

Abstract
Background
Androgen deprivation therapies for the hormone-dependent stages of prostate cancer have become so effective that new forms of chemoresistant tumors are emerging in clinical practice, and require new targeted therapies in the metastatic setting. Yet there are important gaps in our understanding of the relevant transcriptional networks driving this process. Progression from localized to metastatic castration resistant prostate cancer (mCRPC) occurs as a result of accumulated resistance mechanisms that develop upon sustained androgen receptor (AR) suppression. Critical to this progression is the plastic nature by which prostate tumor cells transition from epithelial to mesenchymal states (EMT).
Methods
Here, using prostate cancer cell lines with different AR composition, we systematically manipulated somatic proteins of the Bromodomain and ExtraTerminal (BET) family (BRD2, BRD3 and BRD4) to determine which BET proteins influence EMT. We used the TCGA repository to correlate the expression of individual BET genes with key EMT genes and determined biochemical recurrence in 414 patients and progression free survival in 488 patients.
Results
We found that only BRD4 – and not BRD2 or BRD3 – regulates the expression of SNAI1 and SNAI2, and that the downregulation of these EMT transcription factors significantly increases E-cadherin expression. Furthermore, of the BET genes, only BRD4 correlates with survival outcomes in prostate cancer patients. Moreover, selective degradation of BRD4 protein with MZ1 ablates EMT (transcriptionally and morphologically) induced by TGFß signaling.
Conclusions
Many relapsed/refractory tumors share a neuroendocrine transcriptional signature that had been relatively rare until highly successful antiandrogen drugs like abiraterone and enzalutamide came into widespread use. New therapeutic targets must therefore be developed. Our results identify key EMT genes regulated by BRD4, and offers a novel druggable target to treat mCRPC. BRD4-selective protein degraders offer a promising next generation approach to treat the emerging forms of chemoresistance in advanced prostate cancer.
Keywords: bromodomain, epithelial-mesenchymal transition, epithelial plasticity, prostate cancer, castration-resistant, epigenetic regulators, BET proteins, next-generation BET degrader, BRD4, Snail, Slug, E-cadherin
Introduction
Prostate cancer is the most commonly diagnosed cancer in men and over 90% of prostate cancer-associated mortality is caused by the development of metastases (1,2). Second generation anti-androgen therapies abiraterone and enzalutamide (ADT) remain the standard of care for men with advanced prostate cancer and are initially quite effective in suppressing both androgen production and androgen receptor (AR) signaling, yet disease progression to castration-resistance prostate cancer (CRPC) remains inevitable (2,3). While resistance mechanisms that include AR gene amplification, gain-of-function AR point mutations and complete loss of AR expression are critical for prostate tumor cells to overcome sustained AR suppression, emerging evidence suggests that the transition from an epithelial to mesenchymal state is an additional adaptive evolutionary response that creates an environment suitable for therapeutic resistance (4).
The epithelial-mesenchymal transition (EMT) is a normal biological process active during embryogenesis that is often reactivated in cancer cells (2,5,6). Upon induction, non-motile epithelial cells break away from intracellular tight junctions and acquire a mesenchymal phenotype that creates a highly mobile and invasive cell (2,5,6). On a molecular level, EMT is initiated by several families of transcription factors, including SNAI1 (Snail), SNAI2 (Slug) and ZEB1/2, which repress the expression of E-cadherin (CDH1) and other epithelial genes by docking to several E-boxes in the promoter region (2,5,6). Recent studies demonstrated that derepression of Snail is an adaptive response to AR inhibition and is a critical resistance mechanism in CRPC (4,7). Therefore, the discovery of druggable targets that work by suppressing EMT mediators such as Snail, irrespective of AR status, are highly desirable.
The Bromodomain and ExtraTerminal (BET) family of proteins (BRD2, BRD3, BRD4 and testis-specific BRDT), are epigenetic readers that bind to specific acetylated lysine residues in histones as a means to regulate gene transcription (8). Having been identified as key contributors to the progression of B-cell lymphoma, lung, breast, pancreatic and prostate cancers, intense efforts have been spent towards developing multiple small molecule pan-BET inhibitors and degraders (8). While pan-BET therapies such as JQ1 and ARV-771 effectively downregulate the proto-oncogene c-Myc in variety of cancer models, including CRPC (9–11), evidence continues to suggest that the efficacy for a pan-BET approach is dependent upon the context and the cancer. As we have shown in multiple reports in a variety of breast and prostate cancer models, BET proteins modulate distinct signaling pathways, including those that regulate EMT, and can even have non-overlapping and opposing functions (12–14). Here, we deepen and extend findings in the field to show that only BRD4, and not BRD2 or BRD3, regulates CRPC dissemination, and to show that BRD4 regulates genes critical for the induction of EMT in CRPC, which can be ablated with a BRD4-selective protein degradation approach.
Materials and Methods
Cell Culture
22Rv1 and DU 145 prostate cancer cell lines were cultured in RPMI-1640 medium (Gibco). VCaP prostate cancer cells were cultured in DMEM medium (Gibco). All culture media were supplemented with 10% fetal bovine serum (FBS, Corning) and 1% antibiotics (penicillin/streptomycin, Gibco). Additional information is available in a Supplementary File.
Antibodies and Reagents
The following antibodies were used: anti-BRD2, BRD3 and BRD4 (Bethyl Laboratories), anti-Snail, anti-Slug, anti-E-cadherin, anti-Smad3, anti-β-Actin (Cell Signaling) and anti-phospho-Smad3 (Abcam). Additional information is available in a Supplementary File.
Plasmids, siRNAs and Transfection
Lentivirus-mediated eGFP-BRD4 expressing plasmid (EX-E0102-Lv122) and control vector (EX-NEG-Lv242) were purchased from GeneCopoeia. ON-Targetplus Human BET and Non-Targeting (scramble) SMARTpool siRNAs were purchased from Dharmacon. Additional information is available in a Supplementary File.
Immunoblotting
Cell pellets were lysed in RIPA buffer (50 mmol/L Tris/HCl pH 7.5, 1 mmol/L EDTA, 0.5 mmol/L EGTA, 150 mmol/L NaCl, 0.1% sodium deoxycholate, 0.1% SDS, 1% Triton X-100). Samples containing 25 μg of protein were resolved by SDS-PAGE and transferred to nitrocellulose membranes. Additional information is available in a Supplementary File.
qRT-PCR
Total RNA was extracted using the RNAeasy Kit (Qiagen). Reverse transcription reactions were performed with 1 μg of total RNA with the QuantiTect Reverse Transcription kit (Qiagen). Primer sequences are described in a Supplementary File.
Chromatin Immunoprecipitation
DU 145 cells were treated with 400 nM of either (−)JQ1 or (+)JQ1 for 24 hours, fixed in 0.75% formaldehyde for 10 minutes, quenched with 125 mM glycine for 5 minutes and then lysed for chromatin immunoprecipitation (ChIP) as previously reported (14,15). Additional information and primer sequences are described in a Supplementary File.
