

Abstract
Bromodomain and extra-terminal (BET) family proteins, BRD2-4 and T, are important drug targets; however, the biological functions of each bromodomain remain ill-defined. Chemical probes that selectively inhibit a single BET bromodomain are lacking, although pan inhibitors of the first (D1), and second (D2), bromodomain are known. Here, we develop selective BET D1 inhibitors with preferred binding to BRD4 D1. In competitive inhibition assays we show that our lead compound is 9-33 fold selective for BRD4 D1 over the other BET bromodomains. X-ray crystallography supports a role for the selectivity based on reorganization of a non-conserved lysine and displacement of an additional structured water in the BRD4 D1 binding site relative to our prior lead. Whereas pan-D1 inhibitors displace BRD4 from MYC enhancers, BRD4 D1 inhibition in MM.1S cells is insufficient for stopping Myc expression and may lead to its upregulation. Future analysis of BRD4 D1 gene regulation may shed light on differential BET bromodomain functions.
Keywords: BET bromodomains, Inhibitors, BRD4 D1 selectivity, Structured waters, Myc regulation
Graphical Abstract
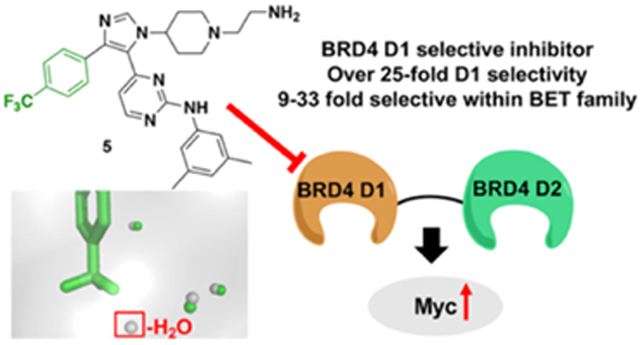
The twin-bromodomain-containing BET proteins, BRD2-4 and T are important drug targets for inflammation, cancer, and heart disease. However, potent and selective inhibitors for a single BET bromodomain are lacking. Here we describe the structure-based design of inhibitors with preferred binding for the first bromodomain of BRD4, and provide design rules for future inhibitors. Cell-based studies identify differential effects relative to pan-BET inhibitors.
Introduction
Epigenetics involves the dynamic modification of DNA, RNA and histone proteins resulting in heritable changes to gene expression.[1] One emerging protein-drug class for epigenetic therapy are bromodomain-containing proteins. Bromodomains are ~110 amino acid structural motifs that function via binding to distinct lysine acylation states, most commonly N-ε-acetylated-lysine on histones at both enhancers and gene promoters.[2,3] Bromodomain and extra-terminal (BET) family proteins, BRD2, 3, 4, and T each contain two N-terminal bromodomains (D1 and D2) and are several of the most heavily studied bromodomain-containing proteins.[4] In the context of disease, histone hyperacetylation at super-enhancer regions and recruitment of BET proteins promotes transcription of pro-survival and proliferative genes such as MYC leading to their malignant role in multiple cancers.[3,5,6] Despite the importance of BET proteins for regulating transcription, the individual contributions of each of their two bromodomains remains unclear. Several mechanisms have been proposed including differential nucleosomal interactions and simultaneous engagement of both acetylated transcription factors and nucleosomes.[7-9] Small molecule inhibitors of individual BET bromodomains will improve our understanding of the molecular mechanisms underlying epigenetic regulation of disease.[10,11]
Selective inhibition of BET bromodomains is difficult due to the high sequence similarity in the acetyl-lysine binding site between each of the eight domains. Overall the D1 bromodomain of BET protein family members are more similar than the individual domains within the same protein (e.g. BRD4 D1:BRD4 D2 = 49% similarity, BRD4 D1:BRD2 D1 = 80% similarity, Fig. 1B).[12] Despite these differences, only three amino acids in the BRD4 D1 and D2 bromodomain binding sites are non-conserved.[13] Given the high sequence similarity amongst BET bromodomains, small molecule inhibitors for a single BET bromodomain are lacking; however, potent pan-D2 inhibitors (e.g ABBV-744, Fig. 1A and GSK046/GSK620) [13,14] and a pan-D1 inhibitor, GSK778[14] were disclosed this year.
Figure 1.

(A) Representative BET bromodomain inhibitors and (B) the sequence similarity in each bromodomain relative to BRD4 D1. Percent similarities relative to BRD4 D1 calculated by PDB Sequence & Structure Alignment. (C) Co-crystal structures of BRD4 D1 with (+)-JQ1, 1, and 6. Structured waters are shown in red (PDB ID: 3MXF, 6MH1, 6WGX).
