

Abstract
β-Arrestin-1 and -2 are intracellular proteins that are able to inhibit signaling via G protein-coupled receptors (GPCRs). However, both proteins can also modulate cellular functions in a G protein-independent fashion. During the past few years, studies with mutant mice selectivity lacking β-arrestin-1 and/or -2 in metabolically important cell types have led to novel insights into the mechanisms through which β-arrestins regulate key metabolic processes in vivo, including whole body glucose and energy homeostasis. The novel information gained from these studies should inform the development of novel drugs, including β-arrestin- or G protein-biased GPCR ligands, that could prove useful for the therapy of several important pathophysiological conditions including type 2 diabetes and obesity.
Keywords: β-Arrestins, G protein-coupled receptors, diabetes, obesity, metabolism, mutant mice
Canonical and non-canonical actions of β-arrestins
G protein-coupled receptors (GPCRs) form a superfamily of cell surface receptors that are activated by a large variety of extracellular ligands including neurotransmitters, hormones, and many other stimuli. About 1/3 of drugs in current clinical act by modulating the activity of specific GPCR subtypes, indicative of the extraordinary therapeutic relevance of this class of receptors [1]. Most ligand-activated GPCRs get phosphorylated by a group of kinases known as GPCR kinases (GRKs). The phosphorylated receptors are then able to bind to a pair of intracellular proteins known as β-arrestin-1 and -2 (alternative nomenclature: arrestin-2 and -3, respectively), thus disrupting receptor/G protein coupling. In addition, β-arrestins act as linker proteins to promote the removal of activated GPCRs from the cell surface, a process referred to as GPCR internalization. Specifically, receptor-associated β-arrestins directly bind clathrin and clathrin adaptor AP2 to promote GPCR internalization via clathrin-coated pits [2, 3]. The ability of β-arrestin-1 and -2 (βarr1 and βarr2, respectively) to terminate GPCR signaling via heterotrimeric G proteins (see Glossary) and to promote GPCR internalization are known as the classical or canonical actions of β-arrestins (Figure 1) [2].
Figure 1.
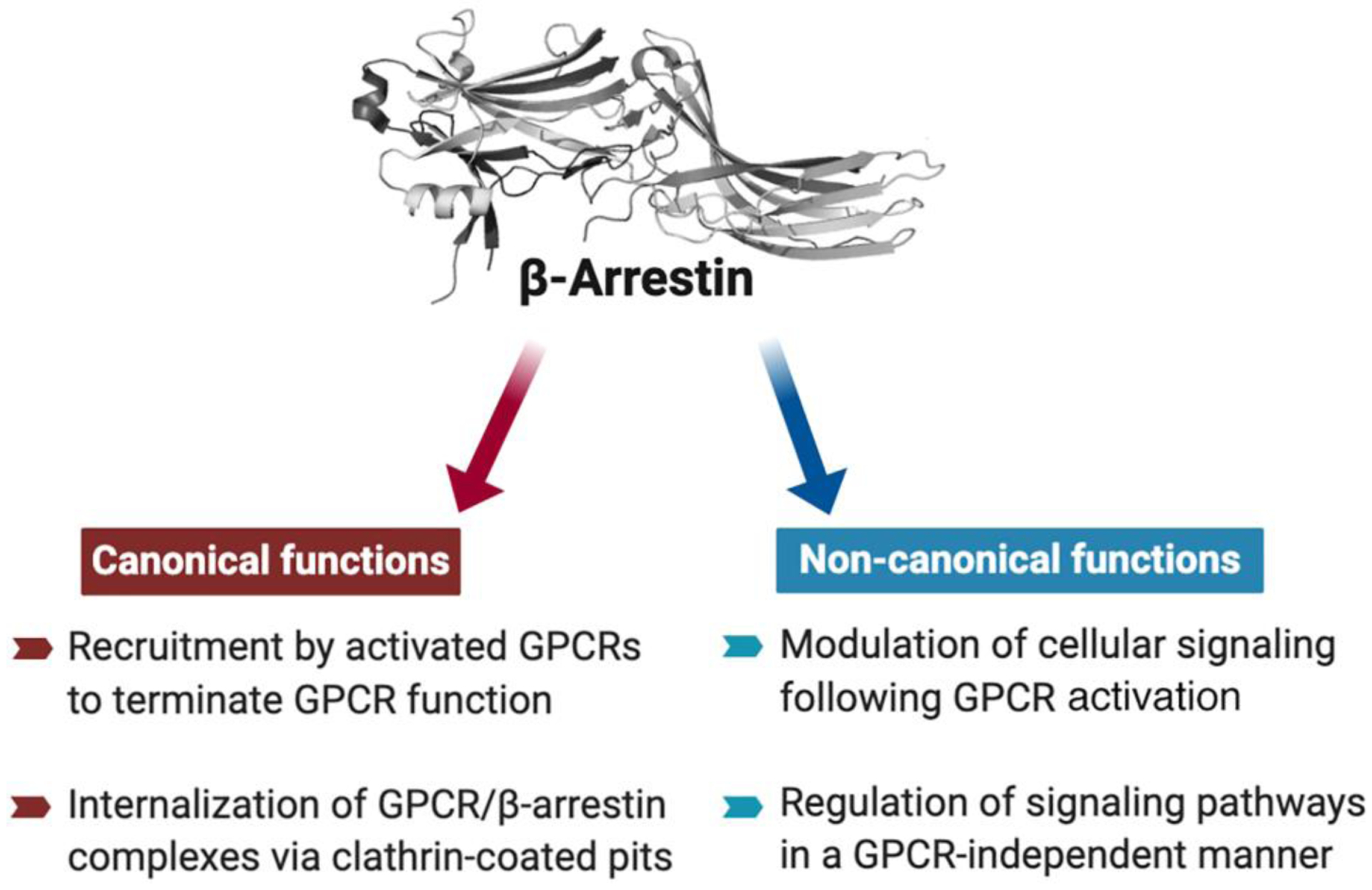
Canonical and non-canonical functions of β-arrestins.
During the past two decades, many studies have shown that β-arrestins can interact with numerous non-receptor binding partners [4–6]. For example, β-arrestins can modulate the activity of c-Src, different members of the three major MAP kinase pathways, cAMP phosphodiesterase, calmodulin, protein phosphatases, ubiquitin ligases, and deubiquitinating enzymes, and many other signaling proteins [4–6]. While many of these novel β-arrestin actions appear to require the interaction of β-arrestins with activated GPCRs, it has become clear that β-arrestins can also modulate cellular signaling in a GPCR-independent fashion (Figure 1) [4, 7]. For example, while β-arrestin-stimulated activation of ERK1/2 usually requires the prior recruitment of β-arrestins by GPCRs, in vitro studies have clearly demonstrated that βarr2 can activate JNK3 in the absence of any GPCRs [4–6]. Interestingly, β-arrestins, in particular βarr1, have also been shown to be present in the nucleus, at least under certain experimental conditions, where they can modulate gene expression patterns [8–11]. These findings indicate that β-arrestins act as key regulators of numerous cytoplasmic and nuclear functions. The signaling roles of βarr1 and βarr2 that extend beyond their ability to mediate GPCR desensitization and internalization are commonly referred to as the non-classical or non-canonical actions of β-arrestins (Figure 1) [4–6]. At this time, our understanding of the physiological relevance of these non-canonical β-arrestin activities remains very limited.
