

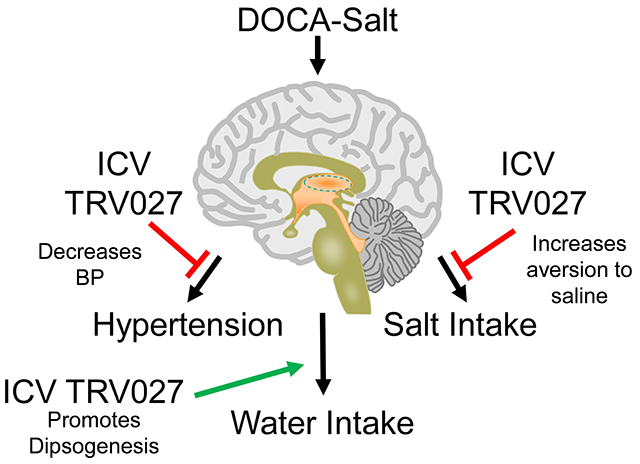
Abstract
Activation of central angiotensin type 1 receptors (AT1R) is required for the increased blood pressure (BP), polydipsia and salt intake in deoxycorticosterone acetate (DOCA)-salt hypertension. TRV120027 (TRV027) is an AT1R-biased agonist that selectively acts through β-arrestin. We hypothesized that intracerebroventricular (ICV) administration of TRV027 would ameliorate the effects of DOCA-salt. In a neuronal cell line, TRV027 induced AT1aR internalization through dynamin and clathrin-mediated endocytosis. We next evaluated the effect of chronic ICV infusion of TRV027 on fluid intake. We measured the relative intake of water versus various saline solutions using a 2-bottle choice paradigm in mice subjected to DOCA with a concomitant ICV infusion of either vehicle, TRV027, or losartan. Sham mice received ICV vehicle without DOCA. TRV027 potentiated DOCA-induced water intake in the presence or absence of saline. TRV027 and losartan both increased the aversion for saline, an effect particularly pronounced for highly aversive saline solutions. ICV Angiotensin II, but not TRV027, increased water and saline intake in the absence of DOCA. In a separate cohort, BP responses to acute ICV injection of vehicle, TRV, or losartan were measured by radiotelemetry in mice with established DOCA-salt hypertension. Central administration of ICV TRV027 or losartan each caused a significant and similar reduction of BP and heart rate. We conclude that administration of TRV027, a selective β-arrestin biased agonist directly into the brain increases aversion to saline and lowers BP in a model of salt-sensitive hypertension. These data suggest that selective activation of AT1R β-arrestin pathways may be exploitable therapeutically.
Keywords: Angiotensin Receptor, Biased Agonist, β-arrestin, Hypertension, Fluid Intake
Graphical Abstract
Introduction
Hypertension is a leading risk factor for disability and premature death from cardiovascular disease.1 Uncontrolled hypertension has a poor prognosis and places the patient at higher risk of target-organ damage leading to heart failure, myocardial infarction, stroke, impaired renal function, and death. On the contrary, intensive lowering of blood pressure (BP) can decrease cardiovascular events and death.2 Body fluid homeostasis and salt ingestion are important determinants of BP regulation.3 Diets high in salt can cause hypertension in salt-sensitive subjects, and lifestyle modification, which includes reduction in salt intake is advocated for patients with hypertension.4 Adherence to antihypertensive treatments remains a problem, particularly for resistant hypertension.5 Thus new therapeutic modalities remain necessary.
The renin-angiotensin system (RAS) is an important regulator of BP and acts through a multitude of renal, vascular, and central mechanisms, among others. The brain contains an intact RAS as it has the capacity for the production and action of angiotensin (Ang) peptides.6 The brain RAS controls cardiovascular and metabolic function by influencing fluid intake and balance and responding to physiological cues that control the sympathetic nervous system. Each of these effects are mediated by specific groups of neurons located in nuclei in the forebrain, hypothalamus and brain stem.7-9 Many of these neurons express Ang II type 1 receptors (AT1R).10 Pharmacological and genetic studies have demonstrated that AT1R in many brain regions are required to mediate the specific outputs controlled by those neurons. For example, AT1R in the subfornical organ are required to mediate the induction of vasopressin release, polydipsia, sodium intake and hypertension in the deoxycorticosterone acetate (DOCA)-salt model of hypertension.11
AT1R are a member of the G protein-coupled receptors (GPCRs) superfamily which exert many of the classical biological effects of Ang II.12 Like other GPCRs, the AT1R exhibit both G-protein and β-arrestin-dependent signaling.13 Classically, β-arrestin mediates desensitization of GPCRs and targets the receptors for internalization.14 Synthetic or biased ligands have been developed for many GPCRs which exclusively target either the G-protein or the β-arrestin pathway.15 For AT1R, it is hypothesized that the canonical signaling through G proteins mediates many of its crucial functions which serve to maintain BP within the physiological range. On the contrary, activation of the β-arrestin pathway mediates a decrease in AT1R functions through desensitization and internalization and thus helps to precisely control the duration of AT1R signals in response to a stimulus. However, the relative roles of the G-protein and β-arrestin pathways in the central neuronal control of BP and its related phenotypes, such as water and salt intake, remains poorly understood.
An agonist which specifically activates the β-arrestin pathway downstream of AT1R might act as a therapeutic drug by reducing some of the detrimental or adverse effects of Ang II signaling, such as cardiac remodeling and vascular dysfunction.16 For example, β-arrestin signaling downstream of the AT1R increases contractility in myocytes independent of G-protein activation, promotes their survival during acute cardiac injury, and causes AT1R internalization.17-20 TRV120027 (TRV027) is a selective AT1R β-arrestin-biased ligand.21 Peripheral administration of TRV027 to rats lowered BP and improved cardiac performance.21 Chronic central TRV027 administration reduced mean arterial BP, increased vagal tone and improved baroreflex sensitivity in spontaneously hypertensive rats.22 In terms of fluid intake, acute central injection of Ang II elevates water intake via Gαq signaling and salt intake via β-arrestin signaling.23 Central β-arrestin stimulation may be responsible for desensitization of the receptor after repeated acute injection of Ang II as it decreased water intake without affecting the Ang II-induced salt intake.24 Here, we investigated how AT1R β-arrestin signaling in the central nervous system affects drinking behavior and BP in mice subjected to DOCA-salt hypertension.
Methods
Top Guidelines:
The data that support the findings of this study are available from the corresponding author upon reasonable request.
Care and Use of Animals:
For all studies, 10-12 weeks old C57BL/6J male mice from Jackson Laboratories (Bar Harbor, ME) were used. For these initial studies only male mice were used because the central mechanisms controlling BP vary during the estrous cycle in females.25 Mice were fed standard laboratory chow (see Supplemental Methods) and tap water ad libitum until we initiated the experimental protocol. All mice were housed at a constant room temperature (24°C) with a 12-hour light/dark cycle (incandescent lights on at 5:00 A.M) in home cages. All experiments were conducted in accordance with the National Institutes of Health “Guide for the Care and Use of Laboratory Animals” and were approved by the University of Iowa and Medical College of Wisconsin Animal Care and Use Committees.
A detailed description of the AT1R internalization assay, surgical preparations, and fluid intake and blood pressure studies are in the Online only Data Supplement.11, 26-28
Statistics:
All data plotted are expressed as mean ± SEM (standard error of the mean). One-way ANOVA was used for the 2-bottle experiment to test for the difference in daily water intake, total fluid intake, saline intake, percentage of total intake from saline, and side preference. Tukey’s test was used for post-hoc analysis. One-way ANOVA with Tukey multiple comparisons procedure was also used for AT1aR internalization studies plasma electrolytes, and tissue weights. Two-way ANOVA with repeated measures was used to test for difference in BP parameters during acute injections. To analyze repeated measures data with missing values, the mixed effects model was used, which is included in the GraphPad Prism 8.0 package. Post-hoc Tukey-Krammer’s test was performed to compare the BP parameter at each time point between each treatment arm and Dunnett post-hoc test was used to compare the BP parameters for each treatment arm at time t(i) compared with the moment of injection t=0. We considered p<0.05 and adjusted p values <0.05 to be the cut-off of statistical significance.
