

SUMMARY
Classic galactosemia (CG) is a rare metabolic disorder that results from profound deficiency of galactose-1-P uridylyltransferase (GALT). Despite early detection by newborn screening and rapid and lifelong dietary restriction of galactose, which is the current standard of care, most patients grow to experience a broad constellation of long-term complications. The mechanisms underlying these complications remain unclear and likely differ by tissue. Here we conducted a pilot study testing the safety and efficacy of GALT gene replacement using our recently-described GALT-null rat model for CG. Specifically, we administered AAV9.CMV.HA-hGALT to 7 GALT-null rat pups via tail vein injection on day 3 of life; 8 GALT-null pups injected with PBS served as the negative control, and 4 GALT+ heterozygous pups injected with PBS served as the positive control. All pups were returned to their nursing mothers, weighed daily, and euthanized for tissue collection two weeks later. Among the AAV9-injected pups in this study, we achieved GALT levels in liver ranging from 64% to 595% wild-type, and in brain ranging from 3% to 42% wild-type. In liver, brain, and blood samples from these animals we also saw a striking drop in galactose, galactitol, and gal-1P. Finally, all treated GALT-null pups showed dramatic improvement in cataracts relative to their mock-treated counterparts. Combined, these results demonstrate that GALT restoration in both liver and brain of GALT-null rats by neonatal tail vein administration using AAV9 is not only attainable but effective.
Keywords: galactosemia, GALT, rat, gene therapy, metabolite, AAV9
1-sentence take-home message:
Here we report that neonatal GALT gene replacement using AAV9 restores GALT activity, normalizes or near-normalizes galactose, galactitol, and gal-1P in both liver and brain, and minimizes cataracts in a GALT-null rat model of classic galactosemia.
INTRODUCTION
Classic galactosemia (CG) is a potentially-lethal autosomal recessive disorder that results from profound deficiency of galactose-1-P uridylyltransferase (GALT), the middle enzyme in the Leloir pathway of galactose metabolism (Figure 1). Early detection by newborn screening and life-long dietary restriction of galactose prevent or resolve the potentially lethal acute symptoms of disease (Berry 2014, Welling et al 2017). However, most infants with CG grow to experience a constellation of long-term developmental and other complications that include cognitive disability, growth delay, difficulties with speech, motor problems, and primary ovarian insufficiency in girls and women, among other challenges. Mechanism remains unclear and may differ by tissue. Despite decades of research, there is no known intervention that prevents or resolves the long-term complications of CG.
Figure 1: The Leloir pathway of galactose metabolism.
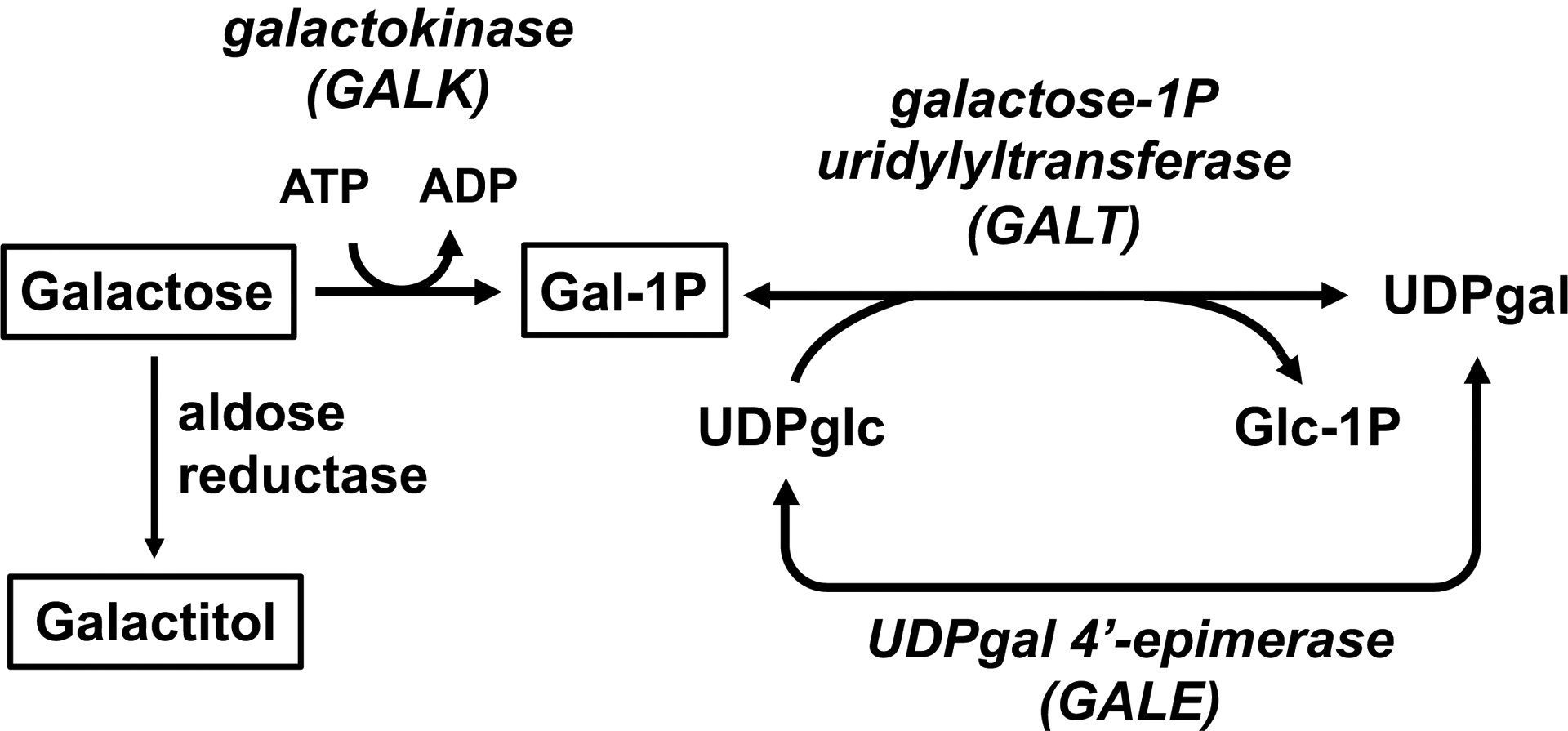
Classic galactosemia (CG) results from profound deficiency of galactose-1-P uridylyltransferase (GALT). The 3 metabolites followed in this study (galactose, galactitol, and gal-1P) are boxed in this figure.
