

Abstract
Herein, we report novel formic acid-mediated rearrangements of dearomatized acylphloroglucinols to access a structurally diverse group of synthetic acylphloroglucinol scaffolds (SASs). Density functional theory (DFT)-optimized orbital and stereochemical analyses shed light on the mechanism of these rearrangements. Products were evaluated by multiplexed activity profiling (MAP), an unbiased platform which determines multiple biological readouts simultaneously at single-cell resolution for markers of cell signaling and can aid in distinguishing genuine activity from assay interference. Using MAP, we identified a number of SASs that suppressed pS6 (Ser235/236), a marker for activation of the mTOR and ERK signaling pathways. These results illustrate how biomimetic synthesis and multiplexed activity profiling can reveal the pharmacological potential of novel chemotypes via diversity-oriented synthesis.
Graphical Abstract

Diverse synthetic acylphloroglucinol scaffolds (SASs) have been produced by unprecedented cyclizations and rearrangements in formic acid, a cation-stabilizing and nucleophilic solvent for alkenes and carbonyls. Multiplexed activity profiling (MAP) was used to evaluate multiple biological readouts at single-cell resolution to decipher true hits from assay interference. The results herein illustrate how biomimetic synthesis combined with MAP can reveal the pharmacological potential of novel chemotypes produced via diversity-oriented synthesis.
Introduction
Polycyclic polyprenylated acylphloroglucinols (PPAPs)1 have played an important role in chemical synthesis2 and pharmacological studies due to their diverse biological activity profiles.3,4,5 PPAPs are classified as “type A” when the acyl group is at the bridgehead position (e.g. garcimultiflorone A (1)) and “type B” when the acyl group is at the α-position of the β-hydroxyenone [e.g. isogarcinol (2), didehydroxyisogarcinol (3), and garcinol (4)].1 With more than 500 members1 discovered to date from a variety of plant species (e.g. Hypericum henryi and Clusia congestiflora),6 most PPAPs are biosynthetically derived from a common dearomatized carbocation 5 involving a combination of enzyme-catalyzed prenylations and carbocation cyclizations (Figure 1a).1,7 However, despite synthetic efforts over the past 20 years,7c,8,9 a synthesis through such a cation was not realized8d,8i,9b until formic acid uniquely promoted the C3-cyclization of substrate (–)-6 to the clusianone core (–)-8a through the related carbocation (–)-7a (Figure 1b).8i Alternatively, the type A PPAP core of nemorosone (–)-8b could be accessed from diastereomer (+)-6 through photocyclization of (+)-7b at C1.8k To date, these biomimetic cyclizations provide the most concise route to type A and B PPAPs.1,2,8
Figure 1.
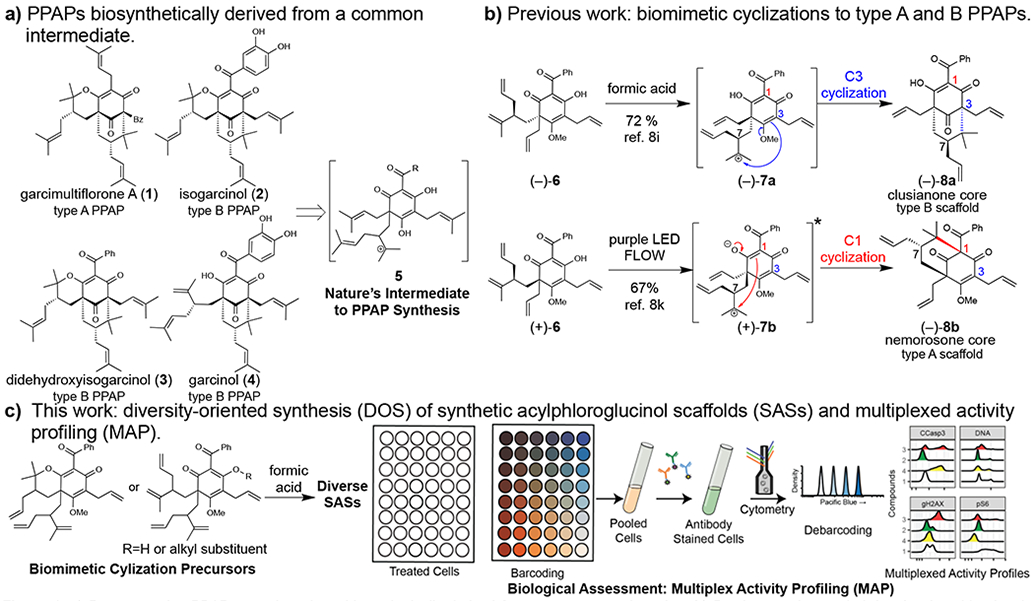
a) Representative PPAP natural products biosynthetically derived from a common carbocation; b) Previous work revealed that formic acid uniquely promoted biomimetic C3-cyclization to the clusianone core, and excited-state intramolecular proton transfer resulted in a biomimetic C1-cyclization to the nemorosone core; c) Biomimetic cyclizations lead to a small library of diverse SASs which were evaluated by MAP for biological activities.
In comparison to other Brønsted acids, formic acid has inherent properties that may be responsible for promoting cyclization to the clusianone core:8i,10 1) formic acid may stabilize carbocations by solvation effects as evidenced by formate adducts of 1,1-disubstituted olefins;10b 2) formate adducts of strained bridgehead ketones may facilitate cyclizations that would otherwise be less feasible.8i,10a,10c In this study, diversity-oriented synthesis (DOS)11 of synthetic acylphloroglucinol scaffolds (SASs) is described using formic acid in comparison to other Brønsted acids for promoting unique cyclizations and rearrangements, and multiplexed-activity profiling (MAP) is used to evaluate activities in diverse biological pathways (Figure 1c).12
MAP and other high-content assays13 have advantages over traditional ‘single-target’ in vitro screening assays14 for evaluating compound libraries. In contrast to in vitro-targeted assays, the MAP approach utilizes multiplexed and orthogonal biological readouts to identify active compounds and provides insight into downstream effects on cell signaling and homeostasis. Moreover, the MAP approach can be adapted to assess specific pathways and mechanistic targets using panels of antibody-based readouts. MAP also reduces the need for secondary counter-screens and allows for rapid identification of ‘true’ hits by including mechanistically validated compound controls that identify non-specific bioactivity and assay interference. Finally, use of fluorescent cell barcoding (FCB)15 for the multiplexed analysis of multiple biological samples16 reduces liquid handling error and consumption of costly fluorophore-conjugated antibodies.
