

Graphical Abstract
Membrane Proteins are Challenging Analytes
Biological membranes formed of amphipathic lipid bilayers provide chemical barriers to separate cells from their external environments and define subcellular compartments. Membrane proteins embedded within these lipid membranes constitute about 30% of the proteome,1,2 and they play key roles in cellular processes such as chemical transport, signal transduction, and enzymatic catalysis. Due to these important roles, membrane proteins represent roughly half of all drug targets.3 Characterizing membrane protein structures is fundamental in elucidating their biochemical mechanisms and developing new therapeutics. However, membrane proteins represent only 2–3% of high-resolution structures in the Protein Data Bank (PDB). These statistics reflect a significant gap in analytical tools to characterize membrane protein structure, function, and interactions, especially in the context of lipid bilayers.
There are several analytical challenges in studying membrane proteins. First, the natural abundance of membrane proteins is typically low, and it can be challenging to overexpress and purify membrane proteins in high yields.4–7 Thus, we need sensitive analytical methods that only require small amounts of sample. Second, the membrane environment is highly heterogeneous and insoluble, and many techniques are not capable of studying membrane proteins in their natural context. Finally, most analytical methods require membrane proteins to be solubilized, but detergents used for solubilization can destabilize membrane proteins and disrupt key protein-lipid interactions.8–11 In other words, membrane proteins have more complex interactions and environmental constraints than soluble proteins due to their amphipathic nature.
To address these challenges, mass spectrometry (MS) is emerging as a critical tool for investigating the structure, dynamics, and interactions of membrane proteins. MS-based methods provide a flexible toolbox that can bridge the gap between high- and low-resolution biophysical techniques and address the unique environmental challenges of membrane proteins.12,13 Structural proteomics methods such as chemical crosslinking, hydrogen-deuterium exchange, and hydroxyl radical foot printing provide unique structural information on inter-residue distances, dynamics, and solvent exposure. Native MS provides direct and label-free detection of protein interactions.
Here, we will review recent developments in native MS of membrane proteins, focusing primarily on new analytical advances from 2018 through 2020. We will organize our discussion from the start of the experiment with solubilization approaches; through instrumental methods such as ionization, activation, and ion mobility; and close with novel experimental designs. For further reading into the specific biological applications of native MS, we refer the reader to several excellent reviews.12–19
What is Native MS?
Native mass spectrometry uses no denaturing ionization conditions, usually with nano-electrospray ionization (nano-ESI), to preserve noncovalent complexes upon transfer from solution to the gas phase for mass analysis.20 Unlike conventional denaturing MS, which often includes organic solvents and low pH, native MS generally uses volatile, aqueous buffers near physiological pH and gentler temperatures and voltages. Preserving noncovalent interactions and quaternary protein structure enables the study of protein-protein interactions, protein-ligand interactions, subunit architecture, and complex stoichiometry. Native MS has been used in a wide variety of applications, ranging from biological nucleosome and ribosome particles to synthetic supramolecular assemblies.21–24
Why use Native MS for Membrane Proteins?
Native MS has emerged as a complementary analytical approach that provides several advantages in studying membrane proteins over classical structural and biophysical techniques. First, native MS is compatible with a wide mass range, spanning from small transmembrane peptides to large multiprotein complexes.25–27 Conventional structural techniques are often limited to specific size ranges, with NMR, X-ray crystallography, and cryo-electron microscopy (EM) best suited for small, medium, and large protein systems, respectively.28–30 Second, native MS is rapid and highly sensitive, requiring only a few microliters of membrane protein at high nanomolar to low micro molar concentrations. Conventional structural biology methods, especially NMR and X-ray crystallography, often require much higher concentrations and amounts. Finally, native MS is label free and thus prevents potential issues with altering the intrinsic properties of membrane proteins or their interaction partners. Spectroscopic techniques such as FRET, SPR, and EPR usually require chemical labels or surface immobilization.31–33
Importantly, native MS is uniquely suited for studying complex biomolecular interactions.34 Conventional structural techniques often rely on averaging many molecules to generate a composite high-resolution structure. However, membrane proteins exist in distinct molecular forms that can vary in covalent modifications, stoichiometry, and conformation. Native MS can help inform on this heterogeneity because it avoids ensemble averaging. Thus, the composition and relative abundances of distinct membrane protein complexes can be simultaneously probed, providing unprecedented chemical specificity and detail in measuring biomolecular interactions. For example, ligand binding can be simultaneously measured for different proteoforms35 or oligomeric states.36 Finally, small molecule interaction partners with ambiguous electron densities in high-resolution techniques can be potentially identified from the shift in mass. Thus, native MS provides a complementary approach for examining heterogeneous, unstructured, and transient interactions that reflect those present in biological membranes.