Studies of patient samples document that following exposure to human breastmilk or cow’s milk-based formula, both of which contain very high levels of galactose (Jenness 1974), infants with CG accumulate dramatically-elevated levels of galactose, galactitol, and gal-1P in blood (Berry 2014) and tissues (Quan-Ma et al 1966). Following dietary restriction of galactose, these metabolite levels drop precipitously, but in most cases do not completely normalize (Welling et al 2017), ostensibly reflecting continued exposure to trace dietary or endogenously produced galactose (Berry et al 1995, Schadewaldt et al 2014). Which, if any, of the galactose metabolites that accumulate in patients lead to most long-term complications in CG remains unknown.
To facilitate studies of mechanism, including studies of metabolites in inaccessible tissues, and to provide an animal model for testing novel interventions, we recently developed and have begun to characterize a GALT-null rat (Rasmussen et al 2020). Our rats demonstrate metabolic perturbations, cataracts, and long-term growth, motor, and cognitive deficits reminiscent of patient outcomes (Rasmussen et al 2020). Using these rats, we have confirmed that different tissues indeed accumulate galactose, galactitol, and gal-1P to different extents and in different proportions, and further, that which metabolite predominates in a given tissue (e.g. liver or brain) can change over time (Rasmussen et al 2020).
Here we applied our GALT-null rats in a small pilot study of GALT gene replacement as a candidate intervention for CG. This study had 3 goals. First, we wanted to test whether we could restore detectable GALT activity to brain as well as liver in rat pups using AAV9 viral vectors administered by tail vein injection during the neonatal period. Second, we wanted to define the relationship between GALT level restored to liver and brain tissues in GALT-null pups and the levels of galactose, galactitol, and gal-1P that would accumulate in those tissues, and in blood. Finally, we wanted to ask if there was any evidence of morbidity associated with over-expression of GALT in rat pups. The results presented here answer each of these questions and lay a foundation for more extensive pre-clinical studies of the safety and efficacy of GALT gene replacement as a candidate intervention for long-term complications in CG.
MATERIALS and METHODS
Viral stock and administration to day 3 rat pups
The viral stock used here was generated from a plasmid (JF1 (pAAV.CMV.HA-hGALT)) assembled and confirmed by Dr. Oskar Laur in the Emory Custom Cloning Core Division (http://www.cores.emory.edu/eigc/). This plasmid included the CMV promoter driving expression of an HA-tagged human GALT transgene that we had previously used successfully in yeast (Elsevier et al 1996). The AAV9 viral stock was prepared by Dr. Xinping Huang in the Emory Viral Vector Core (http://neurology.emory.edu/ENNCF/viral_vector/) and received at a concentration of 5×1013 vg/mL.
For tail vein injections of AAV9 vector at a dose of 5×1013 vg/kg diluted in PBS, or PBS alone (mock), rat pups were anesthetized briefly via isoflurane inhalation and the tail was wiped with betadine. The injection volume (30μL) was administered slowly into the dorsal tail vein using a 30-gauge insulin syringe before gently applying pressure at the injection site until bleeding stopped. Rat pups were returned to the nest with their mother for recovery and monitoring.
Euthanasia, PBS perfusion, and tissue harvest from 17-day old rat pups
Pups were euthanized under deep isoflurane anesthesia at age 17 days, which was 14 days after AAV9 administration. In brief, following anesthesia the chest cavity was opened to expose the heart, and whole blood was withdrawn from the right atrium using a 27G needle into a 2-mL sodium heparin BD Vacutainer tube (#367671) which was inverted 8–10 times and then stored on ice until processing. Next, a 23G needle was inserted into the left ventricle and perfusion with PBS was initiated and continued until the fluid exiting the right atrium was entirely clear.
Tissues to be processed for microscopy were removed, submerged in 4% paraformaldehyde (PFA), and incubated with gentle rocking at 4°C for 24 hours prior to transfer to 70% ethanol and eventual paraffin embedding. The following tissues were collected: right hemisphere of the brain, and liver sections.
Solid tissues to be processed for PCR or quantitative PCR (qPCR), enzyme assays, or metabolite analyses were removed, cut with a razor into small pieces, flash frozen on dry ice, and stored at −80°C until use. The following solid tissues were collected: left hemisphere brain, liver, gonads, heart, and eyes.
Blood was processed as follows shortly after collection: whole blood was transferred to 1 or more 1.5 mL microfuge tubes and spun in an Eppendorf 5415D centrifuge at 2000 × g at 4°C for 15 mins. Supernatant was removed and placed in a clean 1.5 mL screw cap tube and stored at −80°C until use. The red blood cell (RBC) pellet was rinsed with 1 mL of 1X PBS (Corning #21–040-CV) and centrifuged at 2000 × g at 4°C for 15 mins. Supernatant was removed and RBC pellet transferred to a clean 1.5 mL screw cap tube and stored at −80°C until use.
Initial qualitative PCR amplification of human GALT transgene sequence from rat tissue samples
Presence of the AAV9-hGALT construct was first confirmed by standard PCR amplification using the following primers: AAVGALTFwd1, 5’-CGACGTCCCAGACTACGC-3’ and AAVGALTRev1, 5’-CTCCTCGAGCAGACTTTGC-3’. Primer AAVGALTFwd1 binds within the construct sequence encoding the HA tag so as to minimize the chance of nonspecific binding within the rat genome, and primer AAVGALTRev1 binds in a region unique to the hGALT coding sequence. DNA was extracted from pieces of rat liver and brain which had been previously frozen at −80°C. Presence of the anticipated 389 bp amplicon band was confirmed in both liver and brain of all 5 GALT-null AAV9-hGALT-injected animals tested. No amplicon was detected in any of the 3 GALT-null PBS-injected control animals tested.
