

Abstract
The evolving concept that cancer stem cells (CSCs) are the driving element in cancer development, evolution and heterogeneity, has overridden the previous model of a tumor consisting of cells all with similar sequentially acquired mutations and similar potential for renewal, invasion and metastasis. This paradigm shift has focused attention on therapeutically targeting CSCs directly as means of eradicating the disease. In breast cancers, CSCs can be identified by cell surface markers and are characterized by their ability to self-renew and differentiate, resist chemotherapy and radiation, and initiate new tumors upon serial transplantation in xenografted mice. These functional properties of CSCs are regulated by both intracellular and extracellular factors including pluripotency-related transcription factors, intracellular signaling pathways and external stimuli. Several classes of natural products and synthesized compounds have been studied to target these regulatory elements and force CSCs to lose stemness and/or terminally differentiate and thereby achieve a therapeutic effect. However, realization of an effective treatment for breast cancers, focused on the biological effects of these agents on breast CSCs, their functions and signaling, has not yet been achieved. In this review, we delineate the intrinsic and extrinsic factors identified to date that control or promote stemness in breast CSCs and provide a comprehensive compilation of potential agents that have been studied to target breast CSCs, transcription factors and stemness-related signaling. Our aim is to stimulate further study of these agents that could become the basis for their use as stand-alone treatments or components of combination therapies effective against breast cancers.
Keywords: Breast cancer, cancer stem cells, pluripotency, differentiation
1.1. Introduction
Breast cancer is a phenotypically diverse cancer with a large degree of inter- and intra- tumoral genetic and epigenetic heterogeneity. Breast tumors are divided into subtypes based on hormonal receptor status—specifically based on their expression of the estrogen receptor (ER), progesterone receptor (PR), and human epidermal growth factor receptor 2 (HER2). ER and PR are expressed either alone (ER+/PR− or ER−/PR+) or together (ER+/PR+) in a majority of breast carcinomas, and are used as biomarkers and prognostic factors to guide clinical management1. ER+ breast cancers are well differentiated and less aggressive relative to ER- breast cancers. Co-expression of both ER and PR receptors carries better prognosis when compared to ER+/PR− or ER−/PR+ cases2. HER2+ carcinomas, cancers comprising about 25% of all breast cancer cases, feature the most aggressive phenotype among invasive breast cancer3. However, a pathologically complete response often can be achieved from HER2-targeted therapy along with conventional chemotherapy4. Breast carcinomas that do not express ER, PR, or HER2 are referred to as triple negative breast cancers (TNBC) and constitute about 15–20% of breast cancer cases. These are a group of genetically and phenotypically heterogenous tumors with poor prognosis and limited responsiveness to treatment5. Additional functional biomarkers have been investigated for potential implications in diagnosis, treatment, and predictions of drug resistance and prognosis; these include antigen Ki-67 (KI-67; cell proliferation), programmed death-ligand 1 (PD-L1; immune response), HER2Δ16 (drug resistance), and matrix metalloproteinase 9 (MMP-9; invasion and metastasis)6.
Compelling evidence indicates that within a cancer, there is a subpopulation of cells known as tumor-initiating cells (TICs) or cancer stem-like cells (CSCs) that are responsible for the tumor initiation, chemo-/radio-resistance and relapse7, 8. This population is characterized by a stem-cell gene expression signature, drug-resistant phenotype and self-renewal capacity in vitro and in vivo9. CSCs can self-renew through division and give rise to the bulk of tumor cells in the mass through replication and differentiation from the stem cell compartment10, 11. Thus, targeting CSCs can be a promising therapeutic strategy for eradicating breast cancer.
Two models have been proposed to explain the evolution of CSCs12. According to the clonal evolution model, genetic mechanisms are the culprits underlying clonal expansions, with the stepwise acquisition of mutations in single clones culminating in tumor progression. This is followed by selection of more aggressive dominant subclones having a survival advantage and tumorigenic potential13. Meanwhile, the CSC model hypothesizes a role for nongenetic mechanisms as the source of intra-tumoral heterogeneity. In this model, cancers originate from a small subpopulation of tumor cells that can initiate tumorigenesis. CSCs were first identified in acute myeloid leukemia, when a CD34+/CD38− subpopulation of human leukemia cells transplanted into immunocompromised (NOD/SCID) mice, perpetuated the disease and underwent leukemic transformation and differentiation in vivo to form the bulk of the cells phenotypically identifiable as leukemic14. CSCs have now been identified in a variety of cancer types, including breast cancer, colon cancer, melanoma, prostate cancer, lung cancer, and glioblastoma15.
In this review, we examine the known markers identifying and characterizing breast CSCs (BCSCs), the signaling pathways and transcription factors that appear to regulate stemness properties and agents that target them and that might be exploited in treatment of the disease.
1.2. Cancer stem cells
1.2.1. Identification of BCSCs
Stem cell surface markers that are used to isolate BCSCs provide key insights into BCSC biology along with opportunities to develop therapeutics that target them. To date, CSCs in various human cancers have been identified by using one or multiple cell surface markers in fluorescence-activated cell sorting (FACS); measuring functional markers such as aldehyde dehydrogenase 1 (ALDH1) enzyme activity and ATP-binding cassette (ABC) transporter expression; single-cell DNA sequencing; and screening side population cells with the Hoechst-33342 dye exclusion technique15. Identifying, isolating, and characterizing the BCSC populations has so far primarily utilized cell surface markers. In particular, the CD44, CD24, and ALDH1+ markers have become increasingly used to isolate BCSCs, characterize them, and use them as prognostic markers for patients16.
CD44, a non-kinase single-span transmembrane glycoprotein that binds hyaluronan, is involved in controlling cell proliferation, survival, and differentiation; it thus regulates CSC properties including self-renewal, tumor initiation, metastasis, and radio- and chemo-resistance. Alternatively-spliced variants of CD44 play roles in tumor development and progression. CD44 expression is high in BCSCs; its downregulation induces differentiation and sensitizes the cells to chemotherapy17, 18. CD24 is a glycosylphosphatidylinositol-linked cell surface glycoprotein that has been implicated in immunological functions, tumorigenesis, chemoresistance, and metastasis. CD24 expression is low or absent in BCSCs, and its upregulation is associated with poor prognosis in the luminal A and TNBC subtypes19. ALDH1 is a member of group of enzymes that oxidize intracellular aldehydes to carboxylic acids. Its activity is measured by the ALDEFLUOR assay, which assesses nine active isoforms of ALDH; in breast cancer, high ALDH1 activity is associated with stem-like features and chemoresistance. ALDH1+ breast cancers are also characterized by being ER-, EGFRII+ and Ki-67hi 20. Suppression of ALDH1 decreases tumorigenicity and cell migration21.
BCSCs were first isolated from xenografts using a combination of cell surface markers: CD44+/CD24−/low Lin−. The cells with this phenotype are tumorigenic in numbers as low as 100 cells; in contrast, those with different phenotypes failed to form tumors even with tens of thousands of cells7. A high CD44/CD24 ratio is directly correlated with cell proliferation and tumorigenesis, as indicated by increased formation of mammospheres in vitro and xenograft tumors21. In addition, CD44+/CD24− breast cancer cells are enriched for EMT-associated traits, including expression of matrix metalloproteinase 1 (MMP-1), vimentin, and zinc finger E-box binding homeobox 1 (ZEB1); this is suggestive of interplay between EMT and CSC status22. These cells also demonstrate increased expression of the molecular chaperones glucose-regulated protein 78 (GRP78) and 94 (GRP94), which regulate endoplasmic reticulum homeostasis in stem cell development and in invasion of cancer23. Furthermore, the cells exhibit dysregulation of major signaling pathways otherwise involved in the regulation of normal mammary stem cells, such as the Notch, Hedgehog, and Wnt/β-catenin pathways; blockage of these pathways by chemotherapeutic agents inhibits the CSC-like phenotype and tumorigenesis24. In mice, breast cancer cells derived from BRCA1-deficient mammary tumors show increased numbers of CD44+/CD24− and CD133+ cells and increased expression of stem cell-associated genes including Oct4, Notch1, Aldh1, Fgfr1, and Sox125. In the clinical context, the CD44+/CD24− phenotype is associated with resistance to cytostatic agents, grade of malignancy, and patient survival26. Furthermore, CD44+/CD24− BCSCs are resistant to radiation treatment and demonstrate increased expression of Jagged-1, Notch-1, and p-S6K1 (a major downstream regulator of the mTOR pathway)27. The radioresistance of these cells is mediated through upregulation of the checkpoint kinase pathway (CHK); application of the CHK inhibitor, debromohymenialdisine, effectively overcoming the resistance28.
Regarding ALDH as a CSC population marker, Ginestier et al. found that ALDH1 enzymatic activity is high in a subpopulation of breast carcinomas having tumorigenic and self-renewal abilities both in vivo and in vitro29. ALDHhiCD44+ subpopulations of BCSCs are resistant to chemotherapy and radiotherapy and feature increased expression of glutathione-S-transferase pi, p-glycoprotein, and checkpoint kinase 1 (CHK1). Pretreatment of these cell populations with all-trans retinoic acid or the ALDH inhibitor diethylaminobenzaldehyde (DEAB) significantly sensitizes the stem-like breast cancer cells and reduces resistance30. In MCF-7 xenograft tumors, ALDH1A1 (an isoform of ALDH1) promotes tumor angiogenesis by upregulating the retinoic acid/HIF-1α/VEGF signaling pathway, thereby affecting breast cancer progression31. In ALDH1+ BCSCs, the Wnt/β-catenin signaling pathway, known to regulate stem cell niche during development, is dysregulated; downregulation of Wnt expression inhibits the CSC phenotype and suppresses breast cancer metastasis32. In ductal carcinoma in situ (DCIS), expression of ALDH1 along with enhancer of zeste 2 polycomb repressive complex 2 subunit (EZH2), a marker implicated in stem cell maintenance and renewal, is associated with tumor recurrence and progression to invasive breast cancer33.
Studies of invasive breast carcinomas and breast cancer cell lines have shown basal-like tumors to be enriched with CD44+/CD24− and ALDH1+ phenotypes34. Quiescent mesenchymal-like BCSCs are CD44+/CD24− and localize to the tumor periphery, whereas proliferative epithelial-like BCSCs are ALDH1+ and localize in the center35. Table 1 summarizes the BCSC markers, their functions, target genes and relation to tumorigenesis.
Table 1.
Breast cancer stem cell markers, their functions, target genes and tumorigenesis
| Cancer cell surface marker(s)/ Transcription factor | Function in stem cell | Experimental system | Target genes/markers studied | TS/CFA | In vivo study | Ref. |
|---|---|---|---|---|---|---|
| ABCG2+ (CD338) | Tumor initiation | HCC1937 | CD326/EpCAM, CD49f/α6-integrin, CD24, CD10, CD133 | + | Injection of unsorted cells (4×105 and 2×106 cells) | Leccia et al180 |
| ALDH+/hi CD44+ | Chemo-radio resistance | MDA-MB-231 and MDA-MB-468 | GSTP1, P-gp, CHK1, CK8/18/19 | + | NA | Croker et al181 |
| ALDH1+ | Self-renewal; tumor initiation | Cells developed from patient tumors | NA | + | Injection of sorted cells (500 ~ 50,000 cells) | Ginestier et al29 |
| C/EBPδ | Tumor initiation; EMT; metastasis | SUM159, MDA-MB-231, MDA-MB-468, MCF-7, T47D |
IL-6, HIF-1α, CD44, N-cadherin, Vimentin, E-cadherin, Twist and STAT3, Myc, Nanog, and KLF4, OCT4, SOX2, CDH1, FBXW7, NOTCH and NICD | + | Injection of unsorted cells (3×106 or 0.3×106 cells) | Balamurugan et al182 |
| CCR5 | Self-renewal; DNA repair | SUM159, SUM149 and FC-IBC-02 | DNA damage repair genes: FANCB, LIG3, POLE, CRY1; PI3K/Akt signaling; cell survival signaling | + | Injection of sorted cells (4000 cells) | Jiao et al183 |
| CD133+ | Tumor initiation; self-renewal | Cell lines developed from Brca1Δexon11/p53+/− mouse mammary tumors | Expression of stem cell associated genes: OCT4, NOTCH1, ALDH1, FGFR1 and SOX1 | + | Injection of sorted cells (50 ~ 5000 cells) | Wright et al25 |
| CD24+CD29+/CD49f+ | Metastasis; EMT | BRCA-1 mutant mouse derived CSCs | E-cadherin | + | Injection of sorted cells (2×105 cells) | Vassilopoulos et al184 |
| CD24+Thy1+ | Tumor initiation | MMTV-Wnt-1 mouse | CK5, CK14, CK17, NOTCH4, BCL6B | + | Injection of sorted cells (50 ~ 2,000 cells) | Cho et al185 |
| CD29loCD24+CD61+ | Tumor initiation | MMTV-Wnt-1, MMTV-neu and p53 mutant mice | CK14, CK8 | + | NA | Vaillant et al186 |
| CD44+/CD24−/lowLineage− | Tumor initiation | Human breast tumor-derived tumor cells | NA | − | Injection of sorted cells; Limiting dilution (100 ~ 5 × 105 cells) | Al-Hajj et al7 |
| CD44+/CD24−/low/EpCAM+ | Tumor initiation; self-renewal | HMLE cells, MDA-MB-231, MCF-7, MCF10A, SUM149, SUM159, SUM1315 and SUM225 | NA | + | Injection of sorted cells; Limiting dilution (100 ~ 1× 106 cells) | Fillmore et al187 |
| CD44+/CD49fhi/CD133/2hi | Tumor initiation, self-renewal | Human breast cancer tissues | SOX2, BMI-1, NANOG | + | Injection of sorted cells; Limiting dilution (50 ~ 2500 cells) | Meyer et al188 |
| CD44+CD24+/loSSEA-3+ or ESAhiPROCRhiSSEA-3+ | Tumor initiation | MCF-7 and MDA-MB-231 | SSEA-3, Caspase-3, capase-8, caspase-9, caspase-12 SSEA-4, globo-H, β3GalT5 | + | Injection of sorted cells (10 ~ 2500 cells) | Cheung et al189 |
| CD49F+/DLL1hi/DNERhi | Tumor initiation; self-renewal | Human normal mammary stem cells and breast tumors | SERPINB5, TOP2A, CK5, TP63, SOX4, CD24, ADRM1, DNER, DLL1, and JAG1 | + | Injection of sorted cells (500 ~ 1000 cells) | Pece e al190 |
| CD49f+CD61+ | Tumor initiation; self-renewal | MMTV-Her2/neu-induced primary mammary gland tumor | CSC markers: Abcg2, Aldh1, CD133, Gli1 and Tp63; differentiation marker genes: CK5, CK6, CK14, CK18; TGFβ signaling: Pai, Il6, Igfbp3, Foxc2; EMT genes: CDH2, SMA, SNAIL, TWIST1 and ZEB1 | + | Injection of sorted cells (5000 cells) | Lo et al191 |
| CD61(Integrin αvβ3) | Tumor initiation; self-renewal | BT-20, MDA-MB-231and MDA-MB-468 | SLUG | + | Injection of unsorted cells; Limiting dilution (100 ~ 1×105 cells) | Desgrosellier et al192 |
| CD70+ | Self-renewal; EMT; lung-specific metastasis; differentiation | MCF-7, MDA-MB-231 and CN34 | E-cadherin and Vimentin | + | Injection of sorted cells; limiting dilution | Liu et al193 |
| Cx26 | Self-renewal; tumor initiation | Triple negative breast cancer samples; MDA-MB-231 and HCC70 | NANOG, FAK, OCT4, SOX2 | + | Serial dilution injections (8,000 ~ 800,000 cells) | Thiagarajan et al194 |
| CXCR2 | Chemo-radio resistance; tumor initiation | Human breast cancer tissues; 4T1 | ALDH, ABCG2, NOTCH1, SOX2, and NANOG | + | Injection of sorted cells (200 ~ 20,000 cells) | Wang et al195 |
| GD2+ | Self-renewal; tumor initiation; EMT | Transformed HMLE cells and MDA-MB-231 | GD3S; MMPs: MMP2, MMP7 and MMP19; EMT markers: N-cadherin, Vimentin, E-cadherin; stemness markers: CD44, CD24 | + | Injection of sorted cells (1 ~ 10,000 cells) | Battula et al196 |
| Glyoxalase 1 | Self-renewal | MDA-MB-157 and MDA-MB-468 | ALDH1 | + | NA | Tamori et al197 |
| HIF-2α | Self-renewal; chemoresistance | MCF-7 and MDA-MB-231 | Stem cell markers: C-MYC, OCT4, NANOG; Notch pathway related proteins: NOTCHNICD and HEY2; Wnt-pathway related proteins: β-catenin, Axin2 and Survivin | + | Injection of unsorted cells (3 × 106 or 1 × 106 cells) | Yan et al198 |
| Lgr5hi | Self-renewal; tumor initiation; EMT | MCF-7, MDA-MB-231 | EMT markers: E-cadherin, β-catenin, Vimentin, Fibronectin, Snail, slug; Cyclin D1, C-myc, CK14, and CK18, CD44, CD24 | + | Injection of sorted cells (200 ~ 20,000 cells) | Yang et al199 |
| miR-1 | Negative regulator of breast cancer stem cells and EMT | Human breast cancer tissues; MDA-MB-231 | EVI-1; EMT markers: E-cadherin, N-cadherin | − | Injection of sorted cells (5×106 cells) | Wu et al200 |
| miR-221 | Self-renewal | Human breast cancer tissues; T47D and MCF-7 | Stemness genes: NANOG, OCT3/4; β-Catenin, DNMT3b, CD44, CD24, Numb, p53 | + | NA | Roscigno et al201 |
| MUC1+ | Tumor initiation; self-renewal | MCF-7 | ABCG2, CK18, CK19, EpCAM, CD49f | + | NA | Engelmann et al202 |
| Nectin-4+ | EMT; metastasis; self-renewal | MDA-MB-231 | CD44, CD133, PI3K, Akt, β-catenin, E-cadherin, Vimentin | + | Injection of unsorted cells (1×107 cells) | Siddharth et al203 |
| PROCR+/ESA+ | Self-renewal; tumor initiation; EMT | MDA-MB 231 | EMT markers: Vimentin, E-cadherin, SLUG, FOXC2; stem cell markers: ALDH, CD44, CD24, ESA, CD133, CXCR4, ABCG2 | + | Injection of sorted cells; Limiting dilution (100 ~ 2500 cells) | Hwang-Verslues et al204 |
| RUNX1 | Negative regulator of self-renewal and CSC | MCF10AT1, MCF10A, MCF10CA1 and MCF-7 | E-cadherin, Vimentin, FN1, VEGF, MMP13, MMP9, CXCR4, CXCL12, CSC markers: Zeb1, Twist1, CD44, CD24 | + | Injection of unsorted cells (1×106 cells) | Hong et al205 |
| Sca1+ | Self-renewal; tumor initiation; chemoresistance | BALB-neuT mouse | Stem cell markers: Oct-4, CD44, CD29, CD24; differentiation markers: CK14, CK18, CK19, α-SMA | + | Injection of unsorted cells (100 ~ 10,000: cells) | Grange et al206 |
| Syndecan-1 | Self-renewal | SUM149 and SK-BR-3 | CSC markers: CD44, CD24, ALDH; Notch signaling: NOTCH-1, −3, −4, HEY-1; Gli-1, IL-6, IL-8, gp130, STAT3, NFκB, CCL20, EGFR | + | NA | Ibrahim et al207 |
| tDR‐000620 | Predictor of TNBC recurrence | Human patients- derived TNBC samples; MCF-7 and MDA-MB-231 | SOX2, OCT4, ALDH1, CD44, CD24 | + | NA | Feng et al208 |
TS/CFA: Tumor Spheroid (TS) or Colony formation assay (CFA) (+ indicates TS/CFA was used, - indicates TS/CFA was not used); NA: Not available
In addition to cell surface markers, various functional assays are employed in the study of BCSCs; these include the mammary organoid 3D culture model, mammosphere forming assay in serum free medium, and the in vivo injection of FACS-sorted cells in limiting dilutions into immunocompromised mice, with consequent initiation of tumor growth36. Despite the multiplicity of BCSC markers and assays available, universal putative markers have yet to be resolved that can identify specific subpopulations having the most tumorigenic potential in each breast cancer case. Identification of those subpopulations is essential for the development of CSC-targeted therapy and overcoming resistance to chemo- and radio-therapeutic treatments.