We were motivated to develop D1 selective inhibitors within the BET family with a specific focus on BRD4, as BRD4 proteins with impaired D1 function are sufficient for inhibiting chromatin binding.[14,15] However, when pan-D1-selective inhibitors are used over pan-BET inhibitors, divergent biological effects can be observed. In the case of a pan-D1 inhibitor, Olinone, oligodendrocyte progenitor cell differentiation is induced, whereas pan-BET inhibitors inhibit this process.[16] We also reported a pan-D1 inhibitor, 1,4,5-trisubstituted imidazole, 1 (Fig. 1A, formerly V), possessing dual kinase/bromodomain activity.[17] 1 was reported to bind to BET D1 bromodomains with a slight preference for BRD4 D1 (16 and 6-fold selective over BRD2 and BRD3 D1 respectively). Similar to Olinone, its affinity was modest (1.2 μM and 3.4 μM for 1 and Olinone, respectively), while also inhibiting MAP kinase p38α (Kd = 0.47 nM). We attributed part of the BET D1 selectivity to arise from displacement and reorganization of structured water molecules in BRD4 D1 (Fig. 1C). Here, we exploit our deeper access into the bromodomain binding site to displace an additional water. Our new inhibitors have high affinity and are 9-33 fold selective for BRD4 D1 over six additional BET bromodomains, including > 25-fold selective over BET D2 domains. We further show a structure-based design for removing binding to p38α, which was affecting interpretation of the functional effects of BRD4 inhibition[18]. Finally, we conduct a preliminary analysis in BRD4-dependent multiple myeloma cell line MM.1S to test if BRD4 D1 inhibition versus pan-BET inhibition was sufficient to block transcription of a key oncogene MYC. Our work on domain-selective inhibition of BRD4 allow the study of divergent BET bromodomain function and guide future therapeutic designs.
Results and Discussion
We began our investigation through analysis of the co-crystal structure of 1 with BRD4 D1 (PDB: 6MH1). In the case of acetylated histones and acetylated lysine mimetic drugs (e.g. (+)-JQ1), the methyl group of the acetyl groups, points directly into the binding pocket which has up to 6 structured waters (Fig. 1C). In the case of 1, the para-fluorophenyl group binds deeper into the pocket, displacing two water molecules (Fig. 1C).[17] Having access to this unexplored binding pocket, we evaluated the effect of the size of the para substituent to fill the pocket and modulate the structured water network (Table 1, Fig. S3). Three new analogues were synthesized (2-4, Scheme S1). The relative affinities of these inhibitors against BRD4 D1 were tested by competitive inhibition fluorescence anisotropy (FA) assays in which a fluorescein-labeled pan-BET inhibitor, (+)-JQ1 (Fl-JQ1) was used as a tracer. In the retesting of 1, the inhibitory potency was 6-fold weaker than our previous report that used a different tracer.[17] Removing the F atom altogether had minimal effect on potency (2, IC50 = 4.5 μM), whereas a larger methyl group, 3, further improved activity (IC50 = 1.2 μM). However, the most significant effect resulted when a trifluoromethyl group was installed, (4, IC50 = 0.31 μM), leading to a 34-fold enhanced potency relative to 1.
Table 1.
IC50 values of 1 and analogues against BRD4 D1 by fluorescence anisotropy and AlphaScreen. Kinase activity against p38α.
 |
|||||
|---|---|---|---|---|---|
| Cpd | R = | R1 = | BRD4 D1 IC50 by FAa(μM) |
BRD4 D1 IC50 by AlphaScreenb(μM) |
p38α Kdc(nM) |
| (+)-JQ1 | - | - | 0.037 ± 0.008 | 0.051 ± 0.02† | - |
| 1 | F | H | 11 ± 0.8 | 3.8 | 0.47 |
| 2 | H | H | 4.5 ± 0.2 | 2.6 | - |
| 3 | CH3 | H | 1.2 ± 0.2 | 1.1 | - |
| 4 | CF3 | H | 0.31 ± 0.06 | 0.64 | 260 |
| 5 | CF3 | (CH2)2NH2 | 0.13 ± 0.01 | 0.29 | 1900 |
| 6 | CF3 | (CH2)2N(CH3)2 | 0.15 ± 0.03 | 0.20 | - |
| 7 | CF3 | (CH2)2NHCOCH3 | 0.42 ± 0.01 | 1.5 | - |
| 8 | CF3 | (CH2)2NH2 | 1.1 ± 0.2 | 1.3 | >30,000 |
Data represents the mean and standard deviation of three biological replicates.
Data represents the mean of two technical replicates except (+)-JQ1.
Data represents the mean and standard deviation of six biological replicates.
Kd values were determined by KINOMEscan. Data represents the mean of two biological replicates.
