

Abstract
Obesity is associated with systemic inflammation due to macrophage accumulation in adipose tissue (AT). AT macrophages are, therefore, a target for therapeutics to modulate inflammation and prevent comorbidities. Because inflammatory processes have pleiotropic effects throughout the body and are intertwined with metabolic axes, systemic anti-inflammatory therapies are often harmful. We report that targeting AT macrophages using dextran nanocarriers radically alters the pharmacology of anti-inflammatory glucocorticoids, uncoupling the metabolic axis in obese mice. Following a single treatment, expression of inflammatory mediators and markers of inflammatory macrophages decreased with a nearly 20-fold higher potency compared with free drug. As a result, long-term treatment resulted in potent fat mobilization, AT reduction, weight loss, improved glucose tolerance, and altered AT gene expression profiles that led to elevated liver stress. Two weeks after treatment ceased, gene expression of inflammatory mediators in AT remained lower than obese controls, while gene expression related to metabolic function improved. These data demonstrate that nanocarriers show potential for amelioration of obesity-related AT inflammation and metabolic dysfunction, highlighting an important opportunity for nanomedicine to impact chronic metabolic disorders with complex and poorly understood etiology.
Introduction
Obesity is a global health concern due to its causative role in numerous disorders, including high blood pressure, type 2 diabetes, heart disease, stroke, and cancer.1–2 The global prevalence has steadily risen for more than 4 decades, and currently more than 39% of adults are overweight and 13% are obese.3 Adiposity and body mass index are both correlated with the number of macrophages present in adipose tissue (AT), accounting for about 40% of the total AT cell population of obese individuals, compared with 10% in the lean AT state.4 Macrophages exhibit a spectrum of phenotypes, termed polarizations, with radically dissimilar gene expression profiles and behaviours, most broadly categorized as the classically activated M1 state or the alternatively activated M2 state. Macrophages in the obese state are primarily in the M1 state and induce local inflammation in AT, resulting from the secretion of inflammatory molecules such as tumor necrosis factor alpha (TNFα,) interleukin 6 (IL-6), and nitric oxide (NO).5 This localized inflammation correlates with low-grade systemic inflammation, which is thought to be the causative pathologic change that drives the diverse classes of comorbidities.6–7
Over the past two decades, there has been intensive development of pharmacological strategies for clinical treatment of obesity and consequent comorbidities, but there are no broadly effective strategies to date. Given the established role of AT macrophages in disease onset and progression, these cell types are an attractive target for interventions, especially considering their phenotypic plasticity that allows shifts in polarization through exogenous signals.8 However, systemic administration of drugs that target underlying inflammatory processes such as nonsteroidal anti-inflammatory drugs (NSAIDs) and glucocorticoids have not elicited beneficial clinical outcomes due to undesired side effects of gluconeogenesis, increased triglycerides, aberrant fat deposition, immune suppression, and loss of bone density.9 Moreover, systemic modulation of inflammation in general is not desirable because the signaling processes involved exhibit pleiotropic biological functions throughout the body and in different cell types.10–19
Unlike classical small molecule drugs, nanomedicines provide unique pharmacological profiles with tunable pharmacokinetics, biodistribution, and off-target side effects.20 We previously developed a polysaccharide-based nanomedicine to specifically deliver anti-inflammatory small molecules to macrophages by attaching drugs to high molecular weight dextran, which functions as a nanocarrier and binds selectively to the mannose receptor and scavenger receptors on macrophages.21 Dextran has demonstrated selective uptake through receptor-ligand recognition and subsequent endocytosis and drug release by macrophage mannose receptors and class A scavenger receptors, receptors which are upregulated in the classically activated M1 state.22–25 Previously, we reported that 63% of an intraperitoneally (IP) injected dose of dextran distributed to visceral AT (VAT) 24 h after injection and that more than 90% of the targeted cells were CD11b+ myeloid cells. When bound to the anti-inflammatory glucocorticoid receptor (GR) agonist dexamethasone (Dex), the nanocarrier-Dex (ND) conjugate significantly reduced expression of inflammatory markers in AT.21 Here we report that ND drastically alters the pharmacological profile of free dexamethasone (FD) by uncoupling the macrophage processes from GR processes in other cell types. We evaluated the short-term and long-term effects of this therapeutic strategy in a diet induced obesity (DIO) mouse model using transcriptomics, metabolic profiling, and histopathology. We observed that DIO mice treated with a low dose of macrophage-targeted ND resulted in drastically enhanced drug potency. While the altered pharmacokinetics of Dex resulted in adverse liver effects despite the preferential uptake of ND by VAT macrophages, ND significantly improved macrophage and AT function, reduced inflammation, ameliorated metabolic dysfunction, and resulted in dramatic weight loss due to mobilization of AT fat.
Results and discussion
ND targets adipose macrophages and reduces inflammatory gene expression in vitro.
Figure 1a depicts the molecular structure of ND. Dextran is a biocompatible and water-soluble branched polysaccharide with numerous hydroxyl groups. Gel permeation chromatography (GPC) analysis (Figure 1b) reveals the size distribution of ND used in this work with a peak at around 165 min, corresponding to a hydrodynamic diameter of 22 nm based on elution times of size standards. The drug release half-time (t1/2) in phosphate buffered saline (PBS) was approximately 48 h (Figure 1c), which is appropriate for targeted drug release in visceral AT macrophages, which reaches equilibrium in biodistribution after several hours.21 In vitro cultures of macrophages were treated with ND, using either RAW 264.7 monocyte cell lines or primary macrophage cells isolated from bone marrow of lean C57BL/6 mice. These un-polarized M0 cells were treated with lipopolysaccharide (LPS) and/or interferon gamma (IFN-γ) for 24 h to induce polarization to the inflammatory M1 state and treated with ND (500 nM equivalent of FD) to analyze the impact on gene expression. Figure 1d shows example immunofluorescent stains demonstrating expression of the inflammation mediator inducible nitric oxide synthase (NOS2) in M1 cells, with significant reduction after ND treatment. Figure 1e shows the expression of 22 genes related to inflammation measured by qRT-PCR (Fluidigm) after treatment with ND or FD. M1 cells exhibited increased expression of inflammatory genes,26 including TNFα, IL-6, NOS2, and IFN-γ, which were reduced by treatment with either FD or ND. Treatments across a 100-fold range of concentrations were similarly potent for both ND and FD, which is expected due to the equivalent bioavailability of the two drug formulations in an in vitro culture model. Notably, the expression profiles of M1 cells were similar overall after treatment with FD or ND. These similarities validate the capacity for efficient release of functional Dex from the nanocarrier. M0 macrophages did not exhibit substantial change in expression of most inflammatory genes upon treatment with FD or ND, showing selectivity for the inflammatory M1 phenotype. Due to their hyper-phagocytic nature, partly through upregulation of cell surface receptors, M1 macrophages may be able to take dextran up more efficiently than cells in the M0 or M2 states. The data presented here is supported by our in vitro work demonstrating more efficient uptake of dextran in M1 macrophages compared with M2 macrophages.21
Figure 1. Nanocarrier-dexamethasone (ND) characterization, targeting, and impact on inflammatory gene expression in macrophage (MΦ) cells.