1.2.2. Characteristics of BCSCs
CSCs are similar to normal stem or progenitor cells in their ability to self-renew and recapitulate heterogeneity13. Self-renewal is a hallmark of stem cells, in which a stem cell produces two daughter cells with stem cell properties (symmetric division) or one daughter cell with stem cell properties and a second that undergoes differentiation (asymmetric division)37. CSCs express transcription factors (OCT4, NANOG homeobox [NANOG], and SRY-box transcription factor 2 [SOX2]) that are found in early embryonic stem cells. The core stem cell factors regulate pluripotency and self-renewal, and their overexpression is associated with signaling pathways related to malignant transformation, tumorigenicity, tumor progression, relapse, and inhibition of apoptosis38. OCT4, NANOG and SOX2 markers are induced in many cancer types, including breast, prostate, lung, colorectal, and gastrointestinal cancers39. Likewise, normal stem cells and CSCs share common self-renewal signaling pathways including the Notch, Hedgehog, STAT3, and Wnt/β-catenin pathways; all of these are documented as being important signaling cascades in embryonic development and have been shown to contribute to tumorigenesis in multiple types of tumors40. The plausibility of the CSC theory in breast cancer, which hypothesizes that BCSCs are derived from normal progenitor/stem cells, is supported by phenotypic features similar to their lineage-specific normal stem cell counterparts41.
CSCs arise from deregulation of the self-renewal program in stem cells, giving rise to their malignant transformation, or from the dedifferentiation of committed mature cells to acquire CSC-like properties42. In addition to self-renewal, CSCs also display quiescence in response to environmental cues. Thus, while anti-mitotic chemotherapeutic agents have been developed to target proliferating tumor cells, the resident, generally quiescent CSCs remain resistant to chemo- and radio-therapies even at high doses and so are the major cause of relapse—the living evidence of CSC plasticity and the supreme challenge faced by current therapies43.
Ultimately, numerous intrinsic and extrinsic factors regulate CSC traits, including developmental pathways, epigenetics, stem cell transcription factors, epithelial mesenchymal transition (EMT) factors, cell cycle regulation mechanisms, apoptosis pathways, and the tumor microenvironment. All of these factors interact constantly and dynamically regulate CSC survival, proliferation, and metastasis44. As a consequence, CSCs exhibit a spectrum of functional and phenotypic heterogeneity, confirmed by in vitro clonogenic and anchorage-independent growth assays (tumor sphere assays) as well as in vivo limiting dilution xenotransplantation assays45. CSCs constitute only a small proportion (0.01–2%) of the tumor cells in a tumor mass, and isolating and identifying a pure CSC population remains challenging46.
1.3. Major self-renewal pathways in BCSCs
CSC populations are maintained by their self-renewal capacity. The current notion of CSCs states that the self-renewal signaling and transcription factors which regulate growth and maintenance in normal stem cells are dysregulated in BCSCs47. The following section will discuss the major self-renewal pathways in BCSCs.
1.3.1. Notch signaling pathway
Notch is a family of four transmembrane receptors (NOTCH 1–4) that interacts with five ligands: the jagged proteins (JAG1 and JAG2) and the delta-like ligands (DLL1, DLL3, and DLL4)48. While canonical Notch signaling is involved in multiple cellular processes, including embryonic development, stem cell fate determination, apoptosis, cell cycle progression, self-renewal and lineage specific differentiation, non-canonical Notch signaling is associated with immune activation and breast tumorigenesis49. Oncogenic RAS activates NOTCH1 and upregulates the Notch ligand DLL1 along with presenilin-1 through a p38-mediated pathway. There is a correlation between Ras overexpression and upregulation of NOTCH1 in breast carcinomas50. In clinical breast cancer samples, Notch signaling is found to promote BCSCs by inducing expression of sirtuin 2 (SIRT2), leading to deacetylation and activation of ALDH1A151. Notch1 and Notch4 signaling are higher in ESA+/CD44+/CD24low enriched BCSCs. NOTCH1 overexpression in MCF-7 and MCF10A breast cancer cells increased the abundance of the BCSC CD44+/CD24low subpopulation, along with increasing tumor cell invasion and migration. Increased NOTCH1 expression also promotes the EMT phenotype and tumor growth in vivo through crosstalk with STAT3 signaling52.
Notch signaling and expression of its target genes are also elevated in mammosphere-derived stem-like cells. Inhibition of Notch signaling by a γ-secretase inhibitor significantly reduces sphere formation, proliferation and colony formation, and also induces apoptosis53. Likewise, pharmacologic and genetic inhibition reduce stem cell activity in in vitro and tumor formation in vivo54. In CD44+/CD24− mammospheres, the breast tumor suppressor signal peptide, CUB domain and EGF like domain containing 2 (SCUBE2) is overexpressed, with concomitant overexpression of SOX2, OCT4, and NANOG in TNBC. Ectopic expression of SCUBE2 in adherent cells promotes EMT and metastasis by activating Notch signaling and its components55.
Notch4 expression is high in TNBC and is negatively correlated with overall survival56. Notch4+ BCSCs are characterized by increased expression of stemness factors (OCT4, SOX2, NANOG), mammosphere formation in vitro, and tumorigenicity in a serial dilution tumor transplantation xenograft model57. Treating TNBC cells with mTOR inhibitors leads to increased stemness features and greater in vivo tumor initiating capacity. The intrinsic resistance of these cells from TORC1/2 inhibition is driven by their activated Notch1 and FGF1 pathways in association with increased mitochondrial metabolism and FGFR1 signaling. Notably, abrogation of the FGFR-mitochondrial metabolism-Notch1 axis overcomes resistance to TORC1/2 inhibitors by eliminating drug-resistant CSCs58. Meanwhile, JAG1-NOTCH4 receptor activation increases BCSC activity and induces tamoxifen resistance in both patient-derived tumors and xenograft models. Targeting Notch4 reverses the increase in Notch, reducing BCSC activity and improving the tamoxifen resistance59. Thus, in combination with other modalities, targeting the Notch pathway could be a promising strategy for enhancing the effectiveness and sensitivity of breast cancer treatment while simultaneously eradicating BCSCs.
1.3.2. Wnt signaling in BCSCs
The Wnt/Frizzled/β-catenin pathway is an evolutionarily conserved signaling pathway that plays significant roles in embryonic development and tissue homeostasis60. There are 19 Wnt glycoproteins that serve as ligands for the receptors Frizzled (FZD) and LDL receptor related protein 5/6 (LRP5/6)61. Aberrant Wnt signaling is implicated in breast cancers62. Wnt signaling is constitutively activated in basal breast cancer cells, affecting their self-renewal and differentiation63. Regulators of the Wnt signaling pathway, such as lymphoid enhancer-binding factor 1 (LEF1), cyclin D1, β-catenin, and TCF-4 are upregulated in ALDH+ BCSCs. Treating 4T1 BCSCs with Wnt3a ligand induced Wnt/β-catenin signaling and transcriptional activity, while Wnt1 silencing decreased tumor sphere formation and the CD44+/CD24− population in vitro, along with decreasing tumorigenesis and metastasis in xenografts32. Thyroid hormone receptor interactor 6 (TRIP6), an adapter protein involved in regulating the functions of CSCs, enhances stemness in breast cancer cells through activation of the Wnt/β-catenin pathway64. On the converse side, β-catenin silencing has been shown to reduce tumorigenesis in vivo and to suppress cancer stemness in vitro by decreasing the abundance of ALDH+ breast cancer cells and the expression of stemness-related genes, including B lymphoma Mo-MLV insertion region 1 homolog (BMI-1) and MYC proto-oncogene, bHLH transcription factor (c-Myc). In TNBC cells, such silencing also impaired formation of anchorage-independent colonies in soft agar assay and improved chemoresistance65. Treatment of TNBC cells with WNT-targeting pharmacological agents modulates the expression of PD-L1, a ligand for the inhibitory immune checkpoint receptor PD-1, which is highly expressed in the stem cell compartment (ALDH+ or CD44v6-positive) alongside WNT signaling-related genes. This indicates a role of Wnt signaling in TNBC-related immune escape66. The pleiotropic effects of Wnt signaling and its components in breast cancer initiation, progression, and the maintenance of different cancer subtypes remain to be elucidated, and deeper understanding of them is essential for developing BCSC-targeted therapies.
1.3.3. Hedgehog signaling
The Hedgehog (Hh) signaling pathway is involved in animal development and tissue homeostasis and is associated with many solid tumors including pancreatic cancer, lung cancer, breast cancer, basal cell carcinoma, and hematological malignancies. Hh family members include Sonic hedgehog (SHH), Indian hedgehog (IHH), and Desert hedgehog (DHH)67. In cancer, this pathway plays roles in malignant transformation, proliferation, drug resistance, metastasis, and the expansion of cancer stem cells68. Hh signaling is known to drive oncogenesis, specifically resulting from mutations in components of Hh pathway, over-expression of ligands of the Hh pathway, and maintenance of CSC phenotype through regulation of stemness-related genes69. The pathway is significantly upregulated in luminal B and TNBC breast cancer subtypes70. An earlier study in mice showed that overexpression of Gli1 under the MMTV promoter is sufficient to promote development of breast tumors expressing progenitor cell markers71.
In mammospheres, PTCH, SMO, GLI1 and GLI2 are highly expressed, becoming down-regulated upon differentiation. Activation of Hh signaling increases mammosphere forming efficiency (MFE) and size, effects mediated by the polycomb gene BMI-1. Hh signaling is also hyperactivated in the CD44+/CD24−/Lin− BCSC population72. In mammospheres of estrogen receptor-positive MCF-7 breast cancer cells, components of the Hh pathway (PTCH, SMO, GLI1 and GLI2) are highly expressed relative to monolayer cells; treatment with salinomycin, which targets CSCs, induced apoptosis and downregulated target genes of the Hh pathway (c-Myc, Bcl-2, and Snail) in vitro and reduced the tumor growth and expression of PTCH, SMO, GLI1 and GLI2 in xenograft tumors73. In basal-like breast cancer, increased expression of forkhead box C1 (FOXC1), an EMT-associated transcription factor, acts via activation of SMO-independent Hh signaling mediated by GLI2 to enrich CSC properties of the cancer, including ALDH+ cell populations and mammosphere growth. Furthermore, expression of FOXC1 in TNBC cells confers resistance to anti-Hh drugs74. LncRNAs were demonstrated to regulate EMT-associated BCSC stemness through the growth arrest specific 1 (GAS1)-activated lncRNA-Hh pathway. The upregulated Hh signaling increased GLI1, SOX2, and OCT4 expression and MFE in vitro and tumorigenicity in vivo. Silencing lncRNA-Hh reversed these findings75. Hh signaling is also associated with chemoresistance in TNBC. Chemotherapy-induced drug resistance is mediated by GLI1 via upregulation of multidrug resistance protein 1 (MDR1) and breast cancer resistance protein (BCRP)76. Ultimately, activation of the Hh signaling pathway is well-documented as a poor prognostic indicator in both hormone receptor-positive breast cancer and TNBC. However, there are limited Hh-targeted therapies available. Selective inhibition of GLI and other targets might represent an effective strategy for impeding breast cancer development and the activity of cancer stem cells.
1.3.4. TGF-β signaling
The transforming growth factor β (TGF-β) superfamily consists of 42 ligands including TGF-β, activins, Nodal, inhibins, bone morphogenic proteins (BMPs), and growth differentiation factors (GDFs)77. In cancer, TGF-β displays context-dependent dichotomous behaviors, being a tumor suppressor that inhibits cell cycle progression and promotes apoptosis or a tumor promoter that induces EMT and invasion77. Consistent with its tumor suppressor role, constitutive expression of TGF-β1 in mammary epithelial cells of xenografts increased latency of tumor growth and decreased mammary cancer risk78. Similarly, TGF-β reduces the BCSC population and induces luminal differentiation79. Loss of TGF-β-mediated tumor suppression in breast cancer is associated with downregulation of luminal markers and upregulation of basal markers79. In another example, transgenic expression of MMTV-driven dominant-negative TβR2 (DNIIR) in female mice decreased tumor latency and induced spontaneous tumor formation and invasion80. In contrast, mammary epithelial cell-specific expression of TGF-β ligands or TβRs in xenograft tumors promotes lung metastasis, while attenuation of TGF-β signaling decreases metastasis81. These findings suggest a paradoxical role of TGF-β signaling in inhibiting tumor initiation while promoting metastasis.
In immortalized human mammary epithelial cells (HMLE cells), TGF-β1-induced EMT generates stem cell-like cells that express EMT markers and have increased ability to form mammospheres, colonies in soft agar, and xenograft tumors82. Meanwhile, CD44+/CD24− BCSCs generated by TGF-β1-induced EMT are more resistant to radiation compared to their parental cells, mediated by upregulating antioxidant-related genes and reducing activation of death receptor pathways83.
Accumulating evidence has implicated the epigenetic regulation of TGF-β signaling in breast cancer progression84. In TNBC, TGF-β1 inhibits miR-196a-3p and activates its downstream target gene neuropilin-2 to promote metastasis85. Meanwhile, miR-133b and miR-190 have been shown to inhibit TGF-β-induced EMT and metastasis by targeting SMAD2, indicating their roles as tumor suppressors and potential diagnostic biomarkers of breast cancer86. In mouse epithelial NMuMG cells, lncRNA-HIT mediates TGF-β-induced EMT and invasion by targeting E-cadherin; this long noncoding RNA is conserved in humans and elevated in invasive breast cancer. Attenuation of lncRNA-HIT resulted in decreased invasion, migration, and tumor growth87. Overall, due to the complexity of functional switches in TGF-β signaling, specific drugs targeting downstream signaling would be preferable as therapeutics, as they can be utilized without compromising other physiological functions of TGF-β.
1.3.5. STAT3 signaling
The transcription factor signal transducer and activator of transcription (STAT) family consists of seven highly conserved members, STAT1, STAT2, STAT3, STAT4, STAT5a, STAT5b and STAT6; all share structural and functional similarities88. STAT3 is known to contribute to tumor cell proliferation, progression, metastasis, immune suppression, and stem cell self-renewal and maintenance89. STAT3 overexpression is found in more than 40% of breast cancers, mainly in the TNBC subtype. Aberrant activation of STAT3 promotes breast cancer development by deregulating genes implicated in proliferation, angiogenesis, and EMT90. In TNBC, hypoxia induces an increase in the CD44high/CD24low BCSC population and in chemoresistance by activating STAT3 signaling. Genetic knockdown of STAT3 reverses the acquisition of stem-like features, which suggests a significant role of STAT3 in promoting the induction of cancer stemness by hypoxia91.
Cytokines are known risk factors that induce inflammation and promote breast cancer progression. Oncostatin M (OSM), a member of the gp130 family of cytokines, has been implicated in inflammatory functions driving tumor aggressiveness and in increased STAT3 phosphorylation and STAT3-dependent IL-6 production, which promotes breast cancer progression. High expression of OSM correlates with poor breast cancer patient survival92. High levels of another cytokine, IL-35, are associated with poor prognosis in patients. Breast cancer cell-derived IL-35 inhibits conventional T (Tconv) cell proliferation and induces the cells to transform into IL-35-producing induced regulatory T (iTr35) cells by activating STAT1/STAT3, thereby promoting breast cancer progression93.
MiR-124, a tumor suppressor that modulates breast cancer cell proliferation and invasion, is downregulated in breast cancer cells. Overexpression of miR-124 in TNBC decreased STAT3 and suppressed cell proliferation and invasion. Restoration of STAT3 expression reversed miR-124-mediated tumor cell invasion94. Similarly, miR-7 was demonstrated to act as a tumor suppressor by inhibiting breast cancer cell invasion and metastasis, decreasing BCSC populations, and reversing EMT in MCF-7 and MDA-MB-231 cell lines. These miR-7-mediated effects occurred through targeting the oncogene SETDB1, which led to suppression of the downstream target STAT3 as SETDB1 binds to its promoter and regulates its expression95. All told, STAT3 signaling is not simply limited to a role in tumorigenesis but is also important in invoking the immune cell response. STAT3 will be a promising target for breast cancer prevention and therapy.
1.3.6. Other signaling in the regulation of BCSCs
Breast tumorigenesis is driven by aberrant regulation of cell signal transduction pathways owing to the accumulation of genetic and epigenetic changes over time. Apart from the aforementioned pathways, other significant signaling involved in BCSC enrichment and maintenance includes the Hippo, PI3K/Akt/mTOR and BMI-1 pathways16. Dysregulation of any of these individual pathways or of the interplay between them poses a risk of developing breast cancer. In addition, the receptor tyrosine kinase (RTK) class of specialized cell surface receptors respond to environmental cues by relaying appropriate signals in the tumor cell; these include epidermal growth factor receptor (EGFR), platelet derived growth factor receptor (PDGFR), and AXL receptor tyrosine kinase (AXL). RTKs play a multifaceted role in breast cancer development, sharing common downstream pathways such as MAPK, NF-κB, PI3K/Akt, and JAK/STAT signaling; the crosstalk with other key signaling pathways relevant to the regulation of angiogenesis, metastasis, and maintenance of BCSCs. Mutation in or overexpression of RTKs has been observed in different stages of breast cancer to lead to constitutive activation of various signal transductions that promote BCSCs and chemoresistance96.
1.4. Signature of cancer stem cell transcription factors in breast cancer
Pluripotency in embryonic stem cells (ESCs) is regulated by a well-characterized core transcriptional network. The circuitry of this network constitutes major transcription factors of pluripotency, signal transduction machinery, and epigenetic regulators. In human embryonic stem cells, OCT4, NANOG, and SOX2 function as master regulators of pluripotency and self-renewal properties while inhibiting differentiation to control cell fate97. Pluripotency can be induced in adult somatic cells, as evidenced by reprogramming of adult fibroblast cells into pluripotent stem cells with characteristic features of ESCs using the OSKM transcription factors (OCT3/4, SOX2, c-Myc, and Kruppel-like factor 4 [KLF4])98. Astrocytes transduced with the H-ras oncogene or with OSKM factors undergo reprogramming into progenitor cells, resulting in tumorsphere formation. When these tumorspheres are transplanted as xenografts, they form heterogeneous tumors, suggesting an interplay between tumorigenicity and pluripotency42. It can be assumed that CSCs share characteristics with ESCs. The pluripotency transcription factors OCT4, NANOG, and SOX2 are upregulated in human cancers, including breast cancer, glioma, melanoma, and prostate cancer, and their overexpression in tumors is associated with poor differentiation, stem-like phenotype, and inhibition of apoptosis38.
