

Abstract
While mammalian mitochondria are known to possess a robust base excision repair system, direct evidence for the existence of additional mitochondrial DNA repair pathways is elusive. Herein a PCR-based assay was employed to demonstrate that plasmids containing DNA-protein crosslinks are rapidly repaired following electroporation into isolated mammalian mitochondria. Several lines of evidence argue that this repair occurs via homologous recombination. First, DNA-protein crosslinks present on plasmid DNA homologous to the mitochondrial genome were efficiently repaired (21 % repair in three hours), whereas a DNA-protein crosslink present on DNA that lacked homology to the mitochondrial genome remained unrepaired. Second, DNA-protein crosslinks present on plasmid DNA lacking homology to the mitochondrial genome were repaired when they were co-electroporated into mitochondria with an undamaged, homologous plasmid DNA molecule. Third, no repair was observed when DNA-protein crosslink-containing plasmids were electroporated into mitochondria isolated from cells pre-treated with the Rad51 inhibitor B02. These findings suggest that mitochondria utilize homologous recombination to repair endogenous and xenobiotic-induced DNA-protein crosslinks. Consistent with this interpretation, cisplatin-induced mitochondrial DNA-protein crosslinks accumulated to higher levels in cells pre-treated with B02 than in control cisplatin-treated cells. These results represent the first evidence of how spontaneous and xenobiotic-induced DNA-protein crosslinks are removed from mitochondrial DNA.
Keywords: DNA repair, Rad51, Cisplatin, Mitochondria, Homologous recombination, DNA-protein crosslinks
1. Introduction
The earliest recorded descriptions of mitochondria date back to the mid-nineteenth century when the organellar name ‘mitochondrion’ was coined by Carl Benda in 1898, reviewed in [1]. However, it wasn’t until the mid-twentieth century that high-resolution electron micrographs and a more detailed understanding of the function of this organelle evolved [2,3]. This work culminated with the development of the chemiosmotic hypothesis of ATP generation and identification of mitochondria as the ‘powerhouse of the cell’ [4], reviewed in [1,5]. Around this time period, other investigators discovered the existence of DNA within this organelle [6–8]. Subsequently, the mammalian mitochondria genome was determined to be a circular, double-stranded molecule of approximately 16 kilobases that encoded a subset of the proteins present within that organelle as well as genes encoding mitochondrial tRNA and rDNA species [9].
In 1974, Clayton et al. [10] convincingly demonstrated that photo-product adducts induced in mouse L cells were removed from nuclear DNA but not from the mitochondrial genome. This seminal observation led to the hypothesis that this organelle lacked nucleotide excision repair capability, a conclusion verified by numerous investigators [11–14]. These observations supported a general view that the mitochondria were bereft of DNA repair function-a hypothesis consistent with the fact that the mutational rate of mitochondrial DNA is substantially higher than the nuclear genome [15–17]. However, it was subsequently determined that mitochondria possess robust base excision repair (BER) machinery, albeit one entirely dependent on genes encoded within the nuclear genome [18–20]. Thus, it was for many years believed that, while not entirely lacking in DNA repair capacity, mammalian mitochondria lacked most, if not all other types of repair pathways previously characterized in prokaryotes and within nuclei of eukaryotes. More recently, a considerable body of literature has emerged, suggesting that this view may be inaccurate. Studies performed in extracts prepared from mitochondria documented enzymatic activities consistent with the existence of additional DNA repair pathways within that organelle such as non-homologous end-joining and DNA end-binding [21,22], mismatch repair [23] and homologous recombination [24,25]. Finally, investigators have shown that induced mitochondrial DNA double-strand breaks enhance genomic rearrangements, consistent with repair via either end-joining or homologous recombination [26,27].
Despite these latter findings, the hypothesis that mammalian mitochondria possess homologous recombination2 (HR) activity remains somewhat controversial [28,29]. While it is clear that mitochondrial DNA is subject to HR in at least some metazoan species [30], as well as in many fungal and plant species [31], only a limited number of papers have described molecular evidence suggestive of inter and/or intra-molecular recombination of human mitochondrial DNA, both in vivo and in vitro [32–34]. It is noteworthy, however, that the nearly exclusively uni-parental mode of mitochondrial DNA inheritance in humans would severely limit opportunities for genetically distinct mitochondrial genomes to encounter each other [28,35]. Interestingly, Zsurka et al., and Kratysberg et al., have provided clear evidence of mitochondrial HR in cells derived from relatively rare individuals who had paternally-inherited mitochondrial DNA [36,37]. Another potential reason for the relative absence of documented evidence for HR of mitochondrial genomes is evidence that heteroplasmic populations of mitochondrial DNA are separately maintained in distinct populations of mitochondria, creating a physical barrier to recombination between genetically distinct DNA molecules [38,39]. Taken together, these observations are consistent with the hypothesis that HR between identical ‘sister’ genomes, while genetically invisible, may function to repair types of damage not recognized by other mitochondrial DNA repair pathways.
There are several lines of evidence consistent with this hypothesis. First, LeDoux et al. [12] showed that interstrand DNA crosslinks created by cisplatin were repaired in the mitochondria of cultured mammalian cells. Since the base excision repair pathway is not believed capable of repairing DNA crosslinks, this finding strongly supported the interpretation that mammalian mitochondria must possess an additional HR, since it represents a major mechanism through which interstrand DNA crosslinks are repaired in the nucleus [40]. Second, we, and others, have shown that highly purified mitochondrial protein extracts possess the enzymatic machinery needed to complement the HR defect in recA-bacteria [24,25]. Interestingly, expression of a recombinant bacterial RecA protein targeted to the mitochondrial compartment protected mammalian cultured cells from death induced by the DNA-damaging agent bleomycin [41]. Third, numerous studies have documented the presence of DNA repair proteins known to participate in nuclear DNA double-strand break repair pathways within mitochondria. These proteins include known mammalian HR genes such as Rad51, Rad51C, and Xrcc3 [25,42–45]. Taken together, these results are consistent with the interpretation that mammalian mitochondria possess a HR-mediated DNA repair pathway.
Recently, our group developed a quantitative and highly sensitive PCR-based method capable of measuring repair of a variety of lesions present on plasmid DNA molecules [46,47]. These efforts focused on studying nuclear DNA repair and relied on lipofection to introduce substrates into cells. More recently, we determined that DNA-protein crosslink (DPC) substrates electroporated into isolated nuclei also undergo efficient repair (Chesner et al., manuscript in preparation). This finding, together with previous observations showing that electroporation can be used to introduce plasmids into purified mitochondria [21, 48], suggested that this PCR-based assay could be used to study DPC repair in that organelle. This topic is of considerable interest because DPCs have been shown to form on mitochondrial DNA [49,50], and enzymatic trapping of polymerases to DNA due to oxidative damage has been observed in purified protein and mitochondrial extracts from Hela cells [51,52]. There is currently no information regarding the mechanism(s) through which these spontaneous and xenobiotic-induced DPCs are removed from mitochondrial DNA. Experiments studying DPC repair in mammalian cells have indicated a role for the nucleotide excision repair (NER) pathway and HR in the nucleus [40,47,53,54]. Since mitochondria lack a functional NER pathway, we predicted that repair of DPCs present on plasmids electroporated into purified mitochondria would occur via HR. Results presented below convincingly demonstrate that mammalian cells possess a mitochondrial recombinational repair machinery capable of repairing DNA-protein crosslinks.
2. Materials and methods
2.1. Materials
Chemicals and enzymes.
Chemicals were purchased from Sigma Chemical (St. Louis, MO) unless otherwise indicated.
Cell lines.
The human fibrosarcoma cell line HT1080 (CCL-121) was purchased from the American Type Culture Collection, and human embryonic kidney cells expressing the large-T antigen (HEK293T) were a kind gift from Dr. Ashis Basu (University of Connecticut). Human cells were cultured in Dulbecco’s modified Eagle’s media (Life Technologies, Grand Island, NY) supplemented with 9% fetal bovine serum (Atlanta Biologics, Atlanta, GA). Chinese hamster AA8 51D1 cells were a kind gift from Professor Claudia Wiese (Colorado State University) in which the Rad51D gene was knocked out using a gene-targeting vector [55]. Rad51D is a paralog of Rad51 which contributes to the HR pathway in vertebrates [56]. These cells were cultured in alpha minimal essential medium (Life Technologies, Grand Island, NY) supplemented with 9% fetal bovine serum. Chinese hamster lung fibroblast cell line V79 (GM16136) were obtained from the Coriell Institute for Medical Research (Camden, NJ) and cultured in Ham’s F-12 modified essential Eagle’s media (Life Technologies, Grand Island, NY) supplemented with 9% fetal bovine serum. All cells were maintained in a humidified atmosphere of 5% carbon dioxide, 95 % air, at 37 °C.
2.2. Methods
Creation of plasmid DNA repair substrates.
Plasmids containing 8-oxoguanine residues or crosslinks to the human oxoguanine glycosylase (OGG1) were prepared as described by Chesner and Campbell, 2018 [47]. Briefly, single-stranded M13mp18 or recombinant clones derived therefrom were subjected to primer extension using primers harboring an 8-oxoguanine residue (M13 8oxo or M13 8oxo-mito, sequences provided in Table 1). Covalently-closed, circular full-length plasmids were gel purified and used for DNA repair experiments with or without crosslinking to recombinant human oxoguanine glycosylase via borohydride trapping [47]. Plasmids used to assess the influence of a double-strand break in DPC repair were incubated with EcoRI (New England Biolabs) for 30 min prior to electroporation.
Table 1.
Oligodeoxynucleotides (ODNs) used is this study (5’→3’).
| ODN | Sequence | Use |
|---|---|---|
| M13–8oxo | CCGGGTACCGAGCTC(8oxodG) AATTCGTAATCTTGGTCATAGCTG |
Primer extension |
| M13 forward | CGGCTCGTATGTTGTGTG | SSPE-qPCR |
| M13 reverse | GCTGCAAGGCGATTAAGT | SSPE-qPCR |
| M13–8oxo-mito | AGCCACCCCTCACCCACTA(8oxodG)GAT | Primer extension |
| Mito reverse | GGGATCTCATGCTGGAGTTC | SSPE-qPCR |
| M13-mito forward | AAGGGTGGGTAGGTTTGTTG | SSPE-qPCR |
| Human mtDNA tRNA forward | TGGCCATGGGTATGTTGTTA | mtDNA qPCR |
| Human mtDNA tRNA reverse | AAGGGTGGGTAGGTTTGTTG | mtDNA qPCR |
Treatment of HEK293T cells with cisplatin and B02.
HEK293T cells were grown to confluence in 10 cm dishes, media was removed and replaced with 3 mL serum-free Dulbecco’s modified Eagle’s media or in media containing 50μM cisplatin (Aldrich), 5μM B02 (Calbiochem), or 50μM cisplatin and 5μM B02. Cells were incubated for 2 h at 37 °C. The media was removed and low molecular weight DNA was isolated using a Hirt protocol [57] modified as follows: 400 μL of 0.6 % SDS/0.01 M EDTA was added to the cells. After 12 min, the cells were collected using a rubber policeman into a 1.5 mL microcentrifuge tube. NaCl was added to a final concentration of 1 M, the tubes were mixed by gentle inversion, and stored at 4 °C overnight. The samples were sedimented at 16,000 × g for 30 min at 4 °C. The supernatant was kept and sedimented again at 16,000 × g for 30 min at 4 °C. DNA in the supernatant was precipitated by addition of 1/10 vol of 3 M NaOAc and 2 volumes of 100 % ethanol. The samples were incubated on ice for 30 min and subsequently sedimented at 16,000 × g at 4 °C for 10 min. The DNA pellet was washed with 70 % ethanol, air dried, and resuspended in 200 μL of TE buffer.
Quantitation of mitochondrial DPC content.
