

Abstract
DNA helicases have risen to the forefront as genome caretakers. Their prominent roles in chromosomal stability is demonstrated by the linkage of mutations in helicase genes to hereditary disorders with defects in DNA repair, the replication stress response, and/or transcriptional activation. Conversely, accumulating evidence suggests that in cancer cells DNA helicases have a network of pathway interactions such that co-deficiency of certain helicases and their genetically interacting proteins result in synthetic lethality (SL). Such genetic interactions may potentially be exploited for cancer therapies. We will discuss the roles of RECQ DNA helicases in cancer, emphasizing some of the more recent developments in synthetic lethality.
Keywords: helicase, RECQ, synthetic lethality, genetic disease, genomic stability, Werner syndrome, Bloom’s syndrome, Rothmund-Thomson Syndrome, cancer
Importance of DNA Helicases in Tumorigenesis and Promotion of Cancer Cell Proliferation
The last two decades were marked by tremendous interest in anti-cancer treatment strategies based on synthetic lethal (SL) interactions of DNA damage response genes, fueled by the known biological effects of many chemotherapy drugs that inflict genomic DNA damage as a primary mechanism of cancer cell cytotoxicity. A valuable paradigm for this approach was provided by the seminal discovery of Poly [ADP-ribose] polymerase (PARP) inhibitors that selectively kill homologous recombination (HR)-defective (BRCA1 or BRCA2 mutated) tumor cells [1, 2]. We discuss known SL interactions of RECQ helicases that might be exploited to develop novel drug targets and strategies, and to improve the efficacy of current therapeutics in cancers with certain genetic deficiencies.
Mutations in DNA helicase genes may predispose individuals to cancer [3]. The RECQ helicases play a particularly prominent role in genetic disease, with mutations in three RECQ genes (WRN, BLM, RECQL4) linked to distinct hereditary disorders characterized by cancer. There is growing interest that certain DNA helicases are plausible targets for anti-cancer therapy. Over the last three decades, genetic loss-of-function studies conducted with cancer cell lines and in vivo animal models revealed selective dependencies on DNA helicases for tumor cells. Owing to their conserved role in DNA damage repair, rapidly dividing cancer cells may become reliant on RECQ’s for survival [3, 4], as suggested by the increased expression of WRN, BLM and RECQL1 in neoplastic cells (Table 1). Cancer cells which incur replicative lesions at an elevated rate may require specialized DNA helicases to tolerate replicative stress and support their uncontrolled cell growth and division (Figure 1) (Box 1). In addition, the involvement of human RECQ helicases in regulation of gene expression would have a strong influence on cellular proliferative capacity (Box 2).
Table 1.
Experimental Evidence for Pro-Carcinogenic Importance of RecQ Helicases
| RecQ | Experimental Evidence | Refs |
|---|---|---|
| WRN | Upregulation of WRN helicase in transformed human B lymphocyte cells and BCR/ABL-transformed chronic myeloid leukemia cells | [85, 86] |
| Increased WRN expression in hypopharyngeal carcinoma cell lines and tumor tissues | [87] | |
| WRN knockdown results in cell cycle progression defects, DSB accumulation, CFS instability, and reduced survival in hTERT-immortalized human fibroblasts overexpressing Cyclin E or E2F1 protooncogenes | [35] | |
| Replication-associated DNA damage leading to senescence in c-Myc overexpressing hTERT-immortalized human fibroblasts upon WRN depletion | [88] | |
| Genetic depletion of WRN induces DNA damage leading to growth inhibition, apoptosis, or reduced survival in human cancer cell lines (NSCLCa, ovarian adenocarcinoma, endometrial leiomyosarcoma, cervical carcinoma, breast adenocarcinoma, hypopharyngeal carcinoma) and immortalized human fibroblast or epithelial cells | [35, 88– 91] | |
| Pharmacological inhibition of WRN helicase activity induces DNA damage and apoptosis in human cancer cell lines (cervical carcinoma, colorectal carcinoma, adult T-cell leukemia) | [92, 93] | |
| WRN knockdown inhibits tumor growth (NSCLC, hypopharyngeal carcinoma, ovarian adenocarcinoma) in xenograft mouse models | [87, 90, 91] | |
| WRN knockdown potentiates the cytotoxicity of chemotherapeutic drugs (CPT, cisplatin, 5-flurouracil) in cancer cell lines (cervical carcinoma, osteosarcoma) and in xenograft tumor (hypopharyngeal carcinoma) models | [87, 89, 94] | |
| Small molecule WRN helicase inhibition is synergistic with MMC to sensitize cervical carcinoma cells and transformed human fibroblast cells with a defective FA pathway | [70] | |
| BLM | BLM copy number amplification in transformed human B lymphocyte cells | [85] |
| Positive IHC staining for BLM in tumors of lymphoid and epithelial origin | [95] | |
| Increased BLM mRNA and protein expression in cancerous prostate tissues as compared to non-cancerous tissues | [96] | |
| Significant correlation between high BLM mRNA expression and aggressive clinicopathological and molecular phenotypes including high tumor grade, larger tumor size and triple negative phenotypes in METABRICb breast cancer patient cohort; poor survival of breast cancer patients with high BLM mRNA expression; low nuclear and/or high cytoplasmic expression of BLM protein associated with breast cancer aggressiveness | [97] | |
| BLM promotes proliferation and survival of prostate cancer cells by activating AKT (serine/threonine kinase) signaling pathway | [98] | |
| BLM silencing results in elevated DNA damage, reduced proliferation, and apoptosis in prostate carcinoma and osteosarcoma cells and transformed human fibroblasts | [89, 96] | |
| BLM knockdown sensitizes osteosarcoma cells and transformed human fibroblasts to dose-dependent killing by genotoxic drugs (CPT, cisplatin, 5-flurouracil, HU) | [89] | |
| RECQL1 | Increased RECQL1 expression levels in ovarian cancer cells compared to normal cells | [99] |
| Elevated RECQL1 expression in glioblastoma cells compared to normal cells | [100] | |
| RECQL1 copy number amplification in transformed human B lymphocyte cells | [85] | |
| RECQL1 expression is positively correlated with high histologic grade of hepatocellular carcinoma | [101] | |
| 3′-UTR SNP in RECQL1 predicts poor survival in pancreatic cancer patients | [102] | |
| High RECQL1 expression associated with poor prognosis in breast cancer patients | [65] | |
| Reduced migration and invasive phenotypes of breast adenocarcinoma or cervical carcinoma cells upon RECQL1 knockdown | [65] | |
| RECQL1 depletion results in aberrant cell cycle progression, elevated DNA damage, spontaneous SCE, and drug-induced SCE in cervical carcinoma cells | [103] | |
| RECQL1 knockdown results in growth inhibition or reduced survival of the following cancer cell types: hypopharyngeal carcinoma, cervical carcinoma, glioblastoma, hepatocellular carcinoma, pancreatic carcinoma, ovarian carcinoma, osteosarcoma, liver carcinoma; RECQL1 knockdown inhibits xenograft or orthotopic tumor growth (hypopharyngeal carcinoma, hepatocellular carcinoma, NSCLC) in vivo | [87, 94, 100, 101, 103, 104] | |
| RECQL1 depletion sensitized cancer cells (hypopharyngeal carcinoma, glioblastoma, cervical carcinoma, osteosarcoma) to chemotherapeutic agents including CPT, etoposide, HU, temozolomide, and cisplatin 3,18,22,24,25 | [31, 87, 100, 103, 105] | |
Non-small cell lung carcinoma;
Molecular Taxonomy of Breast Cancer International Consortium
Figure 1. Involvement of DNA helicases to promote cancer cell growth and tumor development.