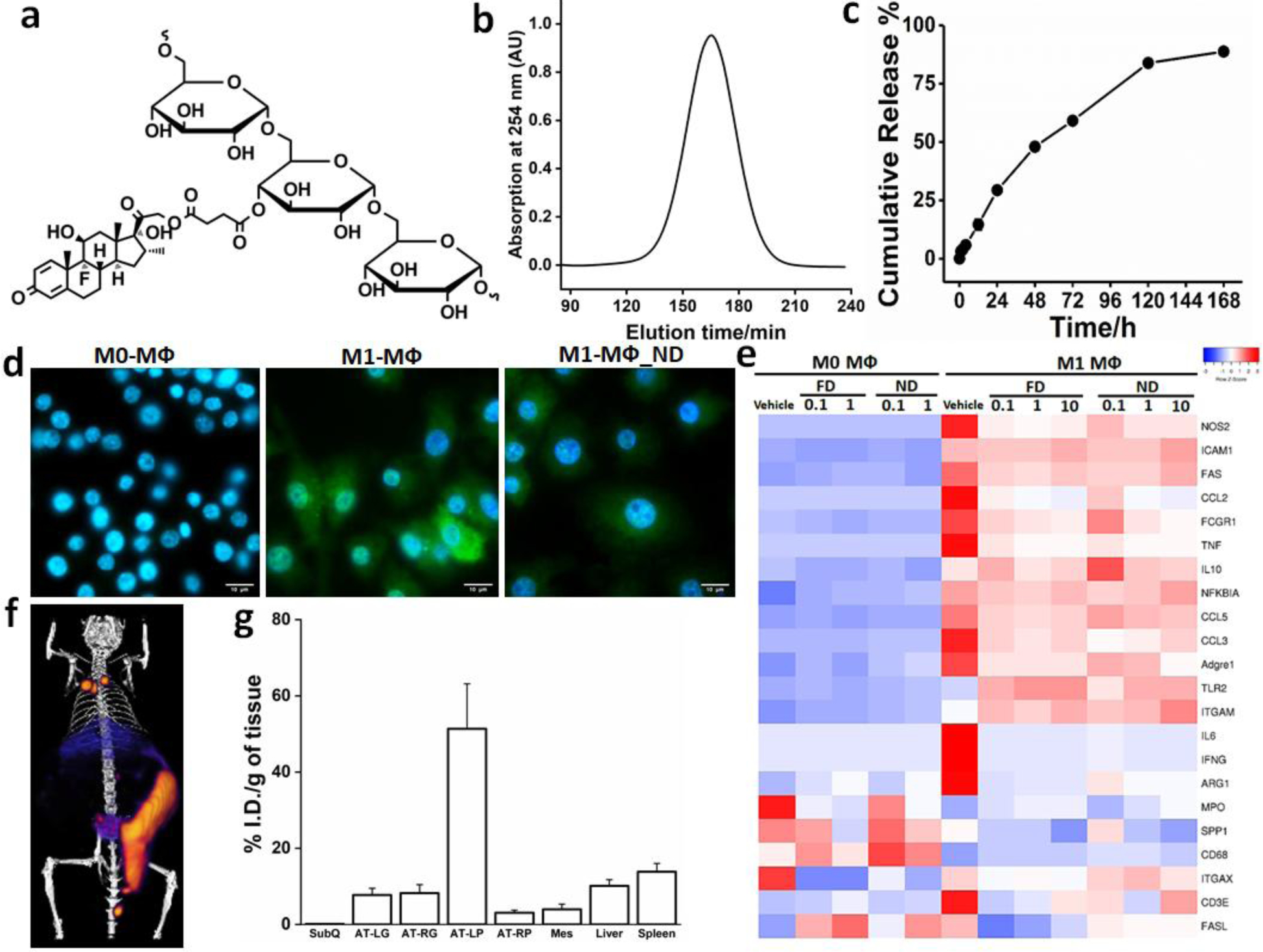
(a) Schematic structure of ND, showing dexamethasone drug conjugated to dextran through a succinate linker. (b) GPC chromatogram with a peak at approximately 165 min, corresponding to a diameter of around 22 nm. (c) Drug release kinetics of ND in PBS; (d) Fluorescence microscopy images of RAW 264.7 cells showing M0 cells, M1 cells, and M1 cells treated with ND, each stained with nuclear dye (Hoechst, blue) and NOS2 (green fluorescence), showing reduction in green fluorescence intensity with ND treatment. (e) Heatmap showing mRNA expression of cultured primary macrophages isolated from bone marrow of lean C57BL/6 mice. Values represent the log2 ratio over housekeeping genes with scaling to obtain relative expression among samples within each gene. Each row represents a specific gene of interest and each column represents the mean of each treatment/control group. Both unstimulated M0 MΦs and M1 MΦs (stimulated with LPS+IFNγ for 24 h) were treated with vehicle (media only), FD, or ND. Doses were 0.1 μM or 1 μM for M0 cells and 0.1 μM, 1 μM, and 10 μM for M1 cells. Treatment of M1 MΦs with ND or FD led to a down-regulation of many genes involved with inflammation. However, the treatments did not exhibit substantial impact on M0 macrophages. (f) Representative PET/CT image of DIO mouse showing efficient targeting of AT by 64Cu-ND. (g) Gamma well counts of 64Cu-ND in subcutaneous (SubQ), left gonadal (AT-LG), right gonadal (AT-RG), left perirenal (AT-LP), right perirenal (AT-RP), and mesenteric (Mes) adipose depots, liver, and spleen. Maximum targeting was to AT-LP. Data are shown as mean ± SE.
Our previous work also demonstrated that IP-injected nanocarrier conjugates selectively targeted macrophages in the VAT in obese mice, with greater ND retention likely due to the greater population of M1 macrophages in VAT of obese animals (as well as larger adipose depots).21 This was verified with the dextran preparation used in this study, wherein 51.3% of the injected dose remained in VAT, resulting in the highest concentration in the perirenal depot on the same side as the injection (left) based on PET/CT and postmortem gamma well counting of radiolabeled conjugates (Figure 1f & 1g). The concentration in the left perirenal depot was 5 times higher than in the liver, the primary site of distribution following intravenous administration.
Acute ND treatment mobilizes stored fat and alters lipid metabolism in obese mice.
Based on the potent down-regulation of inflammatory genes in cultured primary macrophages, we tested the acute effects of ND and FD on physiologic and gene expression outcomes in DIO mice. With the expectation that the dextran nanocarrier would drastically alter the tissue and cell-type distribution of Dex, ND was administered IP to DIO mice in one of three doses (0.1, 0.7, or 5 mg/kg). Twenty-four hours later, blood was collected, the animals were sacrificed, and gene expression analysis and histology were performed on the resected organs. Control groups included DIO mice treated with vehicle (saline) as well as lean mice. ND-treated mice exhibited a dose-dependent increase in circulating triglycerides (Figure 2a), reflecting altered liver lipid metabolism, and non-esterified fatty acids (NEFA) (Figure 2b), reflecting mobilization of stored lipids. Intriguingly, this was not observed for an equivalent dose of FD at the highest dose (5 mg/kg), suggesting that targeting of AT-macrophages resulted in efficient and dose-dependent lipolysis from AT. Apparently, effects of the FD are either substantially lower in potency or systemic activation of GR by Dex has opposing effects on AT lipids. While Dex is known to stimulate lipolysis in isolated adipocytes and AT, systemic Dex in the obese state has the opposite effect, resulting in further fat deposition, likely due to an increase in appetite mediated by the central nervous system.9,27 While increased NEFA were observed concomitantly with a dose-dependent increase in triglycerides, these results were not associated with clear changes in the expression of lipid-metabolizing genes, with an increase in triglyceride synthesis likely due to overwhelming the liver with AT lipolytic products. This suggests that ND selectively isolates the effects of Dex to AT, uncoupling the counteracting processes in distal tissues.
Figure 2. Acute dose of ND alters lipid metabolism and mobilizes stored fat in DIO mice.
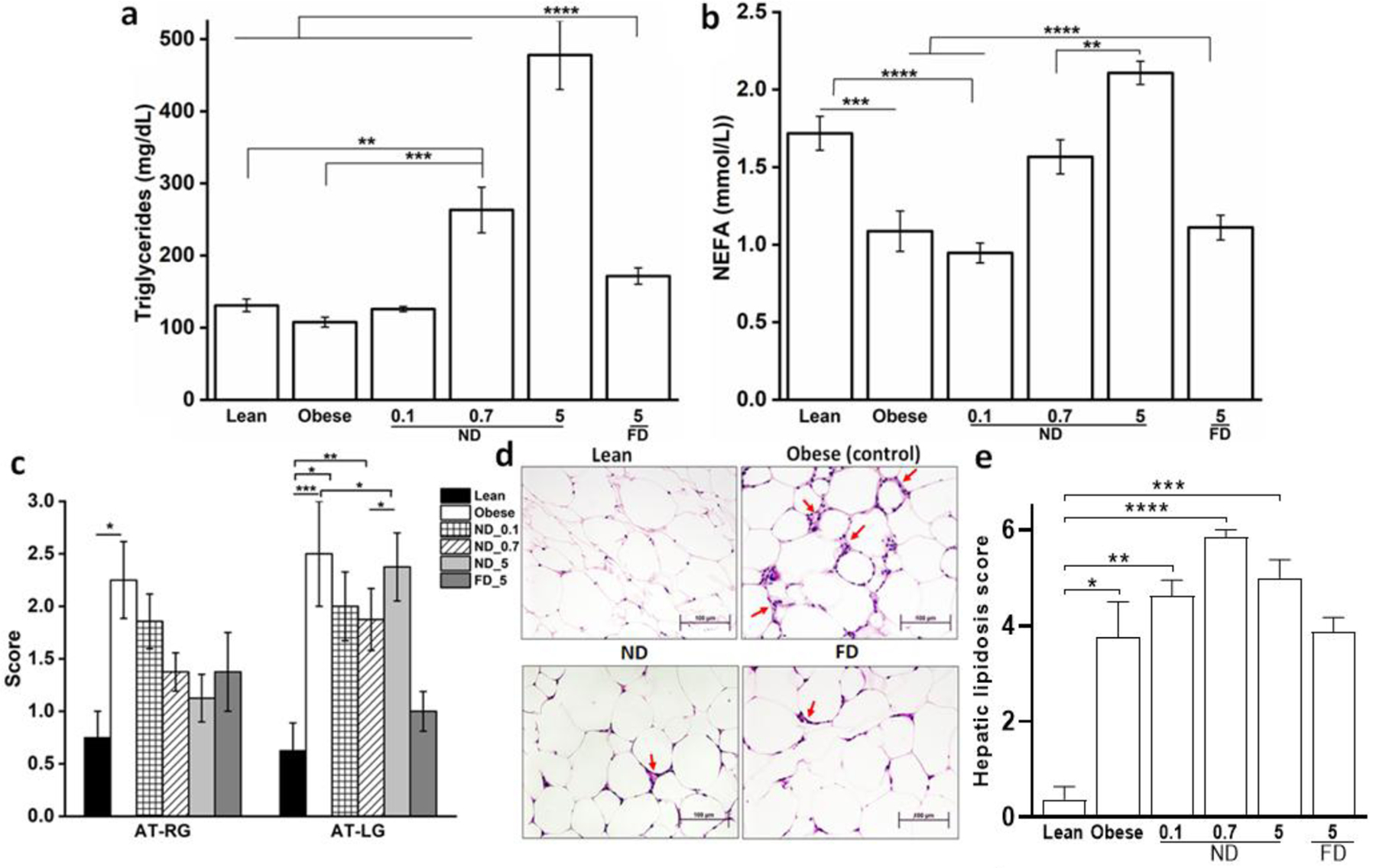
(a) Circulating triglycerides and (b) non-esterified fatty acids (NEFA) in lean mice and obese DIO mice 24 h after indicated treatments of intraperitoneal (IP) injection of ND (0.1, 0.7, or 5 mg/kg) or FD (5 mg/kg). Both triglycerides and NEFA increased in a dose dependent manner with ND treatment. (c) Right and left gonadal AT steatitis scores and (e) hepatic steatosis (lipidosis) scores 24 h after IP saline treatment of lean or obese mice, or of obese mice with ND (0.1, 0.7, or 5 mg/kg), or FD (5 mg/kg). (d) Representative histological images of gonadal AT from lean mice or obese mice (DIO) 24 h after IP saline, or obese mice treated with ND (0.7 mg/kg) or FD (5 mg/kg), showing reduced crown-like structures (red arrows) in response to ND and FD. Data are shown as mean ± SE, n=7–8; *P<0.05; **P<0.01; ***P<0.001; ****P<0.0001; groups were compared using ANOVA with Tukey’s posthoc or Kruskal-Wallis with Dunn’s posthoc.