A Home Away from Home: Membrane Mimetics
Unlike soluble proteins, membrane proteins need some form of membrane mimetic to solubilize the protein prior to analysis. This membrane mimetic environment can be essential for membrane protein function and stability. However, different reconstitution system may not be equally compatible with the analysis method. Membrane mimetics can be a critical part of the native MS experiment and have significant effects on the quality of mass spectra. Thus, we will begin by discussing recent advances in interfacing diverse membrane mimetics with native MS.
Detergent Micelles.
Detergent micelles are the most common membrane mimetic, including for native MS. Following extraction, protein-detergent complexes are purified and exchanged into a volatile buffer that usually contains around one to two times the critical micelle concentration (CMC) of detergent.37 The entire protein-micelle complex is then ionized. Because intact micelle complexes produce highly heterogeneous mass spectra, detergent adducts need to be removed by activation inside the mass spectrometer (Figure 1A). However, there is a delicate balance between adding enough activation for detergent removal and preserving membrane protein interactions.
Figure 1.
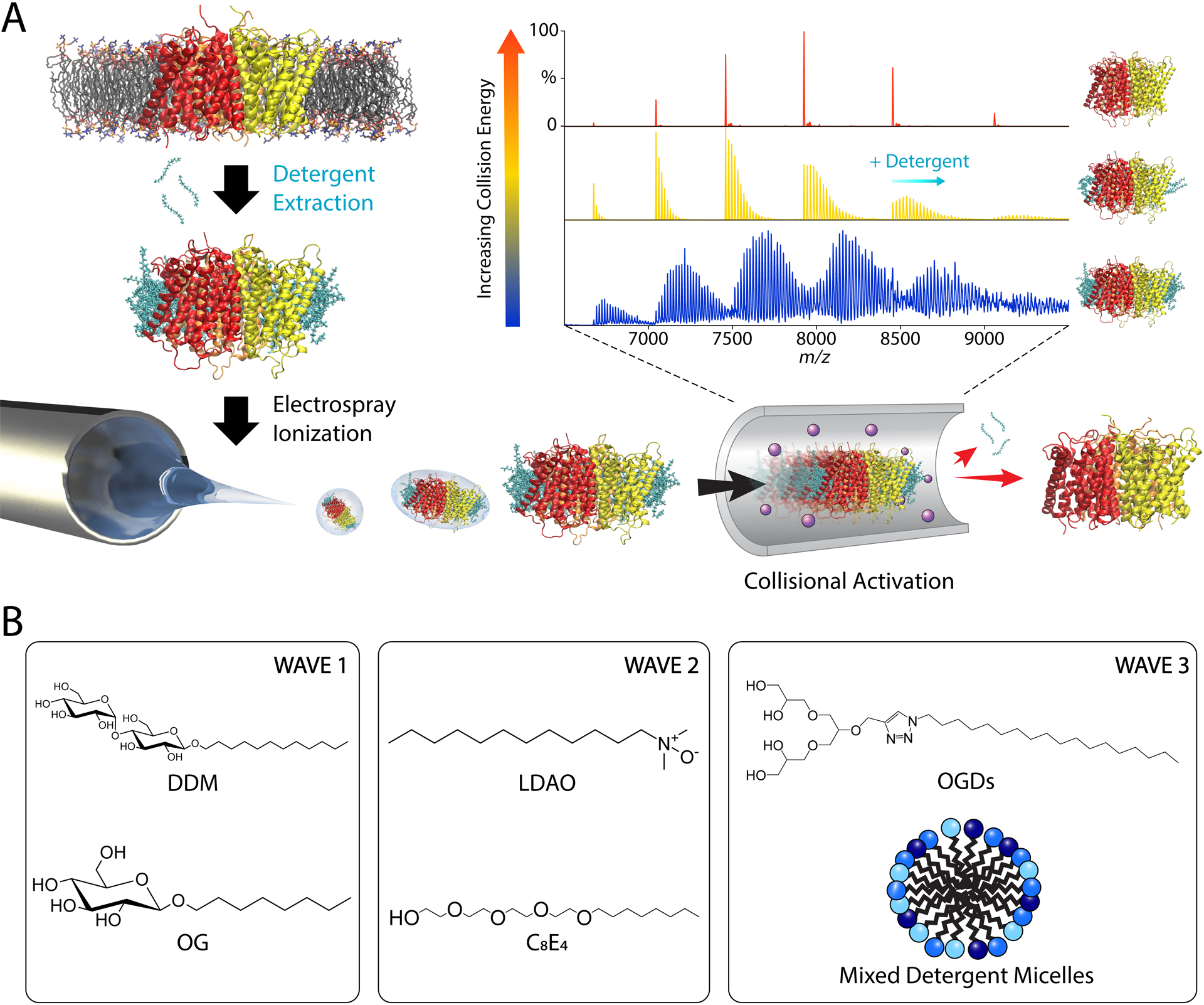
(A) General workflow for conventional native MS of membrane proteins in detergent micelles. Membrane proteins are extracted from membranes using detergents, ionized by ESI inside the micelles, and collisionally activated to remove bound detergents. (B) Common detergents used for native MS grouped in different waves roughly by when they were first applied to native MS.
Detergents must be carefully selected to preserve the native state of the membrane protein and optimize the quality of mass spectra. Non-ionic detergents are often preferable for solubilization of membrane proteins because they are usually less denaturing than ionic detergents.38,39 Non-ionic detergents are also desirable for MS because they minimize ion suppression and are generally easier to remove in the gas phase. Prell and coworkers have recently developed a novel Gabor transform40 data analysis approach that enables measurement of membrane protein oligomeric state in highly adducted Fos-14, a zwitterionic detergent.41,42 However, these Fos-choline micelles can be denaturing for some membrane proteins and should be used carefully.