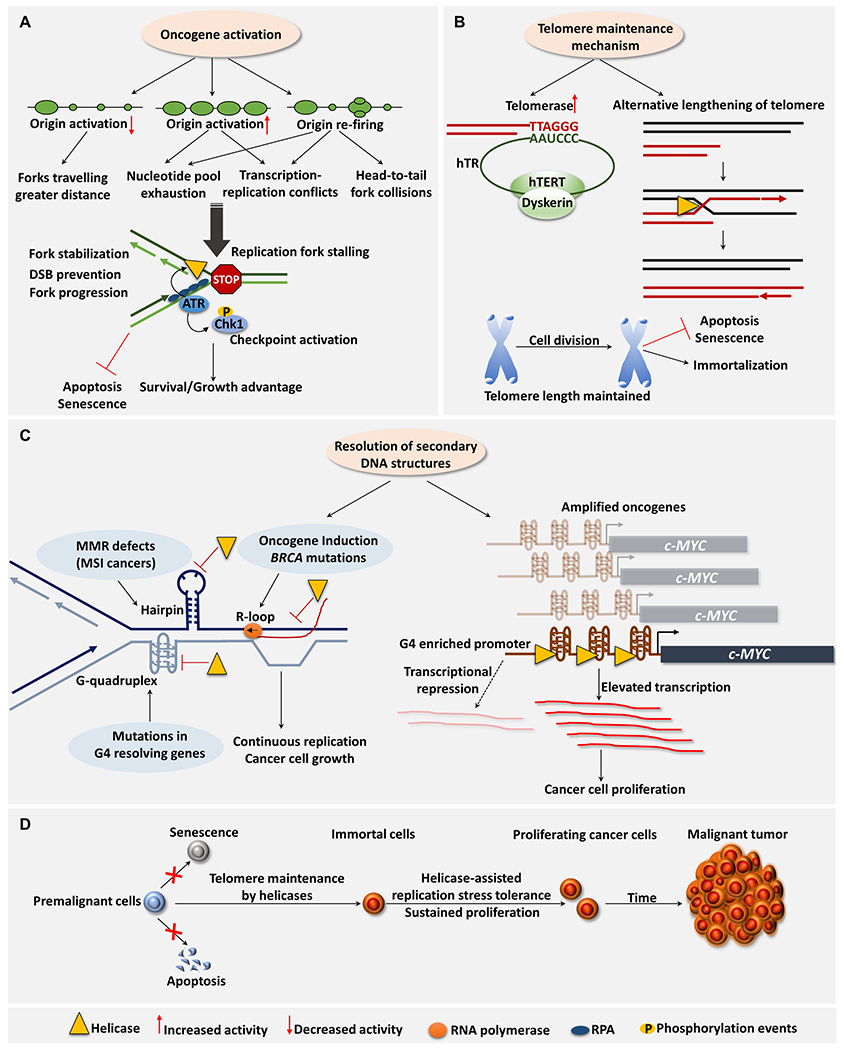
A. Helicases involved in DNA repair and fork stabilization enable cancer cells to cope with elevated replication stress induced upon oncogene activation. Oncogene-induced origin activation results in replication stalling/collapse due to increased inter-origin distance, reduced dNTP pool, increased transcription-replication interference, and head-to-tail fork collisions [106]. ATR-Chk1 pathway activation upon oncogene-induced replication stress promotes cancer cell fitness to evade the DNA damage response (DDR) barrier (e.g., TP53 loss) [107]. ATR phosphorylation of RecQs (e.g., WRN) stabilizes forks, prevents DSB formation, and ensures replication resumption, thereby enabling cancer cell proliferation [35]. B. Cancer cells gain replicative immortality by activating telomere maintenance mechanisms (TMM) including telomerase expression or ALT. In ALT-positive cancer cells, WRN and BLM helicases dissemble HR intermediates, thereby promoting telomere lengthening [108–110]. FANCM cooperates with BLM/BRCA1 to mitigate replication stress at ALT telomeres by resolving non-canonical DNA structures [23]. C. Helicases confer a proliferative advantage to cancer cells by resolving non-canonical DNA structures. Cancer-related genes that undergo somatic copy number amplification (e.g. c-MYC) are characterized by abundant predicted G-quadruplex (G4) forming sequences in their promoter regions [111]. Because promoter G4 stabilization may repress transcription [112]. G4-resolving helicases (e.g., WRN) promote oncogene expression and increase cancer cell proliferation [64]. However, in some cases increased G4 formation is correlated with elevated transcriptional activity [111], suggesting G4 resolution in regulatory regions may repress transcription [59]. D. Helicases enable cancer cells to gain replicative immortality, leading to tumorigenesis.
Box 1. RECQL4 and RECQL5 in Genome Stability Maintenance and Cancer.
The RECQL4 helicase mutated in the cancer-predisposing genetic syndrome Rothmund–Thomson syndrome (RTS) regulates DSB repair by coordinating the DSB repair pathways of HR and NHEJ in a cell-cycle dependent manner [46]. In the S and G2 phase, RECQL4 promotes HR repair by facilitating 5′ end resection by MRN complex and CTIP nuclease at sites of DSBs [47]. Experimental evidence suggests that RECQL4-mediated initial strand resection is required for further extensive resection by BLM/DNA2 and EXO1 nuclease to initiate HR at DSBs [47]. On the other hand, RECQL4 interacts with and stabilizes Ku70/Ku80 at DSBs in the G1 phase of the cell cycle, thereby promoting nonhomologous end-joining (NHEJ) while disfavoring HR [46, 48]. Importantly, in human cancer cells, RECQL4 deficiency triggers RAD52-mediated error-prone single-strand annealing (SSA) activity upon DSB induction [49]. Such regulation of repair pathway choice at DSBs by RECQL4 helicase likely contributes to anticancer therapy resistance as RECQL4-deficient cancer cells, but not nontumorigenic cells, are hypersensitive to ionizing radiation or cisplatin treatment [49]. Further studies are required to determine if RECQL4 coordinates with other RECQs and DNA damage response factors to regulate repair pathway choice. One plausible candidate is WRN which also interacts with Ku70/Ku80 [50] and promotes DSB repair by classical NHEJ while suppressing the error-prone alt-NHEJ pathway [51]. As loss of WRN also compromises HR [52, 53], it will be informative to assess if WRN and RECQL4 genetically or functionally interact during HR-mediated DSB repair in human cancer cells.
RECQL5 maintains genome stability by suppressing deleterious crossover events during HR-mediated repair of DSBs in human mitotic cells. RECQL5-deficient cells exhibit an increased frequency of spontaneous sister chromatid exchange (SCE) [54] which likely results in gross chromosomal rearrangements leading to oncogenic transformation [55]. However, RECQL5-mediated suppression of aberrant recombination is mechanistically distinct from that regulated by BLM helicase [54]. Whereas RECQL5 is believed to disrupt RAD51 nucleoprotein filaments and channel HR repair to less error-prone synthesis-dependent strand annealing (SDSA) [55, 56]. BLM minimizes crossing over through dissolution of HJ intermediates [57]. This non-epistatic functional interaction between RECQL5 and BLM suggests that they can at least partially compensate for each other in suppressing oncogenic crossovers in human cells; however, this remains to be tested. RECQL5 suppresses chromosomal instability in FA-B cells exposed to chemotherapeutic agents that invoke replication-associated DSBs [14]. Accordingly, RECQL5 depletion enhances the sensitivity of FANCB-defective cells to MMC or CPT [14]. Thus, RECQL5 might serve as a target to improve therapy for FA-defective human cancers. In contrast, BLM deficiency suppresses drug-induced chromosomal defects and reverses chemotherapeutic sensitivity in FA cells [14], suggesting divergent functions of BLM and RECQL5 under conditions of replication stress.
Box 2. Regulation of Gene Expression by Multiple RECQ Helicases.
WRN was the first human RECQ helicase in which experimental evidence suggested its role in regulating gene expression [58]. Subsequently, it was reported that both WS and BS cell lines showed altered expression of genes with predicted G4-forming sequences located in the promoters, introns, and downstream regions flanking the transcriptional end site [59]. A potential molecular explanation for these findings is suggested by observations that WRN and BLM efficiently resolve G4 DNA substrates in vitro [60–62]; however, the abundance of other G4-resolving helicases in human cells suggests that the unique versus overlapping roles of these proteins in vivo might be quite complex. Expression profiling demonstrated that mRNA and microRNA associated with cell proliferation/survival and pro-cancer pathways are differentially expressed in BLM-deficient cells [63], suggesting a potential role in the cancer prevalence of BS.
WRN-deficient cells showed down-regulation of genes with predicted G4 motifs at transcriptional start sites and first introns [64]. Like BS, the alterations in gene expression due to WRN deficiency also displayed perturbations in genes important for tumorigenesis. However, the gene expression changes in WS and BS were found to be distinct from one another, suggesting that they are transcriptionally distinct disease states. RECQL1 is also involved in the regulation of expression of genes involved in properties of cancer cells, i.e., cell migration, invasion and metastasis; moreover, loss of RECQL1 correlates with down-regulation of genes with predicted G4 motifs and these genes are enriched for binding to RECQL1, as demonstrated by chromatin immunoprecipitation [65]. However, while RECQL1 can bind G4 DNA in vitro, it poorly resolves G4 substrates that other RECQ helicases (e.g., WRN, BLM) efficiently resolve [66, 67], bringing into question the molecular mechanism whereby RECQL1’s interaction with G4 might operate to regulate G4-associated gene expression. In addition, biochemical studies demonstrate that RECQL5 can also unwind G4 substrates in vitro; however, its activity appears to be significantly weaker than WRN or BLM [68]. Altogether, the evidence suggests that WRN, BLM and RECQL1 regulate gene expression of cancer-associated genes that may be useful for understanding the roles of these helicases in carcinogenesis and value for potential therapeutic targeting.
Synthetic Lethal Interactions of BLM
Bloom’s syndrome (BS) arises from bi-allelic mutations in the BLM gene encoding a RECQ family DNA helicase believed to operate in recombinational DNA repair. BS is a chromosomal instability disorder characterized by a great predisposition to a wide spectrum of cancers. The signature feature of BS is elevated sister chromatid exchange (SCE) [5]. The dedicated function of BLM with a cellular topoisomerase to dissolve four-stranded double Holliday Junction (HJ) DNA structures that arise during homologous recombination (HR) repair or at converging replication forks, as suggested by in vitro studies, provides a molecular mechanism for BLM’s role to preserve chromosomal stability [6]. However, alternative mechanisms exist to deal with elevated SCE when BLM is absent or defective. Typically, the aberrant double HJ or related structure is dealt with by a structure-specific nuclease that cleaves the persistent DNA structure, albeit with consequences for genomic stability. For example, the structure-specific nucleases GEN1, MUS81-EME1 and SLX1-SLX4 modulate SCE formation in BLM-deficient cells [7]. Simultaneous depletion of GEN1 with MUS81 or SLX4 in BLM-deficient cells significantly reduced SCE frequency. Co-depletion of MUS81 with SLX4 did not have an additive effect on SCE frequency, suggesting that MUS81-EME1 and SLX1-SLX4 act in the same pathway to resolve HJs, distinct from the GEN1-mediated pathway. Depletion of GEN1, MUS81, or the nuclease-scaffolding protein SLX4 in BLM-depleted cells reduced cell viability suggesting that BLM exhibits a SL interaction with the structure-specific nucleases GEN1 or MUS81, and possibly SLX1 via its interaction with SLX4 [7].