Low molecular weight DNA isolated from HEK293T cells treated with various drugs as described above was divided in half (100 μL each). One aliquot was stored separately and the other received 5 μL of 10 % SDS and was heated at 65 °C for 10 min. Subsequently, 10.5 μL of 1 M KCl was added and was placed on ice for 10 min. The sample was sedimented for 10 min at 16,000 × g at 4 °C. The supernatant was removed and subjected to centrifugation at 16,000 × g for 10 min at 4 °C. This supernatant, as well as the non-treated sample were diluted 1:100 in TE buffer. Quantitative PCR (qPCR) was performed on 1 μL of each of the diluted samples with primers (100pMol each) specific for human mitochondrial tRNA gene (human mtDNA tRNAf and human mtDNA tRNAr, Table 1) using SYBR Green PCR Master Mix (Invitrogen) in a final volume of 60 μL. Samples were loaded onto a 96-well PCR plate (18 μL/well, in triplicate) and subjected to qPCR using a StepOnePlus real time PCR machine (Applied Biosystems) for 30 cycles using an initial pre-melt of 90 °C followed by 30 cycles of: 90 °C for 15 s and 65 °C for 1 min. The triplicate Ct values for each sample were averaged. Because potassium dodecyl sulfate (KDS) is insoluble, treatment with SDS and KCl will cause proteins (and any covalently linked DNA) to precipitate; qPCR on SDS/KCl treated and non-treated samples provides a measure of the relative amount of mitochondrial DNA that contain DPCs. The values derived are referred to as ‘delta Ct’ or difference in cycle threshold values. These delta Ct values can be used to determine how much mitochondrial DNA was precipitated following SDS/KCl treatment, which in turn is a measurement of the percentage of mitochondrial genomes that harbor cisplatin-induced DPCs, using the formula: 2delta Ct = fold loss of DNA induced by KCl/SDS treatment. For example, if cisplatin treatment induced DPCs in 50 % of mitochondrial genomes, half of mitochondrial DNA would precipitate due to KCl/SDS treatment. As a consequence, the Ct value obtained from the KCl/SDS-treated sample would be one ‘cycle’ higher (21) than that observed in the non-KCl/SDS treated sample. To ensure that this analysis focused on cisplatin-induced mitochondrial DPCs the data presented in Fig. 6 were obtained by subtracting of background DPC levels observed in non-cisplatin treated cells from those observed in cisplatin treated cells.
Fig. 6. Exposure to the Rad51 inhibitor B02 results in enhanced levels of DPC formation in mitochondrial DNA of cells exposed to cisplatin.
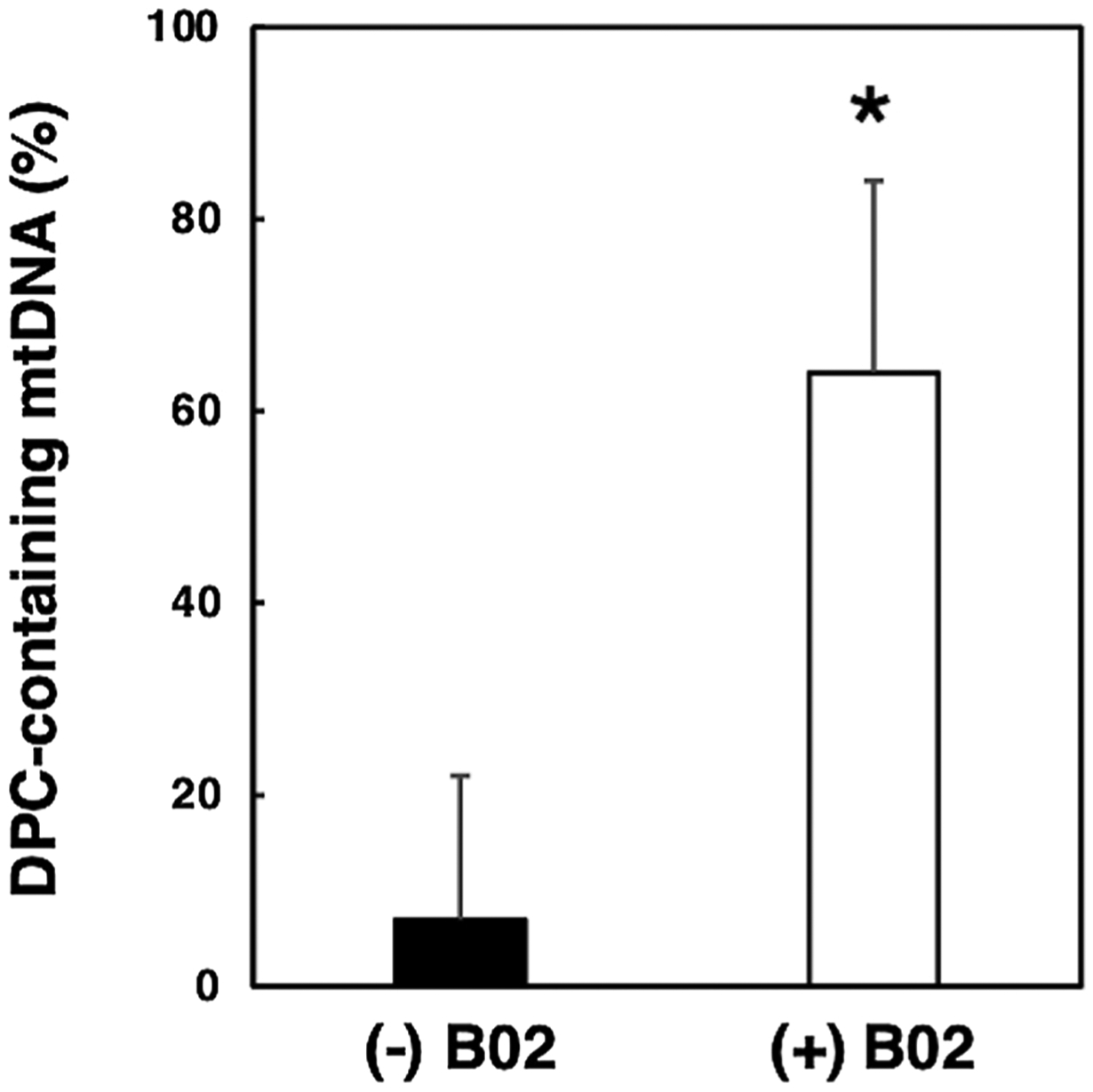
Human HEK293T cells were incubated in the absence or presence of 100 μM cisplatin for two h in the presence or absence of the Rad51 inhibitor B02. Total cellular DNA was isolated and KCl/SDS depletion experiments performed as described in the legend in Fig. 5. Quantitative PCR was performed using a primer pair specific for the human mitochondrial leucine tRNA gene. These data were used to determine the respective percent of mitochondrial DNA (mtDNA) that contained cisplatin-induced DPCs in the absence (−) or presence (+) of the Rad51 inhibitor B02. Results depict mean +/− SEM, N = 5, * P < 0.05, Student t-test.
Mitochondria purification.
Note: cells and reagents were kept cold or on ice at all times during purification. Hamster V79 or human HEK293T cells were grown to confluency in four 15 cm dishes and mitochondria purified as described [58]. HEK293T cells used to examine the involvement of Rad51 in DPC repair on plasmid DNA were treated with 5μM B02 for 1 h at 37 °C prior to mitochondria purification. Cells were scraped into conical tubes and washed twice with 5 mL of buffer containing 1 mM Tris–HCl pH 7.0, 0.13 M NaCl, 5 mM KCl, and 7.5 mM MgCl2 in a table-top centrifuge at 1000 × g for 5 min at room temperature. The cell pellet was resuspended in half the cell volume with 0.1X incubation buffer (IB buffer) consisting of 4 mM Tris–HCl pH 7.4, 2.5 mM NaCl, and 0.5 mM MgCl2 and broken with 10 strokes of a dounce homogenizer. A volume of 10X IB buffer (400 mM Tris–HCl pH 7.4, 250 mM NaCl, and 50 mM MgCl2) equivalent to one-ninth that of the original cell pellet was then added, and the sample transferred to a microcentrifuge tube and spun in a table-top microcentrifuge at 326 × g for 5 min at 4 °C. The supernatant was then transferred to a new tube and sedimented again to completely rid the sample of any unbroken cells and nuclei. To purify mitochondria, the supernatant was spun in a table-top microcentrifuge at 21130 × g for 10 min at 4 °C. The mitochondria were then washed with 0.5 mL of 1X IB buffer (40 mM Tris–HCl pH 7.4, 25 mM NaCl, and 5 mM MgCl2) and sedimented again using the same conditions stated above. Purified mitochondria were then resuspended in the respective buffer for the bicinchoninic acid assay, cytochrome c oxidase assay, or electroporation assay listed below.
Preparation of Nuclear Extract.
HEK293T cells were grown to confluence in four 15 cm dishes. Media was removed, and cells were scraped using a rubber policeman into a chilled 15 mL conical tube. Cells were pelleted at 1000 × g for 5 min in a tabletop centrifuge, and washed in 5 mL Phosphate-buffered saline. Wash was repeated two times. After the final wash, the pellet was resuspended in 1 mL Buffer A (10 mM Tris-HcL, pH 7.4, 10 mM MgCl2, 10 mM KCl). PMSF was added to a final concentration of 1 mM and cells were homogenized with 20 strokes in a loose pestle homogenizer. Nuclei were then subjected to centrifugation at 1100 × g for 8 min at 4 °C. The pellet, which contains the nuclear fraction, was resuspended in 2 mL of Buffer A containing 350 mM NaCl. Protease inhibitors were added to the following final concentrations: Pepstatin, 0.7 μg/mL, leupeptin, 0.1 μg/mL, aproptinin, 1 μg/mL, PMSF, 1 mM. The extracted nuclei were incubated on ice for 1 h, and then centrifuged at 70000 rpm (189,336 × g) in a Beckman TLA 100.3 rotor for 30 min at 2 °C. The clear supernatant was adjusted to 10 % glycerol.
Histone Acetyltransferase Assay.
Histone acetyltransferase (HAT) activity was measured using a kit (Sigma Aldrich, Catalog no. EPI001), following the manufacturer’s recommended protocol. Briefly, 50 μg of nuclear or mitochondrial extract were diluted to a final volume of 40 μL per well in a 96-well plate. For background reading, a sample was prepared with 40 μL water alone. To each well, HAT Substrate I, HAT Substrate II, NADH generating enzyme, and HAT Assay buffer were added. The plate was then incubated at 37 °C for 80 min, and then OD440 was measured in a Tecan Infinite M1000 Promicroplate reader. OD measurements were carried out every 20 min, and the plate was incubated at 37 °C in between measurements. All samples were performed in duplicate. Background reading from buffer and reagents was subtracted from all measurements. HAT activity was determined using the formula HAT activity = (ΔOD/Δt)/ε* protein (ng) where ΔOD/Δt = Change in OD440 per minute, and ε = 37000 M-1cm-1.
Bicinchoninic acid assay (BCA).
Following centrifugation, mitochondria samples were either resuspended in 80 μL of lysis buffer (10 mM Tris–HCl pH 7.4, 10 mM MgCl2, and 10 mM KCl) or water. Diluted albumin (BSA) standards were then prepared in lysis buffer or water according to the manufacturer’s protocol (23225, Thermo Scientific) and 25 μL of each diluted standard was added to a 96-well plate (3595, Costar) along with 25 μL of each sample in either lysis solution or water (loaded in triplicate). The protein concentration was then determined according to the manufacturer’s protocol (23225, Thermo Scientific).
Cytochrome c Oxidase Assay.
After measuring the protein concentration, mitochondria were spun in a table-top microcentrifuge at 21130 × g for 10 min at 4 °C and resuspended in enzyme dilution buffer (10 mM Tris–HCl, pH 7.0, 250 mM sucrose) with or without 1 mM n-Dodecyl β-d-maltoside to a concentration of 0.2 mg/mL. Samples were then incubated at 4 °C for 10 min and absorbance measured at 550 nm using a spectrometer. The absorption of cytochrome c at 550 nm changes with its oxidation state and was recorded over time to determine the ΔA550/minute for each sample. Because n-Dodecyl β-d-maltoside disrupts the mitochondrial membrane, measurement of the rate at which reduced cytochrome c becomes oxidized in the presence or absence of this nonionic detergent can be used to determine the integrity of membranes of isolated mitochondria using the following equation: percent mitochondrial membrane integrity =[(cytochrome C oxidase activity in the presence of 1 mM n-Dodecyl β-d-maltoside-cytochrome c oxidase activity in the absence of n-Dodecyl β-d-maltoside)/(cytochrome c oxidase activity in the presence of n-Dodecyl β-d-maltoside] × 100.
