

Abstract
Cellular therapies first emerged as specialized therapies only available at a few “boutique” centers worldwide. To ensure broad access to these investigational therapies – regardless of geography, demographics, and other factors – more and more academic clinical trials are becoming multi-center. Such trials are typically performed with a centralized manufacturing facility receiving the starting material and shipping the final product, either fresh or cryopreserved, to the patient’s institution for infusion. As these academic multi-center trials increase in number, it is critical to have procedures and training programs in place to allow these sites that are remote from the production facility to successfully participate in these trials and satisfy regulatory compliance and patient safety best practices. Based on the collective experience of the Consortium for Pediatric Cellular Immunotherapy, we summarize the challenges encountered by the institutions in shipping and receiving the starting material and final product as well as preparing the final product for infusion. We also discuss best practices implemented by each of the consortia institutions to overcome these challenges.
Keywords: Cellular therapy, Immunotherapy, Cell Processing, Clinical Trials
Introduction
Cell and gene therapy has been growing rapidly, particularly since the seminal demonstrations of CD19 directed Chimeric Antigen Receptor (CAR) T cell therapy [1, 2]. Although commercial CD19 CAR T therapy is available in many major medical centers, with more than 100 offering Kymriah (https://www.us.kymriah.com/treatment-center-locator/), most experimental cell and gene therapies are available in a limited number of clinical trial sites. These personalized products are manufactured in academic cellular therapy programs (manufacturing site) by their own manufacturing facilities, regulatory teams, and clinical translational investigators. This approach limits the access patients have to these promising therapies due to the complexity of adding additional clinical sites. Indeed, the New York Times published an article in 2017 demonstrating that patients on immunotherapy clinical trials are >90% Caucasian[3], which is not representative of the US population as a whole. While there are a number of suggested causes for this disparity, access to a clinical trial site and the cost associated with traveling to one of these sites are major hurdles. In addition, multicenter clinical trials are essential for pediatric patients given the low prevalence of diseases that are treated.
One way to provide better access to cellular therapy trials is to optimize the ability of academic manufacturing sites to provide personalized cellular therapy products to US and international institutions (remote clinical sites). In this model, the manufacturing is centralized in an academic center, but the clinical trial is multi-institutional. The sponsor of the trial is ultimately responsible for ensuring that participating sites have the resources they need to safely perform the clinical trial, including qualified personnel, training, and procedures. These multi-center clinical trials come with their own challenges, especially related to manufacturing, shipment, storage, and infusion of products from a diverse range of centers, each with their own institutional Standard Operating Procedures (SOP)s and processes. The logistical and quality challenges of providing cellular therapy products to remote clinical sites at multiple academic medical centers when manufacturing operations occur at a limited number of high volume commercial manufacturing sites has been described by others[4, 5]. These authors focused on defining the critical quality attributes and critical process parameters that ensure quality through the entire cycle of providing autologous cellular therapies. Elements that must be optimized include coordination of patient enrollment, scheduling, starting material collection, labelling, shipping, manufacture, storage, and infusion. Here, we extend these earlier reports and describe the formidable challenges when both the clinical site and the manufacturing site are at academic centers based on the experience of the Consortium for Pediatric Cellular Immunotherapy (CPCI). The CPCI is composed of pediatric academic centers from five US Clinical and Translational Science Award (CTSA) hubs focused on accelerating the translation of cellular immunotherapy for pediatric disease. The consortium goals are to expand cGMP manufacturing programs with the capacity to supply cellular products for early phase clinical trials, establish infrastructure to efficiently implement cellular immunotherapy clinical research, and to increase efficiency and reliability of analytic assays to monitor both safety and clinical efficacy of cellular immunotherapy trials for rare pediatric diseases. In doing so, the CPCI seeks to train a cadre of clinical, manufacturing, research, and regulatory teams capable of advancing cellular immunotherapy for a wide range of disorders including cancer, infection and immune tolerance.
As clinical trials evolve from having elaborate cGMP manufacturing capacity, there is still a need for qualified processing laboratories at each remote clinical trial site to ship, receive, and store starting materials and final products. Many remote clinical sites have established stem cell processing laboratories with years of experience with bone marrow, peripheral blood hematopoietic stem cells, and other standard-of-care, minimally manipulated products. Few laboratories have experience with or infrastructure to support more-than-minimally manipulated products (called“351 products” after section 351 of the Public Health Service Act of 1944). Such 351 products often require new equipment (such as an automated thawing device) and procedures, in addition to training beyond typical hematopoietic stem cell processing. For example, these products can have different product containers (vials versus bags), specialized shipping and storage requirements (fresh versus frozen), and may need specialized QC testing on site before product administration; these requirements are not typically required or are more straightforward when processing and distributing minimally manipulated blood banking 361 products. Overall, these differences – especially the variability in container type, size, and requirements – have resulted in non-standardized methods of manufacturing and processing cellular therapy products.