Consistent with the above findings, BLM was found to genetically interact with the nuclease scaffolding protein SLX4, and it was determined that RNAi-mediated depletion of BLM in SLX4 null cells induced SL in the absence of any exogenous DNA damaging agents [8]. The interaction of SLX4 with the structure-specific nucleases XPF, MUS81, or SLX1 in BLM-deficient cells was explored. Genetic complementation with an SLX4 mutant lacking the XPF interaction site could rescue the lethality in BLM-deficient cells whereas a SLX4 mutant lacking the MUS81 or SLX1 interaction site was unable to rescue sensitivity, suggesting that the SLX4-MUS81 or SLX4-SLX1 interactions are crucial for cell viability in the absence of BLM. BLM-deficient cells defective in one of the key HJ resolvases displayed increased nuclear abnormalities due to unprocessed HJs, resulting in dysfunctional mitosis and cell death [8]. The collective evidence suggests that BLM’s ability to catalytically resolve DNA intermediates that arise due to recombination events elicited by DSBs is paramount to the suppression of DNA cleavage by structure-specific nucleases, which has direct consequences for chromosomal instability and survival. Understanding how BLM uniquely takes on its role (apparently distinct from that of WRN which genetically interacts with MUS81 in fork restart ([9]; see below)) will provide important mechanistic insight that may be valuable in delineating BLM’s profound functional capacity to suppress cancer in such a wide variety of tissues.
BLM helicase resides in a complex containing the DNA damage sensor BRCA1 and proteins s of the Fanconi Anemia (FA) core complex also implicated in DNA damage signaling and interstrand cross-link (ICL) repair [10]. The DNA translocase FANCM serves as a bridge between the FA core complex and the BLM-TopIIIα-RMI1-RMI2 complex implicated in double HJ dissolution (mentioned above) and processing of DNA recombinational repair intermediates [11]. This work established a link between FA and BS by mapping protein interaction domains; however, mechanistic details were elusive. Furthermore, BLM genetically interacts with the DNA translocase FANCM in interstrand cross-link (ICL) repair and the replication stress response, as shown in chicken DT40 cells [12]. The FANCM-Histone-Fold-Containing Protein (MHF) complex is important for a replication traverse pathway which enables DNA synthesis to continue past ICLs [13]. FANCM operates with a BLM-RMI2 protein complex in the same pathway to promote replication traverse of ICLs [12]. During replication stress, FANCM recruitment to stalled replication forks is dependent on both BLM helicase activity and BLM protein interaction, suggesting FANCM and BLM orchestrate their functions to activate the FA pathway and traverse the ICL during replicative DNA synthesis. The epistatic interactions between the FA core complex and BLM [12] were further supported by the discovery that BLM and FANCB operate in the same fork recovery pathway [14]. Thus, BLM’s involvement in SL interactions extends beyond genetic factors implicated in DSB metabolism (e.g., structure-specific nucleases) and includes fork remodeling DNA translocases (e.g., FANCM) when replicative DNA synthesis stalls. Given this role, researchers investigated BLM’s genetic interactions that help to preserve specialized ends of chromosomes (telomeres) [15]. In somatic cells, telomeres are shortened over successive cell divisions which can lead to senescence or apoptosis. While telomere shortening can act as a barrier to proliferation and tumorigenesis, most cancer cells reactivate the reverse transcriptase telomerase to elongate their telomeres. Therefore, targeting the telomerase pathway has attracted interest as an anti-cancer strategy [16]. However, 10-15% percent of human cancers use the telomerase-independent and HR-dependent process known as alternate lengthening of telomeres (ALT) to maintain their telomere lengths. ALT activity is most prevalent in mesenchymal bone-, soft tissue- and nervous system-cancers [17], and promotes resistance to telomerase inhibitors [18]. Deciphering the roles of BLM and other DNA helicases in these pathways should be informative for therapeutic purposes.
Based on the importance of telomere metabolism in cellular senescence, it is believed that helicases and their interacting partners play important roles in the ALT pathway. Consistent with its involvement in repair and of DNA breaks and stability of chromosome ends, BLM plays important roles in DSB end-resection [19, 20] and HR at telomeres [21, 22]. In ALT cells, FANCM depletion caused replication stress at telomeres and induced strong BLM focus formation, whereas BLM depletion in FANCM-deficient cells dramatically reduced end-resection at telomeres [23]. BLM depletion also impaired BRCA1 recruitment to damaged telomeres in FANCM-deficient ALT cells. Furthermore, BLM-dependent BRCA1 recruitment to damaged telomeres was shown to be important for end-resection in FANCM-depleted cells. In FANCM-depleted ALT cells, BLM and BRCA1 stimulate end-resection and error-free HR pathway to alleviate telomeric replication stress [23]. Co-depletion of BLM and FANCM led to SL in ALT cells, suggesting that FANCM operates parallel to BLM to preserve telomere ends in ALT cells.
The relationship between FANCM and BLM is complex and depends on replication stress and/or telomere status. BLM depletion in FANCM-knockout (KO) ALT-negative HCT116 cells caused a drastic reduction in viability, suggesting BLM and FANCM display a SL interaction irrespective of ALT status and exogenously induced replication stress [24]. Studies using a HR reporter (HR-Flex) containing an AT-rich sequence (derived from the common fragile sites (CFSs) and prone to form secondary structures) demonstrated that BLM and FANCM cooperatively resolve and faithfully repair secondary DNA structures formed at AT-rich sites. Thus, BLM and FANCM function in at least two non-epistatic pathway(s) to maintain genomic stability: i) CFSs located at non-telomeric AT-rich loci in a manner independent of ALT; ii) chromosome ends in ALT-negative cells. Mechanistic details of the BLM-FANCM genetic interactions operating at these two apparently distinct genomic DNA structures await further studies.
Cancer cells exhibit elevated reactive oxygen species (ROS) due to increased metabolic activities, mitochondrial dysfunction and inflammation [25]. This increases their dependency on pathways that minimize the deleterious effects of ROS [26]. Targeting these pathways can potentially be used as a strategy to sensitize cancer cells. Sgs1, the BLM homolog in budding yeast, is SL with mutation of the sod1 gene encoding Superoxide dismutase (SOD), a key enzyme that helps cells cope with ROS [27]. This eventually led researchers to test if BLM and SOD1 are SL in human cells [28]. SOD1 depletion or pharmacological inhibition of SOD1 by a compound designated lung cancer screen 1 that binds SOD1 and inhibits its enzymatic activity in vitro or ammonium tetrathiomolybdate (a chelator of Cu2+ required for SOD1 activity) in BLM-deficient cells caused elevated DNA damage, apoptosis, and cytotoxicity [28]. Treatment of cells with the ROS scavenger N-acetyl-L-cysteine (NAC) rescued cytotoxicity and restored growth of BLM-deficient cells depleted of SOD1 or treated with the SOD1 inhibitors, suggesting that increased oxidative stress selectively kills BLM-deficient cells. While human cell lines deficient in certain RECQ helicases (e.g., WRN) accumulate oxidative base lesions [29] and display mild sensitivity to agents that induce oxidative stress [30], to our knowledge none of the RECQ-deficient cell lines are reported to be sensitive to depletion or pharmacological inhibition of SOD1, suggesting a potentially unique SL interaction of BLM in this respect.
Synthetic Lethal Interactions of RECQL1
Cells protect stalled forks through activation of distinct genetic pathways which reduce the potency and efficacy of replication stress-inducing cytotoxic drugs such as PARP inhibitors. Understanding fork protective mechanisms and targeting these pathways can serve as a window of opportunity to identify new potential therapeutic targets. An integrated proteomic approach demonstrated that RECQL1 interacts with PARP1, an ADP-ribosylating enzyme important for repair of SSBs and DSBs, and this interaction is strengthened upon PARP1 poly(ADP-ribosyl)ation which occurs upon cellular DNA damage [31]. In vitro biochemical studies showed that RECQL1 catalyzes conversion of regressed (chicken foot-like) fork structures into model replication fork structures, and PARP1 poly (ADP-ribosyl)ation inhibits this fork restoration function of RECQL1. In the presence of a low dose of the Topoisomerase I inhibitor camptothecin (CPT) (50 nM), PARP inhibition caused a significant decrease in the number of regressed forks compared to control cells, whereas RECQL1 depletion caused an increase in reversed forks in the presence or absence of PARP inhibitor. Unlike RECQL1, depletion of WRN or BLM did not alter CPT-induced fork slowing, suggesting that RECQL1 is uniquely important to restart reversed forks; furthermore, PARP1 limits RECQL1-mediated fork reactivation. Thus, RECQL1-dependent fork restart may exert a fork protection mechanism that offers a proliferative advantage to RECQL1-overexpressing tumor cells.