Results
TRV027 induces AT1R internalization
Activation of the β-arrestin pathway by Ang II has been shown to cause receptor internalization.19, 20 To confirm its mechanism of action we first tested if TRV027 could induce AT1aR internalization using the NanoLuc-based HiBiT system.28 The experimental system utilizes an 11 amino acid peptide (HiBit) tag fused to the AT1aR. Binding of the fusion protein to a second subunit (called Large BiT, LgBiT) elaborates a luminescent signal that can be quantified (Figure 1A). These experiments were performed in N43/5 cells, an immortalized cell line which is thought to model hypothalamic agouti-related peptide neurons.27 Both Ang II (p<0.0001) and TRV027 (p<0.0001) induced a significant decrease in surface signal luminescence compared with vehicle suggesting AT1aR internalization in these cells (Figure 1B). The internalization induced by both Ang II and TRV027 was blunted by losartan suggesting they both required AT1aR binding (p=0.002, p=0.003, respectively). Moreover, preincubating cells with dynasore,29 an inhibitor of the GTPase activity required for dynamin-dependent endocytosis significantly blunted AT1aR internalization suggesting it occurs through a mechanism dependent upon dynamin and clathrin-mediated endocytosis (Figure 1C).
Figure 1: AT1aR internalization.
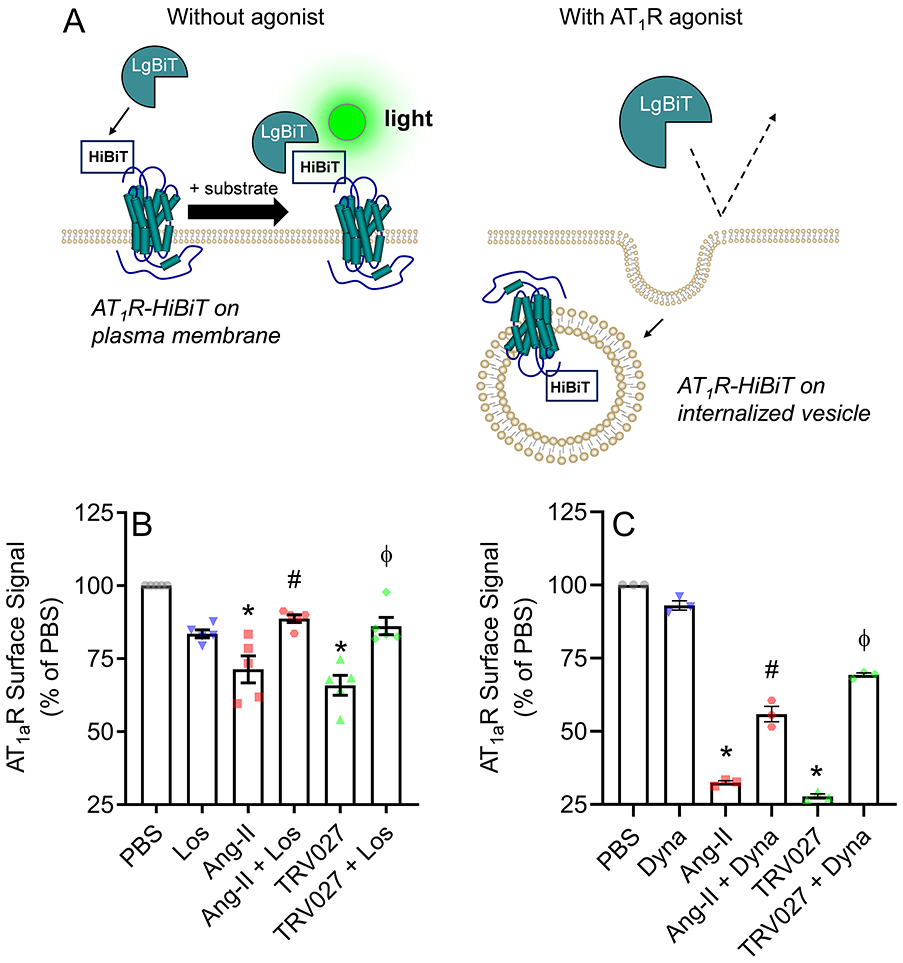
A) Schematic representation of Nano-Luc-based HiBiT system to quantify AT1aR internalization. The coding region of AT1aR was subcloned into a pBiT3.1-N vector to fuse the 11 amino acid peptide tag called HiBiT to AT1aR. Then, immortalized hypothalamic agouti-related peptide N43/5 cells were transfected with AT1aR-tagged HiBiT. When the fusion protein binds to the second subunit called Large BiT (LgBiT) it generates a luminescent signal that can be quantified. Since the LgBiT is membrane-impermeable only HiBiT-tagged AT1aR expressed on the cell surface can generate signal. To determine the percentage of surface AT1aR, the extracellular surface signal luminescence was divided by total luminescence signal, which was obtained by measuring luminescence when cells were lysed. B-C) Hypothalamic agouti-related peptide neuronal N43/5 cells were transfected with AT1aR-tagged HiBiT and the degree of receptor internalization in response to different treatments was calculated by the decrease in surface signal luminescence compared to vehicle control (PBS). B) Cells were treated with either vehicle (PBS), angiotensin II [Ang II] (0.1 μM), or TRV027 (1 μM) with or without losartan (1 μM) pre-incubation. C) Cells were treated with either vehicle, Ang II, or TRV027, with and without dynasore (50 μM) pre-incubation. Three to five independent experiments with 3 technical replicates each were averaged and presented as mean±SEM. Data were analyzed by one-way ANOVA with Tukey’s multiple comparisons procedure. Adjusted p<0.05 was considered significant. *p<0.05 compared with PBS; #p<0.05 compared with Ang II, ϕp<0.05 compared with TRV027. PBS: phosphate-buffered saline; Ang II: Angiotensin II; Los: Losartan; Dyna: Dynasore.
ICV TRV027 alters fluid intake in DOCA-salt mice
To assess the effect of central AT1R-mediated β-arrestin signaling we chronically infused either aCSF, TRV027 or Losartan ICV using a micro-osmotic minipump in mice implanted with a subcutaneous DOCA pellet (Figure 2A). We first allowed the mice to recover for 10 days during which the mice were provided normal chow and drinking water. We then measured water and saline intake in 48-hour blocks in trials containing drinking water or drinking water paired with various saline solutions. There was a 24-hour recovery period between each trial. The protocol employed 4 groups: 1) sham group receiving ICV vehicle (aCSF), 2) DOCA pellet with ICV vehicle, 3) DOCA pellet with ICV TRV027, and 4) DOCA pellet with ICV losartan. In the first trial, we provided the mice with a choice of two drinking water filled burettes. There was no difference in side preference which was approximately 50% between the groups (Figure 2B). DOCA induced a significant increase in daily water intake compared with the sham group (p<0.0001, Figure 2C). The addition of chronic ICV TRV027, but not losartan, further increased water intake compared with DOCA alone (p=0.012).
Figure 2: Central infusion of TRV027 increases water intake.
