

Abstract
HER2 is a well-known oncogenic receptor tyrosine kinase. HER2 gene amplification occurs in about 20% of breast cancer (BC), which leads to overexpression of HER2 protein, known as HER2-positive BC. Inhibitors of HER2 have significantly improved the prognosis of patients with this subset of BC. Since 1998, seven HER2 inhibitors have been developed to treat this disease. However, drug resistance is common and remains a major unresolved clinical problem. Patients typically show disease progression after some time on treatment. This review discusses the complexity and diversified nature of HER2 signaling, the mechanisms of actions and therapeutic activities of all HER2 inhibitors, the roles of HER2 and other signaling proteins in HER2-positive BC resistant to the inhibitors, the non-cell-autonomous mechanisms of drug resistance, and the heterogeneity of tumor HER2 expression. The review presents the concept that drug resistance in HER2-positive BC results primarily from the inability of HER2 inhibitors to deplete HER2. Emerging therapeutics that are promising for overcoming drug resistance are also discussed.
Keywords: Cancer treatment, HER2 inhibitor, PEPDG278D, Receptor tyrosine kinase, Targeted therapy, Therapeutic resistance
1. Introduction
Human epidermal growth factor receptor 2 (HER2), also known as ERBB2 or Neu, is a member of the HER/ERBB family of four closely related receptor tyrosine kinases (RTKs), which also include epidermal growth factor receptor (EGFR)/HER1/ERBB1, HER3/ERBB3, and HER4/ERBB4. These RTKs play important roles in a variety of developmental and physiological processes, but excessive signaling, resulting from overexpression or activating mutation, is oncogenic (Roskoski, 2014; Yarden & Sliwkowski, 2001). HER2 gene amplification occurs in about 20% of breast cancer (BC), which leads to HER2 overexpression, known as HER2-positive BC (Choritz, Busche, Kreipe, & Monitor, 2011; Witton, Reeves, Going, Cooke, & Bartlett, 2003). HER2 immunohistochemistry (IHC) staining scores range from 0, 1+, 2+ to 3+, but only 3+ is considered HER2-positive. In the absence of HER2 amplification, 3+ HER2 IHC staining is rare (Pauletti, Godolphin, Press, & Slamon, 1996). HER2 overexpression renders cells addicted to it (Arpino et al., 2007; Yang, Li, Bhattacharya, & Zhang, 2015) and was a strong predictor of poor disease prognosis before the introduction of HER2-targeted therapies (Slamon et al., 1987; Tandon, Clark, Chamness, Ullrich, & McGuire, 1989). Targeting HER2 has been the linchpin of managing this subset of BC. Seven HER2 inhibitors are currently available for treating HER2-positive BC, including trastuzumab, pertuzumab, trastuzumab emtansine (T-DM1), trastuzumab deruxtecan (DS-8201a), lapatinib, neratinib, and tucatinib. The inhibitors differ in their mechanisms of actions, which will be discussed later. While the inhibitors have significantly improved disease outcome, none of them achieves a cure in the metastatic setting. Primary or acquired resistance is common, and patients typically show disease progression after some time on treatment.
Many clinically relevant drug resistance mechanisms have been shown, including HER2 loss (Mittendorf et al., 2009; Niikura et al., 2012), decreased drug binding to HER2 (Mercogliano et al., 2017; Nagy et al., 2005; Scaltriti et al., 2007), lack of HER2 downregulation (Mohsin et al., 2005; Moulder et al., 2001), compensatory signaling (Gallardo et al., 2012; Zhang et al., 2011), dysregulation of apoptosis and cell cycle (Goel et al., 2016; Scaltriti et al., 2011), and immunodeficiency (Chaganty et al., 2018; Musolino et al., 2008). A number of reviews have addressed drug resistance mechanisms in HER2-positive BC (Arribas, Baselga, Pedersen, & Parra-Palau, 2011; Bailey et al., 2011; D’Amato et al., 2015; Nahta, Yu, Hung, Hortobagyi, & Esteva, 2006; Pohlmann, Mayer, & Mernaugh, 2009; Rexer & Arteaga, 2012). However, the relative importance of these mechanisms is poorly understood, which hampers more effective treatment of the disease and identification of better therapeutic options. This review discusses HER2 oncogenic signaling in HER2-positive BC, the mechanisms of actions of the current HER2 inhibitors and their therapeutic activities, the roles of HER2 and other signaling proteins in drug-resistant HER2-positive BC, the non-cell-autonomous mechanisms of drug resistance, and the heterogeneity of tumor HER2 expression. The review brings forward the concept that drug resistance in HER2-positive BC results primarily from the inability of HER2 inhibitors to deplete HER2. Emerging therapeutic approaches that are promising for overcoming drug resistance in HER2-positive BC are also discussed.
2. Complex and diversified signaling of overexpressed HER2
HER2, like its family members, has an extracellular domain (ECD) for ligand binding, a transmembrane domain, an intracellular tyrosine kinase domain, and a C-terminal tail. Unlike its family members, however, ligand binding is not required for HER2 to function. Crystallographic data show that the ECD of HER2 adopts a fixed conformation that resembles a ligand-activated state, rendering it ready for homo- or hetero-dimerization in the absence of ligand binding (Fig. 1) (Cho et al., 2003; Garrett et al., 2003). Notably, HER3 is also unique, as its kinase activity is greatly reduced due to change in two amino acids (E740H, D815N) (Shi, Telesco, Liu, Radhakrishnan, & Lemmon, 2010), and it relies on transactivation by other RTKs with which it heterodimerizes. HER2-positive BC cell lines and tumors from patients carry 8–66 copies of HER2 gene per cell (Jarvinen et al., 2000; Kao et al., 2009; Tanner et al., 2004). HER2 protein levels in these cells are up to over 400 fold higher than in BC cells without HER2 amplification (Li et al., 2016). In contrast, activating HER2 mutation is relatively uncommon in BC, with an estimated activating HER2 mutation rate of approximately 0.9% in BC (Bose et al., 2013). Approximately 7.1% of HER2-positive BC carry a HER2 mutation but the percentage of activating mutation is not known (Cocco et al., 2018). The oncogenic activity of overexpressed HER2 has been well documented. HER2 amplification or overexpression is correlated with poor outcome in patients (Chia et al., 2008; Tandon et al., 1989; Tovey et al., 2009), although HER2 inhibitors have significantly improved patient survival. Transgenic overexpression of either human HER2 or rodent Neu in mouse mammary tissues induces tumor growth and metastasis (Finkle et al., 2004; Guy et al., 1992). HER2 orchestrates oncogenic signaling mainly from cell surface, along with its variant p95HER2, but it also functions in the nucleus and mitochondria, as described below.
Fig. 1.

Highly diversified oncogenic signaling of HER2 in HER2-positive BC cells. Overexpressed HER2 in cell membrane gives rise to p95HER2 via ECD shedding, forms ligand-independent homodimeric signaling unit, and forms heterodimeric signaling units with a large number of RTKs, many of which have been implicated in drug resistance in HER2-positive BC. The vast signaling network activates various oncogenic signaling pathways. HER2 oncogenic activity is further enhanced by heterodimerization with non-RTK cell surface proteins, including CB2R, MUC1, and MUC4. Overexpressed HER2 also translocates to the nucleus and mitochondria to further diversify its oncogenic signaling. The ovals with numbers in HER2 represent sub-extracellular domains, and the yellow oval represents tyrosine kinase domain. The brown oval indicates tyrosine kinase activation.
2.1. Cell surface HER2
Overexpressed HER2 in cell membrane initiates oncogenic signaling through homodimerization and heterodimerization (Fig. 1). HER2 dimerization is ligand-independent, but ligand binding to other RTKs enhances their association with HER2. HER2 is a preferred dimerization partner with its family members, particularly HER3 (Tzahar et al., 1996). In fact, HER2-HER3 heterodimer is critical for the oncogenic activity of HER2 in cultured cells and mouse xenografts (Holbro et al., 2003; Lee-Hoeflich et al., 2008). Additional 35 RTKs also heterodimerize with HER2 (Fig. 1) (Kennedy et al., 2019). The molecular mechanism of promiscuous heterodimerization of HER2 with other RTKs is not known, but the heterodimers undoubtedly allow HER2 to diversify its signaling extensively and to blunt the impact of HER2 inhibitors. Many RTKs that heterodimerize with HER2 have been implicated in drug resistance and disease progression in HER2-positive BC, including EGFR in a clinical study (DiGiovanna et al., 2005), HER3 in cell lines and mouse xenografts (Watanabe et al., 2019), AXL in cell lines and mouse xenografts including a patient-derived xenograft (Goyette et al., 2018), insulin-like growth factor 1 receptor (IGF1R) in cultured cells (Nahta, Yuan, Zhang, Kobayashi, & Esteva, 2005), fibroblast growth factor receptor 1 (FGFR1) in cell lines and mouse xenografts (Hanker et al., 2017), ephrin type-B receptor 4 (EPHB4) in a clinical and preclinical study (Ding et al., 2020), MET in a clinical and preclinical study (Shattuck, Miller, Carraway, & Sweeney, 2008), as well as macrophage-stimulating protein receptor (MST1R/RON), EPHB2, EPHB3, platelet-derived growth factor receptor beta (PDGFRβ), and rhoptry neck protein 2 (RON2) in a clinical and preclinical study (Alexander et al., 2017). HER2 homodimers and heterodimers undergo auto-phosphorylation and trans-phosphorylation in their kinase domains and C-terminal tails, and the phosphor-tyrosine residues serve as docking sites for various signaling proteins, leading to activation of various signaling pathways, including but not limited to the RAS-RAF-extracellular signal-regulated kinase (ERK) pathway, the SRC-PTEN-phosphoinositide 3-kinase (PI3K)-AKT pathway, and the JAK-signal transducer and activator of transcription 3 (STAT3) pathway (Fig. 1) (Moasser, 2007; Roskoski, 2014; Yarden & Sliwkowski, 2001).
HER2 further enhances its oncogenic activity by interacting with non-RTK cell surface proteins, including cannabinoid receptor type 2 (CB2R), mucin 1 (MUC1), and mucin 4 (MUC4) (Fig. 1). CB2R is a G protein-coupled receptor, and both MUC1 and MUC4 are membrane mucins. In HER2-positive BC cells, CB2R and MUC4 stabilize HER2 (Blasco-Benito et al., 2019; Nagy et al., 2005), and MUC1 activates HER2 (Raina et al., 2014). MUC4 also prevents trastuzumab from binding to HER2 and is an independent predictor of poor disease outcome in patients (Mercogliano et al., 2017; Nagy et al., 2005).
2.2. Nuclear and mitochondrial HER2
HER2 translocates to and functions in the nucleus and mitochondria in HER2-postive BC cells, known as noncanonical functions (Fig. 1), which further extend the oncogenic signaling of HER2. HER2 nuclear translocation and function were recently reviewed (Elizalde, Cordo Russo, Chervo, & Schillaci, 2016). Briefly, full length HER2 is extracted from the membrane and delivered to the nucleus. Nuclear HER2 may contribute to treatment resistance and poor prognosis in HER2-positive BC by 1) rendering resistance of HER2-positive cells to paclitaxel by phosphorylating cell division control 2 (CDC2) at tyrosine 15, 2) stimulating the transcription of cyclooxygenase 2 (COX-2) by binding to its promoter, 3) increasing protein translation and cell growth by binding to and stimulating RNA polymerase 1 and enhancing ribosomal RNA gene transcription, and 4) acting as a STAT3 coactivator, upregulating cyclin D1 and metastasis-promoting microRNA-21, and downregulating metastasis-suppressor programmed cell death 4 (PDCD4).
There appears to be only one study examining mitochondrial HER2. HER2 translocates to mitochondria via mitochondrial HSP70, and mitochondrial HER2 reprograms cellular metabolism from oxidative phosphorylation to aerobic glycolysis (Ding et al., 2012). Cancer cells with higher levels of mitochondrial HER2 is more resistant to trastuzumab (Ding et al., 2012).
2.3. p95HER2
HER2 amplification and overexpression give rise to p95HER2 (90–120 kD), which refers collectively to the carboxy-terminal fragments that remain membrane-bound (Fig. 1). p95HER2 arises via ECD shedding of cell surface HER2 by matrix metalloproteases or translation from internal codons of the gene (Anido et al., 2006; Christianson et al., 1998). p95HER2 has no binding site for trastuzumab and pertuzumab and therefore is insensitive to these agents. p95HER2 is positive in up to 60–80% of HER2-positive BC (Molina et al., 2002; Sperinde et al., 2010). p95HER2 may form homodimers by intermolecular disulfide binding and become constitutively active, and is implicated in trastuzumab resistance and poor prognosis (Arribas et al., 2011). A group of genes involved in metastatic progression were found to be specifically upregulated by p95HER2, such as matrix metalloproteinase 1, angiopoietin-like 4, MET, EPHA2, integrin alpha 2, and interleukin 11 (Arribas et al., 2011). Therefore, p95HER2 diversifies the oncogenic signaling of HER2. However, a study using cultured cells and patient-derived xenografts in mice showed that p95HER2-positive BC cells are effectively targeted by chemotherapy in combination with trastuzumab, presumably due to HER2 stabilization by chemotherapy (Parra-Palau et al., 2014).
