

Abstract
Linker histone H1 proteins bind to nucleosomes and facilitate chromatin compaction1, although their biological functions are poorly understood. Histone H1 (HIST1H1B-E) mutations are highly recurrent in B-cell lymphomas, but their cancer relevance and mechanism are unknown. Here we show that lymphoma-associated H1 alleles are genetic driver mutations in lymphomas. Disruption of H1 function results in profound architectural remodeling of the genome characterized by large-scale, yet focal shifts of chromatin from a compacted, to a relaxed state. This decompaction drives distinct changes in epigenetic states, primarily due to gain of histone H3 lysine 36 dimethylation, and/or loss of repressive H3 lysine 27 trimethylation. These changes unlock expression of stem cell genes that are normally silenced during early development. Loss of H1c and H1e alleles in mice conferred enhanced fitness and self-renewal properties to germinal center B-cells, ultimately leading to aggressive lymphoma with enhanced repopulating potential. Collectively, our data indicate that H1 proteins are normally required to sequester early developmental genes into architecturally inaccessible genomic compartments. We furthermore establish H1 as a bona fide tumor suppressor, whose mutation drives malignant transformation primarily through three-dimensional genome reorganization, followed by epigenetic reprogramming and derepression of developmentally silenced genes.
Linker histones are encoded in humans by ten different genes, five of which (H1A, B, C, D and E) are expressed in a replication-dependent manner. Linker histones act as transcriptional repressors by limiting chromatin accessibility2 and are depleted from actively transcribed domains3. Their functions are mediated directly through condensation of chromatin fiber, or indirectly via: (i) recruitment of transcriptional repressors, or (ii) impaired access of transcriptional activators to core nucleosomes1. A knockout of three H1 isoforms (H1c/d/e) impaired differentiation of mouse embryonic stem cells4, suggesting a role for H1 in epigenetic programming of cellular phenotypes. While a role for mutations in core nucleosomal histones in cancer is well documented, little is known about H1 mutations5,6. Recurrent H1 mutations occur in ~30–40% of diffuse large B-cell lymphomas (DLBCL), ~30 % of follicular lymphomas (FL) and ~50% Hodgkin lymphomas (HL)7–9. These diseases originate from germinal center (GC) B-cells, which arise transiently from resting B-cells during the T-cell dependent humoral immune response10. During the GC reaction, the immunoglobulin loci undergo extensive mutagenesis by activation induced cytosine deaminase (AICDA), and H1 mutant lymphoma alleles manifest AICDA mutation signatures11. While lymphoma H1 mutations are often highly clonal and their mutational landscape suggests loss of function, their effect at the chromatin or functional level has not been defined; although one study of a single H1 mutant reported impaired binding to mononucleosomes7. Homozygous knockout of one or both H1c and H1e, the most commonly mutated isoforms in human lymphoma did not induce a mouse developmental phenotype12. Whether or how H1 isoform dosage or functionality contribute to lymphomagenesis is unknown.
Lymphoma H1 alleles are genetic driver mutations
Examining the TCGA panCancer Atlas we observed that B-cell lymphomas manifest the highest frequency of mutant H1 alleles. 97% of H1 mutant alleles encode missense mutations affecting the globular and C-terminal domains, with H1C and H1E being the most commonly affected isoforms (Extended Data Fig.1a–b). Although H1 mutations occur across DLBCL subtypes, there was significant enrichment for H1 SNVs and focal deletions in the newly defined MCD-DLBCLs13 (Extended Data Fig.1c–f). Analyzing germline-controlled whole genome sequencing profiles from 101 DLBCL patients we observed H1 mutation rates of 8.9% H1B, 24.7% H1C, 11.0% H1D and 42.6% H1E (Extended Data Fig.1g). A rigorous analysis controlling for genomic and epigenomic covariates identified H1C and H1E among the top ten driver mutations (Extended Data Fig.1h). Variant allele frequency varied between 0.2 to 0.4 consistent with clonal heterozygous mutation. There was significant co-occurrence between H1C and H1E, as well as other H1 alleles (Extended Data Fig.1i–j). Further, 85% of H1B-E globular domain (GD) mutations scored as deleterious and affected amino-acids within conserved interaction interfaces including a ASGS motif that directly binds to DNA14 (Extended Data Fig.2a).
Expression of WT or C-terminal mutant mEGFP-tagged H1C in 3T3 cells showed patterns consistent with localization to chromatin, whereas GD mutants affecting the ASGS loop formed extensive nuclear aggregates. In FRAP assays, H1C WT and C-terminal mutants showed similar in vivo dynamics with recovery rates in the order of minutes (Extended Data Fig.2b–c). By contrast, GD mutants recovered rapidly, consistent with failure to incorporate into chromatin. Likewise, using biolayer interferometry we observed that GD mutant H1 manifested higher mononucleosome dissociation constants as compared to WT or C-terminal mutants. Furthermore, Mg2+ precipitation, and atomic force microscopy of 12-mer nucleosome arrays revealed impaired compaction upon loading of C-terminal tail mutant vs wild type control (Extended Data Fig.2d–f). Hence, H1 mutations may result in loss of function through several biochemical mechanisms.
H1c/e deficient GCB-cells manifest increased fitness and disrupted polarity
Quantitative RT-PCR analysis showed that HIST1H1B/C/D/E expression was 2–4 fold higher in GCB-cells than naïve B-cells (Extended Data Fig.3a–b). Given the common co-occurrence of H1C and H1E mutations, we assessed GC formation in H1c−/−;H1e−/− mice, previously reported to have no developmental phenotype2,12. Immunized H1c−/−;H1e−/− mice had no splenomegaly (Extended Data Fig.3c) or disruption of splenic architecture (Fig 1a). However, they manifested enlarged and more abundant GCs, and Ki67+ proliferative cells (Fig.1b–d,Extended Data Fig.3d). There was no observable effect on apoptosis (active Casp3) or DNA damage (γ-H2A.X) (Extended Data Fig.3e,f). The increase in GCB-cells was confirmed by flow cytometry (Extended Data Fig.3g–i). The proportions of other mature and immature B-cells were similar to WT, with minor differences in Ki67+ cells (Extended Data Fig.3j–m). After double immunization, we found no difference in ratios of high vs low affinity NP antibody titers or plasma cells secreting anti-NP immunoglobulins (Extended Data Fig.3n–q). GCs are composed of a dark zone containing proliferative B-cells (centroblasts, CB), and a light zone containing mostly non-dividing B-cells (centrocytes, CC). H1c−/−;H1e−/− mice featured selective increase of CCs (Extended Data Fig.3r). To determine whether H1c/e deficiency endows GCB cells with a fitness advantage we performed mixed bone marrow chimera experiments and observed robust competitive advantage for H1c−/−;H1e−/− at both time points by flow cytometry (Fig.1e–h) and immunofluorescence (Extended Data Fig.3s,t). The competitive advantage was specific to CCs (Fig.1i,j). Administering EdU revealed significant increase of replication of GCB cells, that was specific to CCs (Fig.1k,Extended Data Fig.3u). Therefore H1 deficiency induces increased fitness of GCB-cells manifesting as increased CC self-renewal.
Fig. 1 |. Characterization of H1c−/−/e−/− germinal center B (GCB) cells.
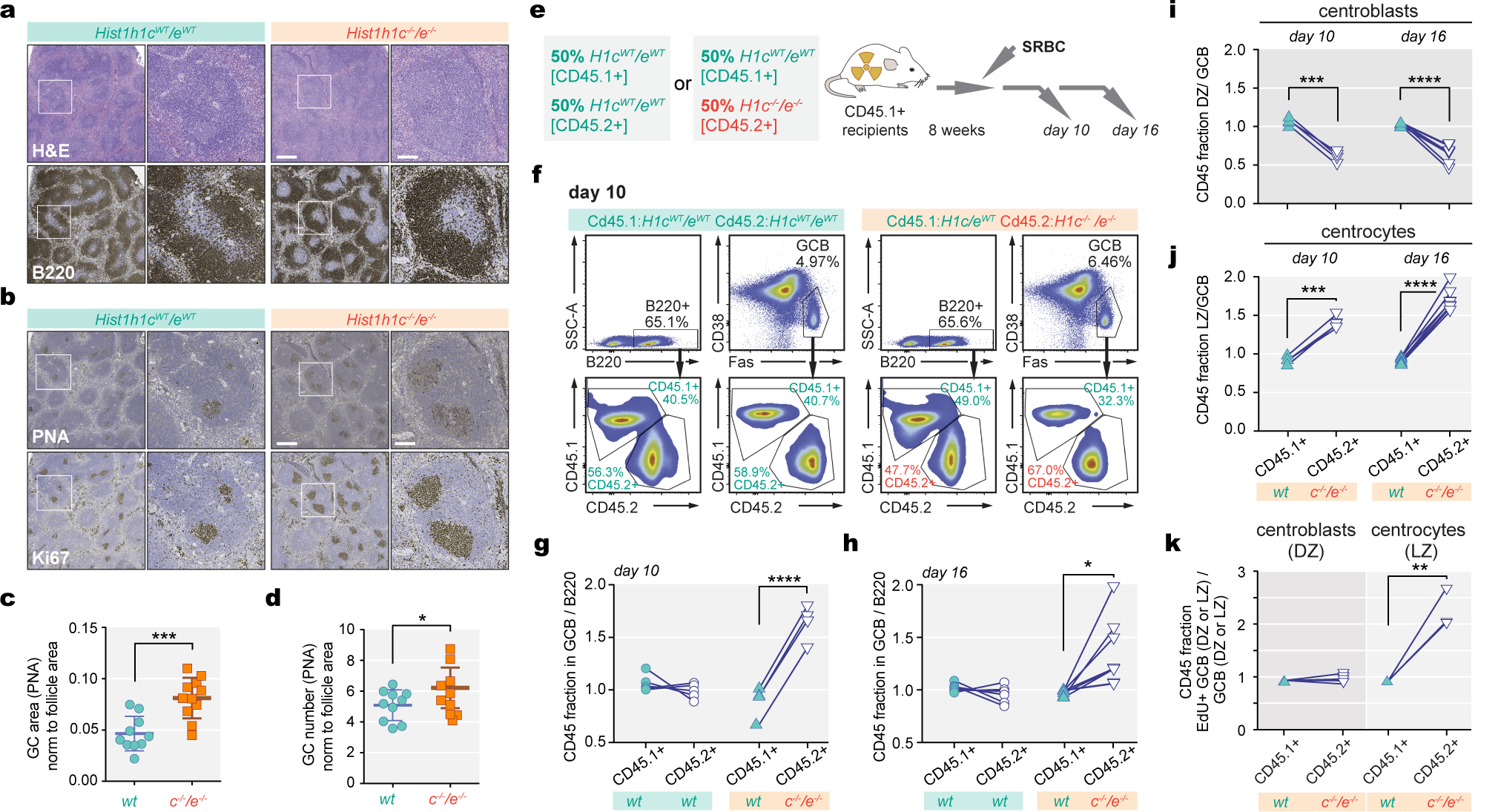
a-b, Spleens of wild-type (WT) and H1c−/−/e−/− mice at day 9 after SRBC immunization stained with H&E and B220, PNA and Ki-67 antibodies. Scale bars: 500 and 100 μm for low high power images, respectively. Images are representative of three independent experiments.c-d, Quantification of GC area (PNA), n=11 per genotype, (***P=0.0004) and number of GCs, n=10 per genotype (*P=0.044) in the spleens of WT and H1c−/−/e−/ mice; mean ± S.D., unpaired t-test. e, Competitive mixed bone marrow chimera scheme. f, Representative flow cytometry plots for relative fractions of CD45.1+ and CD45.2+ within naïve B220+, and GCB-cells at day 10. g-h, Relative (Fas+CD38−) GCB/B220+ ratios for WT (CD45.1+) and H1c−/−/e−/− (CD45.2+) at day 10 (n=4 chimeras, ****P<0.0001) and day 16 (n=7 chimeras, ****P=0.0138, paired t-test). GCB/B220+ ratios in chimeras injected with WT CD45.1+ and WT CD45.2+ at day 10 (n=5) and day 16 (n=7) were unchanged. i, Relative centroblasts/GC B-cell fraction for CD45.1+ and CD45.2+ in mixed chimeras at day 10, ***P=0.0004 (n=4) and day 16, ****P<0.0001 (n=7, paired t-test). j, Relative centrocytes/GCB-cell fraction for CD45.1+ and CD45.2+at day 10, ***P=0.0005 (n=4) or day 16, ****P<0.0001 post-immunization (n=7). k, Relative EdU+ DZ/DZ GC B-cells (n=4) or EdU+ LZ/LZ GCB-cells (n=3, **P= 0.0040, two-sided paired t-test) for WT (CD45.1+ and H1c−/−/e−/− (CD45.2+) fractions 7 days post immunization. Data are mean ± SD.
H1c/e loss induces stem cell transcriptional profiles
RNA-seq in sorted H1c−/−;H1e−/− and WT GCB-cells revealed distinct transcriptional profiles, with 782 differentially expressed genes markedly skewed towards transcriptional activation (Fig.2a and Extended Data Fig.4a). Many upregulated genes were linked to stem cell functionality, including Klf4, Klf5, Meis1, Prdm5, Mycn, Spry2, Hoxa9. Pathway analysis revealed enrichment for i) signatures associated with partially reprogrammed induced pluripotent stem cells (iPSC), adult tissue stem cells, and hematopoietic stem/progenitor cells, ii) direct targets of stem cell transcription factors such as SOX2, NANOG, and SUZ12/PRC2, and iii) genes marked by H3K27me3 in stem and mature hematopoietic cells, including monovalent H3K27me3 in GCB-cells (Fig.2b). H3K27me3 is formed by the PRC2 complex, and is opposed by H3K36me2 mediated by NSD2 and related histone methyltransferases15. Notably, NSD2 is induced in wild-type human and murine GCB-cells, and H1c−/−;H1e−/− GCB-cells strongly upregulated genes activated by NSD2 gain-of-function in B and T cells16 (Fig. 2b,Extended Data Fig.4b,c).
Fig. 2 |. H1c/e deficiency induces stem cell transcriptional profiles in GCB cells.