AT in the obese state is characterized histologically by the presence of crown-like structures (CLS) in which M1 macrophages surround apoptotic adipocytes.28 Treatment with both ND and FD led to a substantial reduction of CLS in gonadal AT (Figure 2c) compared to obese controls, with histological scores of gonadal steatitis, indicating the density of interstitial aggregates of histiocytes or lymphocytes (scoring systems used for histopathological endpoints adapted from29,30 and described in Supplementary information 1.1), improving for both FD and ND treatments (Figure 2d). However, trends were the opposite in liver, in which hepatic steatosis scores increased overall for ND treatment, whereas FD treatment had no effect. This is consistent with the mobilization of stored lipids for the ND group and lack of distal effects from AT in the FD group. These results suggest that at the same dose, ND exhibits a markedly different impact on AT lipid stores compared with FD to shift metabolism toward lipolysis and lipid mobilization.
Acute ND treatment reduces expression of inflammatory genes in adipose tissue.
A characteristic feature of obesity and associated comorbidities is increased AT inflammation mediated by expression of cytokines such as IL-6, TNFα, and MCP-1.31 Fluidigm qRT-PCR was used to measure the expression of 69 genes associated with inflammation, macrophage polarization, lipid metabolism, glucose metabolism, energy balance, and apoptosis in AT depots (left and right gonadal, left and right perirenal, mesenteric, and subcutaneous) and liver tissue. Figure 3a shows results for the left gonadal AT, wherein a single administration of ND resulted in a dose-dependent alteration in the expression of many genes associated with inflammation and metabolism compared with vehicle-treated DIO mice. Similar responses were observed in other adipose depots and liver. As expected, the genes analyzed exhibited major differences in expression in obese and lean AT, as demonstrated in the volcano plot in Figure 3b, reflecting their divergent relationship in the two physiological states in the tissues. In the obese state, there was a marked upregulation of markers of inflammation (TNFα) and lymphocyte infiltration (CD68, ITGAX/CD11c, CD8a, CCR5, CD64/FCGR1, ADGRE1/F4/80, CCL3/MIP-1a), and decreased expression of several metabolic markers (ACACA, UCP3, PCK1, and IRS2). These measurements are consistent with the altered processing of glucose, insulin, and AT lipids that is widely observed with obesity, and is associated with infiltration of inflammatory macrophages.
Figure 3. Acute dose of ND reduces inflammatory gene expression profile in adipose tissue of DIO mice.
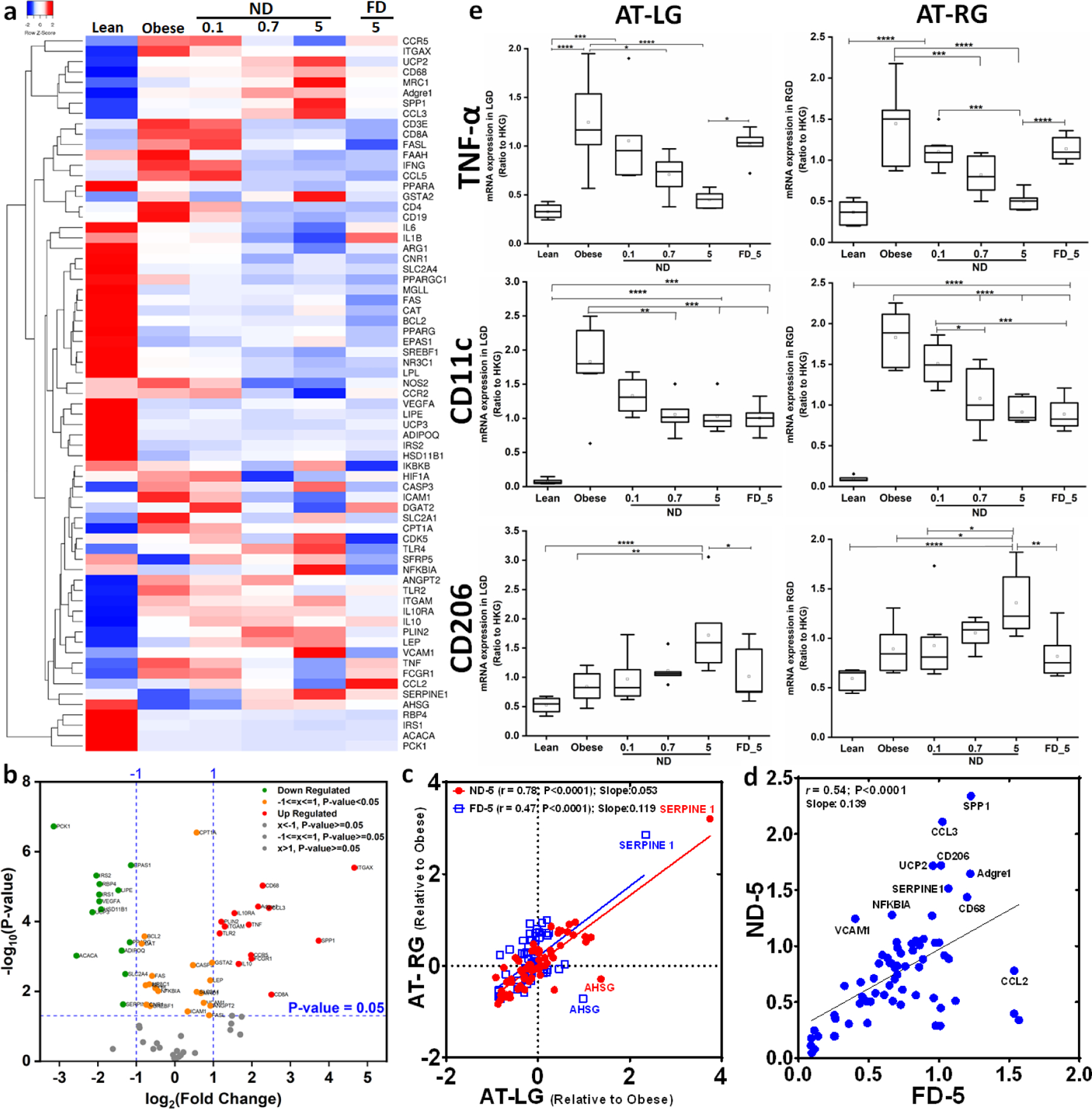
(a) Heatmap of mRNA expression from left gonadal adipose depot of lean mice and DIO mice treated with saline, ND (0.1, 0.7, or 5 mg/kg) or FD (5 mg/kg). Values represent the log2 ratio over housekeeping genes with scaling to obtain relative expression among samples within each gene. Each row represents a specific gene of interest and each column represents mean of each treatment group. The 69 genes in rows are clustered based on correlation of fold change values. All up-regulated genes are colored red, and down-regulated genes are colored blue. (b) Volcano plot of obese (control) vs. lean (control) showing differential expression of genes in obese condition indicating metabolic impairment. (c) Spearman correlations of mRNA expression in right vs. left gonadal AT of DIO mice 24 h after treatment with equivalent dose of ND or FD (5 mg/kg). The values are relative to the value of the obese control group. (d) Gene expression in AT of DIO mice after treatment with ND vs. FD at equivalent dose (5 mg/kg). (e) Dose-dependent down-regulation of TNFα and CD11c mRNA expression and dose-dependent up-regulation of CD206 (mannose receptor; M2 macrophage marker) mRNA expression after treatment with ND. *P<0.05; **P<0.01; ***P<0.001; ****P<0.0001, groups were compared using ANOVA with Tukey’s posthoc; n=7–8; AT-LG = left gonadal adipose tissue; AT-RG = right gonadal adipose tissue.