The ChIP-seq dataset GSE55062 (16) was taken from the NCBI Gene Expression Omnibus database and visualized using Integrative Genomics Viewer (IGV) (17).
Immunocytochemistry
Cells were fixed in absolute methanol for 5 minutes at - 20°C and then permeabilized with PBS, 0.2% Triton X-100 buffer for 10 minutes. After saturation in blocking buffer (0.02% Triton X-100, 2% BSA in PBS) for 30 minutes, permeabilized cells were incubated with primary antibodies and then fluorochrome-conjugated secondary antibodies (diluted in blocking buffer) for 1 hour. Additional information is available in a Supplementary File.
Flow Cytometry
Details are available in a Supplementary File and in Supplementary Table S1.
Kaplan-Meier Analysis
To investigate the correlation between biochemical recurrence-free survival (BCR), progression free survival (PFS) and BRD4, SNAI1, SNAI2 and CDH1 expression, we utilized data from 414 (BCR) and 488 (PFS) prostate cancer patients from the TCGA repository. The normalized expression values of the RNAseq ID 23476 (for BRD4), 6615 (for SNAI1), 6591 (for SNAI2) and 999 (for CDH1) were used. For each gene, we first computed the mean expression and this value was used in the survival analysis. To maximize the sensitivity to detect correlation to survival, each cutoff value between the lower and upper quartiles of expression were evaluated as described previously (14,18). Additional information is available in a Supplementary File. The gene expression data and survival times are listed in Supplementary Tables S2 and S3.
Statistical Analysis
Statistical analyses of the in vitro experiments were performed using Student’s t test or ANOVA as indicated, and were generated by GraphPad Prism software. p < 0.05 was considered statistically significant.
Results
BRD4 regulates transcription factors critical for EMT
We previously established that BRD4 plays a significant role in the regulation of CRPC cell migration and invasion (14). As part of that work we profiled 73 genes involved in pathways important for EMT, and discovered that knockdown of BRD4 significantly downregulated the expression of AHNAK (14). In addition to AHNAK, the findings also suggested that multiple transcription factors known to facilitate transcriptional programs critical for EMT, including members of the Snail family (SNAI1/Snail and SNAI2/Slug)c, were under BET-protein control (14). To investigate whether BET-proteins regulate Snail and Slug expression, we first treated 22Rv1 (AR-H874Y) and DU 145 (AR-null) and cells for 24 hours with the pan-BET inhibitor JQ1 (Fig. 1A–B). We found that JQ1 treatment led to the significant downregulation of both Snail and Slug. Interestingly, we also found that pan-BET inhibition caused a slight reduction in the expression of E-cadherin in both cell lines. Snail and Slug are zinc-finger transcription factors and work to induce EMT by repressing the expression of adhesion molecules like E-cadherin (2,5,6). Therefore, we considered that pan-BET inhibition may obscure the influence that each BET protein has on regulating EMT. To resolve this, we measured the mRNA and protein expression of Snail, Slug and E-cadherin upon depletion of either BRD2, BRD3 and BRD4 in 22Rv1, DU 145 and VCaP (AR-WT/Amp) cells (Fig. 1C–D and Supplementary Fig. S1A–C). In all three cell lines, the knockdown of only BRD4 – and not of BRD2 or BRD3 – significantly reduced the expression of Snail and Slug at both the mRNA and protein level. Intriguingly, E-cadherin expression was significantly increased at the mRNA (VCaP and DU 145) and protein level (DU 145) upon BRD4 depletion, yet only marginally increased under the same conditions in 22Rv1 cells.
Figure 1. BRD4 regulates transcription factors critical for EMT.
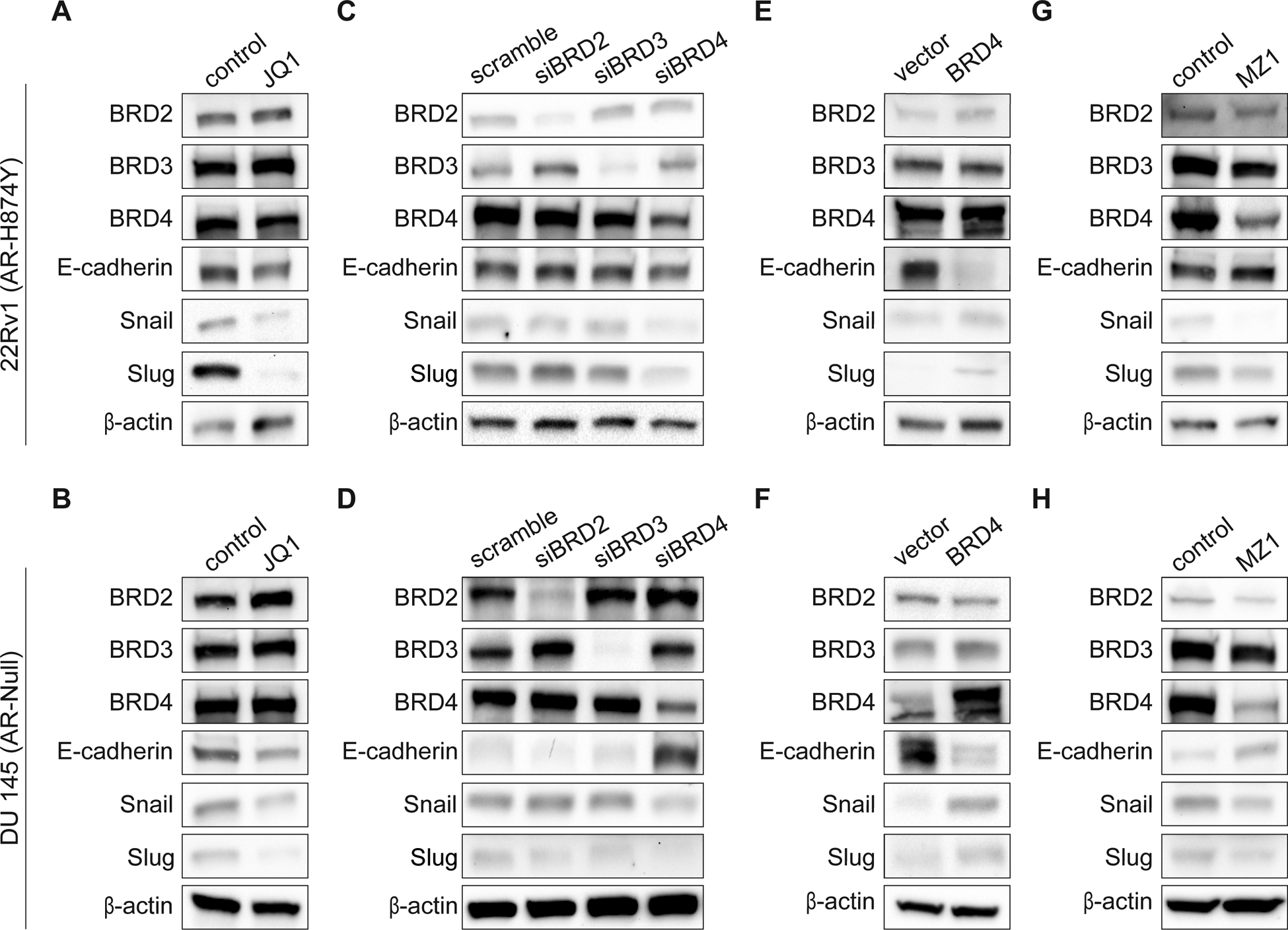
(A-B), Protein expression of BET proteins: BRD2, BRD3 and BRD4, EMT transcription factors: Snail and Slug and EMT marker: E-cadherin in 22Rv1 and DU 145 cells after being treated with either 400 nM of (–)JQ1 or (+)JQ1 for 24 hours. Blots shown are representative of two independent experiments.