Early native MS studies of membrane proteins mostly employed saccharide detergents (Figure 1B), such as n-dodecyl-β-D-maltopyranoside (DDM) and n-octyl-β-D-glucopyranoside (OG), which are heavily used in structural biology.37 Later, Robinson and coworkers pioneered the use of charge-reducing detergents, such as tetraethylene glycol monooctyl ether (C8E4) and lauryldimethylamine-N-oxide (LDAO), for native MS that were easier to dissociate and better preserved membrane protein structure during ionization.8,43 Like C8E4, Triton X-10044 and other detergents with polyethylene glycol hydrophilic head groups are similarly easy to remove and tend to be well suited for native MS. These first- and second-wave detergents continue to be heavily used. However, not all membrane proteins are stable in these detergents, which has driven research towards new detergent strategies.
Recently, Pagel and coworkers developed a new library of modular oligoglycerol detergents (OGDs) for native MS of membrane proteins.45,46 The structure of these OGDs is subdivided into three sections: a polar headgroup, a hydrophobic tail, and a connecting linker group. Each section can be modified to optimize protein purification, retention of protein-lipid interactions, and charge reduction. Notably, OGDs improved purification and native MS analysis of functional neurotensin receptor type 1 (NTSR1), a G-protein coupled receptor (GPCR) especially susceptible to instability from detergents.47 Modification of OGDs with azobenzene enables photoresponsive tuning of charge-reducing properties based on the light-dependent interconversion between trans and cis isomers.48
Another emerging strategy for native MS is mixed micelles with combinations of several detergents. The presence of several detergents may allow membrane proteins to select the optimal detergent interactions within its microenvironment, which may help stabilize challenging membrane proteins. Mixtures of OGD regioisomers generally improved membrane protein isolation and quality of mass spectra.46 With more conventional detergents, NTSR1 was exchanged into mixed DDM, laurylmaltose neopentylglycol (LMNG), and Fos-choline micelles while the adenosine A2A receptor (A2AR) and β1 adrenergic receptor (β1AR) were exchanged into mixed DDM and Fos-choline micelles.49 Optimal mixed micelle compositions vary between proteins and must be determined experimentally.50
It remains to be seen whether these advances in mixed micelles and modular detergents represent a third wave of detergents for native MS, but screening of detergents will continue to be important for finding the optimal conditions for both preserving membrane protein structure and producing quality native mass spectra.37 Having additional detergents and combination strategies will undoubtedly help, especially if some general principles for detergent selection can be discovered.