3. Mechanisms of actions of HER2 inhibitors
The HER2 inhibitors that have been developed for treatment of HER2-positive BC represent three different HER2-targeting strategies, including 1) monoclonal antibodies (trastuzumab and pertuzumab), 2) antibody drug conjugates (T-DM1 and DS-8201a), and 3) tyrosine kinase inhibitors (TKIs) (lapatinib, neratinib and tucatinib). As described below and briefly depicted in Fig. 2, each inhibitor disrupts only a part of the vast HER2 signaling network and may also induce changes that counter its own inhibitory effects.
Fig. 2.

Mechanisms of actions of clinically available HER2 inhibitors. A. Trastuzumab and pertuzumab targets HER2 by binding to ECD subdomains 4 and 2, respectively. Both T-DM1 and DS-8201a retain the therapeutic activities of trastuzumab, but delivery of their chemotherapy payloads requires HER2-mediated internalization and release of their payloads via cleavage by lysosome enzymes. B. Inhibition of tyrosine kinase activities of HER family members by lapatinib, neratinib and tucatinib. The hatched oval in HER3 indicates that its tyrosine kinase is severely impaired.
3.1. Trastuzumab and pertuzumab
Trastuzumab is a fully humanized IgG1 monoclonal antibody. It binds to ECD subdomain 4 of HER2 (Fig. 2A) (Cho et al., 2003). Its therapeutic mechanism is not fully known, but trastuzumab apparently strikes cancer cells using two key strategies. First, via its Fc domain, trastuzumab flags cancer cells to which it binds for destruction by immune cells, such as NK cells and macrophages, known as antibody-dependent cellular cytotoxicity (ADCC) (Clynes, Towers, Presta, & Ravetch, 2000; Toth et al., 2016) and antibody-dependent cellular phagocytosis (Tsao et al., 2019). Second, trastuzumab directly suppresses HER2. By binding to HER2, trastuzumab blocks the proteolytic cleavage of ECD from HER2 by a matrix metalloprotease, thereby preventing formation of p95HER2 (Molina et al., 2001). Trastuzumab also rapidly decouples HER2 from SRC, an oncogenic non-receptor tyrosine kinase, and inactivation of SRC in turn leads to PTEN activation and inhibition of the PI3K-AKT pathway (Nagata et al., 2004). The exact mechanism by which trastuzumab decouples HER2 from SRC is not known, but it may result from conformation change of HER2 upon binding by trastuzumab. Trastuzumab further inhibits the PI3K-AKT pathway by disrupting the HER2-HER3-PI3K complex (Junttila et al., 2009). However, trastuzumab only disrupts ligand-free HER2-HER3-PI3K complex and has no effect on such complex bound by heregulin, a HER3 ligand (Junttila et al., 2009). Trastuzumab in fact stimulates the release of HER ligands from cancer cells through an AKT negative feedback loop and stimulates ligand-mediated heterodimerization and signaling of EGFR, HER3 and HER4 with HER2 (Gijsen et al., 2010).
Trastuzumab may downregulate HER2 in HER2-postive BC cells in vitro (Cuello et al., 2001; Raja et al., 2008), but in trastuzumab-resistant HER2-positive BC cells, it has no effect on HER2 level both in vitro and in vivo (Yang, Li, Bhattacharya, & Zhang, 2019). In a mouse model of HER2-positive BC, where trastuzumab strongly inhibits tumor growth, there is no change in HER2 level in the tumor tissues (Moulder et al., 2001). Moreover, in a neoadjuvant trial of 35 patients with locally advanced HER2-positive BC, trastuzumab induces tumor regression and apoptosis but no change in total HER2 or phosphor-HER2 levels in the tumor tissues (Mohsin et al., 2005). Therefore, tumor inhibition by trastuzumab does not apparently involve HER2 downregulation.
Pertuzumab is also a fully humanized IgG1 monoclonal antibody and binds to subdomain 2 of HER2 ECD (Fig. 2A) (Franklin et al., 2004). Like trastuzumab, pertuzumab elicits ADCC both in vitro and in vivo (Toth et al., 2016). However, it is not clear if pertuzumab also elicits antibody-dependent cellular phagocytosis by tumor associated macrophage. Pertuzumab also efficiently disrupts HER2-HER3 heterodimerization (Franklin et al., 2004), which is consistent with the ECD subdomain 2 of HER2 being essential for dimerization (Cho et al., 2003). Indeed, pertuzumab is more effective than trastuzumab in disrupting the HER2-HER3-PI3K complex, as it disrupts the hetero-complex regardless of heregulin binding, whereas trastuzumab only disrupts ligand-free HER2-HER3-PI3K complex (Franklin et al., 2004; Junttila et al., 2009). Surprisingly, pertuzumab does not inhibit HER2 homodimerization and even increases HER2 phosphorylation (Nami, Maadi, & Wang, 2019). In fact, in HER2-positive BC cells, pertuzumab also stimulates tyrosine phosphorylation of several other RTKs, including MET, neurotrophic receptor tyrosine kinase 1 (NTRK1), MER tyrosine kinase 1 (MERTK1), TIE-2, and RET, and also stimulates the phosphorylation of ERK, while inhibiting the phosphorylation of HER3, IGF1R and insulin receptor (INSR) (Kennedy et al., 2019). Pertuzumab also stimulates binding of adaptor proteins to HER2, including SHC and GRB2, further documenting HER2 activation (Kennedy et al., 2019). Activation of HER2 and other RTKs likely weakens the therapeutic efficacy of pertuzumab. Pertuzumab does not downregulate HER2 expression (Yamashita-Kashima, Shu, Yorozu, Moriya, & Harada, 2017; Yao et al., 2009).
3.2. T-DM1 and DS-8201a
T-DM1 and DS-8201a enable HER2-directed delivery of microtubule destabilizer DM-1 and topoisomerase I inhibitor deruxtecan to cancer cells, respectively. Each trastuzumab molecule is connected via a linker to 3–4 molecules of DM-1 in T-DM1 and 8 molecules of deruxtecan in DS-8201a (Lewis Phillips et al., 2008; Ogitani et al., 2016). Both drugs also retain trastuzumab functions, including induction of ADCC and inhibition of HER2 (Junttila, Li, Parsons, Phillips, & Sliwkowski, 2011; Ogitani et al., 2016). Therefore, the drugs deliver both targeted chemotherapy and antibody therapy. DS-8201a also causes bystander killing (killing of surrounding cells) due to release of its membrane-permeable payload to the extracellular space (Ogitani et al., 2016). However, both drugs require HER2-mediated internalization and then payload release via cleavage by lysosome enzymes (Fig. 2A). Although trastuzumab binding to HER2 triggers internalization of the complex, with the internalized HER2 recycled back to cell membrane (Austin et al., 2004), MUC4, which shields HER2 from trastuzumab, is positive in 60% of HER2-positive BCs (Mercogliano et al., 2017; Nagy et al., 2005). Therefore, MUC4-overexpressing cells may be resistant to T-DM1 and DS-8201a.
3.3. Lapatinib, neratinib, and tucatinib
Phosphor-tyrosine residues in the kinase domains and C-terminal tails of RTKs serve as docking sites for various signaling proteins, leading to activation of various signaling pathways as discussed before. TKIs block oncogenic signaling by inhibiting auto-phosphorylation and trans-phosphorylation of RTKs. Lapatinib is a dual TKI, inhibiting the tyrosine kinase activities of both HER2 and EGFR (Fig. 2B) (Davis et al., 2011). It is a reversible TKI and competes with ATP for the ATP-binding site in the kinase domains (Wood et al., 2004). However, lapatinib upregulates HER3 and induces HER2-HER3 dimerization, which promotes proliferation and limits its own antitumor activity (Claus et al., 2018; J. T. Garrett et al., 2011). Neratinib is a pan-HER TKI, inhibiting the tyrosine kinase activities of all HER family members (Fig. 2B) (Davis et al., 2011). It is an irreversible TKI, covalently binding to a specific cysteine residue (Cys-773 or Cys-805) in the ATP-binding pocket of the RTKs (Wissner & Mansour, 2008). Toxicity is a significant issue for neratinib and is dose-limiting. More than 40% of neratinib-treated patients suffer from grade 3–4 diarrhea, compared to 2% in the control (Chan et al., 2016). In contrast, only 6% of patients treated with lapatinib suffer from grade 3 diarrhea, with no grade 4 diarrhea (Kaufman et al., 2009). Tucatinib is a reversible HER2-specific TKI by competing with ATP for the ATP-binding site in the kinase domain (Fig. 2B) (Kulukian et al., 2020), and is also less toxic than neratinib. For example, in patients receiving a combination treatment regimen with or without tucatinib, grade ≥3 diarrhea is 12.9% or 8.6%, respectively (Murthy et al., 2020). Neratinib and tucatinib are relatively new drugs, approved by the US Food and Drug Administration in 2017 and 2020, respectively, and their potential off-target effects are not known.
4. Clinical activities of HER2 inhibitors
Discussed below are pivotal clinical trials that led to approval of the HER2 inhibitors or the treatment regimens by the US Food and Drug Administration, except for the monotherapy trials of trastuzumab, pertuzumab and lapatinib. The monotherapy trials are included in the discussion here to help understand the therapeutic efficacies of the combination regimens. Key findings are summarized in Table 1. Trastuzumab has been the mainstay treatment for HER2-positive BC, but in patients with metastatic disease treated with trastuzumab as a monotherapy, objective response rate (ORR) is only 26% and median progression-free survival (MPFS) is 3.5–3.8 months (Vogel et al., 2002). Adding trastuzumab to chemotherapy increases ORR from 32% to 50% and MPFS from 4.6 months to 7.4 months (Slamon et al., 2001). Pertuzumab as a monotherapy is largely inactive in patients after progression on trastuzumab, showing ORR and MPFS of only 3.4% and 7.1 weeks, but combining pertuzumab with trastuzumab increases ORR and MPFS to 17.6% and 17.4 weeks (Cortes et al., 2012). Combination of pertuzumab, trastuzumab and docetaxel further increases the therapeutic efficacy, achieving ORR and MPFS of 80.2% and 18.5 months in metastatic disease, compared to 69.3% and 12.4 months in trastuzumab plus docetaxel (Baselga et al., 2012). T-DM1 alone or T-DM1 plus pertuzumab is not superior to trastuzumab plus docetaxel or paclitaxel (Table 1) (Perez et al., 2017). However, T-DM1 is better than lapatinib plus capecitabine. In a phase 3 trial with locally advanced or metastatic disease, T-DM1 achieved ORR and MPFS of 43.6% and 9.6 months, compared to 30.8% and 6.4 months in lapatinib plus capecitabine (Verma et al., 2012). DS-8201a shows significant activity in patients with metastatic disease treated with T-DM1, achieving ORR and MPFS of 60.9% and 16.4 months (Modi et al., 2020). Its superior activity to T-DM1 may be related in part to its bystander killing as mentioned before. Lapatinib as a monotherapy achieves ORR of 39% but MPFS of only 14.6 weeks in patients with relapsed or refractory disease (Kaufman et al., 2009). In another study, lapatinib in combination with capecitabine achieves ORR and MPFS of 22% and 8.4 months in advanced disease, compared to 14% and 4.4 months with capecitabine alone (Geyer et al., 2006). Neratinib efficacy is also modest. In patients who had locally advanced disease, completed neoadjuvant or adjuvant trastuzumab therapy, and then received neratinib or placebo for 1 year, 2-year invasive disease-free survival is 93.9% in the neratinib group but is 91.6% even in the placebo group (Chan et al., 2016). No ORR was reported in the above study. In patients with HER2-positive metastatic BC previously treated with HER2-directed therapies, neratinib in combination with capecitabine achieves ORR and MPFS of 32.8% and 5.6 months, compared to 26.7% and 5.5 months in lapatinib combination with capecitabine (Saura et al., 2020). Tucatinib efficacy as a monotherapy is unknown. In patients previously treated with multiple HER2 inhibitors, adding tucatinib to trastuzumab plus capecitabine increases ORR and MPFS from 22.8% and 5.6 months to 40.6% and 7.8 months, and in patients with brain metastases, adding tucatinib increases MPFS from 5.4 months to 7.6 months (Murthy et al., 2020).
Table 1.