(C-D), Validation of BET depletion by siRNA (25 nmol/L for 72 hours) and protein expression of Snail, Slug and E-cadherin in 22Rv1 and DU 145 cells. Blots shown are representative of three independent experiments.
(E-F), Validation of BRD4 overexpression and protein expression of Snail, Slug and E-cadherin in 22Rv1 and DU 145 cells. Blots shown are representative of three independent experiments.
(G-H), Protein expression of BET proteins: BRD2, BRD3 and BRD4, EMT transcription factors: Snail and Slug and EMT marker: E-cadherin in 22Rv1 or DU 145 cells after being treated with either 0.01% DMSO (control), 10 nM MZ1 (22Rv1) or 100 nM MZ1 (DU 145) for 24 hours. Blots shown are representative of three independent experiments.
To further determine whether BRD4 regulates this EMT gene set, we overexpressed BRD4 in 22Rv1 and DU 145 cells (Fig. 1E–F). Importantly, we found that overexpression of BRD4 in each cell line dramatically increased the expression of Snail and Slug and likewise significantly reduced the expression of E-cadherin (Fig. 1E–F and Supplementary Fig. S2A and S2B). These data support the idea that BRD4 is responsible for driving EMT in CRPC, and therefore any approach that involves targeting BET proteins as a means to repress EMT should focus on only BRD4. Thus, we tested whether MZ1, a BRD4-selective degrader built on Proteolysis Targeted Chimera (PROTAC) technology (19), could repress Snail and Slug while enhance E-cadherin expression (Fig. 1G–H). As predicted, treatment of 22Rv1 and DU 145 cells with a BRD4-selective doses of MZ1 for 24 hours reduced the expression of both Snail and Slug and modestly increased the expression of E-cadherin. Collectively, these results identify BRD4, and not BRD2 or BRD3, as a key transcriptional regulator of EMT, and once again illustrates how the use of pan-BET inhibitors can obscure BET protein functionality.
Selective degradation of BRD4 represses TGFß induced EMT
The induction of EMT in tumor cells is thought to primarily occur through the uptake of secreted soluble factors from nearby stromal cells leading to the activation of tumor cell signaling pathways/EMT transcriptional mediators (2,5,6). Transforming growth factor ß (TGFß) is a multifunctional cytokine and a strong promoter of EMT and metastases in advanced prostate cancer (2,20). To determine whether BRD4 maintains its ability to regulate EMT in the presence of TGFß, we measured the expression of Snail and Slug in 22Rv1 and DU 145 cells treated with either TGFß, MZ1 or MZ1 + TGFß (Fig. 2A–B). Cells stimulated with TGFß for 24 hours showed a significant increase in Snail and Slug expression. More importantly, loss of BRD4 as a result of MZ1 treatment prevented the increase of Snail and Slug that was observed upon TGFß stimulation. Next, we immunostained both cell lines to measure E-cadherin and detect any morphological/phenotypical changes under the aforementioned conditions (Fig. 2C–D). Both 22Rv1 and DU 145 cells are characterized as epithelial and cuboidal in shape (21), and when grown as a monolayer they express moderate levels of E-cadherin at tight junctions as shown in the control cells. As expected, TGFß treated cells had reduced expression of E-cadherin, coupled with a flatter, more spindle-like morphology consistent with a mesenchymal phenotype. Alternatively, cells treated with MZ1 had a significant increase of E-cadherin expression along tight junctions, as observed by their enhanced cuboidal shape. Critically, cells treated with both MZ1 and TGFß maintained their cuboidal shape, indicating that the loss of BRD4 conserves the expression of E-cadherin even in the presence of TGFß. Taken together, these results indicate that degradation of BRD4 successfully prevents the transition of prostate cancer cells from an epithelial state to a mesenchymal state.
Figure 2. Selective degradation of BRD4 represses TGFß induced EMT.
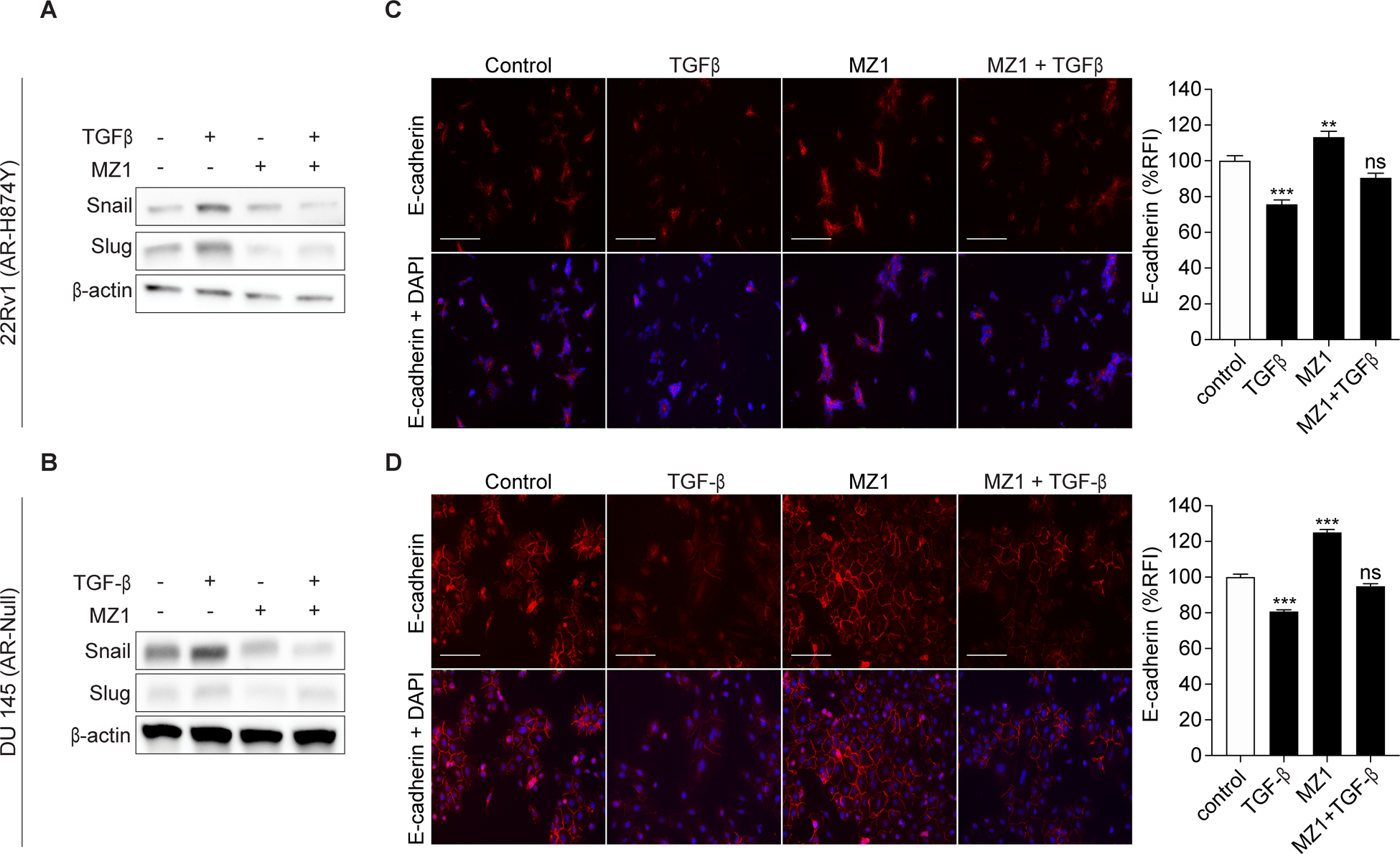
(A-B), Protein expression of Snail and Slug in 22Rv1 and DU 145 cells after being treated with either 0.01% DMSO, 5 ng/mL TGFß, 10 or 100 nM MZ1 or 5 ng/mL TGFß + 10 or 100 nM MZ1 for 24 hours. Blots shown are representative of three independent experiments.