Electroporation of purified mitochondria.
The final mitochondria pellet was resuspended in 50 μL 0.33 M sucrose/10 % glycerol and mixed with 100 ng of an 8-oxoguanine or DPC-containing plasmid (15 μL) (see above). For co-electroporation experiments, 100 ng of DPC-containing DNA was mixed with 25 ng (1 μL) of undamaged M13mp18 (New England Biolabs, Beverly, MA) or 25 ng (1 μL) of pQe30 (Addgene). Note: 1 μL of the DNA mixture was saved and diluted in 500 μL of water to be used as a ‘time 0’ sample for SSPE-qPCR (see section below). Samples were then transferred to a chilled, 1 mm gap cuvette (Fisher Scientific, Waltham, MA) and electroporated using an ECM 630 Electro Cell Manipulator (BTX, Holliston, MA) at a field strength of 1000 V/cm, resistance of 400Ω, and 25μF capacitance. Following electroporation, cuvettes were rinsed with 100 μL of 1X IB buffer + 10 % glycerol (40 mM Tris–HCl pH 7.4, 25 mM NaCl, 5 mM MgCl2, and 10 % glycerol). An additional 900 μL of 1X IB buffer + 10 % glycerol was then added, mixed, and sedimented in a table-top microcentrifuge at 21130 × g for 10 min at 4 °C. The supernatant was then discarded, and the pellet washed three times with 200 μL of 1X IB buffer + 10 % glycerol and sedimented at the same conditions described above. After the washes, the final pellet was resuspended in 50 μL of a solution containing 40 mM Tris–HCl pH 7.4, 25 mM NaCl, 5 mM MgCl2, 10 % glycerol, 1 mM pyruvate, 1 mM ATP (Teknova), 1 mg/mL BSA (New England Biolabs), and 10μM dNTPs (Thermo Scientific) and incubated in a 37 °C water bath for 1, 2, or 4 h. Following incubation, samples were sedimented in a tabletop microcentrifuge at 21130 × g for 10 min at 4 °C. The supernatant was discarded and the pellet washed twice with 200 μL of 1X IB buffer + 10 % glycerol and spun at the same conditions described above [58].
To confirm that DNA repair was occurring within a membrane-bound compartment, the following experiment was performed. Mitochondria that had been electroporated with a repair substrate and permitted to recover, as outlined above, were resuspended in 200 μL DNase buffer containing 66 units DNase (New England Biolabs), 10 % glycerol, 10 mM Tris–HCl pH 8, and 1 mM MgCl2 and incubated in a 37 °C water bath for 30 min. Following incubation, samples were sedimented in a table-top microcentrifuge at 21130 × g for 10 min at 4 °C. The supernatant was discarded and the pellet washed twice with 200 μL of a washing buffer containing 10 % glycerol, 10 mM Tris–HCl pH 7.4, and 1 mM EDTA and sedimented at the same conditions described above [58].
For all other experiments not utilizing DNase, mitochondria were resuspended in 300 μL lysis buffer (0.5 % SDS, 10 mM Tris–HCl pH 7.4, 150 mM NaCl, 1 mM EDTA, and 100 μg proteinase K (New England Biolabs) and incubated in a 37 °C water bath for 15 min. Samples were then mixed with 300 μL of a 50:50 phenol: chloroform mixture. To create the 50:50 phenol: chloroform mixture, equal volumes of buffer-saturated phenol (Invitrogen) and chloroform (Acros) were mixed and spun in a table-top centrifuge at 1000 × g for 5 min at room temperature. The bottom layer of the mixture was then added to the sample, mixed, and spun in a table-top microcentrifuge at 21130 × g for 5 min at room temperature. The top layer was then transferred to a new microcentrifuge tube and ethanol precipitated using 18 μL of 5 M ammonium acetate (final concentration 0.3 M), 12 μL glycogen (Invitrogen, 5 mg/mL), 700 μL of 100 % ethanol, and precipitated at −20 °C overnight.
Purification of nuclei.
Note: cells and reagents were kept cold or on ice at all times during purification. Eight 15 cm dishes of wild-type Chinese hamster lung fibroblasts (V79) were grown to confluency, scraped with a rubber policeman into a 15 mL conical tube, and spun down in a tabletop centrifuge for 5 min at 1000 × g. Cells were washed twice with 5 mL of 1X PBS and resuspended in 2 mL of Buffer A (10 mM Tris–HCl pH 7.4, 10 mM MgCl2, 10 mM KCl, and 1 mM DTT). Resuspended cells were then incubated on ice for 10 15 min and broken with 20 strokes of a dounce homogenizer and ‘loose’ pestle [59]. Broken cells were spun in a table-top microcentrifuge at 1150 × g for 8 min at 4 °C and washed with 0.5 mL of NIB-250 Buffer (15 mM Tris–HCl pH 7.5, 60 mM KCl, 15 mM NaCl, 5 mM MgCl2, 1 mM CaCl2, 250 mM sucrose, and 1 mM DTT) [60]. The resuspended pellet was then spun using the same conditions described above. The final nuclei pellet was resuspended in 800 μl NIB-250 Buffer, divided in half, and mixed with 100 ng of 8-oxo-guanine or DPC-containing plasmid. Samples were then transferred to a chilled, 4 mm gap cuvette (Fisher Scientific, Waltham, MA) and electroporated using a ECM 630 Electro Cell Manipulator (BTX, Holliston, MA) at a field strength of 300 V/cm, resistance of 25Ω, and 950μF capacitance. Following electroporation, samples were transferred to a microcentrifuge tube, spun down using the same conditions as above, and resuspended in 400 μl Incubation Buffer (40 mM Tris–HCl pH 7.4, 25 mM NaCl, 5 mM MgCl2, 10 % glycerol, 1 mM pyruvate, 1 mM ATP (Teknova), 1 mg/mL BSA (New England Biolabs), and 10 μM dNTPs (Thermo Scientific) [58]. Nuclei were incubated in a 37 °C water bath for 2 h and lysed with 0.6 % SDS/0.01 M EDTA (final concentration) at room temperature for 12 min. Chromosomal DNA was then precipitated by adding NaCl to a 1 M final concentration and incubated at 4 °C overnight. The next day, samples were spun in a table-top microcentrifuge at 21130 × g for 30 min at 4 °C. The supernatant was ethanol precipitated overnight at −20 °C and resuspended in 50 μl of water [61]. One microliter of this sample was used to calculate percent repair using SSPE-qPCR as previously described [47].
Quantification of plasmid repair.
Samples recovered from electroporated mitochondria as described were sedimented in a table-top microcentrifuge at 21130 × g for 10 min at 4 °C, allowed to dry, and resuspended in 30 μL of water. For experiments assessing the BER of the 8-oxoguanine lesion, a modification of the technique of Lee et al. [62] was used. Half of the 30 μL sample was mixed with OGG1 (2 pmol) in a buffer containing 100 mM NaCl, 1 mM MgCl2, 20 mM Tris–HCl pH 7.0, and water to reach a final volume of 30 μL and incubated in a 37 °C water bath for 30 min. One microliter of untreated or OGG1-treated samples, as well as ‘time 0’ samples, were subjected to strand-specific primer extension-quantitative PCR (SSPE-qPCR, see [47] for details) to determine the percent of recovered plasmid molecules that had undergone repair, see below. For experiments assessing repair of DPCs the OGG1 treatment was omitted, and one microliter of recovered samples was directly subjected to analysis as outlined below. Briefly, the SSPE-qPCR assay relies on quantitative real-time PCR using primers that flank the site of the lesion. One primer, referred to as ‘Rt’ hybridizes to the damaged strand, and the other primer, referred to as ‘Lt’, hybridizes to the undamaged strand (see Fig. 1). In the absence of repair, extension of the Rt primer is halted at the site of the DNA lesion (either the hOGG1-induced strand break, or the DPC). In contrast, extension of the Lt primer is unimpeded. In order to enhance our ability to detect repair of the damaged strand, eight rounds of strand-specific primer extension (SSPE) are performed using the Rt primer, after which the Lt primer is added, and qPCR performed. In parallel, a control experiment is performed in which qPCR is performed in the absence of any primer extension reactions. Subtracting the Ct (cycle threshold) value for the ‘SSPE-qPCR’ amplification from that ‘qPCR alone’ amplification yields a ‘delta Ct’, or ΔCt value. One then calculates the percentage of DNA repair using the formula: DNA repair = 2 ΔCt/23 × 100 [62,63]. For experiments in which undamaged, homologous donor plasmid was co-electroporated with plasmids containing DPCs, the percent repair was calculated by subtracting the apparent percent repair value of samples recovered from electroporated mitochondria from the 0 h, non-transfected DNA. See text for details.
Fig. 1. Repair of an 8-oxoguanine lesion on plasmid DNA electroporated into purified mitochondria.
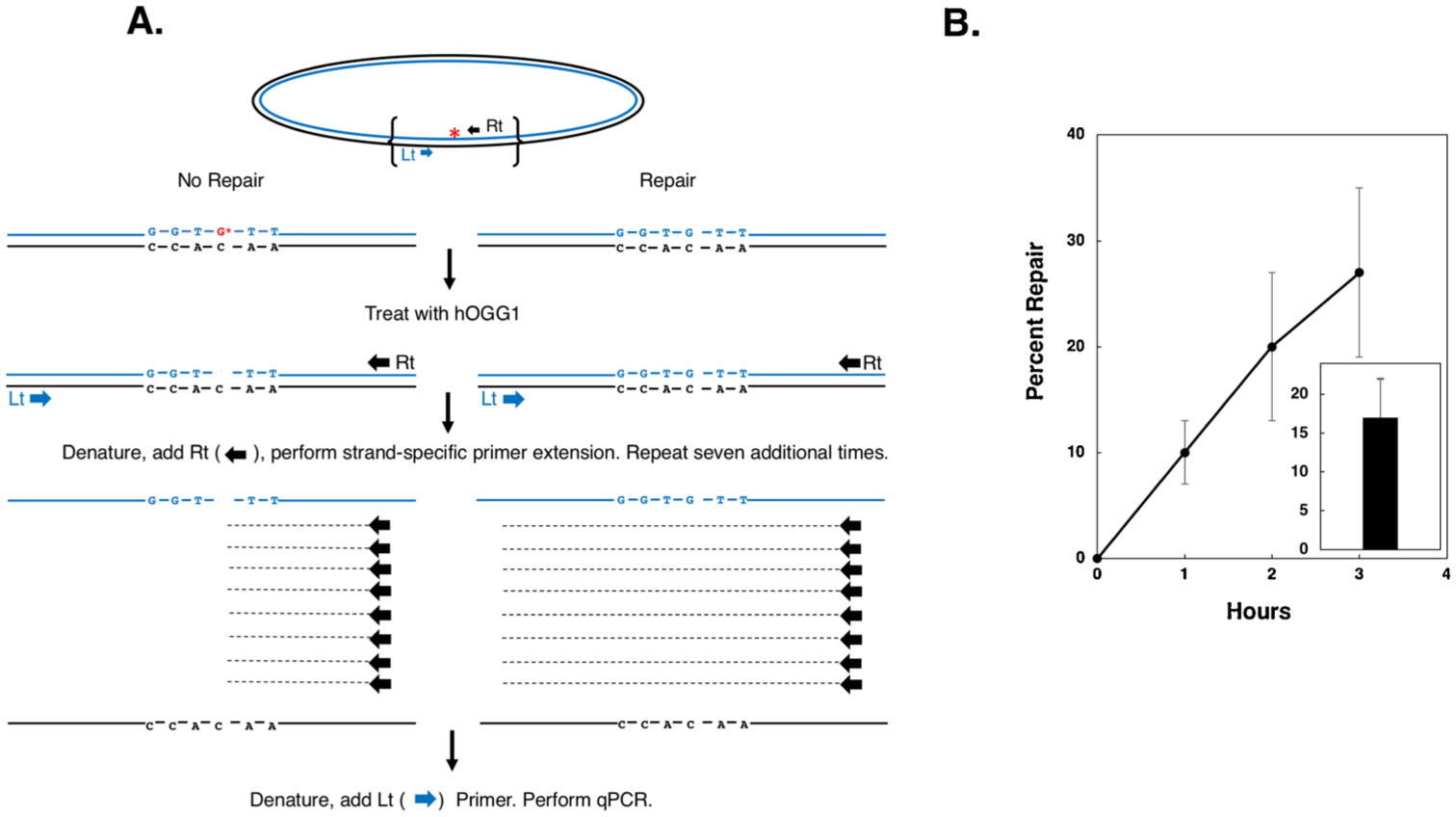
A. Schematic of strand specific-qPCR repair of a plasmid containing a site-specific single 8-oxoguanine residue. Line 1: the plasmid is depicted, with the two complementary strands in blue and black respectively and the 8-oxoguanine residue indicated by a red asterisk. Line 2: the region of the plasmid demarked by brackets in the top line is presented illustrating, on the left, the un-repaired duplex sequence with the 8-oxoguanine residue indicated by a red G*, and on the right, the repaired duplex. Line 3: Depiction of plasmid recovered following electroporation into mitochondria and treated with recombinant human oxoguanine glycosylase (hOGG1). Unrepaired 8-oxoguanine residues are converted to an abasic site with adjacent DNA single strand break. Line 4: depiction of eight sequential rounds of primer extension reactions performed using Taq polymerase and the ‘Rt’ primer. As indicated, the presence of the abasic site/strand break blocks primer extension at the site of the lesion, resulting in incomplete extension products that cannot serve as templates for subsequent amplification utilizing the ‘Lt’ primer. Line 5: The ‘Lt’ primer is added and qPCR performed. B. Plasmid substrates containing a single 8-oxoguanine residue were electroporated into mitochondria purified from HEK293T cells, recovered at 1, 2, and 3 h post-electroporation and BER-mediated repair determined as depicted in (A). Results depict mean percent repair +/− the SEM, N = 3. Inset: Plasmid containing an 8-oxoguanine residue was electroporated into Chinese hamster lung fibroblast V79 cells, recovered 2 h post-electroporation and BER-mediated repair determined. Results depict mean percent repair +/− the SEM, N = 4.