Although these new developments for detergents can improve membrane protein solubilization, stability, and compatibility with native MS, detergents have the potential to disrupt membrane protein interactions.51 This limitation has driven a greater interest in alternative mimetic systems with less dependence on detergent. For example, Hutchison et al. recently developed a new bicelle platform containing lipids cosolubilized by n-dodecyl-β-melibioside to study the oligomerization of transmembrane human amyloid precursor protein (C99).52 Native ion mobility (IM)-MS performed on intact C99 liberated from bicelles showed three oligomer populations, which deviated from other membrane mimetics. This study confirms that lipids can significantly alter the structure and interactions of membrane proteins. Other alternative membrane mimetics have been previously reviewed by Marty et al.53 Here, we will focus on advances in native MS of membrane mimetics over the last few years, especially nanodiscs, lipopolymer nanoparticles, and lipid vesicles.
Nanodiscs.
Nanodiscs are nanoscale lipid bilayers encircled by two copies of amphipathic membrane scaffold protein (MSP).54 Because they contain a lipid bilayer, nanodiscs have been shown to better preserve the stoichiometry and stability of membrane proteins compared with detergent micelles.55,56 Nanodiscs are particularly well-suited for native MS due to their homogeneity and narrow size distribution,53,57 and they remain the only membrane mimetic that is resolvable in its intact form without dissociation.58 The diameter of nanodiscs can be tuned using different MSP variants, enabling a wide range of membrane protein targets to be incorporated.55 Furthermore, nanodisc lipid composition can be tightly controlled during assembly for investigating specific protein-lipid interactions.59,60
Marty and coworkers developed a novel method for tuning the stability of nanodiscs during native MS.58 Instrumental polarity and chemical supercharging agents were used to either stabilize the intact membrane protein-nanodisc complex or destabilize the complex for membrane protein ejection. By stabilizing the membrane protein-nanodisc assembly, the stoichiometries of model membrane proteins ammonium transporter B (AmtB) and aquaporin Z (AqpZ) could be directly measured inside the intact lipid bilayer, a first for membrane proteins by native MS. A similar approach was also used to study the oligomeric states of fragile and polydisperse transmembrane peptide complexes in intact nanodiscs with different lipids.25,26 Building on these methods, we investigated the lipid selectivity of AmtB using binary-lipid nanodiscs comprised of palmitoyl-oleoyl-phosphatidylcholine (POPC) and palmitoyl-oleoyl-phosphatidylglycerol (POPG).61 AmtB was ejected from 50/50 POPC/POPG nanodiscs, and the resulting composition of retained lipids was measured by the average mass of the bound lipids (Figure 2). The average lipid mass was also measured for partially intact and fully intact membrane protein-nanodisc complexes by adjusting the supercharging reagents added prior to ionization. Using these novel methods, we discovered that AmtB was overall selective for POPG lipids but had a few tight POPC binding sites.
Figure 2.