Quantitative real-time PCR to define AAV9 viral genome copy number in tissue samples
We calculated the copy numbers of AAV vector genomes in duplicate liver and brain samples from rats injected with AAV9-hGALT versus PBS using quantitative real-time PCR (qPCR) essentially as described previously (Sanmiguel et al 2019). Host genomic plus episomal viral DNAs were extracted from 25 mg pieces of previously-frozen tissue using the Qiagen DNeasy Blood & Tissue Kit (catalog 69504) following the manufacturer’s protocol. Tissues were incubated with proteinase K for 3 hours, with vortex agitation at the start of each hour. Of 2 biological replicates, one was treated with RNase during isolation, the other was not. Primers were complementary to the hGALT transgene with one primer (5’-CGACGTCCCAGACTACGC-3’) binding to sequence encoding the HA tag and the other (5’-GGATATGCTGATGGTCGTTTG-3’) binding to a region unique to the hGALT open reading frame; the resulting amplicon was 113 bp. To control for any influence of host genomic DNA on the amplification process, standards were prepared by diluting purified AAV9-hGALT plasmid into rat genomic DNA extracted from liver or brain samples from PBS-injected control animals. These dilution standards contained 3 × 10−5, 3 × 10−4, 3 × 10−3, 3 × 10−2, 0.3, 3, and 30 vector genomes per diploid rat genome, respectively. Similarly, we also controlled for the possibility of PCR-inhibitory factors in each test sample by conducting real time amplification of each sample both with and without a known spike of purified vector. In all cases, the spike was detectable at close to the expected level (data not shown).
Real-time amplification was carried out in up to 3 technical replicates per sample using the SYBR Green Master Mix (Applied Biosystems, catalog 4209155) with 40ng of genomic DNA in 25uL reactions, according to the manufacturer’s instructions and using an Applied Biosystems 7900HT thermal cycler. Initial DNA concentration readings were measured by nanodrop, and final values were normalized to DNA concentrations measured by Qubit. Repeat analysis of a subset of samples using Qubit-defined concentration to determine the amount of sample pipetted into the reaction showed very strong correlation with the results of reactions assembled based on nanodrop concentration and later normalized to Qubit concentration. Specific cycling conditions involved an initial denaturation for 10 min at 95°C followed by 40 cycles of 15 sec denaturation at 95°C followed by 60 sec of annealing and extension at 60°C. The results are presented as vector genome copy number per diploid rat genome, also referred to as vg copy number/cell.
Anti-HA immunofluorescence of fixed liver samples
Tissues intended for immunofluorescence were paraffin embedded and sectioned into 5 micron slices onto glass slides. Slides were deparaffinized in Safe Clear and rehydrated through ethanol washes. Following antigen retrieval (DAKO target retrieval X1699) using the steamer method (http://www.kanidis.gr/common/files/ANOSOISTOCHIMIA/DETECTION/ihc_staining_methods_5ed.pdf) with both peroxidase block (3% H2O2) and protein block (DAKO X0909) steps, slides were incubated with anti-HA primary antibody (Cell Signaling Technology) at a 1:1600 dilution in 1X PBS with 0.1% Tween-20 overnight at 4°C. The secondary antibody was Alexa Fluor 488 (Abcam, 1:500). DAPI (Invitrogen) was used as a counterstain at 300 nM to visualize nuclei.
Quantitation of percentage HA positive cells
We collected fluorescent images at both 10x and 20x magnification. For analysis of % HA positive cells in a given image, we used Fiji-Image J imaging software to select 5 random regions of interest (ROIs) from each 10X image. We then used the Cell Counter Plugin from Fiji for manual scoring of each ROI, dividing cells identified by their DAPI-stained nucleus into 4 categories of anti-HA fluorescent signal intensity: bright, moderate, dim, or negative. Counts from the five ROIs were then combined to obtain an approximate total % of HA positive cells for that sample.
Quantitation of GALK, GALT, and GALE activities in samples of liver and brain
Enzyme assays for GALK, GALT, and GALE activities were performed in up to 3 technical replicates, with GALT also tested in biological duplicates, as described previously (Rasmussen et al 2020) with the following modification. Because liver GALT activity in GALT-null animals injected with AAV9-hGALT exceeded normal wild-type levels by up to 595%, the final protein concentration used for GALT assays in these animals ranged from 0.25 – 2 μg protein per 50 μl reaction volume, as was appropriate to remain within the linear range of the assay.
For enzyme assays of brain tissue, the final protein concentration used for GALT assays was 10 μg protein per 50 μl reaction volume in GALT+ (heterozygous) animals, and 40 μg protein per 50 μl reaction volume in GALT-null animals. In GALT-null animals injected with AAV9-hGALT, the final protein concentration was 20 or 40 μg protein per 50 μl reaction volume, depending on the level of activity observed. The final protein concentrations used for GALK and GALE assays in liver was 6 μg protein per 50 μl reaction volume, and in brain was 5 ug protein per 50 μl reaction volume.
Quantitation of galactose, galactitol, and gal-1P in samples of plasma, RBC, liver and brain
Galactose, galactitol, and gal-1P from plasma, RBC, liver, and brain were quantified following the procedure previously described (Rasmussen et al 2020).
RESULTS
GALT gene replacement in neonatal rat pups:
Our first goal was to test whether we could achieve detectable hGALT transgene delivery and expression in both liver and brain of GALT-null pups by intravenous administration of vector. To minimize the risk of unintended genomic disruption by viral integration, and to maximize ability of the vector to cross the blood brain barrier and express transgene in multiple tissues, we used a non-replicating, predominantly non-integrating AAV9 viral vector administered by tail vein injection to 7 GALT-null pups at 5 × 1013 vg/kg. Eight GALT-null pups administered PBS served as the negative control, and 4 GALT+ (heterozygous) pups administered PBS served as the positive control. All pups were 3-days old at the time of injection. Pups were euthanized 2 weeks later and perfused with PBS to remove residual blood from organs prior to harvest. Eyes were photographed and scored for the presence and severity of cataracts as described previously (Rasmussen et al 2020).
To confirm presence of the transgene, we first performed standard qualitative PCR on samples of brain and liver from both treated and control animals using primers specific for the HA-tagged human GALT sequence. As expected, all samples from pups injected with AAV9-hGALT showed amplicons of the expected size and all samples from pups injected with PBS showed no amplicon (data not shown).
To calculate the actual copy number of viral genomes (vg) present in the different animals and tissues we performed real-time qPCR using liver and brain samples derived from all 7 treated pups, plus representative controls (Supplemental Table 1). To calculate actual transgene function, we assayed GALT activity in samples of liver and brain from all 7 AAV9-hGALT-injected pups (Table 1), plus controls, as described previously (Rasmussen et al 2020). As a control for sample integrity, we also tested each protein lysate for both galactokinase (GALK) and UDP-galactose 4’-epimerase activity (GALE, see Figure 1). As expected, the GALK and GALE activity levels in both experimental and control groups were similar (Table 1), and the positive (heterozygous) and negative control cohorts both gave the expected GALT results of approximately 50% and 0% wild-type activity, respectively.