Consistent with in vitro studies of cultured M1 macrophages (Figure 1), there was a reduction in numerous pro-inflammatory macrophage markers (IL-6, FASL, CCR5, CCL2, NOS2, IFNG, FCGR1), with little change in expression of the pan-monocyte lineage marker CD68. However, unlike in vitro studies in which SPP1 and CCL3 decreased and TLR2 increased, the opposite effects occurred in vivo, possibly indicating contributions from other cell types. Also, unlike in vitro studies in which ICAM1, NFKBIA, and CCL5 exhibited little change, there was a substantial decrease in ICAM1 and CCL5, and an increase in NFKBIA in vivo.
For both lean and obese mice, our analysis showed that although IP injections were administered on the left side of the abdomen, mRNA expression patterns of the left and right gonadal AT depots were linearly correlated with each other for each dose of ND and FD (P < 0.0001) with only a few exceptions (Supplementary Figure S1a). Figure 3c shows the fold-change in gene expression for each of the genes relative to control for gonadal fat depots on the left and right sides of the body following a left-side IP injection of ND and FD (5 mg/kg). Notice that the slope of each curve is less than 1, indicating a more potent effect on the left side compared with the right, but still a significant correlation in expression level change, with higher correlation for ND (Spearman r = 0.78) compared with FD (r = 0.47). Data are shown for all doses (0.1, 0.7 and 5 mg/kg P < 0.0001) in Supplementary Figure S1b.
Nanocarrier delivery amplifies anti-inflammatory potency of Dex in adipose.
Although in vitro responses to ND and FD were similar, the responses in vivo were drastically dissimilar due to the unique biodistribution and pharmacokinetic profiles of the two materials. Figure 3d shows the gene expression profile of left gonadal AT after treatment with FD or ND at the same dose. The plot shows that a large number of genes are correlated across the two treatment conditions (r=0.54 for all genes together), while a substantial number of outliers demonstrate key differences between the two therapies. Numerous markers of inflammation were significantly reduced by ND that did not substantially change for FD (TNFα, IL-6, IL-1B, CCR5, CCR2, CCL2, FCGR1, NOS2), while the M2 macrophage marker MRC1 (CD206) increased substantially more with ND treatment. Together, these results show a net change in macrophage phenotype from the M1 to the M2 state that is much more substantial for ND compared with FD.
By comparing across the treatment doses, the markers exhibited a clear dose response with ND across a 50-fold concentration range, with important examples shown in Figure 3e. The majority of these markers, such as TNFα, showed that ND was approximately 20-fold more potent than FD against the inflammatory effects in AT, with the 5 mg/kg FD dose exhibiting similar potency as the 0.1 mg/kg ND dose across the different fat pads as well as the liver (Supplementary Figure S2). However, a subset of inflammatory genes (e.g. CD11c, Figure 3e) yielded similar potency for ND and FD, potentially indicating a disparate effect on dendritic cells compared with macrophages, as CD11c is expressed in both dendritic cells and certain macrophage subpopulations. Mannose receptor (CD206) expression increased with increasing dose of ND across all visceral AT, however it exhibited little change in liver (Figure 3e Supplementary Figure S2). Again, although IP injections were made on the left side, responses were similar in right and left AT.
Long-term ND treatment increases circulating lipids and reduces body weight.
We evaluated the long-term impact of ND on obese mice with repeated treatments. ND was administered to DIO mice every other day by IP injection at a Dex dosage of 0.7 mg/kg, a dose near the measured IC50 for most inflammatory genes after a single treatment. This treatment regimen led to a striking and steady reduction in body weight compared with the control groups while animals remained on a high-fat diet, with weight regain observed upon cessation of ND treatment after 2 wk (Figure 4a). The body weight reduction was due to a loss in fat, as reflected by a total reduction in visceral fat mass via EchoMRI (Supplementary Figure S4a) and postmortem AT weights (Supplementary Figure S4b). This weight loss was intriguing given that systemic administration of glucocorticoids such as Dex are known to cause hyperphagia, increased weight, and insulin resistance.9,32
Figure 4. Long-term treatment of DIO mice with ND reduces body weight, increases circulating triglyceride and non-esterified fatty acid (NEFA) concentrations, alters blood glucose concentrations, and improves gonadal AT mRNA expression of genes associated with glucose and lipid metabolism.
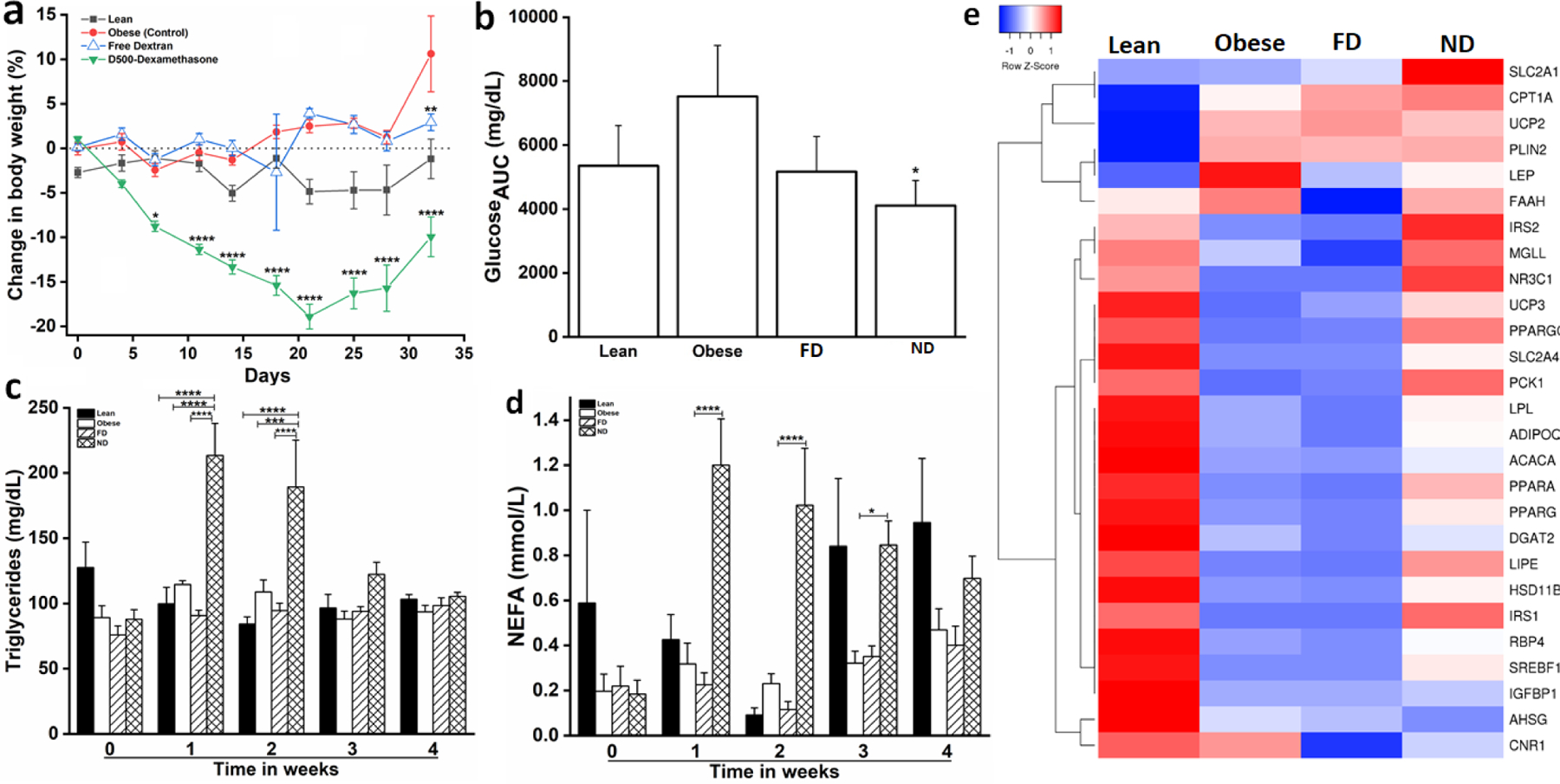
(a) Rapid decline in body weight of mice receiving intraperitoneal (IP) ND (0.7 mg/kg) injections every other day. Lean mice and DIO mice treated with saline (control) or FD maintained or gained body weight. ND treatments ceased at 2 wk of treatment, after which body weight began to increase gradually. (b) Blood glucose concentrations (area under the curve; AUC) following an insulin tolerance test 2 wk after ND (0.7 mg/kg) treatment ceased (wk 4 of study), showing improvement compared to DIO controls. (c) Serum triglycerides and (d) NEFA were greatly increased during IP ND (0.7 mg/kg) treatment, indicating enhanced lipolysis, which declined post-treatment. (e) Heatmap of left gonadal adipose depot mRNA expression of genes involved with lipid and glucose metabolism, showing improved metabolic state after ND intervention; *P<0.05; **P<0.01; ***P<0.001; ****P<0.0001; DIO mice: n= 7–8; lean mice: n=4; groups were compared using ANOVA with Tukey’s posthoc.