1.4.1. OCT4
OCT4, a homeodomain transcription factor of the Pit-Oct-Unc family, is one of the most important transcription factors governing pluripotency99. The human OCT4 gene has three transcript variants (OCT4A, OCT4B, and OCT4B1) and four protein isoforms (OCT4, OCT4B-190, OCT4B-265, and OCT4B-164). Each alternative transcript variant and isoform demonstrates diverse expression patterns and functions100. Distinctive expression patterns of OCT4 variants have been identified in different types of breast cancer: OCT4A and OCT4B are highly expressed in low-grade ductal tumors, whereas OCT4B is overexpressed in lobular type breast cancer. Expression of OCT4 variants is also associated with the expression of ER, PR, HER2 and p53101. Among them, OCT4A is responsible for maintenance of stemness in pluripotent embryonic stem cells100. Ectopic expression of Oct4 in 4T1 mouse breast cancer cells increased tumorsphere formation, expression of stem cell markers such as CD133, CD34, Sca-1, and ALDH1 in vitro, and tumorigenic potential in vivo102. OCT4 controls the expression of target genes by recognizing and binding to DNA regulatory regions through an octamer motif (AGTCAAAT) or by recruiting other transcription factors to regulate a specific set of genes103.
Phenotypically, resistance to chemo- or radiotherapy is among the hallmarks of CSCs. The function of OCT4 in the stemness-mediated resistance of BCSCs to chemotherapy and irradiation is of particular interest in breast cancer. In hormone receptor-positive breast cancer, OCT4 can be used a prognosis indicator for poor clinical outcome and tamoxifen resistance104. Doxorubicin resistant-TNBCs showed increased CSC phenotype along with high expression of signal transducer and activator of transcription 3 (STAT3), OCT4, and c-Myc. Treatment with the STAT3 inhibitor WP1066 decreased phosphorylation of STAT3 and the expression of OCT4 and c-MYC, leading to a reduction in CD44+ BCSC population and restoration of doxorubicin sensitivity105. OCT4 also confers resistance to irradiation by increasing clonogenic survival following irradiation and upregulating interleukin 24 (IL-24) production through STAT3 and NF-κB signaling106.
PD-L1, a T-cell inhibitory molecule with immunomodulatory function, regulates breast cancer stemness by modulating OCT4 and NANOG. In breast cancer, its expression is associated with EMT, chemoresistance, and maintenance of stemness. PD-L1 knockdown inhibits AKT phosphorylation and mTOR activity, with downstream reduction of OCT4 phosphorylation at T235 and therefore of OCT4 activity107. Another regulator of OCT4 is the E3 ubiquitin ligase carboxy terminus of HSP70-interacting protein (CHIP), which was demonstrated to mediate its proteasomal ubiquitination at lysine 284 through microarray analysis of mammospheres derived from MDA-MB-231 and MCF-7 cells. CHIP overexpression decreased OCT4 stability and BCSC populations, while CHIP depletion promoted breast tumor and lung metastasis in xenografts. This finding suggests that CHIP-induced post-translational modification of OCT4 is important in maintenance of BCSCs108.
Although OCT4 is well studied in the context of stemness maintenance, its role in metastasis remains controversial. Overexpression of OCT4 in MDA-MB-231 and 4T1 breast cancer cell lines induced E-cadherin while suppressing cell migration and invasion in vitro and lung metastasis in vivo109. The inhibitory effect of OCT4 on metastasis is mediated through downregulation of Rho family GTPase 1 (RND1) by binding to its promoter region109. In contrast, a previous study from the same group showed downregulation of OCT4 in MCF-7 cells to promote cell migration and invasion by inducing EMT (decreased E-cadherin expression and increased alpha-smooth muscle actin expression)110. Given the multiple regulatory effects of OCT4 on stemness, resistance and metastasis in breast cancer, a better understanding of OCT4 for its interaction and interconnection with other markers and effectors of CSC function is essential.
1.4.2. SOX2
SOX2 is a member of the Sox (SRY-related HMG box) family member of transcription factors with a single high-mobility group DNA-binding domain. It is recognized as a key player in the regulation of early embryonic development, maintenance of undifferentiated ESCs, and cell fate determination, and its expression is dysregulated in several cancer types, including breast, prostate, brain, and lung cancers. SOX2 is additionally involved in tumorigenesis, drug resistance, poor prognosis, and metastasis, indicating a major role in cancer and positioning it as an attractive therapeutic target111. Overexpression of SOX2 in breast cancer cells increased mammosphere formation, while its knockdown suppressed mammosphere formation and delayed tumor formation in xenograft tumor initiation models. Mechanistically, SOX2 overexpression was induced through the activation of a distal enhancer of SOX2 promoter, the same element that natively regulates SOX2 transcription in pluripotent stem cells112. In ER-positive breast cancer patients, SOX2 expression is associated with poor prognosis and endocrine treatment failure, and SOX2 promotes tamoxifen resistance via activation of Wnt signaling113. It also targets SOX9 to regulate luminal progenitor cells and Wnt signaling activity114. In TNBC cases, SOX2 is implicated in BCSC chemoresistance through modulation of TWIST1. Silencing SOX2 increased paclitaxel sensitivity and diminished stemness and TWIST1 expression. This illustrates the significance of SOX2 as a connector between pluripotency, chemoresistance, and the EMT axis115. Likewise, SOX2 knockdown in MCF-7 cells decreased mammosphere formation, CD44+/CD24− subpopulation, ALDH+ population, viability in vitro, and tumorigenicity in vivo113.
1.4.3. NANOG
NANOG is a homeodomain protein found in undifferentiated mammalian ESCs and pluripotent cells. Endogenous Nanog drives ESC self-renewal by maintaining the level of OCT4, which is integral to ESC function. Although Nanog is absent in differentiated cells, its abnormal expression is reported in human cancers including prostate cancer, hepatocellular carcinoma, glioblastoma, colon cancer, and breast cancer. Expression of Nanog is associated with stemness, self-renewal, and tumorigenesis116. When coexpressed with Wnt-1 in the mouse mammary gland, Nanog promotes mammary tumorigenesis and metastasis. Ectopic expression of Nanog in MCF-7 cells enhances colony formation, migration, and invasion in vitro and tumor growth in vivo117. Meanwhile, silencing Nanog reduces colony formation, cell proliferation, and invasion; it furthermore downregulates the cell cycle regulators cyclin D1 and c-Myc, leading to cell cycle arrest at G0/G1118. In BCSCs, Nanog and OCT4 modulate TGF-β-mediated EMT; their induction promoted invasion while knockdown of both inhibited CSC migration in vitro119. In addition, Nanog confers drug resistance in MCF-7 breast cancer cells through STAT3-mediated activation of MDR1120, and in breast ductal carcinoma, its expression has statistically significant relationship with tumor grade, lymph node metastasis, and disease staging121. Tissue microarray analysis revealed that breast cancer patients with strong Nanog expression have significantly lower disease-free survival and overall survival rates than those with weak expression122.
1.4.4. KLF4
KLF4 is a member of the highly conserved Kruppel-like zinc finger transcription factor family, and is one of the four major transcription factors of pluripotency. It plays diverse roles in physiology and disease, with functions in cell cycle regulation, proliferation, apoptosis, differentiation, somatic cell reprogramming, and pluripotency123. KLF4 is differentially expressed in human cancers, and furthermore is bifunctional; it can act as either tumor suppressor or oncogene depending on the tissue, tumor type, and staging123. In breast cancer tissues, its protein expression is correlated with pathological type, histological grade, and lymph node involvement; low-level expression is found in normal breast epithelium, while increased expression is detected in neoplastic cells and prior to invasion124. In estrogen-dependent breast cancer, KLF4 acts as a tumor suppressor by regulating the transcriptional activity of ERα specifically binding to its DNA-binding region and preventing it from binding to estrogen response elements in promoter regions125. It is also self-regulating, in that the isoform KLF4α antagonizes the function of KLF4 and stimulates breast cancer cell proliferation by binding and retaining KLF4 in the cytoplasm, opposing its regulatory activities in the nucleus126. KLF4 is highly expressed in BCSCs from primary mammary tumor and breast cancer cell lines. In the MCF-7 and MDA-MB-231 cell lines, KLF4 knockdown decreased the population of ALDH1+ progenitor cells; it furthermore suppressed cell migration, invasion, and mammosphere formation in vitro and tumorigenesis in vivo127. In BCSCs, KLF4 and the androgen receptor have been demonstrated to mediate stem cell phenotype; this effect is negatively regulated by dual specificity tyrosine phosphorylation regulated kinase 2 (DYRK2), a protein kinase that controls EMT via Snail degradation. Downregulation of DYRK2 promotes KLF4 expression and cancer stem-like properties128.
1.4.5. MYC
MYC is a dimeric transcription factor of the basic helix-loop-helix (bHLH) DNA-binding protein superfamily that regulates a broad range of biological processes such as cell proliferation, differentiation, growth, and apoptosis; it is also implicated in embryonic stem cell self-renewal and pluripotency129. The MYC promoter is a downstream effector target of self-renewal pathways such as the Notch, Wnt, NF-κB and TGF-β signaling pathways130. Of the three MYC family members l-MYC, c-MYC, and n-MYC, the latter two play crucial roles in the maintenance of pluripotency. Co-deletion of both transcription factors in ESCs and in induced pluripotent stem cells (iPSCs) led to destabilization of pluripotency and spontaneous differentiation into primitive endoderm131.
As an important transcription regulator in ESCs, MYC also displays similar regulatory role in CSCs132. In fact, MYC was first recognized as one of the most potent oncogenes, inducing neoplastic transformation of target cells and a wide variety of tumors133. Transient overexpression of MYC in Rat1A cells evoked genomic instability and increased tumorigenicity134. In breast cancer, MYC amplification is associated with disease progression; additionally, its expression is higher in TNBC than in other subtypes. MYC overexpression in the BRCA1-deficient TNBC subtype is associated with poor prognosis135. Meanwhile, targeting MYC in TNBC with triptolide (C1572), a small-molecule natural product, depletes cancer-stem like cells via a proteasome-dependent mechanism136. In combination with MCL1 apoptosis regulator, BCL2 family member (MCL1), MYC promotes chemoresistance of CSCs in TNBC by increasing mitochondrial oxidative phosphorylation and the generation of reactive oxygen species137. Additionally, c-MYC is the effector target of the tumor suppressor gene p53 in mammary stem cells; loss of p53 function is implicated in the development of cancers. In breast tumors, p53 mutation activates c-MYC, leading to maintenance of cancer stemness features and expression of a mitotic gene signature, which correlates with breast cancer aggressiveness and poor prognosis138. Transducing MYC in HMLE cells induces luminal epithelial morphology changes, spheroid formation, and dedifferentiation into progenitor-like states. MYC-driven epigenetic changes are mediated through suppression of lineage-specific transcription factors and activation of de novo enhancers, determined by hyperactivation of the Wnt pathway, which further drives transcriptional activation of oncogenic pathways139.
1.5. BCSCs and therapeutic resistance
Tumor relapse in breast cancer has been attributed to drug-resistant CSCs, and the persistence of CSCs after chemotherapy pinpoints this population as an ‘ultimate target’ that must be eliminated to eradicate cancer. BCSCs share many features of normal stem cells and modulate a multitude of drug resistance mechanisms, including overexpression of drug efflux pumps (e.g. ATP-binding cassette family members ABCG2, P-gp, ABCC1, ABCB5, etc.)140, enhanced DNA repair activity141, increased scavenging of reactive oxygen species142, activation of anti-apoptotic proteins143, and induction of dormancy144, 145. BCSCs exhibit DNA damage repair mechanisms that render them chemo- and radiation-resistant, thus targeting DNA repair pathways is a plausible approach for BCSC-directed therapy141. BCSCs trigger increased expression of free radical scavenging systems at lower ROS levels than do other cells, protecting them from anti-cancer agents. Doxorubicin-dependent CD44+/CD24− BCSCs in MCF-7 cells demonstrate upregulated levels of nuclear factor, erythroid 2 like 2 (NRF2), a key transcription factor that regulates cellular responses to oxidative damage. Specifically, CD44 regulates NRF2 level through p62 expression, and NRF2 activation endows the BCSCs with aggressive phenotype and chemoresistance142.
CSCs activate anti-apoptotic proteins that can withstand cytotoxic agents. Inhibiting these anti-apoptotic proteins (such as Bcl-2) can be a potential therapeutic avenue against chemo-resistance in BCSCs143. Recently, evidence has accumulated for a role of the pro-survival autophagic pathway in BCSC survival and maintenance. Autophagy flux is high in the ALDH+ BCSC population and is essential for tumorigenicity146. This population of BCSCs shows chemoresistance that is enhanced by hypoxia, but the inhibition of autophagy in TNBC can overcome chemoresistance147. Dormant cancer cells can survive an unfavorable microenvironment and undergo reversible growth arrest; furthermore, while in a dormant state, committed tumor cells de-differentiate to become stem-like cells148. Tumor dormancy is characterized by upregulation of autophagic signaling (which maintains the metabolic homeostasis of dormant cancer cells), epigenetic features, stress-lenient signaling, and microenvironmental cues149. In BCSCs, autophagy maintains low-level expression of the glycolysis mediator 6-phosphofructo-2-kinase/fructose-2,6-biphosphatase 3 (PFKFB3) to sustain cellular dormancy. Inactivation of autophagy signaling components re-establishes normal-level PFKFB3 expression, culminating in the reactivation of BCSC self-renewal, tumor aggressiveness, and metastatic outgrowth145. Despite their indirect role in tumor growth, eradicating dormant tumor cells as the source of BCSCs and chemoresistance will offer promising therapeutic implications.
1.6. Potential compounds regulating cancer stem cells and differentiation
Loss of differentiation coupled with uncontrolled proliferation is a hallmark of malignant neoplasms. Differentiation therapy is a therapeutic strategy that re-instates endogenous differentiation programs to induce maturation in tumor cells. Upon differentiation, tumor cells revert back to a non-malignant phenotype, culminating in reduction of proliferation and metastatic potential and upregulation of differentiating markers150. Since chemotherapies target only rapidly-proliferating tumor cells and spare the slowly-dividing population of CSCs, relapse is common. The presence of dedifferentiated CSCs in solid tumors gives rise to their heterogeneous nature with regard to proliferation, metastasis, and relapse after radio- or chemotherapy. A prospective alternative CSC-targeted therapy is to use differentiation-inducing agents to target CSCs and self-renewal signaling, influence the functional hierarchy between tumor cells, and thereby reduce their chemo- and radio-resistance151. A literature search on PubMed in December 2020 yielded 4436 articles on “breast cancer stem cells”, 7758 articles on “breast cancer and differentiation” and 3450 articles on “differentiation inducing agents and cancer” for the past 5 years. From this literature search, Table 2 summarizes differentiation-promoting natural products and synthetic chemicals that have been indicated to target breast cancer stemness signaling. Some potential differentiation-inducing agents for breast cancer including all-trans retinoic acid (ATRA), vitamin D, and histone deacetylase inhibitors (HDACi) are discussed here.
Table 2.