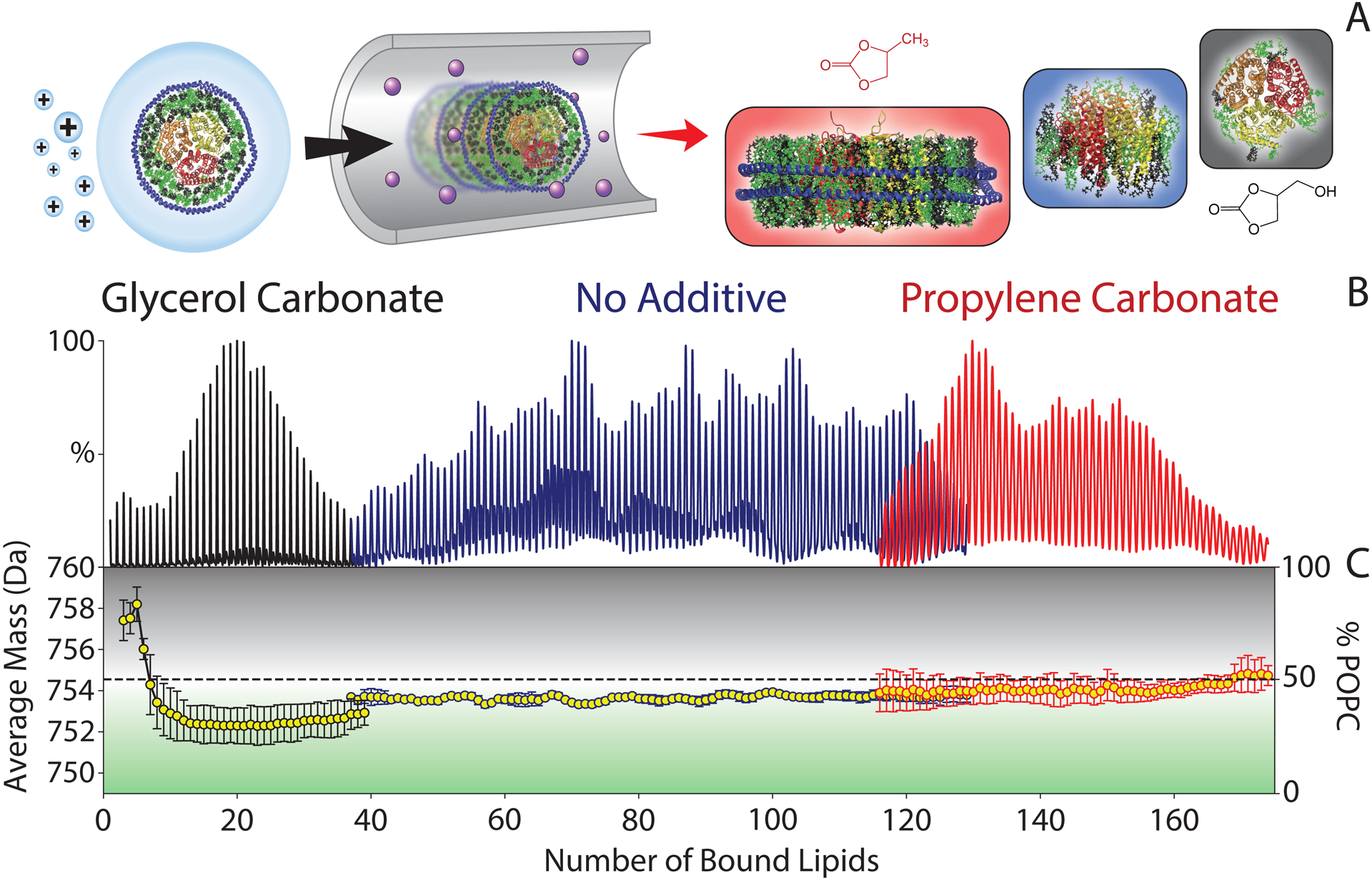
(A) Schematic of native MS of mixed lipid nanodiscs that are preserved by propylene carbonate (red), ejected with a large number of lipids when no additive is present (blue), and ejected with only a few lipids bound by glycerol carbonate (black). (B) Summed deconvolved mass spectra of AmtB in 50/50 POPC/POPG nanodiscs collected with added propylene carbonate, no additive, or glycerol carbonate. Masses are shown as the number of lipids bound to AmtB. (C) The average lipid mass (left axis) and relative mole percentage of POPC (right) for different numbers of bound lipids. Average masses higher than 754.5 Da (dashed line) are enriched in POPC (black), whereas lower masses are enriched in POPG (green). Adapted from Zhang, G.; Keener, J. E.; Marty, M. T. Measuring Remodeling of the Lipid Environment Surrounding Membrane Proteins with Lipid Exchange and Native Mass Spectrometry. Anal. Chem. 2020, 92, 5666–5669 (ref 61). Copyright 2020 American Chemical Society.