Poly(ADP-ribose) chains synthesized by PARP are degraded mainly by poly(ADP-ribose) glycohydrolase (PARG) [32]. It was hypothesized that a PARG inhibitor (PARGi) would prevent degradation of Poly(ADP-ribose) chains, and that PARP1 poly(ADP-ribosyl)ation would sustain its inhibitory effect on RECQL1 and prevent RECQL1-dependent fork restart [33]. Depletion of RECQL1 sensitized Kuramochi ovarian cancer cells to PARGi resulting in elevated DNA damage and reduced proliferation. RECQL1 depletion in the ovarian cancer cells resistant to the PARGi did not show a significant difference in proliferation. These findings suggest that upon sensing replication stress PARP1 activity is initially important for fork stability and fork reversal, and PARP1 inhibits RECQL1-mediated fork restart. When normal conditions are restored, PARG catalyzes degradation of Poly(ADP-ribose) chains and removes the inhibitory effect of PARP1 on RECQL1 by catalyzing the removal of Poly(ADP-ribose) chains after which RECQL1 activity restarts the stalled replication forks. Thus, PARG and RECQL1 work together to restart the stalled replication forks by overcoming the impact of PARP-1 to keep forks regressed. The central role of RECQL1 in preventing nucleolytic degradation of regressed forks may help to inform cancer treatment strategies with the emerging interest in PARG inhibitors that cause fork stalling and replication catastrophe in cancer cells [34].
Synthetic Lethal Interactions of WRN
Like BLM, WRN displays a SL interaction with MUS81 [9]. Replication fork collapse in the absence of WRN engages MUS81 which cleaves collapsed forks, thereby generating DSBs. DSBs generated by MUS81 at stalled forks initiate Rad51-mediated recombination leading to replication restart that ensures cell viability at the expense of compromised genome stability. Consistent with these results, Werner syndrome (WS) cells were found to be hypersensitive to MUS81 knockdown upon treatment with the replication inhibitor hydroxyurea (HU).
Aside from pharmacological induction of replicative stress by an agent such as HU, overexpression of oncogenes such as Cyclin E or E2F1 introduces elevated strain due to heightened DNA synthesis. This led researchers to investigate a putative role of WRN under the latter condition [35]. Cyclin E (a regulatory subunit of cyclin-dependent kinase 2) is known to induce chromosomal instability due to fork slowing, increased origin firing and replication fork reversal [36, 37]. E2F1 is a transcription factor that modulates expression of genes that regulate replication, repair and cell cycle [38]. Murfini et al. showed that WRN is required for fork progression in cells overexpressing Cyclin E or E2F1 [35], suggesting WRN may resolve abnormal DNA structures during oncogene-induced replication stress. WRN down-regulation resulted in excessive chromosomal breakage at CFSs and induced cell death [35]. Co-depletion of WRN and MUS81 caused synthetic sickness in cells experiencing oncogene-induced replication stress, suggesting WRN and MUS81 act in two independent pathways.
Aside from the role of WRN to deal with broken or damaged replication forks, single-molecule DNA fiber studies demonstrated that WRN and Rad51 function in independent pathways to protect nascent DNA strands from MRE11-mediated degradation in response to CPT-induced replication stress [39]. The nascent DNA tracts were shorter in CPT-treated cells deficient in both WRN and Rad51 compared to cells solely deficient in WRN or Rad51 alone, suggesting that WRN and Rad51 additively protect the nascent DNA strands [39]. Together, the results suggest that Rad51 recruitment to collapsed replication forks is crucially important in the absence of WRN. The importance of the WRN-Rad51 genetic interaction was independently evidenced by an RNAi-based SL screen in WRN-deficient cells exposed to HU which revealed that WRN interacts with histone deacetylases HDAC1 and HDAC2 during stalled fork recovery [40]. In addition to heightened HU-sensitivity, loss of HDAC1 amplified defects in fork reactivation and progression in the WRN-depleted fibroblasts exposed to HU. Using iPOND (a technique to identify proteins at active, stalled, and collapsed replication forks), it was observed that Rad51 recruitment to HU-induced stalled forks in the absence of WRN or upon pharmacological inhibition of HDAC1 was reduced [40], suggesting WRN and HDAC1 may act in parallel pathways to recruit Rad51 for the recovery of stalled replication forks.
WRN is engaged with genome caretakers and tumor suppressors to preserve genome homeostasis. Crosstalk between WRN and the tumor suppressor p53 was revealed by genetic studies in mice. Although mutant mice lacking the WRN helicase domain did not show any signs of cancer or accelerated aging typically associated with WS [41], it remained unknown if the outcome of WRN deficiency would be affected by a genetic modifier. Because TP53 is mutated in more than 50% of human cancers [42]. Lombard et al. assessed if p53 loss in WRN mutated mice resulted in WS phenotypes. Their results showed that WRN+/−; TP53−/− mice had an average life span of 149 days, whereas WRN−/−; TP53−/− mice displayed a shorter average life span of 122 days, suggesting a synergistic relationship between p53 and WRN for mortality [41]. WRN deficiency in mice is also modulated by telomere status [43–45], implicating an important role of WRN in telomere metabolism that affects cancer and aging phenotypes.
RECQ helicases as Potential Therapeutic Targets
RECQ helicases are proposed targets for anti-cancer therapy. In recent years, the discovery of small molecule inhibitors of WRN and BLM helicases has proven useful for basic research and suggests a potential avenue for translational studies (Table 1). Specifically, recent developments have elevated WRN helicase to the forefront as a potential target for anti-cancer therapy. We review why WRN might be an ideal candidate for SL approaches to kill certain types of cancers characterized by microsatellite instability (MSI). Given the importance of mismatch repair (MMR) proteins to suppress MSI, a report of WRN’s interaction with MMR proteins [69] may play into its potential involvement in microsatellite-unstable cancers; however, it is still unclear how WRN and MMR proteins interact in nucleic acid metabolism.
WRN and BLM as Pharmacological Targets for Helicase Inhibition in Cell-Based Models
A small molecule (designated NSC 19630) targeting WRN helicase was found to preferentially kill cancer cell lines compared to non-cancerous epithelial and fibroblast cells [92], suggesting a pro-survival function of WRN helicase that can be pharmacologically modulated. Besides, WRN confers resistance of cancer cells grown in culture to agents that induce DNA damage or replication stress. For example, WRN-deficient cells were found to be sensitive to the clinically relevant topoisomerase inhibitor CPT [113]. Evidence suggests that the CPT sensitivity of breast cancer cells is attributed to CPT-induced degradation of WRN protein, implying that WRN status may influence the clinical outcome of topoisomerase inhibitor-based chemotherapy [114]. Previous work demonstrated synergistic killing of cancer cells by sublethal doses of the WRN helicase inhibitor NSC 19630 combined with the topoisomerase-I inhibitor topotecan, G4 stabilizing drug telomestatin, or PARP inhibitor KU0058948 [92]. Inhibition of WRN helicase by a structurally related compound (designated NSC617145) that more potently inhibits WRN helicase activity in vitro potentiated the cytotoxicity of the DNA ICL agent mitomycin C (MMC) in FA mutant cells already known to be defective in ICL repair [70, 115], suggesting that WRN helicase may play a back-up role to the FA pathway for ICL resistance. These observations and others [88, 90, 94] suggest that cancer cells with specific gene defects may rely heavily on WRN for growth and survival. Moroever, exposure of HeLa cells to the WRN helicase inhibitor NSC 617145 induces enrichment of WRN in the chromatin fraction and its proteasomal degradation, suggesting that the compound causes WRN to become more stably bound to genomic DNA and form a potentially toxic protein-DNA lesion [70]; however, further studies are required to substantiate this proposed mechanism of action. Continued progress in the development of potent and pharmacologically active chemical inhibitors of WRN helicase, such as a recently described high-throughput screen [116], should provide useful tools and potentially therapeutic compounds. To our knowledge, no inhibitors of WRN exonuclease activity have been reported.
In addition to WRN, a small molecule inhibitor of BLM with bio-activity in cancer cells has been identified in an in vitro high-throughput BLM helicase activity screen [117]. The BLM helicase inhibitor (designated ML216) caused elevated SCE in human cells, characteristic of BLM-deficient cells [117]. ML216 was found to inhibit cancer cell proliferation and potentiate the cytotoxicity of the DNA polymerase α inhibitor aphidicolin in a BLM-dependent manner. A more potent structural analog of ML216 conferred similar BLM-specific cytotoxicity in cancer cells [118]. Despite these advances, the in vivo efficacy of either the BLM or WRN helicase inhibitors is not known.
WRN as a Synthetic Lethal Target in Microsatellite-Unstable Cancers
WRN helicase has been reported as a potential SL therapeutic target in microsatellite-unstable cancers which may have inactivated tumor suppressor genes. MSI is a hypermutator phenotype that occurs due to impaired repair of mismatches generated during replication of microsatellite repeat regions [71, 72]. Inactivating mutations or epigenetic silencing of mismatch repair (MMR) pathway genes MLH1, MSH2, MSH3, MSH6, and PMS2 give rise to MSI which occurs in >20 types of human cancers with a relatively higher frequency in colon, stomach, endometrial, rectal, esophageal and ovarian carcinomas (~3-28%) [73]. A unique functional genomics approach coupled with systematic computational analysis was utilized to identify and prioritize druggable therapeutic targets in human cancers [74].
WRN was identified from its tractability group as a selective survival dependency in MSI cancer cells. Other RECQ genes (RECQL1, BLM, and RECQL5) were dispensable for MSI cell lines, suggesting a WRN-specific dependency. Downstream loss-of-function assays confirmed SL in WRN-knockout MSI cells but not in microsatellite stable (MSS) lines of the same tissue types. Furthermore, functional rescue experiments using wild type, helicase dead, or exonuclease dead hypomorphic mouse Wrn revealed the loss-of-fitness effect is solely attributed to WRN’s helicase function. Of note, WRN-dependency was significantly correlated with MLH1 promoter hypermethylation, mutations in the MMR gene MSH6, or mutations in the histone methyltransferase gene MLL2 [74]. These observations suggested that MSI cancer cells require WRN helicase activity to cope with or repair detrimental DNA lesions that arise due to defective genome metabolism arising from deficient MMR.