Potential compounds regulating cancer stem cell markers and differentiation in breast cancer
| Agent | Classification | Experimental Model | Significance | Ref |
|---|---|---|---|---|
| Acetaminophen | Synthetic chemical | In vitro, MDA-MB-231 | mRNA level: CK19, AKT2, CD24, and TIMP1↑; MMP2, ALDH1, MMP9, TWIST, NOTCH1, and AKT1↓ Protein level: Vimentin↓/E-cadherin↑, Twist↓ Cell surface marker: CD44hi/CD24low↓, CD44+/CD24+↑ Differentiation induction of CSC: Twist↓, Vimentin↓/E-cadherin↑ |
Afshar et al175 |
| AF38469 | Synthetic chemical |
In vitro, MDA-MB-231, MCF-7; In vivo, MDA-MB-231, MCF-7 xenograft (5–10 µg/day/mouse given in drinking water for 3 weeks) |
Sortilin inhibition, Decreased mammosphere formation, Reduced EMT; In vivo efficacy: breast cancer metastasis and local infiltrative growth↓ |
Rhost et al209 |
| Active Hexose Correlated Compound (AHCC) | Natural product | In vitro, MCF-7 | Decreased mammosphere formation (AHCC alone and in combination with Wasabi) Observation of monocyte-to-macrophage differentiation |
Corradetti et al210 |
| All-trans- retinoic acid (ATRA) | Natural product | In vitro, MDA-MB-231, T47D, ZR75–1, BT549, MCF-7 | RARβ-TET2 complex recruitment, leading to activation RARβ-TET2-miR-200c-PKCζ signaling - increases symmetric commitment and represses asymmetric division in CSC ChIP-seq with RARβ-TET2: co-occupancy in promoters of genes involved in differentiation - RUNX1, BMP6, IKZF1, CAV1 Restoration of normal epithelial like, well-differentiated acinar structure in 3D matrigel ↑ Tamoxifen sensitization |
Wu et al156 |
| In vitro, MCF-7 | Cell markers: CD44+/CD24−, NANOG, Oct3/4↓ Differentiation induction of CSC: involucrin and syndecan↑ Reduced invasiveness and migration Epirubicin sensitization |
Yan et al154 | ||
| Arsenite | Synthetic chemical | In vitro, MDA-MB-231 | Cell surface marker: ICAM-1 expression induction (in combination with Tetrandine) | Yu et al211 |
| Atorvastatin | Synthetic chemical | In vitro, MDA-MB-468, MDA-MB-231 | Gene expression: Hippo, Notch, Wnt↓ Protein level: Yap/Taz protein↓, vimentin↓, E-cadherin↑ Differentiation induction of CSC: CD24+↑ Reduced EMT |
Koohestani mobarhan et al212 |
| β-lapachone | Natural product | In vitro, MDA-MB-231 | NQO1 induction Gene expression: CD44, ALDH1A1, DLGAP5↓ Decreased mammosphere formation |
Kim et al213 |
| BEZ235 | Synthetic chemical | In vitro, HCC1143, SUM149 | PI3K/mTOR inhibition RNAseq and immunofluorescence: CK19hi/Vimhi/ CK14lo |
Risom et al214 |
| In vitro, HCC1143 | Induction of CK19hi/VIMlo/ CK14lo enrichment in HCC1143 cell line. De-enrichment of VIMhi/ CK14lo population |
Risom et al214 | ||
| Bisphenol A | Synthetic chemical | In vitro, MCF10A | ↓BMP4 mediated stem cell maintenance and ↑ myoepithelial differentiation | Clément et al215 |
| BXL0124 | Synthetic chemical | In vitro, SUM159 | mRNA level: OCT4, CD44, LAMA5, NOTCH↓ Differentiation marker: CK14, SMA↑, CK18, CK5↓ Decreased mammosphere formation Differentiation induction of CSC |
Shan et al166 |
| CHM-09 | Synthetic chemical | In vitro, MDA-MB-231 | EGFR Tyrosine kinase inhibitor Differentiation marker: N-cadherin↓, E-cadherin↑ Induction of CSC apoptosis and cell cycle arrest Increased mesenchymal epithelial transition |
Manupati et al216 |
| Citral | Natural product | In vivo, 4T1 cells injected BALB/c mice (50 mg/kg, given orally for 28 days) | In vivo efficacy: primary and secondary xenograft tumor size↓; number of ALDH+ tumor cells↓ | Nigjeh et al217 |
| Curcumin | Natural product | In vitro, MCF-7, MDA-MB-231 | Gene expression: OCT4, NANOG, SOX2↓ Decreased mammosphere formation Differentiation induction of CSC: CD44+/CD24−↓ |
Hu et al218 |
| In vitro, SUM149, MCF10A, MCF-7 | Gene expression: ALDH1A3, PROM1, TP63, ITGA6↓ Decreased mammosphere formation |
Colacino et al219 | ||
| CWP/ICG001 | Small molecule | In vitro, MCF-7, MDA-MB-231 | Induction of Sam68-CBP complex, leading to disruption of CBP/β-catenin Differentiation induction of CSC Increased CSC apoptosis |
Benoit et al220 |
| Dasatinib | Synthetic chemical |
In vitro, paclitaxel-resistant SUM159 |
mRNA level: CDH2, FN1, SNAI1, ZEB1, TP63, SMA↓, E-cadherin↑ protein level: p-Src↓ 3D culture of SUM159. Dasatinib treated group showed formation of round, acinar-like structure Decreased mammosphere formation Differentiation induction of CSC Increased MET |
Tian et al221 |
| Diallyl Trisulfide | Natural product | In vitro, MCF-7, SUM159 | Gene expression level: CD44, ALDH1A1, NANOG, OCT4↓, Wnt/β-catenin signal↓ Reduced CSC viability |
Li et al222 |
| Digitoxin | Natural product | Patient tumors, patient tumor derived xenograft (pretreated cells with 20 nM for 17 days in vitro followed by tail vein injection into NSG mice) |
Increased intracellular calcium, dissociated cell tight junction → altered DNA methylation profile and downregulated target genes of OCT4, SOX2, NANOG, SIN3A In vivo efficacy: delayed metastatic outgrowth over the course of 72 days post-injection |
Gkountela et al223 |
| Disulfiram | Synthetic chemical | In vitro, MDA-MB-231 | Protein level: STAT3, cyclinD, Survivin, ALDHA1↓, Caspase-3↑ Cell surface marker: CD44+/CD24−↓ Increased CSC apoptosis Decreased mammosphere formation |
Kim et al224 |
| Doxorubicin | Synthetic chemical | In vitro, Hs578T | Decreased proliferation, aggregation and mammosphere formation of stem-like cells Affects the balance between self-renewal and differentiation |
Tudoran et al225 |
| EC-70124 | Synthetic chemical | In vitro, HS578T, BT549, MDA-MB-231, HCC3153 | Gene expression: PI3K/mTOR, JAK/STAT ↓ Cell marker: CD44, ALDH1, CD49f, CD133↓ Differentiation induction of CSC Decreased EMT |
Cuenca-López et al226 |
| Efatutazone | Synthetic chemical |
In vitro, MCF10A, MCF10DCIS; In vivo, MCF10DCIS xenograft (30 mg/kg, oral gavage for 3 weeks) |
PPARγ agonist mRNA level: hFABP4, CK8↑, CK6a, CK6b, CK17↓ Differentiation induction of CSC: upregulation of PPARγ responsive genes in epithelial and stromal components; In vivo efficacy: delayed tumor progression; induced differentiation: lipid droplets↑, CD44 and p63 staining↓, CK8 staining↑; FABP4 and PLIN2 mRNA↑ |
Ory et al177 |
| Entinostat | Synthetic chemical |
In vitro, MDA-MB-231, SUM149 and HCC1937; In vivo, MDA-MB-231 xenograft, Entinostat (2.5 mg/kg) 5 days/week (oral), ATRA (5 mg/kg) 5 days/ week (intraperitoneal) and Doxorubicin (2 mg/kg) once a week (intravenous) for 4 weeks |
HDAC inhibition In combination with ATRA and doxorubicin: decreased tumor sphere formation, CD44+/CD24−/EpCam+↓ Induced differentiation markers: RAR-β, ELF3, DHRS3, basal lineage markers (CK5 and CK15), luminal markers (ER, PR) and CK19, epithelial cell-specific genes (CLDN 1, 3, 4, 7), Occludin and E-cadherin↑ and vimentin↓; In vivo efficacy: Tumor-initiating frequencies↓ |
Merino et al227 |
| Flubendazole | Synthetic chemical |
In vitro, MDA-MB-231, BT-549, MCF-7, SK-BR-3; In vivo, MDA-MB-231 xenograft (25 mg/kg/day, given intraperitoneally for 16 days) |
Gene expression: MYC, OCT4, SOX2, NANOG cyclinD1↓ Oil red O staining↑, CD44+/CD24−↓ Decreased mammosphere formation Differentiation induction of CSC: β -catenin, N-cadherin, vimentin↓, CK18↑ Reduced CSC self-renewal; In vivo efficacy: tumorigenicity↓ |
Hou et al178 |
| Graphene Oxide | Synthetic chemical | In vitro, MCF7 | CSC signaling pathway: Wnt, NOTCH, STAT1/3, Nrf2↓ Cell surface marker: CD44+/CD24−↓ Differentiation induction of CSC Decreased mammosphere formation |
Fiorillo et al228 |
| Helichrysetin | Natural product | In vitro, MCF10A, MCF10DCIS, MCF10CA | Differentiation induction of CSC: ID2↓ Decreased mammosphere formation Decreased CSC self-renewal |
Liu et al229 |
| IM-412 | Synthetic chemical | In vitro, MDA-MB-453, MDA-MB-231 | FGFR1/3 inhibition Protein level: Smad2/3, p38/MAPK, Akt, JNK↓ Reduced EMT Differentiation induction of CSC: Inhibition of TGF-β pathway |
Jung et al230 |
| Ivermectin | Synthetic chemical | In vitro, MDA-MB-231 | Gene expression: SOX2, NANOG, OCT4 ↓ Cell surface marker: CD44+/CD24−↓ Decreased CSC viability Decreased CSC self-renewal |
Dominguez-Gomez et al231 |
| K252 | Small molecule | In vitro, MDA-MB-468 | Differentiation induction: ERN1 inhibition Confocal microscopy of CK5 and CK8 expression showing ERN1 and ALPK1 Knockdown induces luminal differentiation (CK5−/ CK8+) in MDA-MB-468 Reduction of colony forming unit of anchorage independent growth of TNBC cell lines Protein level: β-casein↑ |
Strietz et al232 |
| Laminin | Endogenous | In vitro, LM05-E | Gene expression: SOX2, NANOG, OCT4 ↓ Protein level: p-ERK↑ Reduced CSC viability Differentiation induction of CSC Decreased mammosphere formation |
Berardi et al233 |
| Lovastatin | Synthetic chemical | In vitro, MDA-MB-468, MDA-MB-231 | Gene expression: Hippo, Notch, Wnt↓ Protein level: Yap/Taz protein↓, vimentin↓/E-cadherin↑ Differentiation induction of CSC Reduced EMT |
Koohestani mobarhan et al212 |
| Metformin | Synthetic chemical | In vitro, MDA-MB-231, MCF-7 | RNA expression/ Differentiation induction: miRNA-708 ↑, CD47↓ Protein level: CD47↓ |
Tan et al234 |
| Nobiletin | Natural product | In vitro, MCF7 | CD36 inhibition Gene expression: SOX2, OCT4, NANOG↓ Decreased mammosphere formation |
Sp et al235 |
| Ouabain | Natural product | Patient tumors, patient tumor derived xenograft (0.67 mg/kg/day, given intraperitoneally for 3 weeks) |
Increased intracellular calcium, dissociated cell tight junction → altered DNA methylation profile and downregulated target genes of OCT4, SOX2, NANOG, SIN3A In vivo efficacy: spontaneous metastasis formation↓ |
Gkountela et al223 |
| Palbociclib | Small molecule | In vitro, MCF-7, MCF10DCIS | CDK4/6 inhibition Long term suppression of P63 Immunohistochemistry in DCIS Mammosphere formation↓ Differentiation induction of CSC: NELL2↑ |
Kietzman et al236 |
| PD98059 | Synthetic chemical | In vitro, MDA-MB-231 | MAPK inhibition Differentiation marker: N-cadherin↓, E-cadherin↑ Induction of CSC apoptosis and cell cycle arrest Increased MET |
Manupati et al216 |
| Prolactin | Endogenous |
In vitro, SKBR-3 and BT-474; In vivo, SKBR-3 xenograft (0.1 µg/g each second day, given intraperitoneally for 5 weeks) |
mRNA level: ALDHA1, ALDHA3, CD44↓ Cell surface marker: ALDH↓ Decreased mammosphere formation; In vivo efficacy: tumor growth and Ki-67 staining↓ |
Hachim et al237 |
| P123 | Peptide (1.9kDa) | In vitro, CSC cells | BMP signal agonist Cell surface marker: CD44+ population↓, E-cadherin+ population↑in BCSC |
Bosukonda et al238 |
| Quercetin | Natural product | In vitro, MCF-7 | PI3K/Akt/mTOR signal inhibition Decreased mammosphere formation Cell surface marker: CD44+/CD24−↓ |
Li et al239 |
| Quisinostat | Synthetic chemical | In vitro, MDA-MB-231, MDA-MB-468, HCC38, MCF-7 | HDAC Class I and II inhibition Cell surface marker: CD44+/CD24−↓ Decreased CSC viability (in combination with doxorubicin) |
Hii et al240 |
| RANK-Fc | Synthetic chemical | In vivo, MMTV-PyMT transgenic mice (10 mg/kg, subcutaneous, three times a week for 4 weeks (for passage 1) and 2 weeks (for passage 2)) | Luminal epithelial phenotype (TFAP2B, SPDEF, and TFAP2C targets) ↑ In vivo efficacy: tumor initiating-ability↓, secondary mammosphere formation derived from treated tumors↓, tumor cell differentiation↑, lactogenic differentiation↑, Sca1−/lo CSC population↓ |
Yoldi et al241 |
| Resveratrol | Natural product | In vitro, MDA-MB-231 | SIRT1 induction Cell surface marker: CD44+/CD24−↓ Differentiation induction |
Deus et al242 |
| Rosiglitazone | Synthetic chemical | In vitro, Py2T cells derived from MMTV-PyMT tumors and MTfIECad cells derived from MMTV-Neu tumors; In vivo, Py2T xenograft, high dose group: PD98059 (5 mg/kg) and Rosiglitazone (16 mg/kg) daily; low dose group PD98059 (2 mg/kg) and Rosiglitazone (16 mg/kg) daily, given intraperitoneally) for 14 days. | EMT transdifferentiation of breast cancer cells into adipocytes: lipid droplets of fluorescent Nile Red Co-treatment with PD98059 Differentiation induction of CSC: E-cadherin↑; In vivo efficacy: FABP4 and adiponectin expression↑ Development of unilocular lipid droplets in tumor cells Total tumor mass and tumor invasion↓ |
Ishay-Ronen et al243 |
| Salinomycin | Synthetic chemical | In vitro, MCF-7 | Decreased mammosphere formation | Wang et al244 |
| SCH772984 | Synthetic chemical | In vitro, MCF-10A, MDA-MB-231, MDA-MB-436 | ERK inhibition Cell surface marker: ALDH↓ Protein level: p21↑ |
McGrail et al245 |
| Seocalcitol | Synthetic chemical | In vitro, SUM-1315, BT-549, BT-20, SUM-159PT, MDA-MB-468, MFM-223, CAL-148 | Vitamin D receptor signal activation Cell surface marker: ALDH↓ Decreased mammosphere formation Differentiation induction of CSC |
Thakkar et al246 |
| Silibinin | Natural product | In vitro, MDA-MB-468 | Gene expression: CD133, ALDH, C-MYC, NANOG, KLF4, SOX2↓, GATA3, BRCA1↑ Cell surface marker: ALDH+/CD133, ALDH+/CD44, CD133/CD44↓ phenotype of MDA-MB-468 in 2D and 3D culture: increase in size and spindle shape in treatment group. BRCA1 upregulation in 3D culture group with treatment Differentiation induction of CSC Decreased mammosphere forming size |
Abdollahi et al247 |
| Simvastatin | Natural product | In vitro, MDA-MB-468, MDA-MB-231 | Gene expression: Hippo, Notch, Wnt↓ Protein level: Yap/Taz protein↓, vimentin↓/E-cadherin↑ Differentiation induction of CSC Reduced EMT |
Koohestani mobarhan et al212 |
| Trametinib | Synthetic chemical | In vitro, SUM159, HCC1143 | MEK inhibition CK19hi/ CK14hi/VIMlo; Drug Tolerant Progenitor cells enrichment Produced cells with large cytoplasmic volume |
Risom et al214 |
| T315 | Synthetic chemical | In vitro, MDA-MB-231, SUM159; In vivo, SUM159 xenograft (50 mg/kg, twice daily, oral gavage for 11 days) | ILK inhibition, Decreased NOTCH1 signaling, Decreased mammosphere formation; In vivo efficacy: ALDH+ population and mammosphere formation of dispersed tumor cells↓, tumor initiating ability↓ |
Hsu et al248 |
| Vitamin D | Natural product | In vitro, SUM159 | mRNA level: OCT4, CD44, LAMA5, NOTCH↓ Differentiation marker: CK14, SMA↑, CK18, CK5↓ Decreased mammosphere formation Differentiation induction of CSC |
Shan et al166 |
| Withaferin A | Natural product | In vitro, SUM159 and MCF-7; In vivo, MMTV-neu mice (0.1 mg/mouse, three times per week, given intraperitoneally for 28 weeks) | Inhibited self-renewal of BCSC: decrease in mammosphere number Inhibition of stemness markers: ALDH1 and CD44+/CD24−/ESA+↓, Decreased in stemness-related genes: OCT4, SOX2 and NANOG↓; In vivo efficacy: mammosphere number and size↓, ALDH1 activity↓, tumor burden↓ |
Kim et al249 |
| 4a1 | Synthetic chemical | In vitro, MDA-MB-231 | HEXIM induction Protein level: HEXIM1, p27↑, NANOG↓ Nile red staining increased in 4a1 treatment |
Ketchart et al250 |
| 5-aza-2’-deoxycytidine | Synthetic chemical | CD44hi/CD24low expressing CSC isolation from primary malignant breast tumor from patient | mRNA level: p15, p16, BRCA1, BRCA2, p53↑ cell surface marker: CD44+/CD24−↓ protein: ABCG2↓ |
Phan et al251 |
An early success story of differentiation therapy was the use of ATRA as a clinical therapeutic agent. ATRA, an active metabolite of vitamin A, has anti-proliferative, cyto-differentiating and secondary apoptosis-inducing properties and is increasingly used in various tumors such as acute promyelocytic leukemia (APML), breast cancer, bladder cancer and ovarian carcinoma152. In a TNBC xenograft model, combined treatment of ATRA with the epigenetic and chemotherapeutic agents, entinostat and doxorubicin, targets CSCs and induces differentiation by activating ETS transcription factor 1153. ATRA induced differentiation in BCSCs by decreasing the populations of CD44+/CD24−, NANOG-positive and OCT3/4-positive MCF-7 breast cancer cells. ATRA treatment inhibited cell invasion and enhanced the sensitivity of MCF cells to radiation treatment154. In HER2-positive SK-BR-3 and UACC812 cancer cells with co-amplification of ERBB2 and RARA genes, ATRA induces RARα-dependent epithelial differentiation by reorganizing cytoskeletal elements and exerts anti-migratory action by down-regulating EMT-modulator NOTCH1155. ATRA directs the recruitment of RARβ-TET2 complex to epigenetically activate miR-200c that further inhibits PKCζ, a cell polarity protein that dictates asymmetric division of mammalian stem cells, resulting in symmetric division and downregulation of stem cell pool in breast CSCs. ChIP-sequencing analysis showed ATRA enhanced RARβ-TET2 complex co-occupancy in promoters of genes implicated in cell differentiation such as RUNX1, BMP6, IKZF1 and CAV1156.
Vitamin D belongs to a group of fat-soluble secosteroids produced as a result of skin exposure to UV light or obtained from dietary sources such as plants and fish157. Prior studies have demonstrated that the active vitamin D metabolite (1,25D3) and its analogs inhibit breast tumorigenesis in vivo and trigger apoptotic and autophagic cell death in vitro158–160. In addition to its effect on primary breast tumors, vitamin D compounds has demonstrated inhibitory effects on metastasis, achieved through inhibiting EMT161. In MDA-MB-231 breast tumor cells, 1,25D3 induced epithelial marker E-cadherin by CDH1-promoter demethylation, culminating in epithelial differentiation and reduction in tumor progression162. In MCF10DCIS.com xenograft tumors, the Gemini vitamin D analog BXL0124 has been shown to inhibit ductal carcinoma in situ (DCIS) progression to invasive ductal carcinoma (IDC) by maintaining the myoepithelial cell layer and basement membrane163. BXL0124 repressed the expression of CSC marker CD44 at both mRNA and protein levels in MCF10DCIS.com cells via vitamin D receptor (VDR)-dependent mechanism and suppressed the mammary tumor growth in xenografts.164. 1,25D3 and BXL0124 inhibit BCSCs by reducing the CD44+/CD24−/low subpopulation and mammosphere forming efficiency. Treatment of mammospheres with vitamin D compounds targets stem cell phenotype markers (including CD44, CD49f, pNFκB, and c-Notch1) and pluripotency markers (such as OCT4 and KLF4)165. In SUM159 breast cancer cells, 1,25D3 and BXL0124 reduced the self-renewal of mammospheres and suppressed the genes related to pluripotency and Notch signaling. Vitamin D also upregulated myoepithelial differentiating markers including cytokeratin 14 and smooth muscle actin and down-regulated luminal marker, cytokeratin 5166.
When it comes to development and stem cell differentiation, it is well-established that epigenetic regulation plays a significant role. Aberrant epigenetic modifications (including microRNAs and histone modifications) have been implicated in differentiation programs in cancer167; of these, microRNAs provide an appealing target for differentiation therapy. Petrelli et al. showed that miR-100 promotes differentiation in basal-like BCSCs, transforming the basal-like phenotype to luminal type. In basal-like breast cancer, miR-100 inhibits maintenance of BCSCs by targeting the Wnt signaling pathway and polo like kinase 1 (PLK1); conversely, its inhibition induces a stem-like phenotype168. Also of interest in breast cancer is the potential role of HDACi as avenues for differentiation therapy169. Histone acetylation is tightly controlled by histone acetyltransferases and histone deacetylases (HDAC). HDACs are implicated in multiple stages of cancer development, including the regulation of cell cycle regulation, autophagy, apoptosis and angiogenesis170. Aberrant expression of HDACs is associated with solid and hematological malignancies. HDACi can restore the abnormal acetylation status and reactivate the expression of tumor suppressors in cancer cells, inducing differentiation and inhibit tumor progression171. In TNBC cells, a pan-HDACi, Panobinostat, induced E-cadherin and repressed EMT and metastasis by inhibiting ZEB expression172. A low dose of the HDACi abexinostat induces BCSC differentiation in sensitive breast cancer cells, with treated cells exhibiting high expression of luminal and epithelial markers and low expression of mesenchymal markers. Furthermore, abexinostat reduces the BCSC population in patient-derived xenografts expressing low levels of the lncRNA Xist173. HDACi have been evaluated in clinical trials together with other antitumor agents such as primary chemotherapeutic agents, epigenetic-targeted drugs and proteasome inhibitors to improve their efficacy and toxicity.
Other potential compounds of interest that can induce differentiation and target BCSC are acetaminophen, efatutazone and flubendazole. Acetaminophen, an anti-inflammatory drug, was evaluated for its effect on differentiation and tumorigenicity in breast cancer. Treatment of MDA-MB-231 cells with acetaminophen induced morphological changes, decreased CD44+/CD24− and ALDH+ subpopulations, altered markers for differentiation and stemness, and inhibited tumorigenicity. It also increased susceptibility to anti-tumor drugs through suppressing the expression of multidrug efflux pumps. The differentiation-inducing effect of acetaminophen is mediated through the Wnt/β-catenin signaling pathway174. Acetaminophen modulates the expression of EMT-related genes including CK19, TIMP1, MMP2 and TWIST, microRNAs including miR-143 and miR-146a and NOTCH signaling. It reduces the protein levels of Twist and Vimentin, and increases the level of E-cadherin in favor of differentiation. Breast cancer cells treated with acetaminophen showed a significant decrease in in vitro cell migration and an increase in chemo-sensitization175.