To overcome these challenges, we have outlined a number of best practices and examples of how to address practical and logistical obstacles based on our experience as academic manufacturing sites for, or participating in a number of multi-center clinical trials at four different remote clinical sites. Some of these challenges include: logistics of shipping the apheresis product/starting material, receiving, storing, and thawing/infusing the final product, and qualifying/training external cell therapy laboratories for product receipt and infusion. The many steps in the process from apheresis to infusion are shown in Figure 1.
Figure 1.
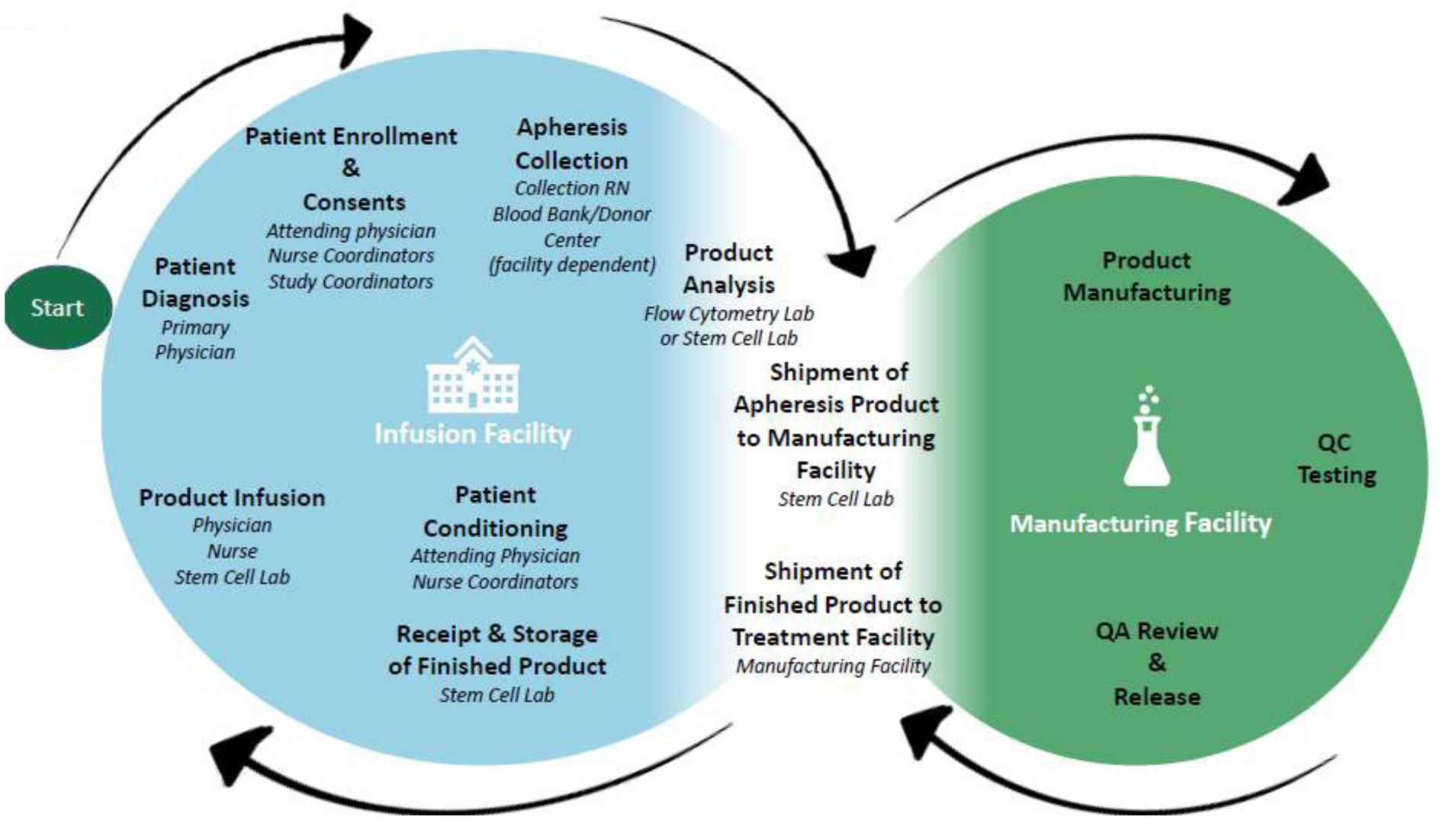
Multi-institutional flow chart for cellular therapy products. The life cycle of autologous cellular therapy products, starting from apheresis collection, moving to the manufacturing laboratory, and finally being shipped to the infusion facility.
Regulations and Standards on Shipment of Starting Materials and Products
We first identified critical steps in the shipment and receiving process and linked them to respective federal regulations or guidances (US Food & Drug Administration) or Foundation for the Accreditation of Cellular Therapy (FACT) standards (Table 1). We note that not all academic manufacturing centers are FACT accredited. Within the CPCI, 2 institutions are FACT accredited, 2 are in the process of obtaining FACT accreditation, and 1 institution has elected to not become FACT accredited in the near future. While FACT accreditation may not be a prerequisite for site qualification, it does provide an added level of confidence that remote clinical sites subscribe to SOPs and policies that conform to federal regulations. If remote clinical sites are not FACT accredited, they may be required to provide additional documentation such as their Quality Plan, training records, and may even undergo an on-site audit or qualification as described later. Further, a gap analysis revealed that regardless of FACT accreditation status, all CPCI institutions meet the requirements listed in Table 1, likely due to the GMP-type nature of the processing that they support. However, requiring FACT or AABB accreditation may become necessary as additional remote clinical sites are added with less experience processing 351 products.