To verify the affinity in an orthogonal assay, we used an AlphaScreen competitive inhibition for BRD4 D1 using a tetraacetylated H4 histone peptide as a more native-like ligand. Due to the low concentration of both protein and histone used in this assay, IC50 values can be used to approximate the Ki of the inhibitor if conditions are appropriately controlled.[19,20] Under our conditions, (+)-JQ1 had a reproducible IC50 comparable to the reported Kd of 50-75 nM (Table 1, Fig. S4).[21,22] Similar to FA, the relative trend of affinity was observed for inhibitors 1-4, with 4 possessing the lowest IC50 value of 0.64 μM. In all cases, IC50 values from AlphaScreen were within 3-fold of the FA IC50 values (Table 1, Fig. S3, S4).
To further improve the affinity and selectivity of our inhibitor, we subsequently targeted one of the three non-conserved residues between BRD4 D1 and D2, namely an Asp144 in BRD4 D1 (His433 in D2) used to develop prior pan-D1 inhibitors.[17] Our co-crystal structure of 1 with BRD4 D1 showed the piperidyl group to be solvent exposed providing a potential vector for attaching polar groups to target the surface-exposed Asp144 (Fig. S2). Several positively charged alkyl amino group were tested; however, only the ethylamino group, 5, and N,N-dimethyl-ethylamino group, 6, led to reasonable potency gains relative to 3 (2.4 and 2.1-fold respectively, Table 1) in the FA assays. Similar effects were confirmed by AlphaScreen (Table 1). To test the effect of a favorable electrostatic interaction, the ethylamino group was acetylated to yield 7. In this case, the IC50 increased by 3.2-fold. This result supports the need for a basic amine attached to the piperidyl group.
Given the ~80-fold increase in potency from 1 to 5, we next sought to evaluate if the selectivity for D1 exhibited by 1 was maintained by 5 using our FA assay. To test the selectivity of these molecules, BRD4 D2 and BRD2 D1 were selected as representative BET D2 and D1 proteins. In both cases, 5 was unable to fully displace Fl-JQ1 from the bromodomains (BRD2 D1, 61% inhibited at 100 μM and BRD4 D2 IC50 > 100 μM, Fig. 2), whereas, pan-BET inhibitor, (+)-JQ1 displaced Fl-JQ1 from both bromodomains. These results demonstrated that while inhibitor potency could be enhanced from 1 to 5, selectivity against both a BET D2 and a BET D1 domain was maintained.
Figure 2. Competitive binding experiments of 5 and (+)-JQ1 by fluorescence anisotropy and a commercial AlphaScreen assay demonstrates selectivity for BRD4 D1.
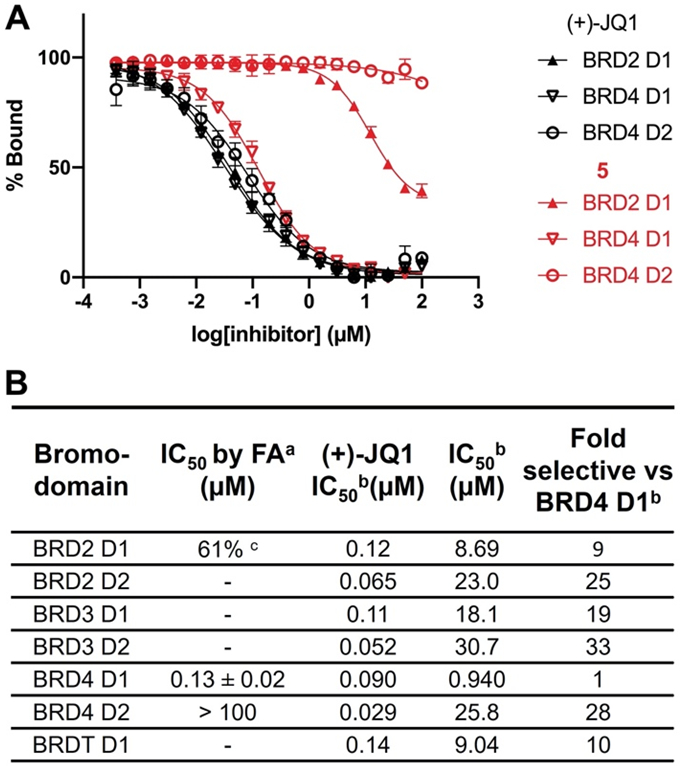
(A) Competitive binding curves of 5 (in red) and (+)-JQ1 (in black) by fluorescence anisotropy against BRD2 D1, BRD4 D1, and BRD4 D2. (B) IC50 values of 5 and (+)-JQ1 by fluorescence anisotropy and a commercial AlphaScreen assay.
a Data represents the mean and SEM of biological replicates.
b IC50 values were determined by a commercial AlphaScreen assay. Data represents the mean of two biological replicates.
c 61% inhibition at 100 μM.