Table 1: GALT, GALK and GALE activities observed in liver and brain samples from control and test animals in this study. Values listed are combined averages across technical replicates for each of 2 biological replicates.
A slide of fixed liver tissue from pup C, stained with anti-HA antiserum, is presented in Supplemental Figure 2.
| brain | brain | brain | |||||
|---|---|---|---|---|---|---|---|
| Positive control GALT+/− pups (PBS-injected) | 62.36 ± 13.69 n=4 | 7.40 ± 1.68 n=4 | 7.50 ± 2.27 n=4 |
4.19 ± 1.38 n=4 |
5.87 ± 1.94 n=4 |
5.13 ± 0.70 n=3 |
|
| Negative control GALT-null pups (PBS-injected) | −0.01 ± 0.50 n=8 | 0.04 ± 0.05 n=8 | 19.70 ± 4.73 n=8 |
5.39 ± 1.40 n=8 |
6.87 ± 3.88 n=8 |
6.10 ± 0.47 n=7 |
|
| GALT-null pups injected with AAV9-hGALT | A | 80.3 (64%) | 0.4 (3 %) | 11.49 ± 3.33 n=7 |
4.82 ± 1.67 n=7 |
5.20 ± 1.16 n=7 |
7.20 ± 1.20 n=7 |
| 89.7 (72%) | 0.4 (2 %) | ||||||
| C | 107.9 (87%)* | 1.1 (7 %) | |||||
| 242.5 (194%) | 4.8 (33%) | ||||||
| E | 256.2 (205%) | 3.2 (22%) | |||||
| 579.0 (464%) | 2.8 (19%) | ||||||
| G | 617.6 (595%) | 6.3 (42%) | |||||
Among the AAV9-injected pups, we observed two clear patterns. First, in each of the 7 pups tested, both AAV vg copy number and GALT activity were strikingly higher in samples of liver than brain (Supplemental Tables 1 and 2). Second, the AAV9-hGALT injected pups showed substantial variability from animal to animal in terms of average AAV genome copy number per cell, which ranged from 4.13 to 73.37 in liver and 0.024 to 0.083 in brain, and also in terms of GALT activity level, which ranged from 64–595% wild-type in liver and 3–42% wild-type in brain (Table 1).
To explore the bases of the apparent variability among animals, we tested both technical and biological replicates of both liver and brain samples from all 7 pups (Supplemental Tables 1 and 2). As expected, we saw very limited variability among technical replicates, but sometimes striking variability among biological replicates, at least in some pups. To be clear, at the time of harvest each PBS-perfused organ was cut into small fragments that were flash-frozen and stored for later use. Tissues to be processed for protein or DNA studies, including biological replicates, therefore represented separate samples that may have derived from different regions of the same organ.
To explore the relationship(s) between vg copy number and GALT activity detected in liver and brain we plotted the averages of technical and biological replicate values calculated for each animal (Supplemental Figure 1, panels A and B). As expected, we observed general positive trends between vg copy number and GALT activity for both liver and brain, with some apparent outliers (pups F and G, see Discussion). We also tested for the presence of a relationship between GALT activity detected in liver and brain (Supplemental Figure 1, panel C) and, as expected, found an apparent correlation, albeit again with some outliers (pups F and G, see Discussion).
Finally, to see what fraction of liver cells demonstrated transgene expression in treated GALT-null pups we performed immunofluorescence using anti-HA antiserum on sections of fixed liver from test versus negative control pups. All samples from test pups showed the presence of HA+ cells; the negative controls did not. In a first attempt to quantify the proportion of HA+ cells we counted stained versus unstained cells in a small set of liver sections from 6 test pups injected with AAV9-hGALT; the averages ranged between about 30–40% stained cells. The results are presented in Supplemental Table 3, and representative treated and untreated tissue sections are presented in Supplemental Figure 2.
Impact of restored GALT activity on galactose metabolites in plasma, RBC, liver, and brain of GALT-null rat pups:
Our second goal was to define the relationship between average GALT activity restored to liver and brain in GALT-null rat pups and the levels of galactose, galactitol, and gal-1P that accumulated in those tissues, and in blood. To address this question, we quantified the levels of all three metabolites in plasma, RBC, liver and brain for all of the rat pups in this study and plotted the values as a function of GALT activity. Specifically, plasma galactose and galactitol, RBC galactose, galactitol, and gal-1P, and liver galactose, galactitol, and gal-1P were each plotted as a function of liver GALT activity, while brain galactose, galactitol, and gal-1P were plotted as a function of brain GALT activity.
The results were striking (Figure 2). With regard to galactose, in plasma, as expected (Rasmussen et al 2020), mock-treated GALT+ pups showed essentially no galactose accumulation and mock-treated GALT-null pups showed a range of high galactose levels (Figure 2). In contrast, all 7 AAV9-hGALT treated GALT-null pups showed near or normalized galactose levels. With minor exception (one outlier pup), this same pattern was repeated in RBC, liver, and brain.
Figure 2: Metabolic impact of restored GALT activity in GALT-null rat pups.