Regulation of glucose and energy homeostasis by β-arrestins
Studies with whole body β-arrestin knockout (KO) mice have shown that β-arrestins regulate numerous physiologic and pathophysiologic processes, highlighting their potential roles as therapeutic targets (see, for example [12]). In this context, the discovery of so-called biased agonists that preferentially activate G protein- vs. β-arrestin-mediated signaling pathways (or vice versa) has opened novel therapeutic perspectives [7, 13–15]. Such biased ligands may be endowed with improved efficacy while causing fewer side effects, depending on whether activation of either G protein- or β-arrestin-mediated signaling is responsible for the desired physiologic/therapeutic outcome [7, 13–15].
Since β-arrestins are ubiquitously expressed, the outcome of metabolic studies using whole-body βarr1- and βarr2-KO mice is difficult to interpret ([12]. Moreover, both βarr1 and βarr2 have been shown to regulate key developmental processes [16–18], further complicating the interpretation of metabolic or other phenotypes exhibited by conventional β-arrestin KO mice.
The recent development of mice harboring floxed βarr1 (Arrb1) or βarr2 (Arrb2) genes has been instrumental in elucidating the metabolic functions of β-arrestins in unprecedented cellular detail [19, 20]. By crossing these newly developed mouse strains with specific Cre driver lines, it has been possible to delete βarr1 or βarr2 in specific cell types that are critical for maintaining euglycemia and proper energy homeostasis. For example, this approach has allowed the inactivation of either βarr1 or βarr2 in a tamoxifen-inducible fashion in pancreatic β-cells of adult mice (for more details, see below) [21, 22].
This review provides a summary of the novel information obtained by analyzing cell type-specific β-arrestin mutant mice, with particular focus on studies that involve the role of β-arrestins in regulating glucose and energy homeostasis. The new insights that have emerged from this work should stimulate the development of novel therapeutic approaches for the treatment of type 2 diabetes (T2D) and related metabolic disorders.
Canonical metabolic functions of βar2: studies with cell type-specific mutant mice
Adipocytes
T2D has emerged as a major threat to human health in most parts of the world, primarily driven by the global obesity epidemic [23, 24]. In the obese state, adipose tissues expand and are infiltrated by macrophages, leading to increased plasma lipid levels and the release of inflammatory adipokines and other factors that impair the ability of various peripheral cell types (adipocytes, hepatocytes, skeletal muscle cells, etc.) to properly respond to insulin. This process, referred to as peripheral insulin resistance, causes impaired glucose homeostasis and can eventually lead to T2D when the amount of insulin released from pancreatic β-cells is no longer able to overcome peripheral insulin resistance [24–27].
In mammals, two major classes of adipocytes can be found, white and brown adipocytes [28–30]. While white adipocytes store excess calories as fat and release fatty acids as a fuel source, mitochondria-rich brown adipocytes are present in dedicated depots and express high levels of uncoupling protein 1 (UCP1) and other thermogenic genes. The major function of brown adipocytes is to burn fatty acids and other substrates to generate heat to maintain body temperature. Brown adipocytes are activated by increased activity of the sympathetic nervous system (e.g. due to cold exposure). Interestingly, a third class of adipocytes, so-called brown-like or ‘beige’ adipocytes that also express UCP-1, can emerge in white adipose tissue (WAT), in particular subcutaneous WAT [28, 29]. This type of adipocytes can be induced after simulation of the sympathetic nervous system. Like brown adipocytes, beige adipocytes are able to convert nutrients into heat. The activity of brown and beige adipocytes can make a significant contribution to whole body energy expenditure [31]. Interestingly, several studies have shown that adult humans harbor both classical brown adipose tissue (BAT) and brown-like or beige adipocytes in WAT. This finding has raised the possibility that this class of adipocytes can be targeted to stimulate energy expenditure to trigger weight loss [31–33].
Recently, Pydi et al. [34] analyzed a novel mutant mouse strain that selectively lacked βarr2 in adipocytes (adipo-βarr2-KO mice). Interestingly, when adipo-βarr2-KO mice were raised on an obesogenic high-fat diet (HFD), they gained considerably less weight and body fat mass than their control littermates. While HFD control mice exhibited hyperglycemia, glucose intolerance, and reduced insulin sensitivity, these metabolic deficits were largely absent in HFD adipo-βarr2-KO mice [34]. Studies with additional mouse models indicated that the metabolic improvements exhibited by HFD adipo-βarr2-KO mice required the browning/beiging of WAT and that the improved metabolic status of HFD adipo-βarr2-KO mice resulted from decreased adiposity caused by enhanced total energy expenditure (TEE) [34].
Mouse adipocytes express dozens of GPCRs [35, 36]. Many studies have focused on the pharmacology and physiology of the Gs-coupled β3-adrenergic receptor (β3-AR), since this receptor subtype is the predominant adrenergic receptor subtype expressed by mouse adipocytes and efficiently stimulates adrenergic agonist-mediated BAT activity [33, 37]. Pydi et al. [34] demonstrated that signaling via adipocyte β3-ARs was increased in adipocytes lacking βarr2 (Figure 2A), suggesting that enhanced activity of adipocyte β3-ARs is responsible for the metabolic improvement observed with HFD adipo-βarr2-KO mice.
Figure 2.
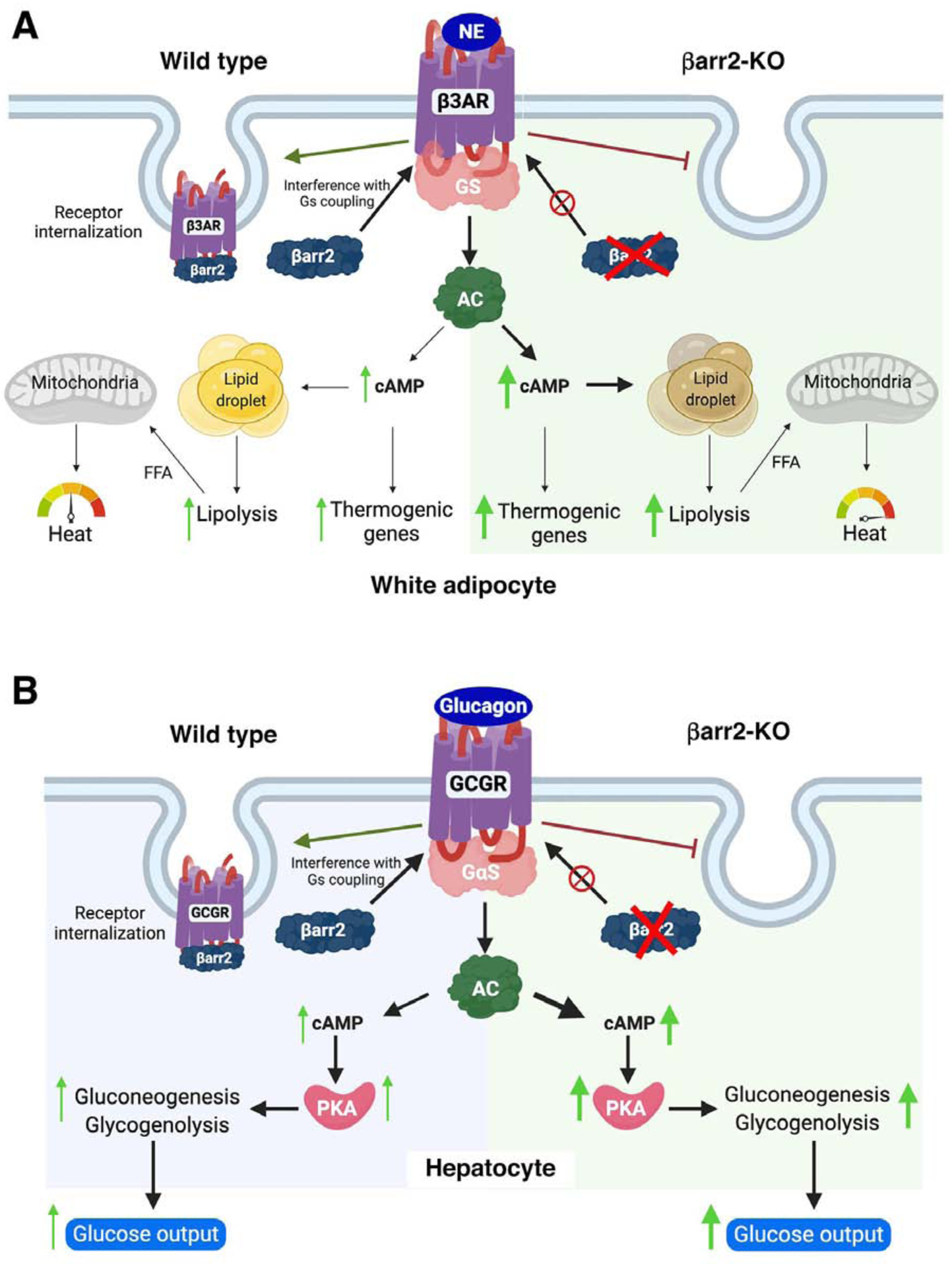
Canonical functions of βarr2 in mouse adipocytes and hepatocytes. A) Adipocytes. In white adipocytes from wild type mice, βarr2 is involved in terminating β3-AR signaling by interacting with activated β3-ARs and promoting receptor internalization. In white adipocytes lacking βarr2, this inhibitory regulation of β3-AR function is absent, resulting in enhanced β3-AR-mediated cellular effects [34]. B) Hepatocytes. In hepatocytes from wild type mice, βarr2 acts as a negative regulator of glucagon receptor (GCGR) signaling. In βarr2-deficient hepatocytes, GCGR signaling is no longer subject to inhibition by βarr2, leading to enhanced glucose output [49]. AC, adenyl cyclase; FFA, free fatty acids; NE, norepinephrine; PKA; protein kinase A.