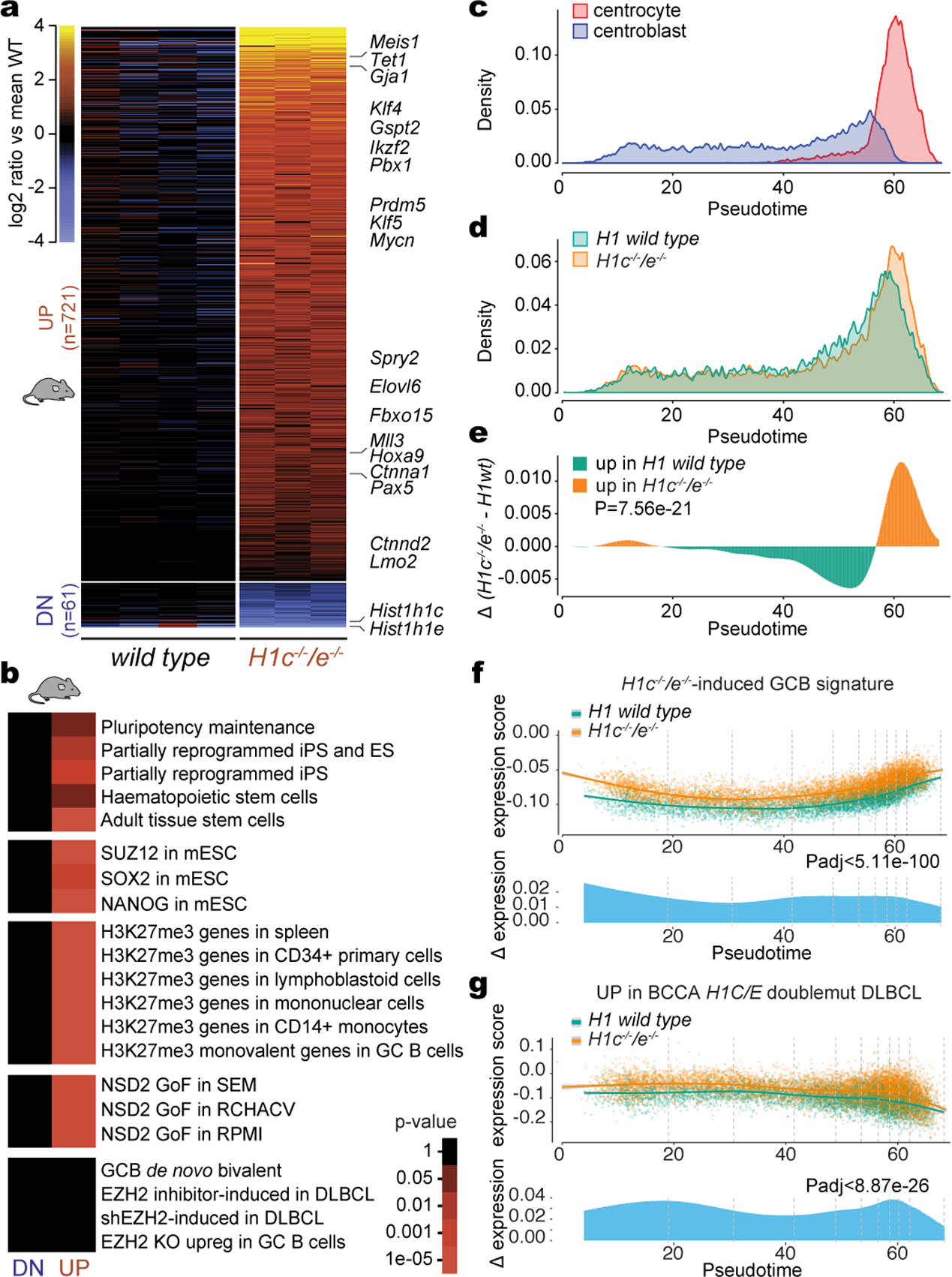
a, Heatmap of differentially expressed genes (FDR <0.05, fold change >1.5) in sorted GCB cells from independent H1c−/−/e−/− (n=3) and WT (n=4) mice. b, Gene pathway enrichment analysis of upregulated and downregulated genes in H1c−/−/e−/− vs WT GC B-cells (hypergeometric mean test). c, Single-cell RNA-seq density plot illustrating the frequency of centroblasts and centrocytes across the Slingshot pseudotime axis. d, Density plot of the frequency of H1c−/−/e−/− (n=9807 cells) and WT (n=6774 cells) GC B-cells across the pseudotime axis. Data are pooled from two independent biological replicates. e, Differential density plot on (d) tested between H1c−/−/e−/− and WT with two-sided Wilcoxon analysis. f-g, Top: Expression of the upregulated H1c−/−/e−/− GC B-cell gene signature (f) and human H1C/E mutant DLBCL gene signature (g) was plotted for each cell on the Y axis with spline curves representing the average for H1c−/−/e−/− and WT cells. Bottom: Differential expression shown as delta spline plot across pseudotime, tested by two-sided Wilcoxon within ten bins of equal cell number (dashed lines).
Among normal immune and hematopoietic cells, only long-term repopulating hematopoietic stem cells were enriched for the H1c−/−;H1e−/− signature, and there was significant enrichment for mesenchymal-like state transitions, and cancer stromal cells (Extended Data Fig.4d,e). Of note, genes repressed by EZH2 in GCB-cells through formation of bivalent chromatin were not de-repressed by loss of H1c/e17. Hence H1c/e deficiency primarily reversed silencing of developmental PRC2 targets (Fig.2b, Extended Data Fig.4f). Examining RNA-seq profiles from H1C/E mutant vs H1 WT DLBCL patients18 we identified 453 significantly differentially expressed genes, again with skewing towards transcriptional upregulation and enrichment for IPSC signatures, H3K27me3 marked genes in hematopoietic cells, and cistromes for NANOG and PRC2 (Extended Data Fig.4g–i).
To determine whether the effect of H1 deficiency on gene expression was emanating from an aberrant GCB sub-population, we performed single-cell RNA-seq in GCB-cells from H1c−/−;H1e−/− and WT mice. Plotting CB and CC across a pseudotime axis (Fig.2c, Extended Data FIg.4j,k), we again observed increased abundance of CC among H1c−/−;H1e−/− GCB-cells (Fig.2d,e). Yet genes upregulated in H1c−/−;H1e−/− GCB-cells and H1C/E mutant DLBCLs were uniformly upregulated across CBs and CCs (Fig. 2f–g) with no novel subpopulations among H1 deficient GCB cells (not shown). Notably, H1c−/−;H1e−/− CC (but not CB) manifested significant upregulation of proliferation genes (Extended Data FIg.4l), consistent with aberrant self-renewal being most evident among centrocytes.
H1c/e loss induces decompaction of developmentally silenced chromatin.
We next performed Hi-C in sorted H1c−/−;H1e−/− and WT GCB-cells. These showed distinct contact profiles, and analyses of chromatin compartment states (c-scores) revealed strong separation between H1c−/−;H1e−/− and control GCB-cells (Extended Data Fig.5a,b). Compartment B chromatin is highly compacted and transcriptionally silent, while compartment A is associated with transcriptionally poised or active chromatin19. We observed extensive, yet focal decompaction affecting 5320 discrete 100 kb chromatin domains, and increased compaction of only 386 regions (Fig.3a,b,Extended Data Fig.5c). Among these, 637 domains shifted entirely from compartment B to compartment A (B-to-A). The remaining shifts consisted of decompaction within respective compartments. (Fig.3a). Focal compartment shifting occurred across all chromosomes, only sparing regions with the most extreme compartment B c-scores that contain gene deserts and pericentric heterochromatin20 (Extended Data Fig.5d,e) . Accordingly, regions that underwent B-to-A decompaction had significantly higher content in genes and CpG islands vs non-shifting B regions (P=0.007 and P=0.0005, respectively). Boundaries of topologically associating domains (TADs) were not significantly affected, but twenty-six TADs manifested significant gain of intra-TAD interactivity (Extended Data Fig.5f) and were enriched for regions with compartment decompaction (Fig.3b). ATAC-Seq profiling revealed 488 differentially accessible peaks, 90% of which manifested gain of accessibility (Extended Data Fig.5g) and significant enrichment within regions that experienced decompaction (Fig.3c), wherein >99% of differentially accessible sites gained accessibility (Extended Data Fig.5h).
Fig. 3 |. H1c/e deficiency induces stem cell transcriptional profiles in GCB cells.
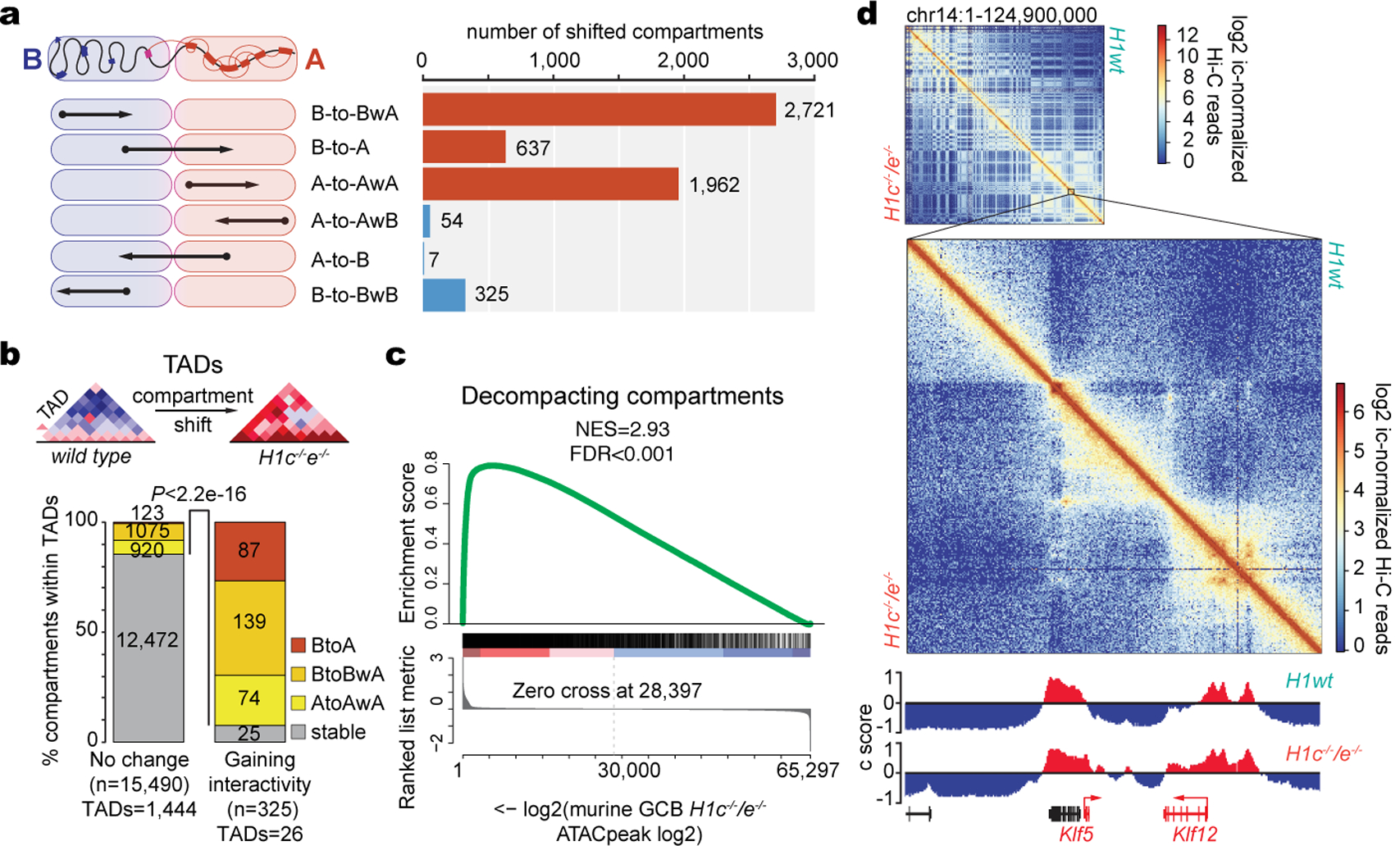
a, Schematic of directionality of shifting within defined compartments (left) and number of shifted compartment regions (100 kb scale) (right), showing decompaction in red and compaction in blue bars. b, Bar graph showing the proportion of stable or decompacting 100 kb compartment bins within TADs gaining (n=26) or non-changing (n=1,444) intra-TAD interactivity in H1c/e deficient GC B-cells. Two sided Fisher’s exact test, P<2.2e-16. c, GSEA analysis of ATAC-seq peaks from decompacting compartments against ranked ATAC-seq peak log2 fold changes in H1c−/−/e−/− vs wild-type H1 GC B-cells. d, Top, contact heat map of chromosome 14, showing WT on the top right and H1c−/−/e−/− GC B-cells in the lower left. Bottom, contact heat map in region containing Klf5 locus. IGV tracks below represent the Eigenvector for compartments A (red) and B (blue) positioned on Chr14:98,000,000–101,000,000.
B-to-A compartment shifts affected genes upregulated in H1 deficient GCB-cells such as Klf5, Meis1, Tusc1, and Spry2 (Fig.3d,Extended Data Fig.5i). Indeed we observed significant upregulation of genes shifting BtoA (n=224, NES=1.73, FDR<0.001), as well as genes decompacting within cognate compartments (Extended Data Fig.5j,k). Decompacted genes were enriched for iPSC reprogramming, mesenchymal-transition states, stem cell transcription factor cistromes, H2K27me3-marked genes in hematopoietic cells, NSD2 gain-of-function induced genes. GCB-specific EZH2 targets were unaffected, which is consistent with a previous report on H1 absence from poised gene promoters21 (Extended Data Fig.5l).
Notably, we observed significant association between genomic domains that decompact during early iPSC reprogramming and those decompacting in H1-deficient GCB-cells (Extended Data Fig.6a–d). Further analysis of the stem cell-associated Klf5 locus using virtual v4C revealed gain of promoter interaction with distal elements shifting BtoA compartments in H1c−/−e−/− GCB-cells, similar to sites that gain interactions in iPSC, also gaining variable degrees of chromatin accessibility (Extended Data Fig.6e). Strikingly, these newly interactive Klf5 sites were significantly enriched for canonical GCB TF consensus motifs OCT2 (P=0.00295) and IRF8 (P=0.00699), suggesting that decompacted genes may become targets of opportunity for GCB-associated TFs. We wondered whether such iPSC-like architectural states would allow H1c/e-deficiency to facilitate stem cell reprogramming. Indeed, OKSM expression in H1c−/−;H1e−/− and WT murine embryonic fibroblasts yielded a three- to four-fold increased efficiency in forming H1c/e-deficient iPSC colonies. (Extended Data Fig.6f–h). These data suggest that these H1 isoforms maintain inactivation of primitive stem cell genes that are silenced during lineage specification and differentiation.
H1 deficiency induces distinct and specific epigenetic states
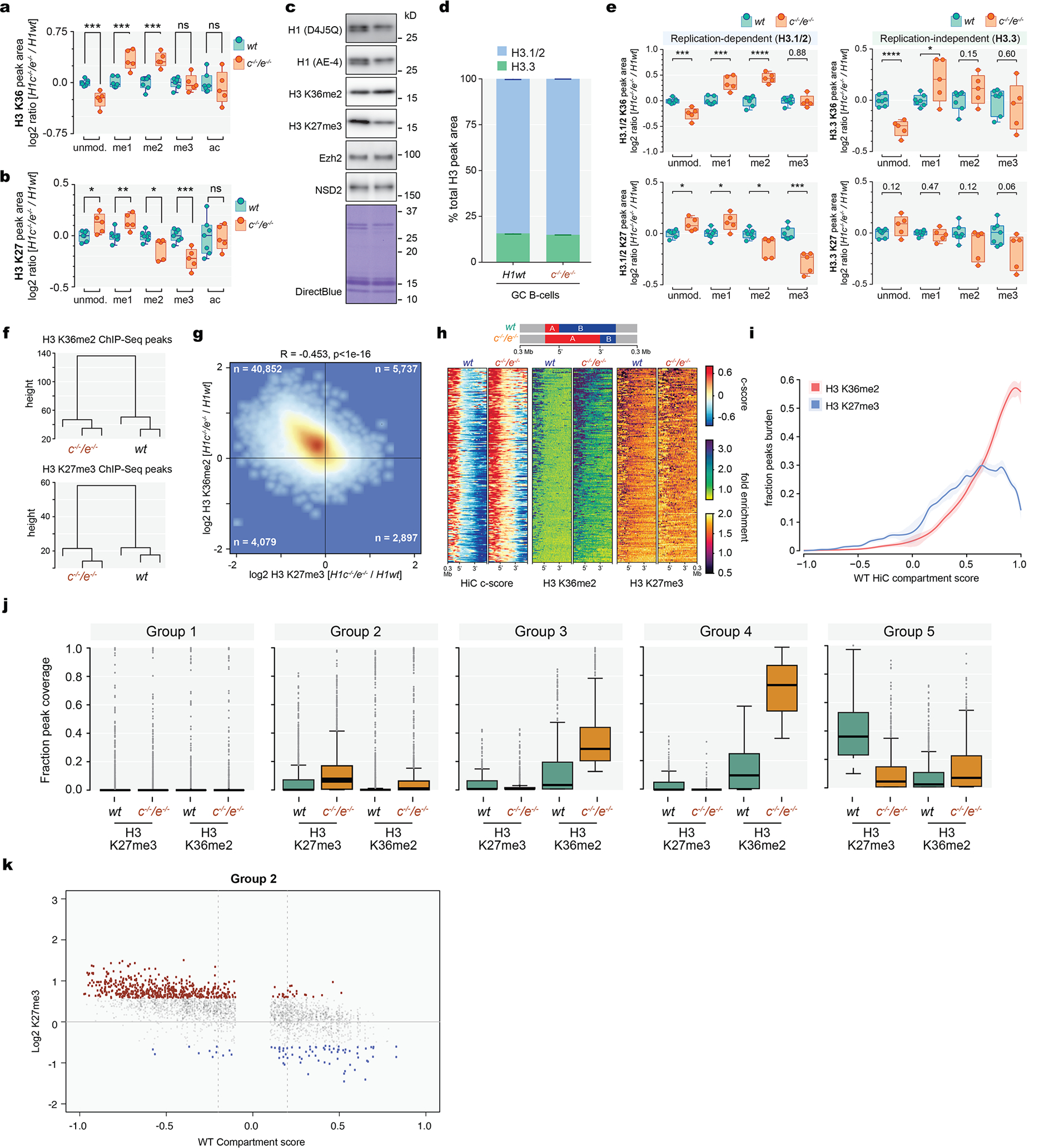
Mass spectrometry-based quantification of histone post-translational modifications in WT and H1c−/−;e−/− GCB-cells revealed significant gain of H3K36me1 and H3K36me2, but not H3K36me3. As H3K36me2 can license chromatin for transcription and antagonize PRC216, we observed significant reduction in H3K27me2/3, but little change in other histone modifications (Extended Data FIg.7a–b). Gain of H3K36me2 and loss of H3K27me3 was confirmed by Western blots, while Ezh2 and Nsd2 levels were unchanged (Extended Data Fig.7c). We found little difference in the relative abundance of H3.3 vs H3.1/2 in H1c−/−;H1e−/− GCB-cells, and similar K36me2 gain and K27me3 loss across H3 variants (Extended Data Fig.7d–e).