Taken together, this indicates that a ND nanocarrier strategy effectively decouples the anti-inflammatory actions of Dex from its metabolic effects. Insulin sensitivity was assessed during wk 4 of the study, with our results indicating that ND treatment improved insulin sensitivity in DIO mice (Figure 4b). Consistent with the acute study (Figure 2), ND treatment increased circulating NEFA (Figure 4d) and triglyceride concentrations (Figure 4c), reflecting mobilization of fat stores from AT and subsequent metabolic activity by the liver.
Not surprisingly, the significant weight loss and metabolic changes observed in ND-treated mice corresponded with dramatic changes in mRNA expression of genes related to lipid and glucose metabolism in left gonadal AT (Figure 4e), with little difference for the obese group compared to the obese group treated with FD alone. The expression of genes involved in lipid metabolism was in line with the elevation in NEFA (Figure 4d). Following ND treatment and rapid weight loss, up-regulated expression of NR3C1, MGLL, IRS1, IRS2, PCK1, PPAR-γ, PPAR-α, PPARGC1A, SREBF1, LIPE, and LPL was observed. PPAR-γ in particular is a critical regulator of macrophage alternate activation, and its down-regulation is protective of AT inflammation.33 The elevation in circulating triglycerides was not associated with drastic changes in the expression of genes involved with lipid metabolism within the liver. While the increase in lipoprotein lipase (LPL) expression in the liver is consistent with greater triglyceride clearance, the increase in circulating triglycerides was a transient effect that normalized in the 2 wk following ND removal (Figure 4c). As such, the potential effects of ND on the expression of lipid metabolizing genes on the liver remains unclear. We also observed an upregulation of genes involved in glucose homeostasis (IGFBP1, SLC2A1, SLC2A4, IRS1, IRS2). Overall, these significant changes in transcription of genes related to metabolism in gonadal AT reflects a global shift toward a phenotype that is expected to reduce obesity comorbidities.
Long-term ND treatment improves inflammatory gene expression.
Throughout the study, peritoneal macrophages were isolated by peritoneal lavage for assessment of the expression of the prototypical pro-inflammatory cytokine IL-6. IL-6 expression was increased in control and FD obese mice compared with lean mice, while ND treatment reduced IL-6 to levels comparable to the lean group (Figure 5a). The left gonadal fat depot of DIO mice similarly exhibited down-regulated expression of IL-6 mRNA after ND treatment (Figure 5b). AT derived IL-6 regulates various metabolic pathways that result in AT-specific lipid release and LPL down-regulation34 and is correlated with weight loss and improved insulin sensitivity.35 After FD treatment, mRNA expression of TNFα, CD11c, and CD206 were greater than lean controls. This was only true for TNFα expression in ND-treated mice. After ND treatment, mRNA expression of CD11c was reduced while changes in TNFα and CD206 were not statistically significant (Figure 5c–e) compared with FD. Furthermore, the percentage of M1-like CD11c+ cells in the CD11b+ fraction was lower in ND treated mice compared to DIO controls, as assessed by flow cytometry after treatment (Figure 5f). CD11c+ macrophages have previously been shown to be critical for recruiting T-cells and activating AT macrophages that contribute to insulin resistance.36 The percentage of M2-like CD206+ cells in the CD11b+ fraction of pooled gonadal and perirenal adipose depots were not significantly different among the obese, dextran, and ND treatment groups (Figure 5g).
Figure 5. Long-term ND treatment in DIO mice reduces mRNA expression of pro-inflammatory cytokines.
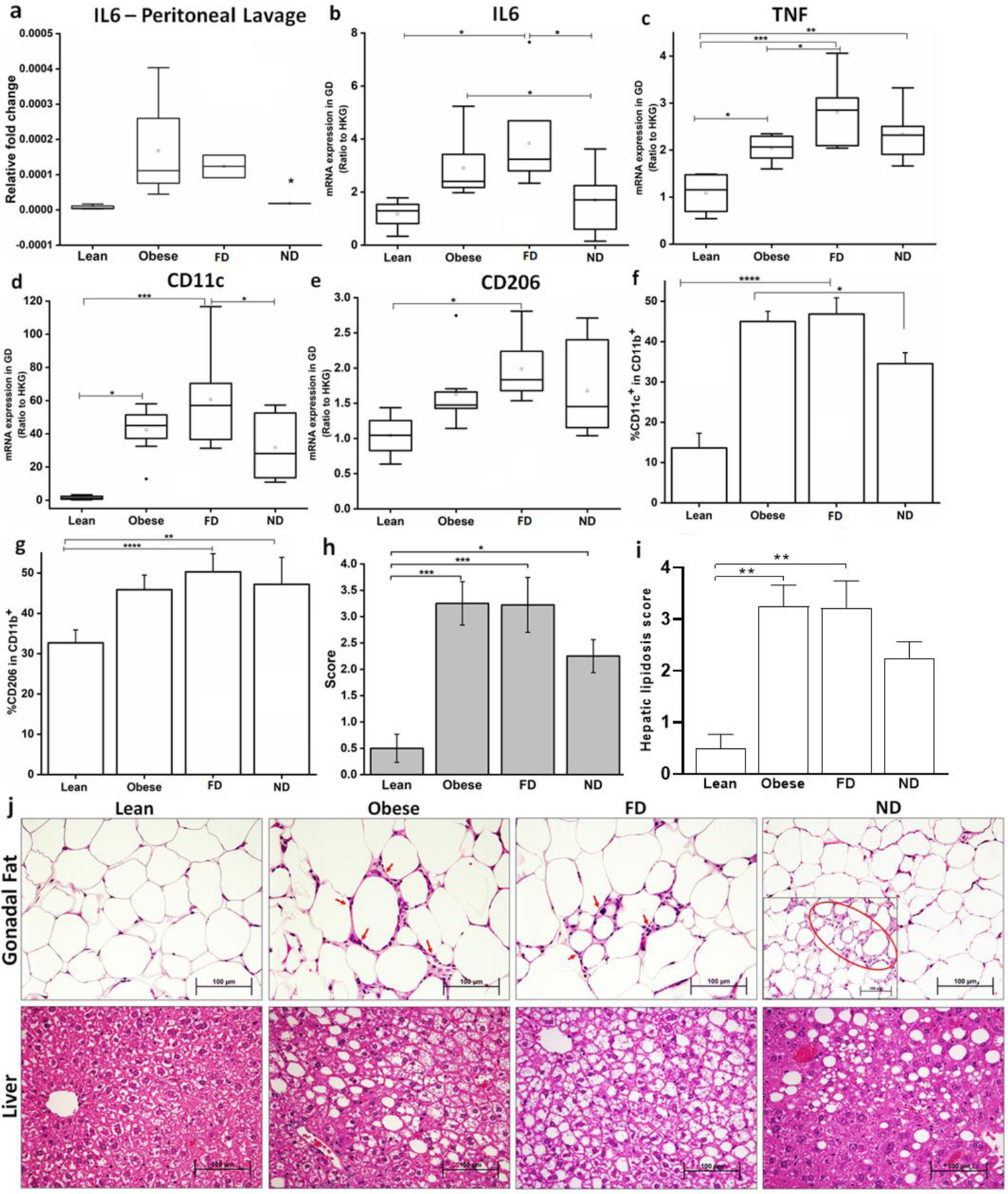
(a) Following long-term ND treatment, DIO mice showed down-regulation of IL-6 expression in macrophages isolated from peritoneal lavage. (b) IL-6 expression in gonadal adipose tissue declined compared to DIO control and FD. (c – e) Expression of CD11c and TNFα were down-regulated, while expression of CD206 (mannose receptor; M2 macrophage marker) was up-regulated in ND-treated mice; (f) CD11c+ myeloid cell populations of pooled gonadal and perirenal adipose tissue samples as measured by flow cytometry, showing a reduction in the M1 macrophage marker after ND treatment (0.7 mg/kg). (g) CD206+ myeloid cell population of pooled gonadal and perirenal adipose by flow cytometry, demonstrating no difference in the M2 macrophage marker after ND treatment (0.7 mg/kg). (h) Gonadal steatitis scores after long-term ND (0.7 mg/kg) treatment. (i) Hepatic steatosis (lipidosis) scores after long-term ND (0.7 mg/kg) treatment. (j) Representative histological micrographs of gonadal fat tissue (top panel) showing improvement in crown-like structure (CLS) density (red arrows). Inset shows part of the adipose depot showing CLS, while part of the fat pad showed absence of CLS after ND (0.7 mg/kg) treatment. The bottom panel shows representative histological micrographs of liver reflecting typical average hepatic steatosis severity scores. First panel: hepatocytes have irregular central pallor typical of hepatocytes in non-fasted rodents; second panel: large (macrovesicular) and smaller (microvesicular) vacuoles are present as well as cytoplasmic pallor in hepatocytes (far lower left); third panel: macro- and microvesicular vacuoles, typical of severe hepatic steatosis in this model; fourth panel: macrovesicular lipid vacuoles predominate affected part of liver. *P<0.05; **P<0.01; ***P<0.001; ****P<0.0001; DIO mice: n= 7–9; lean mice: n=4–8. Data are presented as mean ±SE; groups were compared using ANOVA with Tukey’s posthoc or Kruskal-Wallis with Dunn’s posthoc.