Key clinical trial data of HER2 inhibitors in HER2-positive BC
| Drug | Clinical trial | Disease stage | Prior anti-HER2 treatment | ORR | MPFS in months (M) or weeks (W) |
|---|---|---|---|---|---|
| Trastuzumab (T) 1 | Phase 2 | Metastatic | No | 26% | 3.5–3.8 M |
| Chemotherapy (chemo)*, T 2 | Phase 3 | Metastatic | No | 32% in chemo 50% in chemo + T |
4.6 M in chemo 7.4 M in chemo + T |
| Pertuzumab (P), T 3 | Phase 2 | Metastatic | Progressed on T | 3.4% in P 17.6% in T + P |
7.1 W in P 17.4 W in T + P |
| Docetaxel (D), P, T 4 | Phase 3 | Metastatic | No | 69.3% in T + D 80.2% in T + D + P |
12.4 M in T + D 18.5 M in T + D + P |
| P, T, Taxane**, T-DM1 5 | Phase 3 | Local or metastatic | T and/or L in 31% of patients | 59.7% in T-DM1 64.2% in T-DM1 + P 67.9% in T + taxane |
14.1 M in T-DM1 15.2 M in T-DM1 + P 13.7 M in T + taxane |
| Capecitabine (C), Lapatinib (L), T-DM1 6 | Phase 3 | Local or metastatic | T | 43.6% in T-DM1 30.8% in L + C |
9.6 M in T-DM1 6.4 M in L + C |
| DS-8201a 7 | Phase 2 | Local or metastatic | T-DM1 and/or other HER2 inhibitors | 60.9% | 16.4 M |
| L 8 | Phase 2 | Local or metastatic | T in 75% patients | 39% | 14.6 W |
| C, L 9 | Phase 3 | Local or metastatic | T | 22% (L + C) 14% (C) |
8.4 M (L + C) 4.4 M (C) |
| Neratinib (N), placebo 10 | Phase 3 | Local | T | ORR not reported | MPFS not reported; 2-year invasive disease-free survival: 93.9% (N) and 91.6% (placebo) |
| L, N, C 11 | Phase 3 | Metastatic | T with or without other Her2 inhibitors | 32.8% (N + C) 26.7% (L + C) |
5.6 M (N + C) 5.5 M (L + C) |
| C, T, Tucatinib (Tu) 12 | Phase 3 | Metastatic | Multiple HER2 inhibitors | 40.6% (T + C + Tu) 22.8% (T + C) |
7.8 M (T + C + Tu) 5.6 M (T + C) |
Doxorubicin, epirubicin, cyclophosphamide and/or paclitaxel;
docetaxel or paclitaxel.
Collectively, while HER2 inhibitors provide significant therapeutic benefit to patients and combination therapies often increase such benefit, primary and acquired drug resistance is widespread, and treatment response remains largely temporary. For the three TKIs, there is no clear correlation between their therapeutic efficacies and their HER-targeting specificities. All this is consistent with the notion that HER2 inhibitors are overwhelmed by the vast oncogenic signaling network of HER2.
5. Other signaling proteins implicated in drug resistance in HER2-positive BC
5.1. The PI3K pathway
PI3K pathway activation (activating mutation in the PI3K catalytic subunit PIK3CA and/or PTEN loss) occurs frequently in human HER2-positive BC. One study shows PI3K pathway activation in 47% of 240 patient tumors analyzed, including PIK3CA mutation (exon 9 or exon 20) in 26% of the tumors and PTEN low in 24% of the tumors (Jensen et al., 2012). In preclinical models of HER2-positive BC, mutant PIK3CA or PTEN loss promotes tumor growth and induces resistance to anti-HER2 therapies (Hanker et al., 2013; Nagata et al., 2004). However, clinical data are inconclusive on the role of PI3K and PTEN in HER2-positive BC. There are studies showing that PIK3CA mutation or low PTEN is associated with poor prognosis and treatment resistance (Jensen et al., 2012; Nagata et al., 2004). There are also studies showing that neither PIK3CA mutation nor PTEN status is associated with disease response to trastuzumab or patient survival (Perez et al., 2013; Wang, Liu, Du, Yin, & Lu, 2013). Adding buparlisib, a pan-PI3K inhibitor, to a trastuzumab-based therapy is ineffective and induces significant liver toxicity in a phase 2 trial (Loibl et al., 2017). Also, adding everolimus, an inhibitor of mTOR which is downstream of PI3K, to the combination of trastuzumab and vinorelbine, an anti-microtubule agent, only increases MPFS from 5.8 to 7.0 months (Andre et al., 2014). Collectively, dysregulation of the PI3K pathway may not be a key contributor to drug resistance in HER2-positive BC.
5.2. Cell cycle regulators
Cyclin D1 functions as a regulatory subunit of cyclin-dependent kinase 4 (CDK4) and CDK6. Cyclin D1 plays a critical role in preclinical models of HER2-driven tumorigenesis (Choi et al., 2012; Yu, Geng, & Sicinski, 2001). There is a significant association between high copy number of CCND1 gene which encodes Cyclin D1 and resistance to trastuzumab-based therapy in HER2-positive BC in patients (Goel et al., 2016). In a transgenic model of HER2-driven BC, Cyclin D1-CDK4 mediates resistance to anti-HER2 therapies and CDK4/6 inhibitor abemaciclib is active in trastuzumab-resistant HER2-positive tumors (Goel et al., 2016). However, clinical benefit of abemaciclib in HER2-positive BC is modest. Combination of abemaciclib with trastuzumab and fulvestrant, a selective estrogen receptor degrader, achieves MPFS of 8.3 months in patients previously treated with HER2-targeted therapies, compared to 5.6 months in abemaciclib plus trastuzumab, and 5.7 months in trastuzumab plus investigator’s choice of chemotherapy (Tolaney et al., 2020).
Cyclin E1 encoded by CCNE1 gene regulates cyclin-dependent kinase 2 (CDK2). Chronic treatment of HER2-positive BC cells with trastuzumab leads to trastuzumab resistance and cyclin E1 amplification/overexpression, and forced cyclin E1 overexpression results in resistance to trastuzumab in vitro and in vivo (Scaltriti et al., 2011). CDK2 inhibitor CYC065 strongly inhibits trastuzumab-resistant HER2-positive BC cells in vitro and in vivo and combination of CYC065 with trastuzumab further increases the inhibitory efficacy (Scaltriti et al., 2011). Cyclin E1 is often overexpressed in HER2-positve human tumors (Luhtala, Staff, Tanner, & Isola, 2016; Scaltriti et al., 2011). However, the role of Cyclin E1 in HER2-positive BC patients remains unclear. There are conflicting data as to whether or not Cyclin E1 overexpression confers resistance to trastuzumab or predicts poor prognosis in patients (Luhtala et al., 2016; Scaltriti et al., 2011).
5.3. SRC
Increased SRC activation, i.e., increased phosphorylation rather than increased protein expression, confers resistance of HER2-positive BC cells to trastuzumab in preclinical models and correlates with trastuzumab resistance in patients, whereas targeting SRC sensitizes cells to trastuzumab in vitro and in vivo (Peiro et al., 2014; Zhang et al., 2011). Because SRC is downstream of various RTKs, SRC inhibition may attenuate the signaling of some of the RTKs that contribute to drug resistance in HER2-positive BC. Indeed, SRC inhibition shows encouraging result in patients. In a phase 2 trial of 23 patients with metastatic HER2-positive BC, combination of dasatinib, a SRC inhibitor, with trastuzumab and paclitaxel shows strong efficacy, achieving MPFS of 23.9 months (Ocana et al., 2019). The inhibitory efficacy of this combination regimen seems to be better than that of pertuzumab in combination with trastuzumab and docetaxel described before. However, further evaluation of the combination regimen in a large trial is needed. It is also worth noting that dasatinib is a TKI with a low target specificity, binding and inhibiting many tyrosine and serine/threonine kinases (Lombardo et al., 2004; Rix et al., 2007).
6. Non-cell-autonomous mechanisms of drug resistance in HER2-positive BC
A study using mouse tumor models suggested that trastuzumab efficacy may be curtailed by tissue- and vessel-level barriers to its distribution, such as vascular density and patency (Baker et al., 2018). Vasculogenic mimicry is the formation of microvascular channels by cancer cells to enhance fluid perfusion and contributes to drug resistance (Kirschmann, Seftor, Hardy, Seftor, & Hendrix, 2012). In a study using cell lines and patient tumors, trastuzumab resistance in HER2-positive BC was found to be associated with vasculogenic mimicry (Hori et al., 2019). Trastuzumab induces ADCC by binding to the Fc receptors in immune effector cells. In a study of patients with advanced HER2-positive BC, the FcγRIIIa-158V/F polymorphism was found to be significantly associated with patient response to trastuzumab, with the V/V variant associating with better ORR and MPFS than V/F or F/F (Musolino et al., 2008). A study using cultured cells and mouse tumor models suggested that adipocytes and preadipocytes attenuate induction of ADCC by trastuzumab in HER2-positive BC cells by decreasing interferon-γ secretion from NK cells (Duong et al., 2015). Cancer-associated fibroblasts were also shown to protect HER2-positve BC cells against lapatinib in vitro and in vivo by producing stromal hyaluronan and inducing the PI3K-AKT-mROR pathway in cancer cells (Marusyk et al., 2016; Zervantonakis et al., 2020). A study using cultured cells showed that physical contact of cancer cells with mesenchymal stem cells also induces trastuzumab resistance by activating SRC (Daverey, Drain, & Kidambi, 2015). Tumor-infiltrating lymphocytes (TIL) may also contribute to the response of HER2-positive tumors to HER2 inhibitors. Clinical studies show that presence of TIL is associated with improved outcome of treatment with lapatinib and/or trastuzumab with chemotherapy (Dieci et al., 2019; Salgado et al., 2015). However, another clinical study showed that presence of TIL is associated with improved outcome of treatment with chemotherapy but not chemotherapy plus trastuzumab (Perez et al., 2016). Overall, many non-cell-autonomous tumor microenvironment mechanisms may affect treatment outcome in HER2-positive BC, but the extent to which these mechanisms contribute to drug resistance is unclear.
7. A ligand of HER2 and EGFR overcomes drug resistance in HER2-positive BC
Although the ECD of HER2 was shown to exist in a conformation that resembles a ligand-activated state, and HER2 undergoes homo- or hetero-dimerization in the absence of ligand binding, as mentioned before, we recently found that human peptidase D (PEPD), also known as prolidase, induces HER2 homodimerization and phosphorylation by binding directly to the ECD of HER2 with high affinity (Kd of 7.3 nM) (Yang, Li, & Zhang, 2014). PEPD exists as a homodimer, and each subunit of PEPD binds via its C-terminal sequence to a HER2 molecule (ECD subdomain 3), forming a tetramer complex (Fig. 3A). This is similar to a stem cell factor dimer binding to two KIT molecules (Yuzawa et al., 2007). PEPD also readily binds to preexisting HER2 homodimers (Yang, Li, Bhattacharya, & Zhang, 2019; Yang, Li, & Zhang, 2014). PEPD is a well-known intracellular dipeptidase important for collagen metabolism (Lupi, Tenni, Rossi, Cetta, & Forlino, 2008). However, the enzymatic activity of PEPD plays no role in HER2 binding, as PEPDG278D, a catalytically inactive mutant of PEPD (a single amino acid substitution at #278), shows no difference in binding to HER2. Both PEPD and PEPDG278D also induce EGFR homodimerization and phosphorylation by crosslinking two EGFR molecules (Yang, Li, Bhattacharya, & Zhang, 2015). However, they bind to subdomain 2 of EGFR ECD (Fig. 3A) (Yang, Li, Bhattacharya, & Zhang, 2016a), rather than subdomain 3 seen in HER2, suggesting that different motifs in the PEPD sequence may mediate binding to HER2 and EGFR. In fact, HER2 and EGFR are mutually exclusive in binding to PEPD or PEPDG278D, and PEPD and PEPDG278D disrupt the HER2-EGFR heterodimer (Yang, Li, Bhattacharya, & Zhang, 2015), apparently by forcing homodimerization of HER2 and EGFR. PEPD and PEPDG278D do not bind to HER3 or HER4 (Yang, Li, & Zhang, 2014), and there is no evidence that they bind to other RTKs. Unlike other HER ligands, PEPD and PEPDG278D activate HER2 and EGFR only transiently, because PEPD- or PEPDG278D-bound HER2 and EGFR are efficiently internalized and degraded in lysosomes (Fig. 3A), resulting in marked depletion of HER2 and EGFR in HER2- and/or EGFR-overexpressing cancer cells and strong inhibition of growth of these cells in vitro and in vivo (Yang, Li, & Zhang, 2014; Yang, Li, Bhattacharya, & Zhang, 2015; Yang, Li, Bhattacharya, & Zhang, 2016a; Yang, Li, Bhattacharya, & Zhang, 2019).