Pydi et al. [34] also showed that agonist-induced β3-AR internalization was abolished in cultured mouse adipocytes lacking βarr2 [34], suggesting that βarr2 acts as an inhibitor of β3-AR function in mouse adipocytes (Figure 2A). Selective β3-AR agonists are able to prevent or even reverse obesity and obesity-associated metabolic deficits in several animal models [25, 38, 39], raising the possibility that such drugs might prove useful for therapeutic purposes. Since βarr2 inhibits β3-AR function in adipocytes, G protein-biased β3-AR agonists are predicted to be endowed with increased efficacy and a longer duration of action.
In contrast to mouse adipocytes, human adipocytes preferentially express β1- and β2-ARs and only a small population of β3-ARs [33, 37]. Treatment of human subjects with mirabegron, a β3-AR-selective agonist, resulted in beneficial metabolic effects, including elevated basal metabolic rate, increased insulin sensitivity, and improved glucose tolerance [40–42]. However, the outcome of a recent study [43] suggested that mirabegron-mediated increases in BAT activity in humans are mediated primarily by β2-ARs, due to the limited selectivity of mirabegron in vivo. Clearly, additional studies are needed to clarify these discrepant findings. In any case, the development of highly selective, Gs-biased β3-AR agonists appears to be an attractive goal.
Hepatocytes
Hepatocytes play a central role in the control of whole body glucose and lipid metabolism. The metabolic functions carried out by hepatocytes are regulated by two major hormones released from pancreatic islet, insulin and glucagon [44]. The Gs-coupled glucagon receptor (GCGR) is abundantly expressed by hepatocytes [45]. Hormone-activated GCGRs trigger a cascade of cAMP/PKA-dependent events which ultimately promote gluconeogenesis and glycogen breakdown, thus triggering a pronounced increase in hepatic glucose production (HGP) [45, 46]. Interestingly, enhanced HGP due to unphysiologically high plasma glucagon levels is considered a hallmark of T2D [47, 48].
To explore whether β-arrestins modulate the activity of hepatic GCGRs, Zhu et al. [49] generated and analyzed mice lacking βarr2 or βarr1 selectively in hepatocytes (hep-βarr2-KO and hep-βarr1-KO mice, respectively). The authors found that hep-βarr2-KO mice displayed hyperglycemia and impaired glucose tolerance, as compared to control littermates [49]. In contrast, hep-βarr1-KO mice did not display any noticeable metabolic phenotypes. Treatment of hep-βarr2-KO mice with an anti-GCGR monoclonal antibody restored normal glucose tolerance, strongly suggesting that the metabolic deficits caused by hepatocyte βarr2 deficiency are due to increased hepatic GCGR signaling (Figure 2B). In agreement with this notion, acute glucagon treatment led to significantly greater blood glucose excursions in hep-βarr2-KO mice than in control littermates [49].
Consistent with these in vivo studies, glucagon-induced increases in cAMP levels and glucose output were significantly elevated in βarr2-deficient hepatocytes [49]. In addition, while glucagon treatment of primary hepatocytes prepared from control mice caused rapid GCGR internalization, this effect was not observed with primary hepatocytes lacking βarr2 [49]. Taken together, the in vivo and in vitro data clearly indicate that βarr2 acts as a potent negative regulator of GCGR signaling in hepatocytes, a canonical function of βarr2 (Figure 2B).
Whereas hep-βarr2-KO mice displayed enhanced glucagon sensitivity, transgenic mice selectively over-expressing βarr2 in hepatocytes (hep-βarr2-OE mice) showed the opposite phenotype [49]. Both in vivo and in vitro studies demonstrated that glucagon-stimulated HGP was significantly decreased in hep-βarr2-OE mice [49]. Additional experiments showed that hep-βarr2-OE mice were protected against the metabolic deficits that are usually caused by the consumption of an obesogenic diet [49].
Previous work demonstrated that hepatic βarr2 levels are reduced in humans suffering from T2D [50]. On the basis of this observation, together with the outcome of the study by Zhu et al. [49], it is possible that reduced hepatic βarr2 activity plays a role in the pathophysiology of T2D. Thus, it is conceivable that novel strategies aimed at enhancing the expression or activity of hepatic βarr2 may prove effective in reducing elevated HGP in T2D patients.
Non-canonical metabolic functions of β-arrestins: studies with cell type-specific mutant mice
Adipocytes
In contrast to adipo-βarr2-KO mice, mice that selectively lacked βarr1 in adipocytes (adipo-βarr1-KO mice) displayed pronounced impairments in glucose tolerance and insulin sensitivity when maintained on an obesogenic diet [51]. The lack of adipocyte βarr1 did not interfere with β3-AR function but led to increased expression levels of many myogenic genes in brown adipose tissue (BAT). Importantly, myostatin (Mstn) transcript levels were also increased in BAT lacking βarr1, leading to increased plasma Mstn levels. Both myocytes and brown adipocytes originate from a Myf5-positive cell lineage [52–54], increasing the likelihood that genetic alteration of brown adipocytes can trigger the expression of myogenic genes. While Mstn is well known for its ability to inhibit skeletal muscle growth [55], it can also affect the function of other cell types including brown adipocytes [56, 57].
Additional mechanistic studies, including the use of an Mstn-blocking antibody, indicated that the increase in plasma Mstn levels caused by BAT βarr1 deficiency resulted in impaired insulin signaling in several peripheral tissues, leading to impaired glucose homeostasis in HFD adipo-βarr1-KO mice [51]. A previous study [58] demonstrated that Mstn-dependent signaling can reduce PI3 kinase activity, providing a possible molecular mechanism for the ability of Mstn to suppress insulin signaling. It should be noted in this context that at least two other mutant mouse strains have been described recently that also show increased Mstn release from BAT, leading to significant metabolic changes, including impaired insulin responsiveness [59] and reduced exercise capacity [60].