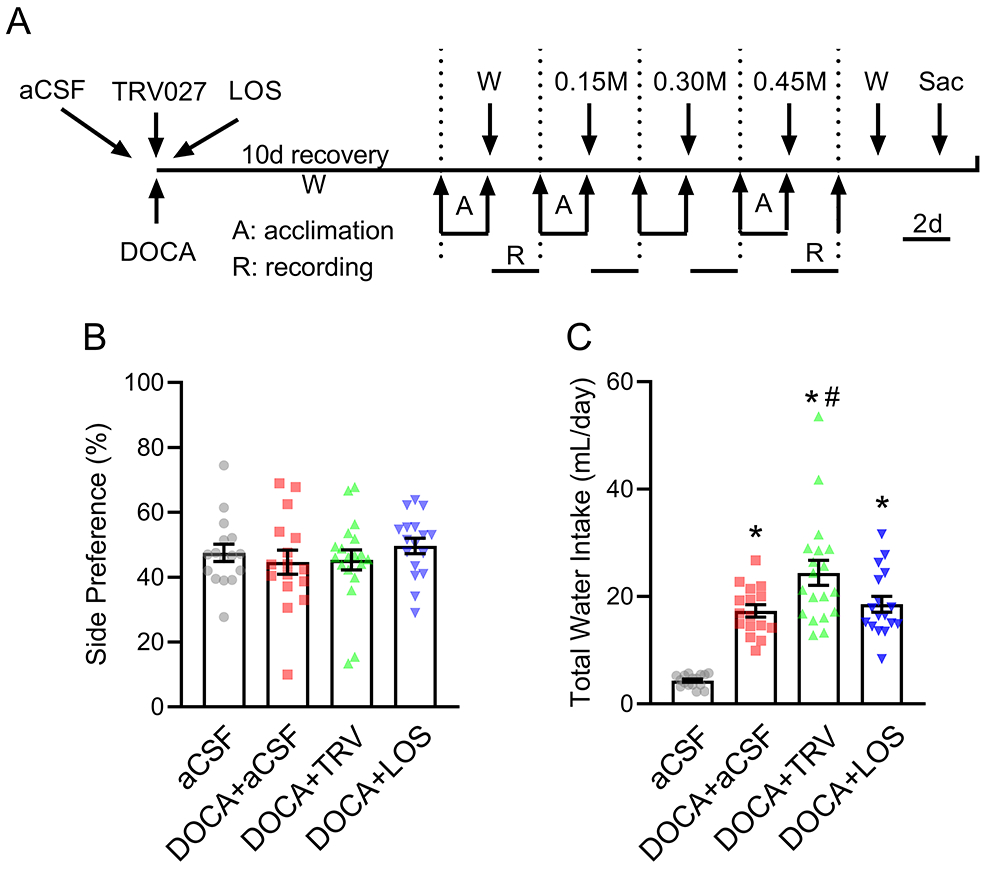
A) Schematic of the experimental protocol to assess drinking behavior. Mice were subjected to subcutaneous implantation of DOCA pellets concomitant with intracerebroventricular (ICV) infusions of either artificial cerebrospinal fluid (aCSF), TRV027, or losartan (LOS). A control group with no DOCA infusion and ICV vehicle infusion was included. Subsequently, mice were subjected to 4 consecutive trials in which mice were presented with a choice between 2 burettes. Each trial consisted of 2 days of acclimation and 2 days of recording. To normalize from the influence of side-bias, the burette positions were interchanged every 24 hours and all drinking data were calculated as the average between 2 days. W: access to water only, 0.15M: access to water vs 0.15M NaCl; 0.30M: access to water vs 0.30M NaCl, 0.45M: access to water vs 0.45M NaCl; Sac: sacrifice. B-C) Side preference (B) and total daily water intake (C) were measured when mice were presented with 2 burettes filled with tap water. Data are expressed as mean±SEM. Data were analyzed by one-way ANOVA with Tukey’s multiple comparisons procedure. *p<0.0001 compared with aCSF; #p<0.05 compared with DOCA+aCSF. N values are: aCSF (n=16), DOCA+aCSF (n=16), DOCA+TRV027 (n=19), DOCA+LOS (n=17). The top data point for DOCA + TRV in panel C was a statistical outlier. However, repeating the analysis by excluding the outlier preserves the statistical significance (p=0.03). Thus, the point was retained for transparency.
In the second trial, mice were presented with a choice of drinking water vs 0.15 M NaCl (Figure 3). Mice treated with DOCA exhibited higher water intake (p<0.0001), saline intake (p<0.0001), total fluid intake (water + saline; p<0.0001), and saline intake as a % of total fluid intake (p<0.024) compared with the sham group. Although DOCA caused an apparent increase in saline intake, the mice did not truly “prefer” saline as the percentage of saline intake compared with total intake did not exceed 50%. Thus, it is more appropriate to conclude that DOCA decreased the aversion to salt, as compared with increased preference for salt. ICV TRV027 infusion trended to increase water intake compared with DOCA alone but this did not reach statistical significance (p=0.077). Neither TRV027 nor losartan affected saline intake or total fluid intake in response to DOCA. DOCA increased the percentage of fluid intake from saline suggesting that DOCA-treated mice were less averse to drinking 0.15 M saline (p=0.024). This aversion to saline was restored to baseline by TRV027 (p=0.002). The data from losartan was equivocal as there was no statistical difference in the percentage of fluid intake from saline compared with either control or DOCA-treated mice (P=0.44 and P=0.45, respectively).
Figure 3: Drinking responses to 0.15 M saline.
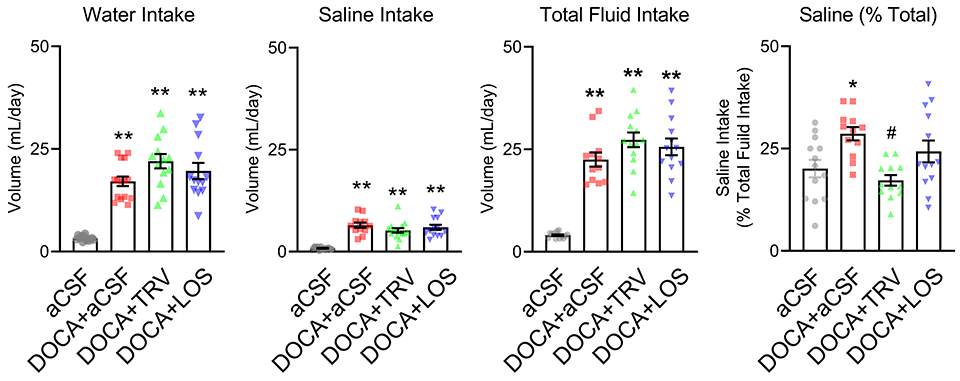
Control and mice subjected to DOCA with concomitant intracerebroventricular infusions of either artificial cerebrospinal fluid (aCSF), TRV027, or losartan (LOS) were presented with a choice of tap water vs 0.15M NaCl (non-aversive saline). Total water intake, total saline intake, total fluid intake (calculated as total water intake plus total saline intake), and percentage of total intake from saline (calculated as total saline intake/total fluid intake x 100) are shown. All data are expressed as mean±SEM. Data were analyzed by one-way ANOVA with Tukey’s multiple comparisons procedure. *p<0.05 and **p<0.0001 compared with aCSF; #p<0.05 compared with DOCA+aCSF. N values are: aCSF (n=13), DOCA+aCSF (n= 15), DOCA+TRV027 (n=13), DOCA+LOS (n=13)
Next, mice were presented with a choice of drinking water vs two different more concentrated (or aversive) saline solutions (0.3 M or 0.45 M NaCl, Figure 4). As before, DOCA-treated mice exhibited an increase in water intake (p<0.0001), saline intake (p<0.0001), total fluid intake (p<0.0001), and the % of total fluid intake from saline (p<0.0001) compared with the sham group. DOCA-treated mice receiving ICV TRV027 exhibited a significantly higher water intake compared with the group that received DOCA with ICV vehicle (p=0.015). This increase in water intake was observed in the 0.3 M saline trial but not the 0.45 M saline trial. Losartan had no effect on water intake induced by DOCA. There was no difference in DOCA-induced total fluid intake in response to either TRV027 or losartan. Both TRV027 and losartan blunted the increase in either 0.3 M (p<0.0001) or 0.45 M (p<0.0001) saline intake induced by DOCA. Consistent with that TRV027 and losartan each markedly blunted the percentage of total fluid intake from saline by DOCA (p<0.0001). Interestingly, TRV027 increased the aversion to 0.3 M saline (p=0.024), and both TRV027 and losartan increased the aversion to 0.45 M saline, even below baseline in the sham group (p=0.001 and 0.013, respectively).
Figure 4: Drinking responses to 0.3M and 0.45M saline.