(C-D), Immunofluorescence images showing the expression of E-cadherin in 22Rv1 and DU 145 cells after being treated with either 0.01% DMSO, 5 ng/mL TGFß, 10 or 100 nM MZ1 or 5 ng/mL TGFß + 10 or 100 nM MZ1 for 24 hours. E-cadherin is stained in red and nuclei is stained in blue with DAPI in the merged images (lower panels). Scale bar, 100 μm. Quantification of E-cadherin immunofluorescence reflected as a percentage of relative fluorescence intensity (%RFI). Bar represents means ± SEM of individual cells (n ≥ 100 (22Rv1) or 350 (DU 145) in all conditions). Results from two independent experiments are shown. Statistical analyses were performed using a one-way ANOVA. Significant differences: ns, nonsignificant, P > 0.05; ***, p < 0.001.
BRD4 mediates SNAI1 and SNAI2 expression through promoter interactions
We next considered that BRD4 may regulate SNAI1 and SNAI2 expression at the receptor level by mediating the activation of Smad3. Likewise, SNAI1 and SNAI2 have previously been shown to be targets of EMT induced through TGFß-Smad3 signaling in prostate cancer (5,22). To test this hypothesis, we measured the phosphorylation of Smad3 in DU 145 cells treated with MZ1 and dosed with TGFß (Fig. 3A). Degradation of BRD4 had no effect on Smad3 phosphorylation at either 30 minutes or 1 hour of TGFß stimulation compared to control cells, implying that the regulation exerted by BRD4 occurs directly at the promoter regions of SNAI1 and SNAI2. To address this, we determined if BRD4 associates with the SNAI1 and SNAI2 promoters by ChIP in DU 145 cells (23,24). We found that BRD4 engages with both the SNAI1 and SNAI2 promoters, and treatment with (+)JQ1 disrupts these interactions (Fig. 3B). Furthermore, we also probed a public repository for ChIP-sequencing data on BRD4 in VCaP cells (GSE55062) (16). BRD4 was detected with RNA Pol II at the promoters for SNAI1 and SNAI2, and notably was displaced upon JQ1 treatment (Fig. 3C). Altogether, these results confirm that BRD4 mediates SNAI1 and SNAI2 expression at the transcriptional level.
Figure 3. BRD4 mediates SNAI1 and SNAI2 expression through promoter interactions.
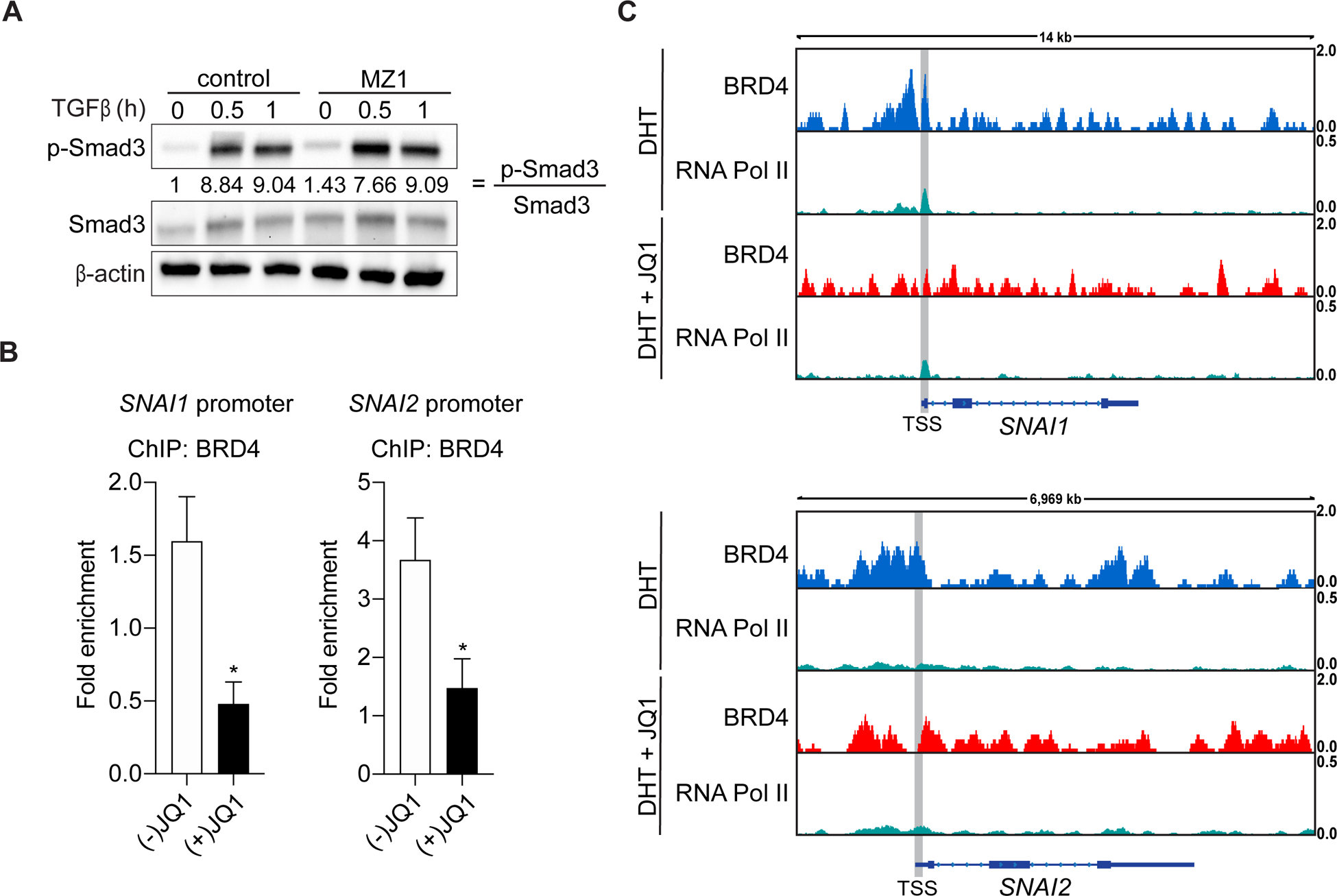
A, Immunoblot of phospho-Smad3 (pSmad3) and Smad3 in DU145 cells treated with either 0.01% DMSO or 100 nM MZ1 for 21 hours, serum starved for 2 hours under the same conditions and then dosed with 5 ng/mL TGFß for the indicated time. The ratio of pSmad3:Smad3 illustrates Smad3 activation and quantifications are relative to control. Blots shown are representative of two independent experiments.