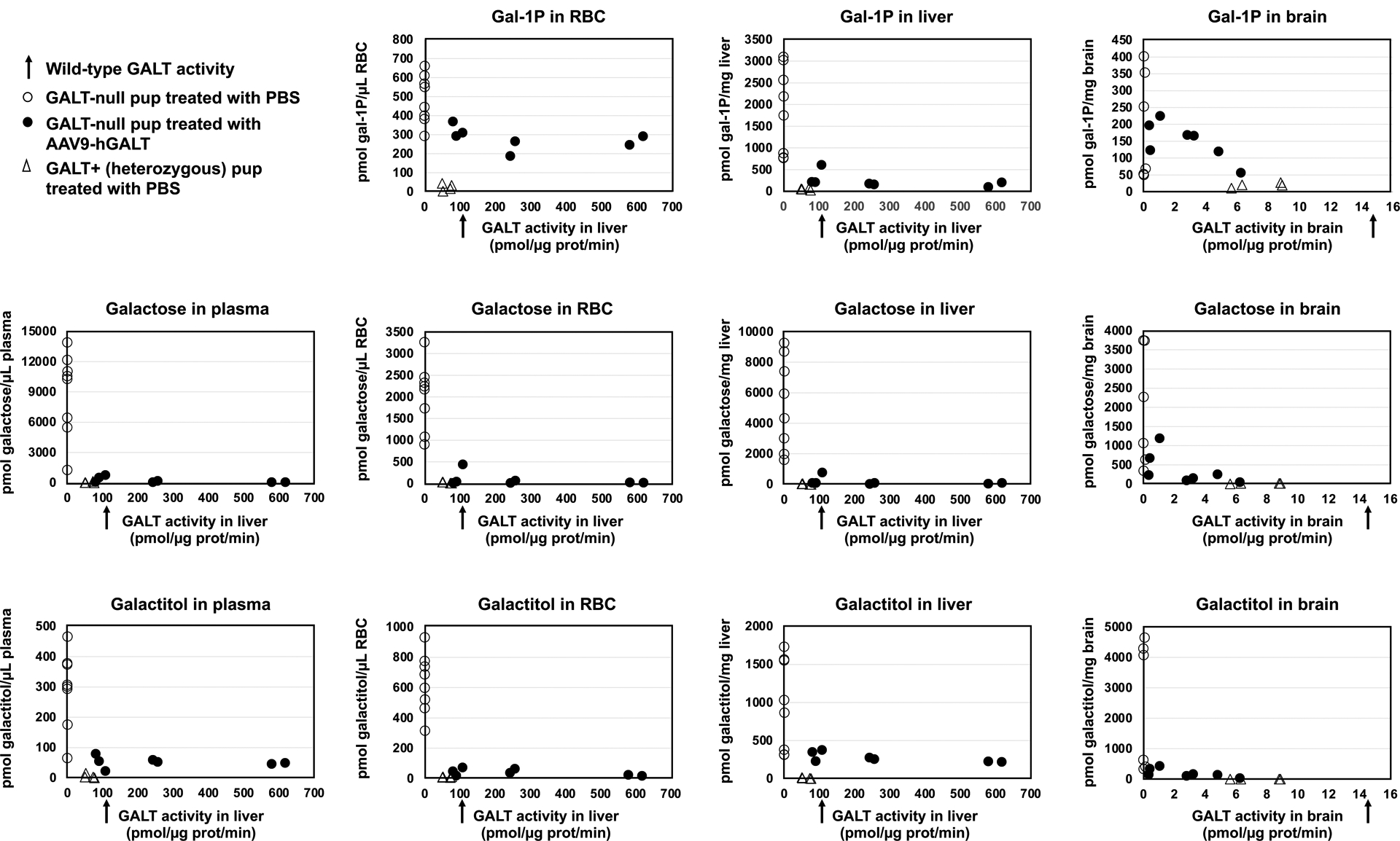
Expression of an hGALT transgene in GALT-null pups normalized, or near-normalized, galactose, galactitol, and gal-1P in all samples tested including plasma, RBC, liver, and brain.
With regard to galactitol, as expected (Rasmussen et al 2020), in all sample types we observed low baseline galactitol levels in all GALT+ (heterozygous) controls and a range of high galactitol levels in all mock-treated GALT-null pups. In all 7 AAV9-hGALT-treated GALT-null pups the plasma galactitol was lowered; however, the pattern was complex. Specifically, galactitol levels were diminished but not to baseline, and the level of residual galactitol was unchanged regardless of whether liver GALT was 64% of wild-type (the lowest in our treated cohort) or 595% of wild-type (the highest in our treated cohort). In liver, the galactitol pattern was very similar to that seen in plasma. In RBC and in brain, galactitol showed a similar pattern but was essentially normalized in samples from the highest GALT-expressing pups.
Finally, with regard to gal-1P, we again saw the expected patterns of very low gal-1P in all GALT+ (heterozygous) controls, and high gal-1P in all mock-treated GALT-null pups (Rasmussen et al 2020). In AAV9-hGALT treated GALT-null pups the pattern was again more complex. In RBC, all treated pups showed similar levels of gal-1P that were at or below the lowest level seen in negative controls, but clearly above the levels seen in positive controls. In liver, the gal-1P levels were near or normalized by AAV9-hGALT treatment, but in brain they were lowered but not fully normalized even at the highest levels of GALT transgene activity. The implications of these complex patterns are considered in the Discussion below.
Over-expression of GALT appears benign in rat pups:
Our third goal for this project was to test whether over-expression of GALT would lead to any detectable negative consequences for rat pups. While our ability to address the question of safety was limited by the size and scope of the pilot study, the short answer is that, at least anecdotally, we saw no evidence of morbidity or mortality associated with either viral transduction or over-expression of GALT. For example, all virus-injected pups gained weight essentially on track with PBS-injected controls. Further, 4 of the 7 GALT-null pups injected with virus expressed levels of GALT in liver that were above wild-type, yet all gained weight on track with their peers and remained apparently well and overtly indistinguishable from their counterparts expressing more “normal” levels of GALT (Supplemental Figure 3). While it is tempting to ask whether GALT replacement rescued the growth phenotype otherwise seen among GALT-null pups (Rasmussen et al 2020), the cohort sizes in this pilot study were insufficient to support a statistically meaningful comparison.
GALT transgene expression diminishes cataract intensity in GALT-null rat pups:
Finally, while testing the impact of GALT transgene expression on most long-term outcomes was beyond the scope of this pilot study, either because of the small numbers of animals tested (insufficient statistical power), or because of the short time-frame (animals euthanized as pups before behavioral testing was possible), we were able to assess the impact of GALT gene replacement on cataract formation using a semi-quantitative approach. As expected (Rasmussen et al 2020), all 8 eyes from the 4 heterozygous pups in this study showed no cataracts, and all 16 eyes from the 8 mock-treated GALT-null pups showed either mild, moderate, or severe cataracts (Figure 3). In contrast, of the 14 eyes from the 7 AAV9-hGALT treated GALT-null pups, about 25% showed no cataract and the remainder showed only mild cataract. This was a small sample, but the difference was unmistakable.
Figure 3: Restored GALT activity minimized cataracts in GALT-null rat pups.