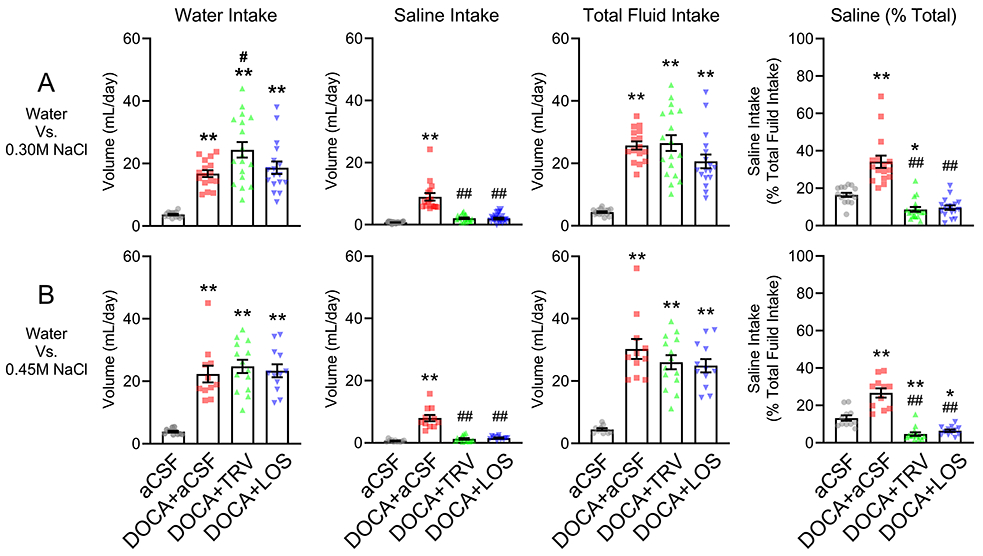
Control and mice subjected to DOCA with concomitant intracerebroventricular infusions of either artificial cerebrospinal fluid (aCSF), TRV027, or losartan (LOS) were presented with a choice of tap water vs 0.30M saline (A) or with a choice of tap water vs 0.45M saline (B). Total water intake, total saline intake, total fluid intake (calculated as total water intake plus total saline intake), and percentage of total fluid intake from saline (calculated as total saline intake/total fluid intake x 100), are shown. All data are expressed as mean±SEM. Data were analyzed by one-way ANOVA with Tukey’s multiple comparisons procedure. *p<0.05 and **p<0.0001 compared with aCSF; #p<0.05 and ##p<0.0001 compared with DOCA+aCSF. N values are: 0.30M saline: aCSF (n=16), DOCA+aCSF (n= 16), DOCA+TRV027 (n=18), DOCA+losartan (n=17). 0.45M saline: aCSF (n=11), DOCA+aCSF (n= 11), DOCA+TRV027 (n=14), DOCA+losartan (n=12).
At the end of the experimental protocol, there was no difference in plasma sodium and potassium between all 4 groups (Figure S1). DOCA induced a significant decrease in body weight compared with sham (P=0.007) (Figure S2A). ICV losartan, but not TRV027, attenuated DOCA-induced body weight decrease (P=0.002 and P=0.11, respectively). DOCA induced an increase in heart weight normalized by body weight (P=0.035) (Figure S2B). ICV TRV027, but not losartan, attenuated DOCA-induced cardiac hypertrophy (P=0.03 and P=0.08, respectively). DOCA-infusion resulted in a significant increase in the relative kidney weight (p<0.0001) and relative liver weight (p<0.0001) compared with the sham group, and ICV TRV027 and ICV losartan had no effect on either parameter (Figure S2C-D).
We next investigated the effect on TRV027 on fluid intake in the absence of DOCA. Three groups of mice underwent the same 2-bottle choice experiment: 1) sham group received ICV vehicle, 2) ICV Ang II and 3) ICV TRV027 infusion. ICV Ang II but not TRV027 elevated water intake when water alone was offered (Figure 5A). As before, there was no change in side preference. ICV Ang II increased water (p<0.0001), saline (p<0.0001), and total fluid (p<0.0001) when a 2-bottle choice of drinking water and either 0.15, 0.3 or 0.45 M saline was offered (Figure 5B). Total fluid intake was similar for all three saline concentrations. However, the relative proportion of water/saline changed with increasing concentrations of saline such that there was an increase in water and decrease in saline intake. Unlike Ang II, ICV TRV027 had no effect on any of these measures.
Figure 5: Effect of ICV TRV027 on water intake in absence of DOCA.
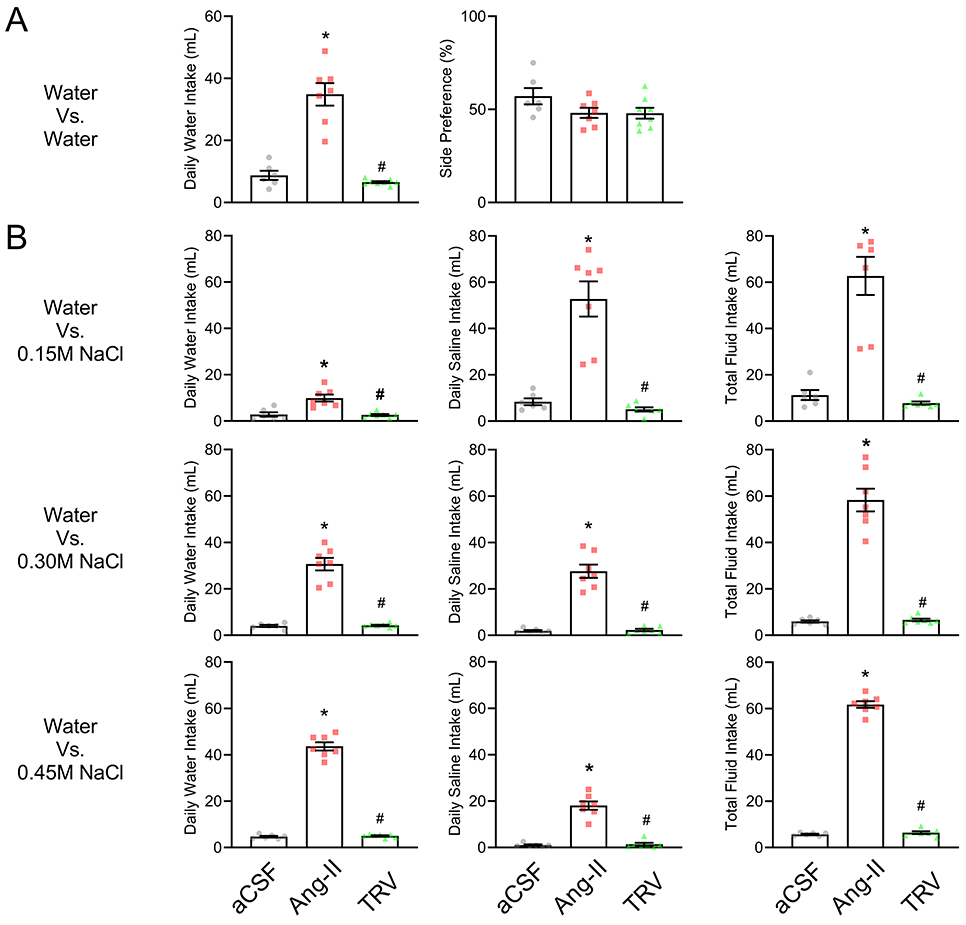
A. The effect of TRV027 on water intake in the absence of DOCA was measured. Three groups of mice underwent 2-bottle choice experiment: 1) sham group received intracerebroventricular (ICV) infusion of vehicle (aCSF), 2) ICV Ang II and 3) ICV TRV027. Total daily water intake (left) and side-bias (right) was measured when mice were presented with 2 burettes filled with tap water. Data are expressed as mean±SEM. Data were analyzed by one-way ANOVA with Tukey’s multiple comparisons procedure. *p<0.05 compared with vehicle. #p<0.05 compared with Ang-II. aCSF(n=6), Ang II (n=7), TRV027(n=8). B. The same three experimental groups were presented with a choice of tap water vs different concentrations of saline. Mice were then presented with a choice of water vs 0.15M NaCl (top), water vs 0.30M NaCl (middle) and water vs 0.45M NaCl (bottom). Panel 1 (left): daily water intake; Panel 2 (center): daily saline intake; Panel 3 (right): daily fluid intake calculated as daily water intake plus daily saline intake. Data are expressed as mean±SEM. Data were analyzed by one-way ANOVA with Tukey’s multiple comparisons procedure. *p<0.05 compared with vehicle. #p<0.05 compared with Ang-II. aCSF(n=6), Ang II (n=7), and TRV027(n=7)
Acute Effect of ICV TRV027 on Blood Pressure
We next evaluated the effect of ICV TRV027 on arterial BP. To exclude changes in BP caused by potential changes in salt intake, we measured BP after established DOCA-salt hypertension. We thus hypothesized that any change in BP over 60 minutes would be unrelated to changes in drinking behavior.