Moreover, Pydi et al. [51] demonstrated that βarr1 can bind to PPARγ in the nucleus of BAT cells and that this interaction leads to reduced transcription of the Mstn gene (Figure 3A), in agreement with previous findings that nuclear βarr1 can inhibit the transcriptional activities of PPARγ [61, 62]. In contrast, the over-expression of βarr1 in adipocytes resulted in mutant mice (adipo-βarr1-OE mice) that exhibited reduced plasma Mstn levels, associated with improved glucose homeostasis when the adipo-βarr1-OE mice were maintained on an obesogenic diet [51]. In sum, these data indicate that βarr1 silences the Mstn gene in BAT and that this βarr1 function is critical for efficient insulin action and the maintenance of euglycemia (Figure 3A). We hypothesize that strategies aimed at increasing the expression or activity of βarr1 in BAT may prove beneficial in patients with impaired glucose homeostasis.
Figure 3.
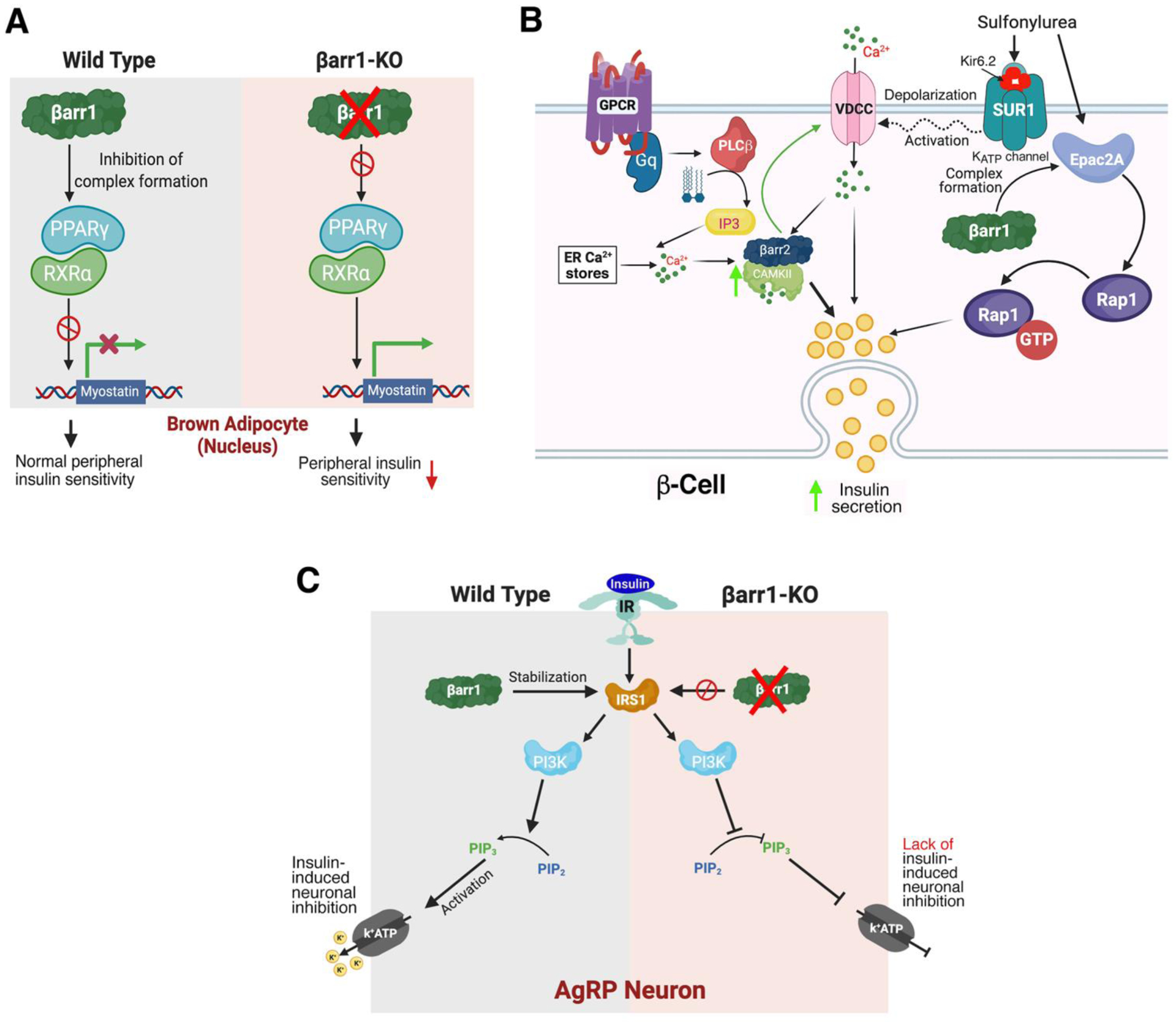
Non-canonical functions of β-arrestins in mouse adipocytes, pancreatic β-cells, and AgRP neurons. A) Adipocytes. In brown adipocytes from wild type mice, βarr1 binding to the PPARγ/RXRα complex is predicted to prevent activation of the myostatin (Mstn) promoter [51] In the absence of βarr1, the PPARγ/RXRα complex can activate the Mstn promoter, leading to increased plasma Mstn levels which cause peripheral insulin resistance [51]. B) β-Cells. The left side of the cartoon shows that βarr2 is required for the proper function of CAMKII in β-cells. βarr2 is predicted to form a complex with CAMKII that stimulates CAMKII activity in β-cells [21]. Activated CAMKII stimulates insulin secretion via phosphorylation of various signaling proteins involved in insulin exocytosis and has been shown to facilitate glucose-dependent calcium influx by acting on VDCCs [89]. Impaired CAMKII activity can fully account for the metabolic impairments observed with beta-βarr2-KO mice [21]. The right side of the cartoon shows that β-cell βarr1 is required for the proper function of most sulphonylurea drugs (SUs). By binding to the SUR1 subunit of the β-cell ATP-gated K+ channel, SU drugs cause channel closure, triggering membrane depolarization, Ca2+ influx through VDCCs, and eventually insulin secretion. Most SUs also stimulate the formation of a βarr1/Epac2a complex, which promotes Rap1-mediated insulin secretion [22]. C) AgRP neurons. In AgRP neurons from wild type mice, insulin induces hyperpolarization and reduced firing frequency by activating a pathway that leads to the opening of KATP channels. In the absence of βarr1, insulin-mediated inhibition of AgRP neurons is abolished, most likely due to reduced stability of IRS1 (IRS1 is stabilized by βarr1 in wild type neurons) [83]. AC, adenylyl cyclase; DAG, diacylglycerol; ER, endoplasmic reticulum; IP3, inositol trisphosphate; IR, insulin receptor; IRS1, insulin receptor substrate 1; PIP2, phosphatidylinositol 4,5-bisphosphate; PIP3, phosphatidylinositol 3,4,5-trisphosphate; PI3K, phosphoinositide 3-kinase; PKA, protein kinase A; PKC, protein kinase C; PPARγ, peroxisome proliferator-activated receptor γ; RXRα, retinoid X receptor α, VDCC, voltage-dependent calcium channel.
Pancreatic β-cells
The release of insulin from pancreatic β-cells is essential for maintaining euglycemia and numerous other key metabolic functions [63]. In T2D, β-cell function deteriorates due to several factors including chronically increased blood glucose and lipid levels, leading to the release of insufficient amounts of insulin [64].