Performing ChIP-RX for H3K36me2 and H3K27me3 indicated a clear difference in the distribution of these marks in H1c−/−;H1e−/− vs WT GCB-cells, yielding 7901 gained vs 33 lost H3K36me2 peaks, and 792 gained vs 4736 lost H3K27me3 peaks, with generally inverse correlation of these marks (Fig.4a,Extended Data Fig.7f,g). Notably, H3K36me2 gain more closely followed B-to-A compartment shifts (Fig.4b, Extended Data Fig.7h). Further analysis of H3K36me2 and H3K27me3 in normal GCB-cells showed that both are generally absent from compartment B, except for H3K27me3 at the least compacted B regions, whereas H3K36me2 increased progressively across compartment A, and H3K27me3 was depleted from the most decompacted regions (Extended Data Fig.7i). Thus, degree of chromatin interactivity is reflected in alternatively demarcated H3K27me3 or H3K36me2 epigenetic states. Indeed hierarchical clustering of these histone marks in regions with compartment decompaction revealed five distinct epigenetic states (Fig. 4c,d,Extended Data Fig.7j): 1) Regions that remain in compartment B, with both marks stably absent. 2) Regions that start in compartment B with low H3K36me2 and H3K27me3, manifesting modest gains with the latter preferentially remaining in compartment B (Extended Data Fig.7k). 3) Regions that start in compartment B or A, undergo moderate gain of H3K26me2 and loss of H3K27me3. 4) Regions that start in compartment A, gain significant H3K36me2 and show modest loss of H3K27me3. 5) A-compartment regions that become highly decompacted and mostly manifest reduction of H3K27me3.
Fig. 4 |. H1c/e deficiency reprograms H3K36me2 and H3K27me3 epigenome trajectories.
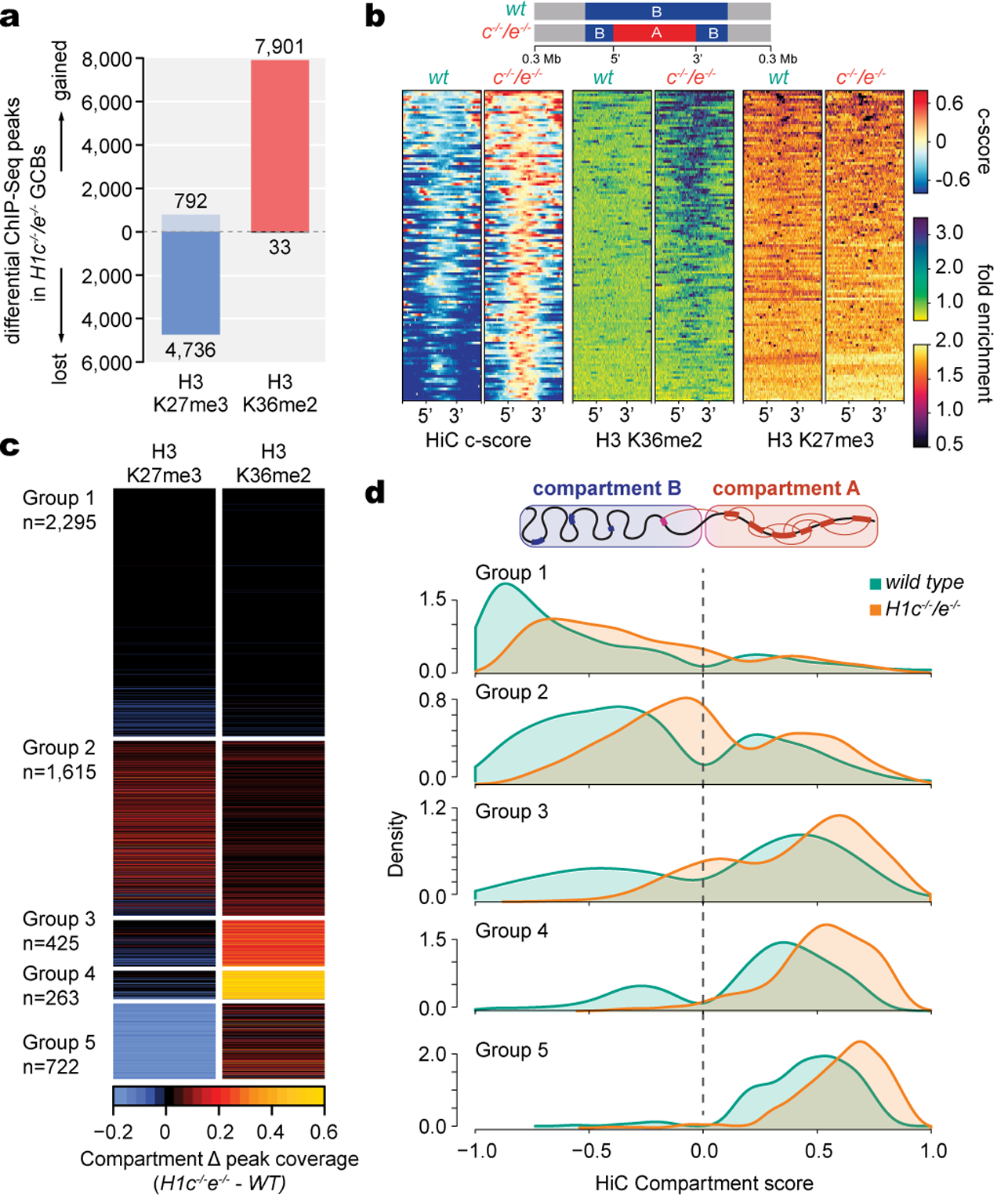
a, Differential ChIP-Seq peaks for H3K36me2 and H3K27me3 between H1c−/−/e−/− and wild-type GC B-cells (FC>1.5). b, Heatmap of compartment score, H3K36me2 and H3K27me3 fold enrichment centered within shifting B to A compartment “islands” (100 kb) and surrounding 300 kb in H1c−/−/e-/ -and wild-type GC B-cells. c, Heatmap of changing fraction peak coverage for H3K27me3 and H3K36me2 in H1c−/−/e−/− compared to wild-type H1 GC B-cells within decompacting compartments (n=5,320), subdivided into the five groups captured by unsupervised hierarchical clustering. d, Density plots showing the distribution of c-scores for shifting compartment groups 1–5 defined in (c) for H1c−/−/e−/−and wild-type H1C GC B-cells.
Genes within epigenetic state groups 3, 4, and 5 manifested significant transcriptional activation, consistent across centroblasts and centrocytes (Extended Data Fig.8a,b). Accordingly, upregulated promoters gained H3K4me3 and H3K27ac marks, primarily found in compartment A (Extended Data Fig.8c,d). In contrast, genes in groups 1 and 2 remained mostly silenced. Profiling of H3K9me2 and H3K9me3 and distribution in normal GCB-cells revealed their confinement to compartment B with H1c−/−e−/− GCB-cells featuring reduction in H3K9me3 but not in H3K9me2 peaks (Extended Data Fig.8e,f). However, regions undergoing decompaction in H1c−/−e−/− GCB-cells manifested significant reduction of both H3K9me2 and H3K9me3 (Extended Data Fig.8g–i). Moreover H3K9me2 and H3K9me3 showed reduction but not complete loss in groups 1 and 2 regions, thus marking compacted territories that are more resistant to full decompaction (Extended Data Fig.8j).
To gain further insight into how H1 abundance influences chromatin compaction, we performed in silico modeling of 50-mer nucleosome arrays22. At higher concentrations, H1 formed a rigid stem and straightened chromatin fibers (Extended Data Fig.9a). These rigid conformations occupied smaller volumes and demonstrated negligible long-range nucleosome contacts. With decreasing H1:nucleosome ratio, fewer DNA linkers were shielded by H1, allowing greater bending and loop formation22 corresponding to enhanced long-range nucleosome/nucleosome contacts (Extended Data Fig.9b–f), mimicking chromatin state changes observed by Hi-C and providing a rationale for the transition states captured experimentally.
Collectively, these data suggest that H1 dosage affects gene expression by: a) sequestering genes within compartment B through dense compaction; b) making less compact compartment-B regions accessible enough to enable PRC2 to form H3K27me3 domains23; c) contributing to the regulation of compartment A genes by establishing chromatin compaction states optimal for specific epigenetic complexes (e.g. PRC2 and NSD2), with regions that lack H3K27me3 at baseline primarily gaining H3K36me2, whereas regions that contain H3K27me3 primarily experienced loss of this mark without substantial gain of H3K36me2. These data suggest a critical functional link between H1 and NSD2, as NSD2 gain-of-function mimics the H1-deficient transcriptional signature in B-cells16, disrupts genomic compartmentalization24, and leads to upregulation of EMT genes25.
H1 loss leads to lymphomas with enhanced self-renewal
We crossed H1c−/−;H1e−/− with VavP-Bcl2 mice to model MCD-DLBCLs (Fig. 5a), since MCDs have the highest BCL2 expression among DLBCLs13. At early timepoints, VavP-Bcl2 mice had intact lymph node architecture albeit with hyperplastic follicles containing foci of small lymphocytes with condensed chromatin. In contrast additional loss of H1c/e yielded disruption of lymph nodes by diffusely infiltrating immunoblastic cells with large nuclei, open chromatin and increased H3K36me2 (Fig.5b,Extended Data Fig.10a,b). VavP-Bcl2;H1c/e-deficient mice also manifested more extensive invasion of liver and lungs by neoplastic Ki67+ B-cells, with T-cell infiltrates as often observed in ABC-DLBCLs26 and more evident expansion of monoclonal B-cell populations (Fig. 5c,Extended Data Fig.10c–f). Long-term observation revealed significantly shorter survival of VavPBcl2;H1c+/−/e+/− and VavPBcl2;H1c−/−/e−/− as compared to VavPBcl2-only mice (Fig.5d). H1c−/−;H1e−/− and H1c+/−;H1e+/−mice not harboring Bcl2 also had shorter survival compared to controls; and at early timepoints manifested lymphoproliferative disease invading extranodal tissues (Extended Data Fig.10g). We noted a trend for greater lethality of the heterozygous condition with two VavP-Bcl2;H1c+/−/e+/− animals already sick from immunoblastic DLBCL at the early timepoint, which did not occur in VavP-Bcl2 or VavP-Bcl2;H1c−/−/e−/− mice (Extended Data Fig.10h). These findings are consistent with H1 mutations being heterozygous in humans, perhaps to avoid loss of isoform-specific functions such as murine H1d uniquely interacting with DNA methyltransferases27,8, which might place homozygous mutants at a relative disadvantage.
Fig. 5. H1c/e deficiency leads to aggressive VavPBcl2 lymphomas.
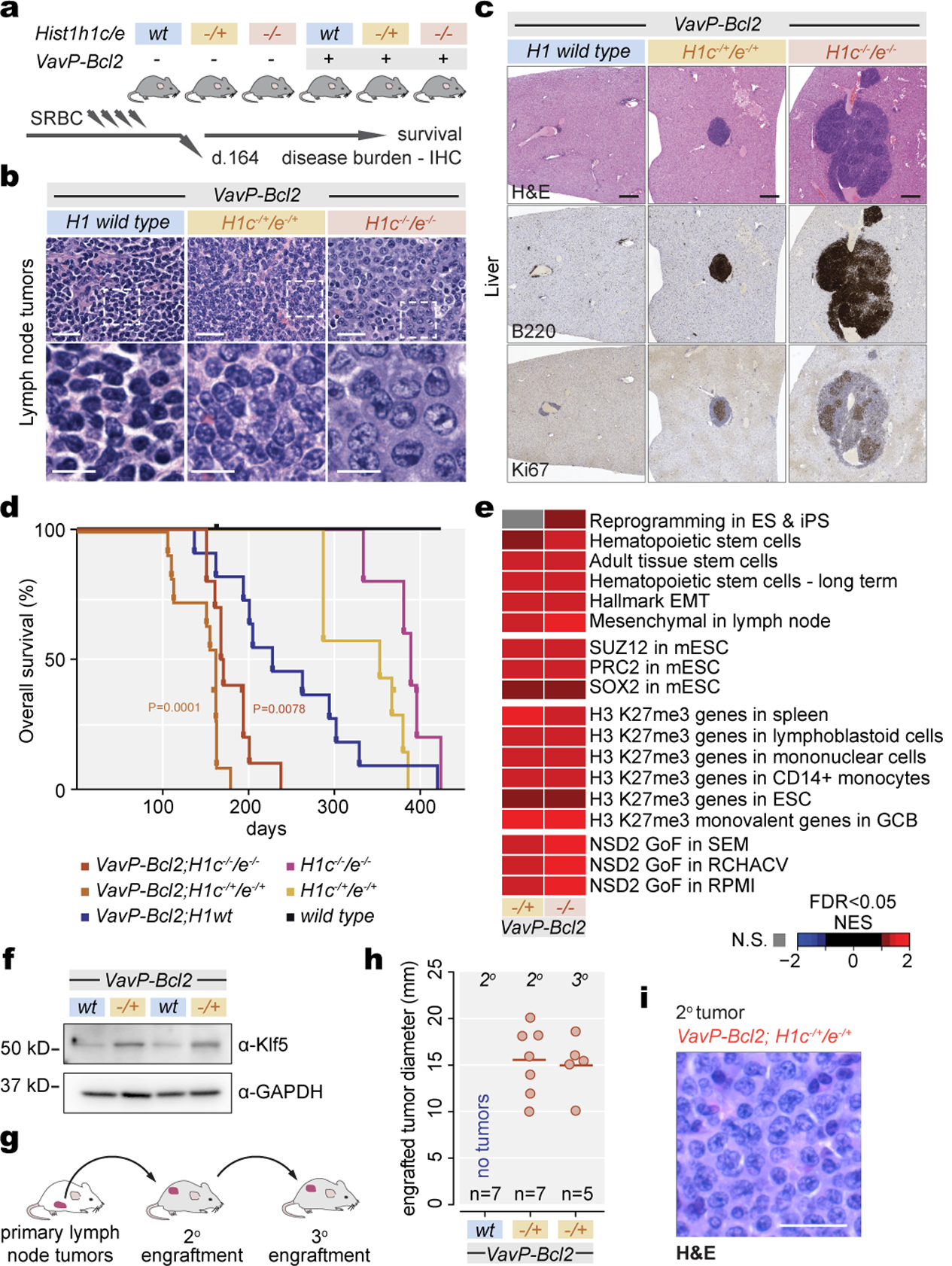
a, Lymphomagenesis experiment scheme. b, IHC images of H&E-stained lymph nodes from animals of indicated genotypes. Scale bars: 25 μm (top) and 10 μm (bottom). Images are representative of two independent experiments. c, Representative IHC images of liver tissue stained for H&E, B220 and Ki67 from animals of indicated genotypes. Scale bar, 100 μm. d, Kaplan-Meier curves depicting overall survival of VavP-Bcl2;H1c−/−/e−/− (n=10), VavP-Bcl2;H1c−/+/e−/+ (n=11), VavP-Bcl2;H1wt (n=11), H1c−/+/e−/+ (n=7), H1c−/−/e−/− (n=5), and WT mice (n=1), assessed by time of death or euthanasia after bone marrow transplantation (BMT). Log-rank test P-value is shown compared to VavP-Bcl2; H1wt control. e, Normalized enrichment score of GSEA on indicated gene sets, using ranked log2 fold change in expression between indicated genotypes vs VavP-Bcl2 control. f, Immunoblot for Klf5 protein from VavP-Bcl2;H1c−/+/e−/+ and control VavP-Bcl2;H1wt B220-enriched lymphoma cells with GAPDH loading control. Sample pairs collected over two independent experiments are shown. g, Tumor engraftment assay scheme. h, Tumor diameter after six weeks in secondary engraftments (n=7 recipients per genotype) and tertiary engraftments of VavP-Bcl2;H1c−/+/e−/+ into seven recipients. i, H&E staining of secondary engrafted VavP-Bcl2;H1c−/+/e−/+ lymphomas. Scale bar, 25 μm. Image is representative of n=7 mice.