Fig. 3.
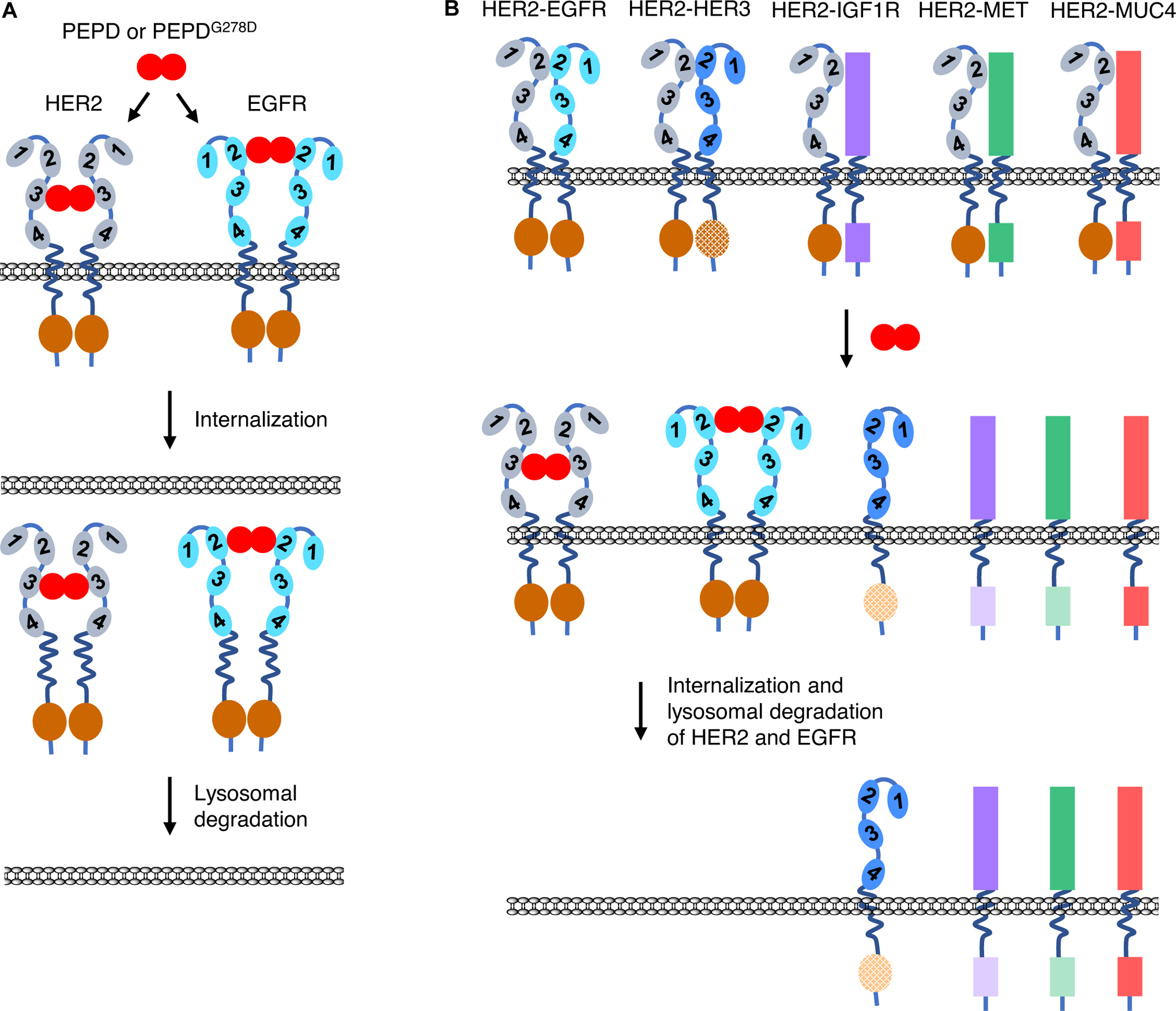
The impact of PEPD or PEPDG278D on HER2, EGFR, and HER2 heterodimers. A. PEPD or PEPDG278D forms a tetramer with HER2 or EGFR by binding to ECD subdomain 3 of HER2 and ECD subdomain 2 of EGFR, which leads to internalization and lysosomal degradation of the RTKs. B. PEPD or PEPDG278D disrupts the heterodimers of HER2-EGFR, HER2-HER3, HER2-IGF1R, HER2-MET, and HER2-MUC4 by forcing HER2 homodimerization and EGFR homodimerization. The PEPD- or PEPDG278D-bound homodimers of HER2 and EGFR are then internalized and degraded. This causes inactivation of all the RTKs. The intracellular kinase domains are indicated with an oval or bar, and the hatched oval in HER3 indicates impaired kinase. The dimmed color in the kinase domains of HER3, IGF1R and MET indicates loss of phosphorylation and inactivation.
Available evidence suggests that PEPD may not modulate HER2 and EGFR in normal cells. First, endogenous PEPD is mainly an intracellular protein, and intracellular PEPD has no effect on HER2 or EGFR (Yang et al., 2013; Yang, Li, & Zhang, 2014). Second, even if PEPD enters the extracellular space, it modulates HER2 and EGFR only if the RTKs are overexpressed, not when they are under-expressed as in normal cells and tissues (Yang, Li, Bhattacharya, & Zhang, 2019). PEPD selectivity for overexpressed HER2 and EGFR is likely due to the requirement for PEPD to form tetramer complexes with the RTKs, as shown in Fig. 3A. Such tetramer may not form if the RTKs are under-expressed and scatter far apart from one another, since protein movement across cell membrane is restricted (Shi, Graber, Baumgart, Stone, & Cohen, 2018).
However, exogenous recombinant PEPD and PEPDG278D show strong antitumor activities. Notably, PEPD and PEPDG278D are degraded in plasma by certain coagulation proteases in vivo, and they are administered to mice along with an anticoagulant (e.g., enoxaparin) to inhibit their degradation (Yang, Li, Bhattacharya, & Zhang, 2015; Yang, Li, Bhattacharya, & Zhang, 2016b; Yang, Li, Bhattacharya, and Zhang, 2019). Described below is the antitumor activity of PEPDG278D in cell and mouse models of HER2-positive BC. We focused on PEPDG278D, because PEPD, not PEPDG278D, was found to stimulate hypoxia-inducible factor 1α (HIF-1α) and its downstream targets, including vascular endothelial growth factor and glucose transporter 1, in the tumor tissues (Yang, Li, Bhattacharya, & Zhang, 2015). The effects of PEPD on HIF-1α and its downstream targets likely stem from the metabolism of imidodipeptides by PEPD and inhibition of HIF-1α degradation by the metabolites (Surazynski et al., 2008). PEPDG278D binds to HER2, disrupts its heterodimerization with all RTKs that have been examined, including HER2-EGFR, HER2-HER3, HER2-IGF1R, and HER2-MET with or without ligand binding and then directs HER2 for lysosomal degradation (Fig. 3B) (Yang, Li, Bhattacharya, & Zhang, 2015; Yang, Li, Bhattacharya, and Zhang, 2019). PEPDG278D also binds to and induces the degradation of EGFR (Fig. 3B) (Yang, Li, Bhattacharya, & Zhang, 2016a). PEPDG278D causes profound loss of both HER2 and EGFR (both expression and phosphorylation), loss of phosphorylation but not expression of all signaling partners analyzed, including HER3, IGF1R, MET, SRC, AKT and ERK (Yang, Li, & Zhang, 2014; Yang, Li, Bhattacharya, & Zhang, 2015; Yang, Li, Bhattacharya, & Zhang, 2016a; Yang, Li, Bhattacharya, and Zhang, 2019). This is accompanied by strong growth inhibition of cancer cells and tumors in vitro and in vivo. PEPDG278D is inactive in cells or tumors lacking the RTKs or expressing them at very low levels (Yang, Li, & Zhang, 2014; Yang, Li, Bhattacharya, & Zhang, 2015; Yang, Li, Bhattacharya, & Zhang, 2016a; Yang, Li, Bhattacharya, and Zhang, 2019). Most interestingly, PEPDG278D induces the aforementioned molecular changes in and causes strong growth inhibition of HER2-positive BC cells and tumors that are resistant to trastuzumab and carry activating PIK3CA mutation, low PTEN, p95HER2, and/or Cyclin E overexpression (Table 2) (Yang, Li, Bhattacharya, and Zhang, 2019). MUC4 is overexpressed in all trastuzumab-resistant cell lines (Table 2), but PEPDG278D binds to and frees HER2 from MUC4 (Fig. 3B) (Yang, Li, Bhattacharya, and Zhang, 2019). These results indicate that trastuzumab-resistant HER2-positive BC cells still require HER2 for survival, despite seemingly compensatory molecular changes. The results also show that PEPDG278D is a promising agent for overcoming drug resistance in HER2-positive BC in patients. Consistent with the inability of PEPDG278D to modulate HER2 and EGFR in normal cells, adverse effects of PEPDG278D have not been detected in mouse studies (Yang, Li, Bhattacharya, & Zhang, 2015; Yang, Li, Bhattacharya, & Zhang, 2016a; Yang, Li, Bhattacharya, and Zhang, 2019).
Table 2.
PEPDG278D inhibits trastuzumab-resistant HER2-positive BC
| Cell line | PIK3CA mutation | PTEN level | p95HER2 expression | Cyclin E1 level | MUC4 level | Inhibition by trastuzumab | Inhibition by PEPDG278D | ||
|---|---|---|---|---|---|---|---|---|---|
| In vitro | In vivo | In vitro | In vivo | ||||||
| BT-474 | E545A | Yes | Low | Yes | Yes | Yes | Yes | ||
| BT-474R2 | E454A | High | High | No | No | Yes | Yes | ||
| JIMT-1 | C420R | Low | High | No | No | Yes | Yes | ||
| HCC-1419 | E545A | Yes | High | No | ND* | Yes | ND | ||
| HCC-1569 | E545A H1047R |
Low | High | No | ND | Yes | ND | ||
| HCC-1954 | E545A | Yes | High | No | ND | Yes | ND | ||
| UACC-893 | E545A H1047R |
low | High | No | ND | Yes | ND | ||
ND, not determined. The data are from Yang, Li, Bhattacharya, & Zhang, 2019, except for expression levels of p95HER2 and Cyclin E1, which were reported in Scaltriti et al., 2011; Sperinde et al., 2010; Parra-Palau et al., 2014.
8. Role of HER2 in drug-resistant HER2-positive BC
The strong inhibitory activity of PEPDG278D against drug-resistant HER2-positive BC, as described above, indicates that HER2 remains a vital oncogenic driver in HER2-positive BC resistant to current HER2 inhibitors. The PEPDG278D results suggest that depleting HER2 may be the most effective HER2-targeting strategy in HER2-positive BC. This concept is consistent with the notion that HER2 depletion may be most effective in shutting down the vast oncogenic signaling network orchestrated by overexpressed HER2 (Fig. 1), including disruption of its homo- and heterodimeric signaling units, reduction of p95HER2, and elimination of nuclear and mitochondrial HER2. Notably, current HER2 inhibitors have little or no effect on HER2 level. The PEPDG278D results also suggest that other molecular changes that have been linked to drug resistance, such as p95HER2, PIK3CA mutation, PTEN loss, SRC activation, and Cyclin E overexpression, may not sustain HER2-positve BC cells if HER2 is depleted. Consistent with the PEPDG278D results described above, HER2 knockdown by siRNA also significantly inhibits various HER2-positive human BC cell lines that show primary resistance or acquired resistance to trastuzumab and/or lapatinib, and a systemically delivered trastuzumab-conjugated nano-construct carrying HER2 siRNA significantly inhibits the growth of trastuzumab-resistant tumors in mice (Gu et al., 2016). Further, downregulation of HER2 by disrupting the HER2-CB2R heteromers also inhibits trastuzumab-resistant HER2-positive cells in vitro and in vivo (Blasco-Benito et al., 2019). The ability of PEPDG278D to induce depletion of both HER2 and EGFR is particularly promising, since EGFR is overexpressed in about 35% of HER2-positive BC tumors and EGFR overexpression is associated with poor prognosis (DiGiovanna et al., 2005; Tsutsui et al., 2003). Indeed, EGFR knockdown by siRNA causes partial growth inhibition of trastuzumab-resistant HER2-positive BC cells (Yang, Li, Bhattacharya, and Zhang, 2019).