PPAPs have antitumor,5a,17 antimicrobial,5d,18 and immunosuppressive activities,5l,5p,19 and their proposed molecular targets include histone acetyltransferase (HAT) enzymes (e.g. PCAF and CBP/P300)5d,5f,20 and calcineurin.5p However, recent studies suggest that many of these bioactivities, identified with in vitro assays at low-to-mid micromolar concentrations, may be the result of assay interference rather than true target inhibition.21 Herein, MAP is used to distinguish genuine activity from assay interference for a diverse collection of SASs produced largely from formic acid-mediated rearrangements (Figure 1c). Previously, we showed that this scalable, flow cytometry-based phenotypic screening system can be applied to fractionated metabolites from extracts for natural product discovery, which is known as multiplexed activity metabolomics (MAM).22
Results and Discussion
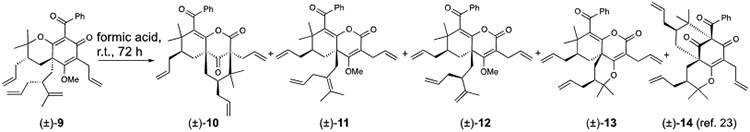
SASs are produced by unique rearrangements in formic acid.
At the outset of our investigation, we discovered that formic acid-promoted rearrangement of pyranodienone substrate (±)-923 led to production of the fused lactones (±)-10, (±)-11, (±)-12, (±)-13, and (±)-14 (Table 1, entry 1), four of which had notable activity (vide infra). Table 1 shows a selection of conditions from an extensive reaction screen to optimize product selectivity for active compounds. All rearrangement products (10-14) deriving from (±)-9 could be obtained in formic acid over 72 h (Table 1, entry 1). After extensive experimentation, we discovered that treatment of (±)-9 with formic acid at 80 °C for 1 h selectively provided the fused lactone (±)-11 in 33% yield without significant formation of byproducts (Table 1, entry 2). Rearrangement of (±)-9 did not proceed in neat TFA but worked well in TFA mixtures with formic acid (Table 1, entries 3-4), suggesting that formic acid may be important for rearrangement. Unexpectedly, lactone (±)-12, the most active compound in this study (IC50 = 0.95 μM, Supplementary Figure S9), could be selectively obtained in 39% yield by reaction of (±)-9 with TFA (20 equiv.) in cyclohexane (Table 1, entry 5), in which case the reaction could be facilitated by trace amounts of water at elevated temperatures. However, all other solvents investigated with TFA and formic acid including ether, acetonitrile, acetic acid, DMF, water, and CH2Cl2 did not provide improved yields of rearrangement products. The diversity of products (10-14) obtained via formic acid-mediated rearrangement of (±)-9 provided insight into the proposed mechanism (Figure 2a). C-cyclization and oxonium-induced ring opening of the pyran to intermediate 15 is supported by the isolation of type A scaffold (±)-14 from the reaction mixture. We propose that addition of formic acid to the strained bridgehead ketone of 15 may lead to formation of hemiketal 16.8i,10a,10c Following cyclization to the bridged oxetane 17, Grob-type fragmentation24 may provide product 12. A concerted or stepwise Grob-type fragmentation process is supported by DFT calculations (see Supplementary S2b). Lactone 12 may also be an intermediate to (±)-10, (±)-11, and (±)-13. The 2-oxo-tetrahydro-2H-chromene core present in 10-13 is also found in PPAP natural products (e.g. theoliolides A and B; mahureones A-E) and a synthetic derivative (Figure 2b).6a,b,d
Table 1.
Select conditions from a Brønsted acid screen for rearrangement of (+)-18 to the bioactive SAS (–)-19.
 | ||||
|---|---|---|---|---|
| Entry | Brønsted Acid | Solvent | Temp/Time | Yield[a] |
| 1 | formic acid[b] | neat | r.t., 72 h | 12% (±)-10 + 5% (±)-11 + 14% (±)-12 11% (±)-13 + 17% (±)-14 |
| 2 | formic acid[b] | neat | 80 °C, 1 h | 33% (±)-11 |
| 3 | formic acid[b]/TFA (5:1) | neat | r.t., 72 h | 18% (±)-10 + 17% (±)-11 + 11% (±)-13 + 10% (±)-14 |
| 4 | formic acid[b]/TFA (1:1) | neat | 10 °C, 72 h | 11% (±)-10 + 21% (±)-11 + 8% (±)-14 |
| 5 | TFA (20 equiv) | CyH | 60 °C, 24 h | 39% (±)-12 |
Isolated yields after purification by silica gel chromatography.
98% formic acid.
Figure 2.

a) Proposed mechanism for rearrangement of (±)-9 in formic acid. b) PPAP natural products and a reported synthetic derivative that possess the 2-oxo-tetrahydro-2H-chromene core found in scaffolds 10, 11, 12, and 13.
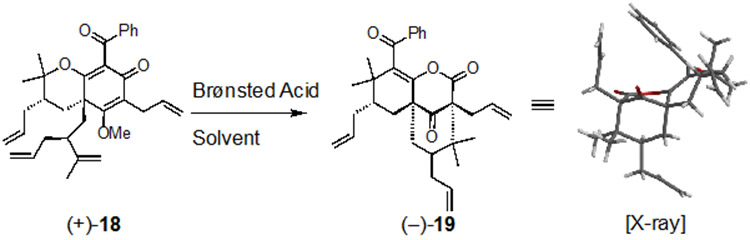
We also found that (+)-18,23 a diastereomer of 9, underwent a similar rearrangement in formic acid providing the bioactive[3.3.1]-bicyclic lactone (−)-19 (a diastereomer of 10) in 9% yield (Table 2, entry 1); its relative stereochemistry was unambiguously confirmed by X-ray crystal structure analysis.16,25 Relative to its diastereomer 9, the product diversity resulting from rearrangement of 18 is significantly decreased. Based on our proposed mechanism, it was not surprising that the rearrangement did not proceed in TFA or HCl (Table 2, entries 4 and 6). Mixtures of formic acid in TFA (1:1) provided SAS (−)-19 in similar yields (Table 2, entries 2 and 3), strongly supporting that formic acid may facilitate its formation. Surprisingly, lactone (−)-19 was obtained in 16% yield from TFA in wet HFIP at 60 °C, in which case the reaction may be facilitated by water at elevated temperatures. We have previously noted that cyclizations of compounds 9 and 18 to [3.3.1]-bicyclic scaffolds do not proceed in dry TFA.23 However, such cyclizations could be achieved in the presence of 1% water which may serve as an appropriate nucleophile for hemiketal formation or contribute to enhanced stability of cationic intermediates leading to these products. The results described in Tables 1 and 2 are in agreement with our proposed mechanism and are supported by stereochemical and DFT-optimized, orbital-alignment analysis (Supplementary Figures S1-S4).16
Table 2.