PPARγ agonists are agents that activate endogenous PPARγ, a member of the nuclear receptor family of ligand-activated transcription factors, with profound effects on cellular differentiation, proliferation and inflammatory response in cancer tissues176. Efatutazone, a high-affinity PPARγ agonist, inhibited MCF10DCIS mammosphere formation and down-regulated Akt phosphorylation. Efatutazone-treated DCIS lesions in xenografts showed less invasive feature with fewer CD44+/p63+ basal progenitor cells and exhibited fat deposition along with mammary epithelial cell differentiation, suggesting that PPARγ agonists can be useful as potential differentiation inducing agent to delay invasive progression in breast cancer177.
Flubendazole, a FDA-approved anthelmintic, is shown to inhibit breast cancer cell proliferation. It exhibits BCSC-targeted effects by inhibiting mammosphere formation and reducing the CD44+/CD24− subpopulation in MDA-MB-231 cells. Flubendazole suppressed the expression of self-renewal genes (OCT4, SOX2, NANOG, CYCLIN D1 and C-MYC) and induced cell differentiation (increasing Oil Red O stain + cells, upregulating epithelial marker Keratin-18 and down-regulating mesenchymal markers – N-cadherin, Vimentin and β-catenin). It also enhanced the chemosensitivity of the breast cancer cells178. These findings demonstrate the novel use of flubendazole as a BCSC-targeted agent with differentiation inducing property.
In addition, knockdown of CD44, a BCSC marker involved in the differentiation, adhesion, and metastasis of cancer cells, sensitized breast cancer cells to doxorubicin or radiation. Its depletion induces BCSCs to differentiate into non-stem-like cells, targeting drug resistance, metastasis, and stem cell-related genes, indicating BCSC marker targeted therapy can modulate differentiation and inhibit breast tumorigenicity at the same time179. Collectively, the above studies illustrate the promise of differentiation agents either as a stand-alone therapy or as part of a combinatorial regimen targeting BCSCs (Fig. 1).
Figure 1. Breast cancer stem cell signaling, transcription factors and agents that target them.
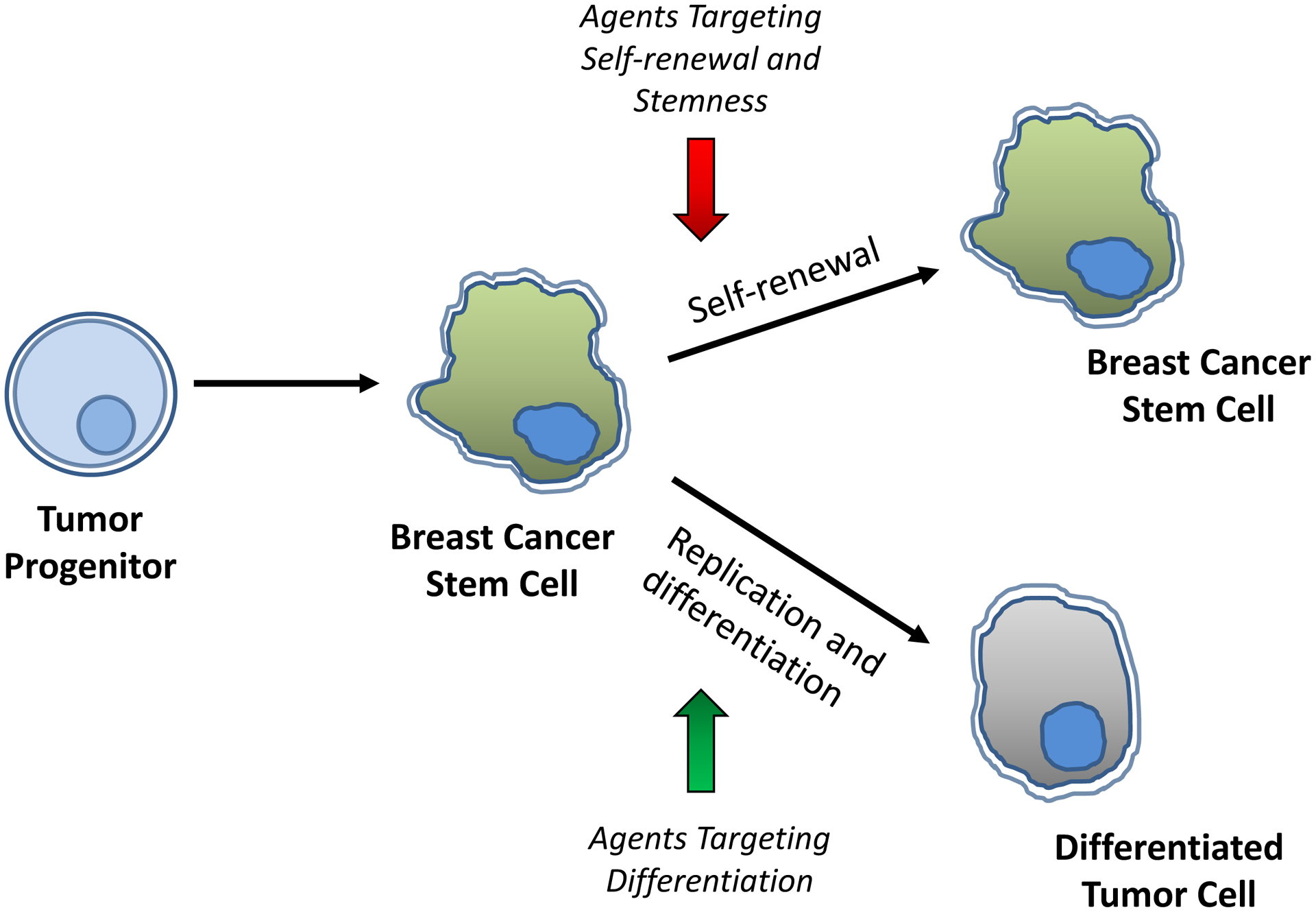
Agents targeting the self-renewal program controlled by transcription factor mediators, OCT4, SOX2, NANOG or KLF4, agents reducing stemness by targeting aberrantly activated signaling pathways involving Notch, Wnt, Hh, STAT3 or TGF-β, and agents inducing differentiation of breast cancer stem cells by reprogramming cells into more differentiated tumor cells are to be exploited in treatment and prevention of breast cancer.
1.7. Conclusion
While there are a fair number of cell surface markers, receptors and ligands, intracellular signaling molecules and transcription factors that identify breast cancer stem cell subpopulations and appear responsible for their stem-like behavior, therapeutic agents that target BCSCs through these elements remain elusive. Likewise, while much in vitro and in vivo evidence indicates that induction of differentiation (or redifferentiation) of CSCs can exert clinically beneficial effects in certain malignancies, this has not yet been achieved effectively in breast cancers. Reducing the properties of stemness that make the CSC compartment resistant to conventional therapy and providing the seeds for recurrence, and inducing the return of those stem cells to their differentiated, somatic origins, could offer improved efficacy in long-term control of the disease.
Financial Support:
This research was supported by the National Institutes of Health grants, R01 AT007036, R01 CA127645, ES005022, New Jersey Health Foundation, Busch Biomedical Grant, and NIEHS/CEED Pilot Grant.
Footnotes
Data Availability: Data sharing is not applicable to this article as no new data were created or analyzed in this study.
Disclosure of Conflict of Interest: “The authors declare no potential conflicts of interest”
References
- 1.Dunnwald LK, Rossing MA, Li CI. Hormone receptor status, tumor characteristics, and prognosis: a prospective cohort of breast cancer patients. Breast Cancer Res. 2007;9(1):R6. doi: 10.1186/bcr1639 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Li Y, Yang D, Yin X, et al. Clinicopathological Characteristics and Breast Cancer-Specific Survival of Patients With Single Hormone Receptor-Positive Breast Cancer. JAMA Netw Open. January 3 2020;3(1):e1918160. doi: 10.1001/jamanetworkopen.2019.18160 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Ross JS, Slodkowska EA, Symmans WF, Pusztai L, Ravdin PM, Hortobagyi GN. The HER-2 receptor and breast cancer: ten years of targeted anti-HER-2 therapy and personalized medicine. Oncologist. April 2009;14(4):320–68. doi: 10.1634/theoncologist.2008-0230 [DOI] [PubMed] [Google Scholar]
- 4.Oh DY, Bang YJ. HER2-targeted therapies - a role beyond breast cancer. Nat Rev Clin Oncol. January 2020;17(1):33–48. doi: 10.1038/s41571-019-0268-3 [DOI] [PubMed] [Google Scholar]
- 5.Yao H, He G, Yan S, et al. Triple-negative breast cancer: is there a treatment on the horizon? Oncotarget. January 3 2017;8(1):1913–1924. doi: 10.18632/oncotarget.12284 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Lee E, Moon A. Identification of Biomarkers for Breast Cancer Using Databases. J Cancer Prev. December 2016;21(4):235–242. doi: 10.15430/jcp.2016.21.4.235 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Al-Hajj M, Wicha MS, Benito-Hernandez A, Morrison SJ, Clarke MF. Prospective identification of tumorigenic breast cancer cells. Proc Natl Acad Sci U S A. April 1 2003;100(7):3983–8. doi: 10.1073/pnas.0530291100 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Gangopadhyay S, Nandy A, Hor P, Mukhopadhyay A. Breast cancer stem cells: a novel therapeutic target. Clinical breast cancer. February 2013;13(1):7–15. doi: 10.1016/j.clbc.2012.09.017 [DOI] [PubMed] [Google Scholar]
- 9.Phi LTH, Sari IN, Yang YG, et al. Cancer Stem Cells (CSCs) in Drug Resistance and their Therapeutic Implications in Cancer Treatment. Stem Cells Int. 2018;2018:5416923. doi: 10.1155/2018/5416923 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Wicha MS. Targeting self-renewal, an Achilles’ heel of cancer stem cells. Nature medicine. Jan 2014;20(1):14–5. doi: 10.1038/nm.3434 [DOI] [PubMed] [Google Scholar]
- 11.Reya T, Morrison SJ, Clarke MF, Weissman IL. Stem cells, cancer, and cancer stem cells. Nature. November 1 2001;414(6859):105–11. doi: 10.1038/35102167 [DOI] [PubMed] [Google Scholar]
- 12.Shackleton M, Quintana E, Fearon ER, Morrison SJ. Heterogeneity in cancer: cancer stem cells versus clonal evolution. Cell. September 4 2009;138(5):822–9. doi: 10.1016/j.cell.2009.08.017 [DOI] [PubMed] [Google Scholar]
- 13.Prasetyanti PR, Medema JP. Intra-tumor heterogeneity from a cancer stem cell perspective. Mol Cancer. February 16 2017;16(1):41. doi: 10.1186/s12943-017-0600-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Bonnet D, Dick JE. Human acute myeloid leukemia is organized as a hierarchy that originates from a primitive hematopoietic cell. Nat Med. July 1997;3(7):730–7. doi: 10.1038/nm0797-730 [DOI] [PubMed] [Google Scholar]
- 15.Sun HR, Wang S, Yan SC, et al. Therapeutic Strategies Targeting Cancer Stem Cells and Their Microenvironment. Front Oncol. 2019;9:1104. doi: 10.3389/fonc.2019.01104 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Crabtree JS, Miele L. Breast Cancer Stem Cells. Biomedicines. July 17 2018;6(3)doi: 10.3390/biomedicines6030077 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Pham PV, Phan NL, Nguyen NT, et al. Differentiation of breast cancer stem cells by knockdown of CD44: promising differentiation therapy. J Transl Med. December 7 2011;9:209. doi: 10.1186/1479-5876-9-209 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Yan Y, Zuo X, Wei D. Concise Review: Emerging Role of CD44 in Cancer Stem Cells: A Promising Biomarker and Therapeutic Target. Stem Cells Transl Med. September 2015;4(9):1033–43. doi: 10.5966/sctm.2015-0048 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Ortiz-Montero P, Liu-Bordes WY, Londoño-Vallejo A, Vernot JP. CD24 expression and stem-associated features define tumor cell heterogeneity and tumorigenic capacities in a model of carcinogenesis. Cancer Manag Res. 2018;10:5767–5784. doi: 10.2147/cmar.S176654 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Zhou L, Sheng D, Wang D, et al. Identification of cancer-type specific expression patterns for active aldehyde dehydrogenase (ALDH) isoforms in ALDEFLUOR assay. Cell Biol Toxicol. April 2019;35(2):161–177. doi: 10.1007/s10565-018-9444-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Li W, Ma H, Zhang J, Zhu L, Wang C, Yang Y. Unraveling the roles of CD44/CD24 and ALDH1 as cancer stem cell markers in tumorigenesis and metastasis. Sci Rep. October 23 2017;7(1):13856. doi: 10.1038/s41598-017-14364-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Xu H, Tian Y, Yuan X, et al. The role of CD44 in epithelial-mesenchymal transition and cancer development. Onco Targets Ther. 2015;8:3783–92. doi: 10.2147/ott.S95470 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Nami B, Ghasemi-Dizgah A, Vaseghi A. Overexpression of molecular chaperons GRP78 and GRP94 in CD44(hi)/CD24(lo) breast cancer stem cells. Bioimpacts. 2016;6(2):105–10. doi: 10.15171/bi.2016.16 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Takebe N, Miele L, Harris PJ, et al. Targeting Notch, Hedgehog, and Wnt pathways in cancer stem cells: clinical update. Nat Rev Clin Oncol. August 2015;12(8):445–64. doi: 10.1038/nrclinonc.2015.61 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Wright MH, Calcagno AM, Salcido CD, Carlson MD, Ambudkar SV, Varticovski L. Brca1 breast tumors contain distinct CD44+/CD24− and CD133+ cells with cancer stem cell characteristics. Breast Cancer Res. 2008;10(1):R10. doi: 10.1186/bcr1855 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Chekhun VF, Lukianova NY, Chekhun SV, et al. Association of CD44(+)CD24(-/low) with markers of aggressiveness and plasticity of cell lines and tumors of patients with breast cancer. Exp Oncol. September 2017;39(3):203–211. [PubMed] [Google Scholar]
- 27.Choi J, Yoon YN, Kim N, et al. Predicting Radiation Resistance in Breast Cancer with Expression Status of Phosphorylated S6K1. Sci Rep. January 20 2020;10(1):641. doi: 10.1038/s41598-020-57496-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Yang ZX, Sun YH, He JG, Cao H, Jiang GQ. Increased activity of CHK enhances the radioresistance of MCF-7 breast cancer stem cells. Oncol Lett. December 2015;10(6):3443–3449. doi: 10.3892/ol.2015.3777 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Ginestier C, Hur MH, Charafe-Jauffret E, et al. ALDH1 is a marker of normal and malignant human mammary stem cells and a predictor of poor clinical outcome. Cell Stem Cell. November 2007;1(5):555–67. doi: 10.1016/j.stem.2007.08.014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Croker AK, Allan AL. Inhibition of aldehyde dehydrogenase (ALDH) activity reduces chemotherapy and radiation resistance of stem-like ALDHhiCD44⁺ human breast cancer cells. Breast Cancer Res Treat. May 2012;133(1):75–87. doi: 10.1007/s10549-011-1692-y [DOI] [PubMed] [Google Scholar]
- 31.Ciccone V, Terzuoli E, Donnini S, Giachetti A, Morbidelli L, Ziche M. Stemness marker ALDH1A1 promotes tumor angiogenesis via retinoic acid/HIF-1α/VEGF signalling in MCF-7 breast cancer cells. J Exp Clin Cancer Res. December 12 2018;37(1):311. doi: 10.1186/s13046-018-0975-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Jang GB, Kim JY, Cho SD, et al. Blockade of Wnt/β-catenin signaling suppresses breast cancer metastasis by inhibiting CSC-like phenotype. Sci Rep. July 23 2015;5:12465. doi: 10.1038/srep12465 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Knudsen ES, Dervishaj O, Kleer CG, Pajak T, Schwartz GF, Witkiewicz AK. EZH2 and ALDH1 expression in ductal carcinoma in situ: complex association with recurrence and progression to invasive breast cancer. Cell Cycle. July 1 2013;12(13):2042–50. doi: 10.4161/cc.25065 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Chekhun SV, Zadvorny TV, Tymovska YO, Anikusko MF, Novak OE, Polishchuk LZ. СD44+/CD24− markers of cancer stem cells in patients with breast cancer of different molecular subtypes. Exp Oncol. March 2015;37(1):58–63. [PubMed] [Google Scholar]
- 35.Liu S, Cong Y, Wang D, et al. Breast cancer stem cells transition between epithelial and mesenchymal states reflective of their normal counterparts. Stem Cell Reports. January 14 2014;2(1):78–91. doi: 10.1016/j.stemcr.2013.11.009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Lombardo Y, de Giorgio A, Coombes CR, Stebbing J, Castellano L. Mammosphere formation assay from human breast cancer tissues and cell lines. J Vis Exp. March 22 2015;(97)doi: 10.3791/52671 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Yoo YD, Kwon YT. Molecular mechanisms controlling asymmetric and symmetric self-renewal of cancer stem cells. J Anal Sci Technol. 2015;6(1):28. doi: 10.1186/s40543-015-0071-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Huilgol D, Venkataramani P, Nandi S, Bhattacharjee S. Transcription Factors That Govern Development and Disease: An Achilles Heel in Cancer. Genes (Basel). October 12 2019;10(10)doi: 10.3390/genes10100794 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Hepburn AC, Steele RE, Veeratterapillay R, et al. The induction of core pluripotency master regulators in cancers defines poor clinical outcomes and treatment resistance. Oncogene. May 2019;38(22):4412–4424. doi: 10.1038/s41388-019-0712-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Aponte PM, Caicedo A. Stemness in Cancer: Stem Cells, Cancer Stem Cells, and Their Microenvironment. Stem Cells Int. 2017;2017:5619472. doi: 10.1155/2017/5619472 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Kreso A, Dick JE. Evolution of the cancer stem cell model. Cell Stem Cell. March 6 2014;14(3):275–91. doi: 10.1016/j.stem.2014.02.006 [DOI] [PubMed] [Google Scholar]
- 42.Friedmann-Morvinski D, Verma IM. Dedifferentiation and reprogramming: origins of cancer stem cells. EMBO Rep. March 2014;15(3):244–53. doi: 10.1002/embr.201338254 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Chen W, Dong J, Haiech J, Kilhoffer MC, Zeniou M. Cancer Stem Cell Quiescence and Plasticity as Major Challenges in Cancer Therapy. Stem Cells Int. 2016;2016:1740936. doi: 10.1155/2016/1740936 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Zhang D, Tang DG, Rycaj K. Cancer stem cells: Regulation programs, immunological properties and immunotherapy. Semin Cancer Biol. October 2018;52(Pt 2):94–106. doi: 10.1016/j.semcancer.2018.05.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Morata-Tarifa C, Jiménez G, García MA, et al. Low adherent cancer cell subpopulations are enriched in tumorigenic and metastatic epithelial-to-mesenchymal transition-induced cancer stem-like cells. Sci Rep. January 11 2016;6:18772. doi: 10.1038/srep18772 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Yang L, Shi P, Zhao G, et al. Targeting cancer stem cell pathways for cancer therapy. Signal Transduct Target Ther. 2020;5:8. doi: 10.1038/s41392-020-0110-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Bai X, Ni J, Beretov J, Graham P, Li Y. Cancer stem cell in breast cancer therapeutic resistance. Cancer Treat Rev. September 2018;69:152–163. doi: 10.1016/j.ctrv.2018.07.004 [DOI] [PubMed] [Google Scholar]
- 48.Bray SJ. Notch signalling in context. Nat Rev Mol Cell Biol. November 2016;17(11):722–735. doi: 10.1038/nrm.2016.94 [DOI] [PubMed] [Google Scholar]
- 49.Ayaz F, Osborne BA. Non-canonical notch signaling in cancer and immunity. Front Oncol. 2014;4:345. doi: 10.3389/fonc.2014.00345 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Weijzen S, Rizzo P, Braid M, et al. Activation of Notch-1 signaling maintains the neoplastic phenotype in human Ras-transformed cells. Nat Med. September 2002;8(9):979–86. doi: 10.1038/nm754 [DOI] [PubMed] [Google Scholar]
- 51.Zhao D, Mo Y, Li MT, et al. NOTCH-induced aldehyde dehydrogenase 1A1 deacetylation promotes breast cancer stem cells. J Clin Invest. December 2014;124(12):5453–65. doi: 10.1172/jci76611 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Zhang X, Zhao X, Shao S, et al. Notch1 induces epithelial-mesenchymal transition and the cancer stem cell phenotype in breast cancer cells and STAT3 plays a key role. Int J Oncol. March 2015;46(3):1141–8. doi: 10.3892/ijo.2014.2809 [DOI] [PubMed] [Google Scholar]
- 53.Grudzien P, Lo S, Albain KS, et al. Inhibition of Notch signaling reduces the stem-like population of breast cancer cells and prevents mammosphere formation. Anticancer Res. October 2010;30(10):3853–67. [PubMed] [Google Scholar]
- 54.Harrison H, Farnie G, Howell SJ, et al. Regulation of breast cancer stem cell activity by signaling through the Notch4 receptor. Cancer Res. January 15 2010;70(2):709–18. doi: 10.1158/0008-5472.Can-09-1681 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Chen JH, Kuo KT, Bamodu OA, et al. Upregulated SCUBE2 expression in breast cancer stem cells enhances triple negative breast cancer aggression through modulation of notch signaling and epithelial-to-mesenchymal transition. Exp Cell Res. September 15 2018;370(2):444–453. doi: 10.1016/j.yexcr.2018.07.008 [DOI] [PubMed] [Google Scholar]