9. HER2-positive vs HER2-negative in BC and intratumor HER2 heterogeneity
It is important to distinguish drug-resistant HER2-positive BC from HER2-positive BC that becomes HER2-negative during treatment or metastasis and therefore is insensitive to anti-HER2 treatment. Chronic treatment of HER2-positive BC cells with trastuzumab, lapatinib or T-DM1 can generate drug-resistant cells with loss of HER2 in vitro and in vivo (Mittendorf et al., 2009; Vicario et al., 2015). Gene copy number may remain unchanged despite HER2 protein loss in cells with acquired resistance to HER2 inhibitors (Vicario et al., 2015). Loss of HER2 occurs in 32% of HER2-positive BCs following trastuzumab-based neoadjuvant therapy, based on fluorescence in situ hybridization (FISH) (Mittendorf et al., 2009). Interestingly, another study shows that chemotherapy, rather than anti-HER2 therapy, is associated with HER2 loss in 24% of patients who have HER2-positive primary BC but subsequently develop HER2-negative metastatic disease (Niikura et al., 2012). Also, nearly 10% of HER2-negative primary BCs develop HER2-positive metastatic disease based on IHC (Lower, Glass, Blau, & Harman, 2009). Therefore, it remains unclear to what extent HER2 loss in patient tumors is induced by anti-HER2 therapies. HER2 discordance between primary and metastatic diseases have been extensively studied (Houssami, Macaskill, Balleine, Bilous, & Pegram, 2011), but no clear factor promoting HER2 discordance was identified (Turner & Di Leo, 2013). It is also unknown whether EGFR compensates for HER2 after HER2-positive BC turns HER2-negative and whether depleting EGFR is effective in this subset of BC. However, it may be important to assess metastatic BC for HER2 overexpression before deciding on anti-HER2 treatment.
According to the American Society of Clinical Oncology and College of American Pathologists guidelines of 2007, HER2-positive BC is defined as 3+ staining by IHC in more than 30% of cancer cells or an average of more than 6 HER2 copies per nucleus without an internal control probe or HER2/CEP17 ratio of more than 2.2 by FISH (Wolff et al., 2007). Intratumor heterogeneity of HER2 expression in HER2-positive BC is well known. Higher HER2 level is associated with better response to combination treatment of trastuzumab with chemotherapy, whereas higher HER2 heterogeneity is associated with worse patient outcome (Lee et al., 2014; Rye et al., 2018). These results are not surprising, since only cancer cells that overexpress and are driven by HER2 may respond to trastuzumab. Notably, this review is focused on BCs that overexpress HER2 but are resistant to trastuzumab and other HER2 inhibitors.
10. Emerging therapeutic approaches
Preclinical studies show that therapeutic strategies aimed at eliminating both HER2 and EGFR may be most promising for overcoming drug resistance in HER2-positive BC. Eliminating HER2 may abolish the vast oncogenic signaling network of HER2, and eliminating both HER2 and EGFR may prevent cancer cells from overcoming HER2 deficit by relying on EGFR. PEPDG278D is a highly promising agent in this regard. HER2-directed nanoparticle delivery of siRNA targeting HER2 or both HER2 and EGFR also warrants investigation. As mentioned before, HER2-directed nanoparticle delivery of HER2 siRNA significantly inhibits the growth of trastuzumab-resistant HER2-positive BC in vitro and in mice. Proteolysis targeting chimeras (PROTAC) has emerged as a promising strategy for targeted degradation of oncoproteins, in which a target protein ligand is tethered to a motif that binds to an E3 ubiquitin ligase. PROTAC causes co-localization of the target protein and the E3 ligase that triggers ubiquitination and subsequent degradation of the target. A lapatinib-based PROTAC degrades both EGFR and HER2 in cultured cancer cells (Burslem et al., 2018). Interestingly, there are also reports that trastuzumab-resistant HER2-positive BC cells are sensitive in vitro and in vivo to inhibition of poly (ADP-ribose) polymerase (Garcia-Parra et al., 2014; Wielgos et al., 2018). However, the molecular mechanism by which inhibition of poly (ADP-ribose) polymerase targets trastuzumab-resistant HER2-positive cells is unclear and additional studies are needed to further evaluate the therapeutic strategy.
Immunotherapy represents a potential alternative approach to targeted degradation of HER2 and EGFR. A p95HER2-T cell bispecific antibody, designed to direct T cells to tumor cells expressing p95HER2, has shown significant therapeutic activity in a preclinical study (Rius Ruiz et al., 2018). This may allow selective killing of p95HER2-positive cancer cells, since p95HER2 is not expressed in normal cells. As mentioned before, p95HER2 is positive in up to 60–80% of HER2-positive BCs. HER2-directed chimeric antigen receptor (CAR)-T cell therapy represents another immunotherapeutic approach to killing drug-resistant HER2 cells without relying on depleting HER2 and EGFR. A phase 1 trial of HER2-directed CAR-T cells in HER2-positive BC is currently being pursued at City of Hope (ClinicalTrial.gov identifier: NCT03696030). p95HER2-directed CAR-T cells may be more interesting, as they may not target normal cells. Immune checkpoint inhibitor therapy has also been evaluated in HER2-positive BC. However, a phase 2 clinical study did not show significant improvement of efficacy by combining T-DM1 with atezolizumab, an anti-PD-1 antibody (Emens et al., 2019).
11. Conclusions
HER2 inhibitors have transformed treatment of HER2-positive BC, but resistance to these agents remains a major unsolved clinical problem. Many resistance mechanisms have been shown, which have guided development of combination treatment strategies. However, the efficacies of rationally designed combination treatments are mixed. Some combination strategies significantly enhance treatment efficacy, such as the triple combination of trastuzumab, pertuzumab and docetaxel. Other combination regimens have marginal or even detrimental effects, such as the combination of trastuzumab with buparlisib. Even with the most effective combination regimen, patients with metastatic disease typically show disease progression after some time on treatment. Many lines of evidence indicate that HER2 remains a critical therapeutic target in drug-resistant HER2-positive BC. Therapeutic strategies aimed at depleting HER2 or depleting both HER2 and EGFR are highly promising for overcoming drug resistance in this disease. Immunotherapies that direct T cells to HER2 or p95HER2 represent promising alternative approach to treating drug-resistant HER2-positive BC.
Acknowledgements
I would like to thank Boyko S. Atanassov of Roswell Park Comprehensive Cancer Center for critical reading of the manuscript and valuable comments. This work was supported in part by the National Cancer Institute (R01CA215093 and R01CA244601) and Roswell Park Alliance Foundation (Developmental Fund).
Abbreviations:
- ADCC
antibody-dependent cellular cytotoxicity
- BC
breast cancer
- CAR-T cell
chimeric antigen receptor T cell
- CB2R
cannabinoid receptor type 2
- CDK4
cyclin-dependent kinase 4
- DS-8201a
trastuzumab deruxtecan
- ECD
extracellular domain
- EGFR
epidermal growth factor receptor
- ERK
extracellular signal-regulated kinase
- FISH
fluorescence in situ hybridization
- HER2
human epidermal growth factor receptor 2
- HIF-1α
hypoxia-inducible factor 1α
- IGF1R
insulin-like growth factor 1 receptor
- IHC
immunohistochemistry
- MPFS
median progression-free survival
- MUC1
mucin 1
- MUC4
mucin 4
- ORR
objective response rate
- PI3K
phosphoinositide 3-kinase
- RTK
receptor tyrosine kinase
- PEPD
peptidase D
- PEPDG278D
a catalytically inactive mutant of PEPD with glycine to aspartic acid mutation at amino acid #278
- PROTAC
proteolysis targeting chimeras
- STAT3
signal transducer and activator of transcription 3
- T-DM1
trastuzumab emtansine
- TIL
tumor-infiltrating lymphocyte
- TKI
tyrosine kinase inhibitor
Footnotes
Conflict of Interest Statement
The author declares that there are no conflicts of interest.
References
- Alexander PB, Chen R, Gong C, Yuan L, Jasper JS, Ding Y, … Wang XF (2017). Distinct receptor tyrosine kinase subsets mediate anti-HER2 drug resistance in breast cancer. The Journal of Biological Chemistry 292, 748–759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andre F, O’Regan R, Ozguroglu M, Toi M, Xu B, Jerusalem G, … Gianni L (2014). Everolimus for women with trastuzumab-resistant, HER2-positive, advanced breast cancer (BOLERO-3): a randomised, double-blind, placebo-controlled phase 3 trial. The Lancet Oncology 15, 580–591. [DOI] [PubMed] [Google Scholar]
- Anido J, Scaltriti M, Bech Serra JJ, Santiago Josefat B, Todo FR, Baselga J, & Arribas J (2006). Biosynthesis of tumorigenic HER2 C-terminal fragments by alternative initiation of translation. The EMBO Journal 25, 3234–3244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arpino G, Gutierrez C, Weiss H, Rimawi M, Massarweh S, Bharwani L, … Schiff R (2007). Treatment of human epidermal growth factor receptor 2-overexpressing breast cancer xenografts with multiagent HER-targeted therapy. Journal of the National Cancer Institute 99, 694–705. [DOI] [PubMed] [Google Scholar]
- Arribas J, Baselga J, Pedersen K, & Parra-Palau JL (2011). p95HER2 and breast cancer. Cancer Research 71, 1515–1519. [DOI] [PubMed] [Google Scholar]
- Austin CD, De Maziere AM, Pisacane PI, van Dijk SM, Eigenbrot C, Sliwkowski MX, … Scheller RH (2004). Endocytosis and sorting of ErbB2 and the site of action of cancer therapeutics trastuzumab and geldanamycin. Molecular Biology of the Cell 15, 5268–5282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bailey TA, Luan H, Clubb RJ, Naramura M, Band V, Raja SM, & Band H (2011). Mechanisms of trastuzumab resistance in ErbB2-driven breast cancer and newer opportunities to overcome therapy resistance. Journal of Carcinogenesis 10, 28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baker JHE, Kyle AH, Reinsberg SA, Moosvi F, Patrick HM, Cran J, … Minchinton AI (2018). Heterogeneous distribution of trastuzumab in HER2-positive xenografts and metastases: role of the tumor microenvironment. Clinical & Experimental Metastasis 35, 691–705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baselga J, Cortes J, Kim SB, Im SA, Hegg R, Im YH, … for the CLEOPATRA Study Group. (2012). Pertuzumab plus trastuzumab plus docetaxel for metastatic breast cancer. The New England Journal of Medicine 366, 109–119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blasco-Benito S, Moreno E, Seijo-Vila M, Tundidor I, Andradas C, Caffarel MM, … Sanchez C (2019). Therapeutic targeting of HER2-CB2R heteromers in HER2-positive breast cancer. Proceedings of the National Academy of Sciences of the United States of America 116, 3863–3872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bose R, Kavuri SM, Searleman AC, Shen W, Shen D, Koboldt DC, … Ellis MJ (2013). Activating HER2 mutations in HER2 gene amplification negative breast cancer. Cancer Discovery 3, 224–237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burslem GM, Smith BE, Lai AC, Jaime-Figueroa S, McQuaid DC, Bondeson DP, … Crews CM (2018). The advantages of targeted protein degradation over inhibition: an RTK case study. Cell Chemical Biology 25, 67–77. doi: 10.1016/j.chembiol.2017.09.009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chaganty BKR, Qiu S, Gest A, Lu Y, Ivan C, Calin GA, … Fan Z (2018). Trastuzumab upregulates PD-L1 as a potential mechanism of trastuzumab resistance through engagement of immune effector cells and stimulation of IFNgamma secretion. Cancer Letters 430, 47–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chan A, Delaloge S, Holmes FA, Moy B, Iwata H, Harvey VJ, … Martin M for the ExteNET Study Group. (2016). Neratinib after trastuzumab-based adjuvant therapy in patients with HER2-positive breast cancer (ExteNET): a multicentre, randomised, double-blind, placebo-controlled, phase 3 trial. The Lancet Oncology 17, 367–377. [DOI] [PubMed] [Google Scholar]