Mice lacking βarr2 in β-cells
The inactivation of βarr2 in pancreatic β-cells of adult mice (beta-βarr2-KO mice) resulted in greatly impaired glucose-stimulated insulin secretion (GSIS) and glucose tolerance when the mutant mice consumed an obesogenic diet [21]. In vitro studies showed that the presence of β-cell βarr2 was required for efficient GSIS and the proper function of L-type Ca2+ channels (LTCCs) [21]. Similar findings were obtained with a human β-cell line (EndoC-β H1 cells) [21].
Additional mechanistic studies revealed that β-cell βarr2 can bind to CAMKII, probably as part of a multi-protein complex, and that this interaction is critical for the proper function of CAMKII [21] (Figure 3B). CAMKII is a Ser/Thr protein kinase that stimulates insulin secretion by activating cellular pathways that promote insulin exocytosis [65, 66]. In agreement with the outcome of the study by Zhu et al. [21], mice with conditional inhibition of CaMKII in β-cells show metabolic phenotypes that closely mimic those displayed by beta-βarr2-KO mice [65]. Interestingly, β-arrestin/CAMKII complexes have also been identified in other tissues or cell types [67, 68].
In contrast to the metabolic deficits observed with beta-βarr2-KO mice, βarr2 over-expression in β-cells resulted in mutant mice (beta-βarr2-OE mice) that were largely protected against the metabolic impairments resulting from the consumption of a calorie-rich diet [21]. Thus, the metabolic profiles displayed by beta-βarr2-KO and beta-βarr2-OE mice suggest that drugs able to increase β-cell βarr2 expression and/or activity may have therapeutic potential for the treatment of T2D.
Mice lacking βarr1 in β-cells
Inactivation of the βarr1 gene in β-cells of adult mice (beta-βarr1-KO mice) had no significant effect on glucose homeostasis and other metabolic parameters when the mutant mice were maintained on a standard chow diet [22]. However, somewhat surprisingly, in vitro and in vivo studies showed that certain sulfonylurea drugs (SUs), including glibenclamide and tolbutamide, showed impairments in their ability to promote insulin release from islets of beta-βarr1-KO mice [22]. SUs are predicted to promote insulin secretion via multiple mechanisms, including binding to the SUR1 subunit of β-cell K+ATP channels and activation of Epac2 [69], a cAMP binding protein that triggers the activation of Rap1 [70]. SU treatment allows Epac2 to activate Rap1, a close relative of the small GTPase Ras, which has the potential to stimulate signaling events that ultimately promote insulin exocytosis [70] (Figure 3B). However, certain SUs, such as gliclazide, are able to stimulate insulin secretion with high efficacy without modulating Epac2 function [71–73].
Barella et al. [22] also demonstrated that purified βarr1 can interact directly with Epac2 and that the formation of βarr1/Epac2 complexes can be stimulated by glibenclamide in mouse β-cells. Moreover, siRNA-mediated knockdown of βarr1 expression in mouse β-cells significantly impaired glibenclamide-dependent stimulation of endogenous Rap1 [22]. These findings indicate that SUs stimulate the formation of βarr1/Epac2 complexes, initiating a series of signaling events that promote insulin exocytosis (Figure 3B). Pharmacological approaches aimed at activating this signaling pathway in β-cells may prove beneficial to stimulate insulin secretion for therapeutic purposes.
Skeletal Muscle
Skeletal muscle (SKM) is the largest insulin-sensitive tissue in the body and functions as the primary site for insulin-dependent glucose removal from the blood [74]. SKM insulin resistance is considered a key deficit driving the onset of T2D [75].
Mice lacking βarr1 selectively in SKM (SKM-βarr1-KO mice) exhibited no significant metabolic deficits, independent of the type of diet that the mice consumed [76]. On the other hand, SKM-βarr2-KO consuming a calorie-rich diet showed mild improvements in glucose and insulin tolerance [76]. In agreement with this finding, insulin-induced Akt phosphorylation was enhanced in SKM lacking βarr2 [76]. Additional studies are needed to elucidate how the absence of βarr2 promotes insulin signaling in SKM.
The β2-adrenergic receptor (β2-AR) is the predominant adrenergic receptor subtype expressed by SKM cells [77]. Previous work [78] has shown that chronic treatment of WT mice with clenbuterol, a selective β2-AR agonist, leads to a significant improvement in glucose tolerance. Meister et al. [76] demonstrated that this beneficial metabolic effect was fully preserved in SKM-βarr1-and -βarr2 single and SKM-βarr1/2 double KO mice. This finding clearly indicates that clenbuterol-mediated improvements in glucose homeostasis are not modulated by SKM β-arrestins.
AgRP neurons
The arcuate nucleus of the hypothalamus (ARC) is of exceptional importance in the neuronal control of energy and glucose homeostasis [79]. Within the ARC, agouti-related peptide (AgRP) neurons represent a neuronal subpopulation that store and release several appetite-inducing agents, including AgRP, neuropeptide Y (NPY), and GABA [79–81]. Moreover, neuronal circuits linked to AgRP neurons are able to regulate peripheral glucose metabolism and carbohydrate utilization independent of the regulation of appetite [59, 82].
Mice selectively lacking βarr2 in AgRP neurons (AgRP-βarr2-KO) did not display any significant metabolic phenotypes, independent of the diet that the mice consumed [83]. On the other hand, AgRP-βarr1-KO mice maintained on a HFD showed pronounced impairments in glucose tolerance and hepatic insulin resistance, associated with liver steatosis and elevated plasma FFA levels [83]. Adipose tissue from HFD AgRP-βarrr1-KO mice showed increased PKA activity, suggesting that the observed increase in plasma FFA levels may be the result of increased lipolysis caused by enhanced sympathetic outflow [83]. Thus, increased lipolytic flux from adipose tissue to the liver may contribute to hepatic lipid accumulation and insulin resistance displayed by HFD AgRP-βarr1-KO mice.
Interestingly, surgical dissection of the hepatic branch of the vagus prevented all metabolic deficits displayed by the HFD AgRP-βarr1-KO mice [83], suggesting that the presence of βarr1 is required for the ability of AgRP neurons to regulate neuronal pathways that promote enhanced vagal outflow to the liver. In agreement with this observation, previous work has shown that insulin and other hormones or nutrients can act on ARC neurons to regulate hepatic glucose fluxes by modulating vagal outflow to the liver [84, 85].
The metabolic impairments displayed by the HFD AgRP-βarr1-KO mice closely mimicked those observed with mice that lacked the insulin receptor selectively in AgRP neurons [86], raising the possibility that βarr1 regulates the responsiveness of AgRP neurons to insulin. Insulin is known to hyperpolarize AgRP neurons (Figure 3C), thus altering neuronal pathways that control glucose homeostasis [59, 86, 87]. Pydi et al. [83] found that insulin was unable to hyperpolarize AgRP neurons lacking βarr1 (Figure 3C). Additional mechanistic studies suggested βarr1 stabilizes IRS-1 via complex formation, thus ensuring proper insulin signaling (also see [88]).
In contrast to HFD AgRP-βarr1-KO mice, mice that over-expressed βarr1 in AgRP neurons (AgRP-βarr1-OE mice) showed striking improvements in glucose homeostasis and insulin sensitivity [83]. These phenotypes were opposite to those observed with HFD AgRP-βarr1-KO mice [83]. Taken together, the metabolic phenotypes displayed by these mutant mice suggest that strategies aimed at increasing the levels and/or activity of βarr1 in AgRP neurons may prove useful to restore impaired glucose homeostasis in T2D and related metabolic disorders.