Table 1.
Shipment/Receiving of Starting Material/Final Products
| Shipping | ||
|---|---|---|
| Activity | Regulations | Standards |
| 21CFR[6, 7] | FACT[8] | |
| Select a qualified courier & qualify transit time | D10.6 | |
| Validate Shipping container | ||
| • Protect integrity of product | 1271.265 | D10.1, D105.1 |
| • Utilize secondary container to prevent leakage | D10.2, D10.5.2 | |
| • Control temperature (refrigerated or frozen) to meet expiration parameters | 1271.265 | D10.3, D10.5, D10.6 |
| Prepare container for shipment | ||
| • Check request orders and confirm with clinical site arrival date and time | D8.2 | |
| • Set and insert temperature logging device | D10.5.2.1 | |
| • Insert study required labels, certificate of analysis, chain of custody documents | 1271.290, 1271.265 | B1.2.1.1* |
| • Insert handling instructions1, including expiration date | D11.1.4.1, C10.5 | |
| • Affix transport labels/stickers | D10.5.1, D10.5.5, D10.5.6, D7.3, D10.9 | |
| • Use tamper evident tape to close shipper | D10.5.4 | |
| Complete post- shipment tasks | ||
| • Receive completed product receipt from site | 1271.290 | |
| • Analyze and document temperature tracking | D10.5.2.2 | |
| • Provide mechanism for customer feedback | 211.098, 1271.320 | |
| Receiving | ||
| Inspect packaging and physical conditions | 1271.265. 211.82 | D11 |
| • Follow instructions for temperature tracking | ||
| Confirm receipt of accompanying documentation | C6 | |
| • Chain of custody | 1271.290 | |
| • Documentation of shipping & transport on public roads | Common Standards Appendix 2 | |
| Verify labeling matches donor/recipient identifiers | 1271.370 | C7 |
| Assign unique identifiers and label materials/documentation | 1271.290 | D7 |
| Confirm donor eligibility (as needed, for allogenic donors) | 1271 subpart C | C6 |
| Quarantine material at appropriate storage temperature to protect safety & improper use | 1271.265, 211.82 | D11 |
| Return documentation | ||
EU regulations require instructions for opening the container, package, and any required manipulation or reconstitution be included as a document accompanying the starting material and/or final product
Immune Effector Accreditation Manual 2017
Interpretation of the requirements in the CFR can be variable. Therefore, FDA has provided extensive guidance on various aspects of cell and gene therapy including packaging. A searchable database of FDA guidance documents is available on their website (https://www.fda.gov/regulatory-information/search-fda-guidance-documents). Therefore, the details of how each of these regulations, guidances, and standards is implemented must be shared between the clinical trial site and the manufacturing site to ensure compliance with best practices.
Qualifying Infusion Sites
Cellular therapy processing facilities across the United States have primarily served as laboratories responsible for minimal manipulation of hematopoietic stem cell products. The processing required for these products requires extensive knowledge and training, but by definition is typically more routine and less variable than processing of more-than-minimal manipulation products.
To qualify potential sites, the CPCI developed a questionnaire that is filled out by the processing lab at remote clinical sites (Figure 1). This questionnaire can be used to ensure that sites are FACT accredited and FDA registered, and have the necessary processes, training, and infrastructure in place to receive, store, and administer products safely. Accreditation from other cell therapy accrediting bodies such as AABB may also be acceptable.
To qualify as a remote clinical site, each site must have SOPs in place which detail receiving and storing the product in a controlled environment, testing the product for sterility and viability, and most importantly, have a procedure to notify the study sponsor and manufacturing site in the event of a positive sterility result after infusion. Once the completed site qualification questionnaire is completed by the remote clinical site it is reviewed by the laboratory management team, including Quality Assurance (QA), the laboratory manager, and the laboratory director to ensure that they understood the procedure and have the necessary training to receive and properly handle the product. More specific details on training can be found in the Training Remote Clinical Sites section below. Additionally, verification of a completed training log confirms all pertinent staff have watched the video and are comfortable with following the procedures.
Finally, each remote clinical site must attest that they will notify the manufacturing site within 24–48 hours (or as described in the protocol) of any positive sterility reports related to the thawed product. This requirement is critical because these issues must be quickly investigated and reported to FDA (if necessary), with expedited notification of the patient’s treating physician, the patient, any other teams involved in the manufacturing.
