholar]
- Chin JW (2014). Expanding and Reprogramming the Genetic Code of Cells and Animals. Annual Review of Biochemistry 83, 379–408. [DOI] [PubMed] [Google Scholar]
- Dalhoff C, Lukinavicius G, Klimasauskas S, and Weinhold E (2006). Direct transfer of extended groups from synthetic cofactors by DNA methyltransferases. Nat. Chem. Biol 2, 31–32. [DOI] [PubMed] [Google Scholar]
- Dang L, White DW, Gross S, Bennett BD, Bittinger MA, Driggers EM, Fantin VR, Jang HG, Jin S, Keenan MC, et al. (2009). Cancer-associated IDH1 mutations produce 2-hydroxyglutarate. Nature 462, 739–744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DeNizio JE, Liu MY, Leddin EM, Cisneros GA, and Kohli RM (2019). Selectivity and Promiscuity in TET-Mediated Oxidation of 5-Methylcytosine in DNA and RNA. Biochemistry 58, 411–421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Engler C, Kandzia R, and Marillonnet S (2008). A One Pot, One Step, Precision Cloning Method with High Throughput Capability. PloS One 3, e3647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- He YF, Li BZ, Li Z, Liu P, Wang Y, Tang Q, Ding J, Jia Y, Chen Z, Li L, et al. (2011). Tet-mediated formation of 5-carboxylcytosine and its excision by TDG in mammalian DNA. Science 333, 1303–1307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ito S, Shen L, Dai Q, Wu SC, Collins LB, Swenberg JA, He C, and Zhang Y (2011). Tet proteins can convert 5-methylcytosine to 5-formylcytosine and 5-carboxylcytosine. Science 333, 1300–1303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iyer LM, Abhiman S, and Aravind L (2011). Natural history of eukaryotic DNA methylation systems. Prog. Mol. Biol. Transl. Sci 101, 25–104. [DOI] [PubMed] [Google Scholar]
- Iyer LM, Zhang D, Burroughs AM, and Aravind L (2013). Computational identification of novel biochemical systems involved in oxidation, glycosylation and other complex modifications of bases in DNA. Nucleic Acids Res 41, 7635–7655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jurkowski TP, and Jeltsch A (2011). On the Evolutionary Origin of Eukaryotic DNA Methyltransferases and Dnmt2. PloS One 6, e28104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim J, Xiao H, Bonanno JB, Kalyanaraman C, Brown S, Tang X, Al-Obaidi NF, Patskovsky Y, Babbitt PC, Jacobson MP, Lee Y, and Almo SC (2013). Structure-guided discovery of the metabolite carboxy-SAM that modulates tRNA function. Nature 498, 123–126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim J, Xiao H, Koh J, Wang Y, Bonanno JB, Thomas K, Babbitt PC, Brown S, Lee Y, and Almo SC (2015). Determinants of the CmoB carboxymethyl transferase utilized for selective tRNA wobble modification. Nucleic Acids Research 43, 4602–4613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kriukienė E, Labrie V, Khare T, Urbanavičiūtė G, Lapinaitė A, Koncevičius K, Li D, Wang T, Pai S, Ptak C, et al. (2013). DNA unmethylome profiling by covalent capture of CpG sites. Nature Communications 4, 2190. [DOI] [PubMed] [Google Scholar]
- Krueger AT, and Kool ET (2009). Redesigning the Architecture of the Base Pair: Toward Biochemical and Biological Function of New Genetic Sets. Chemistry & Biology 16, 242–248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kubiak JM, Culyba MJ, Liu MY, Mo CY, Goulian M, and Kohli RM (2017). A Small-Molecule Inducible Synthetic Circuit for Control of the SOS Gene Network without DNA Damage. ACS Synth. Biol 6, 2067–2076 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu MY, DeNizio JE, and Kohli RM (2016). Quantification of Oxidized 5-Methylcytosine Bases and TET Enzyme Activity. Methods Enzymol. 573, 365–385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lukinavicius G, Lapinaite A, Urbanaviciute G, Gerasimaite R, and Klimasauskas S (2012). Engineering the DNA cytosine-5 methyltransferase reaction for sequence-specific labeling of DNA. Nucleic Acids Res. 40, 11594–11602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malyshev DA, Dhami K, Lavergne T, Chen T, Dai N, Foster JM, Corrêa J, Ivan R, and Romesberg FE (2014). A semi-synthetic organism with an expanded genetic alphabet. Nature 509, 385–388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mehta AP, Li H, Reed SA, Supekova L, Javahishvili T, and Schultz PG (2016a). Replacement of 