Mice were implanted with an ICV cannula and radiotelemeter to measure blood pressure and were subjected to DOCA-salt (Figure 6A). Once hypertension was established, each mouse received an ICV injection of vehicle, losartan or TRV027 in a random manner. All animals exhibited high BP at baseline (t=0) prior to the injections (Figure 6B, Figure S3). There was no difference in BP or heart rate (HR) at baseline (t=0) prior to the injections. To assess the reduction in BP and HR compared to baseline, ΔSystolic BP (SBP), ΔDiastolic BP (DBP), ΔMean BP (MBP) and ΔHR were calculated. Then for each drug, the corresponding Δ was compared to t=0 using a mixed model effect. To assess the difference in response for each drug, the Δ were compared between the 3 groups at each of the timepoints following the injection for 1h. Acute ICV injection of TRV027 or losartan each induced a drop in the SBP, DBP, and MBP from baseline (Figure 6B, Figure S3). This BP reduction was accompanied by a decrease in HR (Figure 6C). There was no change in BP or HR with ICV injection of vehicle.
Figure 6: Effect of acute ICV infusion of TRV027 on systolic blood pressure and heart rate.
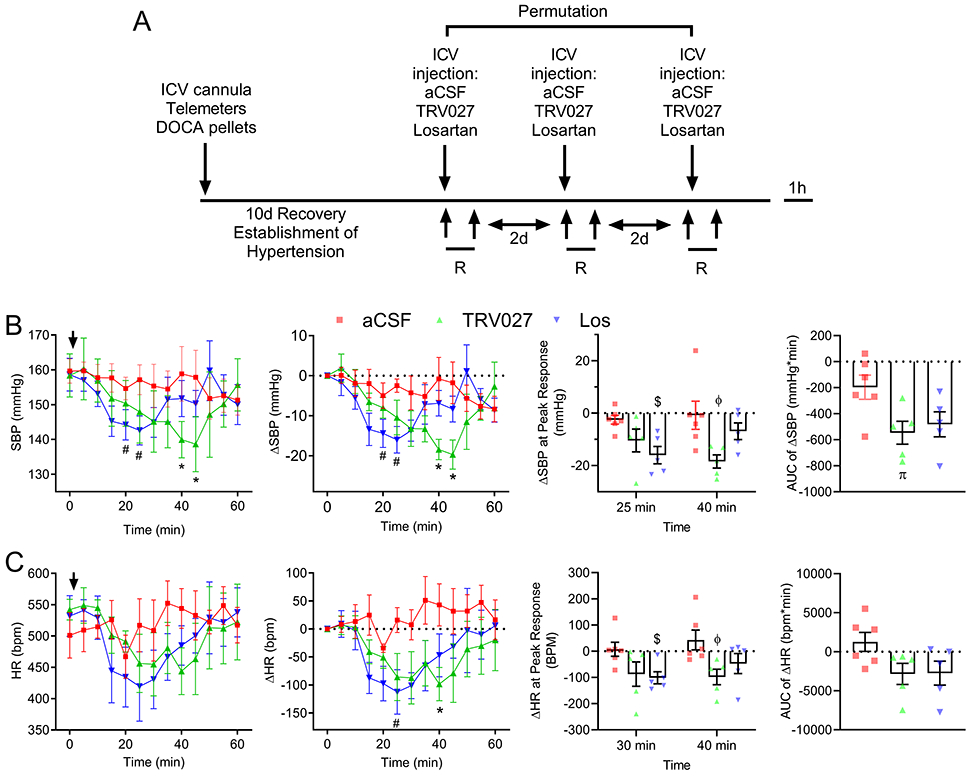
A. Telemetry-instrumented mice were subjected to a surgical implantation of a DOCA pellet and a stainless steel ICV cannula, which facilitates the repeated administration of drugs into the lateral ventricle. Subsequently, baseline blood pressure (BP) and HR were recorded, and mice were tested for the acute responses to a central injection of either vehicle (aCSF), TRV027, or losartan (Los). The systolic, diastolic and mean BP and HR were recorded for the duration of one-hour post-injection. Two days were given for the complete clearance of the preceding drug before a new drug was tested. B-C. Panel 1: SBP (B) and HR (C) were recorded for the duration of one-hour post-injection of either vehicle (aCSF), TRV027, or Los. Data were analyzed by mixed effects model and Dunnett’s multiple comparisons procedure. Panel 2: ΔSBP and ΔHR were calculated by subtracting the baseline SBP and HR at t=0 min from the SBP and HR at any given time t(i). A Mixed effects model with repeated measure and post-hoc Dunnett test was performed to compare the SBP or ΔSBP at each t(i) compared with the control SBP or ΔSBP at t=0 and to compare the HR or ΔHR for each t(i) compared with the control HR or ΔHR at t=0 for each experimental group. Panel 3: Peak responses for TRV027 and Los injections were defined as the drop in ΔSBP or ΔHR that were significantly different from vehicle injection. Peak ΔSBP occurred at 25 min for los and 40 for TRV027 injections, respectively. Peak ΔHR occurred at 30 min for los and 40 min for TRV027 injections. Mixed effects model with post-hoc Tukey-Krammer test was performed to compare the ΔSBP and the ΔHR between TRV027, Los and aCSF injection at each timepoint. Panel 4: The area under the curve (AUC) for ΔSBP or ΔHR are shown. One-way ANOVA with post-hoc Tukey’s test was performed to compare the AUC. Adjusted p<0.05 was considered significant. #p<0.05 compared with Los injection at t=0, *p<0.05 compared with TRV027 at t=0. $p<0.05 compared with aCSF at 30 min; ϕp<0.05 compared with aCSF at 40 min. πp<0.05 compared with aCSF. N values are: aCSF (n=6), TRV027 (n=5), and Los (n=5).
TRV027 injection induced a significant reduction in SBP (ΔSBP) at 40-45 min (p=0.01 and 0.03, respectively) compared with baseline (Figure 6B, panel 2), and compared with vehicle injection (p=0.05 and 0.05, respectively) (Figure 6B, panel 3). Losartan induced a significant reduction in SBP at 25-30 min compared with baseline (p=0.04 and 0.03, respectively)(Figure 6B, panel 2), and at 25 min compared with vehicle (p=0.025)(Figure 6B, panel 3). TRV027 injection induced a significant reduction in HR compared with vehicle injection at 40 min (p=0.04)(Figure 6C, panel 2), while losartan caused a significant reduction in HR at 30 min (p=0.03) compared with vehicle injection (Figure 6C, panel 2). Similarly, TRV027 injection induced a significant reduction in MBP and DBP (Figure S3, panel 2). As measured by the area under the curve, there was a larger SBP response in mice injected with TRV027 compared with losartan (Figure 6B, panel 4). Whereas, the timing of the depressor response to TRV027 and losartan appeared to differ, formal evaluation of the time to nadir on an individual mouse basis revealed no difference in the time to maximal depressor response between TRV027 and losartan (Figure S4).
Discussion
The main conclusions of this study are that central infusion of TRV027, a β-arrestin-biased agonist targeting AT1R: a) prevented the increase in saline intake by increasing the aversion for saline in response to DOCA treatment, particularly when provided with concentrated saline solutions, and b) acutely reduced BP and HR in DOCA-salt hypertensive mice. These data suggest that central β-arrestin stimulation can elicit protective effects in hypertension and influence drinking behavior. The protective effect on modulating arterial BP likely occurred through the autonomic nervous system as there were concomitant decreases in heart rate. One inference from these data is that in AT1R-expressing cells, distinct β-arrestin pathways regulate salt and water intake and BP, which may be exploitable therapeutically.