Select conditions from a Brønsted acid screen for rearrangement of (+)-18 to the bioactive SAS (–)-19.[a]
 | ||||
|---|---|---|---|---|
| Entry | Brønsted Acid | Solvent | Temp/Time | Yield[b] |
| 1 | formic acid[c] | neat | 75 °C, 5.5 h | 9 % |
| 2 | TFA/formic acid[c] (1:1) | neat, | 65 °C, 3 h | 10 % |
| 3 | TFA/formic acid[c] (1:1) | neat | r.t., 4 d | 11 % |
| 4 | TFA | neat | r.t., 8 h | not isolated[d] |
| 5 | TFA (9 equiv) | HFIP[e] | 60 °C, 8 h | 16 % |
| 6 | HCl (4 M) | 1,4-dioxane | r.t., 3.5 h | decomp |
Type B scaffold (+)-36 and O-cyclization product (+)-38 were also produced (see Supporting Information, page S10).
Isolated yields after purification.
98 % formic acid.
Rearrangment not observed.
0.015 M.
Cyclization of substrates lacking a C3-allyl group.
We also investigated formic acid-mediated rearrangements of the corresponding desallyl, dearomatized substrates meso-20a, (±)-20b, and meso-20c to determine how the C3-allyl group affects cyclization (Figure 3).23 Diastereomer meso-20a rearranged to bicyclic lactone (±)-21 in 38% yield (Figure 3a) through a mechanism similar to that described in Figure 2a; its structure and relative stereochemistry was unambiguously confirmed by X-ray crystal structure analysis.16,25 Formic acid-mediated cyclizations of (±)-20b and meso-20c resulted in the type B scaffolds (±)-22, (±)-23, and (±)-24 (Figures 3b and c). Notably, formic acid provided the highest yield of the less thermodynamically stable 7-epi isomer (±)-23.16 Formation of the latter compound may be suppressed by steric interactions in the transition state resulting from an axially-orientated allyl group directed on the concave face of the molecule (Supplementary Figure S4). Relative to the compounds shown in Tables 1 and 2, the absence of a C3-allyl group (Figure 3) may reduce the transition state energy for C-cyclization leading to improved yields of the desallyl-PPAP analogues 21-24.
Figure 3. Cyclizations of meso-20a, (±)-20b, and meso-20c in formic acid.

O-Allyl and O-homoallyl groups facilitate rearrangement to a type A scaffold at elevated temperatures in formic acid.
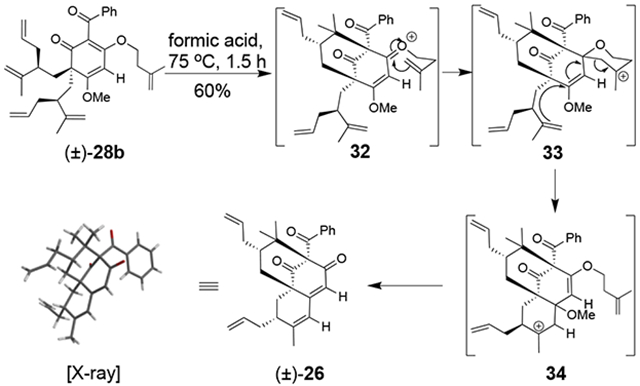
For O-alkylated derivatives of 20, the products of formic acid-promoted rearrangements were influenced by the O-alkyl group. For example, rearrangement to the type A scaffold 26 required an O-allyl or a O-homoallyl alkene (Table 3). This may be attributed to stabilization of an oxonium intermediate through π-π* donation (e.g. 32, Figure 4). Moreover, product 26 was only formed in formic acid at elevated temperatures suggesting that the solvent may also have a stabilizing effect on intermediate cations of 1,1-disubstituted olefins (e.g. 33 and 34, Figure 4).8i,10 Indeed, cyclization of the O-homoallyl substrate (+)-25 in formic acid provided (−)-26 as a single diastereomer in 42% yield (Table 3, entry 1); the relative stereochemistry was unambiguously confirmed by X-ray crystal structure analysis.16,25 The related O-homoallyl substrate (±)-28b led to improved rearrangement yields (60%, Table 3, entry 6). Following protonation of 28b, alkene π-π* donation may stabilize intermediate 32, thereby enabling Prins cyclization to cation 33 (Figure 4).26 In this scenario, premature hydrolysis of the O-alkyl group appears to be prevented allowing for alkene addition to form the proposed carbocation 34, and isomerization of the α-stereocenter results in a more sterically favored conformation by directing the allyl group to the convex face of the molecule. Loss of methanol and the pendant O-isopentenyl group would then afford the type A scaffold 26. In contrast to the rearrangement of 28b in formic acid, we found that TFA led to production of the type B products (±)-22 and (±)-23 in 62% yield (1:1.8 d.r. as determined by 1H NMR analysis, Table 3, entry 4). This result supports our hypothesis that formic acid may have a stabilizing effect on the charged intermediates leading to rearrangement product 26 (Figure 4).8i,10
Table 3.
Cyclization of O-alkylated, dearomatized acylphloroglucinols.
| Entry | Substrate | Conditions[a] | Product | Yield[b] | |
|---|---|---|---|---|---|
| 1 |  |
(+)-25 | formic acid, 75 °C, 1 h | 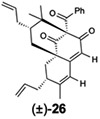 |
42 |
| 2 | 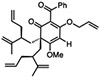 |
(+)-27 | formic acid, 75 “C, 45 min | (±)-26 | 16 |
| 3 | 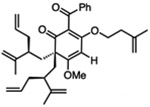 |
(±)-28b | formic acid, 75 °C, 1.5 h | (±)-26 | 60 |
| 4 | 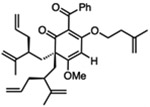 |
(±)-28b | TFA, 75 °C, 1.5 h | 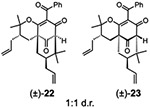 |
62 |
| 5 | 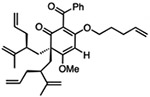 |
(±)-29 | formic acid, 75 °C. 3 h |
(±)-22 + (±)-23 1.8:1 d.r. |
34 |
| 6 | 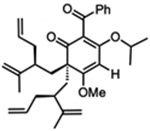 |
(±)-30 | formic acid, 75 °C, 45 min | (±)-26 | 10 |
| 7 | 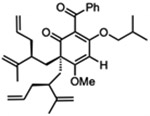 |
(±)-31 | formic acid, 75 °C, 45 min |
(±)-22 + (±)-23 1:1 d.r. |
8 |
98% formic acid.
Isolated yields after purification by silica gel chromatography.
Figure 4. Proposed mechanism for formic acid rearrangement of (±)-28b to type A scaffold (±)-26.