A systematic analysis of publicly available cancer cell line datasets from genome-scale SL screens identified WRN as the top-scoring loss-of-fitness gene in MSI cancers [60]. Unlike the discovery approach by Behan et al. [58], this study sought to identify selective genetic vulnerabilities in MSI cancers by analyzing SL screening data from whole-genome CRISPR-Cas9 (Project Achilles) or RNAi (Project DRIVE) of many cell lines; WRN displayed a consistent selective genetic dependency in MSI cell lines [61, 62]. Consistent with the other finding [58]. RecQ helicases other than WRN were not found to be essential for MSI cancer cells. WRN-dependency of MSI cancer cells was substantiated in competitive cell viability assays. The attenuated rescue of cellular fitness by a helicase-dead WRN mutant in MSI cells confirmed that WRN helicase activity supported growth and viability of cancer cells with MMR deficiency. WRN knockout triggered chromosome instability, DSBs, p53 activation, and cell cycle arrest leading to apoptosis in MSI cells, but not in MSS cells.
Two other research groups reported a WRN-MSI SL interaction. Using a candidate gene approach, researchers sought to identify potential SL interactions of WRN or BLM with DNA damage response genes and found that co-depletion of WRN and the MMR gene MLH1 to be SL [75]. Their observations were validated in multiple MMR-deficient cancer cell lines, indicating that WRN-dependency is a general phenotype of MSI cancer cells. Given the observed WRN-dependency of a subset of cancer cell lines as demonstrated in the Project DRIVE shRNA-based loss-of-function screening dataset [76], genetic biomarkers associated with selective WRN dependency were examined [77]. Integrated analysis of genetic dependency and MSS/MSI status of cancer cell models revealed strong WRN-specific dependency across colorectal, gastric, and endometrial cancer cell lines. Mitotic defects and increased chromosomal aberrations were the underlying source of the WRN-MSI SL. Notably, all four studies described above concluded that WRN helicase but not exonuclease activity is required for survival of MSI tumor cells. Importantly, WRN-specific SL dependency was successfully recapitulated in vivo, showing that inducible knockout or knockdown of WRN significantly attenuated microsatellite unstable colon cancer tumor growth in mouse xenograft models [74, 78].
Although cell-based experimental results suggested that elevated chromosomal instability and DSB accumulation underlie lethality in WRN-knockout microsatellite-unstable cancer cells, the precise molecular mechanism of SL remains obscure. Simplistically, microsatellite-unstable cells may require WRN helicase activity to resolve genomic DNA structures that arise downstream of MMR defects in these cells. However, the question remains as to the identity of these nucleic acid molecules that uniquely require WRN helicase activity for their metabolism. Owing to its DNA branch-migration activity, WRN helicase resolves complex DNA structures (D-loops, HJs) that represent key intermediates of HR, DNA repair, and stalled replication forks [79, 80]. WRN physically and functionally interacts with MMR proteins [69], as well as numerous other DNA replication and repair proteins. Experimental evidence suggests that MMR proteins act in concert with WRN helicase for heteroduplex rejection to suppress recombination between divergent nucleotide sequences [81, 82]. Hypersensitivity of MMR-deficient cancer cells to thymidine which causes dNTP pool imbalance is attributed to HR repair defects [83]. Thus, SL in WRN-depleted MSI cancer cells may be a consequence of the inefficient processing of deleterious HR intermediates due to loss of WRN helicase function. Given the role of WRN helicase to facilitate resolution and replication of DNA hairpins and other non-canonical DNA structures generated by nucleotide repeat sequences, it is plausible that WRN plays a direct role in metabolizing insertion and deletion loops routinely acted upon by MMR proteins [84]. In Figure 2, we propose how WRN might deal with problematic DNA structures arising in microsatellite unstable cancers.
Figure 2. Proposed models illustrating how WRN helicase resolves non-B DNA secondary structures at microsatellite repeats and facilitate DNA synthesis.
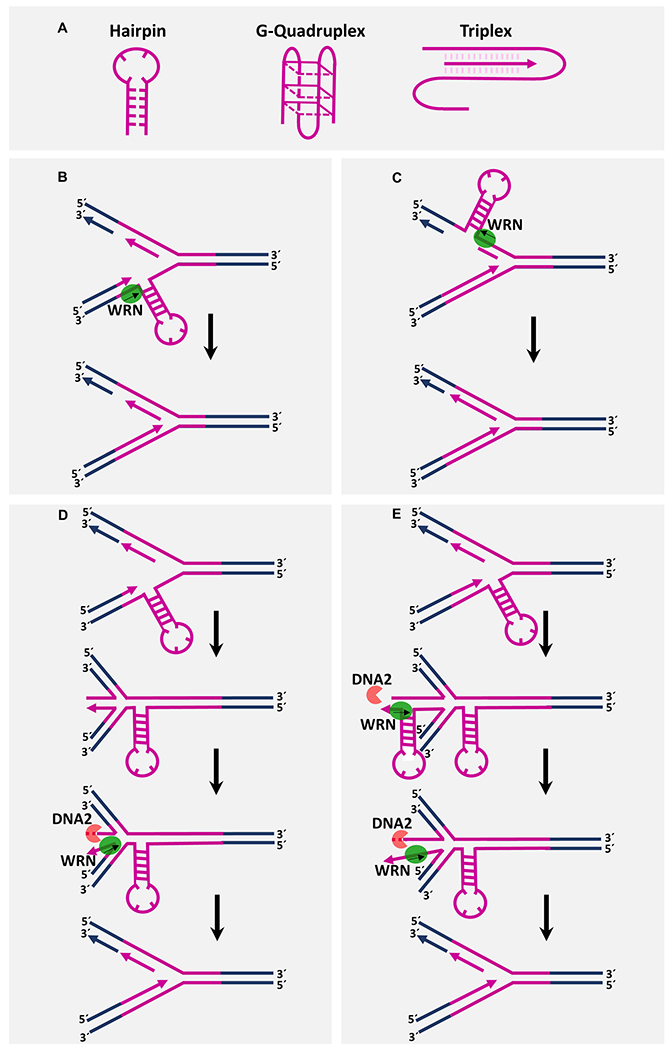
A. Microsatellite-associated non-canonical DNA structures potentially resolved by WRN helicase activity. The microsatellite repeat sequences can adopt various non-B DNA conformations during DNA replication. B and C. Using its helicase activity, WRN resolves secondary structures formed in either the leading (B) or lagging (C) strand and allows smooth DNA synthesis through repeat regions. D. WRN helicase facilitates restart of stalled replication at DNA secondary structures. A replication fork encounters a non-B DNA structure of repeat sequences and regresses to form a four-way “chicken-foot” like structure. Using its 3′-5′ helicase activity, WRN partially unwinds the duplex region of the “middle toe” of the regressed chicken-foot regressed fork, thereby facilitating 5′-3′ nucleolytic processing by DNA2 nuclease. The partially single-stranded regressed fork is reset to an active fork by HR or reverse branch-migration which allows DNA synthesis to continue past the DNA secondary structure element. E. During fork regression caused by DNA secondary structure, the nascently synthesized DNA strand with repeat sequence elements could fold back on itself and form a non-canonical structure within the regressed arm of the reversed fork. WRN helicase may resolve the regressed strand secondary structure to allow DNA2-mediated end resection and fork restart. Repeats sequences are shown in purple.
An important issue is whether impaired MMR solely underpins the WRN-MSI SL. This is particularly relevant because restoration of MMR function by knocking in MLH1 in microsatellite unstable HCT116 cells did not efficiently rescue cell viability upon WRN depletion [74, 75, 77, 78]. Conversely, MLH1-knockout microsatellite-stable SW620 cells did not exhibit a WRN-dependency [74], suggesting that defective MMR may not solely contribute to WRN dependency in microsatellite-unstable tumor cells. In support of this notion, an integrated analysis of MSI frequency and WRN dependency in MMR-defective cancer cell lines revealed that the WRN-dependency of cancer cells depends on the extent of microsatellite mutation events within them [78]. Cancer cell lines derived from lineages with less frequent MSI show diminished WRN dependency [78]. Therefore, it is likely that loss of MMR functions might precede yet unidentified irreversible genetic changes that render microsatellite-unstable tumor cells WRN-dependent. It is plausible that microsatellite unstable cancer cells have a stringent requirement to prevent imperfect (homeologous) recombination between DNA sequences with mismatches, which may involve the aforementioned interaction of WRN with MMR proteins [69]. In addition, WRN helicase may have a primary role to resolve secondary DNA structures formed at tandem repeats during replication (Figure 2).
Lastly, although targeted inhibition of WRN helicase is a promising therapeutic intervention in microsatellite-unstable human cancers, it would be valuable to assess potential confounding factors such as driver gene mutations which influence the therapy response. TP53 mutation status may determine the degree of fitness in microsatellite unstable cancer cells upon WRN perturbation as MSI cells with wild-type p53 exhibited much greater WRN-dependency as compared to those with mutated p53 [78]. This notion is supported by increased activation of p53 in MSI cells upon WRN loss, thereby suggesting a partial but necessary contribution of p53 to WRN dependence [78]. Overall, these studies position WRN helicase as a druggable target in microsatellite unstable cancers but also raise important mechanistic questions and open multiple new avenues of future research.