RNA-seq performed in murine lymphomas showed significant similarity between the VavP-Bcl2;H1c+/−/e+/− and;VavP-Bcl2;H1c−/−/e−/− transcriptional profiles, as well as H1c−/−e−/− GCB-cells (Extended Data Fig.10i,j). Murine H1-deficient lymphomas featured upregulation of many genes including stem cell factors such as Klf5 (Extended Data Fig.10k) which was also induced at the protein level (Fig.5f). We observed significant enrichment for stem cell and mesenchymal transition genes, PRC2 and H3K27me3 target genes, and NSD2 gain-of-function signatures (Fig.5e). The human H1C/E mutant DLBCL signature was significantly enriched in VavPBcl2;H1c+/−/e+/− lymphomas with a trend also seen in VavPBcl2;H1c−/−/e−/− (Extended Data Fig.10l). The common theme of stem cell signatures among murine and human H1 deficient lymphomas prompted us to perform secondary transplantation of lymphoma cells from moribund VavP-Bcl2;H1c+/−/e+/− or VavP-Bcl2-only mice into NOD-SCID recipients (Fig.5g). All mice were sacrificed within six weeks, at which point 100% VavP-Bcl2;H1c+/−/e+/− but no VavP-Bcl2 mice had developed lymphomas (Fig.5h,i) Tertiary transfer into recipient NOD-SCID mice again yielded 100% engraftment of VavP-Bcl2;H1c+/−/e+/− cells (Fig.5h). Hence, loss of H1 endows lymphoma cells with enhanced self-renewal properties consistent with the highly aggressive nature of H1 mutant DLBCL.
In conclusion, our studies point to decompaction of 3D chromatin as the dominant effect of H1 loss of function in GCB-cells. In contrast, compartment shifting was not observed in triple Hist1h1c/d/e knockout embryonic stem cells28. Thus chromatin compartmentalization by H1 may occur specifically during differentiation, consistent with recent findings in conditional H1 triple knockout hematopoietic cells29. H1 isoforms may thus function as key maintenance factors for compaction and epigenetic memory, especially for regions initially marked for repression by PRC2 during lineage specification. We propose a stoichiometric gradient model to explain why GCB-cells are especially sensitive to H1 dosage (Extended Data Fig.10m), due to their extremely rapid proliferative rate10. Reduced H1 dosage in this context could impair maintenance of proper chromatin compartmentalization (and hence “epigenetic memory”) in daughter cells (i.e. centrocytes). The greater inefficiency in generating iPSC from terminally differentiated cells has been attributed to their restrictive epigenetic states30, an effect that our data suggest is reversed by H1 deficiency. Given that stem-like transcriptional programs are linked to cancer31, it is possible that silencing of these through H1-mediated compartmentalization represents a significant barrier to transformation of mature B-cells. The highly recurrent nature of H1 loss of function alleles in lymphoma may reflect the fitness advantage conferred to these mature cells by primitive stem cell programs that are normally sequestered within compartment B.
Extended Data
Extended Data Fig.1 |. Characterization of Hist1H1 allele mutations and deletions in DLBCL.
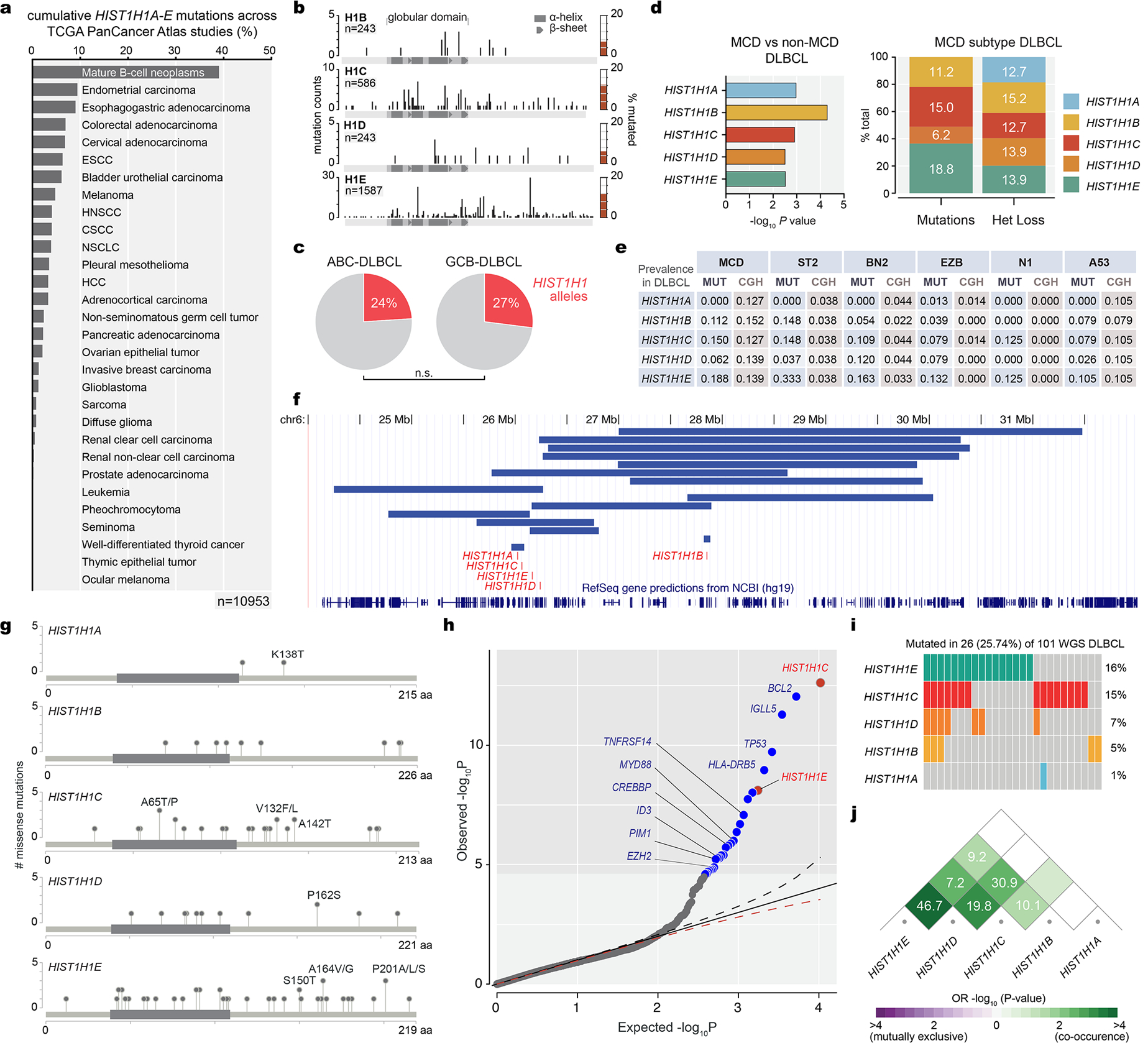
a, A PanCancer Atlas cancer mutation survey was performed using the cBioPortal to search the cumulative mutation frequency of HIST1H1A, HIST1H1B, HIST1H1C, HIST1H1D, HIST1H1E across a total of 10,953 non-redundant patient samples across all cancer types. b, HIST1H1B-E mutation landscape across non-redundant TCGA and BCCA DLBCL samples (number of samples indicated for each H1 isoform); location of mutations is plotted onto protein structure, with bar height corresponding to mutation counts, total frequency of mutations in each H1 isoform is plotted to the right (red bars). c, Prevalence of H1 mutant vs H1 wild-type cases in ABC- and GCB-DLBCL cases (Fisher’s exact test for enrichment in ABC or GCB-DLBCL, P>0.05) d,Prevalence of H1A-E missense mutations and heterozygous loss in MCD subtype DLBCL and their enrichment (-log10 Pvalue) over non-MCD DLBCLs. e, Summary table of frequency of mutations and heterozygous loss of HIST1H1A, HIST1H1B, HIST1H1C, HIST1H1D, HIST1H1E in DLBCL subtypes (MCD, ST2, BN2, EZB, N1, AP53). f, Co-occurrence as odds ratio (OR) and −log10 p-value among HIST1H1 alleles in 101 germline-matched WGS DLBCLs. g, Frequency and location of missense mutations for HIST1H1A, HIST1H1B, HIST1H1C, HIST1H1D, HIST1H1E in 101 germline-controlled DLBCLs. h, Quantile-quantile plot showing the P-values for SNV across 101 germline-matched WGS DLBCLs. Driver analysis derived as probability of mutation count greater than or equal to the observation mutation count under Gamma-Poisson distribution expected P-values for SNVs. Shaded in gray zone contains mutant genes with FDR<0.01, depicted as blue or red dots, several of which are listed in the figure. i, Oncoprint for HIST1H1A, HIST1H1B, HIST1H1C, HIST1H1D, HIST1H1E in 101 germline-matched WGS DLBCLs. j, Co-occurrence as odds ratio (OR) and −log10 p-value among HIST1H1alleles in 101 germline-matched WGS DLBCLs
Extended Data Fig.2|. HIST1H1 mutations are genetic drivers in lymphoma and confer loss of function.
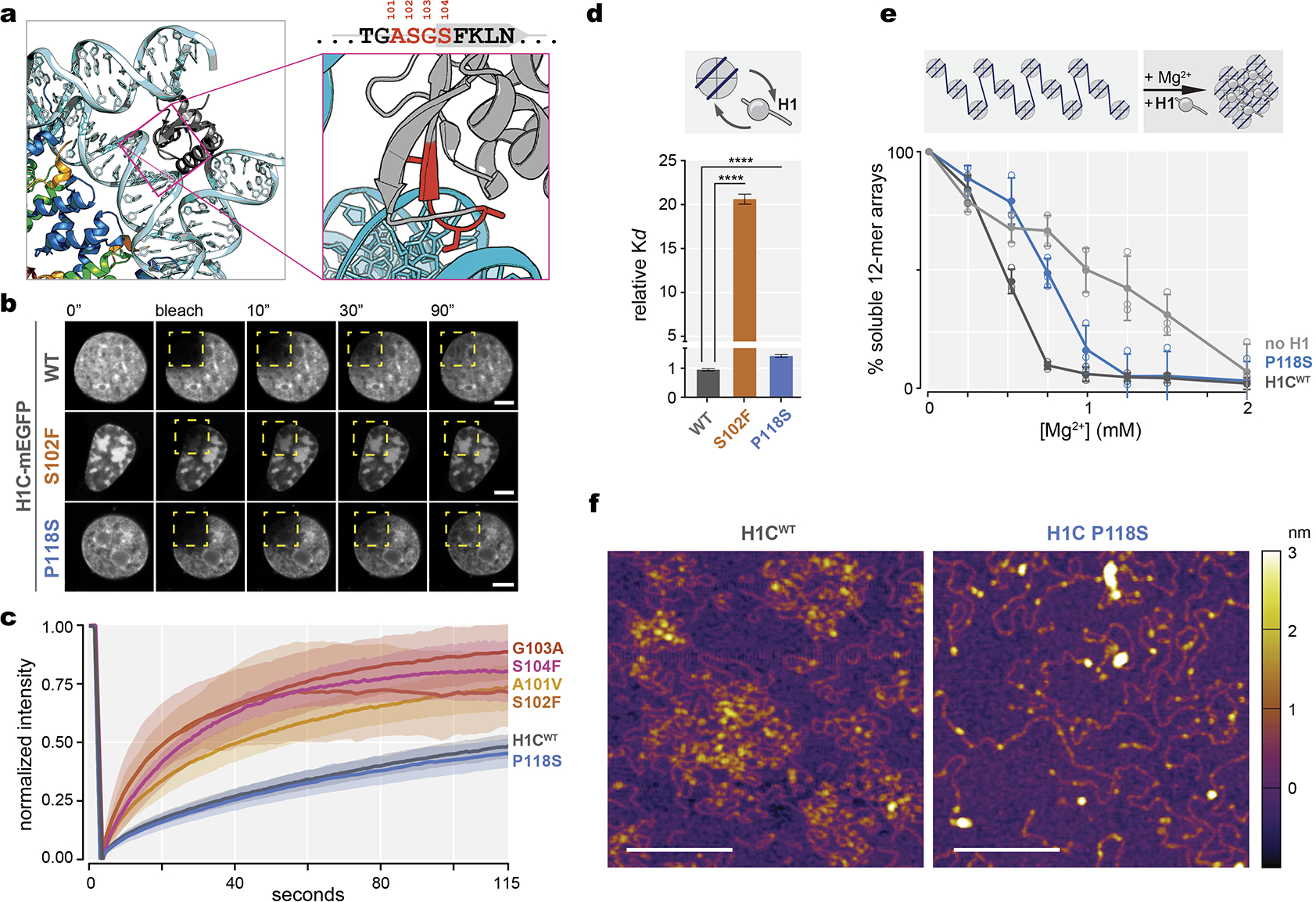
a, Crystal structure of the linker histone globular domain (gray) bound to nucleosome [PDB, 4QLC], with zoomed-in view of ASGS amino acid residues highlighted in red. b, Representative images of fluorescence recovery after photobleaching of ectopically expressed, meGFP-tagged wild-type H1C, S102F, and P118S mutants in 3T3 cells prior to, immediately after, and at 10, 30 and 90 seconds after bleaching the area (yellow dashed square). Scale bars = 5 μm. c, Quantification of normalized intensity as representation of turnover kinetics from (E) for wild-type H1C (n=18), and mutants A101V (n=15), S102F (n=9), S104F (n=10), G103A (n=10), and P118S (n=10) cell measurements, shaded area indicates 95% C.I. Data are pooled from two independent biological experiments.d, Dissociation constant (Kd) of recombinant mutant H1C S102F and P118S compared to WT H1C binding to mononucleosomes determined by biolayer interferometry. Data are mean ± s.e.m (two-sided unpaired t-test, ****P<0.0001). Data are global fit from five concentration measurements. e, Chromatin fiber oligomerization upon serial precipitation by Mg2+ as percent soluble 12-mer arrays was determined for no H1, WT H1C and C-terminal domain P118S mutant. Data are mean ± SD. Data are pooled from three independent biological experiments. f, Atomic force microscopy imaging of chromatin arrays in presence of wild-type H1 and C-terminal domain P118S mutant, scale bars, 200 nm. Images are representative from two independent biological experiments.