Histopathology of left gonadal AT indicated sparse improvement in the formation of CLS. The CLS clusters in the ND treated gonadal fat depot might be a requirement of significant weight loss due to AT weight reduction, requiring the reduction in cell numbers by apoptosis, although this was not specifically assessed. The liver demonstrated an improvement in the steatosis score (Figure 5h & 5i), although gene expression results indicated an overall increase in liver inflammation (TNFα, IL-1β, IL-6, VCAM1, ICAM1) (Supplementary Figure S5). Results examining markers of liver stress (ALT, AST; Supplementary Figure S5) over time indicated that ND treatment caused a large efflux of AT lipolytic products that overwhelmed the liver and induced stress, which improved following ND withdrawal and may have led to improvements in hepatic steatosis scores. These responses correlate with the increased metabolism of lipids and reduced body weight, which likely contributed to the improved insulin sensitivity observed at wk 4 (Figure 4b).
Conclusions
Macrophages are promising targets to reduce the production of inflammatory cytokines and shift AT cell phenotypes to therapeutically impact obesity and its comorbidities. The intrinsic capacity of nanomaterials to target macrophage cells and other phagocytotic cell populations provides a particularly intriguing opportunity to create high-potency interventions for obesity, as pathogenic AT and liver tissues are infiltrated with these cell types in the obese state.38–42 As we previously reported, dextran is an effective nano-sized carrier to deliver dexamethasone to macrophages, allowing tunable pharmacokinetics and biodistribution based on size with efficient solubilization and release of cargo in target tissue.43 Based on our report here, the localization of drug carrier to these cells results in a 20-fold enhancement in potency of Dex compared with free Dex toward gene expression alterations (e.g., TNFα, IL-6) that reflect a global reduction in inflammation (Figure 3a,e). We have also reported high specificity for ND uptake by AT-associated macrophages, with 5-fold greater distribution within AT in obese animals compared with liver distribution and >90% of this dose taken up by macrophages.21 Thus, while we have shown that ND results in AT macrophage-targeted improvements in lipid metabolism at a much higher potency observed by FD, the 20-fold enhancement in potency of Dex may be associated with overwhelming lipolytic products that may result in adverse outcomes in the liver despite reduced hepatic distribution of ND. We expect that this enhanced potency will translate to a higher therapeutic index in a clinical scenario that would allow a reduction in side effects at much lower doses than the 0.7 mg/kg dose tested in this study,44 as Dex alone is a broad-spectrum pharmacological agents with pleiotropic effects on adipocytes, metabolism, endocrine function and inflammation. Similar enhancements in drug potency and side effect attenuation have been observed previously for drug delivery with nanocarriers,45–46 particularly for applications in solid tumors, and our outcomes show that the effects can be extended to metabolic disorders and chronic diseases driven by targets disseminated throughout large tissues. It is further important that this nano-formulation strategy results in a complete shift in pharmacology of the drug, reflected in divergent gene expression profiles after single treatments of ND and FD (Figure 3d). The resulting improvement in AT lipid and glucose metabolism and weight in obese mice (Figure 4) are unlike those of Dex alone, which promotes central adiposity.47 We are unaware of other reports of such a drastic shift in therapeutic effect for a pharmaceutical drug due to nano-formulation, and expect this outcome to inspire the exploration of diverse drug classes for pathologies previously unexplored due to the potential for effects in off-target tissues, such as the liver. Further upcoming studies will focus on determining the dose-response relationship with liver stress, weight loss, and characterization of the specific adipose depots responsible for obesity comorbidities.48
Experimental
Synthesis and characterization of dextran-based dexamethasone conjugates
Reagents and solvents.
Dextran (500 kDa), 4-dimethylaminopyridine, ethylenediaminetetraacetic acid (EDTA), succinic anhydride, anhydrous dimethyl sulfoxide (DMSO), pyridine, N,N’-carbonyldiimidazole and triethylamine were purchased from Sigma Aldrich (St. Louis, MO, USA). Dexamethasone was obtained from Cayman Chemical Company (Ann Arbor, MI, USA) and p-SCN-Bn-NOTA was purchased from Macrocyclics, Inc. (Plano, TX, USA). Amino dextran (500 kDa) was purchased from Molecular Probes (Eugene, OR, USA). Radioactive 64Cu chloride was obtained from Washington University (St. Louis, MO, USA). All other reagents and solvents were obtained from commercial sources and used without further purification.
Synthesis of dexamethasone succinic acid (DSA).
In a reaction flask, dexamethasone (1.0 g, 2.5 mmol) and 4-dimethylaminopyridine (DMAP) were dissolved in pyridine (25 mL). Succinic anhydride (0.77 g, 7.6 mmol) was added to the mixture slowly. The mixture was stirred under a nitrogen atmosphere at room temperature for 14 h. Pyridine was then removed under reduced pressure and water (30 mL) was added to the residue. The mixture was stirred for 15 min, filtered, and washed with water several times. 1H NMR (500 MHz, DMSO-d6, ppm, 298 K): 7.27 (d, J = 10.1 Hz, 1H), 6.21 (dd, J = 10.1, 1.8 Hz, 1H), 5.99 (s, 1H), 5.40 (D, J = 4.1 Hz, 1H), 5.15 (s, 1H), 5.02 (d, J = 17.6 Hz, 1H), 4.78 (d, J = 17.6 Hz, 1H), 4.12 (m, 1H), 2.85 (m, 1H), 2.59 (m, 3H), 2.46 (s, 2H), 2.34 (m, 2H), 2.11 (m, 2H), 1.75 (m, 1H), 1.62 (m, 1H), 1.55 (t, J = Hz, 1H), 1.47 (s, 3H), 1.33 (dt, J = 12.2, 7.7 Hz, 1H), 1.05 (ddd, J = 12.1, 8.0, 3.9 Hz, 1H), 0.86 (s, 3H), 0.77 (d, J = 7.2 Hz, 3H).
Synthesis of dextran-dexamethasone (ND).
In a reaction flask, dextran (100 mg) was dissolved in anhydrous DMSO (10 mL). An anhydrous DMSO solution of DSA (30 mg) and N,N’-carbonyldiimidazole (30 mg) was added to the mixture dropwise. Then triethylamine (170 μL) was added to the mixture, which was stirred at room temperature for 14 h. The mixture was precipitated in cold ethanol to obtain a white solid. The drug loading of ND was determined to be 5.28 wt %.
Synthesis of dextran-NOTA (D-NOTA).
In a reaction flask, amino dextran (10 mg) was dissolved in anhydrous DMSO (10 mL). A solution of triethylamine (10 μL) and p-SCN-Bn-NOTA (0.34 mg) in anhydrous DMSO (1 mL) was then added. After 14 h, the mixture was precipitated to obtain a white solid.
Synthesis of dextran-NOTA-64Cu (D-Rad).
A solution of hot copper chloride was added to a solution of D-NOTA in ammonium acetate. The mixture was incubated with shaking at 37 °C for 45 min before adding cold copper chloride and EDTA to saturate NOTA. Then the mixture was purified using an Amicon filter (molecular weight cutoff 50 kDa) to exchange the solution to PBS. The radiochemical purity was determined to be greater than 95% by thin layer chromatography.
Animals, maintenance and diets
The Institutional animal care and use committee (IACUC) of the University of Illinois at Urbana-Champaign, IL, USA approved all the animal experimentation procedures in this study. All methods were performed in accordance with the United States Public Health Service Policy on Humane Care and Use of Laboratory Animals. C57BL/6J mice were purchased from Jackson Laboratory (Bar Harbor, ME, USA) and fed either a high-fat diet (HFD- 60% ME from fat D12492, Research Diets, Inc., New Brunswick, NJ, USA), or a low-fat diet (LFD- 10% ME from fat D12450J, Research Diets, Inc.), ad libitum, with an adequate and unrestricted supply of water. All animals were housed in a group of 4 in standard shoebox cages under a temperature- and humidity-controlled environment, with a 12-h light-12 h dark cycle. Mice for the experiments were either purchased at 8–10 wk of age and fed for approximately 15–18 wk or purchased at 16–20 wk of age and fed for approximately 4–8 wk to induce obesity and metabolic syndrome.