The observed BRD4 D1 selectivity led us to further assess BET bromodomain selectivity via a commercial AlphaScreen service. We could not control for identical conditions to compare absolute values with our AlphaScreen data, therefore we limited our analysis to a relative comparison of the commercial data. Consistent with our FP data, 5 was selective for BRD4 D1 over BRD4 D2 (28-fold) and displayed similar selectivity against the D2 of BRD2, and 3 (25 and 33-fold respectively). 5 was less selective for BRD4 D1 over other BET D1 domains (9-19 fold) (Fig. 2B, Fig. S5). This D1 selectivity was similar to improved relative to 1. We retested the selectivity in an orthogonal assay with a commercial phage-display assay at DiscoverX and measured a similar selectivity for the closest off-target BRD2 D1 of 8.5-fold (Table S1). 4 was also tested against BRD4 D1 and BRD4 D2 to evaluate if the ethylamino group of 5 improved the selectivity for D1 over D2. In this case, 4 maintained selectivity for BRD4 D1 (Fig. S6). This result indicates that the ethylamino group can improve affinity, but not selectivity.
To more broadly evaluate bromodomain selectivity, we showed that 5 displayed no measurable affinity against six additional bromodomains (p300, BRD1, SMARCA2, SMARCA4, PBRM1(5), and PCAF) that 1 previously inhibited by > 35% (Table S1). During the preparation of this manuscript, Liu et al. reported two BRD4 D1 selective inhibitors with 9-10 fold selectivity for BRD4 D1 over D2 and nanomolar potency. To the best of our knowledge, 5 and these new inhibitors are the first small molecule inhibitors with preferred binding to BRD4 D1 with sub-micromolar potency.[23]
As a final design strategy, we sought to remove the potent p38α binding affinity of our prior inhibitor 1.[17] The p-F phenyl group was originally designed to target a secondary hydrophobic pocket in the kinase. Gallagher et al. showed that having a p-CF3 phenyl group in the kinase binding pocket based on similar analogues led to a 25-fold decrease in IC50 compared with a F atom.[24] We therefore determined the p38α binding affinity for our inhibitors 4 and 5 using a commercial kinase binding assay. In the case of 4, the binding affinity decreased by 550-fold compared with 1. The affinity was further attenuated to micromolar levels with 5. The role of the ethylamino group is unclear. Previously, we mitigated p38α kinase activity using a related scaffold which converted the pyrimidine to a 2,6-disubstituted pyridine and used an exocyclic ether.[18] To further remove p38α kinase activity, molecule 8 was synthesized. In this case, p38α binding could be removed, but at the expense of BRD4 D1 affinity (Table 1). CK1 is a known off-target of p38α inhibitors, [25] for which 5 still inhibits (Fig. S10). Future analogs will be designed to remove affinity for this protein.
We used crystallography to obtain higher-resolution structural information to better understand the molecular basis for the affinity and selectivity of our inhibitors. Although we were unable to co-crystallize 5 with BRD4 D1, a co-crystal structure with 6 was obtained at 1.53 Å resolution (PDB: 6WGX). Similar to 1, the imidazole ring maintains a key hydrogen bond with the conserved Asn140 (Fig. 3A). As designed, the p-CF3 phenyl group of 6 can fill the binding pocket and displaces an additional structured water molecule (Fig. 1C, Fig. 3A). This observation is consistent with computational analysis of the structured water network of BRD4 D1, which indicates higher energy waters in the binding pocket relative to BRD4 D2, and the bromodomains of BRD2 and 3.[26,27]
Figure 3. Structural analysis of 6 and BRD4 D1 interactions.
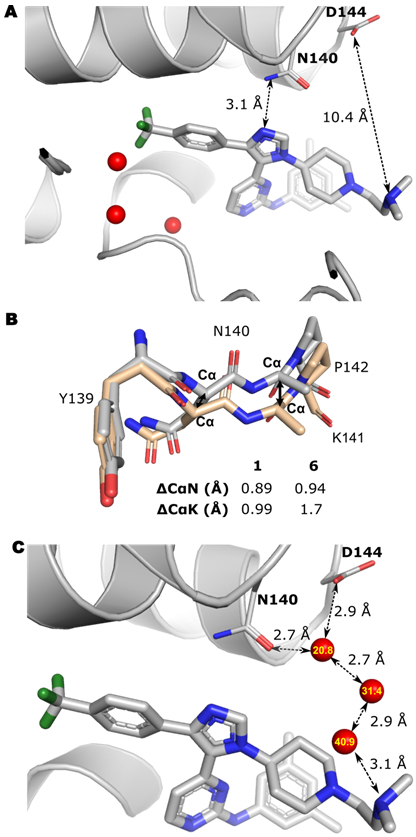
(A) Co-crystal structure of 6 with BRD4 D1. Three structured waters are indicated in red sphere. The imidazole ring forms a conserved hydrogen bond with Asn140. The distance between N, N-dimethyl amino group and Asp144 is indicated. (B) Backbone overlay of BRD4 D1 bound to H4 K5ac,K8ac (PDB: 3UVW, wheat) and BRD4 D1 bound to 1 (PDB:6MH1) or 6 (PBD:6WGX, grey). The Lys141 side chain was removed for clarity. (C) Possible water network bridging 6 to D144. Three waters are indicated in red sphere and B-factor are labeled in yellow, for which higher B-factors indicate less stable waters.