Training at Remote Clinical Sites
After confirming that a remote clinical site is qualified to ship, receive, and store a final cell product or starting material or as a part of the qualification process, the sites typically require protocol specific training. As mentioned above, some of this training can be done during the qualification process. Training remote clinical site staff about collection, storage, and transport to the manufacturing site of starting material and receipt, storage, handling, and preparation of final product for infusion is necessary. Some remote clinical sites may be highly experienced in handling CAR T products or other cell therapy products while others may have little to no experience. The process of handling final products may involve a simple thaw and hand off to the clinical team at the remote clinical site for infusion or may involve processes requiring use of new devices, sampling of final products for additional sterility testing, or other further aseptic product manipulations. There are several ways to conduct training, with advantages and disadvantages to each. As an example, the training process conducted and competency measurement used by a member of the CPCI is to travel to each collaborating remote clinical site with sample shipping materials and SOPs and provide on-site training is summarized in Table 2.
Table 2.
Training and Competency at External Clinical Sites
| Task | Competency Measure | |
|---|---|---|
| Apheresis Product Shipment | Handling of Apheresis Product (visual inspection, preparation of absorbent material and sealing wrap bag, sealing of insulated bubble wrap bag | External site staff demonstrate proficiency without error following onsite training |
| Preparation and placement of completed forms and documentation | External site staff acknowledge understanding of process following demonstration when questioned. | |
| Enabling and placement of temperature logger | External site staff demonstrate proficiency without error following onsite training | |
| Sealing and labeling of shipping container and airway bills | External site staff demonstrate proficiency without error following onsite training | |
| Final Product Receipt and Preparation for infusion | Receipt, inspection of dry shipper container, storage, and documentation of received dry shipper | External site staff acknowledge understanding of process following demonstration |
| Removal and thaw of cell product(s) | External site staff demonstrate proficiency without error after onsite training with an automated thawing device | |
| Recording verification of labels, date and time of thaw, expiration time, and chain of custody | External site staff demonstrate understanding of process following demonstration when questioned | |
| Removal of subject dose from thawed product vial(s) into prepared and labeled syringe(s) | External site staff demonstrate proficiency without error following onsite training using aseptic technique |
On-site training conducted by experienced members of the cell product manufacturing center is strongly recommended for the following reasons: 1) The trainer can provide and document uniform hands-on training for all aspects of the procedures while assessing the competency of staff, particularly in aseptic handling; 2) Differences between facilities, including suitable storage and work areas, personal protective equipment (PPE) requirements, waste disposal, documentation policies, etc. can be assessed and adapted in real-time to accommodate any observed gaps or deficiencies; 3) The trainer is able to address questions from infusion site staffing during the training. Some disadvantages or difficulties of on-site training include having a qualified trainer willing to travel, the travel expenses associated with sending the trainer to external sites, and coordinating and scheduling a date and time that will work for all participants.
Another method utilized by some manufacturing centers is the use of training videos. For example, one CPCI site created a training video to demonstrate the thawing, sampling, preparation, and administration of cellular therapy products at remote clinical sites (Figure 2). Making a training video is relatively inexpensive and videos are easy to distribute to an external site. They can be viewed on demand and provide ongoing training support of new personnel at those sites. This training method does have some limitations. The manufacturing center is unable to assess the expertise and proficiency of the external site staff with procedures depicted in the training video and if there is proprietary information in the video there is no way to control the distribution and viewing of the video. It is also time-consuming to edit the video or re-film as the process evolves.