Non-invasive PET-CT imaging to evaluate adipose targeting
To visualize and assess the in vivo targeting and biodistribution of ND, animals were subjected to PET-CT imaging using a hybrid microPET-SPECT-CT small animal scanner (Inveon, Siemens Healthcare, Siemens Healthcare Diagnostics, Inc., Tarrytown, NY, USA). Twenty-four hours prior to imaging, animals were injected, IP, with a solution of radiolabeled dextran. Each injection contained ~ 100 μCi of dextran-NOTA-64Cu. After 24 h, animals were anesthetized using isofluorane (1–2%) and then positioned supine on the scanner bed. They then underwent a 15-min static PET imaging session with 20% energy window centered at 511 keV, followed by high resolution anatomic CT. After the imaging session, animals were sacrificed, and organs were carefully dissected and collected for biodistribution analysis. The selected organs were weighed and then radioactivity was measured using a gamma well counter (PerkinElmer, Waltham, MA, USA). 64Cu radioactivity was measured and the raw mean counts of each tissue segment was corrected for background activity, radioactive decay and tissue weight, and then converted to injected dose percentage per weight of tissue (% ID/g) using a calibration curve with 64Cu aliquots.
Ex vivo culture of bone marrow macrophages and analysis of M1 macrophage populations
Macrophages were differentiated from bone marrow of lean male adult wild type mice. Under aseptic conditions, femurs and tibia were isolated and marrow was flushed. After dissociation and red blood cell lysis, cells were cultured in DMEM/F-12 with 20 ng/mL macrophage colony-stimulating factor (MCSF R&D Systems) for 7 d. Macrophages were treated with PBS (M0) or 100 ng/mL lipopolysaccharide (LPS) with 50 ng/mL interferon gamma (IFN-γ) (M1), in the presence of vehicle or corresponding doses of free/conjugated dexamethasone. Twenty-four hours later, RNA was isolated with Gene JET RNA purification kit (ThermoFisher Scientific, Waltham, MA, USA). The RNA isolated was quantified for the expression of inflammatory genes using a Fluidigm Biomark HD Real-Time PCR device (Fluidigm Corporation, South San Francisco, CA, USA). Primers were designed and purchased from Fluidigm Deltagene Assays (Fluidigm Corporation, South San Francisco, CA, USA). The primers were pooled at 1 μL per assay for specific target amplifications using Taqman PreAmp Master Mix (Thermo Fisher, San Jose, CA, USA). The fold change in gene expression was analyzed using Fluidigm real-Time PCR analysis software (version 4.1.3).
Therapeutic regimen for acute study
Eight-week-old C57BL/6J mice were purchased and fed either a HFD or LFD, ad libitum for 18 wk with adequate supply of water. Body weight was measured weekly. At 26 wk of age, mice were allotted into the following treatment groups:
Group 1: LFD, treated with saline (lean control), n=8
Group 2: DIO mice – HFD, treated with saline (obese control), n=8
Group 3: DIO mice – HFD, treated with ND at 0.1 mg/kg, n=8
Group 4: DIO mice – HFD, treated with ND at 0.7 mg/kg, n=8
Group 5: DIO mice – HFD, treated with ND at 5 mg/kg, n=8
Group 6: DIO mice – HFD, treated with FD at 5 mg/kg, n=8
All the treatments were administered via IP injections, 24 h before euthanasia by CO2 asphyxiation. Blood was obtained by cardiac puncture for analysis of serum biochemical parameters (AST, ALT, NEFA and triglycerides). The serum chemistry analysis was performed at Comparative Clinical Pathology Services, LLC (Columbia, MO, USA). AT (subcutaneous, gonadal, mesenteric and perirenal fat depots) and liver were dissected and collected for histopathology and qRT-PCR analysis by Fluidigm.
Therapeutic regimen for long-term study
Eleven-week-old C57BL/6J mice were purchased and fed either a HFD or LFD, ad libitum for 15 wk with adequate supply of water. Body weight of the mice were measured weekly. At 26 wk of age, mice were allotted to the following treatment cohorts and groups:
Cohort 1 (for weekly blood sampling):
Mice in this cohort were monitored for variability in serum biochemistry during treatment by collecting blood by cheek bleeding (once weekly for 4 wk).
Group 1: LFD, treated with saline (lean control), n=4
Group 2: DIO mice – HFD, treated with saline (obese control), n=8
Group 3: DIO mice – HFD, treated with free dextran, n=8
Group 4: DIO mice – HFD, treated with ND at 0.7 mg/kg, n=8
Cohort 2 [for weekly glucose tolerance test (GTT), IP lavage, and insulin sensitivity test]:
Mice in this cohort underwent GTT at baseline and then once weekly for 4 wk. IP lavage of these mice was performed at baseline, d 10, and d 20 during treatment to analyze levels of IL-6 mRNA in the isolated macrophages. Insulin sensitivity testing was performed during wk 4.
Group 1: LFD, treated with saline (lean control), n=4
Group 2: DIO mice – HFD, treated with saline (obese control), n=8
Group 3: DIO mice – HFD, treated with free dextran, n=8
Group 4: DIO mice – HFD, treated with ND at 0.7 mg/kg, n=8
All animals in each cohort were weighed to determine their body weight twice weekly up to 5 wk. ND, saline and free dextran were administered via IP injections every other day. The treatment was scheduled for a period of 4 wk unless physiological / behavioral changes were observed during the therapeutic window. All mice remained on a HFD/LFD during treatment. Post-therapy, mice were euthanized by CO2 asphyxiation. Blood was obtained by cardiac puncture for analysis of serum biochemical parameters (AST, ALT, NEFA, and triglycerides), which was performed at Comparative Clinical Pathology Services, LLC (Columbia, MO, USA). AT (subcutaneous, gonadal, mesenteric and perirenal fat depots) and liver were dissected and collected for histopathology and qRT-PCR analysis by Fluidigm.
Intraperitoneal GTT and insulin sensitivity assay.
Intraperitoneal GTT was carried out in DIO and lean mice as described earlier.42 Briefly, mice were fasted overnight (14 h) with unrestricted access to water. They were injected IP with 1 g/kg of glucose (Sigma-Aldrich) solution, prepared using sterile filtered PBS. Plasma blood glucose levels were monitored by tail snipping just before glucose challenge (0 min) and 30, 60, 90 and 120 min post injection using Accu-Check Aviva plus glucometer strips (Roche Diagnostics, Indianapolis, IN, USA). The values were normalized to initial glucose concentration and area under the curve (AUC) was subsequently calculated.
For the insulin sensitivity test, mice in cohort 2 were fasted for 4 h with unrestricted access to water. Mice were injected IP with 0.7 U/kg of insulin (Vetsulin, 40 U/mL, Merck Animal Health, Ames, IA, USA) diluted using sterile filtered PBS. Blood glucose levels were monitored like GTT time points and the calculation of glucose levels was similar to that of GTT.
Intraperitoneal lavage.
After mice were anaesthetized, 2 mL of sterile saline was administered IP and the abdomen was massaged for 5 min. One hundred microliters of peritoneal fluid was isolated and cultured in DMEM/F-12 overnight. RNA was isolated from adherent macrophages with GeneJET RNA purification kit (ThermoFisher Scientific). cDNA libraries were prepared from solutions with equivalent RNA mass using iScript reverse transcription supermix (Bio-Rad Laboratories, Hercules, CA, USA), and gene expression was measured by real-time quantitative PCR with iTaq universal SYBR Green supermix (Bio-Rad Laboratories) on a CFX384 Touch machine (Bio-Rad Laboratories).
Body composition by EchoMRI.
Body compositions of all mice included in the study were determined using an EchoMRITM-100H (EchoMRI LLC, Houston, TX, USA). Before each scan, the instrument was calibrated using a calibration tube. The scans were performed by following protocols provided by the instrument manufacturer. Body composition was determined by scanning for total fat and lean mass at baseline and after 4 wk. Percentages of fat and lean mass were subsequently calculated.
Flow cytometry analysis of macrophage populations in gonadal and perirenal adipose tissue depots.
After mechanical dissociation of adipose tissue depots, tissues were incubated in collagenase (ThermoFisher Scientific) solution for 1–2 h at 37 °C. Cell suspensions were stained for CD11b, CD11c (Becton Dickinson) and CD206 (Biolegend) on ice in fluorescent-activated cell sorter buffer (PBS supplemented with 2% FBS and 1% penicillin streptomycin). Data were acquired on a BD Accuri C6 and were analyzed with Accuri C6 software.
RNA expression analysis in liver and adipose tissue depots by Fluidigm.
For the acute dosing study, total RNA was extracted from liver and from gonadal, perirenal, mesenteric, and subcutaneous fat depots. For the long-term treatment study, the gonadal fat depot and liver were used for RNA extraction. Total RNA from the adipose tissue depots and liver was isolated using RNeasy kits for lipids (Qiagen, Valencia, CA, USA), with DNase digestion (RNase-Free DNase set, Qiagen). The concentration of RNA and its purity were determined using a ND-1000 spectrophotometer (Nanodrop Technologies, Wilmington, DE, USA). cDNA was synthesized using SuperScript III Reverse Transcriptase (Invitrogen, Carlsbad, CA, USA).
Genes related to glucose metabolism, lipid metabolism, inflammation, macrophage polarization, immune cell types, and apoptosis were quantified using a Fluidigm Biomark HD Real-Time PCR instrument. Primers were designed and purchased from Fluidigm Deltagene Assays. The primers were pooled at 1 μL per assay for specific target amplifications using Taqman PreAmp Master Mix. The fold change in gene expression was analyzed using Fluidigm real-Time PCR analysis software (Version 4.1.3).