2′-Deoxycytidine by 2′-Deoxycytidine Analogues in the E. coli Genome. Journal of the American Chemical Society 138, 14230–14233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mehta AP, Li H, Reed SA, Supekova L, Javahishvili T, and Schultz PG (2016b). Replacement of Thymidine by a Modified Base in the Escherichia coli Genome. Journal of the American Chemical Society 138, 7272–7275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller JH (1992). A short course in bacterial genetics: a laboratory manual and handbook for Escherichia coli and related bacteria (Plainview, N.Y.: Cold Spring Harbor Laboratory Press; ). [Google Scholar]
- Mo CY, Manning SA, Roggiani M, Culyba MJ, Samuels AN, Sniegowski PD, Goulian M, and Kohli RM (2016). Systematically Altering Bacterial SOS Activity under Stress Reveals Therapeutic Strategies for Potentiating Antibiotics. mSphere 1, e00163–16 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nabel CS, DeNizio JE, Carroll M, and Kohli RM (2017). DNA Methyltransferases Demonstrate Reduced Activity Against Arabinosylcytosine: Implications for Epigenetic Instability in AML. Biochemistry 56, 2166–2169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nabel CS, Manning SA, and Kohli RM (2012). The Curious Chemical Biology of Cytosine: Deamination, Methylation, and Oxidation as Modulators of Genomic Potential. ACS Chem. Biol 7, 20–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Portela A, and Esteller M (2010). Epigenetic modifications and human disease. Nat. Biotechnol 28, 1057–1068. [DOI] [PubMed] [Google Scholar]
- Sánchez-Romero MA, and Casadesús J (2020). The bacterial epigenome. Nature Reviews Microbiology 18, 7–20. [DOI] [PubMed] [Google Scholar]
- Schutsky EK, DeNizio JE, Hu P, Liu MY, Nabel CS, Fabyanic EB, Hwang Y, Bushman FD, Wu H, and Kohli RM (2018). Nondestructive, base-resolution sequencing of 5-hydroxymethylcytosine using a DNA deaminase. Nat. Biotech 36, 1083–1090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Serebryakova M, Tsibulskaya D, Mokina O, Kulikovsky A, Nautiyal M, Van Aerschot A, Severinov K, and Dubiley S (2016). A Trojan-Horse Peptide-Carboxymethyl-Cytidine Antibiotic from Bacillus amyloliquefaciens. Journal of the American Chemical Society 138, 15690–15698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tahiliani M, Koh KP, Shen Y, Pastor WA, Bandukwala H, Brudno Y, Agarwal S, Iyer LM, Liu DR, Aravind L, and Rao A (2009). Conversion of 5-methylcytosine to 5-hydroxymethylcytosine in mammalian DNA by MLL partner TET1. Science 324, 930–935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang R, Islam K, Liu Y, Zheng W, Tang H, Lailler N, Blum G, Deng H, and Luo M (2013). Profiling Genome-Wide Chromatin Methylation with Engineered Posttranslation Apparatus within Living Cells. Journal of the American Chemical Society 135, 1048–1056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weigele P, and Raleigh EA (2016). Biosynthesis and Function of Modified Bases in Bacteria and Their Viruses. Chemical Reviews 116, 12655–12687. [DOI] [PubMed] [Google Scholar]
- Wilson GG, and Murray NE (1991). Restriction and Modification Systems. Annual Review of Genetics 25, 585–627. [DOI] [PubMed] [Google Scholar]
- Wojciechowski M, Czapinska H, and Bochtler M (2013). CpG underrepresentation and the bacterial CpG-specific DNA methyltransferase M.MpeI. Proceedings of the National Academy of Sciences of the United States of America 110, 105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu Q, Wang K, Wang L, Zhu Y, Zhou G, Xie D, and Yang Q (2016). IDH1/2 Mutants Inhibit TET-Promoted Oxidation of RNA 5mC to 5hmC. PLoS One 11, e0161261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xue JH, Chen GD, Hao F, Chen H, Fang Z, Chen FF, Pang B, Yang QL, Wei X, Fan QQ, et al. (2019). A vitamin-C-derived DNA modification catalysed by an algal TET homologue. Nature 569, 581–585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang X, and Bruice TC (2006). The mechanism of M.HhaI DNA C5 cytosine methyltransferase enzyme: A quantum mechanics/molecular mechanics approach. Proceedings of the National Academy of Sciences of the United States of America 103, 6148–6153. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
This study did not generate any datasets/code.