Concluding remarks and future perspectives
In summary, recent studies with cell type-specific βarr1 and βarr2 mutant mice have shown that the two β-arrestins play critical roles in maintaining euglycemia and energy homeostasis (for a summary, see Table 1). In some cases, the observed metabolic phenotypes were consistent with the classical role of βarr2 to act as a negative regulator of GPCR function. However, many β-arrestin mutant mice exhibited changes in glucose homeostasis and other metabolic processes that involved non-canonical β-arrestin actions, including distinct cytoplasmic signaling pathways and actions in the nucleus to modulate gene transcription. At this time, it remains unclear whether these non-canonical β-arrestin functions require prior recruitment by GPCRs that have not yet been identified. In any case, the multitude of important metabolic processes that are regulated by β-arrestins offer new perspectives for the development of novel classes of therapeutic agents for the treatment of T2D, obesity, and related pathophysiological conditions. Such drugs may include G protein- or β-arrestin-biased agonists or small molecules or peptides that can disrupt or enhance metabolically relevant interactions of βarr1 and βarr2 with key signaling molecules (see Outstanding Questions).
Table 1.
Canonical and Non-Canonical Metabolic Functions of β-Arrestins Revealed by the Use of Cell-Type Specific Mutant Mouse Models
| Cell type | Mutant mouse strain | Key metabolic phenotypes | Likely molecular mechanism(s) | References |
|---|---|---|---|---|
| Phenotypes due to canonical β-arrestin functions | ||||
| Adipocytes | Adipo-βarr2-KO | Decrease in adiposity (HFD mice) Improved glucose homeostasis Relative lack of metabolic deficits in response to HFD feeding | Lack of βarr2-mdeiated inhibition of β3-adrenergic receptor signaling | [34] |
| Hepatocytes | Hep-βarr2-KO | Impaired glucose homeostasis | Lack of βarr2-mediated inhibition of glucagon receptor signaling | [49] |
| Hep-βarr2-OE | Relative lack of metabolic impairments due to HFD feeding | Enhanced βarr2-mediated inhibition of glucagon receptor signaling | [49] | |
| Phenotypes due to non-canonical β-arrestin functions | ||||
| Adipocytes | Adipo-βarr1-KO | Impaired glucose tolerance and insulin sensitivity (HFD mice) | Increases myostatin expression in BAT due to the lack of nuclear βarr1activity | [51] |
| Adipo-βarr1-OE | Improved glucose tolerance and insulin sensitivity (HFD mice) | Reduced myostatin expression in BAT due to enhanced nuclear βarr1 activity | [51] | |
| β-Cells | Beta-βarr2-KO | Impaired insulin release and glucose tolerance (HFD mice) | Impaired CAMKII function due to βarr2 deficiency | [21] |
| Beta-βarr2-OE | Increased insulin secretion and improved glucose tolerance (HFD mice) | Enhanced CAMKII activity (likely mechanism) | [21] | |
| Beta-βarr1-KO | Reduced efficacy of SU drugs to promote insulin secretion | Impaired Epac2 function due to βarr1 deficiency | [22] | |
| Skeletal muscle cells | SKM-βarr2-KO | Slight improvements in glucose and insulin tolerance (HFD mice) | Enhanced insulin-induced Akt activation in SKM due to βarr2 deficiency | [76] |
| AgRP neurons | AgRP-βarr1-KO | Impaired glucose tolerance and insulin sensitivity (HFD mice) | Lack of βarr1 prevents insulin from silencing AgRP neurons | [83] |
| AgRP-βarr1-OE | Improved glucose homeostasis and insulin tolerance | Enhanced insulin sensitivity of AgRP neurons | [83] | |
Abbreviations: SU, sulfonylurea; HFD, high-fat diet; SKM, skeletal muscle; KO, knockout; OE, overexpression.
Outstanding questions.
Are altered β-arrestin levels of clinical relevance in the pathophysiology of T2D and related metabolic disorders?
Are the signaling pathways through which the two β-arrestins regulate key metabolic functions in the mouse conserved in human?
Is it possible to develop therapeutic strategies that selectively enhance or reduce β-arrestin-1 or -2 function in a tissue-specific fashion?
Is it feasible to identify new drugs that increase or decrease the levels of β-arrestin-1 or -2 in certain cell types only?
What are the potential side effects of such novel therapeutic approaches?
Highlights.
Studies with novel mutant mouse models have shown that the two β-arrestins (βarr1 and βarr2) function as key regulators of whole glucose and energy homeostasis by modulating specific signaling pathways in metabolically important cell types, including hepatocytes, adipocytes, pancreatic β-cells, and AgRP neurons of the hypothalamus.
In most cell types examined, βarr1 and βarr2 do not have redundant functions.
β-Arrestins modulate key metabolic processes via canonical (inhibition of GPCR activity) and non-canonical signaling pathways, including modulation of gene expression profiles in the nucleus.
The novel information gained from the analysis of cell-type-specific βarr1 and βarr2 mutant mice should guide the development of novel therapeutic strategies aimed at enhancing or inhibiting βarr1 and/or βarr2 activity for the treatment of type 2 diabetes and related metabolic disorders.
Acknowledgements
Many of the studies reviewed in this article were supported by the Intramural Research Program of the NIH, NIDDK, Bethesda, Maryland, USA. We would like to thank all present and past members of the Wess laboratory who were involved in some the studies summarized in this review. Our special thanks goes to Drs. Robert J. Lefkowitz and Marc G. Caron (Duke University, Durham, NC) for generously sharing their floxed β-arrestin mice with the research community. The s were generated in part by using BioRender. We apologize to all authors whose work we could not discuss because of the short length of this review.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Disclosure Statement
The authors have no financial conflicts of interest to disclose.