Statistical analysis
All statistics were carried out using either SAS (v. 9.4, Cary, NC, USA) or GraphPad Prism (v. 8.3.0, San Diego, CA, USA) to compare means between treatment groups using the appropriate parametric (ANOVA with Tukey’s posthoc) or non-parametric (Kruskal-Wallis with Dunn’s posthoc) statistical tests. Data are presented as mean ± standard error (SE) and a p<0.05 was considered statistically significant unless indicated otherwise.
Supplementary Material
Acknowledgements
This research was supported by the UIUC College of Agricultural, Consumer, and Environmental Sciences (ACES) Future Interdisciplinary Research Explorations (FIRE) program and the National Institutes of Health (R01DK112251).
Footnotes
Electronic Supplementary Information (ESI) available: The Supporting Information file contains methods for histological evaluation and culturing of RAW cells, as well as Supplementary Figures S1 to S4.
Conflicts of interest
There are no conflicts to declare.
References
- 1.Messerli FH, Christie B, DeCarvalho JG, Aristimuno GG, Suarez DH, Dreslinski GR, and Frohlich ED, Arch. Intern. Med 1981, 141, 81. [DOI] [PubMed] [Google Scholar]
- 2.Tai ES, Lau TN, Ho SC, Fok AC, and Tan CE, Int. J. Obes. Relat. Metab. Disord 2000, 24, 751. [DOI] [PubMed] [Google Scholar]
- 3.Nuertey BD, Alhassan AI, Nuertey AD, Mensah IA, Adongo V, Kabutey C, Addai J, and Biritwum RB, BMC Obes. 2017, 4, 26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Weisberg SP, McCann D, Desai M, Rosenbaum M, Leibel RL, and Ferrante AW Jr., J. Clin. Invest 2003, 112, 1796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Lumeng CN, Bodzin JL, and Saltiel AR, J. Clin. Invest 2007, 117, 175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Carr KA, Daniel TO, Lin H, and Epstein LH, Curr. Drug Abuse Rev 2011, 4, 190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Lumeng CN, Deyoung SM, Bodzin JL, and Saltiel AR, Diabetes 2007, 56, 16. [DOI] [PubMed] [Google Scholar]
- 8.Morris DL, Singer K, and Lumeng CN, Curr. Opin. Clin. Nutr. Metab. Care 2011, 14, 341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Gounarides JS, Korach-Andre M, Killary K, Argentieri G, Turner O, and Laurent D, Endocrinology 2008, 149, 758. [DOI] [PubMed] [Google Scholar]
- 10.Rocha VZ and Folco EJ, Int. J. Inflam 2011, 2011, 529061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Veiseh O, Tang BC, Whitehead KA, Anderson DG, and Langer R, Nat. Rev. Drug Discov 2015, 14, 45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ma L, Kohli M, and Smith AM, ACS Nano 2013, 7, 9518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Xu X, Ho W, Zhang X, Bertrand N, and Farokhzad O, Trends Mol. Med 2015, 21, 223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Choi CH, Alabi CA, Webster P, and Davis ME, Proc. Natl. Acad. Sci. U.S.A 2010, 107, 1235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Cho K, Wang X, Nie S, Chen ZG, and Shin DM, Clin. Cancer Res 2008, 14, 1310. [DOI] [PubMed] [Google Scholar]
- 16.Ballester M, Jeanbart L, de Titta A, Nembrini C, Marsland BJ, Hubbell JA, and Swartz MA, Sci. Rep 2015, 5, 14274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Jacobs A, Culligan D, and Bowen D, Contrib. Nephrol 1991, 88, 266. [DOI] [PubMed] [Google Scholar]
- 18.Loh KP, Ho D, Chiu GNC, Leong DT, Pastorin G, and Chow EK, Adv. Mater 2018, 30, e1802368. [DOI] [PubMed] [Google Scholar]
- 19.Glassman PM and Muzykantov VR, J. Pharmacol. Exp. Ther 2019, 370, 570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Ventola CL, P.T 2017, 42, 742. [PMC free article] [PubMed] [Google Scholar]
- 21.Ma L, Liu TW, Wallig MA, Dobrucki IT, Dobrucki LW, Nelson ER, Swanson KS, and Smith AM, ACS Nano 2016, 10, 6952. [DOI] [PubMed] [Google Scholar]
- 22.Prigozy TI, Sieling PA, Clemens D, Stewart PL, Behar SM, Porcelli SA, Brenner MB, Modlin RL, and Kronenberg M, Immunity 1997, 6, 187. [DOI] [PubMed] [Google Scholar]
- 23.Sallusto F, Cella M, Danieli C, and Lanzavecchia A, J. Exp. Med 1995, 182, 389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Tan MC, Mommaas AM, Drijfhout JW, Jordens R, Onderwater JJ, Verwoerd D, Mulder AA, van der Heiden AN, Scheidegger D, Oomen LC, et al. , Eur. J. Immunol 1997, 27, 2426. [DOI] [PubMed] [Google Scholar]
- 25.Yang M, Ding J, Feng X, Chang F, Wang Y, Gao Z, Zhuang X, and Chen X, Theranostics 2017, 7, 97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Lauterbach MA and Wunderlich FT, Pflugers Arch. 2017, 469, 385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Xu C, He J, Jiang H, Zu L, Zhai W, Pu S, and Xu G, Mol. Endocrinol 2009, 23, 1161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Murano I, Barbatelli G, Parisani V, Latini C, Muzzonigro G, Castellucci M, and Cinti S, J. Lipid Res 2008, 49, 1562. [DOI] [PubMed] [Google Scholar]
- 29.Chen YJ, Wallig MA, and Jeffery EH, J. Nutr 2016, 146, 542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Paik J, Fierce Y, Treuting PM, Brabb T, and Maggio-Price L, J. Nutr 2013, 143, 1240. [DOI] [PubMed] [Google Scholar]
- 31.Ellulu MS, Patimah I, Khaza’ai H, Rahmat A, and Abed Y, Arch. Med. Sci 2017, 13, 851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Poggioli R, Ueta CB, Drigo RA, Castillo M, Fonseca TL, and Bianco AC, Obesity 2013, 21, E415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Subramanian V and Ferrante AW Jr., Nestle Nutr. Workshop Ser. Pediatr. Program 2009, 63, 151. [DOI] [PubMed] [Google Scholar]
- 34.Eder K, Baffy N, Falus A, and Fulop AK, Inflamm. Res 2009, 58, 727. [DOI] [PubMed] [Google Scholar]
- 35.Bastard JP, Jardel C, Bruckert E, Blondy P, Capeau J, Laville M, Vidal H, and Hainque B, J. Clin. Endocrinol. Metab 2000, 85, 3338. [DOI] [PubMed] [Google Scholar]
- 36.Wu H, Perrard XD, Wang Q, Perrard JL, Polsani VR, Jones PH, Smith CW, and Ballantyne CM, Arterioscler. Thromb. Vasc. Biol 2010, 30, 186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Hu G, Guo M, Xu J, Wu F, Fan J, Huang Q, Yang G, Lv Z, Wang X, and Jin Y, Front. Immunol 2019, 10, 1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Gustafson HH, Holt-Casper D, Grainger DW, and Ghandehari H, Nano Today 2015, 10, 487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Rattan R, Bhattacharjee S, Zong H, Swain C, Siddiqui MA, Visovatti SH, Kanthi Y, Desai S, Pinsky DJ, and Goonewardena SN, Bioorg. Med. Chem 2017, 25, 4487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Weissleder R, Nahrendorf M, and Pittet MJ, Nat. Mater 2014, 13, 125. [DOI] [PubMed] [Google Scholar]
- 41.Miller MA, Zheng YR, Gadde S, Pfirschke C, Zope H, Engblom C, Kohler RH, Iwamoto Y, Yang KS, Askevold B, Kolishetti N, Pittet M, Lippard SJ, Farokhzad OC, and Weissleder R, Nat. Commun 2015, 6, 8692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Ma X, Pham VT, Mori H, MacDougald OA, Shah YM, and Bodary PF, PLoS One 2017, 12, e0179889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Singh AP, Biswas A, Shukla A, and Maiti P, Signal Transduct. Target Ther 2019, 4, 33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Shae D, Becker KW, Christov P, Yun DS, Lytton-Jean AKR, Sevimli S, Ascano M, Kelley M, Johnson DB, Balko JM, and Wilson JT, Nat. Nanotechnol 2019, 14, 269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Koshy ST, Cheung AS, Gu L, Graveline AR, and Mooney DJ, Adv. Biosyst 2017, 1, 1600013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Lee MJ, Pramyothin P, Karastergiou K, and Fried SK, Biochim. Biophys. Acta 2014, 1842, 473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Aouadi M, Tencerova M, Vangala P, Yawe JC, Nicoloro SM, Amano SU, Cohen JL, and Czech MP, Proc. Natl. Acad. Sci. U.S.A 2013, 110, 8278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Ladiges WC, Knoblaugh SE, Morton JF, Korth MJ, Sopher BL, Baskin CR, MacAuley A, Goodman AG, LeBoeuf RC, Katze MG, Diabetes 2005, 54, 1074. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.