A second amino acid difference in the binding site between BET D1 and D2 is the residue next to the conserved Asn; Lys vs Pro, respectively, leading to a more flexible backbone for D1. We analyzed a select set of pan-BET inhibitors, including a 1,4,5-trisubstituted triazole analogue of 1 co-crystallized with BRD4 D1, and compared the backbone Cα-Cα differences at Asn140 and Lys141 relative to a H4 histone co-crystal structure for reference (Table S3). The movement of the Cα of Asn140 ranged from 0.36-0.87 Å (mean = 0.69 Å), and the Cα movement for Lys141 ranged from 0.25-1.3 Å (mean 0.98 Å). Alternatively, for our three D1 selective 1,4,5-trisubstituted imidazoles (1, IV, and 6, Fig. S13), we found a larger movement for the Cα of Asn140 of 0.89-0.94 Å (mean = 0.92 Å, Fig. 3B). The largest displacement was observed for 6 at the Cα of Lys141, with a 1.7 Å displacement with values ranging from 0.99-1.7 Å (mean = 1.5 Å, Fig. 3B, Table S3). Gilan et al. attributed part of their pan-D1 selectivity through reorganization of Lys141.[14]
A final analysis shows the non-conserved Asp144 residue remained 10 Å away from the ethylamino group of 6. Additional unfit electron density could be accounted for suggesting a flexible ethylamino side-chain (Fig. S12).However, we also note a structured water molecule that forms a bridging hydrogen-bond between D144 and N140 (Fig. 3C). The ethylamino group of 6, may engage this water molecule through a network of two additional waters. Targeting this structured water has recently been proposed by Wellaway et al. to help drive D1 selectivity.[28] New inhibitor designs based on our inhibitor scaffold would need to target this water directly to improve affinity and selectivity. From this structural analysis, we attribute our inhibitor selectivity to at least two key differences in structured water displacement and backbone reorganization of the binding site and highlight a potential third mechanism through engaging a new structured water molecule bridging non-conserved D144 and N140.
Having developed a series of compounds with BRD4 D1 affinity and selectivity distinct from other pan-BET, pan-D1 or pan-D2 inhibitors, we evaluated their cellular activity. MM.1S multiple myeloma cells are highly BRD4-dependent due to super-enhancer regulation of MYC.[3] As such, antiproliferation EC50 values correspond well to in vitro potency for pan-BET inhibitors under our assay conditions (e.g. (+)-JQ1 = 0.12 μM, Fig. S14A). We evaluated whether BRD4 D1 inhibitors have similar effects as pan-BET inhibitors. While compounds 4, 5, and 6 reduced cellular proliferation, the EC50 for growth inhibition was in the low micro-molar range (2.2-4.6 μM, Fig. S14A); higher than the in vitro potency. A comparison of salt and free-base forms of 5 gave similar EC50 values, suggesting the observed anti-proliferative activity cannot be solely attributed to a counterion effect (Fig. S14B).
Cellular target engagement of BRD4 was tested to help explain why our compounds had higher than expected MM.1S EC50 values. We conducted thermal stability profiling in an isothermal dose-response format using an established CETSA assay.[29] After a 1-hour incubation, a dose-dependent stabilization of BRD4 was observed with compounds 4, 5, 6 and control (+)-JQ1 (Fig. 4A). In contrast to our antiproliferation experiments, BRD4 stabilization was observed at submicromolar concentrations similar to our biochemical potency for BRD4 D1. This result shows that target engagement and cell permeability cannot explain the lack of cell activity for 4-6. An unexplained finding for inhibitors 5 and 6 at concentrations exceeding 1 μM, was an increased stabilization of BRD4 over a saturating concentration of (+)-JQ1 at 5 μM.
Figure 4. Target engagement, cellular permeability, and transcriptional effects of 4, 5, and 6 in MM.1S cells.