Extended Data Fig. 3 |. H1c−/−/e−/−mature B-cells manifest normal development in spleen and bone marrow.
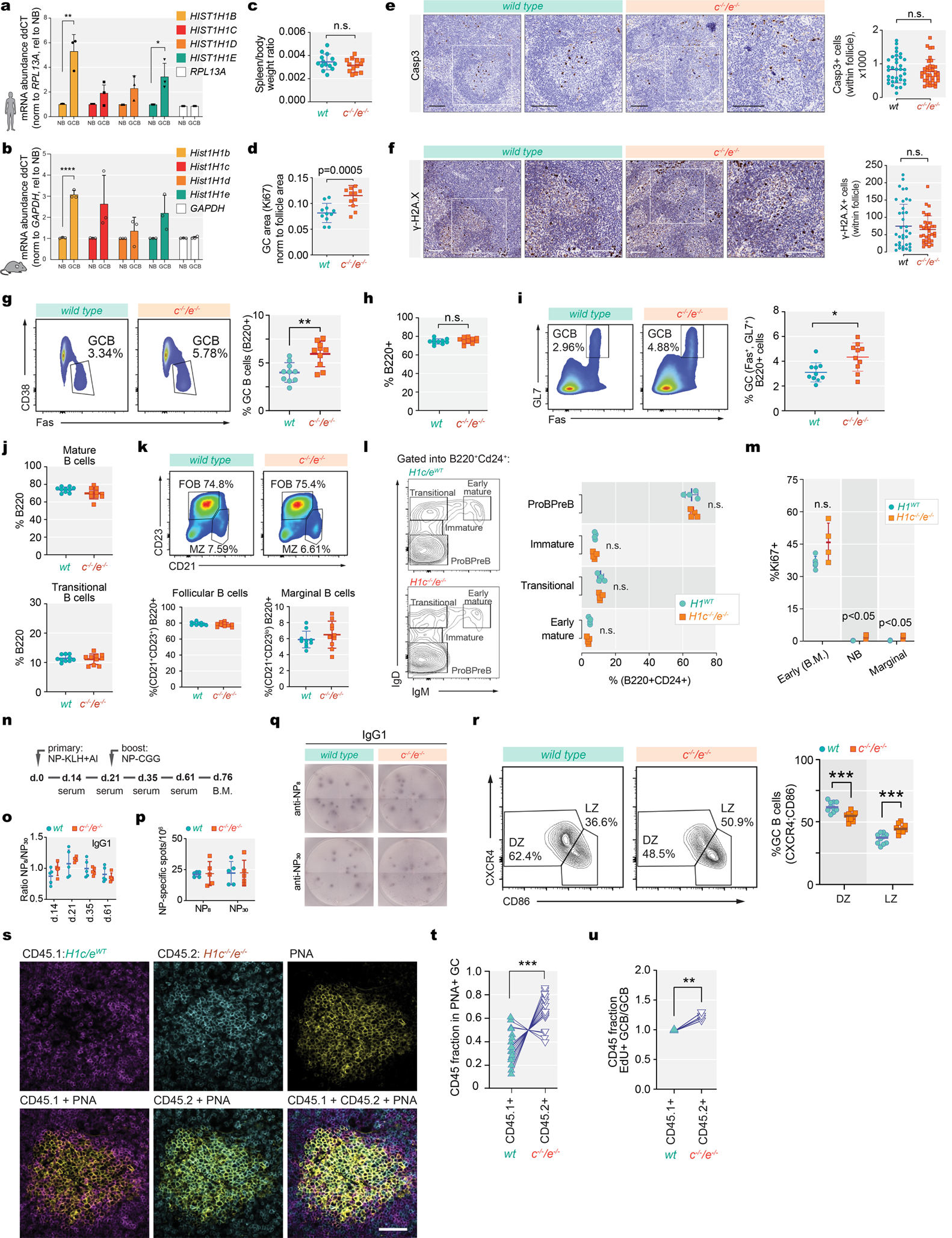
a, Human Hist1H1B-E mRNA normalized to RPL13A in GC B cells relative to Naïve B cells (HIST1H1B, **P=0.004; HIST1H1E, **P=0.027), isolated from three independent human tonsil specimens. Data are mean ± sd, two-sided unpaired t-tests. b, Mouse Hist1h1b-e mRNA levels normalized to GAPDH in sorted GC B-cells (n=3) relative to Naïve B-cells(n=3) (HIST1H1B, **P<0.0001). Data are mean ± sd, two-sided unpaired t-tests. c, Quantification of spleen/body weight ratios of two-months old H1c−/−/e−/− (n=13) and wild-type littermate control (n=14) mice. Data are pooled from two independent experiments. d, Quantification of GC area (Ki67 staining) in the spleens of H1c−/−/e−/−(n=10) and WT (n=10) mice. ***P=0.0005. Data are mean ± sd, .two-sided unpaired t-tests. e-f, Immunohistochemistry images of spleen sections of cleaved Casp3 (e) and gamma-H2AX (f) staining and quantification (right) of positively stained follicular cells from H1c−/−/e−/− (n=3) and littermate wild-type H1 control (n=3) mice immunized with SRBC and sacrificed 10d post immunization. Scale bars, 100 μm. P<0.05, two-sided unpaired t-tests. Data are mean ± sd. g, Flow cytometry analysis and quantification of (Fas+CD38−) GC B cells within total B-cells from H1c−/−/e−/−and WT mice (n=10 per genotype). Two-sided unpaired t-tests,**P=0.0018. Data are mean ± sd. h, Quantification of flow cytometry %B220+ of splenocytes in H1c−/−/e−/− (n=10) and WT (n=10) mice 9 days post SRBC immunization. P<0.05,two-sided unpaired t-tests. Data are mean ± sd. i, Flow cytometry analysis and quantification of GC B-cells (Fas+GL7+) from H1c−/−/e−/− (n=10) and WT (n=10) mice. Two-sided unpaired t-tests, *P=0.041.Data are mean ± sd. j, Flow cytometry analysis and quantification of mature B-cells (B220+IgD+IgM+) and transitional B-cells (B220+IgDintIgM+) in spleens from H1c−/−/e−/−(n=10) and WT (n=10) mice. P<0.05, two-sided unpaired t-tests. Data are mean ± sd. k, Flow cytometry quantification of follicular B cells (B220+D23+CD21+) and marginal zone B cells (B220+D23loCD21+) in spleens from H1c−/−/e−/−(n=10) and WT (n=10) mice. P<0.05, two-sided unpaired t-tests.Data are mean ± sd. l, Flow cytometry analysis gated on B220+CD24+ and quantification of ProPreB (IgM−IgD−), Immature (IgM−IgDlo), Transitional (IgD+IgM−), and Early Mature (IgD+IgM+) B cells in bone marrow of H1c−/−/e−/−(n=4) and WT (n=5). P<0.05, two-sided unpaired t-tests. Data are mean ± sd. m, Percentage of Ki67+early B-cells (B220+CD24+) in bone-marrow of H1c−/−/e−/− (n=4) and WT H1 (n=5) mice, as well as naive B-cells (***P=0.0004)and marginal zone B-cells (***P=0.001) in the spleens of H1c−/−/e−/− (n=5) and WT H1 (n=5) mice. n, Schematic diagram of primary NP-KLH and secondary immunization 21 days after with NP-CGG. o, Ratio between high (NP8) and low (NP30) affinity NP-specific IgG1 antibody titers in sera of H1c−/−/e−/− (n=5) and WT (n=5) mice by ELISA. P<0.05, two-sided unpaired t-testsData are mean ± sd. p, ELISPOT quantification of NP-specific (anti-NP8and anti-NP30) IgG1-secreting cells from the bone marrow of H1c−/−/e−/− (n=5) and WT (n=5) mice. P<0.05, two-sided unpaired t-test. Data are mean ± sd. Data are representative from two independent experiments. q, Representative images of anti-NP8 and anti-NP30 96-well ELISPOT. r, Flow cytometry analysis and quantification of centroblasts within dark zone (DZ) (CXCR4+CD86−), ***P=0.0002 and centrocytes within light zone (LZ) (CXCR4−CD86+), ***P=0.0002 within GCB cells from H1c−/−/e−/−(n=10) and WT (n=10) mice. Two-sided unpaired t-test, ***P=0.0002. Data are representative from three independent experiments. s, Immunofluorescence confocal microscopy images of GCs at day 7 post immunization in mixed chimeras. Scale bar = 50 μm. Images are representative of two independent experiments. t, Quantification based on (s) the fraction of PNA+CD45.1 or CD45.2 cells (17 GCs, n=3 mice). Two-sided paired t-test, ***P= 0.0004. u, Relative EdU+ GC B-cell/GC B-cell fraction for WT CD45.1+and H1c−/−/e−/− CD45.2+at day 7 post immunization (n=4 chimeras). Two-sided paired t-test, **P= 0.0065. Data are representative from two independent experiments.
Extended Data. 4 |. H1c/e deficiency induces stem cell transcriptional profiles in GCB cells and DLBCL.
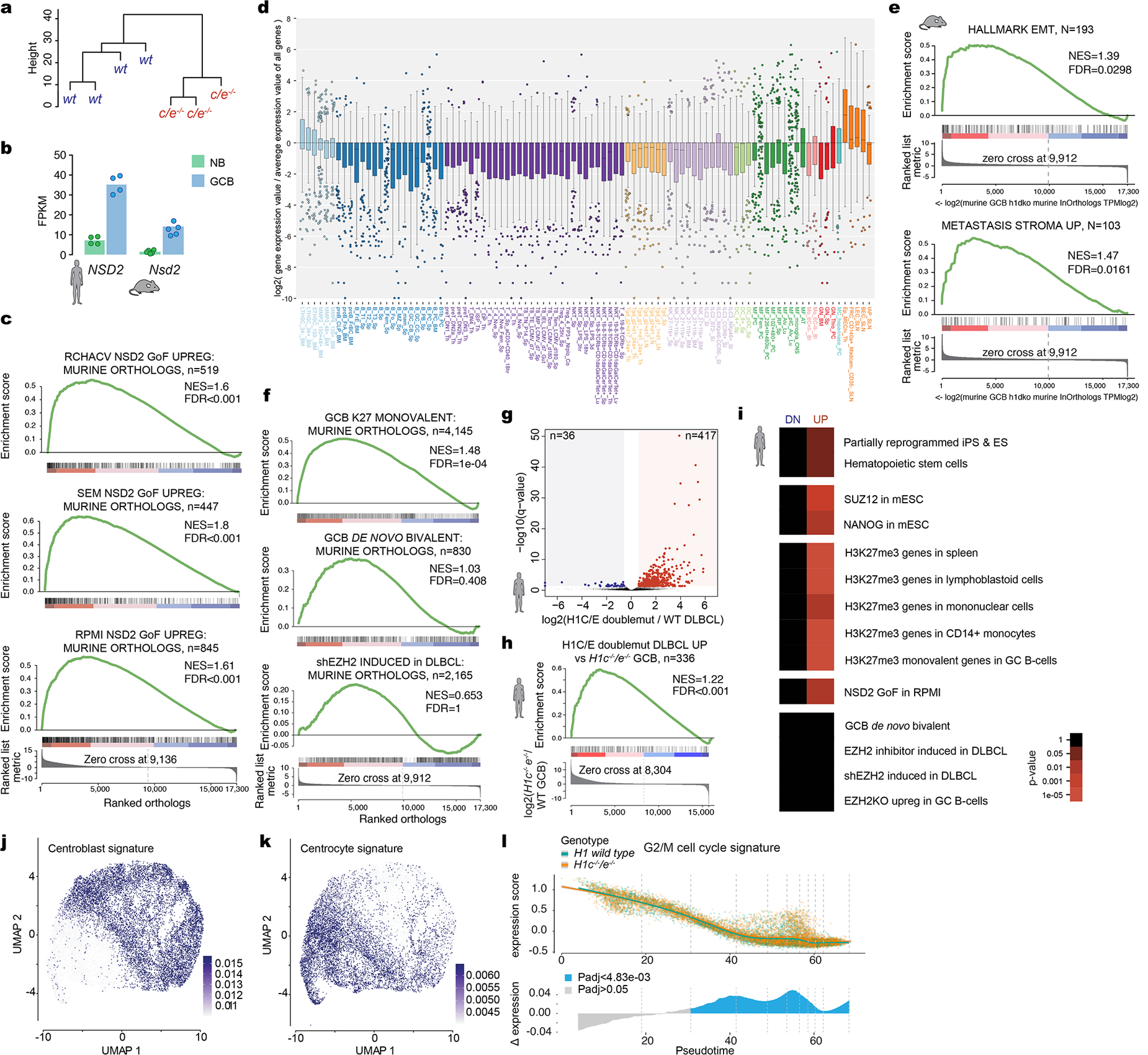
a, Unsupervised hierarchical clustering analysis of RNA-seq data from sorted H1c−/−/e−/−and wild-type H1 GCB cells, based on genes in the top 90thpercentile variability.b, FPKM expression of NSD2from human and mouse naïve B and GCB cells RNA-seq profiles. c, GSEA analysis of genes linked to NSD2 gain-of-function mutation in three cell lines (RCHACV, SEM, RPMI) against ranked murine H1c−/−/e−/−GC B cell expression changes. d, Boxplot of log2 relative gene expression normalized to average expression value of all genes from top 200 differentially upregulated H1c/e-deficient signature against ImmGen database. Boxplot center represents median, bounds of box are 1stand 3rdquartile and whiskers extend out 1.5*interquartile range from the box. e, GSEA analysis with indicated gene sets, using ranked log2 fold change in expression betweenH1c/e-deficient and wild-type murine GC B cells. f, GSEA analysis of gene sets linked to EZH2 against ranked murine H1c−/−/e−/−GC B cell expression changes. g, Volcano plot showing differentially expressed genes comparing H1C/E double mutant (n=18) vs H1 wild type DLBCL patients (n=237) (FDR <0.05, fold change >1.5). Red field denotes upregulated and blue field downregulated genes. h, GSEA analysis withH1C/E double mutant vs H1 wild-type DLBCL patient unregulated genes, using ranked log2 fold-change changes in murine GCH1c−/−/e-/−. i, Gene pathway enrichment analysis of upregulated and downregulated genes from (g), hypergeometric mean test. j-k, Sorted H1c−/−e−/−or WT GC B-cells (n=2 per genotype) were subjected to droplet based (10x) single cell RNA-seq. Centroblasts (j) and centrocytes (k) were defined based on enrichment for centroblast and centrocyte signature profiles, respectively projected onto the UMAP distribution of cells. l, Top:Expression of G2M cell cycle proliferation gene signature was plotted for each cell on the Y axis with spline curves representing the average for H1c−/−/e−/−and WT cells. Bottom: Differential expression is shown as delta spline plot (blue) across pseudotime and tested by two-sided Wilcoxon rank-sum within ten bins of equal cell number (dashed lines).
Extended Data Fig. 5 |. HiC Compartment analysis and integration with ATAC-seq inH1c−/−/e−/−and wild-type H1 GCB-cells.