When O-allyl substrate (±)-27 was heated in formic acid, (±)-26 was only produced in 16% yield along with undesired O-cyclization products (Table 3, entry 2). Based on the proposed mechanism, a lower rearrangement yield was expected as the terminal alkene of 27 may be less electron donating to the resulting oxonium intermediate relative to the 1,1-disubstituted alkene in 28b. In comparison, the O-pentenyl substrate (±)-29 only provided type B scaffolds (Table 3, entry 5). Substrates (±)-30 and (±)-31 (Table 3, entries 6 and 7) with O-alkyl groups that lack allyl or homoallyl alkenes resulted in lower yields of (±)-26 and largely afforded undesired type B and O-cyclization products. Since 28b provided the best yield of rearrangement product 26, formic acid-promoted cyclizations of its diastereomers (meso-28a and meso-28c) were also investigated (Figure 5). Type A product (±)-26 was obtained in 40% yield from meso-28a by a mechanism similar to that proposed in Figure 4.16 Cyclization of meso-28c provided the type B scaffold (±)-24 along with the oxycyclization product (±)-35. It was not surprising that rearrangement to 26 did not occur for the case of meso-28c as the transition state would experience unfavorable steric interactions from the axially-oriented C7-allyl group directed on its concave face (as shown for the formation of (±)-10 in Supplementary Figure S4b).16 Taken together, these transformations resulted in diverse SASs with unexplored biological activities through novel cyclization and rearrangement pathways.
Figure 5. Cyclizations of meso-28a and meso-28c in formic acid.

MAP captures known mechanisms of model compounds.
Due to the interesting activities of isogarcinol (2) and garcinol (4), the diverse SASs from this study merited a broad and unbiased investigation of their potential biological activities.8i,16,23 We utilized MAP to perform comparative assays of these compounds in the context of reported activities for 2 and 4. MAP is multiplexed in two distinct ways: 1) multiple biological readouts are assessed simultaneously at single-cell resolution using panels of fluorophore-conjugated antibodies, and 2) the assay utilizes fluorescent cell barcoding (FCB) to enable robust large-scale screening of multiple treatment conditions, including different compounds, doses, or timepoints. For biological evaluation of the novel SASs produced, the primary panel included markers for apoptosis and homeostasis, such as cleaved caspase 3 (CCasp3),27 DNA damage (γH2AX),28 ERK/mTOR signaling [phopsho-S6, (pS6)],29 and cell cycle progression (DNA content and phospho-histone H3).30 To address one of the most frequently reported mechanisms proposed for PPAP bioactivity, we also assessed HAT inhibition using acetyl histone-specific antibodies against H3K9Ac and H3K18Ac as representative substrates of PCAF and CBP/P300, respectively, in combination with a readout for total histone H3 content.31 Unless otherwise stated, compounds were tested in Jurkat cells (human T-cell leukemia) for 16 h at both 1 and 10 μM to establish dose-dependent bioactivity and prioritize compounds for EC50 analysis.
Eight control compounds were included in the screening panel alongside the synthetic SAS library to provide internal biological validation of the assay. Four of these controls were selected to maximally perturb the chosen activity markers. Additional controls included two PPAPs (isogarcinol (2) and garcinol (4), Figure 1a) for comparison to the structurally related SASs and two synthetic HAT inhibitors (C64632 and A48533). Phenotypes induced by these control compounds (Figure 6) were consistent with their established mechanistic targets and uses as chemical probes. For example, staurosporine effectively induced apoptosis in ~90% of the cells, etoposide increased relative γH2AX via induction of double-stranded breaks, rapamycin reduced S6 phosphorylation via inhibition of mTOR, and nocodazole led to an accumulation of cells in G2/M phase.
Figure 6. Multiplexed Bioactivity Assessment of the SAS library with Reference Compounds.
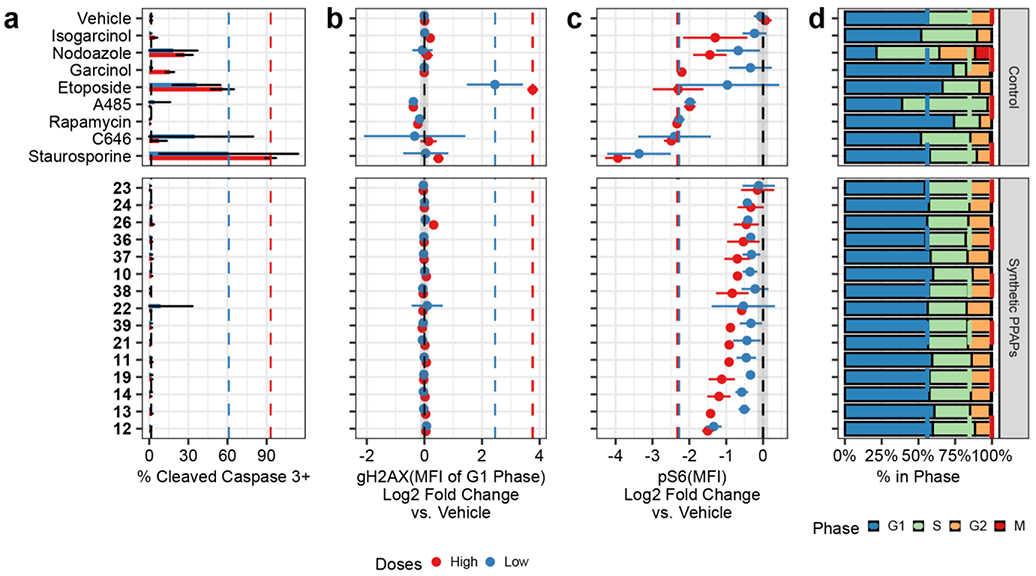

Jurkat cells were treated for 16 h at two doses (high - 10 μM, low - 1 μM), with the exception of rapamycin (10 nM and 1 nM) and staurosporine (1 μM and 100 nM). Each panel represents an average of 3 replicates of a single marker [channel]: a) Casp3 as a marker of apoptosis [PE]; b) γH2AX as a marker of DNA damage [PerCPCy5.5], c) pS6 [Ser235/236] as a marker of mTOR/ERK signaling [Alexa Fluor 647]; and d) Cell cycle distribution, assessed using biaxial gating on DNA content [YO-PRO-1] and phospho-histone H3 (Ser28) [PE-Cy7]. Median fluorescence intensity (MFI) of γH2AX and pS6 were normalized to the average of the two vehicle-treated wells. The error bars represent the 95% confidence interval for each point estimate. The dashed lines in panels A-C show the vehicle-treated wells (black) and the reference compounds for each channel at the high (red) and low (blue) doses. e) SASs evaluated.