KCl/SDS precipitation of mitochondrial DNA treated with cisplatin.
Twenty 15 cm dishes of HT1080 cells were grown to confluency. Five mL of a 5 mM cisplatin stock was mixed with serum-free media in a total volume of 250 mL. Growth media was then removed and replaced with 25 mL of serum-free media with or without drug and incubated at 37 °C for 3 h. Following incubation, cells were washed with 5 mL of 1X PBS, scraped into conical tubes, and sedimented in a table-top centrifuge at 1000 × g for 5 min at room temperature. Cell pellets were then resuspended in 4 mL isolation buffer (0.3 M mannitol, 0.1 % BSA, 0.2 mM EDTA, and 10 mM HEPES adjusted to a final pH of 7.4) and lysed in a glass dounce homogenizer (Wheaton) for 5 strokes with the loose pestle and 5 strokes with the tight pestle. Samples were then divided into 1.5 mL microcentrifuge tubes and sedimented at 1000 × g in a table-top microcentrifuge for 10 min at 4 °C. The supernatant was transferred to a new tube and spun in a table-top microcentrifuge at 14000 × g for 15 min at 4 °C to pellet the mitochondria [64]. Mitochondrial DNA was purified using alkaline lysis with SDS and resuspended in 40 μL TE (pH 8.0) containing 20 μg/mL RNase A [65]. Each sample was then divided in half and subjected to KCl/SDS precipitation. One microliter of 10 % SDS (final concentration 0.5 %) was added to one tube and omitted from the other, and both samples were incubated at 65 °C for 10 min. Two microliters of 1 M KCl (100 mM final concentration) was added to the sample containing SDS and both samples were incubated on ice for 5 min and then sedimented in a table-top microcentrifuge at 21130 × g for 10 min at 4 °C to precipitate any protein-bound DNA [61]. The supernatants (containing mitochondrial DNA that does not contain any DPCs) was then resolved on a 0.8 % agarose gel at 60 V for 2 h, stained with ethidium bromide (0.5 μg/mL) for 30 min and destained in water for 30 min. Gels were imaged for 0.3 s on a Thermo Scientific myECL Imager and DNA bands quantified using ImageJ. ImageJ creates density histograms of each DNA band permitting one to quantitate the relative amount of mitochondrial DNA present in the treated versus the un-treated samples. These values (depicted in Fig. 5 in arbitrary units) can be used to calculate the percent of mitochondrial genomes that harbor one or more DPCs (and were thus precipitated upon treatment with KCl/SDS), using the formula: Percent of mitochondrial genomes containing one or more DPC=[1-(amount of mitochondrial DNA present in KCl/SDS-treated sample/amount of mitochondrial DNA present in un-treated sample)] × 100.
Fig. 5. Formation of cisplatin-induced DNA-protein crosslinks in human cells.
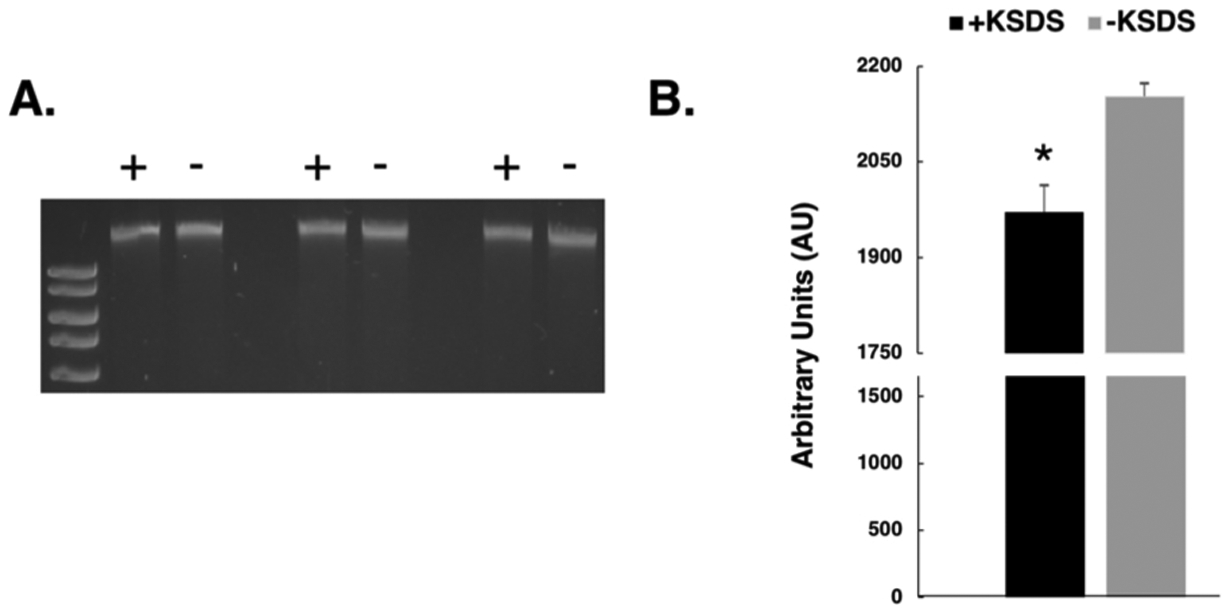
Human HT1080 cells were treated with 100μM cisplatin for three hours and mitochondrial DNA purified as described in methods. A. Purified DNA was divided in equal aliquots which were incubated in the presence (+) or absence (−) of KCl/SDS and DPC-containing DNA sedimented by centrifugation as described in the methods section. Soluble material was electrophoretically resolved on a 0.8 % agarose gel in triplicate and stained with ethidium bromide. B. Scanning densitometry was performed on the gel to determine the relative amount of mitochondrial DNA present in the KCl/SDS treated (black bar) and untreated samples (grey bar). Results depict the mean amount of DNA present +/− SEM, N = 3. *, P < 0.05, Student t-test.
Cloning of a 475bp human mitochondrial insert into M13mp18.
A 475 base pair KpnI/SacI fragment of human mitochondrial DNA was ligated into a modified M13mp18 plasmid containing a kanamycin resistance gene [47] that has been digested with these same enzymes, at a vector to insert ratio of 1:5. One microliter of the ligation mixture was transformed into NEB Turbo electrocompetent bacteria (New England Biolabs) and plated on LB agar plates with kanamycin (50 μg/mL). Individual clones containing the mitochondrial insert were screened by restriction digest analysis of double-stranded DNA. Single-stranded virion DNA was subsequently purified, primer extended with an 8-oxoguanine-containing oligonucleotide (M13–8oxo-mito, Table 1) and crosslinked as described above. Crosslinked plasmids containing the human mitochondrial insert (M13-Mito) were electroporated as described above into purified mitochondria from the HEK293T cells in the presence or absence of an undamaged donor plasmid and repair quantified after three hours using SSPE-qPCR.
3. Results
3.1. Purified mitochondria are intact and functional
Given abundant evidence that mammalian mitochondria possess a robust base excision repair machinery, we first set out to determine whether plasmid substrates containing the BER substrate 8-oxoguanine (Supplemental Fig. 1A) electroporated into isolated mammalian mitochondria would be subject to efficient repair. To perform this experiment, mitochondria were isolated from wild-type V79 hamster cells and electroporated with 100 ng of double-stranded M13mp18 DNA containing a single 8-oxoguanine residue (Table 1). Post electroporation, mitochondria were washed three times and incubated for one, two, or three hours at 37 °C in buffer containing ATP, BSA, and dNTPs. After this incubation, plasmid DNA was recovered and 8-oxoguanine repair efficiency measured using a modified PCR-based, DNA repair assay [62]. To further assess the membrane integrity of the mitochondria following purification, we measured the activity of the inner mitochondrial membrane marker enzyme cytochrome c oxidase before and after lysis. Cytochrome c oxidase activity measurements performed on three separate preparations indicated that approximately 72 % of the purified mitochondria were intact (Table 2). Enzyme marker analysis revealed that the purified mitochondria possess negligible levels of histone acetyltransferase activity (Supplementary Fig. 3), suggesting they are nearly devoid of contaminating nuclear proteins.
Table 2. Measurement of the outer membrane integrity of purified mitochondria.
Mitochondria were purified from human HEK293T cells and membrane integrity determined by measuring cytochrome c oxidase activity in the presence and absence of the detergent n-Dodecyl β-d-maltoside, as described in the methods section. Results depict mean percent repair +/− the SEM, N = 3.
| Abs550 +Detergent | Abs550 −Detergent | % Membrane Integrity |
|---|---|---|
| 0.156 | 0.048 | 69 |
| 0.126 | 0.03 | 76 |
| 0.102 | 0.03 | 71 |
We have previously used an assay, referred to as the Strand Specific Primer Extension-quantitative real-time PCR (SSPE-qPCR, depicted schematically in Fig. 1A and described in the methods section), to examine repair of plasmids bearing a variety of different types of DNA damage transfected into wild-type and repair-deficient mammalian cells [46,47]. As the cartoon in Fig. 1A indicates, primer extension of a repaired substrate generates eight (or 23) additional strands of DNA that can subsequently be amplified during the qPCR phase of the assay. In contrast, treatment of unrepaired plasmid with oxoguanine glycosylase results in the formation of an abasic site with an adjacent DNA strand break (Supplemental Fig. 1B) which blocks DNA polymerase during the eight rounds of primer extension. In order to calculate the percentage of recovered plasmid molecules that have undergone repair, two reactions are performed; one in which the strand specific primer extension (SSPE) step is omitted but qPCR is performed, and the other in which both the SSPE and the qPCR steps are performed. The Ct or cycle threshold values are determined for each of the two respective qPCR reactions, and the latter is subtracted from the former to generate a ‘delta Ct’ (ΔCt). Percent repair is then calculated using the formula: DNA repair = 2 ΔCt/23 × 100. Results from these experiments showed that 8-oxoguanine-containing plasmids were repaired at near linear kinetics in mitochondria purified from HEK293T cells (y = 9.1x+0.6, R2 = 0.9935, Fig. 1B). An analogous series of experiments were performed using mitochondria purified from Chinese hamster lung fibroblast V79 cells in which repair was measured two h post-electroporation. As the results in Fig. 1B, inset reveal, BER repair activity in mitochondria obtained from hamster-derived cells was essentially identical to that observed in mitochondria derived from human cells (17 % vs 20 % repair). To rule out the possibility that repair observed was due to contaminating nuclear or cytosolic enzymes, mitochondria were incubated with DNase prior to plasmid recovery. Results from these experiments showed no inhibition of repair in the presence of DNase (data not shown). This finding that the electroporated 8-oxoguanine-containing plasmid gained access to intact mitochondria, from which the DNase was excluded, is consistent with the interpretation that the repair was catalyzed by mitochondrial enzymes.