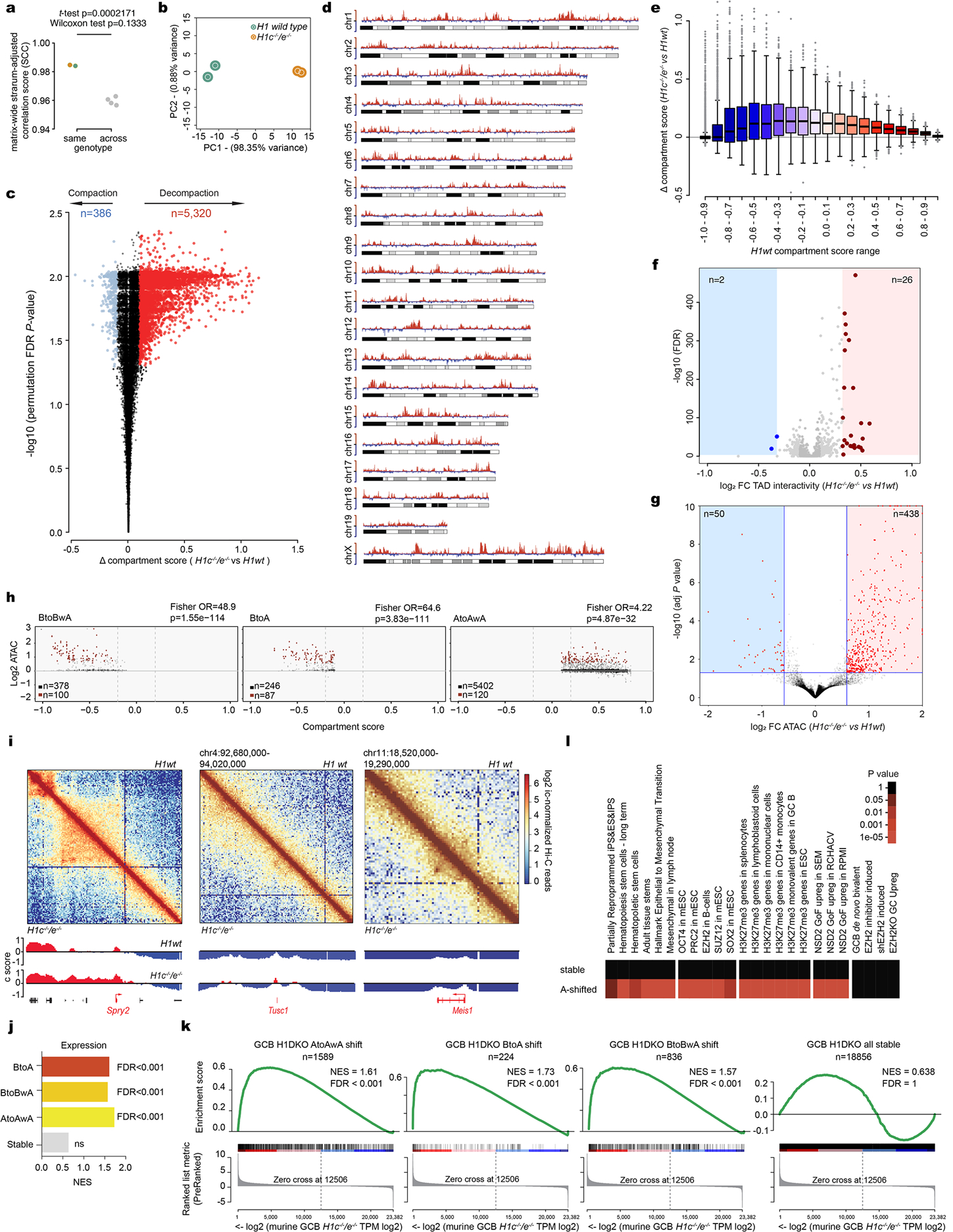
a, Genome-wide correlation score (Stratum adjusted correlation score SCC) of HiC matrices within same genotype (***P=0.0002171, two-sided unpaired t-test), and across genotypes (P<0.05, two-sided Wilcoxon test). b, PCA of compartment bins processed with Hi-C bench at a resolution of 100kb from Hi-C replicates for H1c−/−/e−/−and WT GCB-cells. c, Volcano plot showing significant compartment score shifts in H1c−/−/e−/−murine GC B-cells based on delta compartment score and −log10 (permutation FDR P-value): Decompacted (red) compartment bins n= 5320 and compacted (blue) compartment bins (n=386). d, Delta compartment score (H1c−/−/e−/− vs wild-type) across mouse chromosomes (positive y axis is in red for decompacting loci and negative y-axis is blue for compacting loci) plotted as ideograms. e, Boxplot of delta compartment score (H1c−/−/e−/− vs wild-type) across compartment score range (−1.0 to 1.0) separated into 0.1 bins. No statistical evaluation was derived for this graph. f, Volcano plot showing TADs with significant gain of intra-TAD interactivity (n=26, red) and reduction of intra-TAD interactivity (n=2, blue; two-sided unpaired t-test FDR-adjusted P-value<0.05,fold-change>1.5). g, Volcano plot showing significant ATAC-seq peaks gaining accessibility (n=438) and losing accessibility (n=53; two-sided unpaired t-test FDR-adjusted p-value<0.05, fold-change>1.5). h, Scatter plot showing ATAC-Seq peaks log2FC betweenH1c−/−/e−/− vs WT GCB-cells in decompacting compartments versus WT compartment score: B-to-BwA (left), B-to-A (middle), and A-toAwA (right). Peaks gaining accessibility (FC>1.5, Padj<0.05) are marked in red.Estimated odds ratios and p-values were calculated using Fisher’s Exact Test. Although ATACseq peaks are more prevalent in AtoAwA compartment (right), increased accessibility is more enriched in BtoBwA and BtoA compartments. I, HiC contact maps of regions surrounding Spry (left), Tusc1 (middle) and Meis1(right) genes. The top of each square shows H1 wild-type contacts and the bottom of each square those in H1c−/−/e−/−GC B-cells.Heatmaps represent the Pearson correlation of interactions in wild-type H1 and H1c−/−/e−/−GCB-cells. Bottom tracks represent the Eigenvector (PC1) for compartments A and B in red and blue, respectively and show the position of genes within these loci. j-k,GSEA analysis of genes shifting to A compartments (BtoA, BtoBwA, AtoAwA) or genes contained in stable compartments using ranked log2 gene expression in H1c−/−/e−/−GCB-cells. (NES and FDR values as implemented by GSEA). l, Gene pathway enrichment analysis of genes in decompacting and stable compartments in H1c−/−/e−/−vs wild-type GC B-cells (Hypergeometric mean test).
Extended Data Fig. 6 |. 3D changes due to H1c/e deficiency in GCB-cells recapitulate decompaction during iPS differentiation.
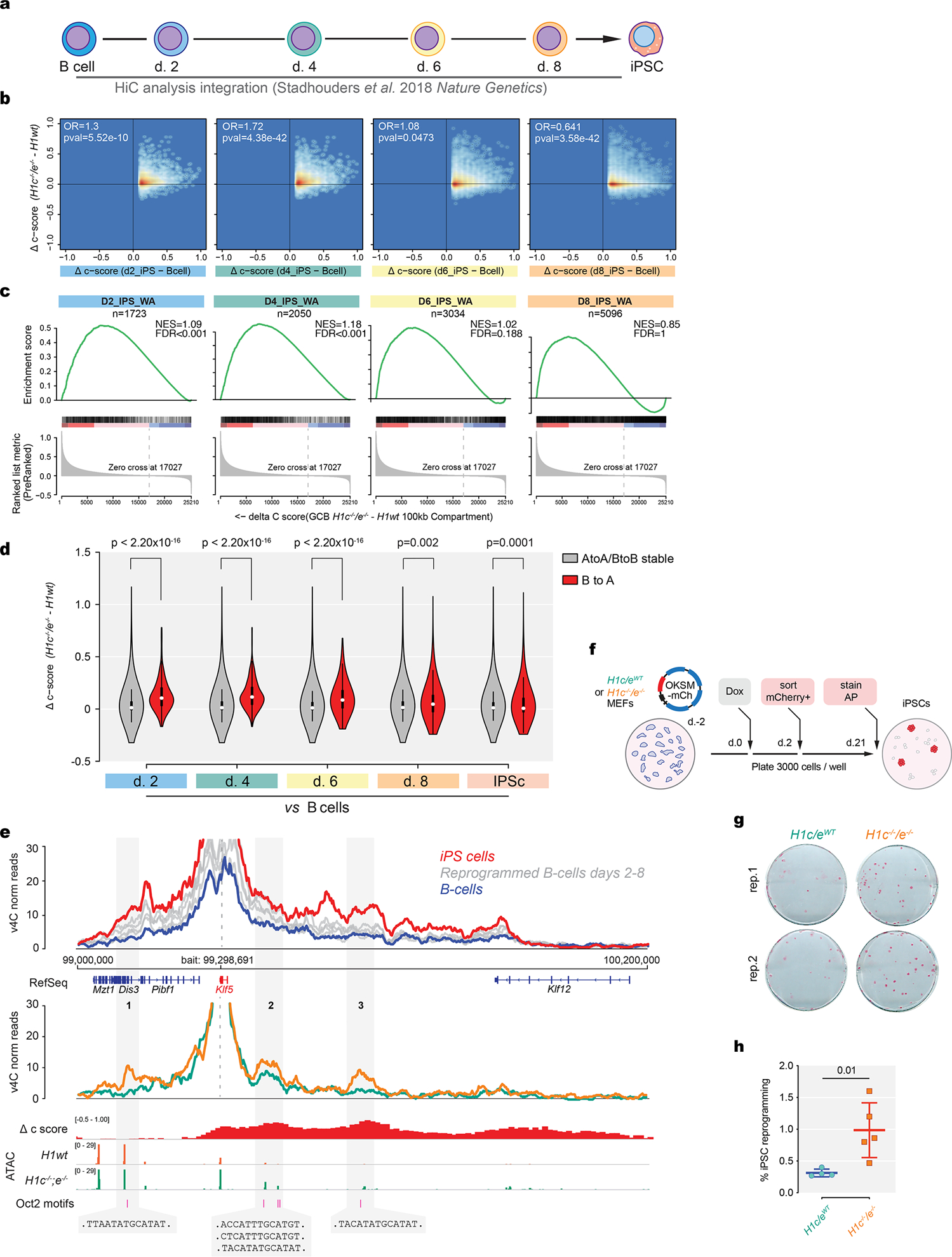
a,Schematic of B-cell differentiation to iPS study (Stadhouders et al., 2018) with time-points for HiC analysis. b, Correlation plots comparing shifting to A compartment scores in iPS (Day 2, Day 4, Day 6, Day 8) compared to control B-cells versus compartment score changes in H1c/e-deficient GC B-cells. Estimated odds ratios and p-values were calculated using Fisher’s exact test.c, GSEA analysis of shifting to A compartments in iPS (Day 2, Day 4, Day 6, Day 8) against ranked delta compartment scores derived from H1c/e-deficient minus wild-type H1 murine GC B-cells (NES and FDR values as implemented by GSEA). d, Violin plots comparing stable and shifting B to A compartments during iPS differentiation (Day 2, Day 4, Day 6, Day 8 and fully undifferentiated) to the estimated delta compartment score (c-score) due to H1c/e deficiency of those same regions in GC B-cells. (Day 2, P<2.2×10−16; Day 4, P<2.2×10−16; Day 6, P=0.002; Day 8, 0.0001; estimated with two-sided Wilcoxon test). Boxplot center represents median, bounds of box are 1st and 3rd quartile and whiskers extend out 1.5*interquartile range from the box. e, Virtual 4C analyses on KLF5 locus (chr14: 99,000,000–100,200,000) anchored on KLF5 promoter for (top) B-cell reprogramming (blue) to iPS (red) with 4 time intermediate states (gray) from Stadhouders et al.study as well as GCB-cells (bottom) (H1c−/−/e−/−and wild-type H1). IgV tracks below comprise delta compartment score and ATAC-seq signal in H1c−/−/e−/−vs WT GCB-cells. Gained HiC interactions in H1c−/−/e−/−compared to WT GCB-cells (shaded in gray 1–3: pval=0.04; pval=0.059; pval=0.02 respectively, two-sided unpaired t-test) have Oct2 motif sequences as shown. f, Schematic of experimental setup with H1c−/−/e−/− or littermate WT H1 mouse embryonic fibroblasts. g, Representative images of alkaline phosphatase (AP) stained H1c−/−/e−/− and wild type H1 iPSC day 21 colonies. h,Percent iPSC reprogramming efficiency of H1c−/−/e−/− (n=5 transfections on 2 biological replicates) and WT (n=4 transfections on 2 biological replicates) mouse embryonic fibroblasts determined as the ratio of AP+ colonies to the number of seeded mCherry+cells.P=0.01,two-sided unpaired t-tests.Data are mean ± sd. Data are representative from three independent experiments.
Extended Data Fig. 7. Altered H3K36me2 and H3K27me3 distribution in H1c/e-deficient GCB-cells.