B, DU 145 cells treated with either 400 nM of (–)JQ1 or (+)JQ1 for 24 hours and then harvested for ChIP. BRD4 interacts with the SNAI1 and SNAI2 promoters and is displaced upon exposure to (+)JQ1. Results from two independent experiments are shown. Statistical analyses were performed using the Student’s t test. Significant differences: ns, nonsignificant, P > 0.05; *.
C, Publicly available ChIP-seq datasets performed on DHT or DHT + JQ1-treated VCaP cells were analyzed for the binding of BRD4 and RNA Pol II at the SNAI1 and SNAI2 locus. The analyzed datasets are accessible on the GEO platform (GSE55062 (16)). Data visualization was performed using Integrative Genomics Viewer (IGV).
BRD4 expression correlates with SNAI1 and survival outcomes in prostate cancer patients
The individual expression of BRD4, Snail, Slug and E-cadherin have all previously been shown to independently associate with prostate cancer disease states and patient outcomes (25–27). Since we determined that BRD4, and not BRD2 or BRD3 expression correlates with SNAI1, SNAI2 and CDH1 in our cell lines, we considered whether these genes would associate in the same manner in prostate cancer patients and likewise influence survival outcomes. Using the TCGA repository, we compared the expression of BRD2, BRD3 and BRD4 to SNAI1 and found that only BRD4 had a positive correlation with the gene (Fig. 3A (28)). Next, we performed a meta-analysis of 414 (BCR) and 488 (PFS) patients with adenocarcinoma of the prostate, and determined that BRD4, SNAI1, SNAI2 and CDH1 expression correlates with BCR-free survival and Progression-free survival (Fig. 3B). High expression of BRD4, SNAI1, SNAI2 and low expression of CDH1 significantly associated with a shorter time to BCR in prostate cancer patients (HR = 1.72, log-rank p = 0.047). Likewise, the same expression profile correlated with time to PFS (HR = 1.95, log-rank p = 0.0019). Notably, when substituting either BRD2 or BRD3 for BRD4, or when removing BRD4 from this gene set, the expression profile no longer significantly associated with time to BCR (Supplementary Fig. S3). Overall, these results demonstrate that the expression of BRD4, but not BRD2 or BRD3, significantly correlates with this EMT gene set and dramatically influences survival outcomes in prostate cancer patients.
Discussion
Our results show that BRD4, and not BRD2 or BRD3, positively regulates key transcriptional mediators of EMT in multiple prostate cancer cell lines of varying AR composition (Fig. 1 and Supplementary Fig. S1–S2). These findings build upon and support our previous work which showed that selective targeting of BRD4 dramatically impedes prostate cancer cell migration and invasion (14). The discoveries outlined throughout this study were realized only after knocking down each BET family member, as treatment of prostate cancer cells with JQ1 revealed a critically different EMT expression profile (Fig. 1A–D). Whereas silencing or overexpressing BRD4 reduced/enhanced the expression of Snail and Slug, and conversely enhanced/reduced the expression of E-cadherin, JQ1 consistently downregulated the expression of all three genes (Fig. 1A–F). These results imply that the collective inhibition of BRD2, BRD3 and BRD4 turns on or off multiple off-target transcriptional networks, given that multiple studies have previously shown that E-cadherin expression is inversely correlated with Snail and Slug expression (2,4,5). This concept was confirmed when treating cells with MZ1 produced a comparable EMT expression profile to cells that were treated with siRNA specific to BRD4 (Fig. 1G–H).
Because MZ1 phenocopied the EMT expression profile that was generated when using siRNA against BRD4, it was important to determine whether MZ1 could prevent prostate cancer cells from transitioning to a mesenchymal state when exposed to conditions that mimicked the tumor microenvironment (Fig. 2) (2,20). Remarkably, MZ1 prevented Snail and Slug expression from increasing in TGFß stimulated prostate cancer cells (Fig. 2A–B). By limiting the expression of Snail and Slug under TGFß stimulation, prostate cancer cell architecture remained undisturbed. Evidence to support this finding was robust, as E-cadherin expression remained strong along tight junctions when exposed to MZ1 + TGFß (Fig. 2C–D). We confirmed that the regulation of Snail and Slug by BRD4 occurs at the transcriptional level, as ChIP analysis showed BRD4 localization at both the SNAI1 and SNAI2 promoters (Fig. 3B–C). Moreover, the loss of BRD4 did not impact Smad3 activation (Fig. 3A). These results were unexpected given that we previously showed that BRD4 directly regulates AHNAK, and AHNAK has been shown to serve as a necessary scaffolding protein for TGFß-Smad3 phosphorylation (29). Therefore, we can now say that the loss of BRD4 disengages key transcriptional machinery at BRD4-specific promoters, many of which directly influence EMT, including SNAI1, SNAI2 and AHNAK. This disruption enhances the expression of certain epithelial markers such as E-cadherin, which shifts prostate cancer cells into an elevated epithelial state and severely impairs cell motility and invasiveness (14).
Finally, we also show that only BRD4 positively correlates with SNAI1 as well as survival outcomes in patients with advanced prostate cancer (Fig. 4A–B and Supplementary Fig. S3). The BRD4-SNAI1 relationship is critical for EMT and metastasis across diverse malignancies (30), as recently reported for gastric (31) and lung cancers (32). Our findings illustrate once again the paramount significance of BRD4, compared to BRD2 and BRD3, in the context of prostate cancer progression. Yet, the overwhelming majority of BET protein therapeutic agents in clinical trials, including studies that have mCRPC patients, are pan-BET inhibitors (33). Considering the functional opposition of BRD2 and BRD4 in other transcriptional contexts important for EMT, such as in ER+ breast cancer, pan-BET inhibitors carry unappreciated dangers in cancer clinical trials (34).
Figure 4. BRD4 expression correlates with SNAI1 and survival outcomes in prostate cancer patients.