Concluding Remarks
Since the complete sequencing of the human genome announced in the early 2000’s (https://web.ornl.gov/sci/techresources/Human_Genome/project/journals.shtml#2003), the field of personalized medicine has accelerated in its efforts toward diagnosis, prevention strategies, and cures of genetic diseases and cancer. We propose that helicases are rising stars as genome caretakers, but there is much to learn (see Outstanding Questions box). Resistance of tumors to drugs that exploit apparent SL is a valid concern, as clinical trials for other targets (such as the PARP inhibitors) are modified for improvement and to determine efficacy. Based on the apparently strong reliance of cancer cells on RECQ helicases to deal with the accumulation of replicative lesions in rapidly dividing cells, it seems that the focus should be on the most proliferative cancers, keeping in mind the potential cytotoxicity to surrounding normal cells. In the case of WRN, it will be important to determine the precise DNA structure(s) that WRN acts upon to deal with MSI, as this may yield insight to targeting WRN’s unappreciated role(s) in other cancers. Further development of reliable and robust diagnostics will provide insight to which cancers will be best to target BLM or RECQL1.
Outstanding Questions Box.
How are the RecQ helicases unique in their pathway functions to suppress chromosomal instability or promote carcinogenesis?
What are the precise replicative lesions that elicit an increased reliance on DNA helicases for cancer cells to proliferate and survive despite the elevated DNA damage?
Can the catalytic activity of DNA helicases be modulated pharmacologically in vivo in a manner that could be used for therapeutic strategies to combat cancer?
What is the molecular mechanism for the synthetic lethality of WRN helicase deficiency in microsatellite unstable cancer cells?
Is WRN a druggable target in microsatellite unstable cancers?
By what mechanisms do helicases in addition to WRN determine fitness in cancer cells characterized by specific molecular defects other than microsatellite instability?
Research studies such as those summarized here will continue to advance the field’s appreciation and knowledge of how RECQ helicases and other DNA damage response proteins have become leading actors in genome metabolism and potentially represent targets for cancer therapy. Indeed, the multiple connections of RECQ helicases with other “druggable” pathways might be useful to identify specific interactors that might be more easily targeted. DNA damage checkpoint signaling pathways mediated by the protein kinases ATR and ATM engage in crosstalk with RECQs and other DNA repair or replication stress response proteins at multiple levels [86], suggesting an opportunity to exploit their putative SL. Development in biomarkers that assess the efficacy of drugs that target such factors, as well as clinical trials in progress, suggest that anti-cancer therapies may make significant advances in the near future.
Highlights.
Cancer cells rely on RecQ helicases to deal with replicative stress so that they can proliferate
Genetic interactions of WRN, BLM, and RECQL1 suggest novel paradigms for anti-cancer therapy
Targeted small molecule inhibition of DNA helicase activity is useful for basic research and may be a potential avenue for translational studies
WRN helicase is an emerging synthetic lethal therapeutic target in microsatellite-unstable cancers
DNA helicases are gaining traction in the field of personalized medicine
Acknowledgments
This work was supported by the Intramural Research Program, National Institute on Aging, NIH.
Glossary
- alternative lengthening of telomeres (ALT)
a telomerase-independent but recombination-dependent process/mechanism to maintain telomere length that is prominent in certain types of cancer cells
- double-strand break repair
cellular mechanisms whereby the two ends of a DNA double helix in which both strands are severed are rejoined; the three major pathways of double-strand break repair are non-homologous end-joining, microhomology medicate end-joining, and homologous recombination.
- fork regression
the initial step in fork remodeling when replicative DNA synthesis is stalled by DNA damage or perturbation to the double-stranded DNA ahead of the replication fork; by enabling fork reversal, fork regression can suppress double-strand breaks and genomic instability, provided that the subsequent steps of the process occur faithfully
- G-quadruplex (G4)
a secondary structure in DNA or RNA that forms in sequences that are rich in guanine. Distinct from the DNA double helix, G4 is characterized by a guanine tetra-helical structure in which stabilized by an alternate form of hydrogen bonding. The guanine tetrads can from one, two or four strands. G4 structures are believed to modulate processes such as replication and transcription and are structural components of specialized regions of the genome such as guanine-rich telomeres.
- helicase
an enzyme that catalytically unwinds structured nucleic acids by disrupting complementary hydrogen bonds in a reaction driven by the hydrolysis of nucleoside triphosphate
- Holliday Junction
mobile junction between four strands of DNA that represents an intermediate in genetic recombination and important for genomic stability
- microsatellite instability
change in a short, repeated sequence of DNA distinguishable from what was inherited; found in many types of cancer and may be a consequence of replication errors that fail to be corrected
- reactive oxygen species
chemically reactive molecules and free radicals derived from molecular oxygen that can damage macromolecules and elicit cellular signaling pathways
- RECQ
family of amino acid sequence-related helicase enzymes initially identified in bacteria but known to exist in eukaryotes as well; in human there are five different RECQ helicases
- replication traverse
DNA synthesis catalyzed by DNA polymerase and components of the replisome past a DNA lesion (e.g., interstrand cross-link) thought to inhibit helicase-catalyzed duplex DNA unwinding and typically stall the replication machinery
- RNA interference
classically a biological mechanism whereby gene silencing is achieved through the creation of a double-stranded RNA molecule that either deters transcription or causes RNA degradation; technical advances enabled researcher to silence specific genes in a range of organisms, which can be a useful tool for studying gene function
- small molecule inhibitor
a chemical compound usually of less than 500 Daltons that inhibit a specific function or interaction of protein; small molecule inhibitors are emerging in medicinal cancer therapeutic strategies
- synthetic lethality
relationship of two genes that when mutated disallow viability of the cells, whereas mutation of only one in the pair is still compatible with viability
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Bryant HE et al. (2005) Specific killing of BRCA2-deficient tumours with inhibitors of poly(ADP-ribose) polymerase. Nature 434 (7035), 913–7. [DOI] [PubMed] [Google Scholar]
- 2.Farmer H et al. (2005) Targeting the DNA repair defect in BRCA mutant cells as a therapeutic strategy. Nature 434 (7035), 917–21. [DOI] [PubMed] [Google Scholar]
- 3.Brosh RM Jr. (2013) DNA helicases involved in DNA repair and their roles in cancer. Nat Rev Cancer 13 (8), 542–58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Datta A and Brosh RM Jr. (2018) New Insights Into DNA Helicases as Druggable Targets for Cancer Therapy. Front Mol Biosci 5, 59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Chaganti RS et al. (1974) A manyfold increase in sister chromatid exchanges in Bloom’s syndrome lymphocytes. Proc Natl Acad Sci U S A 71 (11), 4508–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Wu L and Hickson ID (2003) The Bloom’s syndrome helicase suppresses crossing over during homologous recombination. Nature 426 (6968), 870–4. [DOI] [PubMed] [Google Scholar]
- 7.Sarbajna S et al. (2014) Roles of SLX1-SLX4, MUS81-EME1, and GEN1 in avoiding genome instability and mitotic catastrophe. Genes Dev 28 (10), 1124–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Garner E et al. (2013) Human GEN1 and the SLX4-associated nucleases MUS81 and SLX1 are essential for the resolution of replication-induced Holliday junctions. Cell Rep 5 (1), 207–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Franchitto A et al. (2008) Replication fork stalling in WRN-deficient cells is overcome by prompt activation of a MUS81-dependent pathway. J Cell Biol 183 (2), 241–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Meetei AR et al. (2003) A multiprotein nuclear complex connects Fanconi anemia and Bloom syndrome. Mol Cell Biol 23 (10), 3417–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Deans AJ and West SC (2009) FANCM connects the genome instability disorders Bloom’s Syndrome and Fanconi Anemia. Mol Cell 36 (6), 943–53. [DOI] [PubMed] [Google Scholar]
- 12.Ling C et al. (2016) Bloom syndrome complex promotes FANCM recruitment to stalled replication forks and facilitates both repair and traverse of DNA interstrand crosslinks. Cell Discov 2, 16047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Huang J et al. (2013) The DNA translocase FANCM/MHF promotes replication traverse of DNA interstrand crosslinks. Mol Cell 52 (3), 434–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kim TM et al. (2015) RECQL5 and BLM exhibit divergent functions in cells defective for the Fanconi anemia pathway. Nucleic Acids Res 43 (2), 893–903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.de Lange T (2004) T-loops and the origin of telomeres. Nat Rev Mol Cell Biol 5 (4), 323–9. [DOI] [PubMed] [Google Scholar]
- 16.Shay JW (2016) Role of Telomeres and Telomerase in Aging and Cancer. Cancer Discov 6 (6), 584–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Henson JD and Reddel RR (2010) Assaying and investigating Alternative Lengthening of Telomeres activity in human cells and cancers. FEBS Lett 584 (17), 3800–11. [DOI] [PubMed] [Google Scholar]