- Chia S, Norris B, Speers C, Cheang M, Gilks B, Gown AM, …Gelmon K (2008). Human epidermal growth factor receptor 2 overexpression as a prognostic factor in a large tissue microarray series of node-negative breast cancers. Journal of Clinical Oncology 26, 5697–5704. [DOI] [PubMed] [Google Scholar]
- Cho HS, Mason K, Ramyar KX, Stanley AM, Gabelli SB, Denney DW Jr., & Leahy DJ (2003). Structure of the extracellular region of HER2 alone and in complex with the Herceptin Fab. Nature 421, 756–760. [DOI] [PubMed] [Google Scholar]
- Choi YJ, Li X, Hydbring P, Sanda T, Stefano J, Christie AL, … Sicinski P (2012). The requirement for cyclin D function in tumor maintenance. Cancer Cell 22, 438–451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choritz H, Busche G, Kreipe H, & On behalf of the Study Group HER2 Monitor. (2011). Quality assessment of HER2 testing by monitoring of positivity rates. Virchows Archiv 459, 283–289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Christianson TA, Doherty JK, Lin YJ, Ramsey EE, Holmes R, Keenan EJ, & Clinton GM (1998). NH2-terminally truncated HER-2/neu protein: relationship with shedding of the extracellular domain and with prognostic factors in breast cancer. Cancer Research 58, 5123–5129. [PubMed] [Google Scholar]
- Claus J, Patel G, Autore F, Colomba A, Weitsman G, Soliman TN, … Parker PJ (2018). Inhibitor-induced HER2-HER3 heterodimerisation promotes proliferation through a novel dimer interface. Elife 7, e32271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clynes RA, Towers TL, Presta LG, & Ravetch JV (2000). Inhibitory Fc receptors modulate in vivo cytotoxicity against tumor targets. Nature Medicine 6, 443–446. [DOI] [PubMed] [Google Scholar]
- Cocco E, Javier Carmona F, Razavi P, Won HH, Cai Y, Rossi V, … Scaltriti M (2018). Neratinib is effective in breast tumors bearing both amplification and mutation of ERBB2 (HER2). Science Signaling 11, eaat9773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cortes J, Fumoleau P, Bianchi GV, Petrella TM, Gelmon K, Pivot X, … Baselga J (2012). Pertuzumab monotherapy after trastuzumab-based treatment and subsequent reintroduction of trastuzumab: activity and tolerability in patients with advanced human epidermal growth factor receptor 2-positive breast cancer. Journal of Clinical Oncology 30, 1594–1600. [DOI] [PubMed] [Google Scholar]
- Cuello M, Ettenberg SA, Clark AS, Keane MM, Posner RH, Nau MM, … Lipkowitz S (2001). Down-regulation of the erbB-2 receptor by trastuzumab (herceptin) enhances tumor necrosis factor-related apoptosis-inducing ligand-mediated apoptosis in breast and ovarian cancer cell lines that overexpress erbB-2. Cancer Research 61, 4892–4900. [PubMed] [Google Scholar]
- D’Amato V, Raimondo L, Formisano L, Giuliano M, De Placido S, Rosa R, & Bianco R (2015). Mechanisms of lapatinib resistance in HER2-driven breast cancer. Cancer Treatment Reviews 41, 877–883. [DOI] [PubMed] [Google Scholar]
- Daverey A, Drain AP, & Kidambi S (2015). Physical intimacy of breast cancer cells with mesenchymal stem cells elicits trastuzumab resistance through Src activation. Scientific Reports 5, 13744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davis MI, Hunt JP, Herrgard S, Ciceri P, Wodicka LM, Pallares G, … Zarrinkar PP (2011). Comprehensive analysis of kinase inhibitor selectivity. Nature Biotechnology 29, 1046–1051. [DOI] [PubMed] [Google Scholar]
- Dieci MV, Conte P, Bisagni G, Brandes AA, Frassoldati A, Cavanna L, … Guarneri V (2019). Association of tumor-infiltrating lymphocytes with distant disease-free survival in the ShortHER randomized adjuvant trial for patients with early HER2+ breast cancer. Annals of Oncology 30, 418–423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DiGiovanna MP, Stern DF, Edgerton SM, Whalen SG, Moore D 2nd, & Thor AD (2005). Relationship of epidermal growth factor receptor expression to ErbB-2 signaling activity and prognosis in breast cancer patients. Journal of Clinical Oncology 23, 1152–1160. [DOI] [PubMed] [Google Scholar]
- Ding Y, Liu Z, Desai S, Zhao Y, Liu H, Pannell LK, … Tan M (2012). Receptor tyrosine kinase ErbB2 translocates into mitochondria and regulates cellular metabolism. Nature Communications 3, 1271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ding J, Yao Y, Huang G, Wang X, Yi J, Zhang N, … Cheng H (2020). Targeting the EphB4 receptor tyrosine kinase sensitizes HER2-positive breast cancer cells to Lapatinib. Cancer Letters 475, 53–64. [DOI] [PubMed] [Google Scholar]
- Duong MN, Cleret A, Matera EL, Chettab K, Mathe D, Valsesia-Wittmann S, … Dumontet C (2015). Adipose cells promote resistance of breast cancer cells to trastuzumab-mediated antibody-dependent cellular cytotoxicity. Breast Cancer Research 17, 57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elizalde PV, Cordo Russo RI, Chervo MF, & Schillaci R (2016). ErbB-2 nuclear function in breast cancer growth, metastasis and resistance to therapy. Endocrine-Related Cancer 23, T243–T257. [DOI] [PubMed] [Google Scholar]
- Emens LA, Esteva FJ, Beresford M, Saura C, De Laurentiis M, Skim SB, … Loi S (2019). Overall survival (OS) in KATE2, a phase II study of programmed death ligand 1 (PD-L1) inhibitor atezolizumab (atezo)+trastuzumab emtansine (T-DM1) vs placebo (pbo)+T-DM1 in previously treated HER2+ advanced breast cancer (BC). Annals of Oncology 30 (Supplement 5), v104–v142. [Google Scholar]
- Finkle D, Quan ZR, Asghari V, Kloss J, Ghaboosi N, Mai E, … Erickson S (2004). HER2-targeted therapy reduces incidence and progression of midlife mammary tumors in female murine mammary tumor virus huHER2-transgenic mice. Clinical Cancer Research 10, 2499–2511. [DOI] [PubMed] [Google Scholar]
- Franklin MC, Carey KD, Vajdos FF, Leahy DJ, de Vos AM, & Sliwkowski MX (2004). Insights into ErbB signaling from the structure of the ErbB2-pertuzumab complex. Cancer Cell 5, 317–328. [DOI] [PubMed] [Google Scholar]
- Gallardo A, Lerma E, Escuin D, Tibau A, Munoz J, Ojeda B, … Peiro G (2012). Increased signalling of EGFR and IGF1R, and deregulation of PTEN/PI3K/Akt pathway are related with trastuzumab resistance in HER2 breast carcinomas. British Journal of Cancer 106, 1367–1373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garcia-Parra J, Dalmases A, Morancho B, Arpi O, Menendez S, Sabbaghi M, … Albanell J (2014). Poly (ADP-ribose) polymerase inhibition enhances trastuzumab antitumour activity in HER2 overexpressing breast cancer. European Journal of Cancer 50, 2725–2734. [DOI] [PubMed] [Google Scholar]
- Garrett TP, McKern NM, Lou M, Elleman TC, Adams TE, Lovrecz GO, … Ward CW (2003). The crystal structure of a truncated ErbB2 ectodomain reveals an active conformation, poised to interact with other ErbB receptors. Molecular Cell 11, 495–505. [DOI] [PubMed] [Google Scholar]
- Garrett JT, Olivares MG, Rinehart C, Granja-Ingram ND, Sanchez V, Chakrabarty A, … Arteaga CL (2011). Transcriptional and posttranslational up-regulation of HER3 (ErbB3) compensates for inhibition of the HER2 tyrosine kinase. Proceedings of the National Academy of Sciences of the United States of America 108, 5021–5026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Geyer CE, Forster J, Lindquist D, Chan S, Romieu CG, Pienkowski T, … Cameron D (2006). Lapatinib plus capecitabine for HER2-positive advanced breast cancer. The New England Journal of Medicine 355, 2733–2743. [DOI] [PubMed] [Google Scholar]
- Gijsen M, King P, Perera T, Parker PJ, Harris AL, Larijani B, & Kong A (2010). HER2 phosphorylation is maintained by a PKB negative feedback loop in response to anti-HER2 herceptin in breast cancer. PLoS Biology 8, e1000563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goel S, Wang Q, Watt AC, Tolaney SM, Dillon DA, Li W, … Zhao JJ (2016). Overcoming therapeutic resistance in HER2-positive breast cancers with CDK4/6 inhibitors. Cancer Cell 29, 255–269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goyette MA, Duhamel S, Aubert L, Pelletier A, Savage P, Thibault MP, … Cote JF (2018). The receptor tyrosine kinase AXL is required at multiple steps of the metastatic cascade during HER2-positive breast cancer progression. Cell Reports 23, 1476–1490. [DOI] [PubMed] [Google Scholar]
- Gu S, Hu Z, Ngamcherdtrakul W, Castro DJ, Morry J, Reda MM, … Yantasee W (2016). Therapeutic siRNA for drug-resistant HER2-positive breast cancer. Oncotarget 7, 14727–14741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guy CT, Webster MA, Schaller M, Parsons TJ, Cardiff RD, & Muller WJ (1992). Expression of the neu protooncogene in the mammary epithelium of transgenic mice induces metastatic disease. Proceedings of the National Academy of Sciences of the United States of America 89, 10578–10582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hanker AB, Garrett JT, Estrada MV, Moore PD, Ericsson PG, Koch JP, … Arteaga CL (2017). HER2-overexpressing breast cancers amplify FGFR signaling upon acquisition of resistance to dual therapeutic blockade of HER2. Clinical Cancer Research 23, 4323–4334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hanker AB, Pfefferle AD, Balko JM, Kuba MG, Young CD, Sanchez V, … Arteaga CL (2013). Mutant PIK3CA accelerates HER2-driven transgenic mammary tumors and induces resistance to combinations of anti-HER2 therapies. Proceedings of the National Academy of Sciences of the United States of America 110, 14372–14377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holbro T, Beerli RR, Maurer F, Koziczak M, Barbas CF 3rd, & Hynes NE (2003). The ErbB2/ErbB3 heterodimer functions as an oncogenic unit: ErbB2 requires ErbB3 to drive breast tumor cell proliferation. Proceedings of the National Academy of Sciences of the United States of America 100, 8933–8938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hori A, Shimoda M, Naoi Y, Kagara N, Tanei T, Miyake T, … Noguchi S (2019). Vasculogenic mimicry is associated with trastuzumab resistance of HER2-positive breast cancer. Breast Cancer Research 21, 88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Houssami N, Macaskill P, Balleine RL, Bilous M, & Pegram MD (2011). HER2 discordance between primary breast cancer and its paired metastasis: tumor biology or test artefact? Insights through meta-analysis. Breast Cancer Research and Treatment 129, 659–674. [DOI] [PubMed] [Google Scholar]
- Jarvinen TA, Tanner M, Rantanen V, Barlund M, Borg A, Grenman S, & Isola J (2000). Amplification and deletion of topoisomerase IIalpha associate with ErbB-2 amplification and affect sensitivity to topoisomerase II inhibitor doxorubicin in breast cancer. American Journal of Pathology 156, 839–847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jensen JD, Knoop A, Laenkholm AV, Grauslund M, Jensen MB, Santoni-Rugiu E, … Ewertz M (2012). PIK3CA mutations, PTEN, and pHER2 expression and impact on outcome in HER2-positive early-stage breast cancer patients treated with adjuvant chemotherapy and trastuzumab. Annals of Oncology 23, 2034–2042. [DOI] [PubMed] [Google Scholar]
- Junttila TT, Akita RW, Parsons K, Fields C, Lewis Phillips GD, Friedman LS, … Sliwkowski MX (2009). Ligand-independent HER2/HER3/PI3K complex is disrupted by trastuzumab and is effectively inhibited by the PI3K inhibitor GDC-0941. Cancer Cell 15, 429–440. [DOI] [PubMed] [Google Scholar]
- Junttila TT, Li G, Parsons K, Phillips GL, & Sliwkowski MX (2011). Trastuzumab-DM1 (T-DM1) retains all the mechanisms of action of trastuzumab and efficiently inhibits growth of lapatinib insensitive breast cancer. Breast Cancer Research and Treatment 128, 347–356. [DOI] [PubMed] [Google Scholar]
- Kao J, Salari K, Bocanegra M, Choi YL, Girard L, Gandhi J, … Pollack JR (2009). Molecular profiling of breast cancer cell lines defines relevant tumor models and provides a resource for cancer gene discovery. PLoS One 4, e6146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaufman B, Trudeau M, Awada A, Blackwell K, Bachelot T, Salazar V, … Johnston S (2009). Lapatinib monotherapy in patients with HER2-overexpressing relapsed or refractory inflammatory breast cancer: final results and survival of the expanded HER2+ cohort in EGF103009, a phase II study. Lancet Oncology 10, 581–588. [DOI] [PubMed] [Google Scholar]