References
- 1.Sriram K and Insel PA (2018) G Protein-Coupled Receptors as Targets for Approved Drugs: How Many Targets and How Many Drugs? Mol Pharmacol 93 (4), 251–258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Pierce KL and Lefkowitz RJ (2001) Classical and new roles of beta-arrestins in the regulation of G-protein-coupled receptors. Nat Rev Neurosci 2 (10), 727–33. [DOI] [PubMed] [Google Scholar]
- 3.Tian X et al. (2014) β-arrestins and G protein-coupled receptor trafficking. Handb Exp Pharmacol 219, 173–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Gurevich VV and Gurevich EV (2019) Plethora of functions packed into 45 kDa arrestins: biological implications and possible therapeutic strategies. Cell Mol Life Sci 76 (22), 4413–4421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Peterson YK and Luttrell LM (2017) The Diverse Roles of Arrestin Scaffolds in G Protein-Coupled Receptor Signaling. Pharmacol Rev 69 (3), 256–297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Ahn S et al. (2020) SnapShot: β-Arrestin Functions. Cell 182 (5), 1362–1362.e1. [DOI] [PubMed] [Google Scholar]
- 7.Gurevich VV and Gurevich EV (2020) Biased GPCR signaling: Possible mechanisms and inherent limitations. Pharmacol Ther, 107540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Kang J et al. (2005) A nuclear function of beta-arrestin1 in GPCR signaling: regulation of histone acetylation and gene transcription. Cell 123 (5), 833–47. [DOI] [PubMed] [Google Scholar]
- 9.Tao Y et al. (2015) Astroglial beta-Arrestin1-mediated Nuclear Signaling Regulates the Expansion of Neural Precursor Cells in Adult Hippocampus. Sci Rep 5, 15506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Mo W et al. (2008) Nuclear beta-arrestin1 functions as a scaffold for the dephosphorylation of STAT1 and moderates the antiviral activity of IFN-gamma. Mol Cell 31 (5), 695–707. [DOI] [PubMed] [Google Scholar]
- 11.Hoeppner CZ et al. (2012) Identification of a nuclear localization sequence in beta-arrestin-1 and its functional implications. J Biol Chem 287 (12), 8932–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Zhao J and Pei G (2013) Arrestins in metabolic regulation. Prog Mol Biol Transl Sci 118, 413–27. [DOI] [PubMed] [Google Scholar]
- 13.Luttrell LM et al. (2015) Fulfilling the Promise of “Biased” G Protein-Coupled Receptor Agonism. Mol Pharmacol 88 (3), 579–88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Smith JS et al. (2018) Biased signalling: from simple switches to allosteric microprocessors. Nat Rev Drug Discov 17 (4), 243–260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Wootten D et al. (2018) Mechanisms of signalling and biased agonism in G protein-coupled receptors. Nat Rev Mol Cell Biol 19 (10), 638–653. [DOI] [PubMed] [Google Scholar]
- 16.Wilbanks AM et al. (2004) Beta-arrestin 2 regulates zebrafish development through the hedgehog signaling pathway. Science 306 (5705), 2264–7. [DOI] [PubMed] [Google Scholar]
- 17.Kovacs JJ et al. (2009) Arrestin development: emerging roles for beta-arrestins in developmental signaling pathways. Dev Cell 17 (4), 443–58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Philipp M et al. (2013) The role of arrestins in development. Prog Mol Biol Transl Sci 118, 225–42. [DOI] [PubMed] [Google Scholar]
- 19.Urs NM et al. (2016) Distinct cortical and striatal actions of a beta-arrestin-biased dopamine D2 receptor ligand reveal unique antipsychotic-like properties. Proc Natl Acad Sci U S A 113 (50), E8178–e8186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kim J et al. (2018) beta-arrestin 1 regulates beta2-adrenergic receptor-mediated skeletal muscle hypertrophy and contractility. Skelet Muscle 8 (1), 39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Zhu L et al. (2017) beta-arrestin-2 is an essential regulator of pancreatic beta-cell function under physiological and pathophysiological conditions. Nat Commun 8, 14295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Barella LF et al. (2019) beta-Cell-intrinsic beta-arrestin 1 signaling enhances sulfonylurea-induced insulin secretion. J Clin Invest 130, 3732–3737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Ghaben AL and Scherer PE (2019) Adipogenesis and metabolic health. Nat Rev Mol Cell Biol 20 (4), 242–258. [DOI] [PubMed] [Google Scholar]
- 24.Gonzalez-Muniesa P et al. (2017) Obesity. Nat Rev Dis Primers 3, 17034. [DOI] [PubMed] [Google Scholar]
- 25.Kusminski CM et al. (2016) Targeting adipose tissue in the treatment of obesity-associated diabetes. Nat Rev Drug Discov 15 (9), 639–60. [DOI] [PubMed] [Google Scholar]
- 26.Saltiel AR and Olefsky JM (2017) Inflammatory mechanisms linking obesity and metabolic disease. J Clin Invest 127 (1), 1–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Guh DP et al. (2009) The incidence of co-morbidities related to obesity and overweight: a systematic review and meta-analysis. BMC Public Health 9, 88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Harms M and Seale P (2013) Brown and beige fat: development, function and therapeutic potential. Nat Med 19 (10), 1252–63. [DOI] [PubMed] [Google Scholar]
- 29.Cohen P and Spiegelman BM (2015) Brown and Beige Fat: Molecular Parts of a Thermogenic Machine. Diabetes 64 (7), 2346–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Scherer PE (2019) The many secret lives of adipocytes: implications for diabetes. Diabetologia 62 (2), 223–232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Wang W and Seale P (2016) Control of brown and beige fat development. Nat Rev Mol Cell Biol 17 (11), 691–702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Rosen ED and Spiegelman BM (2014) What we talk about when we talk about fat. Cell 156 (1–2), 20–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Chen KY et al. (2020) Opportunities and challenges in the therapeutic activation of human energy expenditure and thermogenesis to manage obesity. J Biol Chem 295 (7), 1926–1942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Pydi SP et al. (2019) Adipocyte beta-arrestin-2 is essential for maintaining whole body glucose and energy homeostasis. Nat Commun 10 (1), 2936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Amisten S et al. (2015) An atlas of G-protein coupled receptor expression and function in human subcutaneous adipose tissue. Pharmacol Ther 146, 61–93. [DOI] [PubMed] [Google Scholar]
- 36.Regard JB et al. (2008) Anatomical profiling of G protein-coupled receptor expression. Cell 135 (3), 561–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Ceddia RP and Collins S (2020) A compendium of G-protein-coupled receptors and cyclic nucleotide regulation of adipose tissue metabolism and energy expenditure. Clin Sci (Lond) 134 (5), 473–512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Cannon B and Nedergaard J (2004) Brown adipose tissue: function and physiological significance. Physiol Rev 84 (1), 277–359. [DOI] [PubMed] [Google Scholar]
- 39.Collins S and Surwit RS (2001) The beta-adrenergic receptors and the control of adipose tissue metabolism and thermogenesis. Recent Prog Horm Res 56, 309–28. [DOI] [PubMed] [Google Scholar]
- 40.O’Mara AE et al. (2020) Chronic mirabegron treatment increases human brown fat, HDL cholesterol, and insulin sensitivity. J Clin Invest 130, 2209–2219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Finlin BS et al. (2020) The beta3-adrenergic receptor agonist mirabegron improves glucose homeostasis in obese humans. J Clin Invest 130, 2319–2331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Cypess AM et al. (2015) Activation of human brown adipose tissue by a beta3-adrenergic receptor agonist. Cell Metab 21 (1), 33–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Blondin DP et al. (2020) Human Brown Adipocyte Thermogenesis Is Driven by β2-AR Stimulation. Cell Metab 32 (2), 287–300.e7. [DOI] [PubMed] [Google Scholar]
- 44.Lin HV and Accili D (2011) Hormonal regulation of hepatic glucose production in health and disease. Cell Metab 14 (1), 9–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Cho YM et al. (2012) Targeting the glucagon receptor family for diabetes and obesity therapy. Pharmacol Ther 135 (3), 247–78. [DOI] [PubMed] [Google Scholar]
- 46.Estall JL and Drucker DJ (2006) Glucagon and glucagon-like peptide receptors as drug targets. Curr Pharm Des 12 (14), 1731–50. [DOI] [PubMed] [Google Scholar]