3.2. Repair of DPC-containing plasmids electroporated into mitochondria
We next generated a DPC substrate by trapping recombinant human oxoguanine glycosylase onto the 8-oxoguanine-containing plasmid described above by incubating these two molecules together in the presence of borohydride (Supplemental Fig. 1C) [46,47]. Unlike the 8-oxoguanine substrate, the DPC substrate does not need to be processed prior to analysis via SSPE-qPCR because the DPC lesion blocks polymerase-mediated primer extension. We subsequently used the strategy described above to examine the repair efficiency of DPC-containing plasmids that had been electroporated into purified mammalian mitochondria and subsequently recovered. Despite numerous efforts, we failed to detect any evidence for repair of this DPC substrate (data not shown). Our previous results [46,47], as well as that of others [53] have indicated that DPC substrates transfected into mammalian cells undergo efficient repair via the NER pathway, a mechanism absent from the mitochondria. Because we have more recently determined that mammalian nuclear DPC repair also occurs via a recombinational mechanism (Chesner et al., manuscript in preparation), we hypothesized that DPCs present on a plasmid containing homology to the mitochondrial genome would be more efficient substrates for repair. To test this hypothesis, we generated a DPC repair substrate with the lesion present within a 475 base pair fragment homologous to the human mitochondrial genome that had been cloned into M13mp18 (Table 1). This substrate, referred to as pM13 Mito (Fig. 2A), was electroporated into isolated mitochondria, incubated for two or three h, and analyzed using the SSPE-qPCR assay described above. In contrast to experiments using a plasmid DPC substrate lacking homology to the mitochondrial genome, in which no repair was observed, we determined that approximately 20 % of the recovered pM13 Mito plasmid DNA had undergone DPC repair by three hpost-electroporation (Fig. 2B).
Fig. 2. DPC-containing plasmids are repaired via HR in purified mitochondria.
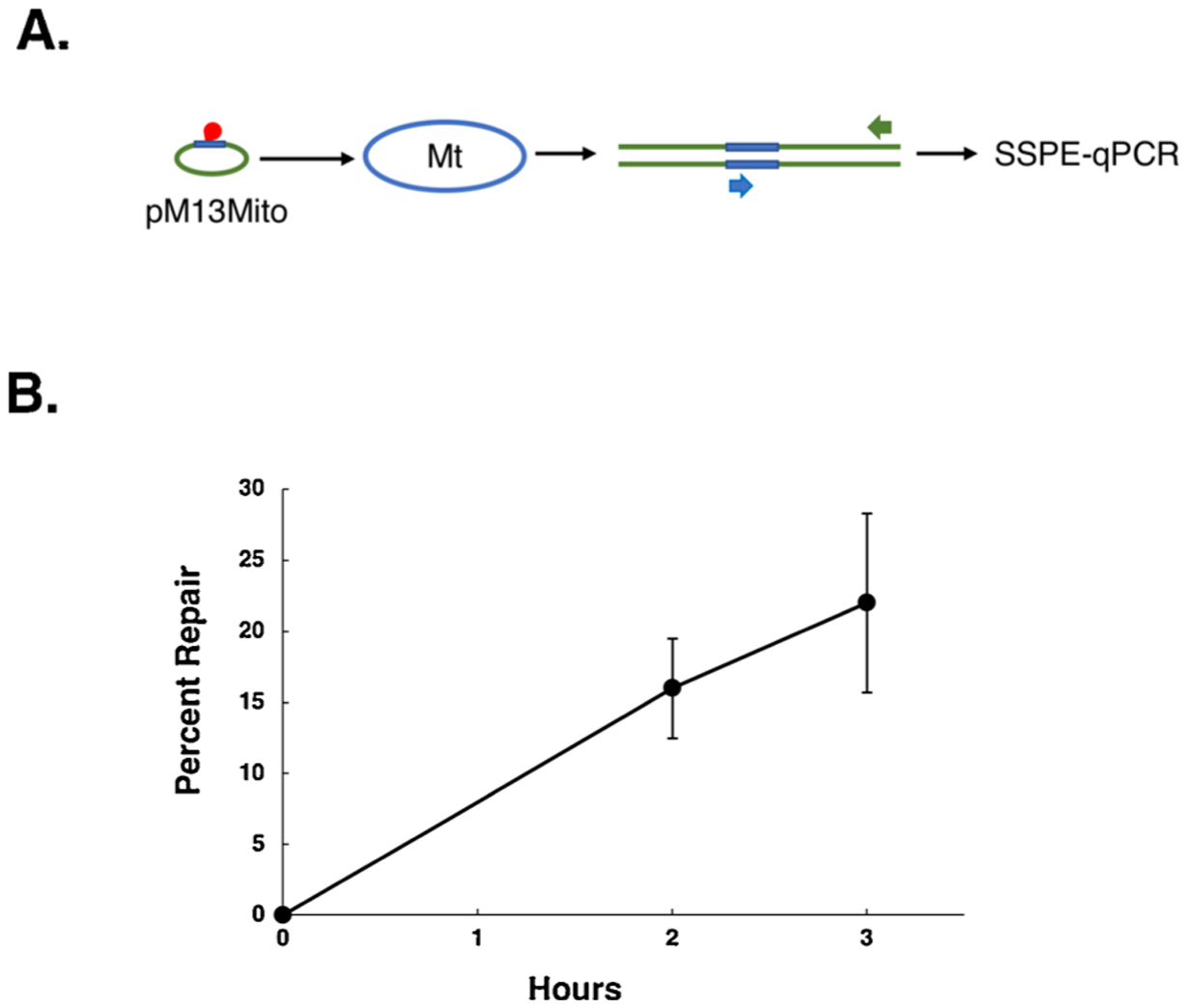
A. Schematic representation of mitochondrial DPC repair. Plasmid pM13 Mito (depicted by green oval with blue arc) harboring a DPC (indicated in red) within the portion of pM13 Mito that is homologous to the mitochondrial genome (depicted in blue) is electroporated into purified mitochondria (indicated by blue oval), recovered, and subjected to SSPE-qPCR using a pair of primers (indicated by Blue and Green arrows) that, respectively, hybridize to mitochondrial DNA- and M13-specific portions of the plasmid. B. Plasmid pM13 Mito containing a single DPC residue was electroporated into mitochondria purified from human HEK293T cells, recovered following a two- or three-hincubation period, and analyzed by SSPE-qPCR as depicted in (A). Results depict mean percent repair +/− the SEM, N = 2 and 7 (two-hour and three-hour incubation, respectively).
The most likely interpretation of these results is that mammalian mitochondrial DPC repair is catalyzed via a recombinational mechanism. To further test this hypothesis, we performed the experiments depicted in Fig. 3A, in which DPC-bearing dsM13 plasmids were electroporated into mammalian mitochondria along with undamaged, non-homologous plasmid, or with undamaged, homologous dsM13plasmid. We performed parallel experiments utilizing mitochondria isolated from either Chinese hamster lung fibroblast-derived V79 cells, or human HEK293T cells. As the results in Fig. 3B illustrate, we failed to observe DPC repair in experiments in which the undamaged plasmid was not homologous to the DPC-containing dsM13 plasmid. In contrast, co-electroporation of an undamaged, homologous plasmid resulted in detectable DPC repair in mitochondria isolated from either human (9%) or hamster (11 %) cell lines. The results presented in Fig. 3B were obtained from analysis of plasmids recovered following a two-h post-electroporation incubation period. To extend these findings, an additional series of experiments were performed in which DPC-containing dsM13 was co-electroporated into mitochondria from HEK293T cells along with an undamaged, homologous donor and plasmid DNA recovered at various time points. The results from these experiments, depicted in Fig. 3C, reveal a near-linear time course of repair (y =3.2571x+1.8, R2 = 0.91) over the four-h time course examined.
Fig. 3. Inter-plasmid recombinational repair of DPCs in purified mitochondria.
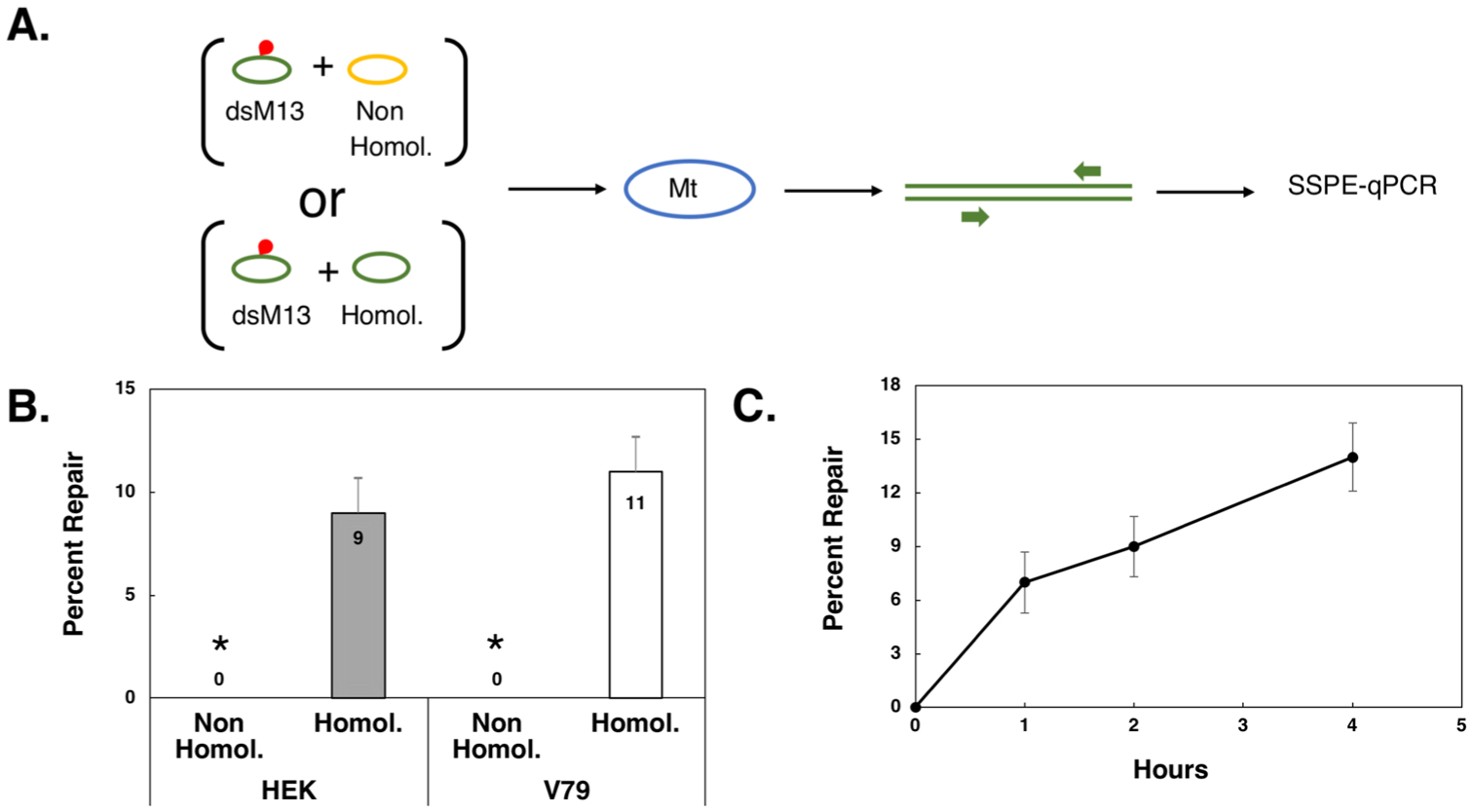
A. Schematic representation of experimental design. Double-stranded M13 (dsM13, green oval) harboring a DPC (indicated in red) was co-electroporated with a non-homologous plasmid (yellow oval) or homologous, undamaged dsM13 DNA (green oval) into purified mitochondria (blue oval), recovered, and subjected to SSPE-qPCR using a primer pair (indicated by green arrows) that flanked the site of the DPC. B. Plasmid DNA that had been coelectroporated into mitochondria purified from human HEK293T (HEK) or Chinese hamster lung fibroblast V79 (V79) cells as depicted in (A) was purified following a two-h incubation and the percent of DPC repair determined using SSPE-qPCR. Results depict mean percent repair +/− SEM, N = 6. * P < 0.05, Student t-test. C. DPC-containing dsM13 was coelectroporated with undamaged dsM13 into mitochondria purified from HEK293T cells and a time course of mitochondrial DPC repair performed at the indicated time points. Results depict mean percent repair, +/− SEM, N = 3.