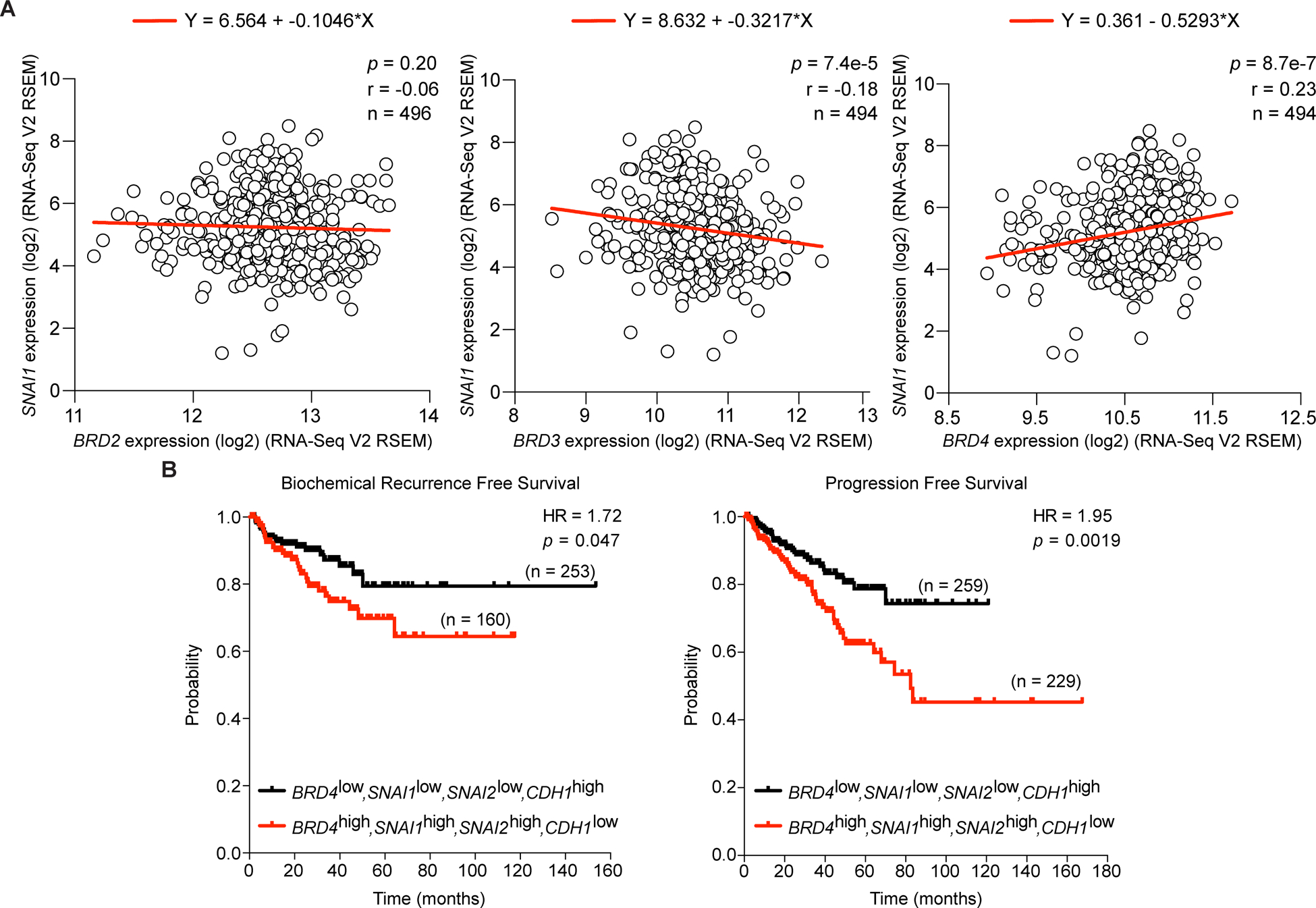
A, Comparison of BRD2, BRD3, BRD4 and SNAI1 expression in prostate cancer patient clinical samples by linear regression. Spearman rank correlation, p value and total number of individuals indicated.
B, Kaplan-Meier curve of biochemical recurrence-free survival and progression free survival of 414 (BCR) and 488 (PFS) prostate cancer patients was calculated from the TCGA database. Patients were segregated into cohorts with ‘low expression’ (BRD4low, SNAI1low, SNAI2low and CDH1high) and ‘high expression’ (BRD4high, SNAI1high, SNAI2high and CDH1low). Hazard ratio and p value are indicated.
In diverse cancer types, including prostate cancer, EMT defines a transcriptional phenotype associated with lineage plasticity, increased cancer stem-like cell formation and the emergence of chemoresistance. An unsurprising consequence of the clinical success of antiandrogen drugs like abiraterone and enzalutamide is the increased prevalence in the clinic of novel forms of advanced, AR-independent prostate cancer with characteristic genomic and transcriptional patterns (35), such as greater lineage plasticity, EMT and neuroendocrine signatures (36,37). An effective response to these clinical shifts demands creativity and resourcefulness to identify and develop alternative therapies for emergent, chemoresistant tumors. However, progress will continue to be hampered without properly defining which BET proteins are responsible for regulating biological processes like EMT and metastasis across both AR-competent and AR-deficient models of CRPC. In previous studies that focused on targeting BET proteins as a means for treating CRPC, success was measured by determining how well a pan-BET inhibitor or degrader diminished pro-proliferative markers like c-Myc (10,11,38). While c-Myc is a well-established BET target gene (39,40) and known driver of prostate cancer cell proliferation (41), survival (41) and EMT (42), its attenuation by JQ1 is limited to AR-competent CRPC cell lines (16). By not identifying relevant BET proteins and targets across both AR-competent and AR-deficient CRPC cell lines, investigators continue to give the false impression that BET proteins are only viable for targeting in AR-competent settings (43). In summary, our findings (Fig. 5) underscore the need for targeted BRD4-selective small molecules like MZ1 and warrant further exploration as a means for treating patients with mCRPC.
Figure 5. Visual overview of BRD4’s role in regulating EMT in CRPC.