Upon stimulation, AT1R signals via G proteins for a finite period of time after which they dissociate from G proteins and are internalized following their phosphorylation and interaction with β-arrestins.30 We showed that TRV027 caused internalization of AT1aR and this was mediated by dynamin-dependent process likely involving clathrin-mediated endocytosis because the internalization was blocked by dynasore, a potent inhibitor of endocytic pathways known to depend on dynamin.29 Thus, like many other cell types, β-arrestin stimulation leads to AT1aR internalization in N43/5 cells. These cells are embryonic mouse hypothalamic cells which exhibit a gene signature typical of agouti-related peptide neurons and also endogenously express AT1aR and other RAS receptors including AT1bR, AT2R and MasR.31
One of the first observations we made was that TRV027 increased total water intake in mice treated with DOCA. This suggests that β-arrestin signaling can have a modulatory effect on water intake in the context of DOCA. Interestingly however, TRV027 did not increase water intake when administered to mice in the absence of DOCA. Similar to this, an analog of Ang II (Sar1,Ile4, Ile8) known as SII, which has been reported to act independent of G protein activation, failed to stimulate water intake when injected into the third ventricle of rats.32, 33 This leads us to conclude that activation of β-arrestin in AT1R-containing cells does not stimulate water intake in the absence of another stimulus such as DOCA suggesting it is more effective in high RAS states. Recall, DOCA-salt is considered as high brain RAS activity state even though circulating RAS is depressed.26 Whether ICV TRV027 at higher doses can increase water intake in absence of DOCA stimulation is yet to be determined.
Previous studies reported that SII increased saline intake to a similar extent as Ang II in rats.23 In our study, whereas central Ang II strongly stimulated saline intake, TRV027 did not. Indeed, at least in the presence of aversive saline solutions, TRV027 markedly decreased saline intake and increased aversion to saline in mice treated with DOCA. The effect of TRV027 was very similar to losartan. The prime difference between the effects of TRV027 and losartan was that losartan failed to stimulate water intake in these studies. Our data suggest that TRV027 may have two separate actions, first to augment the effects of DOCA on water intake, and second to mimic the effects of losartan on saline intake and preference. We caution that this does not mean that TRV027 is acting as an AT1R antagonist. However, it is notable that central infusion of SII blunted the dipsogenic effect of Ang II.33 Clearly, future studies are required to assess if the differences in drinking behavior between mice treated with ICV TRV027 and losartan and are due primarily to β-arrestin signaling.
TRV027 potentiated water intake and simultaneously decreased saline intake particularly when mice were provided 0.3 M saline. The potentiated water intake is likely to be β-arrestin mediated as it was not observed in mice treated with losartan, which behaves as an antagonist for both pathways. It is also possible that ICV TRV027 could be increasing water intake by stimulating a separate receptor. Thus far though there is no evidence that TRV027 can activate receptors other than AT1R, although it has been reported that both TRV027 and SII, but not losartan, can interfere with the mechanotransduction through an AT1R/B2 receptor heterodimer.34 Previous studies suggest that acute central Ang II injection elevates water intake through protein kinase C (PKC) and salt intake through mitogen‐activated protein (MAP) kinase.23 This would be consistent with activation of water intake by a Gαq-mediated pathway whereas salt intake could be mediated by β-arrestin pathway, which is known to engage MAP kinases including extracellular signal-regulated kinases 1 and 2 (ERK1/2).35 This is also consistent with the importance of PKC in mediating the increase in fluid intake in mice over-expressing the RAS in neurons in the central nervous system.36 Our study could be considered in conflict with previous observations using SII as the biased agonist because in our study TRV027 potentiated DOCA-induced water intake and attenuated salt intake. There are several possible explanations. First, TRV027-induced water intake occurred in the context of a chronic infusion of DOCA, which activates both AT1R and mineralocorticoid receptors in the brain whereas, the previous studies utilizing SII were in models in which only AT1R was stimulated. Given the interaction between Ang II and aldosterone signaling is well documented it is plausible that β-arrestin activation could lead to different outcomes depending on the status of mineralocorticoid receptors.37 Moreover, SII and TRV027 were shown to activate subtly different downstream efforts and thus are not functionally equivalent.38
We have previously showed that selective production of Ang II in the subfornical organ (SFO) is sufficient to increase water and saline intake.8 Moreover, prevention of excess Ang II production specifically in the SFO of mice overexpressing the RAS in the brain reduces water and salt intake.39 Our studies show that AT1R neurons in the SFO are required for the DOCA-salt-induced increase in water and salt intake.11 Optogenetic studies suggest that water and salt intake are regulated in part by distinct AT1R-expressing neurons in the SFO which differentially project to the organum vasculosum of the lamina terminalis and ventral part of the bed nucleus of the stria terminalis, respectively.40 Interestingly, there was much diminished staining for c-fos in the anteroventral wall of the third ventricle, the region controlling fluid intake, when comparing central injection of SII with Ang II.33 Future studies employing mice carrying conditional alleles of AT1R or β-arrestin 1 and 2 will help assess the regional- and cell-specificity of TRV027 action.
In our study, acute infusion of TRV027 decreased BP to the same extent as losartan. When viewed across the cohort of mice studied, it appeared as if the peak changes differed temporally, but when examined as time to nadir on an individual animal basis, that difference was not significant. These data suggest that central β-arrestin stimulation is protective against hypertension but exerts little or no effect on BP in the absence of an activated brain RAS status. It is important to recognize that the BP studies were performed with acute TRV027 administration. We did this because the data from the drinking study suggested that chronic central administration of TRV027 may prevent hypertension through an effect on salt intake. Indeed, both DOCA and salt are required to mediate the full pressor response.41 Moreover, these acute changes in BP suggest an autonomic, rather than humoral output from the brain. Indeed, the prominent bradycardic response evoked by the central administration of TRV027 may indicate an influence of the β-arrestin signaling on the sympathetic nerve activity. Consistent with this, it was reported that overexpression of β-arrestin 1 or β-arrestin 2 in the rostral ventrolateral medulla reduced BP and sympathetic outflow in hypertensive rats and downregulated AT1R.42 Similarly, chronic central TRV027 infusion reduced BP, vagal tone and improved baroreflex sensitivity in spontaneous hypertensive rats but had no effect on normotensive controls.22
Perspectives
Intravenous administration of TRV027 is well tolerated in humans.43 Unfortunately, clinical trial data showed that peripheral TRV027 did not exhibit a clinical benefit in acute heart failure.44 However, it was successful in decreasing the adverse events in a subgroup of patients with concomitant hypertension.45 That central administration of TRV027 attenuated salt intake and high BP under conditions of elevated brain RAS status implies that the clinical uses of TRV027 must be reevaluated. The detrimental augmentation of the sympathetic tone is observed in salt-sensitive hypertension, obesity-induced hypertension (and selective leptin resistance), and chronic heart disease and all these have been linked to an elevated status of brain RAS activity.46, 47 Thus, this study might serve as a foundation to justify the design of novel therapeutic strategies targeting AT1R β-arrestin signaling in the central nervous system to treat conditions where an overactivation of the brain RAS exists.
Supplementary Material
Novelty and Significance.
What is New?
Angiotensin receptors trigger both G-protein and β-arrestin signaling pathways.
TRV027, a compound that selectively modulates only the β-arrestin pathway augmented water and blunted saline intake when infused in the brain of mice treated with DOCA.
Moreover, acute administration of TRV027 in the brain induced a potent and transient depressor and bradycardic response in hypertensive mice.
What is Relevant?
Given the dramatic increase in salt consumption and prevalence of hypertension in the modern society, the use of TRV027 could be an appealing strategy to safely treat hypertension.
Summary
Central infusion of TRV027 reduces blood pressure and increases aversion to saline in the DOCA-salt model of hypertension. These data suggest that selective activation of AT1R β-arrestin pathways may be exploitable therapeutically.
Acknowledgments
The authors gratefully acknowledge the technical assistance of Ko-Ting Lu, Kelsey Wackman and Debbie Davis with surgical procedures and animal care.
Sources of Funding
This work was supported through research grants from the National Institutes of Health (NIH) to C.D.S. (HL084207, HL144807), J.L.G. (HL134850), S.S.K. (HL132351) and grants from the American Heart Association to C.D.S. (15SFRN23480000), J.L.G. (18EIA33890055), G.D. (19POST34380239) P.N. was funded with training awards from the NIH for this study (2T32HL007121041, 4T32DK007690, and 5T32HL134643).
Footnotes
Disclosures
CDS is a member of a Scientific Advisory Board for Ionis Pharmaceuticals. His contributions to that board are unrelated to the content of this manuscript.