- 18.Ivancich M et al. (2017) Treating Cancer by Targeting Telomeres and Telomerase. Antioxidants (Basel) 6 (1). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Gravel S et al. (2008) DNA helicases Sgs1 and BLM promote DNA double-strand break resection. Genes Dev 22 (20), 2767–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Nimonkar AV et al. (2011) BLM-DNA2-RPA-MRN and EXO1-BLM-RPA-MRN constitute two DNA end resection machineries for human DNA break repair. Genes Dev 25 (4), 350–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Barefield C and Karlseder J (2012) The BLM helicase contributes to telomere maintenance through processing of late-replicating intermediate structures. Nucleic Acids Res 40 (15), 7358–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Lillard-Wetherell K et al. (2004) Association and regulation of the BLM helicase by the telomere proteins TRF1 and TRF2. Hum Mol Genet 13 (17), 1919–32. [DOI] [PubMed] [Google Scholar]
- 23.Pan X et al. (2017) FANCM, BRCA1, and BLM cooperatively resolve the replication stress at the ALT telomeres. Proc Natl Acad Sci U S A 114 (29), E5940–E5949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Wang H et al. (2018) BLM prevents instability of structure-forming DNA sequences at common fragile sites. PLoS Genet 14 (11), e1007816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Ishikawa K et al. (2008) ROS-generating mitochondrial DNA mutations can regulate tumor cell metastasis. Science 320 (5876), 661–4. [DOI] [PubMed] [Google Scholar]
- 26.Anastasiou D et al. (2011) Inhibition of pyruvate kinase M2 by reactive oxygen species contributes to cellular antioxidant responses. Science 334 (6060), 1278–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Pan X et al. (2006) A DNA integrity network in the yeast Saccharomyces cerevisiae. Cell 124 (5), 1069–81. [DOI] [PubMed] [Google Scholar]
- 28.Sajesh BV and McManus KJ (2015) Targeting SOD1 induces synthetic lethal killing in BLM- and CHEK2-deficient colorectal cancer cells. Oncotarget 6 (29), 27907–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Das A et al. (2007) The human Werner syndrome protein stimulates repair of oxidative DNA base damage by the DNA glycosylase NEIL1. J Biol Chem 282 (36), 26591–602. [DOI] [PubMed] [Google Scholar]
- 30.Szekely AM et al. (2005) Werner protein protects nonproliferating cells from oxidative DNA damage. Mol Cell Biol 25 (23), 10492–506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Berti M et al. (2013) Human RECQ1 promotes restart of replication forks reversed by DNA topoisomerase I inhibition. Nat Struct Mol Biol 20 (3), 347–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Bonicalzi ME et al. (2005) Regulation of poly(ADP-ribose) metabolism by poly(ADP-ribose) glycohydrolase: where and when? Cell Mol Life Sci 62 (7-8), 739–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Pillay N et al. (2019) DNA Replication Vulnerabilities Render Ovarian Cancer Cells Sensitive to Poly(ADP-Ribose) Glycohydrolase Inhibitors. Cancer Cell 35 (3), 519–533 e8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Slade D (2020) PARP and PARG inhibitors in cancer treatment. Genes Dev 34 (5-6), 360–394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Murfuni I et al. (2013) The WRN and MUS81 proteins limit cell death and genome instability following oncogene activation. Oncogene 32 (5), 610–20. [DOI] [PubMed] [Google Scholar]
- 36.Jones RM et al. (2013) Increased replication initiation and conflicts with transcription underlie Cyclin E-induced replication stress. Oncogene 32 (32), 3744–53. [DOI] [PubMed] [Google Scholar]
- 37.Neelsen KJ et al. (2013) Oncogenes induce genotoxic stress by mitotic processing of unusual replication intermediates. J Cell Biol 200 (6), 699–708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Ren B et al. (2002) E2F integrates cell cycle progression with DNA repair, replication, and G(2)/M checkpoints. Genes Dev 16 (2), 245–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Su F et al. (2014) Nonenzymatic role for WRN in preserving nascent DNA strands after replication stress. Cell Rep 9 (4), 1387–401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Kehrli K et al. (2016) Class I Histone Deacetylase HDAC1 and WRN RECQ Helicase Contribute Additively to Protect Replication Forks upon Hydroxyurea-induced Arrest. J Biol Chem 291 (47), 24487–24503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Lombard DB et al. (2000) Mutations in the WRN gene in mice accelerate mortality in a p53-null background. Mol Cell Biol 20 (9), 3286–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Hussain SP and Harris CC (1998) Molecular epidemiology of human cancer: contribution of mutation spectra studies of tumor suppressor genes. Cancer Res 58 (18), 4023–37. [PubMed] [Google Scholar]
- 43.Chang S et al. (2004) Essential role of limiting telomeres in the pathogenesis of Werner syndrome. Nat Genet 36 (8), 877–82. [DOI] [PubMed] [Google Scholar]
- 44.Du X et al. (2004) Telomere shortening exposes functions for the mouse Werner and Bloom syndrome genes. Mol Cell Biol 24 (19), 8437–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Laud PR et al. (2005) Elevated telomere-telomere recombination in WRN-deficient, telomere dysfunctional cells promotes escape from senescence and engagement of the ALT pathway. Genes Dev 19 (21), 2560–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Lu H et al. (2017) Cell cycle-dependent phosphorylation regulates RECQL4 pathway choice and ubiquitination in DNA double-strand break repair. Nat Commun 8 (1), 2039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Lu H et al. (2016) RECQL4 Promotes DNA End Resection in Repair of DNA Double-Strand Breaks. Cell Rep 16 (1), 161–173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Shamanna RA et al. (2014) RECQ helicase RECQL4 participates in non-homologous end joining and interacts with the Ku complex. Carcinogenesis 35 (11), 2415–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Kohzaki M et al. (2020) Human RECQL4 represses the RAD52-mediated single-strand annealing pathway after ionizing radiation or cisplatin treatment. International Journal of Cancer 146 (11), 3098–3113. [DOI] [PubMed] [Google Scholar]
- 50.Cooper MP et al. (2000) Ku complex interacts with and stimulates the Werner protein. Genes Dev 14 (8), 907–12. [PMC free article] [PubMed] [Google Scholar]
- 51.Shamanna RA et al. (2016) WRN regulates pathway choice between classical and alternative non-homologous end joining. Nat Commun 7, 13785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Chen L et al. (2003) WRN, the protein deficient in Werner syndrome, plays a critical structural role in optimizing DNA repair. Aging Cell 2 (4), 191–9. [DOI] [PubMed] [Google Scholar]
- 53.Khadka P et al. (2015) Differential and Concordant Roles for Poly(ADP-Ribose) Polymerase 1 and Poly(ADP-Ribose) in Regulating WRN and RECQL5 Activities. Mol Cell Biol 35 (23), 3974–89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Hu Y et al. (2005) Recql5 and Blm RecQ DNA Helicases Have Nonredundant Roles in Suppressing Crossovers. Molecular and Cellular Biology 25 (9), 3431–3442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Hu Y et al. (2007) RECQL5/Recql5 helicase regulates homologous recombination and suppresses tumor formation via disruption of Rad51 presynaptic filaments. Genes Dev 21 (23), 3073–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Paliwal S et al. (2014) Human RECQ5 helicase promotes repair of DNA double-strand breaks by synthesis-dependent strand annealing. Nucleic Acids Res 42 (4), 2380–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Wu L et al. (2005) The HRDC domain of BLM is required for the dissolution of double Holliday junctions. Embo j 24 (14), 2679–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Balajee AS et al. (1999) The Werner syndrome protein is involved in RNA polymerase II transcription. Mol Biol Cell 10 (8), 2655–68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Johnson JE et al. (2010) Altered gene expression in the Werner and Bloom syndromes is associated with sequences having G-quadruplex forming potential. Nucleic Acids Res 38 (4), 1114–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Fry M and Loeb LA (1999) Human werner syndrome DNA helicase unwinds tetrahelical structures of the fragile X syndrome repeat sequence d(CGG)n. J Biol Chem 274 (18), 12797–802. [DOI] [PubMed] [Google Scholar]
- 61.Mohaghegh P et al. (2001) The Bloom’s and Werner’s syndrome proteins are DNA structure-specific helicases. Nucleic Acids Res 29 (13), 2843–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Sun H et al. (1998) The Bloom’s syndrome helicase unwinds G4 DNA. J Biol Chem 273 (42), 27587–92. [DOI] [PubMed] [Google Scholar]
- 63.Nguyen GH et al. (2014) Regulation of gene expression by the BLM helicase correlates with the presence of G-quadruplex DNA motifs. Proc Natl Acad Sci U S A 111 (27), 9905–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Tang W et al. (2016) The Werner syndrome RECQ helicase targets G4 DNA in human cells to modulate transcription. Hum Mol Genet 25 (10), 2060–2069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Li XL et al. (2014) Identification of RECQ1-regulated transcriptome uncovers a role of RECQ1 in regulation of cancer cell migration and invasion. Cell Cycle 13 (15), 2431–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Popuri V et al. (2008) The Human RecQ helicases, BLM and RECQ1, display distinct DNA substrate specificities. J Biol Chem 283 (26), 17766–76. [DOI] [PubMed] [Google Scholar]