The finding that a DPC present on a plasmid lacking homology to the mitochondrial genome was not repaired while an analogous lesion present on a plasmid containing homology was repaired, strongly supports the interpretation that the mitochondrial DPC repair occurs via a homologous recombinational mechanism. It is noteworthy that the requirement for a homologous donor plasmid distinguishes the mitochondrial repair activity described in these experiments from those observed in transfected, intact cells [47] or in electroporated, isolated nuclei (Supplemental Fig. 2). In both those systems, efficient DPC repair does not require the presence of an undamaged, homologous donor DNA molecule. Collectively, these results further support the interpretation that the repair activity observed following mitochondrial electroporation is not due to contaminating nuclear repair proteins, and is, indeed, catalyzed by mitochondrial DNA repair proteins.
We hypothesized that mitochondrial recombinational repair of DPCs was likely to occur via a mechanism broadly similar to that observed in the nucleus. We, therefore, performed additional experiments to test a number of predictions based on the Szostak double-strand break repair model of HR [66]. A defining feature of this model is that homologous recombination is initiated by a DNA double-strand break within the ‘recipient’ molecule [67]. Evidence showing that targeted DNA double-strand breaks produced by the restriction endonuclease I-SceI induce HR at chromosomal loci within mammalian somatic cells [68,69] supports this model. Therefore, we reasoned that repair substrates with a double-strand break adjacent to the DPC site would be repaired more efficiently than their counterpart molecules lacking a double-strand break. To test this hypothesis, we performed two parallel sets of experiments in which DPC-containing plasmids were electroporated into isolated mitochondria. In one set of experiments, the DPC-containing plasmid was identical to that used in the experiments described above. In the other set of experiments, the DPC-bearing plasmid had an EcoRI-induced DNA double-strand break adjacent to the site of the DPC (Table 1). In both sets of experiments, the DPC-containing plasmids were co-electroporated into mitochondria in the presence of an undamaged, donor plasmid. A total of five independent experiments were performed, three using mitochondria purified from human HEK293T cells, and two using mitochondria purified from Chinese hamster V79 cells. Data presented in Fig. 4A illustrate that the presence of a double-strand break enhanced DPC repair efficiency (measured two h post-electroporation) by approximately two-fold over that seen in control experiments in which the DPC-containing plasmid did not have a double-strand break. Analysis of the pooled data from these experiments revealed that induced double-strand breaks significantly increased DPC repair in mitochondria isolated from mammalian cells (18 % vs 10 %, P < 0.05).
Fig. 4. Characterization of mitochondrial DPC repair.
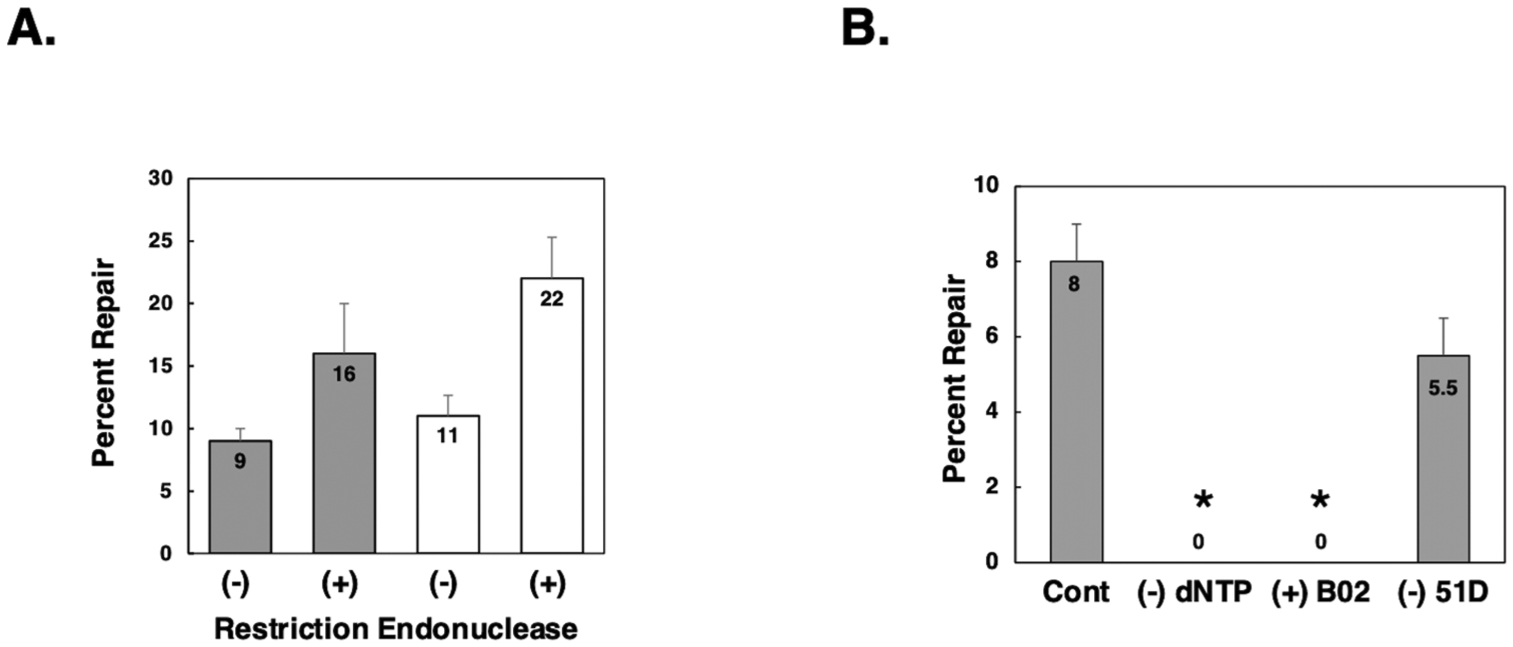
A: DPC-containing dsM13 that was undigested (−) or restriction digested with EcoR (+) was co-electroporated with homologous, undamaged dsM13 DNA into mitochondria purified from HEK293T (grey bars) or V79 (white bars) cells and repair efficiency determined as described above. The results depict the mean percent repair +/− SEM, N = 5. B: DPC-containing dsM13 DNA was co-electroporated in the presence of homologous, undamaged dsM13 into mitochondria purified from human HEK293T cells as described above, with the following modifications as noted: (Cont) normal control experiment; (−) dNTP deoxynucleotide triphosphates were omitted from the incubation buffer; (+) B02 experiments were performed in mitochondria that had been purified from HEK293T cells pretreated with the Rad51 inhibitor B02 (5μM) for one h prior to organelle isolation; (−) 51D experiments were performed in mitochondria isolated from Chinese hamster ovary cells in which the Rad51D gene had been inactivated. Values depict mean percent repair, N = 3. *, P < 0.05, Student t-test.
The Szostak model of homologous recombination postulates that 3’ protruding single-stranded DNA from the recipient strand invades the donor strand and primes de novo DNA synthesis. We, therefore, predicted that repair of electroporated plasmid DPC substrates would be dependent on the presence of deoxynucleoside triphosphates in the incubation buffer. To test this hypothesis, we performed parallel DPC repair experiments. In one set of experiments, dNTPs were omitted from the buffer in which the electroporated mitochondria were incubated, and in the other set, we performed ‘standard’ experiments, using incubation buffer that contained dNTPs. As the results in Fig. 4B illustrate, mitochondrial DPC repair was 100 % dependent on the presence of exogenous dNTPs.
The Rad51 protein is known to play a key role in homologous pairing and strand transfer of nuclear DNA [70]. A number of investigators have shown the Rad51 protein is present in mammalian mitochondria [25, 42–45]. Consequently, we performed a series of experiments in which mitochondrial DPC repair was assayed using mitochondria prepared from cells that had been incubated in the presence or absence of the Rad51 inhibitor B02 for one hour prior to organelle isolation. As is depicted in Fig. 4B, we failed to detect any evidence of DPC repair in three independent experiments. Both the inhibitory effects of B02 treatment and the omission of dNTPs from the incubation buffer had significant effects on mitochondrial DPC repair (P < 0.05).
We next examined DPC repair activity in mitochondria isolated from Chinese hamster ovary cells deficient in the Rad51D gene. We observed that, as in the case of wild-type cells, DPC repair in the Rad51D-deficient clones was entirely dependent on the presence of co-electroporated homologous, donor plasmid DNA. When a non-homologous plasmid was co-electroporated with the DPC-containing plasmid, no repair was detected (not shown); while, in contrast, co-electroporation of a homologous, donor plasmid resulted in levels of DPC repair that were not significantly different from those detected in wild-type hamster cells (Fig. 4B). While these data are not sufficient to definitively rule out a role for the Rad51D protein in mitochondrial homologous recombinational repair, they strongly support this interpretation. It is noteworthy that work published by Mishra et al. [43], documented the presence of Rad51 as well as Rad51 paralogs Rad51C and Xrcc3 in mammalian mitochondria and concluded that the other Rad51 paralogs (including Rad51D) were not present.
3.3. Repair of drug-induced mitochondrial DPCs
The above experiments examined repair of exogenous DNA electroporated in mitochondria, however, we wanted to more directly explore the role of HR in repairing xenobiotic-induced lesions on mitochondrial, genomic DNA. To do this, we first examined the formation of mitochondrial DPCs following cellular exposure to the crosslinking agent cisplatin. Human HEK293T cells were incubated in growth media containing 100μM cisplatin for 3 h and mitochondrial DNA isolated. To estimate the relative amount of mitochondrial DNA that harbored cisplatin-induced DPCs, we exploited the finding that potassium dodecyl sulfate (KDS) selectively precipitates DPC-containing DNA while leaving protein-free DNA soluble [61]. Mitochondrial DNA recovered from cisplatin-treated cells was divided into two identical aliquots, KCl and SDS were added to one (see methods for details) and then both were subjected to centrifugation at ~21 K × g for 5 min. The soluble material from three independent cisplatin-treatment experiments was resolved by agarose gel electrophoresis and stained with ethidium bromide (Fig. 5A). The respective amounts of mitochondrial DNA present in the KCl/SDS treated (+) samples and the non-KCl/SDS-treated (−) samples were determined by scanning densitometry as described in methods (Fig. 5B). The amount present in the former was then divided by that present in the latter to determine the percentage of mitochondrial DNA that was depleted, i.e. precipitated due to the presence of DPCs. This analysis revealed that KCl/SDS treatment resulted in an 8% depletion in the mitochondrial DNA from cells treated with cisplatin, consistent with the interpretation that slightly less than 10 % of mitochondrial genomes derived from cisplatin-treated cells harbored one or more DPCs.