Although markers were selected to evaluate orthogonal biological pathways, the phenotypes induced by control compounds revealed both known and novel cross-talk that could otherwise confound interpretation of individual markers if assessed in isolation. For example, DNA replication stress is known to induce a DNA damage response in cycling cells, which leads to a relative increase in median γH2AX for both S and G2 phase cells over G1 phase cells.34 This crosstalk is evident in cells treated with compounds that perturb cell cycle progression. Specifically, nocodazole increased γH2AX, while G1 arresting compounds paradoxically decrease γH2AX relative to vehicle-treated cells (Supplementary Figure S8). To account for this phenomenon, γH2AX intensity was normalized to vehicle treated cells in each phase of the cell cycle. This result suggested that etoposide, consistent with its reported mechanism, was the only DNA-damaging compound in our library at the tested doses, while the SASs did not exhibit notable DNA damage activity.
SASs induce selective suppression of pS6.
Consistent with previous reports,5a,17 garcinol (4) exhibited modest antiproliferative cytotoxicity at 10 μM: ~20% of cells were positive for the apoptosis marker CCasp3 (Figure 6a) and the fraction of cells in G1 phase increased (Figure 6d), which is consistent with cell cycle arrest. In addition, isogarcinol (2) and garcinol (4) also decreased basal pS6 at Ser235/236, which indicates inhibition of mTOR/ERK signaling.29 These results are consistent with their reported immunosuppressive activities.5l Although the bioactivities exhibited by 2 and 4 were dose-dependent (Figure 6), the perturbation was not specific to any one phenotypic marker, suggesting that their bioactivities are relatively non-specific. Five rearrangement products (11-14 and 19) selectively exhibited the pS6 suppression phenotype with minimal cytotoxicity relative to garcinol (4). This phenotype was not specific to the Jurkat model system, as the same effect was observed in KG-1 (acute myeloid leukemia) cells treated in a similar manner (Supplementary Figure S10). The antibody used the pS6 readout specific for Ser235/236 phosphorylation, which is downstream of both the mitogenic mTOR and ERK pathways (Supplementary Figure S10). To determine whether mTOR or ERK was responsible for the observed phenotype, a subset of the SAS library was assayed for suppression of pS6 at Ser240/244, a site that is downstream of mTOR, but not ERK.29b This secondary panel revealed pS6 was suppressed at both Ser235/236 and Ser240/244, suggesting that suppression of mTOR signaling is a component of the phenotype induced by SASs (Figure 6 and Supplementary Figure S10).
The compounds depicted in Figures 6a-d are ordered from least to most active in the measured suppression of pS6 (Ser235/236) at 10 μM. The compound library was not designed for an exhaustive study of SAS structure-activity relationships (SAR), but some general SAR trends are readily evident from MAP data. Notably, compounds 12 [IC50 = 0.95 μM] and 13 [IC50 = 1.0 μM] bearing the 2-oxo-tetrahydro-2H-chromene core (cf. Figure 2) were more active than isogarcinol (2) [IC50 = 12 μM] but lack the [3.3.1]-bicyclic core that is characteristic of many PPAPs (Supplementary Figure S9). Four of the five active compounds (11-13 and 19, Figure 6e) have fused 6-6 lactone ring systems, and compound 14 is a type A scaffold. More importantly, all of the SASs in this study (11-14 and 19) resulted from the same formic acid-promoted rearrangement pathway described in Figure 2a originating from pyranodienones 9 and 18.
In contrast to isogarcinol (2) and garcinol (4), SASs lack catecholic hydroxy groups, which have been linked to their cytotoxicity.5f Additionally, catechols are known “pan-assay interference” (PAINS)35 functional groups that contribute to non-specific hits in screening assays. Consistent with this model, 2 and 4 exhibited cytotoxicity relative to vehicle-treated wells (Figure 6a), while the catechol-lacking SASs showed minimal cytotoxicity at the tested doses.
PPAPs are nonspecific HAT modulators.
Garcinol (4) and C646 were originally identified as HAT inhibitors in in vitro assays using recombinant HATs.36 However, recent reports suggest that the activity of these compounds may represent assay interference rather than targeted inhibition.21 To test this hypothesis, cells exposed to compounds were also stained for antibodies against acetyl-histone (H3K9Ac, H3K18Ac) and total histone (H3) content (Figure 7). Examining each marker separately, we found that isogarcinol (2), garcinol (4), C646, and A485 suppressed H3K9 and H3K18 acetyl content while the SASs did not appear to influence histone acetylation. However, the cytotoxic compounds etoposide and staurosporine, which are not thought to act directly on HATs, suppressed histone acetylation to an equal or greater extent compared to C646 and 4. In an attempt to account for this discrepancy, cells were stained for total histone H3, which should not be influenced by HAT inhibition. When H3K9Ac/H3K18Ac is compared to total histone H3 content, we observe the loss of acetyl-histone H3 accompanied by a proportional loss of total histone H3. One exception to this pattern is A485, a selective p300/CBP inhibitor33 that suppresses H3K18Ac without significantly affecting total H3 or H3K9Ac content (Figure 7). The correlation between acetyl and total histone for cytotoxic compounds is consistent with a non-specific loss of histone epitopes rather than a specific loss of one histone mark and emphasizes the importance of considering total histone content when assessing specific histone eptiopes.37 Though the mechanism of this non-specific histone loss is unknown, the observed loss of total and acetyl-histone content with cytotoxic compound treatment suggests that apoptosis leads to loss of histones,38 or otherwise alters the accessibility of histone epitopes in a manner that confounds antibody-based detection of histone marks. This leads us to conclude, in agreement with recent reports,21 that the tested PPAPs and SASs, including isogarcinol (2) and garcinol (4), are not direct inhibitors of HAT-induced acetylation at concentrations below 10 μM.
Figure 7. Assessment of histone acetylation in treated Jurkat cells. Jurkat cell treated as in Figure 1, stained with antibodies against total H3 (PE), H3K9Ac (Alexa Fluor 488), or H3K18Ac (Alexa Fluor 647).
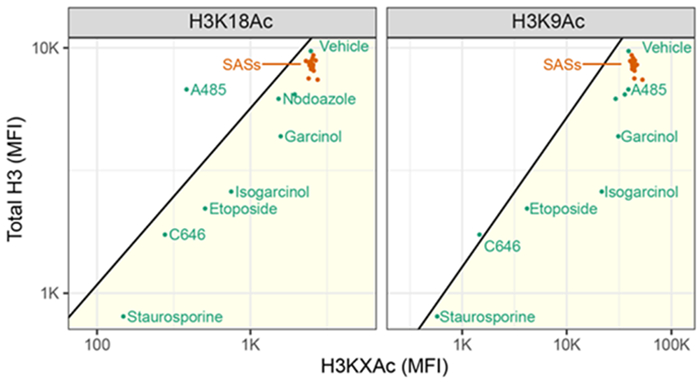
Median fluorescence intensity (MFI) for each channel was calculated and averaged over three replicates which were plotted as points, with reference compounds in green and SASs in orange. Compounds below the diagonal exhibited a loss of total H3 greater than or equal to the loss of acetyl-histone H3, indicative of non-specific histone loss. See the Supplementary Information (Supplementary Figure S7).