References
- 1.Forouzanfar MH, Liu P, Roth GA, Ng M, Biryukov S, Marczak L, Alexander L, Estep K, Hassen Abate K, Akinyemiju TF, Ali R, Alvis-Guzman N, Azzopardi P, Banerjee A, Barnighausen T, Basu A, Bekele T, Bennett DA, Biadgilign S, Catala-Lopez F, Feigin VL, Fernandes JC, Fischer F, Gebru AA, Gona P, Gupta R, Hankey GJ, Jonas JB, Judd SE, Khang YH, Khosravi A, Kim YJ, Kimokoti RW, Kokubo Y, Kolte D, Lopez A, Lotufo PA, Malekzadeh R, Melaku YA, Mensah GA, Misganaw A, Mokdad AH, Moran AE, Nawaz H, Neal B, Ngalesoni FN, Ohkubo T, Pourmalek F, Rafay A, Rai RK, Rojas-Rueda D, Sampson UK, Santos IS, Sawhney M, Schutte AE, Sepanlou SG, Shifa GT, Shiue I, Tedla BA, Thrift AG, Tonelli M, Truelsen T, Tsilimparis N, Ukwaja KN, Uthman OA, Vasankari T, Venketasubramanian N, Vlassov VV, Vos T, Westerman R, Yan LL, Yano Y, Yonemoto N, Zaki ME and Murray CJ. Global Burden of Hypertension and Systolic Blood Pressure of at Least 110 to 115 mm Hg, 1990-2015. JAMA. 2017;317:165–182. [DOI] [PubMed] [Google Scholar]
- 2.Group SR, Wright JT Jr., Williamson JD, Whelton PK, Snyder JK, Sink KM, Rocco MV, Reboussin DM, Rahman M, Oparil S, Lewis CE, Kimmel PL, Johnson KC, Goff DC Jr., Fine LJ, Cutler JA, Cushman WC, Cheung AK and Ambrosius WT. A Randomized Trial of Intensive versus Standard Blood-Pressure Control. N Engl J Med. 2015;373:2103–2116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Hall JE, Granger JP, do Carmo JM, da Silva AA, Dubinion J, George E, Hamza S, Speed J and Hall ME. Hypertension: physiology and pathophysiology. Comprehensive Physiology. 2012;2:2393–2442. [DOI] [PubMed] [Google Scholar]
- 4.Calhoun DA, Jones D, Textor S, Goff DC, Murphy TP, Toto RD, White A, Cushman WC, White W, Sica D, Ferdinand K, Giles TD, Falkner B and Carey RM. Resistant hypertension: diagnosis, evaluation, and treatment. A scientific statement from the American Heart Association Professional Education Committee of the Council for High Blood Pressure Research. Hypertension. 2008;51:1403–1419. [DOI] [PubMed] [Google Scholar]
- 5.Durand H, Hayes P, Morrissey EC, Newell J, Casey M, Murphy AW and Molloy GJ. Medication adherence among patients with apparent treatment-resistant hypertension: systematic review and meta-analysis. J Hypertens. 2017;35:2346–2357. [DOI] [PubMed] [Google Scholar]
- 6.Nakagawa P and Sigmund CD. How Is the Brain Renin-Angiotensin System Regulated? Hypertension. 2017;70:10–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Grobe JL, Grobe CL, Beltz TG, Westphal SG, Morgan DA, Xu D, de Lange WJ, Li H, Sakai K, Thedans DR, Cassis LA, Rahmouni K, Mark AL, Johnson AK and Sigmund CD. The Brain Renin-Angiotensin System Controls Divergent Efferent Mechanisms to Regulate Fluid and Energy Balance. Cell Metabolism. 2010;12:431–442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Coble JP, Cassell MD, Davis DR, Grobe JL and Sigmund CD. Activation of the renin-angiotensin system, specifically in the subfornical organ is sufficient to induce fluid intake. Am J Physiol Regul Integr Comp Physiol. 2014;307:R376–R386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Ito S, Komatsu K, Tsukamoto K, Kanmatsuse K and Sved AF. Ventrolateral medulla AT1 receptors support blood pressure in hypertensive rats. Hypertension. 2002;40:552–559. [DOI] [PubMed] [Google Scholar]
- 10.Gonzalez AD, Wang G, Waters EM, Gonzales KL, Speth RC, Van Kempen TA, Marques-Lopes J, Young CN, Butler SD, Davisson RL, Iadecola C, Pickel VM, Pierce JP and Milner TA. Distribution of angiotensin type 1a receptor-containing cells in the brains of bacterial artificial chromosome transgenic mice. Neuroscience. 2012;226:489–509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Hilzendeger AM, Cassell MD, Davis DR, Stauss HM, Mark AL, Grobe JL and Sigmund CD. Angiotensin type 1a receptors in the subfornical organ are required for deoxycorticosterone acetate-salt hypertension. Hypertension. 2013;61:716–722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Forrester SJ, Booz GW, Sigmund CD, Coffman TM, Kawai T, Rizzo V, Scalia R and Eguchi S. Angiotensin II Signal Transduction: An Update on Mechanisms of Physiology and Pathophysiology. Physiol Rev. 2018;98:1627–1738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Smith JS, Lefkowitz RJ and Rajagopal S. Biased signalling: from simple switches to allosteric microprocessors. Nat Rev Drug Discov. 2018;17:243–260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Lohse MJ, Benovic JL, Codina J, Caron MG and Lefkowitz RJ. beta-Arrestin: a protein that regulates beta-adrenergic receptor function. Science. 1990;248:1547–1550. [DOI] [PubMed] [Google Scholar]
- 15.Wei H, Ahn S, Shenoy SK, Karnik SS, Hunyady L, Luttrell LM and Lefkowitz RJ. Independent beta-arrestin 2 and G protein-mediated pathways for angiotensin II activation of extracellular signal-regulated kinases 1 and 2. Proc Natl Acad Sci USA. 2003;100:10782–10787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Tan L, Yan W, McCorvy JD and Cheng J. Biased Ligands of G Protein-Coupled Receptors (GPCRs): Structure-Functional Selectivity Relationships (SFSRs) and Therapeutic Potential. Journal of Medicinal Chemistry. 2018;61:9841–9878. [DOI] [PubMed] [Google Scholar]
- 17.Kim KS, Abraham D, Williams B, Violin JD, Mao L and Rockman HA. beta-Arrestin-biased AT1R stimulation promotes cell survival during acute cardiac injury. American Journal of Physiology: Heart and Circulatory Physiology. 2012;303:H1001–H1010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Rajagopal K, Whalen EJ, Violin JD, Stiber JA, Rosenberg PB, Premont RT, Coffman TM, Rockman HA and Lefkowitz RJ. Beta-arrestin2-mediated inotropic effects of the angiotensin II type 1A receptor in isolated cardiac myocytes. Proc Natl Acad Sci USA. 2006;103:16284–16289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Anborgh PH, Seachrist JL, Dale LB and Ferguson SS. Receptor/beta-arrestin complex formation and the differential trafficking and resensitization of beta2-adrenergic and angiotensin II type 1A receptors. Molecular Endocrinology. 2000;14:2040–2053. [DOI] [PubMed] [Google Scholar]
- 20.Zhang J, Ferguson SS, Barak LS, Menard L and Caron MG. Dynamin and beta-arrestin reveal distinct mechanisms for G protein-coupled receptor internalization. The Journal of Biological Chemistry. 1996;271:18302–18305. [DOI] [PubMed] [Google Scholar]
- 21.Violin JD, DeWire SM, Yamashita D, Rominger DH, Nguyen L, Schiller K, Whalen EJ, Gowen M and Lark MW. Selectively engaging beta-arrestins at the angiotensin II type 1 receptor reduces blood pressure and increases cardiac performance. The Journal of Pharmacology and Experimental Therapeutics. 2010;335:572–579. [DOI] [PubMed] [Google Scholar]