The data in Fig. 5 illustrate that cisplatin-treated cells accumulate xenobiotic-induced mitochondrial DPCs. We reasoned that if HR repair removed these lesions, cells exposed to cisplatin in the presence of the Rad51 inhibitor B02 would harbor higher levels of mitochondrial DPCs than cells exposed to cisplatin alone. While the results presented in Fig. 5 demonstrate the viability of using scanning densitometry to directly assess mitochondrial DPC levels, we wished to pursue a more sensitive PCR-based strategy. Thus, we used qPCR to quantify the amount of DNA remaining in the supernatant of untreated and KCl/SDS-treated samples. Using the analysis outlined in the methods section, we determined that approximately 7% of mitochondria recovered from cisplatin-treated cells harbored one or more DPCs (Fig. 6). In contrast, when the cisplatin-treated cells were simultaneously exposed to the Rad51 inhibitor B02, we observed that the number of mitochondrial genomes removed by KCl/SDS precipitation increased by nearly 10-fold, from 7% to 64 % (Fig. 6). These data are consistent with the hypothesis that mammalian cells use a Rad51-dependent recombinational pathway to repair xenobiotic-induced DNA damage.
4. Discussion
For decades it was believed that mammalian mitochondria possessed a rudimentary DNA repair machinery solely comprised of the BER pathway. In recent years, a considerable body of data has emerged, suggesting that this view is inaccurate, and that additional DNA repair pathways such as mismatch repair, non-homologous end-joining, and homologous recombination may also function in this organelle [21–25], reviewed in [18,71–74]. This evolving insight, combined with our interest in cellular repair of DPCs, led us to ask whether mammalian mitochondria were able to repair these lesions. The results described in this report confirm that mitochondria isolated from mammalian cells are able to repair DPC lesions present on electroporated plasmid substrates. While abundant evidence from ours and other laboratories indicate that the DPCs are repaired in the nuclei of mammalian cells via both the NER and HR pathways [40,47,53,54], the results described herein indicate that HR represents the predominant pathway through which mitochondria repair DPCs. This conclusion is consistent with current models that mammalian mitochondria lack NER activity. Our data further reveal that in addition to catalyzing inter-plasmid recombinational-mediated repair of DPCs, endogenous mitochondrial DNA is capable of engaging with the organellar recombinational machinery to repair DPCs present within plasmid sequences homologous to the mitochondrial genome. These results are consistent with the interpretation that homologous recombination is used to repair DPCs within cellular mitochondrial DNA. Further support for this model came from our finding that treatment of cells with the Rad51 inhibitor B02 resulted in a greater than six-fold increase in cisplatin-induced mitochondrial DPC levels over those seen in cells treated with cisplatin alone. Taken together, our results provide compelling support for the conclusion that mammalian mitochondria possess a Rad51-dependent homologous recombinational repair pathway that is used to repair endogenous and xenobiotic-induced mitochondrial DPCs. A recent review article highlighted the fact that mitochondrial DNA repair enzymes are susceptible to becoming covalently crosslinked to the mitochondrial genome [75], and it is tempting to speculate that the repair activity described herein is utilized to repair these lesions. In addition to DPC repair, it is likely that this mitochondrial homologous recombinational repair machinery is also capable of repairing other types of DNA lesions. Dahal et al. [25], showed that DNA double-strand breaks stimulated HR-like activity present in mitochondrial protein extracts, suggesting that mitochondrial DNA double-strand break repair is likely to occur via a recombinational mechanism. Such an interpretation is consistent with the presence of recombinant mitochondrial genomes that have previously been detected in culture cells [32]. Indeed, a more recent paper has confirmed the presence of Holliday junctions in replicating mitochondrial DNA of mammalian cells in culture [76], suggesting that HR represents a mechanism through which stalled replication forks are restarted.
Because nuclear recombinational repair in mammalian cells also displays dependence on other Rad51 paralogs, we investigated the frequency of DPC repair in cells in which the Rad51D gene was inactivated. Interestingly, we observed no apparent difference in the relative frequency with which wild-type and Rad51D KO cells repaired DPC-containing plasmids. We have not yet examined the influence of inactivation/downregulation of other Rad51 paralogs on mitochondrial DPC repair; however, it is noteworthy that a publication by Mishra et al. determined that the Rad51C/Xrcc3 plays an essential role in maintaining the integrity of the mammalian mitochondrial genome [43]. It is therefore conceivable that the phenotype these authors observed in Rad51C-deficient cells reflects deficient mitochondrial recombinational repair of spontaneous DNA damage in that organelle.
In addition to clearly establishing the existence of mitochondrial homologous recombinational repair in mammalian cells, the model we have developed provides a unique opportunity that can be exploited to gain a substantially deeper understanding of the mechanism of recombinational repair of DNA damage in mammalian cells. To our knowledge, there are no previous reports of a mammalian cell-free system that supports homologous recombination. We believe the system we have developed combines the respective advantages inherent in studying homologous recombination in an intact cell system, with those associated with studies using simplified cell extract-based systems. We are able to introduce precisely defined DNA lesions that are subject to recombinational repair within an intact biological system. The results herein, and in a previous study [47], demonstrate that we can precisely manipulate the nature of the repair substrate and are also able to alter levels of key cofactors such as dNTPs, etc. While the studies described herein relied on a highly sensitive PCR-based assay, we note that the efficiency of repair catalyzed within the mitochondrial system, combined with the ability to isolate large quantities of purified mitochondria, indicate that it will be possible to pursue more biochemical approaches to dissect the details of the reaction mechanism of mitochondrial recombinational repair. It will be of interest to determine the identity of other proteins required to carry out mitochondrial repair of DPCs, as well as to explore the range of DNA lesions, other than DPCs, that are also subject to recombinational repair in mammalian mitochondria. There will be of particular interest to identify the molecular mechanism through which DNA containing DPC lesions is recognized, and whether (and if so how) the protein is proteolytically processed as part of the repair mechanism. It is clear that mitochondrial DNA damage contributes to the cytotoxic, mutagenic, and even therapeutic effects of a variety of classes of chemical agents. We envision that the system described in this study represents an important tool that can be utilized to expand our knowledge of how mammalian cells recognize and respond to the DNA damage these agents induce within the mitochondrial genome.
5. Conclusions
In conclusion, we have provided the first evidence for repair of DNA-protein crosslinks in mammalian mitochondria. DPCs present on exogenous plasmid DNA electroporated into purified mitochondria or formed by the crosslinking agent cisplatin were repaired via a homologous recombinational mechanism that was dependent on the Rad51 protein. Using a qPCR-based assay to measure repair, we were able to manipulate key factors such as sequence homology, presence of a double-strand break, and absence of dNTPs to precisely measure differences in DPC repair. Importantly, this research presents a sensitive system that could be used in future research to investigate the repair of other xenobiotic-induced mitochondrial adducts such as interstrand crosslinks and double-strand breaks.
Supplementary Material
Acknowledgments
We acknowledge Professor Claudia Wiese (University of Colorado) for providing CHO cells, and the Natalia Tretyakova (University of Minnesota) and Ashis Basu (University of Connecticut) labs for their support and technical advice during the early and intermediate stages of this work, and Hai Dang Nguyen University of Minnesota) for helpful editorial assistance.
Funding
This work was funded by the National Institutes of Health (ES023350). Lisa N. Chesner and Maram Essawy were supported by Training Grant5T32HL007741. Funding for open access charge: National Institutes of Health.
Footnotes
Declaration of Competing Interest
The authors report no declarations of interest.
Appendix A. Supplementary data
Supplementary material related to this article can be found, in the online version, at doi:https://doi.org/10.1016/j.dnarep.2020.103026.
Homologous Recombination; HR, Base Excision Repair; BER, DNA-Protein Crosslink; DPC, Nucleotide Excision Repair; NER, Oxoguanine Glycosylase; OGG.
References
- [1].Pagliarini DJ, Rutter J, Hallmarks of a new era in mitochondrial biochemistry, Genes Dev. 27 (2013) 2615–2627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Palade GE, The fine structure of mitochondria, Anat. Rec 114 (1952) 427–451. [DOI] [PubMed] [Google Scholar]
- [3].Palade GE, The organization of living matter, Proc. Natl. Acad. Sci. U. S. A 52 (1964) 613–634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Mitchell P, Moyle J, Respiration-driven proton translocation in rat liver mitochondria, Biochem. J 105 (1967) 1147–1162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Ernster L, Schatz G, Mitochondria: a historical review, J. Cell Biol 91 (1981) 227s–255s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Nass MM, The circularity of mitochondrial DNA, Proc. Natl. Acad. Sci. U.S.A 56 (1966) 1215–1222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Nass S, Nass MM, Intramitochondrial fibers with DNA characteristics. II. Enzymatic and other hydrolytic treatments, J. Cell Biol 19 (1963) 613–629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Schatz G, Haslbrunner E, Tuppy H, Deoxyribonucleic acid associated with yeast mitochondria, Biochem. Biophys. Res. Commun 15 (1964) 127–132. [DOI] [PubMed] [Google Scholar]
- [9].Anderson S, Bankier AT, Barrell BG, de Bruijn MH, Coulson AR, Drouin J, Eperon IC, Nierlich DP, Roe BA, Sanger F, Schreier PH, Smith AJ, Staden R, Young IG, Sequence and organization of the human mitochondrial genome, Nature 290 (1981) 457–465. [DOI] [PubMed] [Google Scholar]
- [10].Clayton DA, Doda JN, Friedberg EC, The absence of a pyrimidine dimer repair mechanism in mammalian mitochondria, Proc. Natl. Acad. Sci 71 (1974) 2777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Clayton DA, Doda JN, Friedberg EC, Absence of a pyrimidine dimer repair mechanism for mitochondrial DNA in mouse and human cells, Basic Life Sci. 5b (1975) 589–591. [DOI] [PubMed] [Google Scholar]
- [12].LeDoux SP, Wilson GL, Beecham EJ, Stevnsner T, Wassermann K, Bohr VA, Repair of mitochondrial DNA after various types of DNA damage in Chinese hamster ovary cells, Carcinogenesis 13 (1992) 1967–1973. [DOI] [PubMed] [Google Scholar]
- [13].Pascucci B, Versteegh A, van Hoffen A, van Zeeland AA, Mullenders LH, Dogliotti E, DNA repair of UV photoproducts and mutagenesis in human mitochondrial DNA, J. Mol. Biol 273 (1997) 417–427. [DOI] [PubMed] [Google Scholar]
- [14].Waters R, Moustacchi E, The fate of ultraviolet-induced pyrimidine dimers in the mitochondrial DNA of Saccharomyces cerevisiae following various post-irradiation cell treatments, Biochim. Biophys. Acta 366 (1974) 241–250. [DOI] [PubMed] [Google Scholar]
- [15].Brown WM, George M, Wilson AC, Rapid evolution of animal mitochondrial DNA, Proc. Natl. Acad. Sci. U.S.A 76 (1979) 1967–1971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Howell N, Kubacka I, Mackey DA, How rapidly does the human mitochondrial genome evolve? Am. J. Hum. Genet 59 (1996) 501–509. [PMC free article] [PubMed] [Google Scholar]
- [17].Saitou N, Ueda S, Evolutionary rates of insertion and deletion in noncoding nucleotide sequences of primates, Mol. Biol. Evol 11 (1994) 504–512. [DOI] [PubMed] [Google Scholar]