Conclusion
Formic acid-promoted rearrangements of dearomatized acylphloroglucinol substrates resulted in a diverse collection of synthetic acylphloroglucinol scaffolds (SASs) which were evaluated for activities in diverse biological pathways using multiplexed activity profiling (MAP). MAP evaluates several markers of apoptosis to distinguish genuine activity from assay interference that is commonly associated with PPAPs. Rearrangements described in this work were facilitated by formic acid which has an inherent ability to stabilize carbocations in solution and form adducts with strained carbonyls.8i,10 Formic acid-mediated rearrangements of 9, 18, and meso-20c produced a diverse group of SASs, five of which (11-14 and 19) displayed notable biological activity. The absence of a C3-allyl group reduced product diversity but improved rearrangement and cyclization yields. Furthermore, we discovered that a pendant O-homoallyl group was critical for rearrangement to the type A scaffold (±)-26 which may result from oxonium stabilization by alkene π-π* donation. The most active analogues in this study derived from a single formic acid-promoted rearrangement pathway, and two of these compounds (12 and 13) were more active than the natural product isogarcinol (Figure 6).23
MAP was effectively used as an unbiased platform to assess the bioactivities of SASs which revealed that 11-14 and 19 suppressed pS6 (Ser235/236) with minimal cytotoxicity relative to garcinol (4). As pathways leading to pS6 are central to the adaptive immune response, metabolism, and the mTOR signaling pathway, further study of SASs in target-identification campaigns and as immunosuppressants is warranted. We found that isogarcinol (2) and garcinol (4) resulted in non-specific inhibition of HAT activity in vivo, but did induce non-specific changes in measured histone and acetyl-histone content, which could account for previous reports of HAT activity. Our study highlights the combination of controlled synthetic diversity with multiplexed activity analysis across a broad panel of cell status markers to facilitate thorough exploration of the potential chemical activity space around both privileged and novel scaffolds. Further studies to employ diversity-oriented synthesis/reaction discovery approaches to produce novel scaffolds for evaluation by MAP are currently in progress and will be reported in due course.
Supplementary Material
Acknowledgements
We thank the National Institutes of Health for research (R35 GM118173, U01TR002625, R01 GM092218, and R01 CA226833) and training support (T32 GM065086, T32 GM007347, F30 CA236131). Work at the BU-CMD was supported by NIH R24 Grant GM-111625. We thank Dr. Jeffrey Bacon (Boston University) for X-ray crystal structure analyses, NMR (CHE-0619339) and MS (CHE-0443618) facilities at Boston University are supported by the NSF. Flow Cytometry experiments were performed in the VMC Flow Cytometry Shared Resource. The VMC Flow Cytometry Shared Resource is supported by the Vanderbilt Ingram Cancer Center (P30 CA68485) and the Vanderbilt Digestive Disease Research Center (DK058404). A485 was supplied by the Structural Genomics Consortium under an Open Science Trust Agreement: http://www.thesgc.org.
Footnotes
Supporting information for this article is given via a link at the end of the document.
References
- [1].For a recent review on PPAPs, see: Yang XW, Grossman RB, Xu G, Chem. Rev 2018, 118, 3508–3558. [DOI] [PubMed] [Google Scholar]
- [2].a) For select chemical syntheses of PPAPs, see: Simpkins NS, Taylor JD, Weller MD, Hayes CJ, Synlett 2010, 639–643 [Google Scholar]; b) Garnsey MR, Lim D, Yost JM, Coltart DM, Org. Lett 2010, 12, 5234–5237 [DOI] [PubMed] [Google Scholar]; c) Biber N, Möws K, Plietker B, Nat. Chem 2011, 3, 938–942 [DOI] [PubMed] [Google Scholar]; d) Uwamori M, Saito A, Nakada M, J. Org. Chem 2012, 77, 5098–5107 [DOI] [PubMed] [Google Scholar]; e) Sparling BA, Moebius DC, Shair MD, J. Am. Chem. Soc 2013, 135, 644–647 [DOI] [PubMed] [Google Scholar]; f) Lindermayr K, Plietker B, Angew. Chem. Int. Ed 2013, 52, 12183–12186 [DOI] [PubMed] [Google Scholar]; g) Horeischi F, Biber N, Plietker B, J. Am. Chem. Soc 2014, 136, 4026–4030. [DOI] [PubMed] [Google Scholar]; h) Bellavance G, Barriault L, Angew. Chem. Int. Ed 2014, 53, 6701–6704 [DOI] [PubMed] [Google Scholar]; i) Ting CP, Maimone TJ, J. Am. Chem. Soc 2015, 137, 10516–10519 [DOI] [PubMed] [Google Scholar]; j) Bellavance G, Barriault L, J. Org. Chem 2018, 83, 7215–7230 [DOI] [PubMed] [Google Scholar]; k) Wang L, Sun L, Wang X, Wu R, Zhou H, Zheng C, Xu H, Org. Lett 2019, 21, 8075–8079 [DOI] [PubMed] [Google Scholar]; l) Shen X, Ting CP, Xu G, Maimone TJ, Nat. Commun 2020, 11, 508–515; [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].a) Dal Piaz F, Tosco A, Eletto D, Piccinelli AL, Moltedo O, Franceschelli S, Sbardella G, Remondelli P, Rastrelli L, Vesci L, Pisano C, De Tommasi N, ChemBioChem 2010, 11, 818–827 [DOI] [PubMed] [Google Scholar]; b) Zhu L, Qi J, Chiao CY, Zhang Q, Porco JA Jr., Faller DV, Dai Y, Int. J. Oncol 2014, 45, 2128–2136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].a) Ravindra KC, Selvi BR, Arif M, Reddy BA, Thanuja GR, Agrawal S, Pradhan SK, Nagashayana N, Dasgupta D, Kundu TK, J. Biol. Chem 2009, 284, 24453–24464 [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Sell TS, Belkacemi T, Flockerzi V, Beck A, Sci. Rep 2014, 4, 7500–7511 [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Zhang H, Zhang D-D, Lao Y-Z, Fu W-W, Liang S, Yuan Q-H, Yang L, Xu H-X, J. Nat. Prod 2014, 77, 1700–1707. [DOI] [PubMed] [Google Scholar]