- Kennedy SP, Han JZR, Portman N, Nobis M, Hastings JF, Murphy KJ, … Croucher DR (2019). Targeting promiscuous heterodimerization overcomes innate resistance to ERBB2 dimerization inhibitors in breast cancer. Breast Cancer Research 21, 43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kirschmann DA, Seftor EA, Hardy KM, Seftor RE, & Hendrix MJ (2012). Molecular pathways: vasculogenic mimicry in tumor cells: diagnostic and therapeutic implications. Clinical Cancer Research 18, 2726–2732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kulukian A, Lee P, Taylor J, Rosler R, de Vries P, Watson D, … Peterson S (2020). Preclinical activity of HER2-selective tyrosine kinase inhibitor tucatinib as a single agent or in combination with trastuzumab or docetaxel in solid tumor models. Molecular Cancer Therapeutics 19, 976–987. [DOI] [PubMed] [Google Scholar]
- Lee-Hoeflich ST, Crocker L, Yao E, Pham T, Munroe X, Hoeflich KP, … Stern HM (2008). A central role for HER3 in HER2-amplified breast cancer: implications for targeted therapy. Cancer Research 68, 5878–5887. [DOI] [PubMed] [Google Scholar]
- Lee HJ, Seo AN, Kim EJ, Jang MH, Suh KJ, Ryu HS, … Park SY (2014). HER2 heterogeneity affects trastuzumab responses and survival in patients with HER2-positive metastatic breast cancer. American Journal of Clinical Pathology 142, 755–766. [DOI] [PubMed] [Google Scholar]
- Lewis Phillips GD, Li G, Dugger DL, Crocker LM, Parsons KL, Mai E, … Sliwkowski MX (2008). Targeting HER2-positive breast cancer with trastuzumab-DM1, an antibody-cytotoxic drug conjugate. Cancer Research 68, 9280–9290. [DOI] [PubMed] [Google Scholar]
- Li JY, Perry SR, Muniz-Medina V, Wang X, Wetzel LK, Rebelatto MC, … Coats SR (2016). A biparatopic HER2-targeting antibody-drug conjugate induces tumor regression in primary models refractory to or ineligible for HER2-targeted therapy. Cancer Cell 29, 117–129. [DOI] [PubMed] [Google Scholar]
- Loibl S, de la Pena L, Nekljudova V, Zardavas D, Michiels S, Denkert C, … Loi S (2017). Neoadjuvant buparlisib plus trastuzumab and paclitaxel for women with HER2+ primary breast cancer: A randomised, double-blind, placebo-controlled phase II trial (NeoPHOEBE). European Journal of Cancer 85, 133–145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lombardo LJ, Lee FY, Chen P, Norris D, Barrish JC, Behnia K, … Borzilleri RM (2004). Discovery of N-(2-chloro-6-methyl- phenyl)-2-(6-(4-(2-hydroxyethyl)- piperazin-1-yl)-2-methylpyrimidin-4- ylamino)thiazole-5-carboxamide (BMS-354825), a dual Src/Abl kinase inhibitor with potent antitumor activity in preclinical assays. Journal of Medicinal Chemistry 47, 6658–6661. [DOI] [PubMed] [Google Scholar]
- Lower EE, Glass E, Blau R, & Harman S (2009). HER-2/neu expression in primary and metastatic breast cancer. Breast Cancer Research and Treatment 113, 301–306. [DOI] [PubMed] [Google Scholar]
- Luhtala S, Staff S, Tanner M, & Isola J (2016). Cyclin E amplification, over-expression, and relapse-free survival in HER-2-positive primary breast cancer. Tumour Biology 37, 9813–9823. [DOI] [PubMed] [Google Scholar]
- Lupi A, Tenni R, Rossi A, Cetta G, & Forlino A (2008). Human prolidase and prolidase deficiency: an overview on the characterization of the enzyme involved in proline recycling and on the effects of its mutations. Amino Acids 35, 739–752. [DOI] [PubMed] [Google Scholar]
- Marusyk A, Tabassum DP, Janiszewska M, Place AE, Trinh A, Rozhok AI, … Polyak K (2016). Spatial proximity to fibroblasts impacts molecular features and therapeutic sensitivity of breast cancer cells influencing clinical outcomes. Cancer Research 76, 6495–6506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mercogliano MF, De Martino M, Venturutti L, Rivas MA, Proietti CJ, Inurrigarro G, … Schillaci R (2017). TNFalpha-induced mucin 4 expression elicits trastuzumab resistance in HER2-positive breast cancer. Clinical Cancer Research 23, 636–648. [DOI] [PubMed] [Google Scholar]
- Mittendorf EA, Wu Y, Scaltriti M, Meric-Bernstam F, Hunt KK, Dawood S, … Gonzalez-Angulo AM (2009). Loss of HER2 amplification following trastuzumab-based neoadjuvant systemic therapy and survival outcomes. Clinical Cancer Research 15, 7381–7388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moasser MM (2007). The oncogene HER2: its signaling and transforming functions and its role in human cancer pathogenesis. Oncogene 26, 6469–6487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Modi S, Saura C, Yamashita T, Park YH, Kim SB, Tamura K, … Investigators DE-B (2020). Trastuzumab deruxtecan in previously treated HER2-positive breast cancer. The New England Journal of Medicine 382, 610–621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mohsin SK, Weiss HL, Gutierrez MC, Chamness GC, Schiff R, Digiovanna MP, … Chang JC (2005). Neoadjuvant trastuzumab induces apoptosis in primary breast cancers. Journal of Clinical Oncology 23, 2460–2468. [DOI] [PubMed] [Google Scholar]
- Molina MA, Codony-Servat J, Albanell J, Rojo F, Arribas J, & Baselga J (2001). Trastuzumab (herceptin), a humanized anti-Her2 receptor monoclonal antibody, inhibits basal and activated Her2 ectodomain cleavage in breast cancer cells. Cancer Research 61, 4744–4749. [PubMed] [Google Scholar]
- Molina MA, Saez R, Ramsey EE, Garcia-Barchino MJ, Rojo F, Evans AJ, … Clinton GM (2002). NH(2)-terminal truncated HER-2 protein but not full-length receptor is associated with nodal metastasis in human breast cancer. Clinical Cancer Research 8, 347–353. [PubMed] [Google Scholar]
- Moulder SL, Yakes FM, Muthuswamy SK, Bianco R, Simpson JF, & Arteaga CL (2001). Epidermal growth factor receptor (HER1) tyrosine kinase inhibitor ZD1839 (Iressa) inhibits HER2/neu (erbB2)-overexpressing breast cancer cells in vitro and in vivo. Cancer Research 61, 8887–8895. [PubMed] [Google Scholar]
- Murthy RK, Loi S, Okines A, Paplomata E, Hamilton E, Hurvitz SA, … Winer EP (2020). Tucatinib, trastuzumab, and capecitabine for HER2-positive metastatic breast cancer. The New England Journal of Medicine 382, 597–609. [DOI] [PubMed] [Google Scholar]
- Musolino A, Naldi N, Bortesi B, Pezzuolo D, Capelletti M, Missale G, … Ardizzoni A (2008). Immunoglobulin G fragment C receptor polymorphisms and clinical efficacy of trastuzumab-based therapy in patients with HER-2/neu-positive metastatic breast cancer. Journal of Clinical Oncology 26, 1789–1796. [DOI] [PubMed] [Google Scholar]
- Nagata Y, Lan KH, Zhou X, Tan M, Esteva FJ, Sahin AA, … Yu D (2004). PTEN activation contributes to tumor inhibition by trastuzumab, and loss of PTEN predicts trastuzumab resistance in patients. Cancer Cell 6, 117–127. [DOI] [PubMed] [Google Scholar]
- Nagy P, Friedlander E, Tanner M, Kapanen AI, Carraway KL, Isola J, & Jovin TM (2005). Decreased accessibility and lack of activation of ErbB2 in JIMT-1, a herceptin-resistant, MUC4-expressing breast cancer cell line. Cancer Research 65, 473–482. [PubMed] [Google Scholar]
- Nahta R, Yu D, Hung MC, Hortobagyi GN, & Esteva FJ (2006). Mechanisms of disease: understanding resistance to HER2-targeted therapy in human breast cancer. Nature Clinical Practice Oncology 3, 269–280. [DOI] [PubMed] [Google Scholar]
- Nahta R, Yuan LX, Zhang B, Kobayashi R, & Esteva FJ (2005). Insulin-like growth factor-I receptor/human epidermal growth factor receptor 2 heterodimerization contributes to trastuzumab resistance of breast cancer cells. Cancer Research 65, 11118–11128. [DOI] [PubMed] [Google Scholar]
- Nami B, Maadi H, & Wang Z (2019). The effects of pertuzumab and its combination with trastuzumab on HER2 homodimerization and phosphorylation. Cancers 11, 375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Niikura N, Liu J, Hayashi N, Mittendorf EA, Gong Y, Palla SL, … Ueno NT (2012). Loss of human epidermal growth factor receptor 2 (HER2) expression in metastatic sites of HER2-overexpressing primary breast tumors. Journal of Clinical Oncology 30, 593–599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ocana A, Gil-Martin M, Antolin S, Atienza M, Montano A, Ribelles N, … Ruiz-Borrego M (2019). Efficacy and safety of dasatinib with trastuzumab and paclitaxel in first line HER2-positive metastatic breast cancer: results from the phase II GEICAM/2010–04 study. Breast Cancer Research and Treatment 174, 693–701. [DOI] [PubMed] [Google Scholar]
- Ogitani Y, Aida T, Hagihara K, Yamaguchi J, Ishii C, Harada N, … Agatsuma T (2016). DS-8201a, a novel HER2-targeting ADC with a novel DNA topoisomerase I inhibitor, demonstrates a promising antitumor efficacy with differentiation from T-DM1. Clinical Cancer Research 22, 5097–5108. [DOI] [PubMed] [Google Scholar]
- Parra-Palau JL, Morancho B, Peg V, Escorihuela M, Scaltriti M, Vicario R, … Arribas J (2014). Effect of p95HER2/611CTF on the response to trastuzumab and chemotherapy. Journal of the National Cancer Institute 106, dju291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pauletti G, Godolphin W, Press MF, & Slamon DJ (1996). Detection and quantitation of HER-2/neu gene amplification in human breast cancer archival material using fluorescence in situ hybridization. Oncogene 13, 63–72. [PubMed] [Google Scholar]
- Peiro G, Ortiz-Martinez F, Gallardo A, Perez-Balaguer A, Sanchez-Paya J, Ponce JJ, … Lerma E (2014). Src, a potential target for overcoming trastuzumab resistance in HER2-positive breast carcinoma. British Journal of Cancer 111, 689–695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perez EA, Ballman KV, Tenner KS, Thompson EA, Badve SS, Bailey H, & Baehner FL (2016). Association of stromal tumor-infiltrating lymphocytes with recurrence-free survival in the N9831 adjuvant trial in patients with early-stage HER2-positive breast cancer. JAMA Oncology 2, 56–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perez EA, Barrios C, Eiermann W, Toi M, Im YH, Conte P, … Ellis P (2017). Trastuzumab emtansine with or without pertuzumab versus trastuzumab plus taxane for human epidermal growth factor receptor 2-positive, advanced breast cancer: primary results from the phase III MARIANNE study. Journal of Clinical Oncology 35, 141–148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perez EA, Dueck AC, McCullough AE, Chen B, Geiger XJ, Jenkins RB, … Reinholz MM (2013). Impact of PTEN protein expression on benefit from adjuvant trastuzumab in early-stage human epidermal growth factor receptor 2-positive breast cancer in the North Central Cancer Treatment Group N9831 trial. Journal of Clinical Oncology 31, 2115–2122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pohlmann PR, Mayer IA, & Mernaugh R (2009). Resistance to trastuzumab in breast cancer. Clinical Cancer Research 15, 7479–7491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raina D, Uchida Y, Kharbanda A, Rajabi H, Panchamoorthy G, Jin C, … Kufe D (2014). Targeting the MUC1-C oncoprotein downregulates HER2 activation and abrogates trastuzumab resistance in breast cancer cells. Oncogene 33, 3422–3431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raja SM, Clubb RJ, Bhattacharyya M, Dimri M, Cheng H, Pan W, … Band H (2008). A combination of trastuzumab and 17-AAG induces enhanced ubiquitinylation and lysosomal pathway-dependent ErbB2 degradation and cytotoxicity in ErbB2-overexpressing breast cancer cells. Cancer Biology & Therapy 7, 1630–1640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rexer BN, & Arteaga CL (2012). Intrinsic and acquired resistance to HER2-targeted therapies in HER2 gene-amplified breast cancer: mechanisms and clinical implications. Critical Reviews in Oncogenesis 17, 1–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rius Ruiz I, Vicario R, Morancho B, Morales CB, Arenas EJ, Herter S, … Arribas J (2018). p95HER2-T cell bispecific antibody for breast cancer treatment. Science Translational Medicine 10, eaat1445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rix U, Hantschel O, Durnberger G, Remsing Rix LL, Planyavsky M, Fernbach NV, … Superti-Furga G (2007). Chemical proteomic profiles of the BCR-ABL inhibitors imatinib, nilotinib, and dasatinib reveal novel kinase and nonkinase targets. Blood 110, 4055–4063. [DOI] [PubMed] [Google Scholar]