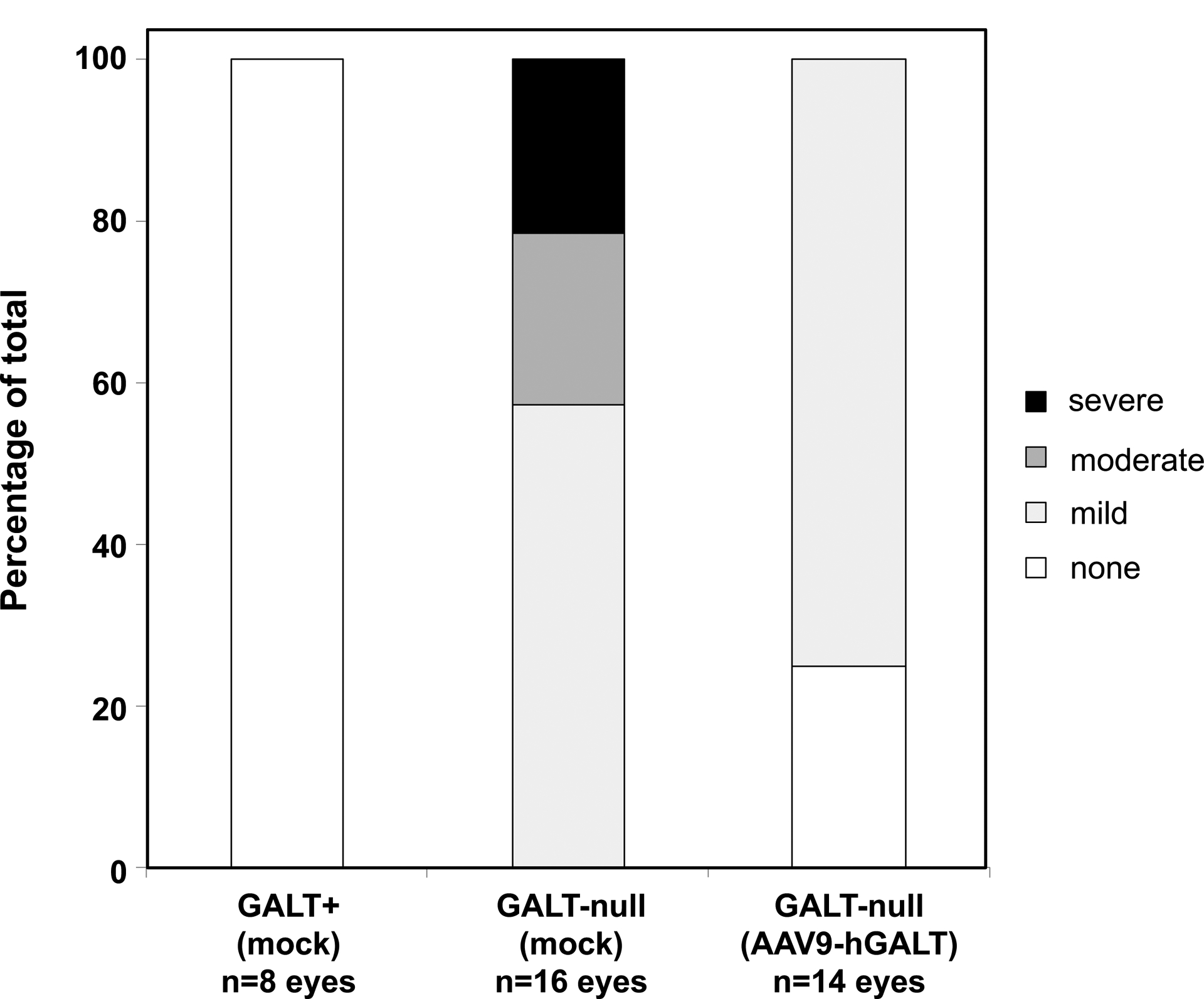
As a group, the 7 GALT-null pups treated with AAV9-hGALT in this study demonstrated clear improvement of cataracts relative to their mock-treated counterparts.
DISCUSSION
This study had three goals: (1) to test whether we could achieve GALT transgene expression in brain, as well as liver, following intravenous administration of AAV9-hGALT vector in GALT-null rat pups, (2) to define how much GALT activity would be needed to normalize galactose metabolites in these pups, and (3) to learn whether GALT over-expression might be toxic to the pups. To the limits of the present study, we have now achieved all 3 goals. First, we clearly demonstrated strong GALT activity in both liver and brain following single tail vein injection of AAV9-hGALT in pups, and with some outliers (discussed below), the levels of GALT activity observed showed the expected trends with viral genome copy number. Second, the GALT activity levels we achieved normalized, or near-normalized, most galactose metabolites in blood, liver, and brain of the test animals. Finally, we saw no evidence of toxicity in rat pups over-expressing GALT by up to almost 6-fold in liver.
GALT expression in liver and brain:
As presented in Table 1, we achieved a range of GALT activities in both liver and brain samples from the GALT-null pups that received AAV9-hGALT. Expression in liver ranged from 64–595% wild-type, and in brain from 3–42% wild-type, and there was an apparent positive relationship between the 2 (Supplemental Figure 1, panel C). The difference we observed between liver and brain GALT transgene activity is fully consistent with prior reports from other investigators using AAV9 to deliver other transgenes into rodents by intravenous injection (Pan et al 2019, Zincarelli et al 2008). Possible explanations include preferential perfusion of the liver and limited permeability of the blood-brain barrier, even to serotype AAV9, among other reasons.
We cannot explain why the expression range among individual pups was so large, as all ostensibly received the same dose from the same viral stock administered at the same age and by the same hands. That said, tail vein injection into neonatal rat pups is not trivial and it is likely that some injections were technically more successful than others. Further, our GALT-null rats are outbred, so it is also possible that background differences between individual pups may have made some more receptive than others. Studies of viral genome copy number revealed the expected trends with GALT activity in both organs, albeit with some outliers.
Specifically, at least 2 pups appeared as apparent outliers in graphs comparing vg copy number and GALT activity in liver and brain (Supplemental Figure 1, panels A and B): pups F and G. Pup F demonstrated far more GALT activity in liver than expected given the corresponding AAV9 vg/cell result, and pup G demonstrated far more GALT activity in brain than we would have expected given the corresponding AAV9 vg/cell result. It is interesting to note that while both of these pups showed no exceptional technical variability in either tissue or assay type, pup F showed by far the greatest biological variability (50-fold) in qPCR studies of liver, and pup G showed the greatest biological variability (>5-fold) in qPCR studies of brain (Supplemental Table 1). Pup G also showed the greatest biological variability in GALT activity assays of brain (Supplemental Table 3; almost 5-fold). To be clear, like biological replicates, GALT activity and qPCR studies were conducted on separate biological samples derived from the same organ. The “noise” in our data is therefore consistent with prior studies documenting that transduction following intravenous injection of AAV can vary substantially from region to region in an organ (Gray et al 2011, Stone et al 2008).