Figure 2.
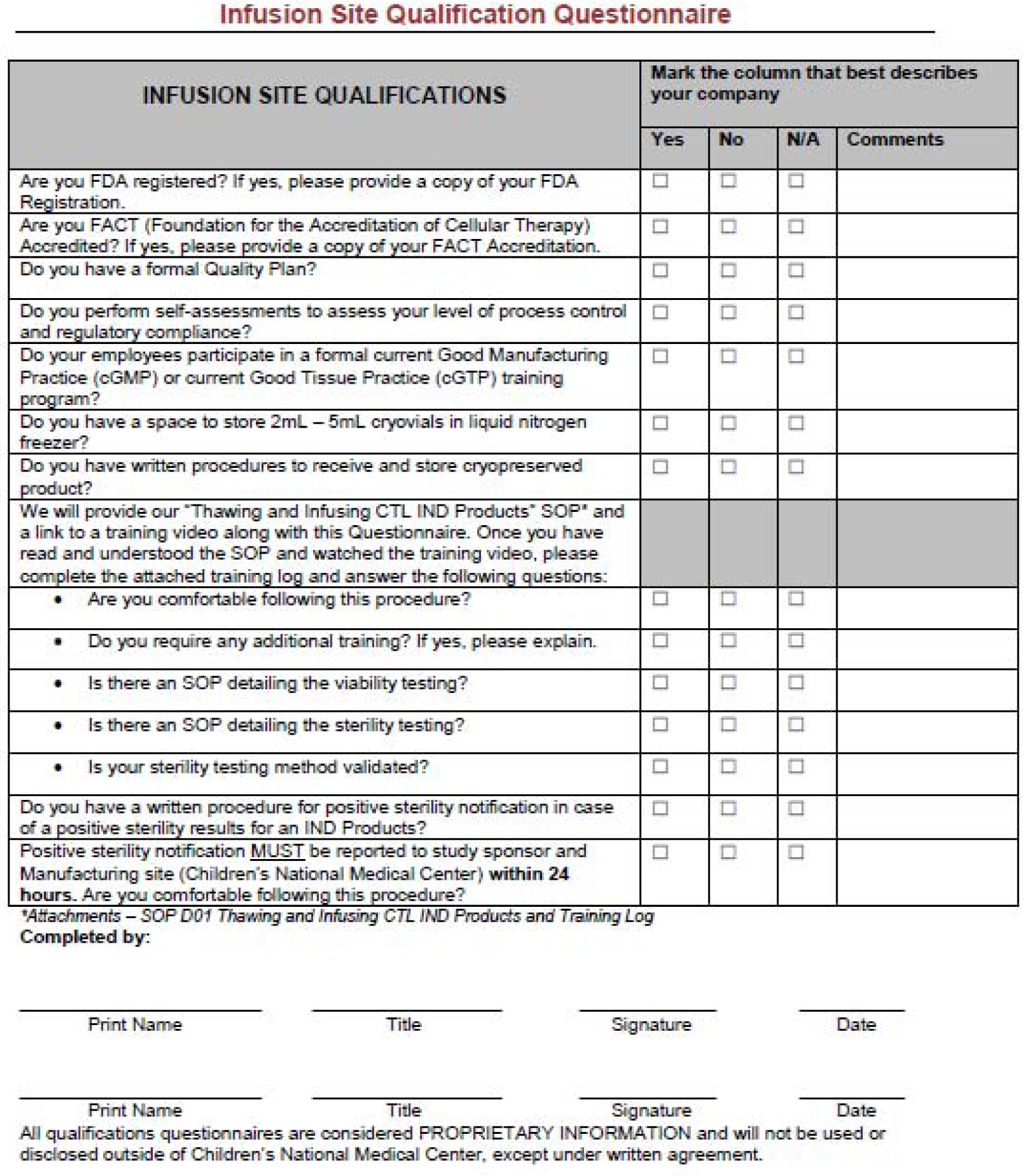
Qualification Questionnaire for Infusion Sites. An example questionnaire that is used to qualify cell therapy sites for infusion.
Although not as rigorous of an assessment as in-person training, a written assessment can be given as a measure of competence. This method also does not allow for or address infrastructural, procedural, and policy differences between facilities and requires the external staff to determine how to adapt. Using this approach, the manufacturing center relies upon documentation, rather than first-hand experience to determine the effectiveness of training on site staff.
In addition to on-site or video training, a handbook or SOP for final product preparation for infusion is recommended to codify all procedural steps, address frequently asked questions, provide contact information of the appropriate manufacturing center staff in case a question or issue arises during preparation for infusion, instruct sites on documentation and return of forms as well as preparation of shipping containers to be sent back to the manufacturing center or shipping vendor. Whenever possible, the inclusion of photos depicting key steps should be included.
Whatever method(s) are deployed for external site training, it is critical that dry runs with the external site are performed as a final step. These practice runs can identify issues not apparent during training. For example, the external site might discover an inability to download the cryologger software to local PCs due to configuration or local information technology policies. Identification and resolution of such issues prior to having a cell product in hand is key to smooth operations at an external site.
Shipping of Starting Materials
After collection from a patient, starting materials, such as apheresis product or whole blood, must be adequately labeled, packaged, and shipped to the manufacturing site. Personnel at sites participating in a multi-center clinical trial must be adequately trained on trial-wide standard operating procedures (SOPs) to ensure the correct packing and shipping of starting materials. Procedures should include confirmation of all addresses and labeling requirements before the package leaves the shipping site. Pre-printed shipping labels, provided by the trial, may also help to minimize errors. Effective and prompt communication between the collection site (remote clinical site) and the manufacturing site is likewise essential to ensure that the manufacturing site is ready to receive and process the material when it arrives. Therefore, each site’s trial coordinators must understand the importance of 1) prompt communication with the manufacturing site about patient enrollment and schedules, 2) completion of all chain of custody documentation and, 3) packing of material into the shipping container according to common SOPs. In our experience, a common source of manufacturing deviations is inadequate labelling or incorrect packing of starting materials by local trial coordinators or shipping personnel. Such errors can lead to more serious deviations such as out of specification temperature readings due to incorrect placement of temperature probes or cold packs during packing.
There are a number of courier services capable of transporting cryopreserved or fresh cell products, including legacy logistics companies like FedEx, more focused logistics companies like World Courier and US Xpress, and finally cell-therapy specific courier services like CryoPort and QuickStat. The courier service should be selected by the manufacturing site and must take into account the specific requirements of a trial. For example, trials that require shipments and deliveries over the weekend or on holidays must use a courier service that is available during those times. Likewise, some services can navigate the vagaries of international shipments and relevant customs barriers (e.g. shipments to and from Canada) while others cannot. When travel distances are short, for example between two affiliated institutions in the same metro area or even in the same state, personal transport of starting materials by clinical trial staff may be faster and more reliable than using a courier service. Even so, there may be liability concerns associated with this approach and these should be considered appropriately. Additionally, some courier services are willing to participate in completing chain of custody documentation and others will use only their proprietary tracking systems to document when packages are picked up and delivered. FDA regulations must always be followed, but some states also have specific requirements and registrations that must be completed prior to shipping to the state.