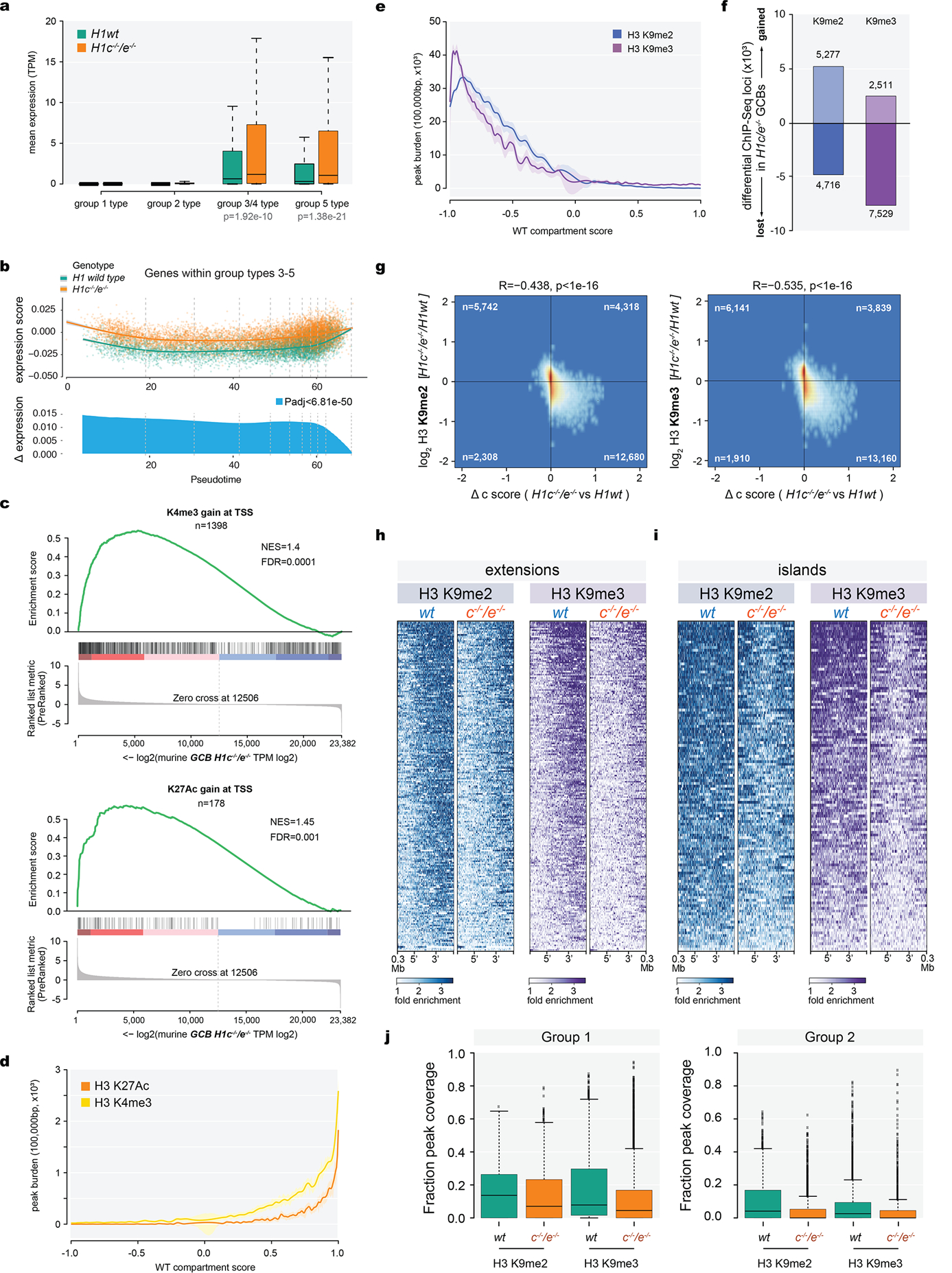
a-b, Mass spectrometry of H3 K36 (a) and K27 (b) post-translational modifications, log2-transformed and normalized to average wild type peak area from H1c−/−/e−/−(n=5) and WT (n=7) acid-extracted samples from GCB-cells; two-sided unpaired t-test: K36 unmod, ***P=0.0005; K36me1, ***P=0.0003; K36me2 ***P=0.0002; K36me3, P=0.93; K36ac, P=0.56; K27unmod, *P=0.0157; K27me1, **P=0.0072; K27me2, *P=0.0175; K27me3, ***P=0.0007; K27ac, P=0.9337. box plots show median and 25thto 75thpercentile, whiskers indicate data range; Data are representative from two independent experiments. c, Immunoblots for H1 (D4J5Q and AE-4 antibodies), H3K36me2, H3K27me3, EZH2, and NSD2 from sorted wild-type and H1c−/−/e−/−GC B-cells. Direct blue stain is included as loading control. A representative image of at least three experiments is shown. Uncropped gels are shown in Supplementary Figure 1. d, Mass spectometry-based relative abundance of H3.1/.2 (replication-dependent) and H3.3 (replication-independent) isoforms, shown as average % total peak area of H3 K27-K36 peptide containing H3.3-specific S31, in acid-extracted histones from WT (H3.3, 15.76%, n=7) and H1c−/−/e−/− (H3.3, 15.07%, n=5) GCB-cells; two-sided unpaired t-test, P=0.0004, Data are mean ± SD. e, Mass spectrometry of H3 K36 (top) and K27 (bottom) post-translational modifications across H3.1/2 (left) and H3.3 (right) isoforms, log2-transformed and normalized to average wild type peak area from samples acid-extracted from wild type (n=7) and H1c−/−/e−/−(n=5) GC B-cells; Two-sided unpaired t-test: H3.1/2 K36 unmod, ***P=0.0005; H3.1/2 K36me1, ***P=0.0003; H3.1/2 K36me2, ****P<0.0001; H3.1/2 K36me2, P=0.88; H3.1/2 K27unmod, *P=0.0100; H3.1/2 K27me1, *P=0.0162; H3.1/2 K27me2, *P=0.0129; H3.1/2 K27me3, ***P=0.0002; H3.3 K36unmod, ****P<0.0001; H3.3 K36me1, *P=0.036; H3.3 K36me2, P=0.15; H3.3 K36me3, P=0.5974; H3.3 K27unmod, P=0.1187; H3.3 K27me1, P=0.4743; H3.3 K27me2, P=0.1199; H3.3 K27me3, P=0.0628. Box plots show median and 25thto 75thpercentile, whiskers indicate data range. f, Unsupervised hierarchical clustering analysis of ChIP-seq data for H3K27me3 and H36me2 in biological triplicates from sorted H1c−/−/e−/−and wild-type H1GC B-cells.g, Genome wide correlation plot of log2 fold (H1c−/−/e−/−vs wild-type H1) change of normalized reads within ChIP-Seq peak union for H3K36me2 and H3K27me3. (Pearson correlation coefficient R=−0.453, P<1e-16). h, Heatmap of HiC compartment score, H3K36me2 and H3K27me3 centered within shifting B to A compartments (100kb) and surrounding 300kb for H1c−/−/e−/−and wild-type GC B-cells for compartment “extensions” (top). i, Fraction of ChIP-Seq peak coverage (H3K36me2 in red and H3K27me3 in blue) within 100kb compartments across HiC compartment score (X axis, −1 to 1) for WT GCB-cells. Cubic smoothing spline of data is presented with error bars as shaded regions indicate 99% confidence intervals. j, Fraction peak (H3K27me3 and H3K36me2) coverage of regions within shifting compartment groups 1–5 in H1c−/−/e−/−and wild-type H1 GC B-cells. Paired Wilcoxon test, Group 1: H3K27me3, P<1e-16; H3K36me2, P=0.597;Group 2: H3K27me3, P<1e-16; H3K36me2, P<1e-16; Group 3: H3K27me3, P=3.38e-13; H3K36me2, P<1e-16;Group 4: H3K27me3, P=8.71e-15; H3K36me2, P<1e-16;Group 5: H3K27me3, P<1e-16; H3K36me2, P<1e-16. Boxplot center represents median, bounds of box are 1stand 3rdquartile and whiskers extend out 1.5*interquartile range from the box. k,Scatter plot of H3K27me3 peak log2 fold change(H1c/e deficient compared to WT GCB-cells) versus WT compartment score for decompacting group 2. Gain of H3K27me3 (red dots) largely occurred within regions shifting from compartment B while loss of H3K72me3 (blue dots) were more prevalent within regions shifting compartments from compartment A.
Extended Data Fig. 8 |. Changes in activation marks H3K4me3 and H3K27ac and repressive marks H3K9me2 and H3K9me3 in H1-deficient GCB-cells are associated with compartment B decompaction.
a, Genes defined as type Groups 3 and 4 (n=108 genes) and Group 5 (n=152 genes) manifest transcriptional activation and significant upregulation in H1c−/−/e−/−GCB-cells compared to WT GCB-cells. Paired Wilcoxon test. Boxplot center represents median, bounds of box are 1stand 3rdquartile and whiskers extend out 1.5*interquartile range from the box. b, Top: Expression of the genes defined as type Groups 3–5 was plotted for each cell on the Y axis. Average expression is represented by the different colored spline curves for each genotype as indicated. Bottom: Differential expression between H1c−/−/e−/− and wild type cells were represented as the delta spline plot across pseudotime. Cells are divided by pseudotime into bins of equal cell number (gray vertical dashed lines) and tested for signature enrichment compared to WT GCB-cells by two-sided Wilcoxon (P < 6.81−50). c, Fraction of histone peak coverage for H3K4me3 and H3K27Ac within 100kb compartments across HiC compartment score (X axis) for wild-type H1 GC B-cells. Cubic smoothing spline of data is presented with error bars as shaded regions indicate 99% confidence intervals. d, GSEA with genes marked with gain in H3K4me3 (top) or H3K27Ac (bottom) on their promoters (TSS +/−500 bp) using the ranked log2 fold-change in murine H1c−/−/e−/−GCB-cells. e, Fraction of histone peak coverage (for H3K9me2 and H3K9me3) within 100kb compartments across HiC compartment score (X axis) for wild-type H1 GC B-cells. Cubic smoothing spline of data is presented with error bars as shaded regions indicate 99% confidence intervals. f, CUT&RUN peaks for H3K9me2 and H3K9me3 show altered abundance between H1c−/−/e−/− compared to WT GC B-cells (FC>1.5, n=5277 gained and n=4717 lost H3K9me2 and n=2511 gained and n=7529 lost H3K9me3 peaks). g, Genome wide correlation plot of log2 fold H3K9me2 (left) and H3K9me3 (right) (H1c−/−/e−/−vs wild-type H1) peaks versus delta compartment score (H1c−/−/e−/−- wild-type H1). Both H3K9me2 and H3K9me3 changes were largely loss and were anti-correlated with compartment decompaction (Pearson correlation coefficient R=−0.438, P<1e-16 and R=−0.543, P<1e-16, respectively). h-i, Heatmaps of H39me2 and H3k9me3 centered within shifting B to A compartments (100kb) and surrounding 300kb for H1c−/−/e−/−and WT GCB-cells for compartment “extensions” (h) and “islands” (i). j, Fraction peak (H3K9me2 and H3K9me3) coverage of regions within shifting compartment groups 1–2 in H1c−/−/e−/−and WT GCB-cells. Paired Wilcoxon test, Group 1: H3K9me2, P<1e-16, H3K9me3, P<1e-16; Group 2: H3K9me2, P<1e-16; H3K9me3, P<1e-16 ; Boxplot center represents median, bounds of box are 1st and 3rdquartile and whiskers extend out 1.5*interquartile range from the box.
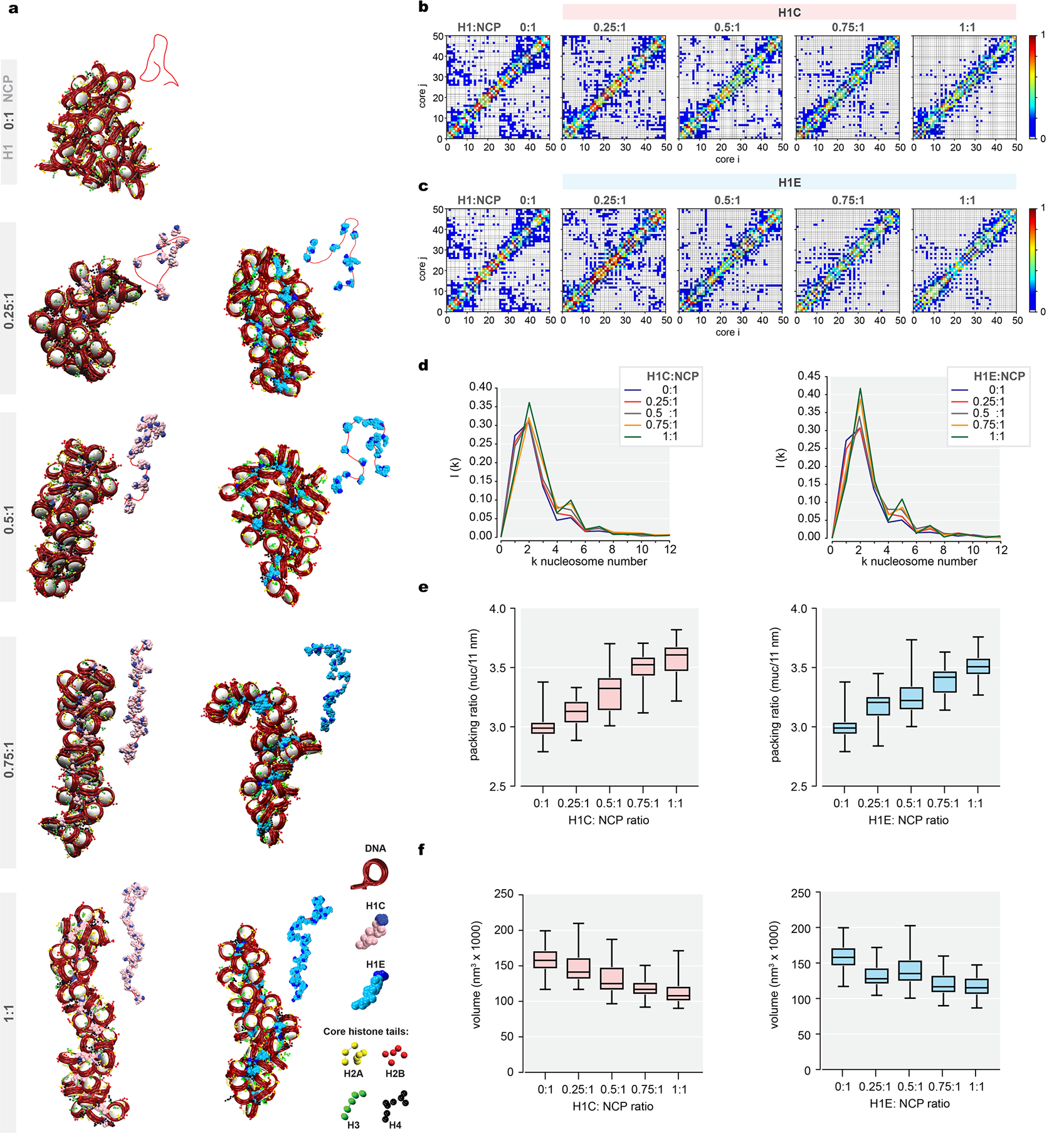
Extended Data Fig.9. Linker histone incorporation reduces interactivity of chromatin fiber.
a, Representative equilibrated configurations of 50-nucleosome chromatin fibers obtained in silico in absence of H1 and in presence of 0.25, 0.5, 0.75, and 1 H1 per nucleosome - H1C (left) and H1E (right). Fiber contour (in red) on which the H1 mean positions are shown is shown on the top right of each model. Color key shows DNA, linker histone, H2A, H2B, H3, and H4 tails. b-c, Contact maps for the 1000-configuration ensembles obtained from left to right, in absence of H1 and in presence of 0.25, 0.50, 0.75, and 1 H1 per nucleosome, H1C in (b) and H1E in (c). d, The nucleosome/nucleosome interaction patterns, or a one-dimensional decomposition of each contact map shown in (b) and (c), indicate the dominant zigzag pattern of the fiber (i+/−2) and increase of long-range interactions as the H1C (left) or H1E (right) density decreases. e, Packing ratio calculated as the number of nucleosomes contained in 11 nm of fiber for systems without H1 and in presence of 0.25, 0.50, 0.75, and 1 H1 per nucleosome (left, H1C and right, H1E). n=1000 chromatin ensemble configurations for each H1 per nucleosome ratio. Ordinary one-way ANOVA for multiple comparisons, ****P<0.0001. Boxplot center represents median, bounds of box are 1stand 3rdquartile and whiskers extend out 1.5*interquartile range from the box. f, Volume of chromatin fibers calculated assuming a cylindrical shape for systems with no H1 and in presence of 0.25, 0.5, 0.75, and 1 H1 molecule per nucleosome (left, H1C and right, H1E), n=1000 chromatin ensemble configurations for each H1 per nucleosome ratio. Ordinary one-way ANOVA for multiple comparisons, ****P<0.0001. Boxplot center represents median, bounds of box are 1st and 3rd quartile and whiskers extend out 1.5*interquartile range from the box.
Extended Data Fig.10 |. H1c/e loss leads to aggressive Vav-PBcl2 lymphomas with DLBCL-like morphology.