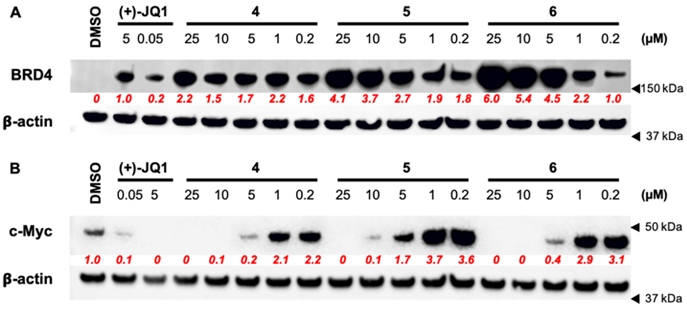
(A) Cellular target engagement of BRD4 and cell permeability of 4, 5, 6 by isothermal dose-response stability profiling after 1 h. treatment of MM.1S cells. Quantified densitometry relative to 5 μM (+)-JQ1 intensity levels shown in red. (B) Representative western blot of expressed c-Myc levels after 8 h. treatment of MM.1S cells. Quantified densitometry relative to untreated (DMSO) intensity levels in red. Full images of representative blots shown in Figure S15.
Pan-BET bromodomain inhibitors have widely shown efficacy downregulating MYC in hematological cancers, which is thought to be a main mechanism for cellular growth inhibition.[6] Here, we were surprised to find elevated c-Myc protein levels relative to DMSO treated cells at submicromolar concentrations of compounds 4-6. Elevated c-Myc protein levels were not observed with control (+)-JQ1 (Fig. 4B). Downregulation of c-Myc was observed when compound concentrations were >10-fold above their respective biochemical IC50 values, at concentrations comparable to the MM.1S antiproliferation EC50. Together these results highlight the dependence of this cell line on c-Myc expression.
While biochemical studies with protein mutants or pan-D1 inhibitors show BRD4 D1 inhibition is sufficient to displace BRD4 from chromatin,[14,15,30] our data suggest BRD4 D1 inhibition alone may be insufficient for decreasing c-Myc protein levels in MM.1S cells. Although a decrease in c-Myc is observed at high compound concentrations, given our moderate 9-33 fold selectivity for BRD4 D1 and residual p38α activity, we cannot rule out effects from inhibiting other BET bromodomains or kinases such as CK1, or drug efflux, although we have reported on structurally related but less selective inhibitors which do not show these effects.[17,18,31] Reanalyzing the data of Raux et al., who reported a BRD4 D1 selective inhibitor with micromolar affinity (Kd = 1.4 μM),[32] we note a similar trend of high antiproliferation EC50 values (27 μM), and a Myc response to inhibitor that first increases and then finally is suppressed by 50 μM inhibitor. Due to the established role of D1 in maintaining chromatin occupancy of BRD4,[14] it would be of interest to evaluate how chromatin occupancy for all BET proteins correlates with c-Myc expression in the presence of a BRD4 D1 inhibitor and to compare how overall BET expression levels are altered as a result.[33] The effects of our inhibitors on Myc expression have only been studied in one cell line and merit further assessment. While the inhibitors of Liu et al. were not used to evaluate Myc expression, these inhibitors effectively inhibited inflammatory gene expression in human small airway epithelial cells, demonstrating a contextual dependence for studying the effects of selective BET inhibitors.[23] These results support a broader cellular investigation of the transcriptional effects of selective BRD4 D1 inhibition.
Conclusion
In conclusion, we describe new structure-activity-relationship data, which has led to a selective BRD4 D1 small molecule inhibitor with submicromolar affinity. We rationalize this high level of selectivity to arise from at least two mechanisms: flexibility in a D1-conserved YNKP motif and displacement of structured-waters in the acetyl-lysine binding site of BRD4 D1, where an additional water can be displaced relative to our previous reports.[17] This binding mode is distinct from the binding mode of the BRD4 D1 inhibitors described by Liu et al.[23] With a useful set of tool compounds in hand, we have conducted a preliminary investigation on how a BRD4 D1 selective inhibitor can regulate oncogene expression in Myc-sensitive MM.1S cells. In contrast to the current literature, showing in many cases, pan-BD1 inhibitors phenocopy pan-BET inhibitors,[14] our results reveal an increase in Myc expression at concentrations mirroring biochemical potency, followed by downregulation at concentrations where other BET bromodomains may be inhibited. This effect is similar to data reported by Raux et al. using a different chemical scaffold. [32] The origins of these effects are currently under investigation.
Supplementary Material
Acknowledgements
We would like to dedicate this manuscript in honor of François Diederich who inspired a generation of scientists for tackling problems in molecular recognition. We gratefully acknowledge support for this research from the Masonic Cancer Center at the University of Minnesota, (W.C.K.P and D.A.H) the NIH (R35-GM118047 to H.A. and R01-GM110129 to D.A.H), the American Heart Association (15SDG25710427 to W.C.K.P.). J.A.J. was supported by a NIH Biotechnology training grant (T32-GM008347-23) and A.D. was supported by the NIH chemistry-biology interface training grant (T32-GM008700). This work is based on research conducted at the NE-CAT beamlines at the Advanced Photon Source, which are supported by the NIH (P30-GM124165). The Pilatus 6M detector on 24-ID-C beamline is funded by a NIH-ORIP HEI grant (S10-RR029205). We thank staff at the NE-CAT beamlines for assistance in data collection, Dr. Peter Ycas for help preparing the His9-BRD4 D1 plasmid, and Dr. Brian Van Ness for providing MM.1S cells.