Selection of a shipping container must accommodate the size of the product, the temperature at which it is shipped, and desired features such as built-in continuous temperature monitoring and GPS tracking of the shipper. FACT common standards (C10)[9] set out requirements for shipping starting materials. In particular, these standards require that the material be placed in a sealed secondary container to prevent leaks, that the temperature be controlled and continuously monitored, and that the container be validated for the shipping task. Packaging of starting materials commonly involves several different materials inside a shipper including a sealed biohazard bag, an absorbent sheet, cold packs, padding/bubble wrap, and an additional outer container (such as a cardboard box).
Common commercially available shippers include the Credo Series 4–248, manufactured by Pelican, evo 2–8°C Smart Shipper and evo CRT (15–25°C) Smart Shipper, manufactured by SAVSU Technologies, and STP-320, manufactured by Saf-T-Pak / Inmark. At one Consortium site, the approach to validation includes testing to determine how long the shipper maintains an acceptable internal temperature when exposed to extreme temperatures. The goal is to show that the shipper can maintain the desired temperature for up to 36 hours, since shipping from any location in North America is generally completed in this timeframe. To perform these validations, staff placed sample starting material in different primary containers (such as vacutainers or a blood bag) in the shipper and then placed the shipper in an environment at different temperatures: −20 to −30°C, 0 to +8°C, +18 to +25°C, and +32 to +37°C. Staff then recorded the temperature at intervals using a probe next to the sample material to identify the point at which the temperature is out of range. In addition to validation at specified external temperatures, performance qualification was done to test the shipper under conditions comparable to the shipment of starting material. Performance qualification involves shipping sample material to different locations and determining if the material stays within the desired temperature range. An example of data collected from such a performance qualification for the evo CRT Smart Shipper is shown in Table 3 below.
Table 3.
Qualification of a Cell Transport Container
| Acceptance Criteria: Payload Temperature Range of +15.0°C to +25.0°C for 36 Hours | ||||||
|---|---|---|---|---|---|---|
| Test Start Date | Shipped from San Francisco to: | Evo I.D.# | Unique Shipment I.D.# | Temp. Range over 36 Hrs | Time in Transit (Hrs) | Disposition |
| 9/13/17 | Arlington Heights, IL | 2235 | 796 | 20.8°C to 21.4°C | 23:21 | PASS |
| 9/13/17 | Denver, CO | 2452 | 795 | 20.8°C to 21.8°C | 24:04 | PASS |
| 9/13/17 | Albuquerque, NM | 2172 | 794 | 20.3°C to 20.9°C | 24:45 | PASS |
Given the many potential points of failure during shipping of starting material, regular tracking and trending of deviations using paper-based and in-person audits of the packing and shipping process are essential components of a quality assurance program. Repeated out-of-temperature deviations in particular should alert the manufacturing site to potential problems with the starting material shipping process.
Receiving the Final Product at the remote clinical sites
Selecting a date
In the experience of the Consortium, there is typically little flexibility when choosing a shipment date for receipt of cellular therapy products. Usually, the clinical research team works with the attending physicians to determine the optimal date of delivery and infusion. The clinical research team then contacts the remote clinical site’s processing laboratory to confirm staff are available for receiving the delivery. It is critical that the remote clinical site’s clinical team the manufacturing site, and the remote clinical site’s processing lab communicate effectively to ensure seamless delivery and infusion of the product to the patient. Across the consortium, it is common to avoid Friday delivery. Most laboratories are not open on weekends, so should there be a delay in shipping, or a lost item, there would not be staff available to receive the product or investigate lost shipments.
It is important that the type of shipper and study storage requirements be factored into receipt. For some studies, the product is required to remain in the shipper until time of infusion. If the study is using a Cryoport shipper, the shipper is typically validated for seven days from when the liquid nitrogen charge occurred. Once charged by Cryoport, it is shipped to the manufacturing site for product packing and then shipped to the remote clinical site’s processing laboratory so that by the time the shipper is dispensed from the manufacturing site it has already been charged for two days and only five days of the validated charge remain. For these types of shipments, one Consortium site, for example, usually coordinates to have the product delivered on the day of infusion. If the shipper is coming via a company such as Fedex, it may go through the hospital receiving dock. The product will then get routed through the normal mail delivery process which can delay receipt and infusion, especially on days that the product arrives same day as infusion. To more efficiently handle the receipt, the remote clinical site implemented a process to ensure that all deliveries are processed through the receiving dock. Once the itinerary is determined, and tracking information is received, laboratory staff forward that information to the receiving dock and send a reminder the day before delivery. The receiving dock is then able to expedite the processing and delivery of the shipper. If the shipment is to be guaranteed for a 10:00am delivery, the receiving dock can then have it to the laboratory by 10:30am. During this time, laboratory staff are able to prepare paperwork, check calculations, call for pre-medication of the patient and be ready for an 11am infusion, the preferred infusion time of many remote clinical sites.