- 56.Wang JW, Wei XL, Dou XW, Huang WH, Du CW, Zhang GJ. The association between Notch4 expression, and clinicopathological characteristics and clinical outcomes in patients with breast cancer. Oncol Lett. June 2018;15(6):8749–8755. doi: 10.3892/ol.2018.8442 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Zhou L, Wang D, Sheng D, et al. NOTCH4 maintains quiescent mesenchymal-like breast cancer stem cells via transcriptionally activating SLUG and GAS1 in triple-negative breast cancer. Theranostics. 2020;10(5):2405–2421. doi: 10.7150/thno.38875 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Bhola NE, Jansen VM, Koch JP, et al. Treatment of Triple-Negative Breast Cancer with TORC1/2 Inhibitors Sustains a Drug-Resistant and Notch-Dependent Cancer Stem Cell Population. Cancer Res. January 15 2016;76(2):440–52. doi: 10.1158/0008-5472.Can-15-1640-t [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Simões BM, O’Brien CS, Eyre R, et al. Anti-estrogen Resistance in Human Breast Tumors Is Driven by JAG1-NOTCH4-Dependent Cancer Stem Cell Activity. Cell Rep. September 29 2015;12(12):1968–77. doi: 10.1016/j.celrep.2015.08.050 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Wang J, Sinha T, Wynshaw-Boris A. Wnt signaling in mammalian development: lessons from mouse genetics. Cold Spring Harb Perspect Biol. May 1 2012;4(5)doi: 10.1101/cshperspect.a007963 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Eubelen M, Bostaille N, Cabochette P, et al. A molecular mechanism for Wnt ligand-specific signaling. Science. August 17 2018;361(6403)doi: 10.1126/science.aat1178 [DOI] [PubMed] [Google Scholar]
- 62.Koval A, Katanaev VL. Dramatic dysbalancing of the Wnt pathway in breast cancers. Sci Rep. May 9 2018;8(1):7329. doi: 10.1038/s41598-018-25672-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Monteiro J, Gaspar C, Richer W, et al. Cancer stemness in Wnt-driven mammary tumorigenesis. Carcinogenesis. January 2014;35(1):2–13. doi: 10.1093/carcin/bgt279 [DOI] [PubMed] [Google Scholar]
- 64.Zhao X, Jiang C, Xu R, Liu Q, Liu G, Zhang Y. TRIP6 enhances stemness property of breast cancer cells through activation of Wnt/β-catenin. Cancer Cell Int. 2020;20:51. doi: 10.1186/s12935-020-1136-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Xu J, Prosperi JR, Choudhury N, Olopade OI, Goss KH. β-Catenin is required for the tumorigenic behavior of triple-negative breast cancer cells. PLoS One. 2015;10(2):e0117097. doi: 10.1371/journal.pone.0117097 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Castagnoli L, Cancila V, Cordoba-Romero SL, et al. WNT signaling modulates PD-L1 expression in the stem cell compartment of triple-negative breast cancer. Oncogene. May 2019;38(21):4047–4060. doi: 10.1038/s41388-019-0700-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Briscoe J, Thérond PP. The mechanisms of Hedgehog signalling and its roles in development and disease. Nat Rev Mol Cell Biol. July 2013;14(7):416–29. doi: 10.1038/nrm3598 [DOI] [PubMed] [Google Scholar]
- 68.Sari IN, Phi LTH, Jun N, Wijaya YT, Lee S, Kwon HY. Hedgehog Signaling in Cancer: A Prospective Therapeutic Target for Eradicating Cancer Stem Cells. Cells. November 10 2018;7(11)doi: 10.3390/cells7110208 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Cochrane CR, Szczepny A, Watkins DN, Cain JE. Hedgehog Signaling in the Maintenance of Cancer Stem Cells. Cancers. September 2015;7(3):1554–85. doi: 10.3390/cancers7030851 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Riaz SK, Khan JS, Shah STA, et al. Involvement of hedgehog pathway in early onset, aggressive molecular subtypes and metastatic potential of breast cancer. Cell Commun Signal. January 8 2018;16(1):3. doi: 10.1186/s12964-017-0213-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Fiaschi M, Rozell B, Bergström A, Toftgård R. Development of mammary tumors by conditional expression of GLI1. Cancer Res. June 1 2009;69(11):4810–7. doi: 10.1158/0008-5472.Can-08-3938 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Liu S, Dontu G, Mantle ID, et al. Hedgehog signaling and Bmi-1 regulate self-renewal of normal and malignant human mammary stem cells. Cancer Res. June 15 2006;66(12):6063–71. doi: 10.1158/0008-5472.Can-06-0054 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.He M, Fu Y, Yan Y, et al. The Hedgehog signalling pathway mediates drug response of MCF-7 mammosphere cells in breast cancer patients. Clinical science (London, England : 1979). November 2015;129(9):809–22. doi: 10.1042/cs20140592 [DOI] [PubMed] [Google Scholar]
- 74.Han B, Qu Y, Jin Y, et al. FOXC1 Activates Smoothened-Independent Hedgehog Signaling in Basal-like Breast Cancer. Cell Rep. November 3 2015;13(5):1046–58. doi: 10.1016/j.celrep.2015.09.063 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Zhou M, Hou Y, Yang G, et al. LncRNA-Hh Strengthen Cancer Stem Cells Generation in Twist-Positive Breast Cancer via Activation of Hedgehog Signaling Pathway. Stem Cells. January 2016;34(1):55–66. doi: 10.1002/stem.2219 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Riobo-Del Galdo NA, Lara Montero Á, Wertheimer EV. Role of Hedgehog Signaling in Breast Cancer: Pathogenesis and Therapeutics. Cells. April 25 2019;8(4)doi: 10.3390/cells8040375 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Zarzynska JM. Two faces of TGF-beta1 in breast cancer. Mediators Inflamm. 2014;2014:141747. doi: 10.1155/2014/141747 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Boulanger CA, Smith GH. Reducing mammary cancer risk through premature stem cell senescence. Oncogene. April 26 2001;20(18):2264–72. doi: 10.1038/sj.onc.1204312 [DOI] [PubMed] [Google Scholar]
- 79.Tang B, Yoo N, Vu M, et al. Transforming growth factor-beta can suppress tumorigenesis through effects on the putative cancer stem or early progenitor cell and committed progeny in a breast cancer xenograft model. Cancer Res. September 15 2007;67(18):8643–52. doi: 10.1158/0008-5472.Can-07-0982 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Gorska AE, Jensen RA, Shyr Y, Aakre ME, Bhowmick NA, Moses HL. Transgenic mice expressing a dominant-negative mutant type II transforming growth factor-beta receptor exhibit impaired mammary development and enhanced mammary tumor formation. Am J Pathol. October 2003;163(4):1539–49. doi: 10.1016/s0002-9440(10)63510-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Xie F, Ling L, van Dam H, Zhou F, Zhang L. TGF-β signaling in cancer metastasis. Acta Biochim Biophys Sin (Shanghai). January 1 2018;50(1):121–132. doi: 10.1093/abbs/gmx123 [DOI] [PubMed] [Google Scholar]
- 82.Mani SA, Guo W, Liao MJ, et al. The epithelial-mesenchymal transition generates cells with properties of stem cells. Cell. May 16 2008;133(4):704–15. doi: 10.1016/j.cell.2008.03.027 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Konge J, Leteurtre F, Goislard M, et al. Breast cancer stem cell-like cells generated during TGFβ-induced EMT are radioresistant. Oncotarget. May 4 2018;9(34):23519–23531. doi: 10.18632/oncotarget.25240 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Suriyamurthy S, Baker D, Ten Dijke P, Iyengar PV. Epigenetic Reprogramming of TGF-β Signaling in Breast Cancer. Cancers (Basel). May 24 2019;11(5)doi: 10.3390/cancers11050726 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Chen Y, Huang S, Wu B, et al. Transforming growth factor-β1 promotes breast cancer metastasis by downregulating miR-196a-3p expression. Oncotarget. July 25 2017;8(30):49110–49122. doi: 10.18632/oncotarget.16308 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Yu Y, Luo W, Yang ZJ, et al. miR-190 suppresses breast cancer metastasis by regulation of TGF-β-induced epithelial-mesenchymal transition. Mol Cancer. March 6 2018;17(1):70. doi: 10.1186/s12943-018-0818-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Richards EJ, Zhang G, Li ZP, et al. Long non-coding RNAs (LncRNA) regulated by transforming growth factor (TGF) β: LncRNA-hit-mediated TGFβ-induced epithelial to mesenchymal transition in mammary epithelia. J Biol Chem. March 13 2015;290(11):6857–67. doi: 10.1074/jbc.M114.610915 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Ma JH, Qin L, Li X. Role of STAT3 signaling pathway in breast cancer. Cell Commun Signal. February 28 2020;18(1):33. doi: 10.1186/s12964-020-0527-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Yu H, Lee H, Herrmann A, Buettner R, Jove R. Revisiting STAT3 signalling in cancer: new and unexpected biological functions. Nat Rev Cancer. November 2014;14(11):736–46. doi: 10.1038/nrc3818 [DOI] [PubMed] [Google Scholar]
- 90.Banerjee K, Resat H. Constitutive activation of STAT3 in breast cancer cells: A review. Int J Cancer. June 1 2016;138(11):2570–8. doi: 10.1002/ijc.29923 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Soleymani Abyaneh H, Gupta N, Alshareef A, Gopal K, Lavasanifar A, Lai R. Hypoxia Induces the Acquisition of Cancer Stem-like Phenotype Via Upregulation and Activation of Signal Transducer and Activator of Transcription-3 (STAT3) in MDA-MB-231, a Triple Negative Breast Cancer Cell Line. Cancer Microenviron. December 2018;11(2–3):141–152. doi: 10.1007/s12307-018-0218-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Tawara K, Scott H, Emathinger J, et al. HIGH expression of OSM and IL-6 are associated with decreased breast cancer survival: synergistic induction of IL-6 secretion by OSM and IL-1β. Oncotarget. March 12 2019;10(21):2068–2085. doi: 10.18632/oncotarget.26699 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Hao S, Chen X, Wang F, et al. Breast cancer cell-derived IL-35 promotes tumor progression via induction of IL-35-producing induced regulatory T cells. Carcinogenesis. December 31 2018;39(12):1488–1496. doi: 10.1093/carcin/bgy136 [DOI] [PubMed] [Google Scholar]
- 94.Shi P, Chen C, Li X, Wei Z, Liu Z, Liu Y. MicroRNA‑124 suppresses cell proliferation and invasion of triple negative breast cancer cells by targeting STAT3. Mol Med Rep. May 2019;19(5):3667–3675. doi: 10.3892/mmr.2019.10044 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Zhang H, Cai K, Wang J, et al. MiR-7, inhibited indirectly by lincRNA HOTAIR, directly inhibits SETDB1 and reverses the EMT of breast cancer stem cells by downregulating the STAT3 pathway. Stem Cells. November 2014;32(11):2858–68. doi: 10.1002/stem.1795 [DOI] [PubMed] [Google Scholar]
- 96.Butti R, Das S, Gunasekaran VP, Yadav AS, Kumar D, Kundu GC. Receptor tyrosine kinases (RTKs) in breast cancer: signaling, therapeutic implications and challenges. Mol Cancer. February 19 2018;17(1):34. doi: 10.1186/s12943-018-0797-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Wang Z, Oron E, Nelson B, Razis S, Ivanova N. Distinct lineage specification roles for NANOG, OCT4, and SOX2 in human embryonic stem cells. Cell Stem Cell. April 6 2012;10(4):440–54. doi: 10.1016/j.stem.2012.02.016 [DOI] [PubMed] [Google Scholar]
- 98.Takahashi K, Yamanaka S. Induction of pluripotent stem cells from mouse embryonic and adult fibroblast cultures by defined factors. Cell. August 25 2006;126(4):663–76. doi: 10.1016/j.cell.2006.07.024 [DOI] [PubMed] [Google Scholar]
- 99.Shi G, Jin Y. Role of Oct4 in maintaining and regaining stem cell pluripotency. Stem Cell Res Ther. December 14 2010;1(5):39. doi: 10.1186/scrt39 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Wang X, Dai J. Concise review: isoforms of OCT4 contribute to the confusing diversity in stem cell biology. Stem Cells. May 2010;28(5):885–93. doi: 10.1002/stem.419 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Soheili S, Asadi MH, Farsinejad A. Distinctive expression pattern of OCT4 variants in different types of breast cancer. Cancer Biomark. 2017;18(1):69–76. doi: 10.3233/cbm-160675 [DOI] [PubMed] [Google Scholar]
- 102.Kim RJ, Nam JS. OCT4 Expression Enhances Features of Cancer Stem Cells in a Mouse Model of Breast Cancer. Lab Anim Res. June 2011;27(2):147–52. doi: 10.5625/lar.2011.27.2.147 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Herr W, Cleary MA. The POU domain: versatility in transcriptional regulation by a flexible two-in-one DNA-binding domain. Genes Dev. July 15 1995;9(14):1679–93. doi: 10.1101/gad.9.14.1679 [DOI] [PubMed] [Google Scholar]
- 104.Gwak JM, Kim M, Kim HJ, Jang MH, Park SY. Expression of embryonal stem cell transcription factors in breast cancer: Oct4 as an indicator for poor clinical outcome and tamoxifen resistance. Oncotarget. May 30 2017;8(22):36305–36318. doi: 10.18632/oncotarget.16750 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Cheng CC, Shi LH, Wang XJ, et al. Stat3/Oct-4/c-Myc signal circuit for regulating stemness-mediated doxorubicin resistance of triple-negative breast cancer cells and inhibitory effects of WP1066. Int J Oncol. July 2018;53(1):339–348. doi: 10.3892/ijo.2018.4399 [DOI] [PubMed] [Google Scholar]
- 106.Kim JY, Kim JC, Lee JY, Park MJ. Oct4 suppresses IR‑induced premature senescence in breast cancer cells through STAT3- and NF‑κB-mediated IL‑24 production. Int J Oncol. July 2018;53(1):47–58. doi: 10.3892/ijo.2018.4391 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Almozyan S, Colak D, Mansour F, et al. PD-L1 promotes OCT4 and Nanog expression in breast cancer stem cells by sustaining PI3K/AKT pathway activation. Int J Cancer. October 1 2017;141(7):1402–1412. doi: 10.1002/ijc.30834 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Cho Y, Kang HG, Kim SJ, et al. Post-translational modification of OCT4 in breast cancer tumorigenesis. Cell Death Differ. November 2018;25(10):1781–1795. doi: 10.1038/s41418-018-0079-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Shen L, Qin K, Wang D, et al. Overexpression of Oct4 suppresses the metastatic potential of breast cancer cells via Rnd1 downregulation. Biochim Biophys Acta. November 2014;1842(11):2087–95. doi: 10.1016/j.bbadis.2014.07.015 [DOI] [PubMed] [Google Scholar]
- 110.Hu J, Qin K, Zhang Y, et al. Downregulation of transcription factor Oct4 induces an epithelial-to-mesenchymal transition via enhancement of Ca2+ influx in breast cancer cells. Biochem Biophys Res Commun. August 12 2011;411(4):786–91. doi: 10.1016/j.bbrc.2011.07.025 [DOI] [PubMed] [Google Scholar]
- 111.Wuebben EL, Rizzino A. The dark side of SOX2: cancer - a comprehensive overview. Oncotarget. July 4 2017;8(27):44917–44943. doi: 10.18632/oncotarget.16570 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Leis O, Eguiara A, Lopez-Arribillaga E, et al. Sox2 expression in breast tumours and activation in breast cancer stem cells. Oncogene. March 15 2012;31(11):1354–65. doi: 10.1038/onc.2011.338 [DOI] [PubMed] [Google Scholar]
- 113.Piva M, Domenici G, Iriondo O, et al. Sox2 promotes tamoxifen resistance in breast cancer cells. EMBO Mol Med. January 2014;6(1):66–79. doi: 10.1002/emmm.201303411 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Domenici G, Aurrekoetxea-Rodríguez I, Simões BM, et al. A Sox2-Sox9 signalling axis maintains human breast luminal progenitor and breast cancer stem cells. Oncogene. April 2019;38(17):3151–3169. doi: 10.1038/s41388-018-0656-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Mukherjee P, Gupta A, Chattopadhyay D, Chatterji U. Modulation of SOX2 expression delineates an end-point for paclitaxel-effectiveness in breast cancer stem cells. Sci Rep. August 23 2017;7(1):9170. doi: 10.1038/s41598-017-08971-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Jeter CR, Yang T, Wang J, Chao HP, Tang DG. Concise Review: NANOG in Cancer Stem Cells and Tumor Development: An Update and Outstanding Questions. Stem Cells. August 2015;33(8):2381–90. doi: 10.1002/stem.2007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Lu X, Mazur SJ, Lin T, Appella E, Xu Y. The pluripotency factor nanog promotes breast cancer tumorigenesis and metastasis. Oncogene. May 15 2014;33(20):2655–64. doi: 10.1038/onc.2013.209 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Han J, Zhang F, Yu M, et al. RNA interference-mediated silencing of NANOG reduces cell proliferation and induces G0/G1 cell cycle arrest in breast cancer cells. Cancer Lett. August 1 2012;321(1):80–8. doi: 10.1016/j.canlet.2012.02.021 [DOI] [PubMed] [Google Scholar]
- 119.Wang D, Lu P, Zhang H, et al. Oct-4 and Nanog promote the epithelial-mesenchymal transition of breast cancer stem cells and are associated with poor prognosis in breast cancer patients. Oncotarget. November 15 2014;5(21):10803–15. doi: 10.18632/oncotarget.2506 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Bourguignon LY, Peyrollier K, Xia W, Gilad E. Hyaluronan-CD44 interaction activates stem cell marker Nanog, Stat-3-mediated MDR1 gene expression, and ankyrin-regulated multidrug efflux in breast and ovarian tumor cells. J Biol Chem. June 20 2008;283(25):17635–51. doi: 10.1074/jbc.M800109200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Emadian Saravi O, Naghshvar F, Torabizadeh Z, Sheidaei S. Immunohistochemical Expression of Nanog and Its Relation with Clinicopathologic Characteristics in Breast Ductal Carcinoma. Iran Biomed J. May 2019;23(3):184–9. doi: 10.29252/.23.3.184 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Nagata T, Shimada Y, Sekine S, et al. Prognostic significance of NANOG and KLF4 for breast cancer. Breast Cancer. January 2014;21(1):96–101. doi: 10.1007/s12282-012-0357-y [DOI] [PubMed] [Google Scholar]
- 123.Zhang J, Li G, Feng L, Lu H, Wang X. Krüppel-like factors in breast cancer: Function, regulation and clinical relevance. Biomed Pharmacother. March 2020;123:109778. doi: 10.1016/j.biopha.2019.109778 [DOI] [PubMed] [Google Scholar]