- [18].Bogenhagen DF, Repair of mtDNA in vertebrates, Am. J. Hum. Genet 64 (1999) 1276–1281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Bogenhagen DF, Pinz KG, Perez-Jannotti RM, Enzymology of mitochondrial base excision repair, Prog. Nucleic Acid Res. Mol. Biol 68 (2001) 257–271. [DOI] [PubMed] [Google Scholar]
- [20].Prakash A, Doublie S, Base excision repair in the mitochondria, J. Cell. Biochem 116 (2015) 1490–1499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Lakshmipathy U, Campbell C, Double strand break rejoining by mammalian mitochondrial extracts, Nucleic Acids Res. 27 (1999) 1198–1204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Coffey G, Lakshmipathy U, Campbell C, Mammalian mitochondrial extracts possess DNA end-binding activity, Nucleic Acids Res. 27 (1999) 3348–3354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Mason PA, Matheson EC, Hall AG, Lightowlers RN, Mismatch repair activity in mammalian mitochondria, Nucleic Acids Res. 31 (2003) 1052–1058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Thyagarajan B, Padua RA, Campbell C, Mammalian mitochondria possess homologous DNA recombination activity, J. Biol. Chem 271 (1996) 27536–27543. [DOI] [PubMed] [Google Scholar]
- [25].Dahal S, Dubey S, Raghavan SC, Homologous recombination-mediated repair of DNA double-strand breaks operates in mammalian mitochondria, Cell. Mol. Life Sci 75 (2018) 1641–1655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Bacman SR, Williams SL, Moraes CT, Intra- and inter-molecular recombination of mitochondrial DNA after in vivo induction of multiple double-strand breaks, Nucleic Acids Res. 37 (2009) 4218–4226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Srivastava S, Moraes CT, Double-strand breaks of mouse muscle mtDNA promote large deletions similar to multiple mtDNA deletions in humans, Hum. Mol. Genet 14 (2005) 893–902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Elson JL, Andrews RM, Chinnery PF, Lightowlers RN, Turnbull DM, Howell N, Analysis of European mtDNAs for recombination, Am. J. Hum. Genet 68 (2001) 145–153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Howell N, mtDNA recombination: what do in vitro data mean? Am. J. Hum. Genet 61 (1997) 19–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Ladoukakis ED, Zouros E, Direct evidence for homologous recombination in mussel (Mytilus galloprovincialis) mitochondrial DNA, Mol. Biol. Evol 18 (2001) 1168–1175. [DOI] [PubMed] [Google Scholar]
- [31].Barr CM, Neiman M, Taylor DR, Inheritance and recombination of mitochondrial genomes in plants, fungi and animals, New Phytol. 168 (2005) 39–50. [DOI] [PubMed] [Google Scholar]
- [32].Holt IJ, Dunbar DR, Jacobs HT, Behaviour of a population of partially duplicated mitochondrial DNA molecules in cell culture: segregation, maintenance and recombination dependent upon nuclear background, Hum. Mol. Genet 6 (1997) 1251–1260. [DOI] [PubMed] [Google Scholar]
- [33].Kajander OA, Karhunen PJ, Holt IJ, Jacobs HT, Prominent mitochondrial DNA recombination intermediates in human heart muscle, EMBO Rep. 2 (2001) 1007–1012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].D’Aurelio M, Gajewski CD, Lin MT, Mauck WM, Shao LZ, Lenaz G, Moraes CT, Manfredi G, Heterologous mitochondrial DNA recombination in human cells, Hum. Mol. Genet 13 (2004) 3171–3179. [DOI] [PubMed] [Google Scholar]
- [35].Howell N, Kubacka I, Keers SM, Turnbull DM, Chinnery PF, Co-segregation and heteroplasmy of two coding-region mtDNA mutations within a matrilineal pedigree, Hum. Genet 116 (2005) 28–32. [DOI] [PubMed] [Google Scholar]
- [36].Zsurka G, Kraytsberg Y, Kudina T, Kornblum C, Elger CE, Khrapko K, Kunz WS, Recombination of mitochondrial DNA in skeletal muscle of individuals with multiple mitochondrial DNA heteroplasmy, Nat. Genet 37 (2005) 873–877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Kraytsberg Y, Schwartz M, Brown TA, Ebralidse K, Kunz WS, Clayton DA, Vissing J, Khrapko K, Recombination of human mitochondrial DNA, Science 304 (2004) 981. [DOI] [PubMed] [Google Scholar]
- [38].Jacobs HT, Lehtinen SK, Spelbrink JN, No sex please, we’re mitochondria: a hypothesis on the somatic unit of inheritance of mammalian mtDNA, Bioessays 22 (2000) 564–572. [DOI] [PubMed] [Google Scholar]
- [39].Gilkerson RW, Schon EA, Hernandez E, Davidson MM, Mitochondrial nucleoids maintain genetic autonomy but allow for functional complementation, J. Cell Biol 181 (2008) 1117–1128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Nakano T, Katafuchi A, Matsubara M, Terato H, Tsuboi T, Masuda T, Tatsumoto T, Pack SP, Makino K, Croteau DL, Van Houten B, Iijima K, Tauchi H, Ide H, Homologous recombination but not nucleotide excision repair plays a pivotal role in tolerance of DNA-protein cross-links in mammalian cells, J. Biol. Chem 284 (2009) 27065–27076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Paul R, Dalibart R, Lemoine S, Lestienne P, Expression of E coli RecA targeted to mitochondria of human cells, Mutat. Res 486 (2001) 11–19. [DOI] [PubMed] [Google Scholar]
- [42].Jin ZL, Kim NH, RAD51 maintains chromosome integrity and mitochondrial distribution during porcine oocyte maturation in vitro, J. Reprod. Dev 63 (2017) 489–496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].Mishra A, Saxena S, Kaushal A, Nagaraju G, RAD51C/XRCC3 facilitates mitochondrial DNA replication and maintains integrity of the mitochondrial genome, Mol. Cell. Biol 38 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [44].Sage JM, Gildemeister OS, Knight KL, Discovery of a novel function for human Rad51: maintenance of the mitochondrial genome, J. Biol. Chem 285 (2010) 18984–18990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [45].Sage JM, Knight KL, Human Rad51 promotes mitochondrial DNA synthesis under conditions of increased replication stress, Mitochondrion 13 (2013) 350–356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [46].Chesner LN, Campbell C, A simple, rapid, and quantitative assay to measure repair of DNA-protein crosslinks on plasmids transfected into mammalian cells, J. Vis. Exp 5 (2018). LID - 10.3791/57413 [doi]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [47].Chesner LN, Campbell C, A quantitative PCR-based assay reveals that nucleotide excision repair plays a predominant role in the removal of DNA-protein crosslinks from plasmids transfected into mammalian cells, DNA Repair 62 (2018) 18–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [48].Collombet JM, Wheeler VC, Vogel F, Coutelle C, Introduction of plasmid DNA into isolated mitochondria by electroporation. A novel approach toward gene correction for mitochondrial disorders, J. Biol. Chem 272 (1997) 5342–5347. [DOI] [PubMed] [Google Scholar]
- [49].Borisov AM, Guliaeva NA, Rasskazova EA, Ploskonosova II, Gaznev AI, [DNA-protein cross-links in nuclei and mitochondria of tissue cells from rats of various age exposed to gamma-radiation], Radiats. Biol. Radioecol 44 (2004) 377–382. [PubMed] [Google Scholar]
- [50].Sordet O, Khan Qasim A., Pommier Yves, Apoptotic topoisomerase I-DNA complexes induced by oxygen radicals and mitochondrial dysfunction, Cell Cycle 9 (2004) 1095–1097. [PubMed] [Google Scholar]
- [51].Quinones JL, Demple B, When DNA repair goes wrong: BER-generated DNA-protein crosslinks to oxidative lesions, DNA Repair 44 (2016) 103–109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [52].Quinones JL, Thapar U, Yu K, Fang Q, Sobol RW, Demple B, Enzyme mechanism-based, oxidative DNA-protein cross-links formed with DNA polymerase beta in vivo, Proc. Natl. Acad. Sci. U.S.A 112 (2015) 8602–8607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [53].Baker DJ, Wuenschell G, Xia L, Termini J, Bates SE, Riggs AD, O’Connor TR, Nucleotide excision repair eliminates unique DNA-protein cross-links from mammalian cells, J. Biol. Chem 282 (2007) 22592–22604. [DOI] [PubMed] [Google Scholar]
- [54].Nakano T, Morishita S, Katafuchi A, Matsubara M, Horikawa Y, Terato H, Salem AM, Izumi S, Pack SP, Makino K, Ide H, Nucleotide excision repair and homologous recombination systems commit differentially to the repair of DNA-protein crosslinks, Mol. Cell 28 (2007) 147–158. [DOI] [PubMed] [Google Scholar]
- [55].Hinz JM, Tebbs RS, Wilson PF, Nham PB, Salazar EP, Nagasawa H, Urbin SS, Bedford JS, Thompson LH, Repression of mutagenesis by Rad51D-mediated homologous recombination, Nucleic Acids Res. 34 (2006) 1358–1368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [56].Reh WA, Nairn RS, Lowery MP, Vasquez KM, The homologous recombination protein RAD51D protects the genome from large deletions, Nucleic Acids Res. 45 (2017) 1835–1847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [57].Hirt B, Selective extraction of polyoma DNA from infected mouse cell cultures, J. Mol. Biol 26 (1967) 365–369. [DOI] [PubMed] [Google Scholar]
- [58].Yoon YG, Koob MD, Efficient cloning and engineering of entire mitochondrial genomes in Escherichia coli and transfer into transcriptionally active mitochondria, Nucleic Acids Res. 31 (2003) 1407–1415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [59].Jessberger R, Berg P, Repair of deletions and double-strand gaps by homologous recombination in a mammalian in vitro system, Mol. Cell. Biol 11 (1991) 445–457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [60].Klein JC, Bleeker MJ, Saris CP, Roelen HC, Brugghe HF, van den Elst H, van der Marel GA, van Boom JH, Westra JG, Kriek E, et al. , Repair and replication of plasmids with site-specific 8-oxodG and 8-AAFdG residues in normal and repair-deficient human cells, Nucleic Acids Res. 20 (1992) 4437–4443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [61].Zhitkovich A, Costa M, A simple, sensitive assay to detect DNA-protein crosslinks in intact cells and in vivo, Carcinogenesis 13 (1992) 1485–1489. [DOI] [PubMed] [Google Scholar]
- [62].Lee HW, Lee HJ, Hong CM, Baker DJ, Bhatia R, O’Connor TR, Monitoring repair of DNA damage in cell lines and human peripheral blood mononuclear cells, Anal. Biochem 365 (2007) 246–259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [63].Schmittgen TD, Livak KJ, Analyzing real-time PCR data by the comparative C(T) method, Nat. Protoc 3 (2008) 1101–1108. [DOI] [PubMed] [Google Scholar]
- [64].Lakshmipathy U, Campbell C, Mitochondrial DNA ligase III function is independent of Xrcc1, Nucleic Acids Res. 28 (2000) 3880–3886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [65].Sambrook J, Russell DWDW, Laboratory CSH, Molecular Cloning : a Laboratory Manual, 3rd. ed, Cold Spring Harbor Laboratory, 2001. [Google Scholar]
- [66].Szostak JW, Orr-Weaver TL, Rothstein RJ, Stahl FW, The double-strand-break repair model for recombination, Cell 33 (1983) 25–35. [DOI] [PubMed] [Google Scholar]
- [67].Bee L, Fabris S, Cherubini R, Mognato M, Celotti L, The efficiency of homologous recombination and non-homologous end joining systems in repairing double-strand breaks during cell cycle progression, PLoS One 8 (2013), e69061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [68].Rouet P, Smih F, Jasin M, Expression of a site-specific endonuclease stimulates homologous recombination in mammalian cells, Proc. Natl. Acad. Sci. U.S.A 91 (1994) 6064–6068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [69].Rouet P, Smih F, Jasin M, Introduction of double-strand breaks into the genome of mouse cells by expression of a rare-cutting endonuclease, Mol. Cell. Biol 14 (1994) 8096–8106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [70].Haber JE, DNA Repair: The Search for Homology, Bioessays 40 (2018), e1700229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [71].Croteau DL, Stierum RH, Bohr VA, Mitochondrial DNA repair pathways, Mutat. Res. Repair 434 (1999) 137–148. [DOI] [PubMed] [Google Scholar]
- [72].Stein A, Sia EA, Mitochondrial DNA repair and damage tolerance, Front. Biosci. (Landmark Ed) 22 (2017) 920–943. [DOI] [PubMed] [Google Scholar]
- [73].Zinovkina LA, Mechanisms of mitochondrial DNA repair in mammals, Biochemistry Mosc. 83 (2018) 233–249. [DOI] [PubMed] [Google Scholar]
- [74].Chen XJ, Mechanism of homologous recombination and implications for aging-related deletions in mitochondrial DNA, Microbiol. Mol. Biol. Rev 77 (2013) 476–496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [75].Caston RA, Demple B, Risky repair: DNA-protein crosslinks formed by mitochondrial base excision DNA repair enzymes acting on free radical lesions, Free Radic. Biol. Med 107 (2017) 146–150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [76].Torregrosa-Muñumer R, Hangas A, Goffart S, Blei D, Zsurka G, Griffith J, Kunz WS, Pohjoismäki JLO, Replication fork rescue in mammalian mitochondria, Sci. Rep 9 (2019) 8785. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.