Although same-day infusions are possible as outlined above, most laboratories prefer the cells to arrive the day before infusion, which gives ample time to review all the paperwork and double check calculations; however, as mentioned, most laboratories do not have complete control over the scheduling. A consortium remote clinical site process laboratory once had a product arrive with an incorrect dose on the study dosing form. The dosing directions were followed, which led to an incorrect dose being infused, per the study form. Since then, a calculation verification system has been implemented as a part of the standard processing worksheet. It should be noted that some studies may not give enough pertinent information to complete the dose confirmation. Thus, early deliveries are strongly recommended to ensure ample time to review all the documents, prevent deviations, and allow time for staff to clarify any questions with the sponsor.
Some studies allow for products to be relocated to the liquid nitrogen storage tank of the remote clinical site’s processing laboratory. In this case, the laboratory requires the arrival date, type of storage container (vials, cassettes) and number of products for receipt. Staff can verify that space is available (a FACT standard) and store the product indefinitely, until infusion or until product expiration. On rare occasions, products will be shipped fresh. In this case, the product is likely be received on the day of infusion.
Processing laboratories typically have limited involvement in scheduling of infusions beyond confirming if staff are available to receive the shipment. To best prepare for upcoming shipments and infusions, staff attend weekly referral meetings with the clinical site team for updates about patient status, product delivery dates and proposed infusion dates. The clinical team of the remote clinical site also provides the laboratory with a final ship date, all the corresponding shipping information, and the infusion date. Although management and supervisors are typically responsible for coordinating patient processing and scheduling shipments, email groups have been created that included entire laboratories on all notifications. This group email is used for all scheduling information, so laboratory and clinical teams are aware of upcoming deliveries and infusions. These emails help disseminate information, prevent bottlenecks, and create redundancies for when staff take vacation, are sick, or are too busy to respond. Some laboratories in the Consortium also have staff available at night, so if inquiries come in on the group email, the response time is quicker than just having one contact for studies.
Some protocols require the remote clinical site to download temperature monitoring information once the product is removed. Prior to receipt, confirm with the manufacturing site which software is needed for downloading temperature monitoring. As mentioned earlier, mock runs are great way to test software and data logger access prior to receiving a real patient product.
After the product has been removed, the dewar/shipper will need to be returned. The shipper can be returned by Fedex (ground) or by a courier, depending on the protocol. If the courier is going to take the shipper after it is emptied, you will need to determine if they will be waiting for you to unpack, or if they will return at a scheduled time. Dewars may be old with locks that do not securely close the lid. We utilize zip ties to keep the lids closed in cases where the fastener is loose.
Chain of Custody
According to the new FACT and Immune Effector Cell (IEC) standards[10], the chain of custody (COC) for immune effector cells must travel with the product from collection through infusion. In the experience of the Consortium, few shipping companies have a designated COC form designed to meet this new standard. If COC documentation is not adequate each site should create a form and work with each sponsor to implement an acceptable compliant process. This form should be prepared prior to arrival of the product and would then start the COC at product receipt. The courier would sign for release of the product to the cell process laboratory. If there is more than one type of product in the delivery (eg. CD8+ and CD4+ cells), two COC forms would be prepared, one for each product/infusion.
For COC traveling with the product from collection to infusion (across multiple sites), it is important to know where the document is, and ensure the courier signs before they leave. COC can be found in plastic pockets on outside of delivery container or in a sleeve inside the container.
Forms
The manufacturing sites have forms that must be completed to document receipt and infusion. If possible, it is a best practice to get copies of those forms before product delivery (should be available during training or mock runs). As discussed earlier, it is best practice to conduct a mock run for delivery (either with manufacturing site, or on your own). An additional consideration when filling out forms from sponsors is to use the specified dating practice as some sponsors have requested European dating.
Infusion of Product
Each protocol has specific instructions for product infusion. Is the product thawed at bedside? Is the product thawed and reconstituted in the laboratory? Is the product infused fresh? While these questions are common and should be addressed prior to the mock shipment, unanticipated situations do occur in practice. To identify how common deviations occur during these multi-center clinical trials, data were analyzed from 3 consortium institutions with experience shipping manufactured products to external clinical sites. In total there were 5 shipping and/or handling deviations out of 129 shipments (3.9%). Of these deviations, no more than 2 happened at any one manufacturing site and despite the deviations from procedure, solutions were identified that allowed for all products to be infused. While a 3.9% incident occurrence is low, especially for 3 unique manufacturing sites, it does illustrate that shipping and handling deviations will occur and will require real-time attention and mitigation strategies, even from experienced manufacturing sites interacting with highly trained remote clinical sites.