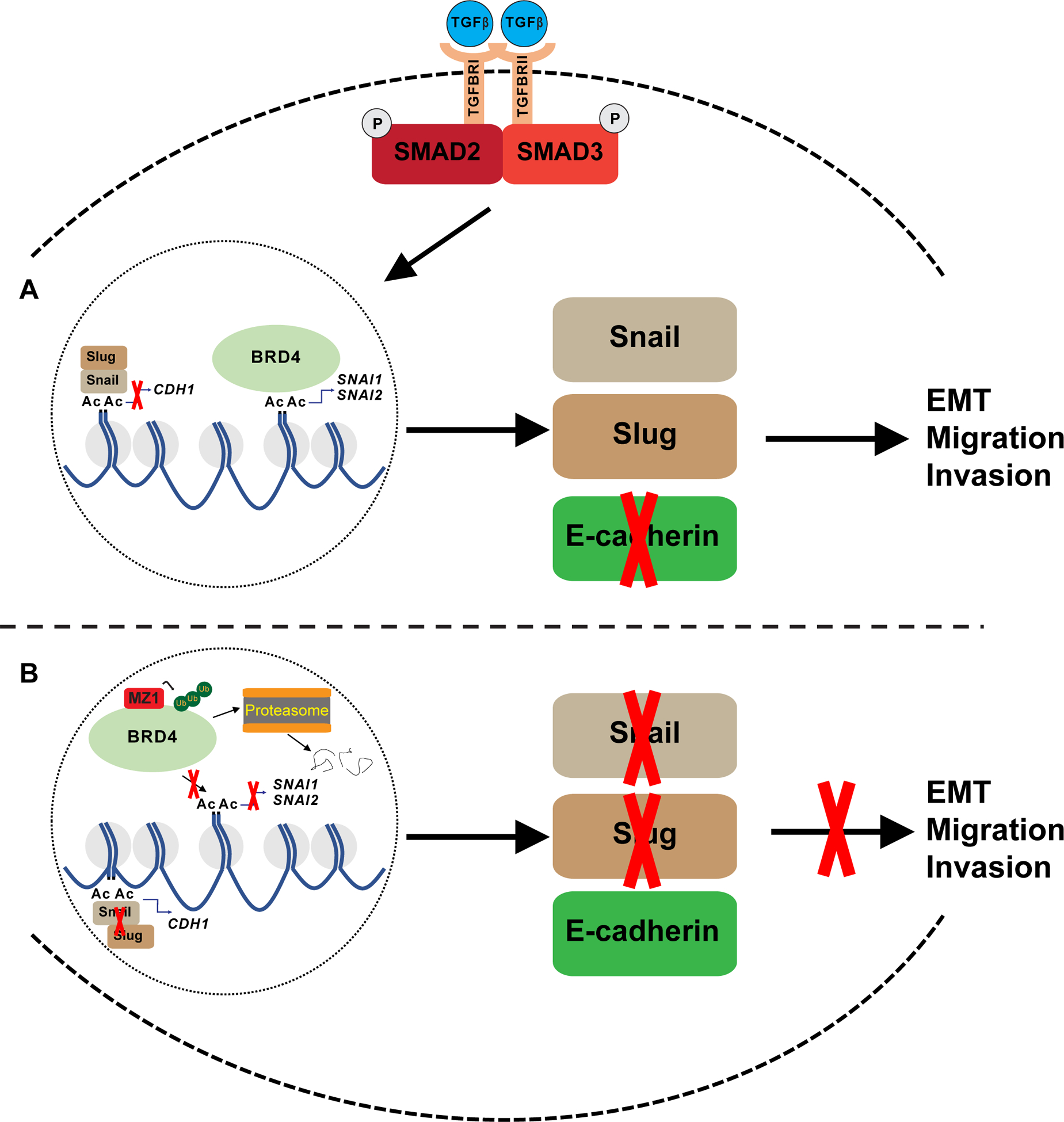
A, Graphic highlights BRD4 regulation of Snail and Slug expression through its interaction with the SNAI1 and SNAI2 promoter. Snail and Slug are shown to facilitate migration and invasion through its ability to inhibit E-cadherin expression. TGFß-Smad3 signaling works through BRD4 and drives EMT.
B, Illustration showing MZ1 selectively binding too and degrading BRD4 through polyubiquitination and proteasome-dependent degradation. As a result, BRD4 is unable to recruit co-activator proteins to the SNAI1 and SNAI2 promoters and carry out transcription of these genes. Lack of Snail and Slug enhances E-cadherin expression, and as a result, EMT and cell migration and invasion are inhibited.
Supplementary Material
Acknowledgements
We thank Dr. Alessio Ciulli for providing MZ1 and the Boston University-Boston Medical Center Flow Cytometry and Cellular Imaging Core Facilities for technical assistance. J. Shafran is supported by a T32 training grant, ‘Research Training in Immunology’ from NIAID (5T32AI007309-30, PI Kepler). The funders had no role in study design, data collection and analysis, preparation of the manuscript or decision to publish.
Grant support: This study was supported by grants from the National Institutes of Health (DK090455, U01CA182898, R01CA222170; GV Denis; T32AI007309; TB Kepler) and the Shipley Prostate Cancer Research Center.
Footnotes
Conflicts of interest: The authors state that they have no conflicts of interest to disclose.
Supplementary information: A file of supplementary methods, figures and data is provided.
References
- 1.Siegel RL, Miller KD, Jemal A. Cancer statistics, 2019. CA Cancer J Clin 2019;69:7–34 [DOI] [PubMed] [Google Scholar]
- 2.Das R, Gregory PA, Hollier BG, Tilley WD, Selth LA. Epithelial plasticity in prostate cancer: principles and clinical perspectives. Trends Mol Med 2014;20:643–51 [DOI] [PubMed] [Google Scholar]
- 3.Rice MA, Malhotra SV, Stoyanova T. Second-Generation Antiandrogens: From Discovery to Standard of Care in Castration Resistant Prostate Cancer. Front Oncol 2019;9:801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Miao L, Yang L, Li R, Rodrigues DN, Crespo M, Hsieh JT, et al. Disrupting Androgen Receptor Signaling Induces Snail-Mediated Epithelial-Mesenchymal Plasticity in Prostate Cancer. Cancer Res 2017;77:3101–12 [DOI] [PubMed] [Google Scholar]
- 5.Smith BN, Odero-Marah VA. The role of Snail in prostate cancer. Cell Adh Migr 2012;6:433–41 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Lim J, Thiery JP. Epithelial-mesenchymal transitions: insights from development. Development 2012;139:3471–86 [DOI] [PubMed] [Google Scholar]
- 7.Sun Y, Wang BE, Leong KG, Yue P, Li L, Jhunjhunwala S, et al. Androgen deprivation causes epithelial-mesenchymal transition in the prostate: implications for androgen-deprivation therapy. Cancer Res 2012;72:527–36 [DOI] [PubMed] [Google Scholar]
- 8.Belkina AC, Denis GV. BET domain co-regulators in obesity, inflammation and cancer. Nat Rev Cancer 2012;12:465–77 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Filippakopoulos P, Qi J, Picaud S, Shen Y, Smith WB, Fedorov O, et al. Selective inhibition of BET bromodomains. Nature 2010;468:1067–73 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Raina K, Lu J, Qian Y, Altieri M, Gordon D, Rossi AM, et al. PROTAC-induced BET protein degradation as a therapy for castration-resistant prostate cancer. Proc Natl Acad Sci U S A 2016;113:7124–9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Gao L, Schwartzman J, Gibbs A, Lisac R, Kleinschmidt R, Wilmot B, et al. Androgen receptor promotes ligand-independent prostate cancer progression through c-Myc upregulation. PLoS One 2013;8:e63563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Andrieu G, Tran AH, Strissel KJ, Denis GV. BRD4 Regulates Breast Cancer Dissemination through Jagged1/Notch1 Signaling. Cancer Res 2016;76:6555–67 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Andrieu GP, Denis GV. BET Proteins Exhibit Transcriptional and Functional Opposition in the Epithelial-to-Mesenchymal Transition. Mol Cancer Res 2018;16:580–6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Shafran JS, Andrieu GP, Gyorffy B, Denis GV. BRD4 Regulates Metastatic Potential of Castration-Resistant Prostate Cancer through AHNAK. Mol Cancer Res 2019;17:1627–38 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Belkina AC, Nikolajczyk BS, Denis GV. BET protein function is required for inflammation: Brd2 genetic disruption and BET inhibitor JQ1 impair mouse macrophage inflammatory responses. J Immunol 2013;190:3670–8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Asangani IA, Dommeti VL, Wang X, Malik R, Cieslik M, Yang R, et al. Therapeutic targeting of BET bromodomain proteins in castration-resistant prostate cancer. Nature 2014;510:278–82 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Robinson JT, Thorvaldsdottir H, Winckler W, Guttman M, Lander ES, Getz G, et al. Integrative genomics viewer. Nat Biotechnol 2011;29:24–6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Budczies J, Klauschen F, Sinn BV, Gyorffy B, Schmitt WD, Darb-Esfahani S, et al. Cutoff Finder: a comprehensive and straightforward Web application enabling rapid biomarker cutoff optimization. PLoS One 2012;7:e51862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Zengerle M, Chan KH, Ciulli A. Selective Small Molecule Induced Degradation of the BET Bromodomain Protein BRD4. ACS Chem Biol 2015;10:1770–7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Cao Z, Kyprianou N. Mechanisms navigating the TGF-beta pathway in prostate cancer. Asian J Urol 