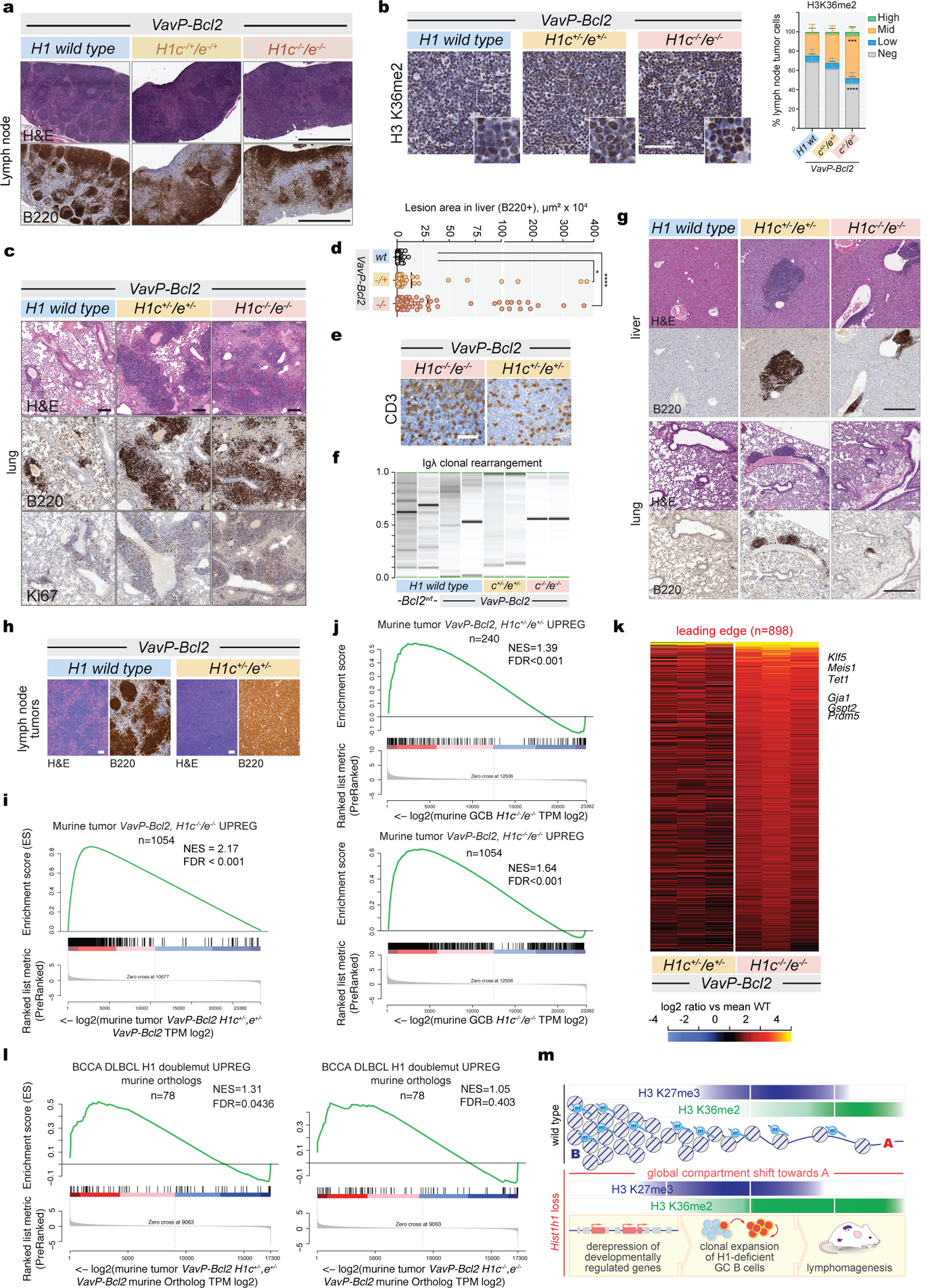
a, Immunohistochemistry images of lymph node stained for H&E and B220 from VavP-Bcl2;H1c−/−/e−/−, VavP-Bcl2;H1c−/+/e−/+, and VavP-Bcl2;H1 wild type animals at day 164. Scale bar, 1 mm. Images are representative of n=11 mice per genotype examined over 2 independent experiments. b, Representative immunohistochemistry stains of lymphomatous VavP-Bcl2;H1c−/−/e−/−, VavP-Bcl2;H1c−/+/e−/+, and VavP-Bcl2;H1 wild type lymph nodes stained with H3K36me2 ,and quantification of intensity (binned as high, mid, low and negative). Scale bar, 50 μm. Tissue derived from 3 animals per genotype with 4 tumor lymph nodes each. Data are mean±sd. two-sided unpaired t-test. c, Immunohistochemistry images of lung tissue stained for H&E, B220 and Ki67 from VavP-Bcl2;H1c−/−/e−/−, VavP-Bcl2;H1c−/+/e−/+, and VavP-Bcl2;H1 wild type animals at day 164. Scale bar, 100 μm. Images are representative of n=11 mice per genotype examined over 2 independent experiments. d, Quantification of B220 lesion areas in liver tissue (Fig. 5c) from VavP-Bcl2;H1c−/−/e−/− (****P<0.0001) and VavP-Bcl2;H1c−/+/e−/+ (*P=0.0308) compared to VavP-Bcl2 (n=7 mice per genotype, mean±SD; two-sided unpaired t-tests). e, Immunohistochemistry stains for CD3 from VavP-Bcl2;H1c−/−/e−/− and VavP-Bcl2;H1c−/+/e−/+ lymphomas. Scale bar, 50 μm. Images are representative of n=4 mice per genotype examined over 2 independent experiments. f, PCR for Igλ clonal rearrangement to report on tumor clonality of B220+ cells from VavP-Bcl2;H1c−/−/e−/−, VavP-Bcl2;H1c−/+/e−/+, VavP-Bcl2; and WT mice at day 164. g, Immunohistochemistry stains for H&E and B220 in liver and lung tissues from H1c−/−/e−/−, H1c−/+/e−/+, and WT mice at day 164. Scale bar, 500 μm. Images are representative of n=6 mice per genotype examined over two independent experiments. h, Immunohistochemistry images of lymph node tissue stained for H&E and B220 from VavP-Bcl2;H1c−/+/e−/+ and VavP-Bcl2; H1 wild type mice. Images are representative of n=4 mice per genotype examined over two independent experiments. Scale bars, 100 μm. i, GSEA with the VavP-Bcl2;H1c−/−/e−/− vs VavP-Bcl2 lymphoma upregulated gene set ranked against log2 fold-change changes from murine VavP-Bcl2;H1c−/+/e−/+ vs VavP-Bcl2. j, Top: GSEA with genes upregulated in VavP-Bcl2;H1c−/+/e−/+ vs VavP-Bcl2 lymphomas using the ranked log2 fold-change in murine H1c−/−/e−/− GC B-cells. Bottom: GSEA with genes upregulated in VavP-Bcl2;H1c−/−/e−/− vs VavP-Bcl2 mice using the ranked log2 fold-change in murine H1c−/−/e−/− GC B-cells. k, Heatmap showing differential expression of leading edge genes (n=898) from VavP-Bcl2;H1c−/−/e−/− and VavP-Bcl2;H1c−/+/e−/+ lymphomas. l, GSEA with human DLBCL H1C/E double mutant upregulated genes, against the ranked log2 fold-change gene expression profiles of murine VavP-Bcl2;H1c−/+/e−/+ (left) and VavP-Bcl2;H1c−/−/e−/− (right) lymphomas. m, Summary model depicting chromatin as contiguous B-to-A space, with H3 K27 and K36 methylations occupying distinct compartments within. Loss of H1 results in global shift of compartment interactivity towards A, with both H3 K27 and K36 methylations shifting into ectopic regions. Most compacted regions devoid of either K27 or K36 methylation appear largely protected from H1 loss. Biological effects of H1 loss in GC B-cells are summarized below.
Supplementary Material
ACKNOWLEDGEMENTS
E.C., A.M. and C.D.A. are funded through NIH/NCI R01 CA234561 and STARR I9-A9-062. AM and AT are funded by NIH/NCI P01 CA229086. Research in CDA lab is also supported by the NCI P01 CA196539 Leukemia and Lymphoma Society (LLS-SCOR 7006-13), The Rockefeller University and St Jude Children’s Research Hospital Collaborative of Chromatin Regulation in Pediatric Cancer, and Robertson Therapeutic Development Fund. A.M. is also funded by NIH/NCI R35 CA220499, LLS TRP 6572, LLS SCOR 7012 ,the Follicular Lymphoma Consortium, the Samuel Waxman Cancer Research Foundation and the Chemotherapy Foundation. JDL, AM and CDA are funded by LLS SCOR 17403-19 and JDL is funded by R01 CA195732 and The Samuel Waxman Cancer Research Foundation. NY is funded by CDMRP (CA181397). Research in EpiCypher is supported by R44 DE029633 and R44 GM116584. AAS was funded by the Damon Runyon Cancer Research Foundation (DRG-2185-14). A.S. is funded through GM116143. Histone proteomics work was performed at Northwestern Proteomics, generously supported by NCI CCSG P30 CA060553 awarded to the Robert H Lurie Comprehensive Cancer Center, instrumentation award (S10OD025194) from NIH Office of Director, and the National Resource for Translational and Developmental Proteomics supported by P41 GM108569. The in-silico modeling work was supported by award R35-GM122562 and Philip Morris and Philip Morris International award to T.S. The authors thank the Weill Cornell Medicine’s Laboratory of Comparative Pathology, the Epigenomics Core, the Flow Cytometry Core Facility, the Optical Microscopy Core, Rockefeller University Genomics Resource Center, Rockefeller University Bio-Imaging Resource Center, and New York University Langone Health’s Genome Technology Center.
Footnotes
Data availability
All sequencing data that support the findings of this study have been deposited in the National Center for Biotechnology Information Gene Expression Omnibus (GEO) accession number GSE143293. All other data that supported the finding of this study are available from the corresponding author upon request.
CONFLICT OF INTEREST
A.M. has research funding from Janssen Pharmaceuticals and Daiichi Sankyo, has consulted for Epizyme and Constellation and is on the advisory board for KDAC Pharma. NLK is a consultant for Thermo Fisher Scientific. C.D.A is a co-founder of Chroma Therapeutics and Constellation Pharmaceuticals, and a Scientific Advisory Board member of EpiCypher. EpiCypher is a commercial developer of the CUTANA® CUT&RUN platform. M.I. has received consultancy fees from Novartis Venture Fund outside of the scope of the work.
References
- 1.Fyodorov DV, Zhou BR, Skoultchi AI & Bai Y Emerging roles of linker histones in regulating chromatin structure and function. Nat Rev Mol Cell Biol 19, 192–206, doi: 10.1038/nrm.2017.94 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Fan Y et al. Histone H1 depletion in mammals alters global chromatin structure but causes specific changes in gene regulation. Cell 123, 1199–1212, doi: 10.1016/j.cell.2005.10.028 (2005). [DOI] [PubMed] [Google Scholar]
- 3.Cao K et al. High-resolution mapping of h1 linker histone variants in embryonic stem cells. PLoS Genet 9, e1003417, doi: 10.1371/journal.pgen.1003417 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Zhang Y et al. Histone h1 depletion impairs embryonic stem cell differentiation. PLoS Genet 8, e1002691, doi: 10.1371/journal.pgen.1002691 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Nacev BA et al. The expanding landscape of ‘oncohistone’ mutations in human cancers. Nature 567, 473–478, doi: 10.1038/s41586-019-1038-1 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Bennett RL et al. A Mutation in Histone H2B Represents a New Class of Oncogenic Driver. Cancer Discov 9, 1438–1451, doi: 10.1158/2159-8290.CD-19-0393 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Okosun J et al. Integrated genomic analysis identifies recurrent mutations and evolution patterns driving the initiation and progression of follicular lymphoma. Nat Genet 46, 176–181, doi: 10.1038/ng.2856 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Li H et al. Mutations in linker histone genes HIST1H1 B, C, D, and E; OCT2 (POU2F2); IRF8; and ARID1A underlying the pathogenesis of follicular lymphoma. Blood 123, 1487–1498, doi: 10.1182/blood-2013-05-500264 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Reichel J et al. Flow sorting and exome sequencing reveal the oncogenome of primary Hodgkin and Reed-Sternberg cells. 125, 1061–1072, doi: 10.1182/blood-2014-11-610436 (2015). [DOI] [PubMed] [Google Scholar]
- 10.Mesin L, Ersching J & Victora GD Germinal Center B Cell Dynamics. Immunity 45, 471–482, doi: 10.1016/j.immuni.2016.09.001 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Chapuy B et al. Molecular subtypes of diffuse large B cell lymphoma are associated with distinct pathogenic mechanisms and outcomes. Nat Med 24, 679–690, doi: 10.1038/s41591-018-0016-8 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Fan Y et al. H1 linker histones are essential for mouse development and affect nucleosome spacing in vivo. Mol Cell Biol 23, 4559–4572 (2003). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Wright GW et al. A Probabilistic Classification Tool for Genetic Subtypes of Diffuse Large B Cell Lymphoma with Therapeutic Implications. Cancer Cell 37, 551–568 e514, doi: 10.1016/j.ccell.2020.03.015 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Zhou BR et al. Structural Mechanisms of Nucleosome Recognition by Linker Histones. Mol Cell 59, 628–638, doi: 10.1016/j.molcel.2015.06.025 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Lu C et al. Histone H3K36 mutations promote sarcomagenesis through altered histone methylation landscape. Science 352, 844–849, doi: 10.1126/science.aac7272 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Swaroop A et al. An activating mutation of the NSD2 histone methyltransferase drives oncogenic reprogramming in acute lymphocytic leukemia. Oncogene 38, 671–686, doi: 10.1038/s41388-018-0474-y (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Beguelin W et al. EZH2 is required for germinal center formation and somatic EZH2 mutations promote lymphoid transformation. Cancer Cell 23, 677–692, doi: 10.1016/j.ccr.2013.04.011 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Ortega-Molina A et al. The histone lysine methyltransferase KMT2D sustains a gene expression program that represses B cell lymphoma development. Nat Med 21, 1199–1208, doi: 10.1038/nm.3943 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Eagen KP Principles of Chromosome Architecture Revealed by Hi-C. Trends Biochem Sci 43, 469–478, doi: 10.1016/j.tibs.2018.03.006 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Rao SS et al. A 3D map of the human genome at kilobase resolution reveals principles of chromatin looping. Cell 159, 1665–1680, doi: 10.1016/j.cell.2014.11.021 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Izzo A et al. The genomic landscape of the somatic linker histone subtypes H1.1 to H1.5 in human cells. Cell Rep 3, 2142–2154, doi: 10.1016/j.celrep.2013.05.003 (2013). [DOI] [PubMed] [Google Scholar]
- 22.Bascom GD, Myers CG & Schlick T Mesoscale modeling reveals formation of an epigenetically driven HOXC gene hub. Proc Natl Acad Sci U S A 116, 4955–4962, doi: 10.1073/pnas.1816424116 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kim JM et al. Linker histone H1.2 establishes chromatin compaction and gene silencing through recognition of H3K27me3. Sci Rep 5, 16714, doi: 10.1038/srep16714 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Lhoumaud P et al. NSD2 overexpression drives clustered chromatin and transcriptional changes in a subset of insulated domains. Nat Commun 10, 4843, doi: 10.1038/s41467-019-12811-4 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Ezponda T et al. The histone methyltransferase MMSET/WHSC1 activates TWIST1 to promote an epithelial-mesenchymal transition and invasive properties of prostate cancer. Oncogene 32, 2882–2890, doi: 10.1038/onc.2012.297 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Mulder TA, Wahlin BE, Osterborg A & Palma M Targeting the Immune Microenvironment in Lymphomas of B-Cell Origin: From Biology to Clinical Application. Cancers (Basel) 11, doi: 10.3390/cancers11070915 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Yang SM, Kim BJ, Norwood Toro L & Skoultchi AI H1 linker histone promotes epigenetic silencing by regulating both DNA methylation and histone H3 methylation. Proc Natl Acad Sci U S A 110, 1708–1713, doi: 10.1073/pnas.1213266110 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Geeven G et al. Local compartment changes and regulatory landscape alterations in histone H1-depleted cells. Genome Biol 16, 289, doi: 10.1186/s13059-015-0857-0 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Wilcockson M et al. H1 linker histones regulate the balance of repressive and active chromatin domains via localized genomic compaction. Nature In Press (2020) [Google Scholar]
- 30.Eminli S et al. Differentiation stage determines potential of hematopoietic cells for reprogramming into induced pluripotent stem cells. Nat Genet 41, 968–976, doi: 10.1038/ng.428 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Wong DJ et al. Module map of stem cell genes guides creation of epithelial cancer stem cells. Cell Stem Cell 2, 333–344, doi: 10.1016/j.stem.2008.02.009 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.