- 124.Song X, Xing YM, Wu W, et al. Expression of Krüppel-like factor 4 in breast cancer tissues and its effects on the proliferation of breast cancer MDA-MB-231 cells. Exp Ther Med. May 2017;13(5):2463–2467. doi: 10.3892/etm.2017.4262 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Akaogi K, Nakajima Y, Ito I, et al. KLF4 suppresses estrogen-dependent breast cancer growth by inhibiting the transcriptional activity of ERalpha. Oncogene. August 13 2009;28(32):2894–902. doi: 10.1038/onc.2009.151 [DOI] [PubMed] [Google Scholar]
- 126.Ferralli J, Chiquet-Ehrismann R, Degen M. KLF4α stimulates breast cancer cell proliferation by acting as a KLF4 antagonist. Oncotarget. July 19 2016;7(29):45608–45621. doi: 10.18632/oncotarget.10058 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Yu F, Li J, Chen H, et al. Kruppel-like factor 4 (KLF4) is required for maintenance of breast cancer stem cells and for cell migration and invasion. Oncogene. May 5 2011;30(18):2161–72. doi: 10.1038/onc.2010.591 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Mimoto R, Imawari Y, Hirooka S, Takeyama H, Yoshida K. Impairment of DYRK2 augments stem-like traits by promoting KLF4 expression in breast cancer. Oncogene. March 30 2017;36(13):1862–1872. doi: 10.1038/onc.2016.349 [DOI] [PubMed] [Google Scholar]
- 129.Fagnocchi L, Zippo A. Multiple Roles of MYC in Integrating Regulatory Networks of Pluripotent Stem Cells. Front Cell Dev Biol. 2017;5:7. doi: 10.3389/fcell.2017.00007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Xu J, Chen Y, Olopade OI. MYC and Breast Cancer. Genes Cancer. June 2010;1(6):629–40. doi: 10.1177/1947601910378691 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Chappell J, Dalton S. Roles for MYC in the establishment and maintenance of pluripotency. Cold Spring Harb Perspect Med. December 1 2013;3(12):a014381. doi: 10.1101/cshperspect.a014381 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Kim J, Woo AJ, Chu J, et al. A Myc network accounts for similarities between embryonic stem and cancer cell transcription programs. Cell. October 15 2010;143(2):313–24. doi: 10.1016/j.cell.2010.09.010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Sheiness D, Fanshier L, Bishop JM. Identification of nucleotide sequences which may encode the oncogenic capacity of avian retrovirus MC29. J Virol. November 1978;28(2):600–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Felsher DW, Bishop JM. Transient excess of MYC activity can elicit genomic instability and tumorigenesis. Proc Natl Acad Sci U S A. March 30 1999;96(7):3940–4. doi: 10.1073/pnas.96.7.3940 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Fallah Y, Brundage J, Allegakoen P, Shajahan-Haq AN. MYC-Driven Pathways in Breast Cancer Subtypes. Biomolecules. July 11 2017;7(3)doi: 10.3390/biom7030053 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Yang A, Qin S, Schulte BA, Ethier SP, Tew KD, Wang GY. MYC Inhibition Depletes Cancer Stem-like Cells in Triple-Negative Breast Cancer. Cancer Res. December 1 2017;77(23):6641–6650. doi: 10.1158/0008-5472.Can-16-3452 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Lee KM, Giltnane JM, Balko JM, et al. MYC and MCL1 Cooperatively Promote Chemotherapy-Resistant Breast Cancer Stem Cells via Regulation of Mitochondrial Oxidative Phosphorylation. Cell Metab. October 3 2017;26(4):633–647.e7. doi: 10.1016/j.cmet.2017.09.009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Santoro A, Vlachou T, Luzi L, et al. p53 Loss in Breast Cancer Leads to Myc Activation, Increased Cell Plasticity, and Expression of a Mitotic Signature with Prognostic Value. Cell Rep. January 15 2019;26(3):624–638.e8. doi: 10.1016/j.celrep.2018.12.071 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Poli V, Fagnocchi L, Fasciani A, et al. MYC-driven epigenetic reprogramming favors the onset of tumorigenesis by inducing a stem cell-like state. Nat Commun. March 9 2018;9(1):1024. doi: 10.1038/s41467-018-03264-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Crawford RR, Potukuchi PK, Schuetz EG, Schuetz JD. Beyond Competitive Inhibition: Regulation of ABC Transporters by Kinases and Protein-Protein Interactions as Potential Mechanisms of Drug-Drug Interactions. Drug Metab Dispos. May 2018;46(5):567–580. doi: 10.1124/dmd.118.080663 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Weeden CE, Asselin-Labat ML. Mechanisms of DNA damage repair in adult stem cells and implications for cancer formation. Biochim Biophys Acta Mol Basis Dis. January 2018;1864(1):89–101. doi: 10.1016/j.bbadis.2017.10.015 [DOI] [PubMed] [Google Scholar]
- 142.Ryoo IG, Choi BH, Ku SK, Kwak MK. High CD44 expression mediates p62-associated NFE2L2/NRF2 activation in breast cancer stem cell-like cells: Implications for cancer stem cell resistance. Redox Biol. July 2018;17:246–258. doi: 10.1016/j.redox.2018.04.015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Hu Y, Yagüe E, Zhao J, et al. Sabutoclax, pan-active BCL-2 protein family antagonist, overcomes drug resistance and eliminates cancer stem cells in breast cancer. Cancer Lett. June 1 2018;423:47–59. doi: 10.1016/j.canlet.2018.02.036 [DOI] [PubMed] [Google Scholar]
- 144.Ji X, Lu Y, Tian H, Meng X, Wei M, Cho WC. Chemoresistance mechanisms of breast cancer and their countermeasures. Biomed Pharmacother. June 2019;114:108800. doi: 10.1016/j.biopha.2019.108800 [DOI] [PubMed] [Google Scholar]
- 145.La Belle Flynn A, Calhoun BC, Sharma A, Chang JC, Almasan A, Schiemann WP. Autophagy inhibition elicits emergence from metastatic dormancy by inducing and stabilizing Pfkfb3 expression. Nat Commun. August 14 2019;10(1):3668. doi: 10.1038/s41467-019-11640-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Gong C, Bauvy C, Tonelli G, et al. Beclin 1 and autophagy are required for the tumorigenicity of breast cancer stem-like/progenitor cells. Oncogene. May 2 2013;32(18):2261–72, 2272e.1–11. doi: 10.1038/onc.2012.252 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Bousquet G, El Bouchtaoui M, Sophie T, et al. Targeting autophagic cancer stem-cells to reverse chemoresistance in human triple negative breast cancer. Oncotarget. May 23 2017;8(21):35205–35221. doi: 10.18632/oncotarget.16925 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Shekhani MT, Jayanthy AS, Maddodi N, Setaluri V. Cancer stem cells and tumor transdifferentiation: implications for novel therapeutic strategies. Am J Stem Cells. 2013;2(1):52–61. [PMC free article] [PubMed] [Google Scholar]
- 149.Rossari F, Zucchinetti C, Buda G, Orciuolo E. Tumor dormancy as an alternative step in the development of chemoresistance and metastasis - clinical implications. Cell Oncol (Dordr). April 2020;43(2):155–176. doi: 10.1007/s13402-019-00467-7 [DOI] [PubMed] [Google Scholar]
- 150.Xu WP, Zhang X, Xie WF. Differentiation therapy for solid tumors. J Dig Dis. April 2014;15(4):159–65. doi: 10.1111/1751-2980.12122 [DOI] [PubMed] [Google Scholar]
- 151.Jin X, Jin X, Kim H. Cancer stem cells and differentiation therapy. Tumour Biol. October 2017;39(10):1010428317729933. doi: 10.1177/1010428317729933 [DOI] [PubMed] [Google Scholar]
- 152.Siddikuzzaman, Guruvayoorappan C, Berlin Grace. All trans retinoic acid and cancer. Immunopharmacol Immunotoxicol. June 2011;33(2):241–9. doi: 10.3109/08923973.2010.521507 [DOI] [PubMed] [Google Scholar]
- 153.Merino VF, Nguyen N, Jin K, et al. Combined Treatment with Epigenetic, Differentiating, and Chemotherapeutic Agents Cooperatively Targets Tumor-Initiating Cells in Triple-Negative Breast Cancer. Cancer research. April 01 2016;76(7):2013–24. doi: 10.1158/0008-5472.can-15-1619 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Yan Y, Li Z, Xu X, et al. All-trans retinoic acids induce differentiation and sensitize a radioresistant breast cancer cells to chemotherapy. BMC Complement Altern Med. March 31 2016;16:113. doi: 10.1186/s12906-016-1088-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Zanetti A, Affatato R, Centritto F, et al. All-trans-retinoic Acid Modulates the Plasticity and Inhibits the Motility of Breast Cancer Cells: ROLE OF NOTCH1 AND TRANSFORMING GROWTH FACTOR (TGFβ). J Biol Chem. July 17 2015;290(29):17690–709. doi: 10.1074/jbc.M115.638510 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Wu MJ, Kim MR, Chen YS, Yang JY, Chang CJ. Retinoic acid directs breast cancer cell state changes through regulation of TET2-PKCζ pathway. Oncogene. June 1 2017;36(22):3193–3206. doi: 10.1038/onc.2016.467 [DOI] [PMC free article] [PubMed] [Google Scholar] [Research Misconduct Found]
- 157.Bikle DD. Vitamin D and bone. Curr Osteoporos Rep. June 2012;10(2):151–9. doi: 10.1007/s11914-012-0098-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Colston KW, Chander SK, Mackay AG, Coombes RC. Effects of synthetic vitamin D analogues on breast cancer cell proliferation in vivo and in vitro. Biochem Pharmacol. August 18 1992;44(4):693–702. doi: 10.1016/0006-2952(92)90405-8 [DOI] [PubMed] [Google Scholar]
- 159.Høyer-Hansen M, Bastholm L, Mathiasen IS, Elling F, Jäättelä M. Vitamin D analog EB1089 triggers dramatic lysosomal changes and Beclin 1-mediated autophagic cell death. Cell Death Differ. October 2005;12(10):1297–309. doi: 10.1038/sj.cdd.4401651 [DOI] [PubMed] [Google Scholar]
- 160.Simboli-Campbell M, Narvaez CJ, Tenniswood M, Welsh J. 1,25-Dihydroxyvitamin D3 induces morphological and biochemical markers of apoptosis in MCF-7 breast cancer cells. J Steroid Biochem Mol Biol. July 1996;58(4):367–76. doi: 10.1016/0960-0760(96)00055-6 [DOI] [PubMed] [Google Scholar]
- 161.Wilmanski T, Barnard A, Parikh MR, et al. 1α,25-Dihydroxyvitamin D Inhibits the Metastatic Capability of MCF10CA1a and MDA-MB-231 Cells in an In Vitro Model of Breast to Bone Metastasis. Nutr Cancer. October 2016;68(7):1202–9. doi: 10.1080/01635581.2016.1213868 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162.Lopes N, Carvalho J, Durães C, et al. 1Alpha,25-dihydroxyvitamin D3 induces de novo E-cadherin expression in triple-negative breast cancer cells by CDH1-promoter demethylation. Anticancer Res. January 2012;32(1):249–57. [PubMed] [Google Scholar]
- 163.Wahler J, So JY, Kim YC, et al. Inhibition of the transition of ductal carcinoma in situ to invasive ductal carcinoma by a Gemini vitamin D analog. Cancer Prev Res (Phila). June 2014;7(6):617–26. doi: 10.1158/1940-6207.Capr-13-0362 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164.So JY, Lee HJ, Smolarek AK, et al. A novel Gemini vitamin D analog represses the expression of a stem cell marker CD44 in breast cancer. Mol Pharmacol. March 2011;79(3):360–7. doi: 10.1124/mol.110.068403 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165.Wahler J, So JY, Cheng LC, Maehr H, Uskokovic M, Suh N. Vitamin D compounds reduce mammosphere formation and decrease expression of putative stem cell markers in breast cancer. J Steroid Biochem Mol Biol. April 2015;148:148–55. doi: 10.1016/j.jsbmb.2014.10.016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166.Shan NL, Wahler J, Lee HJ, et al. Vitamin D compounds inhibit cancer stem-like cells and induce differentiation in triple negative breast cancer. J Steroid Biochem Mol Biol. October 2017;173:122–129. doi: 10.1016/j.jsbmb.2016.12.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167.Boland MJ, Nazor KL, Loring JF. Epigenetic regulation of pluripotency and differentiation. Circ Res. July 7 2014;115(2):311–24. doi: 10.1161/circresaha.115.301517 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168.Petrelli A, Carollo R, Cargnelutti M, et al. By promoting cell differentiation, miR-100 sensitizes basal-like breast cancer stem cells to hormonal therapy. Oncotarget. February 10 2015;6(4):2315–30. doi: 10.18632/oncotarget.2962 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169.Damaskos C, Garmpis N, Valsami S, et al. Histone Deacetylase Inhibitors: An Attractive Therapeutic Strategy Against Breast Cancer. Anticancer Res. January 2017;37(1):35–46. doi: 10.21873/anticanres.11286 [DOI] [PubMed] [Google Scholar]
- 170.Suraweera A, O’Byrne KJ, Richard DJ. Combination Therapy With Histone Deacetylase Inhibitors (HDACi) for the Treatment of Cancer: Achieving the Full Therapeutic Potential of HDACi. Front Oncol. 2018;8:92. doi: 10.3389/fonc.2018.00092 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171.Li Y, Seto E. HDACs and HDAC Inhibitors in Cancer Development and Therapy. Cold Spring Harb Perspect Med. October 3 2016;6(10)doi: 10.1101/cshperspect.a026831 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 172.Rhodes LV, Tate CR, Segar HC, et al. Suppression of triple-negative breast cancer metastasis by pan-DAC inhibitor panobinostat via inhibition of ZEB family of EMT master regulators. Breast Cancer Res Treat. June 2014;145(3):593–604. doi: 10.1007/s10549-014-2979-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 173.Salvador MA, Wicinski J, Cabaud O, et al. The histone deacetylase inhibitor abexinostat induces cancer stem cells differentiation in breast cancer with low Xist expression. Clin Cancer Res. December 1 2013;19(23):6520–31. doi: 10.1158/1078-0432.Ccr-13-0877 [DOI] [PubMed] [Google Scholar]
- 174.Takehara M, Hoshino T, Namba T, Yamakawa N, Mizushima T. Acetaminophen-induced differentiation of human breast cancer stem cells and inhibition of tumor xenograft growth in mice. Biochem Pharmacol. May 1 2011;81(9):1124–35. doi: 10.1016/j.bcp.2011.02.012 [DOI] [PubMed] [Google Scholar]
- 175.Afshar E, Hashemi-Arabi M, Salami S, Peirouvi T, Pouriran R. Screening of acetaminophen-induced alterations in epithelial-to-mesenchymal transition-related expression of microRNAs in a model of stem-like triple-negative breast cancer cells: The possible functional impacts. Gene. June 20 2019;702:46–55. doi: 10.1016/j.gene.2019.02.106 [DOI] [PubMed] [Google Scholar]
- 176.Michalik L, Desvergne B, Wahli W. Peroxisome-proliferator-activated receptors and cancers: complex stories. Nat Rev Cancer. January 2004;4(1):61–70. doi: 10.1038/nrc1254 [DOI] [PubMed] [Google Scholar]
- 177.Ory V, Kietzman WB, Boeckelman J, et al. The PPARγ agonist efatutazone delays invasive progression and induces differentiation of ductal carcinoma in situ. Breast Cancer Res Treat. May 2018;169(1):47–57. doi: 10.1007/s10549-017-4649-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- 178.Hou ZJ, Luo X, Zhang W, et al. Flubendazole, FDA-approved anthelmintic, targets breast cancer stem-like cells. Oncotarget. March 20 2015;6(8):6326–40. doi: 10.18632/oncotarget.3436 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 179.Van Phuc P, Nhan PL, Nhung TH, et al. Downregulation of CD44 reduces doxorubicin resistance of CD44CD24 breast cancer cells. Onco Targets Ther. 2011;4:71–8. doi: 10.2147/ott.S21431 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 180.Leccia F, Del Vecchio L, Mariotti E, et al. ABCG2, a novel antigen to sort luminal progenitors of BRCA1- breast cancer cells. Mol Cancer. September 12 2014;13:213. doi: 10.1186/1476-4598-13-213 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 181.Croker AK, Allan AL. Inhibition of aldehyde dehydrogenase (ALDH) activity reduces chemotherapy and radiation resistance of stem-like ALDHhiCD44(+) human breast cancer cells. Breast Cancer Res Treat. May 2012;133(1):75–87. doi: 10.1007/s10549-011-1692-y [DOI] [PubMed] [Google Scholar]
- 182.Balamurugan K, Mendoza-Villanueva D, Sharan S, et al. C/EBPdelta links IL-6 and HIF-1 signaling to promote breast cancer stem cell-associated phenotypes. Oncogene. September 27 2018;doi: 10.1038/s41388-018-0516-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 183.Jiao X, Velasco-Velazquez MA, Wang M, et al. CCR5 Governs DNA Damage Repair and Breast Cancer Stem Cell Expansion. Cancer research. April 1 2018;78(7):1657–1671. doi: 10.1158/0008-5472.Can-17-0915 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 184.Vassilopoulos A, Chisholm C, Lahusen T, Zheng H, Deng CX. A critical role of CD29 and CD49f in mediating metastasis for cancer-initiating cells isolated from a Brca1-associated mouse model of breast cancer. Oncogene. November 20 2014;33(47):5477–82. doi: 10.1038/onc.2013.516 [DOI] [PubMed] [Google Scholar]
- 185.Cho RW, Wang X, Diehn M, et al. Isolation and molecular characterization of cancer stem cells in MMTV-Wnt-1 murine breast tumors. Stem Cells. February 2008;26(2):364–71. doi: 10.1634/stemcells.2007-0440 [DOI] [PubMed] [Google Scholar]
- 186.Vaillant F, Asselin-Labat ML, Shackleton M, Forrest NC, Lindeman GJ, Visvader JE. The mammary progenitor marker CD61/beta3 integrin identifies cancer stem cells in mouse models of mammary tumorigenesis. Cancer Res. October 1 2008;68(19):7711–7. doi: 10.1158/0008-5472.Can-08-1949 [DOI] [PubMed] [Google Scholar]
- 187.Fillmore CM, Kuperwasser C. Human breast cancer cell lines contain stem-like cells that self-renew, give rise to phenotypically diverse progeny and survive chemotherapy. Breast Cancer Res. 2008;10(2):R25. doi: 10.1186/bcr1982 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 188.Meyer MJ, Fleming JM, Lin AF, Hussnain SA, Ginsburg E, Vonderhaar BK. CD44posCD49fhiCD133/2hi defines xenograft-initiating cells in estrogen receptor-negative breast cancer. Cancer research. June 1 2010;70(11):4624–33. doi: 10.1158/0008-5472.Can-09-3619 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 189.Cheung SK, Chuang PK, Huang HW, et al. Stage-specific embryonic antigen-3 (SSEA-3) and β3GalT5 are cancer specific and significant markers for breast cancer stem cells. Proc Natl Acad Sci U S A. January 26 2016;113(4):960–5. doi: 10.1073/pnas.1522602113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 190.Pece S, Tosoni D, Confalonieri S, et al. Biological and molecular heterogeneity of breast cancers correlates with their cancer stem cell content. Cell. January 8 2010;140(1):62–73. doi: 10.1016/j.cell.2009.12.007 [DOI] [PubMed] [Google Scholar]