Learning from the experiences above and additional experiences from within the consortium, we have listed examples of incidents that have occurred within the consortium and how they were addressed:
A fresh product arrived in a single port bag for infusion. The port was not compatible with institutional supplies, and the nursing staff was unable to spike the bag at bedside. The Principal Investigator and laboratory medical director were engaged and they immediately called the manufacturing site and discussed a resolution. Cell processing staff took the bag back to the cell processing laboratory. Staff attached a coupler and syringe and transferred the product to a new infusion bag. The original bag was rinsed with Plasmalyte and added to the product for infusion. The entire product was returned to the unit for infusion with no further issues. It is best practice at first product administration to have the cell processing technician stay on the unit until infusion has begun successfully. It is also helpful to have the PI available and manufacturing contact numbers on hand. This exemplifies the importance of practice runs using mock materials specific to the product/trial.
A product arrived in multiple vials. The infusion dosing form instructed staff to thaw two vials to achieve the dose. Once the thaw was completed and reconstituted, a cell count determined the dose was below the prescribed dose (or study mandated dose). This was a first run, and the manufacturing had not accounted for cell loss during thaw and transfer (there was no rinse as part of the protocol). The staff immediately contacted the manufacturing site for advice and were instructed to thaw an additional vial to achieve the prescribed dose.
A product was shipped in a transport container and the sponsor requested that the temperature data that was tracked during shipment be downloaded by the site and sent back to the sponsor. Despite a mock shipment being requested, it was never performed and when the site went to download the data from the data logger, it was determined that special software and a micro-USB adapter were required to download the data, which was purchased for future products.
Based on our experience as a consortium supporting multi-center clinical trials, we offer the following suggestions related to successful shipping and receiving:
Perform mock shipments until all parties are satisfied and comfortable with new products and clinical trials. Make these mock shipments as real as possible.
Be prepared for things to go wrong
Have extra staff available for product receipt on new protocols
Have the contact information for responsible individuals at the manufacturing and distribution site readily available for each shipment and ensure that someone will be available at all times
Track all shipments closely and in real-time
Conclusion
Expanding the number of clinical sites participating in cellular immunotherapy clinical trials is crucial given the rarity of many pediatric diseases and in providing access to these promising therapies to a larger and more diverse pediatric population. Specialized manufacturing sites are highly experienced in regulatory guidelines and producing cell therapy products, and physicians at clinical sites are equipped to handle patient care. However, experience handling and shipping of starting material and final product manipulation is often overlooked during the planning stages of a clinical trial. We stress that shipping and handling is complex and critical to the success of a trial and should be considered early and often in trial planning. Indeed, having representation by the manufacturing site, the CR team, and the remote clinical site when planning new trials would be helpful. Our multi-institutional Consortium has described shipping and handling best practices which meet regulatory guidelines, ensure operational effectiveness, provide proper training, and highlight problems and incidents encountered and approaches taken to overcome these challenges.
Acknowledgements:
Research reported in this publication was supported by the National Center For Advancing Translational Sciences of the National Institutes of Health under Award Number U01TR002487. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
We thank Liz Gruber who serves as the Program Coordinator for the CPCI and the CPCI steering committee for reviewing the manuscript. We thank Amy L. Putnam and Angela Lares for collecting data on shipper qualification.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Conflict of Interest Statement:
PJH has patents related to cellular therapies and is a co-founder and consultant of Mana Therapeutics and serves on their board of directors and serves on the board of directors of the Foundation for the Accreditation of Cellular Therapy (FACT).
References
- 1.Porter DL, et al. , Chimeric Antigen Receptor–Modified T Cells in Chronic Lymphoid Leukemia. New England Journal of Medicine, 2011. 365(8): p. 725–733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Grupp SA, et al. , Chimeric Antigen Receptor–Modified T Cells for Acute Lymphoid Leukemia. New England Journal of Medicine, 2013. 368(16): p. 1509–1518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.In Cancer Trials, Minorities Face Extra Hurdles .
- 4.Levine BL, et al. , Global Manufacturing of CAR T Cell Therapy. Molecular Therapy - Methods & Clinical Development, 2017. 4: p. 92–101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Chen LN, et al. , How do I structure logistic processes in preparation for outsourcing of cellular therapy manufacturing? Transfusion, 2019. 59(8): p. 2506–2518. [DOI] [PubMed] [Google Scholar]
- 6.21 CFR PART 1271 Human Cells, Tissues, and Cellular and Tissue-Based Products. U. S. Food and Drug Administration. [Google Scholar]
- 7.21 CFR PART 211 Current Good Manufacturing Parctice for Finisahed Pharamaceuticals. U. S. Food and Drug Administration. [Google Scholar]
- 8.FACT-JACIE International Standards for Hematopoietic Cellular Therapy Product Collection, Processing, and Administration. 2018, Foundation for the Accreditation of Cellular Therapy. [Google Scholar]
- 9.FACT Common Standards for Cellular Therapies. 2019, Foundation for the Accreditation of Cellular Therapy. [Google Scholar]
- 10.FACT Standards for Immune Effector Cells Accreditation Manual. 2018, Foundation for the Accreditation of Cellular Therapy. [Google Scholar]