- [5].a) For discussions on the bioactivity of isogarcinol (2) and related compounds, see: Matsumoto K, Akao Y et al. , Biol. Pharm. Bull 2003, 26, 569–571 [DOI] [PubMed] [Google Scholar]; b) Hong J, Kwon SJ et al. , Free Radic. Biol. Med 2007, 42, 1211–1221 [DOI] [PubMed] [Google Scholar]; c) Sarli V, Giannis A, Chem. Biol 2007, 14, 605–606 [DOI] [PubMed] [Google Scholar]; d) Mantelingu K, Reddy BA et al. , Chem. Biol 2007, 14, 645–657 [DOI] [PubMed] [Google Scholar]; e) Marti G, Eparvier V et al. , Phytochemistry 2009, 70, 75–85 [DOI] [PubMed] [Google Scholar]; f) Arif M, Pradhan SK et al. , J. Med. Chem 2009, 52, 267–277 [DOI] [PubMed] [Google Scholar]; g) Marti G, Eparvier V, Litaudon M, Grellier P, Guéritte F, Molecules 2010, 15, 7106–7114 [DOI] [PMC free article] [PubMed] [Google Scholar]; h) Tian Z, Shen J et al. , PloS One 2011, 6, e21370–e21375 [DOI] [PMC free article] [PubMed] [Google Scholar]; i) Baliga MS, Bhat HP, Pai RJ, Boloor R, Palatty PL, Food Res. Int. 2011, 44, 1790–1799 [Google Scholar]; j) Tchakam PD, Lunga PK et al. , BMC Complementent Altern. Med. 2012, 12, 136–143 [DOI] [PMC free article] [PubMed] [Google Scholar]; k) Cao S, Cen J, CN103275048A, June 18, 2013 [Google Scholar]; l) Cen J, Shi M, Yang Y et al. , PloS one 2013, 8, e66503–e66512 [DOI] [PMC free article] [PubMed] [Google Scholar]; m) Kuete V, Tchakam PD et al. , Phytomedicine 2013, 20, 528–536 [DOI] [PubMed] [Google Scholar]; n) Fu Y, Zhou H, Wang M, Cen J, Wei Q, J. Agric. Food Chem 2014, 62, 4127–4134 [DOI] [PubMed] [Google Scholar]; o) Hongxi X, Kaikai S, Zhijie D, Hong Z, CN103536588A, October 25, 2013 [Google Scholar]; p) Cen J, Wang M et al. , Biochimie 2015, 111, 119–124 [DOI] [PubMed] [Google Scholar]; q) Park G, Tan J et al. , J. Biol. Chem 2016, 291, 3531–3540 [DOI] [PMC free article] [PubMed] [Google Scholar]; r) Bridi H, de Carvalho Meirelles G, von Poser GL, Phytochemistry 2018, 155, 203–232. [DOI] [PubMed] [Google Scholar]
- [6].a) Möws K, Schürmann M, Preut H, Plietker B, Acta Cryst. 2009, E65, o2139 [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Sparling BA, Tucker JK, Moebius DC, Shair MD, Org. Lett 2015, 17, 3398–3401 [DOI] [PubMed] [Google Scholar]; c) Yang X-W, Li M-M, et al. , J. Nat. Prod 2015, 78, 885–895 [DOI] [PubMed] [Google Scholar]; d) Nguyen L-T, Lai NTDT et al. , Tetrahedron Lett. 2016, 57, 2737–2741. [Google Scholar]
- [7].a) For discussions on PPAP biosynthesis, see: Rao AR, Venkatswamy G, Pendse D, Tetrahedron Lett. 1980, 21, 1975–1978 [Google Scholar]; b) Cuesta-Rubio O, Velez-Castro H, Frontana-Uribe BA, Cárdenas J, Phytochemistry 2001, 57, 279–283 [DOI] [PubMed] [Google Scholar]; c) Raikar SB, Nuhant P, Delpech B, Marazano C, Eur. J. Org. Chem 2008, 1358–1369 [DOI] [PubMed] [Google Scholar]; d) Zhou K, Wunsch C, Dai J, Li SM, Org. Lett 2017, 19, 388–391. [DOI] [PubMed] [Google Scholar]
- [8].a) For biomimetic syntheses of PPAPs, see: Tsukano C, Siegel DR, Danishefsky SJ, Angew. Chem. Int. Ed 2007, 46, 8840–8844 [DOI] [PubMed] [Google Scholar]; b) Qi J, Porco JA Jr., J. Am. Chem. Soc 2007, 129, 12682–12683 [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Zhang Q, Mitasev B, Qi J, Porco JA Jr., J. Am. Chem. Soc 2010, 132, 14212–14215 [DOI] [PMC free article] [PubMed] [Google Scholar]; d) Qi J, Beeler AB, Zhang Q, Porco JA Jr., J. Am. Chem. Soc 2010, 132, 13642–13644 [DOI] [PMC free article] [PubMed] [Google Scholar]; e) Zhang Q, Porco JA Jr., Org. Lett 2012, 14, 1796–1799 [DOI] [PMC free article] [PubMed] [Google Scholar]; f) George JH, Hesse MD, Baldwin JE, Adlington RM, Org. Lett 2010, 12, 3532–3535 [DOI] [PubMed] [Google Scholar]; g) Simpkins NS, Weller MD, Tetrahedron Lett. 2010, 51, 4823–4826 [Google Scholar]; h) Pepper HP, Lam HC, Bloch WM, George JH, Org. Lett 2012, 14, 5162–5164 [DOI] [PubMed] [Google Scholar]; i) Boyce JH, Porco JA Jr., Angew. Chem. Int. Ed 2014, 53, 7832–7837 [DOI] [PMC free article] [PubMed] [Google Scholar]; j) Pepper HP, Tulip SJ, Nakano Y, George JH, J. Org. Chem 2014, 79, 2564–2573 [DOI] [PubMed] [Google Scholar]; k) Wen S, Boyce JH, Kandappa SK, Sivaguru J, Porco JA Jr., J. Am. Chem. Soc 2019, 141, 11315–11321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].a) For biomimetic cyclizations of dearomatized acylphloroglucinols, see: Mitasev B, Porco JA Jr., Org. Lett 2009, 11, 2285–2288 [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Couladouros EA, Dakanali M, Demadis KD, Vidali VP, Org. Lett 2009, 11, 4430–4433. [DOI] [PubMed] [Google Scholar]
- [10].a) For discussions on the properties of formic acid, see: Schaefer JP, J. Am. Chem. Soc 1960, 82, 4091–4094 [Google Scholar]; b) Gibson HW, Chem. Rev 1969, 69, 673–692 [Google Scholar]; c) Pawar DM, Cain-Davis D, Noe EA, J. Org. Chem 2007, 72, 2003–2007. [DOI] [PubMed] [Google Scholar]