- Roskoski R Jr. (2014). The ErbB/HER family of protein-tyrosine kinases and cancer. Pharmacological Research 79, 34–74. [DOI] [PubMed] [Google Scholar]
- Rye IH, Trinh A, Saetersdal AB, Nebdal D, Lingjaerde OC, Almendro V, … Russnes HG (2018). Intratumor heterogeneity defines treatment-resistant HER2+ breast tumors. Molecular Oncology 12, 1838–1855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salgado R, Denkert C, Campbell C, Savas P, Nuciforo P, Aura C, … Loi S (2015). Tumor-infiltrating lymphocytes and associations with pathological complete response and event-free survival in HER2-positive early-stage breast cancer treated with lapatinib and trastuzumab: a secondary analysis of the NeoALTTO trial. JAMA Oncology 1, 448–454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saura C, Oliveira M, Feng YH, Dai MS, Chen SW, Hurvitz SA, … for the NALA Investigators. (2020). Neratinib plus capecitabine versus lapatinib plus capecitabine in HER2-positive metastatic breast cancer previously treated with >/= 2 HER2-directed regimens: phase III NALA rrial. Journal of Clinical Oncology 38, doi: 10.1200/JCO.20.00147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scaltriti M, Eichhorn PJ, Cortes J, Prudkin L, Aura C, Jimenez J, … Baselga J (2011). Cyclin E amplification/overexpression is a mechanism of trastuzumab resistance in HER2+ breast cancer patients. Proceedings of the National Academy of Sciences of the United States of America 108, 3761–3766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scaltriti M, Rojo F, Ocana A, Anido J, Guzman M, Cortes J, … Baselga J (2007). Expression of p95HER2, a truncated form of the HER2 receptor, and response to anti-HER2 therapies in breast cancer. Journal of the National Cancer Institute 99, 628–638. [DOI] [PubMed] [Google Scholar]
- Shattuck DL, Miller JK, Carraway KL 3rd, & Sweeney C (2008). Met receptor contributes to trastuzumab resistance of Her2-overexpressing breast cancer cells. Cancer Research 68, 1471–1477. [DOI] [PubMed] [Google Scholar]
- Shi Z, Graber ZT, Baumgart T, Stone HA, & Cohen AE (2018). Cell membranes resist flow. Cell 175, 1769–1779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi F, Telesco SE, Liu Y, Radhakrishnan R, & Lemmon MA (2010). ErbB3/HER3 intracellular domain is competent to bind ATP and catalyze autophosphorylation. Proceedings of the National Academy of Sciences of the United States of America 107, 7692–7697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Slamon DJ, Clark GM, Wong SG, Levin WJ, Ullrich A, & McGuire WL (1987). Human breast cancer: correlation of relapse and survival with amplification of the HER-2/neu oncogene. Science 235, 177–182. [DOI] [PubMed] [Google Scholar]
- Slamon DJ, Leyland-Jones B, Shak S, Fuchs H, Paton V, Bajamonde A, … Norton L (2001). Use of chemotherapy plus a monoclonal antibody against HER2 for metastatic breast cancer that overexpresses HER2. The New England Journal of Medicine 344, 783–792. [DOI] [PubMed] [Google Scholar]
- Sperinde J, Jin X, Banerjee J, Penuel E, Saha A, Diedrich G, … Lipton A (2010). Quantitation of p95HER2 in paraffin sections by using a p95-specific antibody and correlation with outcome in a cohort of trastuzumab-treated breast cancer patients. Clinical Cancer Research 16, 4226–4235. [DOI] [PubMed] [Google Scholar]
- Surazynski A, Donald SP, Cooper SK, Whiteside MA, Salnikow K, Liu Y, & Phang JM (2008). Extracellular matrix and HIF-1 signaling: the role of prolidase. International Journal of Cancer 122, 1435–1440. [DOI] [PubMed] [Google Scholar]
- Tandon AK, Clark GM, Chamness GC, Ullrich A, & McGuire WL (1989). HER-2/neu oncogene protein and prognosis in breast cancer. Journal of Clinical Oncology 7, 1120–1128. [DOI] [PubMed] [Google Scholar]
- Tanner M, Kapanen AI, Junttila T, Raheem O, Grenman S, Elo J, … Isola J (2004). Characterization of a novel cell line established from a patient with Herceptin-resistant breast cancer. Molecular Cancer Therapeutics 3, 1585–1592. [PubMed] [Google Scholar]
- Tolaney SM, Wardley AM, Zambelli S, Hilton JF, Troso-Sandoval TA, Ricci F, … Andre F (2020). Abemaciclib plus trastuzumab with or without fulvestrant versus trastuzumab plus standard-of-care chemotherapy in women with hormone receptor-positive, HER2-positive advanced breast cancer (monarcHER): a randomised, open-label, phase 2 trial. Lancet Oncology 21, 763–775. [DOI] [PubMed] [Google Scholar]
- Toth G, Szoor A, Simon L, Yarden Y, Szollosi J, & Vereb G (2016). The combination of trastuzumab and pertuzumab administered at approved doses may delay development of trastuzumab resistance by additively enhancing antibody-dependent cell-mediated cytotoxicity. MAbs 8, 1361–1370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tovey SM, Brown S, Doughty JC, Mallon EA, Cooke TG, & Edwards J (2009). Poor survival outcomes in HER2-positive breast cancer patients with low-grade, node-negative tumours. British Journal of Cancer 100, 680–683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsao LC, Crosby EJ, Trotter TN, Agarwal P, Hwang BJ, Acharya C, … Hartman ZC (2019). CD47 blockade augmentation of trastuzumab antitumor efficacy dependent on antibody-dependent cellular phagocytosis. JCI Insight 4, e131882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsutsui S, Ohno S, Murakami S, Kataoka A, Kinoshita J, & Hachitanda Y (2003). Prognostic value of the combination of epidermal growth factor receptor and c-erbB-2 in breast cancer. Surgery 133, 219–221. [DOI] [PubMed] [Google Scholar]
- Turner NH, & Di Leo A (2013). HER2 discordance between primary and metastatic breast cancer: assessing the clinical impact. Cancer Treatment Reviews 39, 947–957. [DOI] [PubMed] [Google Scholar]
- Tzahar E, Waterman H, Chen X, Levkowitz G, Karunagaran D, Lavi S, … Yarden Y (1996). A hierarchical network of interreceptor interactions determines signal transduction by Neu differentiation factor/neuregulin and epidermal growth factor. Molecular and Cellular Biology 16, 5276–5287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Verma S, Miles D, Gianni L, Krop IE, Welslau M, Baselga J, … Group ES (2012). Trastuzumab emtansine for HER2-positive advanced breast cancer. The New England Journal of Medicine 367, 1783–1791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vicario R, Peg V, Morancho B, Zacarias-Fluck M, Zhang J, Martinez-Barriocanal A, … Arribas J (2015). Patterns of HER2 gene amplification and response to anti-HER2 therapies. PLoS One 10, e0129876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vogel CL, Cobleigh MA, Tripathy D, Gutheil JC, Harris LN, Fehrenbacher L, … Press M (2002). Efficacy and safety of trastuzumab as a single agent in first-line treatment of HER2-overexpressing metastatic breast cancer. Journal of Clinical Oncology 20, 719–726. [DOI] [PubMed] [Google Scholar]
- Wang Y, Liu Y, Du Y, Yin W, & Lu J (2013). The predictive role of phosphatase and tensin homolog (PTEN) loss, phosphoinositol-3 (PI3) kinase (PIK3CA) mutation, and PI3K pathway activation in sensitivity to trastuzumab in HER2-positive breast cancer: a meta-analysis. Current Medical Research and Opinion 29, 633–642. [DOI] [PubMed] [Google Scholar]
- Watanabe S, Yonesaka K, Tanizaki J, Nonagase Y, Takegawa N, Haratani K, … Nakagawa K (2019). Targeting of the HER2/HER3 signaling axis overcomes ligand-mediated resistance to trastuzumab in HER2-positive breast cancer. Cancer Medicine 8, 1258–1268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wielgos ME, Zhang Z, Rajbhandari R, Cooper TS, Zeng L, Forero A, … Yang ES (2018). Trastuzumab-resistant HER2(+) breast cancer cells retain sensitivity to poly (ADP-ribose) polymerase (PARP) inhibition. Molecular Cancer Therapeutics 17, 921–930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wissner A, & Mansour TS (2008). The development of HKI-272 and related compounds for the treatment of cancer. Arch Pharm Chemistry in Life Science 341, 465–477. [DOI] [PubMed] [Google Scholar]
- Witton CJ, Reeves JR, Going JJ, Cooke TG, & Bartlett JM (2003). Expression of the HER1–4 family of receptor tyrosine kinases in breast cancer. The Journal of Pathology 200, 290–297. [DOI] [PubMed] [Google Scholar]
- Wolff AC, Hammond ME, Schwartz JN, Hagerty KL, Allred DC, Cote RJ, … American Society of Clinical Oncology/College of American Pathologists. (2007). American Society of Clinical Oncology/College of American Pathologists guideline recommendations for human epidermal growth factor receptor 2 testing in breast cancer. Archives of Pathology & Laboratory Medicine 131, 18–43. [DOI] [PubMed] [Google Scholar]
- Wood ER, Truesdale AT, McDonald OB, Yuan D, Hassell A, Dickerson SH, … Shewchuk L (2004). A unique structure for epidermal growth factor receptor bound to GW572016 (Lapatinib): relationships among protein conformation, inhibitor off-rate, and receptor activity in tumor cells. Cancer Research 64, 6652–6659. [DOI] [PubMed] [Google Scholar]
- Yamashita-Kashima Y, Shu S, Yorozu K, Moriya Y, & Harada N (2017). Mode of action of pertuzumab in combination with trastuzumab plus docetaxel therapy in a HER2-positive breast cancer xenograft model. Oncology Letters 14, 4197–4205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang L, Li Y, Ding Y, Choi KS, Kazim AL, & Zhang Y (2013). Prolidase directly binds and activates epidermal growth factor receptor and stimulates downstream signaling. The Journal of Biological Chemistry 288, 2365–2375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang L, Li Y, & Zhang Y (2014). Identification of prolidase as a high affinity ligand of the ErbB2 receptor and its regulation of ErbB2 signaling and cell growth. Cell Death and Disease 5, e1211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang L, Li Y, Bhattacharya A, & Zhang Y (2015). Inhibition of ERBB2-overexpressing tumors by recombinant human prolidase and its enzymatically inactive mutant. EBioMedicine, 2, 396–405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang L, Li Y, Bhattacharya A, & Zhang Y (2016a). Dual inhibition of ErbB1 and ErbB2 in cancer by recombinant human prolidase mutant hPEPD-G278D. Oncotarget 7, 42340–42352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang L, Li Y, Bhattacharya A, & Zhang Y (2016b). A plasma proteolysis pathway comprising blood coagulation proteases. Oncotarget 7, 40919–40938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang L, Li Y, Bhattacharya A, & Zhang Y (2019). A recombinant human protein targeting HER2 overcomes drug resistance in HER2-positive breast cancer. Science Translational Medicine 11, eaav1620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yao E, Zhou W, Lee-Hoeflich ST, Truong T, Haverty PM, Eastham-Anderson J, … Hoeflich KP (2009). Suppression of HER2/HER3-mediated growth of breast cancer cells with combinations of GDC-0941 PI3K inhibitor, trastuzumab, and pertuzumab. Clinical Cancer Research 15, 4147–4156. [DOI] [PubMed] [Google Scholar]
- Yarden Y, & Sliwkowski MX (2001). Untangling the ErbB signalling network. Nature Reviews Molecular Cell Biology 2, 127–137. [DOI] [PubMed] [Google Scholar]
- Yu Q, Geng Y, & Sicinski P (2001). Specific protection against breast cancers by cyclin D1 ablation. Nature, 411, 1017–1021. [DOI] [PubMed] [Google Scholar]
- Yuzawa S, Opatowsky Y, Zhang Z, Mandiyan V, Lax I, & Schlessinger J (2007). Structural basis for activation of the receptor tyrosine kinase KIT by stem cell factor. Cell 130, 323–334. [DOI] [PubMed] [Google Scholar]
- Zervantonakis IK, Poskus MD, Scott AL, Selfors LM, Lin JR, Dillon DA, … Brugge JS (2020). Fibroblast-tumor cell signaling limits HER2 kinase therapy response via activation of MTOR and antiapoptotic pathways. Proceedings of the National Academy of Sciences of the United States of America 117, 16500–16508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang S, Huang WC, Li P, Guo H, Poh SB, Brady SW, … Yu D (2011). Combating trastuzumab resistance by targeting SRC, a common node downstream of multiple resistance pathways. Nature Medicine 17, 461–469. [DOI] [PMC free article] [PubMed] [Google Scholar]