- 47.D’Alessio D (2011) The role of dysregulated glucagon secretion in type 2 diabetes. Diabetes Obes Metab 13 Suppl 1, 126–32. [DOI] [PubMed] [Google Scholar]
- 48.Unger RH and Cherrington AD (2012) Glucagonocentric restructuring of diabetes: a pathophysiologic and therapeutic makeover. J Clin Invest 122 (1), 4–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Zhu L et al. (2017) Hepatic beta-arrestin 2 is essential for maintaining euglycemia. J Clin Invest 127 (8), 2941–2945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Luan B et al. (2009) Deficiency of a beta-arrestin-2 signal complex contributes to insulin resistance. Nature 457 (7233), 1146–9. [DOI] [PubMed] [Google Scholar]
- 51.Pydi SP, J S, Barella LF, Zhu L, Sakamoto W, Meister J, Wang L, Lu H, Cui Y, Gavrilova O, Wess J (2020) Beta-arrestin-1 suppresses myogenic reprogramming of brown fat to maintain euglycemia. Science Advances 6 (23). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Seale P et al. (2008) PRDM16 controls a brown fat/skeletal muscle switch. Nature 454 (7207), 961–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Kajimura S et al. (2009) Initiation of myoblast to brown fat switch by a PRDM16-C/EBP-beta transcriptional complex. Nature 460 (7259), 1154–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Timmons JA et al. (2007) Myogenic gene expression signature establishes that brown and white adipocytes originate from distinct cell lineages. Proc Natl Acad Sci U S A 104 (11), 4401–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Lee SJ and McPherron AC (1999) Myostatin and the control of skeletal muscle mass. Curr Opin Genet Dev 9 (5), 604–7. [DOI] [PubMed] [Google Scholar]
- 56.Fournier B et al. (2012) Blockade of the activin receptor IIb activates functional brown adipogenesis and thermogenesis by inducing mitochondrial oxidative metabolism. Mol Cell Biol 32 (14), 2871–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Braga M et al. (2013) Inhibition of in vitro and in vivo brown fat differentiation program by myostatin. Obesity (Silver Spring) 21 (6), 1180–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Ji M et al. (2008) Myostatin induces p300 degradation to silence cyclin D1 expression through the PI3K/PTEN/Akt pathway. Cell Signal 20 (8), 1452–8. [DOI] [PubMed] [Google Scholar]
- 59.Steculorum SM et al. (2016) AgRP Neurons Control Systemic Insulin Sensitivity via Myostatin Expression in Brown Adipose Tissue. Cell 165 (1), 125–138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Kong X et al. (2018) Brown Adipose Tissue Controls Skeletal Muscle Function via the Secretion of Myostatin. Cell Metab 28 (4), 631–643 e3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Zhuang LN et al. (2011) Beta-arrestin-1 protein represses adipogenesis and inflammatory responses through its interaction with peroxisome proliferator-activated receptor-gamma (PPARgamma). J Biol Chem 286 (32), 28403–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Wang C et al. (2016) beta-arrestin-1 contributes to brown fat function and directly interacts with PPARalpha and PPARgamma. Sci Rep 6, 26999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Roden M and Shulman GI (2019) The integrative biology of type 2 diabetes. Nature 576 (7785), 51–60. [DOI] [PubMed] [Google Scholar]
- 64.Eizirik DL et al. (2020) Pancreatic β-cells in type 1 and type 2 diabetes mellitus: different pathways to failure. Nat Rev Endocrinol 16 (7), 349–362. [DOI] [PubMed] [Google Scholar]
- 65.Dadi PK et al. (2014) Inhibition of pancreatic beta-cell Ca2+/calmodulin-dependent protein kinase II reduces glucose-stimulated calcium influx and insulin secretion, impairing glucose tolerance. J Biol Chem 289 (18), 12435–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Yamamoto H et al. (2003) New aspects of neurotransmitter release and exocytosis: involvement of Ca2+/calmodulin-dependent phosphorylation of synapsin I in insulin exocytosis. J Pharmacol Sci 93 (1), 30–4. [DOI] [PubMed] [Google Scholar]
- 67.Xiao K et al. (2007) Functional specialization of beta-arrestin interactions revealed by proteomic analysis. Proc Natl Acad Sci U S A 104 (29), 12011–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Mangmool S et al. (2010) beta-Arrestin-dependent activation of Ca(2+)/calmodulin kinase II after beta(1)-adrenergic receptor stimulation. J Cell Biol 189 (3), 573–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Shibasaki T et al. (2014) Cooperation between cAMP signalling and sulfonylurea in insulin secretion. Diabetes Obes Metab 16 Suppl 1, 118–25. [DOI] [PubMed] [Google Scholar]
- 70.Shibasaki T et al. (2007) Essential role of Epac2/Rap1 signaling in regulation of insulin granule dynamics by cAMP. Proc Natl Acad Sci U S A 104 (49), 19333–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Zhang CL et al. (2009) The cAMP sensor Epac2 is a direct target of antidiabetic sulfonylurea drugs. Science 325 (5940), 607–10. [DOI] [PubMed] [Google Scholar]
- 72.Hinke SA (2009) Epac2: a molecular target for sulfonylurea-induced insulin release. Sci Signal 2 (85), pe54. [DOI] [PubMed] [Google Scholar]
- 73.Takahashi T et al. (2013) Antidiabetic sulfonylureas and cAMP cooperatively activate Epac2A. Sci Signal 6 (298), ra94. [DOI] [PubMed] [Google Scholar]
- 74.Stump CS et al. (2006) The metabolic syndrome: role of skeletal muscle metabolism. Ann Med 38 (6), 389–402. [DOI] [PubMed] [Google Scholar]
- 75.DeFronzo RA and Tripathy D (2009) Skeletal muscle insulin resistance is the primary defect in type 2 diabetes. Diabetes Care 32 Suppl 2, S157–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Meister J et al. (2019) Metabolic effects of skeletal muscle-specific deletion of beta-arrestin-1 and -2 in mice. PLoS Genet 15 (10), e1008424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Lynch GS and Ryall JG (2008) Role of beta-adrenoceptor signaling in skeletal muscle: implications for muscle wasting and disease. Physiol Rev 88 (2), 729–67. [DOI] [PubMed] [Google Scholar]
- 78.Sato M et al. (2014) Improving type 2 diabetes through a distinct adrenergic signaling pathway involving mTORC2 that mediates glucose uptake in skeletal muscle. Diabetes 63 (12), 4115–29. [DOI] [PubMed] [Google Scholar]
- 79.Morton GJ et al. (2014) Neurobiology of food intake in health and disease. Nat Rev Neurosci 15 (6), 367–78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Krashes MJ et al. (2011) Rapid, reversible activation of AgRP neurons drives feeding behavior in mice. J Clin Invest 121 (4), 1424–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Krashes MJ et al. (2013) Rapid versus delayed stimulation of feeding by the endogenously released AgRP neuron mediators GABA, NPY, and AgRP. Cell Metab 18 (4), 588–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Cavalcanti-de-Albuquerque JP et al. (2019) Regulation of substrate utilization and adiposity by Agrp neurons. Nat Commun 10 (1), 311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Pydi SP, C Z, He Z, Barella LF, Pham J, Cui Y, Oberlin DJ, Egritag HE, Urs N, Gavrilova O, Schwartz GJ, Buettner C, Williams KW, Wess J (2020) Beneficial metabolic role of β-arrestin-1 expressed by AgRP neurons. Science Advances 6 (23), 10.1126/sciadv.aaz1341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Pocai A et al. (2005) Hypothalamic K(ATP) channels control hepatic glucose production. Nature 434 (7036), 1026–31. [DOI] [PubMed] [Google Scholar]
- 85.Pocai A et al. (2005) A brain-liver circuit regulates glucose homeostasis. Cell Metab 1 (1), 53–61. [DOI] [PubMed] [Google Scholar]
- 86.Konner AC et al. (2007) Insulin action in AgRP-expressing neurons is required for suppression of hepatic glucose production. Cell Metab 5 (6), 438–49. [DOI] [PubMed] [Google Scholar]
- 87.Huang Y et al. (2018) PI3K is integral for the acute activity of leptin and insulin in arcuate NPY/AgRP neurons in males. J Endocr Soc 2 (6), 518–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Usui I et al. (2004) beta-arrestin-1 competitively inhibits insulin-induced ubiquitination and degradation of insulin receptor substrate 1. Mol Cell Biol 24 (20), 8929–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Hudmon A et al. (2005) CaMKII tethers to L-type Ca2+ channels, establishing a local and dedicated integrator of Ca2+ signals for facilitation. J Cell Biol 171 (3), 537–47. [DOI] [PMC free article] [PubMed] [Google Scholar]