- [11].a) For reviews on diversity-oriented synthesis (DOS), see: Schrieber SL, Science 2000, 287, 1964–1969 [DOI] [PubMed] [Google Scholar]; b) Galloway WRJD, Isidro-Llobet A, Spring DR, Nat. Commun 2010, 1, 1–13 [DOI] [PubMed] [Google Scholar]; c) O’Connor CJ, Beckmann HSG, Spring DR, Chem. Soc. Rev 2012, 41, 4444–4456 [DOI] [PubMed] [Google Scholar]; d) Yi S, Varun BV, Choi Y, Park SB, Front. Chem 2018, 6, 1–8 [DOI] [PMC free article] [PubMed] [Google Scholar]; e) Pavlinov I, Gerlach EM, Aldrich LN, Org. Biomol. Chem 2019, 17, 1608–1623 [DOI] [PubMed] [Google Scholar]; f) Gerry CJ, Schreiber SL, Curr. Opin. Chem. Biol 2020, 56, 1–9. [DOI] [PubMed] [Google Scholar]
- [12].For an alternative study investigating single-output activities of PPAP analogs, see: Lin X, Tian D, Fu Y, Li Y, Huang L, Gu W, Song J, Li Y, Ben-David Y, Wen M, Eur. J. Med. Chem 2019, 162, 765–780. [DOI] [PubMed] [Google Scholar]
- [13].a) Feng Y, Mitchison TJ, Bender A, Young DW, Tallarico JA, Nat. Rev. Drug Discovery 2009, 8, 567–578 [DOI] [PubMed] [Google Scholar]; b) Kurita KL, Glassey E, Linington RG, Proc. Natl. Acad. Sci. U.S.A 2015, 112, 11999–12004 [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Melillo B, Zoller J, Hua BK, Verho O, Borghs JC, Nelson SD, Maetani M, Wawer MJ, Clemons PA, Schreiber SL, J. Am. Chem. Soc 2018, 140, 11784–11790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Khurana V, Tardiff DF, Chung CY, Lindquist S, Nat. Rev. Neurol 2015, 11, 339–350. [DOI] [PubMed] [Google Scholar]
- [15].Krutzik PO, Nolan GP, Nat. Methods 2006, 3, 361–368. [DOI] [PubMed] [Google Scholar]
- [16].Please see the Supporting Information for complete experimental details and mechanistic studies.
- [17].Pan MH, Chang WL, Lin-Shiau SY, Ho CT, Lin JK, Agr J. Food Chem. 2001, 49, 1464–1474. [DOI] [PubMed] [Google Scholar]
- [18].Jeffers V, Gao H et al. , Antimicrob. Agents Chemother 2016, 60, 2164–2170 [DOI] [PMC free article] [PubMed] [Google Scholar]; Guttroff C, Baykal A, Wang H, Popella P, Kraus F, Biber N, Krauss S, Götz F, Plietker B, Angew. Chem. Int. Ed 2017, 56, 15852–15856. [DOI] [PubMed] [Google Scholar]
- [19].a) Koeberle A, Northoff H, Werz O, Biochem. Pharmacol 2009, 77, 1513–1521Y. [DOI] [PubMed] [Google Scholar]; b) Fu Y, Zhou H, Wang M, Cen J, Wei Q, J. Agric. Food Chem 2014, 62, 4127–4134. [DOI] [PubMed] [Google Scholar]
- [20].a) Nishino T, Wang C, Mochizuki-Kashio M, Osawa M, Nakauchi H, Iwama A, PloS One 2011, 6, e24298–e24306 [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Collins HM, Abdelghany MK et al. , BMC Cancer 2013, 13, 37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Dahlin JL, Nelson KM et al. , Nat. Commun 2017, 8, 1527–1540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Earl DC, Ferrell PB Jr., Leelatian N, Froese JT, Reisman BJ, Irish JM, Bachmann BO, Nat. Commun 2018, 9, 39–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Boyce JH, Eschenbrenner-Lux V, Porco JA Jr., J. Am. Chem. Soc 2016, 138, 14789–14797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].a) Grob CA, Baumann W, Helv. Chim. Acta 1955, 38, 594–610 [Google Scholar]; b) Prantz K, Mulzer J, Chem. Rev 2010, 110, 3741–3766. [DOI] [PubMed] [Google Scholar]
- [25].CCDC 1909310, 1909311, and 1909309, (19, 21, and 26,) contain the supplementary crystallographic data for this paper. These data can be obtained free of charge from The Cambridge Crystallographic Data Centre.
- [26].Snider BB, Rodini DJ, Van Straten J, J. Am. Chem. Soc 1980, 102, 5872–5880. [Google Scholar]
- [27].Nicholson DW, Ali A, Thornberry NA, et al. , Nature 1995, 376, 37–43. [DOI] [PubMed] [Google Scholar]
- [28].Rogakou EP, Nieves-Neira W, Boon C, Pommier Y, Bonner WM, J. Biol. Chem 2000, 275, 9390–9395. [DOI] [PubMed] [Google Scholar]
- [29].Ferrari S, Bandi HR, Hofsteenge J, Bussian BM, Thomas G, J. Biol. Chem 1991, 266, 22770–22775 [PubMed] [Google Scholar]; Pende M, Um SH, Mieulet V, Sticker M, Goss VL, Mestan J, Mueller M, Fumagalli S, Kozma SC, Thomas G, Mol. Cell. Biol 2004, 24, 3112–3124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Goto H, Tomono Y, Ajiro K, Kosako H, Fujita M, Sakurai M, Okawa K, Iwamatsu A, Okigaki T, Takahashi T, Inagaki M, J. Biol. Chem 1999, 274, 25543–25549. [DOI] [PubMed] [Google Scholar]
- [31].Jin Q, Yu LR, Wang L, Zhang Z, Kasper LH, Lee JE, Wang C, Brindle PK, Dent SY, Ge K, EMBO J. 2011, 30, 249–262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Bowers EM, Yan G et al. , Chem. Biol 2010, 17, 471–482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Lasko LM, Jakob CG, Edalji RP et al. , Nature 2017, 550, 128–132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].MacPhail SH, Banáth JP, Yu Y, Chu E, Olive PL, Radiat. Res 2003, 159, 759–767. [DOI] [PubMed] [Google Scholar]
- [35].Baell JB, Holloway GA, J. Med. Chem 2010, 53, 2719–2740. [DOI] [PubMed] [Google Scholar]
- [36].Balasubramanyam K, Altaf M, Varier RA, Swaminathan V, Ravindran A, Sadhale PP, Kundu TK, J. Biol. Chem 2004, 279, 33716–33726. [DOI] [PubMed] [Google Scholar]
- [37].Cheung P, Vallania F et al. , Cell 2018, 173, 1385–1397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Wu D, Ingram A et al. , J. Biol. Chem 2002, 277, 12001–12008. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.