Footnotes
Institute and/or researcher Twitter usernames: @UMNChemistry, @UMN_MedChem, @fewill26
Contributor Information
Huarui Cui, Department of Chemistry, University of Minnesota-Twin Cities, 207 Pleasant St. SE, Minneapolis, MN 55455 (USA).
Anand Divakaran, Department of Medicinal Chemistry, University of Minnesota-Twin Cities, 2231 6th St. SE, Minneapolis, MN 55455 (USA).
Anil K. Pandey, Department of Chemistry, University of Minnesota-Twin Cities, 207 Pleasant St. SE, Minneapolis, MN 55455 (USA)
Jorden A. Johnson, Department of Chemistry, University of Minnesota-Twin Cities, 207 Pleasant St. SE, Minneapolis, MN 55455 (USA)
Huda Zahid, Department of Chemistry, University of Minnesota-Twin Cities, 207 Pleasant St. SE, Minneapolis, MN 55455 (USA).
Zachariah J. Hoell, Department of Chemistry, University of Minnesota-Twin Cities, 207 Pleasant St. SE, Minneapolis, MN 55455 (USA)
Mikael O. Ellingson, Department of Chemistry, University of Minnesota-Twin Cities, 207 Pleasant St. SE, Minneapolis, MN 55455 (USA)
Ke Shi, Department of Biochemistry, Molecular Biology, and Biophysics, University of Minnesota-Twin Cities, 321 Church St. SE, Minneapolis, MN 55455 (USA).
Hideki Aihara, Department of Biochemistry, Molecular Biology, and Biophysics, University of Minnesota-Twin Cities, 321 Church St. SE, Minneapolis, MN 55455 (USA).
Daniel A. Harki, Department of Medicinal Chemistry, University of Minnesota-Twin Cities, 2231 6th St. SE, Minneapolis, MN 55455 (USA)
William C.K. Pomerantz, Department of Chemistry, University of Minnesota-Twin Cities, 207 Pleasant St. SE, Minneapolis, MN 55455 (USA); Department of Medicinal Chemistry, University of Minnesota-Twin Cities, 2231 6th St. SE, Minneapolis, MN 55455 (USA)
References
- [1].Jenuwein T, Allis CD, Science (80-. ) 2001, 293, 1074 LP – 1080. [DOI] [PubMed] [Google Scholar]
- [2].Filippakopoulos P, Picaud S, Mangos M, Keates T, Lambert JP, Barsyte-Lovejoy D, Felletar I, Volkmer R, Müller S, Pawson T, et al. , Cell 2012, 149, 214–231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Lovén J, Hoke HA, Lin CY, Lau A, Orlando DA, Vakoc CR, Bradner JE, Lee TI, Young RA, Cell 2013, 153, 320–334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Filippakopoulos P, Knapp S, Nat. Rev. Drug Discov 2014, 13, 337–356. [DOI] [PubMed] [Google Scholar]
- [5].Brown JD, Lin CY, Duan Q, Griffin G, Federation AJ, Paranal RM, Bair S, Newton G, Lichtman AH, Kung AL, et al. , Mol. Cell 2014, 56, 219–231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Delmore JE, Issa GC, Lemieux ME, Rahl PB, Shi J, Jacobs HM, Kastritis E, Gilpatrick T, Paranal RM, Qi J, et al. , Cell 2011, 146, 904–917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Miller TCR, Simon B, Rybin V, Grötsch H, Curtet S, Khochbin S, Carlomagno T, Müller CW, Nat. Commun 2016, 7, 13855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Huang B, Yang X-D, Zhou M-M, Ozato K, Chen L-F, Mol. Cell. Biol 2009, 29, 1375–1387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Shi J, Wang Y, Zeng L, Wu Y, Deng J, Zhang Q, Lin Y, Li J, Kang T, Tao M, et al. , Cancer Cell 2014, 25, 210–225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Belkina AC, Denis GV, Nat. Rev. Cancer 2012, 12, 465–477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Arrowsmith CH, Bountra C, Fish PV, Lee K, Schapira M, Nat. Rev. Drug Discov 2012, 11, 384–400. [DOI] [PubMed] [Google Scholar]
- [12].Nakamura Y, Umehara T, Nakano K, Moon KJ, Shirouzu M, Morita S, Uda-Tochio H, Hamana H, Terada T, Adachi N, et al. , J. Biol. Chem 2007, 282, 4193–4201. [DOI] [PubMed] [Google Scholar]
- [13].Faivre EJ, McDaniel KF, Albert DH, Mantena SR, Plotnik JP, Wilcox D, Zhang L, Bui MH, Sheppard GS, Wang L, et al. , Nature 2020, 578, 306–310. [DOI] [PubMed] [Google Scholar]