2015;2:11–8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Schwarze SR, Fu VX, Desotelle JA, Kenowski ML, Jarrard DF. The identification of senescence-specific genes during the induction of senescence in prostate cancer cells. Neoplasia 2005;7:816–23 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Medici D, Hay ED, Olsen BR. Snail and Slug promote epithelial-mesenchymal transition through beta-catenin-T-cell factor-4-dependent expression of transforming growth factor-beta3. Mol Biol Cell 2008;19:4875–87 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Li D, Sun H, Sun WJ, Bao HB, Si SH, Fan JL, et al. Role of RbBP5 and H3K4me3 in the vicinity of Snail transcription start site during epithelial-mesenchymal transition in prostate cancer cell. Oncotarget 2016;7:65553–67 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Li N, Dhar SS, Chen TY, Kan PY, Wei Y, Kim JH, et al. JARID1D Is a Suppressor and Prognostic Marker of Prostate Cancer Invasion and Metastasis. Cancer Res 2016;76:831–43 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Gravdal K, Halvorsen OJ, Haukaas SA, Akslen LA. A switch from E-cadherin to N-cadherin expression indicates epithelial to mesenchymal transition and is of strong and independent importance for the progress of prostate cancer. Clin Cancer Res 2007;13:7003–11 [DOI] [PubMed] [Google Scholar]
- 26.Ware KE, Somarelli JA, Schaeffer D, Li J, Zhang T, Park S, et al. Snail promotes resistance to enzalutamide through regulation of androgen receptor activity in prostate cancer. Oncotarget 2016;7:50507–21 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Welti J, Sharp A, Yuan W, Dolling D, Nava Rodrigues D, Figueiredo I, et al. Targeting Bromodomain and Extra-Terminal (BET) Family Proteins in Castration-Resistant Prostate Cancer (CRPC). Clin Cancer Res 2018;24:3149–62 [DOI] [PubMed] [Google Scholar]
- 28.Cerami E, Gao J, Dogrusoz U, Gross BE, Sumer SO, Aksoy BA, et al. The cBio cancer genomics portal: an open platform for exploring multidimensional cancer genomics data. Cancer Discov 2012;2:401–4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Sohn M, Shin S, Yoo JY, Goh Y, Lee IH, Bae YS. Ahnak promotes tumor metastasis through transforming growth factor-beta-mediated epithelial-mesenchymal transition. Sci Rep 2018;8:14379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Stemmler MP. PCAF, ISX, and BRD4: a maleficent alliance serving lung cancer malignancy. EMBO Rep 2020;21:e49766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Qin ZY, Wang T, Su S, Shen LT, Zhu GX, Liu Q, et al. BRD4 Promotes Gastric Cancer Progression and Metastasis through Acetylation-Dependent Stabilization of Snail. Cancer Res 2019;79:4869–81 [DOI] [PubMed] [Google Scholar]
- 32.Wang LT, Liu KY, Jeng WY, Chiang CM, Chai CY, Chiou SS, et al. PCAF-mediated acetylation of ISX recruits BRD4 to promote epithelial-mesenchymal transition. EMBO Rep 2020;21:e48795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Alqahtani A, Choucair K, Ashraf M, Hammouda DM, Alloghbi A, Khan T, et al. Bromodomain and extra-terminal motif inhibitors: a review of preclinical and clinical advances in cancer therapy. Future Sci OA 2019;5:FSO372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Andrieu G, Belkina AC, Denis GV. Clinical trials for BET inhibitors run ahead of the science. Drug Discov Today Technol 2016;19:45–50 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Abida W, Cyrta J, Heller G, Prandi D, Armenia J, Coleman I, et al. Genomic correlates of clinical outcome in advanced prostate cancer. Proc Natl Acad Sci U S A 2019;116:11428–36 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Aggarwal R, Huang J, Alumkal JJ, Zhang L, Feng FY, Thomas GV, et al. Clinical and Genomic Characterization of Treatment-Emergent Small-Cell Neuroendocrine Prostate Cancer: A Multi-institutional Prospective Study. J Clin Oncol 2018;36:2492–503 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Beltran H, Hruszkewycz A, Scher HI, Hildesheim J, Isaacs J, Yu EY, et al. The Role of Lineage Plasticity in Prostate Cancer Therapy Resistance. Clin Cancer Res 2019;25:6916–24 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Wyce A, Degenhardt Y, Bai Y, Le B, Korenchuk S, Crouthame MC, et al. Inhibition of BET bromodomain proteins as a therapeutic approach in prostate cancer. Oncotarget 2013;4:2419–29 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Delmore JE, Issa GC, Lemieux ME, Rahl PB, Shi J, Jacobs HM, et al. BET bromodomain inhibition as a therapeutic strategy to target c-Myc. Cell 2011;146:904–17 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Mertz JA, Conery AR, Bryant BM, Sandy P, Balasubramanian S, Mele DA, et al. Targeting MYC dependence in cancer by inhibiting BET bromodomains. Proc Natl Acad Sci U S A 2011;108:16669–74 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Fan L, Peng G, Sahgal N, Fazli L, Gleave M, Zhang Y, et al. Regulation of c-Myc expression by the histone demethylase JMJD1A is essential for prostate cancer cell growth and survival. Oncogene 2016;35:2441–52 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Amatangelo MD, Goodyear S, Varma D, Stearns ME. c-Myc expression and MEK1-induced Erk2 nuclear localization are required for TGF-beta induced epithelial-mesenchymal transition and invasion in prostate cancer. Carcinogenesis 2012;33:1965–75 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Faivre EJ, McDaniel KF, Albert DH, Mantena SR, Plotnik JP, Wilcox D, et al. Selective inhibition of the BD2 bromodomain of BET proteins in prostate cancer. Nature 2020;578:306–10 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.