Considering that the presence of even cryptic GALT activity minimizes or prevents long-term complications in CG patients (Berry 2014, Ryan et al 2012, Ryan et al 2013, Spencer et al 2013), and that we were able to achieve as much as 42% wild-type GALT activity in brain using our current approach, we consider the results presented here extremely promising. We should also note that the transgene expression levels reported here represent animals harvested at a single time point -- 2 weeks after viral injection -- so expression may not yet have peaked, but also may have eventually diminished over time, at least in highly mitotic tissues (Zincarelli et al 2008).
Relationship between GALT activity and galactose metabolites in blood, liver, and brain:
While it remains unclear which, if any, of the galactose metabolites typically monitored in patients with CG, or in animal models, actually cause most of the long-term complications observed, it was reassuring to see that even the lowest levels of GALT transgene expression achieved here were sufficient to normalize, or near-normalize, most galactose metabolites in blood, liver, and brain (Figure 2).
Galactose seemed the easiest metabolite to normalize and gal-1P the most difficult, perhaps because gal-1P is strictly intracellular, limiting the ability of transduced cells to cross-correct their non-transduced neighbors. From the immunofluorescence studies of liver (Supplemental Figure 3), it was clear that, at least at the 2-week time point, while many liver cells in the treated pups were expressing GALT transgene, many also were not.
One of the perplexing observations from our study was that, in some tissues, gal-1P or galactitol persisted above baseline even in animals expressing more than wild-type levels of GALT. Prime examples included gal-1P in RBC, and galactitol in plasma and liver. To be clear, the levels of these metabolites seen in GALT-null pups expressing transgene were well below the levels seen in mock-treated GALT-null pups, but they were not fully normalized regardless of how much GALT activity was present. One possible explanation for the gal-1P result in RBC is that most of the RBC in circulation at the time of euthanasia may have matured before transgene expression turned on, and once gal-1P formed in those cells it was “stuck.” Galactose and galactitol in the same RBC might have been able to exit the cells to be taken up and metabolized by other cells, or excreted in urine.
The elevated baseline of galactitol seen in plasma and liver despite strong GALT activity in liver is more difficult to explain, but might reflect a limited ability of these compartments to rid themselves of galactitol once it has formed (e.g. before AAV9-hGALT injection, or after injection but before the GALT transgene turned on). Future studies, beyond the scope of this pilot, will be required to determine whether the elevated galactitol levels seen in plasma and liver, or the elevated gal-1P baseline seen in RBC at 2 weeks post-AAV9-hGALT injection, will diminish over time.
GALT over-expression:
When planning a gene-replacement approach to intervention for CG, or any condition, two of the key questions that must be addressed are “How much transgene expression do we need?” and “Is too much a bad thing?” Our studies of metabolite levels in treated and control tissues represent a first step toward asking the first question. Our finding that even rat pups demonstrating 585% wild-type GALT activity in liver remained apparently well represents one step toward asking the second question. In both cases, the answers suggest that gene replacement may be both an effective and lenient approach to intervention for long-term complications in CG.
Limitations and implications:
While compelling, this study also had clear limitations, the most notable being small cohort sizes, a short time frame that prevented testing long-term behavioral outcomes, and a single time point assessed following viral administration. We also did not test transduction efficiency or biodistribution of our AAV9 vector in wild-type rats, so we cannot say whether GALT-deficiency in our test animals might have impacted either process. Given that galactose plays a role in the entry of AAV9 into cells (Bell et al 2012) this is a reasonable possibility that will be tested in future studies.
That we have not yet characterized sufficient numbers of GALT-null rats to define whether or not they exhibit deficits in liver function, brain histology, or a number of other potentially important outcomes also means that we were not able to test whether the GALT gene replacement achieved here might have rescued those potential outcomes. We also looked at only a small number of tissue types that, while relevant for long-term outcomes in CG, may or may not represent other tissues in terms of transduction efficiency. Larger, more extensive studies will be required to test the generalizability of the results reported here.
The work reported here also has important implications for the feasibility of future clinical studies addressing gene replacement as a candidate intervention for CG. To be clear, the data reported here demonstrate that GALT restoration to both liver and brain achieved by intravenous administration of AAV9-hGALT is both possible and metabolically effective, at least in neonatal rats. However, we do not yet know how long the effect will last, or if it will be sufficient to prevent cognitive and other long-term deficits. Larger, longer term studies will be required to address those questions. Of note, a recent report by Balakrishnan and colleagues (Balakrishnan et al 2020) also tested GALT restoration as an approach to intervention, but those authors used a mouse rather than a rat model of CG, and mRNA-based rather than viral-based therapy. As reported, Balakrishnan and colleagues introduced their mRNA either by single or repeated IV injections into 8-week old (adult) mice, or by intraperitoneal injection into neonates. The relative level of GALT activity achieved, the longevity of the effect from a single dose, and the ability of the restored GALT activity to lower gal-1P levels in recipient animals were all measured and, as reported, were markedly less effective in that study than the corresponding results reported here.
Finally, that we were able to minimize or prevent cataracts in GALT-null rat pups with AAV9-hGALT gene replacement suggests that transgene expression may have initiated well before the 2-week time point at which the pups were harvested in our study. Future time-course studies will be required to clarify this point. Nonetheless, that we were able to see clear impact on cataracts also raises the compelling possibility that this type of intervention might be sufficient to minimize or prevent at least some of the other long-term deficits seen in GALT-null rats, and by extension, perhaps eventually, in patients.
Supplementary Material
Supplemental Figure 1: Relationship between viral genome copy number detected by quantitative real-time PCR and level of GALT achieved in liver and brain samples from experimental pups in this study. (A) Average AAV9-hGALT viral genomes per cell in liver versus average GALT activity achieved in liver. (B) Average AAV9-hGALT viral genomes per cell detected in brain versus average GALT activity achieved in brain. (C) Average GALT activity detected in liver versus brain. Each value plotted represents the average of both technical and biological replicates for that tissue and animal.
Supplemental Figure 2: Immunofluorescence showing expression of HA-hGALT in liver from a rat pup administered AAV9-HA-hGALT as a neonate and euthanized 2 weeks later. Immunofluorescence staining of fixed liver tissue from rat pup C shows clear anti-HA signal above background in about 30% of visible cells.