- [14].Gilan O, Rioja I, Knezevic K, Bell MJ, Yeung MM, Harker NR, Lam EYN, Chung C, Bamborough P, Petretich M, et al. , Science (80-. ) 2020, 8455. [Google Scholar]
- [15].Philpott M, Rogers CM, Yapp C, Wells C, Lambert JP, Strain-Damerell C, Burgess-Brown NA, Gingras AC, Knapp S, Müller S, Epigenetics and Chromatin 2014, 7, 14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Gacias M, Gerona-Navarro G, Plotnikov AN, Zhang G, Zeng L, Kaur J, Moy G, Rusinova E, Rodriguez Y, Matikainen B, et al. , Chem. Biol 2014, 21, 841–854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Divakaran A, Talluri SK, Ayoub AM, Mishra NK, Cui H, Widen JC, Berndt N, Zhu JY, Carlson AS, Topczewski JJ, et al. , J. Med. Chem 2018, 61, 9316–9334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Carlson AS, Cui H, Divakaran A, Johnson JA, Brunner RM, Pomerantz WCK, Topczewski JJ, ACS Med. Chem. Lett 2019, 10, 1296–1301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Pomerantz WCK, Johnson JA, Ycas PD, in (Ed.: Mai A), Springer International Publishing, Cham, 2020, pp. 287–337. [Google Scholar]
- [20].Ycas PD, Zahid H, Chan A, Olson N, Johnson JA, Talluri S, Schönbrunn E, Pomerantz W, Org. Biomol. Chem 2020, DOI 10.1039/D0OB00506A. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Filippakopoulos P, Qi J, Picaud S, Shen Y, Smith WB, Fedorov O, Morse EM, Keates T, Hickman TT, Felletar I, et al. , Nature 2010, 468, 1067–1073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Mishra NK, Urick AK, Ember SWJ, Schonbrunn E, Pomerantz WC, ACS Chem. Biol 2014, 9, 2755–2760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Liu Z, Chen H, Wang P, Li Y, Wold EA, Leonard G, Joseph S, Brasier AR, Tian B, Zhou J, et al. , J. Med. Chem 2020, DOI 10.1021/acs.jmedchem.0c00035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Gallagher TF, Seibel GL, Kassis S, Laydon JT, Blumenthal MJ, Lee JC, Lee D, Boehm JC, Fier-Thompson SM, Abt JW, et al. , Bioorganic Med. Chem 1997, 5, 49–64. [DOI] [PubMed] [Google Scholar]
- [25].Shanware NP, Williams LM, Bowler MJ, Tibbetts RS, BMB Rep. 2009, 42, 142–147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Aldeghi M, Ross GA, Bodkin MJ, Essex JW, Knapp S, Biggin PC, Commun. Chem 2018, 1, DOI 10.1038/s42004-018-0019-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Bharatham N, Slavish PJ, Young BM, Shelat AA, J. Mol. Graph. Model 2018, 81, 197–210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Wellaway CR, Bamborough P, Bernard SG, Chung CW, Craggs PD, Cutler L, Demont EH, Evans JP, Gordon L, Karamshi B, et al. , J. Med. Chem 2020, DOI 10.1021/acs.jmedchem.0c00566. [DOI] [PubMed] [Google Scholar]
- [29].Jafari R, Almqvist H, Axelsson H, Ignatushchenko M, Lundbäck T, Nordlund P, Molina DM, Nat. Protoc 2014, 9, 2100–2122. [DOI] [PubMed] [Google Scholar]
- [30].Tyler DS, Vappiani J, Cañeque T, Lam EYN, Ward A, Gilan O, Chan YC, Hienzsch A, Rutkowska A, Werner T, et al. , Science (80-. ) 2017, 356, 1397–1401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Penas C, Maloof ME, Stathias V, Long J, Tan SK, Mier J, Fang Y, Valdes C, Rodriguez-Blanco J, Chiang CM, et al. , Nat. Commun 2019, 10, 1–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Raux B, Voitovich Y, Derviaux C, Lugari A, Rebuffet E, Milhas S, Priet S, Roux T, Trinquet E, Guillemot JC, et al. , J. Med. Chem 2016, 59, 1634–1641. [DOI] [PubMed] [Google Scholar]
- [33].Raina K, Lu J, Qian Y, Altieri M, Gordon D, Rossi AMK, Wang J, Chen X, Dong H, Siu K, et al. , Proc. Natl. Acad. Sci. U. S. A 2016, 113, 7124–7129. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.