- 191.Lo PK, Kanojia D, Liu X, et al. CD49f and CD61 identify Her2/neu-induced mammary tumor-initiating cells that are potentially derived from luminal progenitors and maintained by the integrin-TGFβ signaling. Oncogene. May 24 2012;31(21):2614–26. doi: 10.1038/onc.2011.439 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 192.Desgrosellier JS, Lesperance J, Seguin L, et al. Integrin alphavbeta3 drives slug activation and stemness in the pregnant and neoplastic mammary gland. Developmental cell. August 11 2014;30(3):295–308. doi: 10.1016/j.devcel.2014.06.005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 193.Liu L, Yin B, Yi Z, et al. Breast cancer stem cells characterized by CD70 expression preferentially metastasize to the lungs. Breast Cancer. November 2018;25(6):706–716. doi: 10.1007/s12282-018-0880-6 [DOI] [PubMed] [Google Scholar]
- 194.Thiagarajan PS, Sinyuk M, Turaga SM, et al. Cx26 drives self-renewal in triple-negative breast cancer via interaction with NANOG and focal adhesion kinase. Nature communications. 2018;9(1):578. doi: 10.1038/s41467-018-02938-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 195.Wang Y, Tu L, Du C, et al. CXCR2 is a novel cancer stem-like cell marker for triple-negative breast cancer. OncoTargets and therapy. 2018;11:5559–5567. doi: 10.2147/ott.S174329 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 196.Battula VL, Shi Y, Evans KW, et al. Ganglioside GD2 identifies breast cancer stem cells and promotes tumorigenesis. J Clin Invest. June 2012;122(6):2066–78. doi: 10.1172/jci59735 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 197.Tamori S, Nozaki Y, Motomura H, et al. Glyoxalase 1 gene is highly expressed in basal-like human breast cancers and contributes to survival of ALDH1-positive breast cancer stem cells. Oncotarget. November 23 2018;9(92):36515–36529. doi: 10.18632/oncotarget.26369 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 198.Yan Y, Liu F, Han L, et al. HIF-2α promotes conversion to a stem cell phenotype and induces chemoresistance in breast cancer cells by activating Wnt and Notch pathways. J Exp Clin Cancer Res. October 19 2018;37(1):256. doi: 10.1186/s13046-018-0925-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 199.Yang L, Tang H, Kong Y, et al. LGR5 Promotes Breast Cancer Progression and Maintains Stem-Like Cells Through Activation of Wnt/β-Catenin Signaling. Stem Cells. October 2015;33(10):2913–24. doi: 10.1002/stem.2083 [DOI] [PubMed] [Google Scholar]
- 200.Wu L, Wang T, He D, Li X, Jiang Y. miR-1 inhibits the proliferation of breast cancer stem cells by targeting EVI-1. OncoTargets and therapy. 2018;11:8773–8781. doi: 10.2147/ott.S188836 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 201.Roscigno G, Quintavalle C, Donnarumma E, et al. MiR-221 promotes stemness of breast cancer cells by targeting DNMT3b. Oncotarget. January 5 2016;7(1):580–92. doi: 10.18632/oncotarget.5979 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 202.Engelmann K, Shen H, Finn OJ. MCF7 side population cells with characteristics of cancer stem/progenitor cells express the tumor antigen MUC1. Cancer Res. April 1 2008;68(7):2419–26. doi: 10.1158/0008-5472.Can-07-2249 [DOI] [PubMed] [Google Scholar]
- 203.Siddharth S, Goutam K, Das S, et al. Nectin-4 is a breast cancer stem cell marker that induces WNT/β-catenin signaling via Pi3k/Akt axis. Int J Biochem Cell Biol. August 2017;89:85–94. doi: 10.1016/j.biocel.2017.06.007 [DOI] [PubMed] [Google Scholar]
- 204.Hwang-Verslues WW, Kuo WH, Chang PH, et al. Multiple lineages of human breast cancer stem/progenitor cells identified by profiling with stem cell markers. PloS one. December 21 2009;4(12):e8377. doi: 10.1371/journal.pone.0008377 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 205.Hong D, Fritz AJ, Finstad KH, et al. Suppression of Breast Cancer Stem Cells and Tumor Growth by the RUNX1 Transcription Factor. Molecular cancer research : MCR. December 2018;16(12):1952–1964. doi: 10.1158/1541-7786.Mcr-18-0135 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 206.Grange C, Lanzardo S, Cavallo F, Camussi G, Bussolati B. Sca-1 identifies the tumor-initiating cells in mammary tumors of BALB-neuT transgenic mice. Neoplasia. December 2008;10(12):1433–43. doi: 10.1593/neo.08902 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 207.Ibrahim SA, Gadalla R, El-Ghonaimy EA, et al. Syndecan-1 is a novel mole cular marker for triple negative inflammatory breast cancer and modulates the cancer stem cell phenotype via the IL-6/STAT3, Notch and EGFR signaling pathways. journal article. March 07 2017;16(1):57. doi: 10.1186/s12943-017-0621-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- 208.Feng W, Li Y, Chu J, et al. Identification of tRNA-derived small noncoding RNAs as potential biomarkers for prediction of recurrence in triple-negative breast cancer. 2018;7(10):5130–5144. doi:doi: 10.1002/cam4.1761 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 209.Rhost S, Hughes É, Harrison H, et al. Sortilin inhibition limits secretion-induced progranulin-dependent breast cancer progression and cancer stem cell expansion. Breast Cancer Res. November 20 2018;20(1):137. doi: 10.1186/s13058-018-1060-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 210.Corradetti B, Vaiasicca S, Mantovani M, Virgili E, Bonucci M, Hammarberg Ferri I. Bioactive Immunomodulatory Compounds: A Novel Combinatorial Strategy for Integrated Medicine in Oncology? BAIC Exposure in Cancer Cells. Integr Cancer Ther. Jan-Dec 2019;18:1534735419866908. doi: 10.1177/1534735419866908 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 211.Yu B, Yuan B, Kiyomi A, et al. Differentiation induction of human breast cancer cells by arsenite in combination with tetrandrine. Am J Transl Res. 2019;11(12):7310–7323. [PMC free article] [PubMed] [Google Scholar]
- 212.Koohestanimobarhan S, Salami S, Imeni V, Mohammadi Z, Bayat O. Lipophilic statins antagonistically alter the major epithelial-to-mesenchymal transition signaling pathways in breast cancer stem-like cells via inhibition of the mevalonate pathway. J Cell Biochem. September 6 2018;doi: 10.1002/jcb.27544 [DOI] [PubMed] [Google Scholar]
- 213.Kim DW, Cho JY. NQO1 is Required for β-Lapachone-Mediated Downregulation of Breast-Cancer Stem-Cell Activity. Int J Mol Sci. November 30 2018;19(12)doi: 10.3390/ijms19123813 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 214.Risom T, Langer EM, Chapman MP, et al. Differentiation-state plasticity is a targetable resistance mechanism in basal-like breast cancer. Nat Commun. September 19 2018;9(1):3815. doi: 10.1038/s41467-018-05729-w [DOI] [PMC free article] [PubMed] [Google Scholar]
- 215.Clément F, Xu X, Donini CF, et al. Long-term exposure to bisphenol A or benzo(a)pyrene alters the fate of human mammary epithelial stem cells in response to BMP2 and BMP4, by pre-activating BMP signaling. Cell Death Differ. January 2017;24(1):155–166. doi: 10.1038/cdd.2016.107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 216.Manupati K, Dhoke NR, Debnath T, et al. Inhibiting epidermal growth factor receptor signalling potentiates mesenchymal-epithelial transition of breast cancer stem cells and their responsiveness to anticancer drugs. Febs j. June 2017;284(12):1830–1854. doi: 10.1111/febs.14084 [DOI] [PubMed] [Google Scholar]
- 217.Nigjeh SE, Yeap SK, Nordin N, Rahman H, Rosli R. In Vivo Anti-Tumor Effects of Citral on 4T1 Breast Cancer Cells via Induction of Apoptosis and Downregulation of Aldehyde Dehydrogenase Activity. Molecules. September 5 2019;24(18)doi: 10.3390/molecules24183241 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 218.Hu C, Li M, Guo T, et al. Anti-metastasis activity of curcumin against breast cancer via the inhibition of stem cell-like properties and EMT. Phytomedicine. May 2019;58:152740. doi: 10.1016/j.phymed.2018.11.001 [DOI] [PubMed] [Google Scholar]
- 219.Colacino JA, McDermott SP, Sartor MA, Wicha MS, Rozek LS. Transcriptomic profiling of curcumin-treated human breast stem cells identifies a role for stearoyl-coa desaturase in breast cancer prevention. Breast Cancer Res Treat. July 2016;158(1):29–41. doi: 10.1007/s10549-016-3854-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 220.Benoit YD, Mitchell RR, Risueño RM, et al. Sam68 Allows Selective Targeting of Human Cancer Stem Cells. Cell Chem Biol. July 20 2017;24(7):833–844.e9. doi: 10.1016/j.chembiol.2017.05.026 [DOI] [PubMed] [Google Scholar]
- 221.Tian J, Raffa FA, Dai M, et al. Dasatinib sensitises triple negative breast cancer cells to chemotherapy by targeting breast cancer stem cells. Br J Cancer. December 2018;119(12):1495–1507. doi: 10.1038/s41416-018-0287-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 222.Li X, Meng Y, Xie C, et al. Diallyl Trisulfide inhibits breast cancer stem cells via suppression of Wnt/β-catenin pathway. J Cell Biochem. May 2018;119(5):4134–4141. doi: 10.1002/jcb.26613 [DOI] [PubMed] [Google Scholar]
- 223.Gkountela S, Castro-Giner F, Szczerba BM, et al. Circulating Tumor Cell Clustering Shapes DNA Methylation to Enable Metastasis Seeding. Cell. January 10 2019;176(1–2):98–112.e14. doi: 10.1016/j.cell.2018.11.046 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 224.Kim YJ, Kim JY, Lee N, et al. Disulfiram suppresses cancer stem-like properties and STAT3 signaling in triple-negative breast cancer cells. Biochem Biophys Res Commun. May 13 2017;486(4):1069–1076. doi: 10.1016/j.bbrc.2017.03.164 [DOI] [PubMed] [Google Scholar]
- 225.Tudoran O, Soritau O, Balacescu L, et al. Regulation of stem cells-related signaling pathways in response to doxorubicin treatment in Hs578T triple-negative breast cancer cells. Mol Cell Biochem. November 2015;409(1–2):163–76. doi: 10.1007/s11010-015-2522-z [DOI] [PubMed] [Google Scholar]
- 226.Cuenca-López MD, Serrano-Heras G, Montero JC, et al. Antitumor activity of the novel multi-kinase inhibitor EC-70124 in triple negative breast cancer. Oncotarget. September 29 2015;6(29):27923–37. doi: 10.18632/oncotarget.4736 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 227.Merino VF, Nguyen N, Jin K, et al. Combined Treatment with Epigenetic, Differentiating, and Chemotherapeutic Agents Cooperatively Targets Tumor-Initiating Cells in Triple-Negative Breast Cancer. Cancer Res. April 1 2016;76(7):2013–2024. doi: 10.1158/0008-5472.Can-15-1619 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 228.Fiorillo M, Verre AF, Iliut M, et al. Graphene oxide selectively targets cancer stem cells, across multiple tumor types: implications for non-toxic cancer treatment, via “differentiation-based nano-therapy”. Oncotarget. February 28 2015;6(6):3553–62. doi: 10.18632/oncotarget.3348 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 229.Liu Y, Pandey PR, Sharma S, et al. ID2 and GJB2 promote early-stage breast cancer progression by regulating cancer stemness. Breast Cancer Res Treat. May 2019;175(1):77–90. doi: 10.1007/s10549-018-05126-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 230.Jung SY, Yi JY, Kim MH, et al. IM-412 inhibits the invasion of human breast carcinoma cells by blocking FGFR-mediated signaling. Oncol Rep. November 2015;34(5):2731–7. doi: 10.3892/or.2015.4249 [DOI] [PubMed] [Google Scholar]
- 231.Dominguez-Gomez G, Chavez-Blanco A, Medina-Franco JL, et al. Ivermectin as an inhibitor of cancer stem‑like cells. Mol Med Rep. February 2018;17(2):3397–3403. doi: 10.3892/mmr.2017.8231 [DOI] [PubMed] [Google Scholar]
- 232.Strietz J, Stepputtis SS, Preca BT, et al. ERN1 and ALPK1 inhibit differentiation of bi-potential tumor-initiating cells in human breast cancer. Oncotarget. December 13 2016;7(50):83278–83293. doi: 10.18632/oncotarget.13086 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 233.Berardi DE, Raffo D, Todaro LB, Simian M. Laminin Modulates the Stem Cell Population in LM05-E Murine Breast Cancer Cells through the Activation of the MAPK/ERK Pathway. Cancer Res Treat. October 2017;49(4):869–879. doi: 10.4143/crt.2016.378 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 234.Tan W, Tang H, Jiang X, et al. Metformin mediates induction of miR-708 to inhibit self-renewal and chemoresistance of breast cancer stem cells through targeting CD47. J Cell Mol Med. September 2019;23(9):5994–6004. doi: 10.1111/jcmm.14462 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 235.Sp N, Kang DY, Kim DH, et al. Nobiletin Inhibits CD36-Dependent Tumor Angiogenesis, Migration, Invasion, and Sphere Formation Through the Cd36/Stat3/Nf-Kb Signaling Axis. Nutrients. June 15 2018;10(6)doi: 10.3390/nu10060772 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 236.Kietzman WB, Graham GT, Ory V, et al. Short- and Long-Term Effects of CDK4/6 Inhibition on Early-Stage Breast Cancer. Mol Cancer Ther. December 2019;18(12):2220–2232. doi: 10.1158/1535-7163.Mct-19-0231 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 237.Hachim IY, López-Ozuna VM, Hachim MY, Lebrun JJ, Ali S. Prolactin hormone exerts anti-tumorigenic effects in HER-2 overexpressing breast cancer cells through regulation of stemness. Stem Cell Res. October 2019;40:101538. doi: 10.1016/j.scr.2019.101538 [DOI] [PubMed] [Google Scholar]
- 238.Bosukonda A, Carlson WD. Harnessing the BMP signaling pathway to control the formation of cancer stem cells by effects on epithelial-to-mesenchymal transition. Biochem Soc Trans. February 8 2017;45(1):223–228. doi: 10.1042/bst20160177 [DOI] [PubMed] [Google Scholar]
- 239.Li X, Zhou N, Wang J, et al. Quercetin suppresses breast cancer stem cells (CD44(+)/CD24(−)) by inhibiting the PI3K/Akt/mTOR-signaling pathway. Life Sci. March 1 2018;196:56–62. doi: 10.1016/j.lfs.2018.01.014 [DOI] [PubMed] [Google Scholar]
- 240.Hii LW, Chung FF, Soo JS, Tan BS, Mai CW, Leong CO. Histone deacetylase (HDAC) inhibitors and doxorubicin combinations target both breast cancer stem cells and non-stem breast cancer cells simultaneously. Breast Cancer Res Treat. February 2020;179(3):615–629. doi: 10.1007/s10549-019-05504-5 [DOI] [PubMed] [Google Scholar]
- 241.Yoldi G, Pellegrini P, Trinidad EM, et al. RANK Signaling Blockade Reduces Breast Cancer Recurrence by Inducing Tumor Cell Differentiation. Cancer Res. October 1 2016;76(19):5857–5869. doi: 10.1158/0008-5472.Can-15-2745 [DOI] [PubMed] [Google Scholar]
- 242.Deus CM, Serafim TL, Magalhães-Novais S, et al. Sirtuin 1-dependent resveratrol cytotoxicity and pro-differentiation activity on breast cancer cells. Arch Toxicol. March 2017;91(3):1261–1278. doi: 10.1007/s00204-016-1784-x [DOI] [PubMed] [Google Scholar]
- 243.Ishay-Ronen D, Diepenbruck M, Kalathur RKR, et al. Gain Fat-Lose Metastasis: Converting Invasive Breast Cancer Cells into Adipocytes Inhibits Cancer Metastasis. Cancer Cell. January 14 2019;35(1):17–32.e6. doi: 10.1016/j.ccell.2018.12.002 [DOI] [PubMed] [Google Scholar]
- 244.Wang T, Narayanaswamy R, Ren H, Torchilin VP. Combination therapy targeting both cancer stem-like cells and bulk tumor cells for improved efficacy of breast cancer treatment. Cancer Biol Ther. June 2 2016;17(6):698–707. doi: 10.1080/15384047.2016.1190488 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 245.McGrail DJ, Lin CC, Dai H, et al. Defective Replication Stress Response Is Inherently Linked to the Cancer Stem Cell Phenotype. Cell Rep. May 15 2018;23(7):2095–2106. doi: 10.1016/j.celrep.2018.04.068 [DOI] [PubMed] [Google Scholar]
- 246.Thakkar A, Wang B, Picon-Ruiz M, Buchwald P, Ince TA. Vitamin D and androgen receptor-targeted therapy for triple-negative breast cancer. Breast Cancer Res Treat. May 2016;157(1):77–90. doi: 10.1007/s10549-016-3807-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- 247.Abdollahi P, Ebrahimi M, Motamed N, Samani FS. Silibinin affects tumor cell growth because of reduction of stemness properties and induction of apoptosis in 2D and 3D models of MDA-MB-468. Anticancer Drugs. June 2015;26(5):487–97. doi: 10.1097/cad.0000000000000205 [DOI] [PubMed] [Google Scholar]
- 248.Hsu EC, Kulp SK, Huang HL, et al. Function of Integrin-Linked Kinase in Modulating the Stemness of IL-6-Abundant Breast Cancer Cells by Regulating γ-Secretase-Mediated Notch1 Activation in Caveolae. Neoplasia. June 2015;17(6):497–508. doi: 10.1016/j.neo.2015.06.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 249.Kim SH, Singh SV. Mammary cancer chemoprevention by withaferin A is accompanied by in vivo suppression of self-renewal of cancer stem cells. Cancer Prev Res (Phila). July 2014;7(7):738–47. doi: 10.1158/1940-6207.Capr-13-0445 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 250.Ketchart W, Yeh IJ, Zhou H, et al. Induction of HEXIM1 activities by HMBA derivative 4a1: Functional consequences and mechanism. Cancer Lett. August 28 2016;379(1):60–9. doi: 10.1016/j.canlet.2016.05.029 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 251.Phan NL, Trinh NV, Pham PV. Low concentrations of 5-aza-2’-deoxycytidine induce breast cancer stem cell differentiation by triggering tumor suppressor gene expression. Onco Targets Ther. 2016;9:49–59. doi: 10.2147/ott.S96291 [DOI] [PMC free article] [PubMed] [Google Scholar]