Supplemental Figure 3: GALT activity in liver versus body mass of pups at age 17 days. Rat pups expressing up to almost 6-fold the wild-type level of GALT in liver showed no evidence of impaired growth relative to controls or their counterparts expressing more normal levels of GALT.
ACKNOWLEDGMENTS
We are grateful to numerous colleagues for their contributions to this project. We specifically thank Dr. Michael Castle for his priceless guidance on everything AAV, Drs. Oskar Laur and Xinping Huang and their colleagues in the Emory Integrated Cores for generating the AAV vector and viral stock used in this study, and Dr. Lyra Griffiths, also from the Emory Integrated Cores, for performing the quantitative PCR required to calculate viral genome copy number in tissue samples. We also thank all members of the Fridovich-Keil lab for constant support, and the many professionals working in the Emory Division of Animal Resources and the Emory IACUC without whom this work could not have been conducted. This work was supported in part by grants from the National Institutes of Health R01DK107900 and R21HD092785 (both to JLFK), and in part by the Emory Integrated Genomics Core (EIGC), which is subsidized by the Emory University School of Medicine and is one of the Emory Integrated Core Facilities.
Footnotes
Competing interests: All of the authors declare that they have no competing interests.
Ethics approval: This manuscript does not include any human subjects work. All animal work was performed with approval of the Emory IACUC (Protocol PROTO201700095; PI: JL Fridovich-Keil) and oversight of the Emory Division of Animal Resources.
REFERENCES
- Balakrishnan B, An D, Nguyen V, DeAntonis C, Martini PGV, Lai K (2020) Novel mRNA-Based Therapy Reduces Toxic Galactose Metabolites and Overcomes Galactose Sensitivity in a Mouse Model of Classic Galactosemia. Mol Ther 28: 304–312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bell CL, Gurda BL, Van Vliet K, Agbandje-McKenna M, Wilson JM (2012) Identification of the galactose binding domain of the adeno-associated virus serotype 9 capsid. J Virol 86: 7326–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berry GT, Nissim I, Lin ZP, Mazur AT, Gibson JB, Segal S (1995) Endogenous Synthesis of Galactose in Normal Men and Patients with Hereditary Galactosemia. Lancet 346: 1073–1074. [DOI] [PubMed] [Google Scholar]
- Elsevier JP, Wells L, Quimby BB, Fridovich-Keil JL (1996) Heterodimer formation and activity in the human enzyme galactose-1-phosphate uridylyltransferase. Proc Natl Acad Sci U S A 93: 7166–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gray SJ, Matagne V, Bachaboina L, Yadav S, Ojeda SR, Samulski RJ (2011) Preclinical differences of intravascular AAV9 delivery to neurons and glia: a comparative study of adult mice and nonhuman primates. Mol Ther 19: 1058–69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jenness R (1974) Proceedings: Biosynthesis and composition of milk. J Invest Dermatol 63: 109–18. [DOI] [PubMed] [Google Scholar]
- Pan X, Sands SA, Yue Y, Zhang K, LeVine SM, Duan D (2019) An Engineered Galactosylceramidase Construct Improves AAV Gene Therapy for Krabbe Disease in Twitcher Mice. Hum Gene Ther 30: 1039–1051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quan-Ma R, Wells HJ, Wells WW, Sherman FE, Egan TJ (1966) Galactitol in the tissues of a galactosemic child. Am J Dis Child 112: 477–8. [DOI] [PubMed] [Google Scholar]
- Rasmussen SA, Daenzer JMI, MacWilliams JA et al. (2020) A galactose-1-phosphate uridylyltransferase-null rat model of classic galactosemia mimics relevant patient outcomes and reveals tissue-specific and longitudinal differences in galactose metabolism. J Inherit Metab Dis 43: 518–528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ryan EL, DuBoff B, Feany MB, Fridovich-Keil JL (2012) Mediators of a long-term movement abnormality in a Drosophila melanogaster model of classic galactosemia. Dis Model Mech 5: 796–803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ryan EL, Lynch ME, Taddeo E, Gleason TJ, Epstein MP, Fridovich-Keil JL (2013) Cryptic residual GALT activity is a potential modifier of scholastic outcome in school age children with classic galactosemia. J Inherit Metab Dis 36: 1049–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanmiguel J, Gao G, Vandenberghe LH (2019) Quantitative and Digital Droplet-Based AAV Genome Titration. Methods Mol Biol 1950: 51–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schadewaldt P, Kamalanathan L, Hammen HW, Kotzka J, Wendel U (2014) Endogenous galactose formation in galactose-1-phosphate uridyltransferase deficiency. Arch Physiol Biochem 120: 228–39. [DOI] [PubMed] [Google Scholar]
- Spencer JB, Badik JR, Ryan EL et al. (2013) Modifiers of ovarian function in girls and women with classic galactosemia. J Clin Endocrinol Metab 98: E1257–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stone D, Liu Y, Li ZY et al. (2008) Biodistribution and safety profile of recombinant adeno-associated virus serotype 6 vectors following intravenous delivery. J Virol 82: 7711–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Welling L, Bernstein LE, Berry GT et al. (2017) International clinical guideline for the management of classical galactosemia: diagnosis, treatment, and follow-up. J Inherit Metab Dis 40: 171–176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zincarelli C, Soltys S, Rengo G, Rabinowitz JE (2008) Analysis of AAV serotypes 1–9 mediated gene expression and tropism in mice after systemic injection. Mol Ther 16: 1073–80. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplemental Figure 1: Relationship between viral genome copy number detected by quantitative real-time PCR and level of GALT achieved in liver and brain samples from experimental pups in this study. (A) Average AAV9-hGALT viral genomes per cell in liver versus average GALT activity achieved in liver. (B) Average AAV9-hGALT viral genomes per cell detected in brain versus average GALT activity achieved in brain. (C) Average GALT activity detected in liver versus brain. Each value plotted represents the average of both technical and biological replicates for that tissue and animal.
Supplemental Figure 2: Immunofluorescence showing expression of HA-hGALT in liver from a rat pup administered AAV9-HA-hGALT as a neonate and euthanized 2 weeks later. Immunofluorescence staining of fixed liver tissue from rat pup C shows clear anti-HA signal above background in about 30% of visible cells.
Supplemental Figure 3: GALT activity in liver versus body mass of pups at age 17 days. Rat pups expressing up to almost 6-fold the wild-type level of GALT in liver showed no evidence of impaired growth relative to controls or their counterparts expressing more normal levels of GALT.