- 22.Carvalho-Galvao A, Ogunlade B, Xu J, Silva-Alves CRA, Mendes-Junior LG, Guimaraes DD, Cruz JC, Queiroz TM, Balarini CM, Braga VA, Filipeanu CM, Lazartigues E and de Franca-Silva MDS. Central administration of TRV027 improves baroreflex sensitivity and vascular reactivity in spontaneously hypertensive rats. Clinical Science (Lond). 2018;132:1513–1527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Daniels D, Mietlicki EG, Nowak EL and Fluharty SJ. Angiotensin II stimulates water and NaCl intake through separate cell signalling pathways in rats. Exp Physiol. 2009;94:130–137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Vento PJ and Daniels D. Mitogen-activated protein kinase is required for the behavioural desensitization that occurs after repeated injections of angiotensin II. Exp Physiol. 2012;97:1305–1314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Hinojosa-Laborde C, Chapa I, Lange D and Haywood JR. Gender differences in sympathetic nervous system regulation. Clinical and Experimental Pharmacology & Physiology. 1999;26:122–126. [DOI] [PubMed] [Google Scholar]
- 26.Grobe JL, Buehrer BA, Hilzendeger AM, Liu X, Davis DR, Xu D and Sigmund CD. Angiotensinergic signaling in the brain mediates metabolic effects of deoxycorticosterone (DOCA)-salt in C57 mice. Hypertension. 2011;57:600–607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Hernandez-Caceres MP, Toledo-Valenzuela L, Diaz-Castro F, Avalos Y, Burgos P, Narro C, Pena-Oyarzun D, Espinoza-Caicedo J, Cifuentes-Araneda F, Navarro-Aguad F, Riquelme C, Troncoso R, Criollo A and Morselli E. Palmitic Acid Reduces the Autophagic Flux and Insulin Sensitivity Through the Activation of the Free Fatty Acid Receptor 1 (FFAR1) in the Hypothalamic Neuronal Cell Line N43/5. Front Endocrinol. 2019;10:176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Schwinn MK, Machleidt T, Zimmerman K, Eggers CT, Dixon AS, Hurst R, Hall MP, Encell LP, Binkowski BF and Wood KV. CRISPR-Mediated Tagging of Endogenous Proteins with a Luminescent Peptide. ACS Chem Biol. 2018;13:467–474. [DOI] [PubMed] [Google Scholar]
- 29.Macia E, Ehrlich M, Massol R, Boucrot E, Brunner C and Kirchhausen T. Dynasore, a cell-permeable inhibitor of dynamin. Dev Cell. 2006;10:839–850. [DOI] [PubMed] [Google Scholar]
- 30.Gaborik Z, Szaszak M, Szidonya L, Balla B, Paku S, Catt KJ, Clark AJ and Hunyady L. Beta-arrestin- and dynamin-dependent endocytosis of the AT1 angiotensin receptor. Mol Pharmacol. 2001;59:239–247. [DOI] [PubMed] [Google Scholar]
- 31.Sapouckey SA, Morselli LL, Deng G, Patil CN, Balapattabi K, Oliveira V, Claflin KE, Gomez J, Pearson NA, Potthoff MJ, Gibson-Corley KN, Sigmund CD and Grobe JL. Exploration of cardiometabolic and developmental significance of angiotensinogen expression by cells expressing the leptin receptor or agouti-related peptide. Am J Physiol Regul Integr Comp Physiol. 2020;318:R855–R869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Holloway AC, Qian H, Pipolo L, Ziogas J, Miura S, Karnik S, Southwell BR, Lew MJ and Thomas WG. Side-chain substitutions within angiotensin II reveal different requirements for signaling, internalization, and phosphorylation of type 1A angiotensin receptors. Mol Pharmacol. 2002;61:768–777. [DOI] [PubMed] [Google Scholar]
- 33.Daniels D, Yee DK, Faulconbridge LF and Fluharty SJ. Divergent behavioral roles of angiotensin receptor intracellular signaling cascades. Endocrinology. 2005;146:5552–5560. [DOI] [PubMed] [Google Scholar]
- 34.Quitterer U, Fu X, Pohl A, Bayoumy KM, Langer A and AbdAlla S. Beta-Arrestin1 Prevents Preeclampsia by Downregulation of Mechanosensitive AT1-B2 Receptor Heteromers. Cell. 2019;176:318–333 e19. [DOI] [PubMed] [Google Scholar]
- 35.Wei H, Ahn S, Shenoy SK, Karnik SS, Hunyady L, Luttrell LM and Lefkowitz RJ. Independent beta-arrestin 2 and G protein-mediated pathways for angiotensin II activation of extracellular signal-regulated kinases 1 and 2. Proc Natl Acad Sci USA. 2003;100:10782–10787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Coble JP, Johnson RF, Cassell MD, Johnson AK, Grobe JL and Sigmund CD. Activity of PKC-alpha within the Subfornical Organ is Necessary for Fluid Intake due to Brain Angiotensin. Hypertension. 2014;64:141–148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Shade RE, Blair-West JR, Carey KD, Madden LJ, Weisinger RS and Denton DA. Synergy between angiotensin and aldosterone in evoking sodium appetite in baboons. Am J Physiol Regul Integr Comp Physiol. 2002;283:R1070–R1078. [DOI] [PubMed] [Google Scholar]
- 38.Santos GA, Duarte DA, Parreiras ESLT, Teixeira FR, Silva-Rocha R, Oliveira EB, Bouvier M and Costa-Neto CM. Comparative analyses of downstream signal transduction targets modulated after activation of the AT1 receptor by two beta-arrestin-biased agonists. Front Pharmacol. 2015;6:131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Sakai K, Agassandian K, Morimoto S, Sinnayah P, Cassell MD, Davisson RL and Sigmund CD. Local production of angiotensin II in the subfornical organ causes elevated drinking. J Clin Invest. 2007;117:1088–1095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Matsuda T, Hiyama TY, Niimura F, Matsusaka T, Fukamizu A, Kobayashi K, Kobayashi K and Noda M. Distinct neural mechanisms for the control of thirst and salt appetite in the subfornical organ. Nature Neuroscience. 2017;20:230–241. [DOI] [PubMed] [Google Scholar]
- 41.O’Donaughy TL and Brooks VL. Deoxycorticosterone acetate-salt rats: hypertension and sympathoexcitation driven by increased NaCl levels. Hypertension. 2006;47:680–685. [DOI] [PubMed] [Google Scholar]
- 42.Sun JC, Liu B, Zhang RW, Jiao PL, Tan X, Wang YK and Wang WZ. Overexpression of ß-Arrestin1 in the Rostral Ventrolateral Medulla Downregulates Angiotensin Receptor and Lowers Blood Pressure in Hypertension. Frontiers in Physiology. 2018;9:297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Soergel DG, Subach RA, Cowan CL, Violin JD and Lark MW. First clinical experience with TRV027: pharmacokinetics and pharmacodynamics in healthy volunteers. Journal of Clinical Pharmacology. 2013;53:892–899. [DOI] [PubMed] [Google Scholar]
- 44.Pang PS, Butler J, Collins SP, Cotter G, Davison BA, Ezekowitz JA, Filippatos G, Levy PD, Metra M, Ponikowski P, Teerlink JR, Voors AA, Bharucha D, Goin K, Soergel DG and Felker GM. Biased ligand of the angiotensin II type 1 receptor in patients with acute heart failure: a randomized, double-blind, placebo-controlled, phase IIB, dose ranging trial (BLAST-AHF). European Heart Journal. 2017;38:2364–2373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Cotter G, Davison BA, Butler J, Collins SP, Ezekowitz JA, Felker GM, Filippatos G, Levy PD, Metra M, Ponikowski P, Teerlink JR, Voors AA, Senger S, Bharucha D, Goin K, Soergel DG and Pang PS. Relationship between baseline systolic blood pressure and long-term outcomes in acute heart failure patients treated with TRV027: an exploratory subgroup analysis of BLAST-AHF. Clinical research in cardiology: 2018;107:170–181. [DOI] [PubMed] [Google Scholar]
- 46.Acelajado MC, Pisoni R, Dudenbostel T, Dell’Italia LJ, Cartmill F, Zhang B, Cofield SS, Oparil S and Calhoun DA. Refractory hypertension: definition, prevalence, and patient characteristics. Journal of Clinical Hypertension. 2012;14:7–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Deng G and Grobe JL. The renin-angiotensin system in the arcuate nucleus controls resting metabolic rate. Current Opinion in Nephrology and Hypertension. 2019;28:120–127. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.