- 67.Wu Y et al. (2008) FANCJ helicase defective in Fanconia anemia and breast cancer unwinds G-quadruplex DNA to defend genomic stability. Mol Cell Biol 28 (12), 4116–28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Budhathoki JB et al. (2016) A Comparative Study of G-Quadruplex Unfolding and DNA Reeling Activities of Human RECQ5 Helicase. Biophys J 110 (12), 2585–2596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Saydam N et al. (2007) Physical and functional interactions between Werner syndrome helicase and mismatch-repair initiation factors. Nucleic Acids Res 35 (17), 5706–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Aggarwal M et al. (2013) Werner syndrome helicase has a critical role in DNA damage responses in the absence of a functional fanconi anemia pathway. Cancer Res 73 (17), 5497–507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Kunkel TA and Erie DA (2015) Eukaryotic Mismatch Repair in Relation to DNA Replication. Annu Rev Genet 49, 291–313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Lipkin SM et al. (2000) MLH3: a DNA mismatch repair gene associated with mammalian microsatellite instability. Nat Genet 24 (1), 27–35. [DOI] [PubMed] [Google Scholar]
- 73.Cortes-Ciriano I et al. (2017) A molecular portrait of microsatellite instability across multiple cancers. Nat Commun 8, 15180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Behan FM et al. (2019) Prioritization of cancer therapeutic targets using CRISPR-Cas9 screens. Nature 568 (7753), 511–516. [DOI] [PubMed] [Google Scholar]
- 75.Kategaya L et al. (2019) Werner Syndrome Helicase Is Required for the Survival of Cancer Cells with Microsatellite Instability. iScience 13, 488–497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.McDonald ER 3rd et al. (2017) Project DRIVE: A Compendium of Cancer Dependencies and Synthetic Lethal Relationships Uncovered by Large-Scale, Deep RNAi Screening. Cell 170 (3), 577–592.e10. [DOI] [PubMed] [Google Scholar]
- 77.Lieb S et al. (2019) Werner syndrome helicase is a selective vulnerability of microsatellite instability-high tumor cells. Elife 8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Chan EM et al. (2019) WRN helicase is a synthetic lethal target in microsatellite unstable cancers. Nature 568 (7753), 551–556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Opresko PL et al. (2009) The Werner syndrome helicase/exonuclease processes mobile Dloops through branch migration and degradation. PLoS One 4 (3), e4825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Sharma S et al. (2006) Mechanisms of RecQ helicases in pathways of DNA metabolism and maintenance of genomic stability. Biochem J 398 (3), 319–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Goldfarb T and Alani E (2005) Distinct roles for the Saccharomyces cerevisiae mismatch repair proteins in heteroduplex rejection, mismatch repair and nonhomologous tail removal. Genetics 169 (2), 563–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Sugawara N et al. (2004) Heteroduplex rejection during single-strand annealing requires Sgs1 helicase and mismatch repair proteins Msh2 and Msh6 but not Pms1. Proc Natl Acad Sci U S A 101 (25), 9315–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Mohindra A et al. (2002) Defects in homologous recombination repair in mismatch-repair-deficient tumour cell lines. Hum Mol Genet 11 (18), 2189–200. [DOI] [PubMed] [Google Scholar]
- 84.Kamath-Loeb AS et al. (2001) Interactions between the Werner syndrome helicase and DNA polymerase delta specifically facilitate copying of tetraplex and hairpin structures of the d(CGG)n trinucleotide repeat sequence. J Biol Chem 276 (19), 16439–46. [DOI] [PubMed] [Google Scholar]
- 85.Kawabe T et al. (2000) Differential regulation of human RecQ family helicases in cell transformation and cell cycle. Oncogene 19 (41), 4764–72. [DOI] [PubMed] [Google Scholar]
- 86.Slupianek A et al. (2011) BCR/ABL stimulates WRN to promote survival and genomic instability. Cancer Res 71 (3), 842–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Arai A et al. (2011) RECQL1 and WRN proteins are potential therapeutic targets in head and neck squamous cell carcinoma. Cancer Res 71 (13), 4598–607. [DOI] [PubMed] [Google Scholar]
- 88.Robinson K et al. (2009) c-Myc accelerates S-phase and requires WRN to avoid replication stress. PLoS One 4 (6), e5951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Mao FJ et al. (2010) The human WRN and BLM RecQ helicases differentially regulate cell proliferation and survival after chemotherapeutic DNA damage. Cancer Res 70 (16), 6548–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Moser R et al. (2012) MYC-driven tumorigenesis is inhibited by WRN syndrome gene deficiency. Mol Cancer Res 10 (4), 535–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Opresko PL et al. (2007) Role for the Werner syndrome protein in the promotion of tumor cell growth. Mech Ageing Dev 128 (7-8), 423–36. [DOI] [PubMed] [Google Scholar]
- 92.Aggarwal M et al. (2011) Inhibition of helicase activity by a small molecule impairs Werner syndrome helicase (WRN) function in the cellular response to DNA damage or replication stress. Proc Natl Acad Sci U S A 108 (4), 1525–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Moles R et al. (2016) WRN-targeted therapy using inhibitors NSC 19630 and NSC 617145 induce apoptosis in HTLV-1-transformed adult T-cell leukemia cells. J Hematol Oncol 9 (1), 121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Futami K et al. (2007) Increased chemotherapeutic activity of camptothecin in cancer cells by siRNA-induced silencing of WRN helicase. Biol Pharm Bull 30 (10), 1958–61. [DOI] [PubMed] [Google Scholar]
- 95.Turley H et al. (2001) The distribution and expression of the Bloom’s syndrome gene product in normal and neoplastic human cells. Br J Cancer 85 (2), 261–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Qian X et al. (2017) RecQ helicase BLM regulates prostate cancer cell proliferation and apoptosis. Oncol Lett 14 (4), 4206–4212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Arora A et al. (2016) Clinicopathological and prognostic significance of RECQL5 helicase expression in breast cancers. Carcinogenesis 37 (1), 63–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Chen K et al. (2019) Bloom Syndrome Protein Activates AKT and PRAS40 in Prostate Cancer Cells. Oxid Med Cell Longev 2019, 3685817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Sanada S et al. (2013) RECQL1 DNA repair helicase: a potential therapeutic target and a proliferative marker against ovarian cancer. PLoS One 8 (8), e72820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Mendoza-Maldonado R et al. (2011) The human RECQ1 helicase is highly expressed in glioblastoma and plays an important role in tumor cell proliferation. Mol Cancer 10, 83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Futami K et al. (2010) RecQL1 DNA repair helicase: A potential tumor marker and therapeutic target against hepatocellular carcinoma. Int J Mol Med 25 (4), 537–45. [DOI] [PubMed] [Google Scholar]
- 102.Li D et al. (2016) RECQ1 A159C Polymorphism Is Associated With Overall Survival of Patients With Resected Pancreatic Cancer: A Replication Study in NRG Oncology Radiation Therapy Oncology Group 9704. Int J Radiat Oncol Biol Phys 94 (3), 554–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Sharma S and Brosh RM Jr. (2007) Human RECQ1 is a DNA damage responsive protein required for genotoxic stress resistance and suppression of sister chromatid exchanges. PLoS One 2 (12), e1297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Futami K et al. (2008) Anticancer activity of RecQL1 helicase siRNA in mouse xenograft models. Cancer Sci 99 (6), 1227–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Popuri V et al. (2012) RECQ1 is required for cellular resistance to replication stress and catalyzes strand exchange on stalled replication fork structures. Cell Cycle 11 (22), 4252–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Hills SA and Diffley JF (2014) DNA replication and oncogene-induced replicative stress. Curr Biol 24 (10), R435–44. [DOI] [PubMed] [Google Scholar]
- 107.Bartek J et al. (2012) Thresholds of replication stress signaling in cancer development and treatment. Nat Struct Mol Biol 19 (1), 5–7. [DOI] [PubMed] [Google Scholar]
- 108.Bhattacharyya S et al. (2009) Telomerase-associated protein 1, HSP90, and topoisomerase IIalpha associate directly with the BLM helicase in immortalized cells using ALT and modulate its helicase activity using telomeric DNA substrates. J Biol Chem 284 (22), 14966–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Mendez-Bermudez A et al. (2012) The roles of WRN and BLM RecQ helicases in the Alternative Lengthening of Telomeres. Nucleic Acids Res 40 (21), 10809–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Temime-Smaali N et al. (2008) Topoisomerase IIIalpha is required for normal proliferation and telomere stability in alternative lengthening of telomeres. Embo j 27 (10), 1513–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Hänsel-Hertsch R et al. (2016) G-quadruplex structures mark human regulatory chromatin. Nat Genet 48 (10), 1267–72. [DOI] [PubMed] [Google Scholar]
- 112.Siddiqui-Jain A et al. (2002) Direct evidence for a G-quadruplex in a promoter region and its targeting with a small molecule to repress c-MYC transcription. Proc Natl Acad Sci U S A 99 (18), 11593–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Pichierri P et al. (2000) Werner’s syndrome lymphoblastoid cells are hypersensitive to topoisomerase II inhibitors in the G2 phase of the cell cycle. Mutat Res 459 (2), 123–33. [DOI] [PubMed] [Google Scholar]
- 114.Shamanna RA et al. (2016) Camptothecin targets WRN protein: mechanism and relevance in clinical breast cancer. Oncotarget 7 (12), 13269–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Aggarwal M et al. (2013) Targeting an Achilles’ heel of cancer with a WRN helicase inhibitor. Cell Cycle 12 (20), 3329–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Sommers JA et al. (2019) A high-throughput screen to identify novel small molecule inhibitors of the Werner Syndrome Helicase-Nuclease (WRN). PLoS One 14 (1), e0210525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Nguyen GH et al. (2013) A small molecule inhibitor of the BLM helicase modulates chromosome stability in human cells. Chem Biol 20 (1), 55–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Rosenthal AS et al. (2013) Synthesis and SAR studies of 5-(pyridin-4-yl)-1,3,4-thiadiazol-2-amine derivatives as potent inhibitors of Bloom helicase. Bioorg Med Chem Lett 23 (20), 5660–6. [DOI] [PMC free article] [PubMed] [Google Scholar]