Recent native MS studies have also used nanodiscs to study challenging membrane protein targets. Debruycker et al. used nanodiscs to resolve lipid binding to LmrP, a multidrug transporter of the major facilitator superfamily, at lower activation levels compared to DDM micelles.62 Ro et al. observed that nanodiscs provided improved retention of copper ions and more efficient subunit ejection compared to Triton X-100 micelles for top-down proteomics of a metalloenzyme.63 Nanodiscs were also used to study the oligomerization of a methyltransferase which retained activity in nanodiscs but not in detergent micelles.64
In addition to conventional nanodiscs made with MSP belts, alternative lipoprotein and lipopeptide nanodiscs have also emerged as potential membrane mimetics. Peptidiscs have amphipathic peptides as scaffolds rather than MSP, and they were recently used by Carlson et al. to help confirm the stoichiometry of several membrane proteinpeptidisc complexes.65 Picodiscs made with saposin A proteins as the belt have also proven useful for measuring interactions between soluble proteins and glycolipids.66 There is significant exciting work on nanodiscs that is beyond the scope of this review, and we refer the reader to recent reviews14,67 and other studies.68–70 Overall, the homogeneity of nanodiscs enables unique native MS studies of membrane protein-lipid interactions inside intact lipid bilayers.
SMALPs.
Although nanodiscs provide a more native lipid environment for membrane proteins, they still rely on detergent for initial solubilization from the membrane. Styrene maleic acid lipid particles (SMALPs) are membrane mimetics that do not require any detergent.71,72 SMA-copolymers can be used to directly solubilize membrane proteins from their native environment in the absence of detergent, yielding SMALPs comprised of lipids that surrounded membrane proteins in the natural lipid bilayer. However, both the SMALP and SMA-copolymer belt are highly heterogeneous, so the intact complex cannot be resolved by native MS, unlike the more homogeneous MSP nanodiscs.58 Thus, membrane proteins must be ejected from the SMALP, but this has proven difficult with conventional collision-induced dissociation (CID), potentially due to the stability of the overall complex, the charge of the polymer belt, or difficulty resolving heterogeneous ejected fragments.
Morgner and coworkers demonstrated the first application of SMALPs for native MS, which used laser-induced liquid bead ion desorption (LILBID) to successfully ionize SMALPs and estimate the oligomeric state of embedded membrane proteins.73 By adjusting the power of the mid-IR laser, membrane proteins could be transferred to the gas phase either embedded in SMALPs or ejected as dissociated monomers. The peak shapes corresponding to SMALP complexes were broad and unresolved, but the polymer and lipid content could be estimated using the number of transmembrane helices for the embedded protein due to the unique low charge states produced by LILBID. Hesketh et al. recently developed a method for exchanging membrane proteins from SMALPs to amphipols or DDM, but native MS spectra had similarly limited resolution.74
In summary, SMALPs provide a promising vehicle for capturing the native lipid environment around membrane proteins but will require additional method development to overcome the inherent limitations in interfacing heterogeneous SMALPs with native MS. Potentially, supercharging reagents that have been used for ejecting membrane proteins from MSP nanodiscs58 or new activation methods will enable more experiments using SMALPs to deliver membrane proteins for native MS.
Lipid Vesicles.
Robinson and coworkers developed an exciting new method, sonication of lipid vesicles for MS (SoLVe-MS), that uses lipid vesicles derived directly from native membranes (Figure 3).75,76 Here, large membrane fragments isolated from cells by ultracentrifugation are sonicated to produce smaller liposomes with the same endogenous lipids and membrane proteins. In addition to better reflecting the natural membrane environment, SoLVe-MS examines endogenous membrane proteins and does not require purification and overexpression. High activation levels throughout the instrument are used to break apart the liposomes and remove adducts. One limitation is that the heterogeneity of protein assemblies creates significant challenges for assigning the masses.77 Small molecule MS, lipid omics, proteomics, and molecular dynamics (MD) simulations were necessary to assign proteins and their interaction partners more confidently. As instrumentation improves, both in sensitivity and ability to do multilevel activation, native MS of membrane proteins in liposomes will continue to advance and enable analysis of less abundant endogenous membrane protein complexes. Overall, these new developments in membrane mimetics enable new experiments that were not previously possible, expanding the toolbox for studying the interactions of membrane proteins in more natural contexts.
Figure 3.
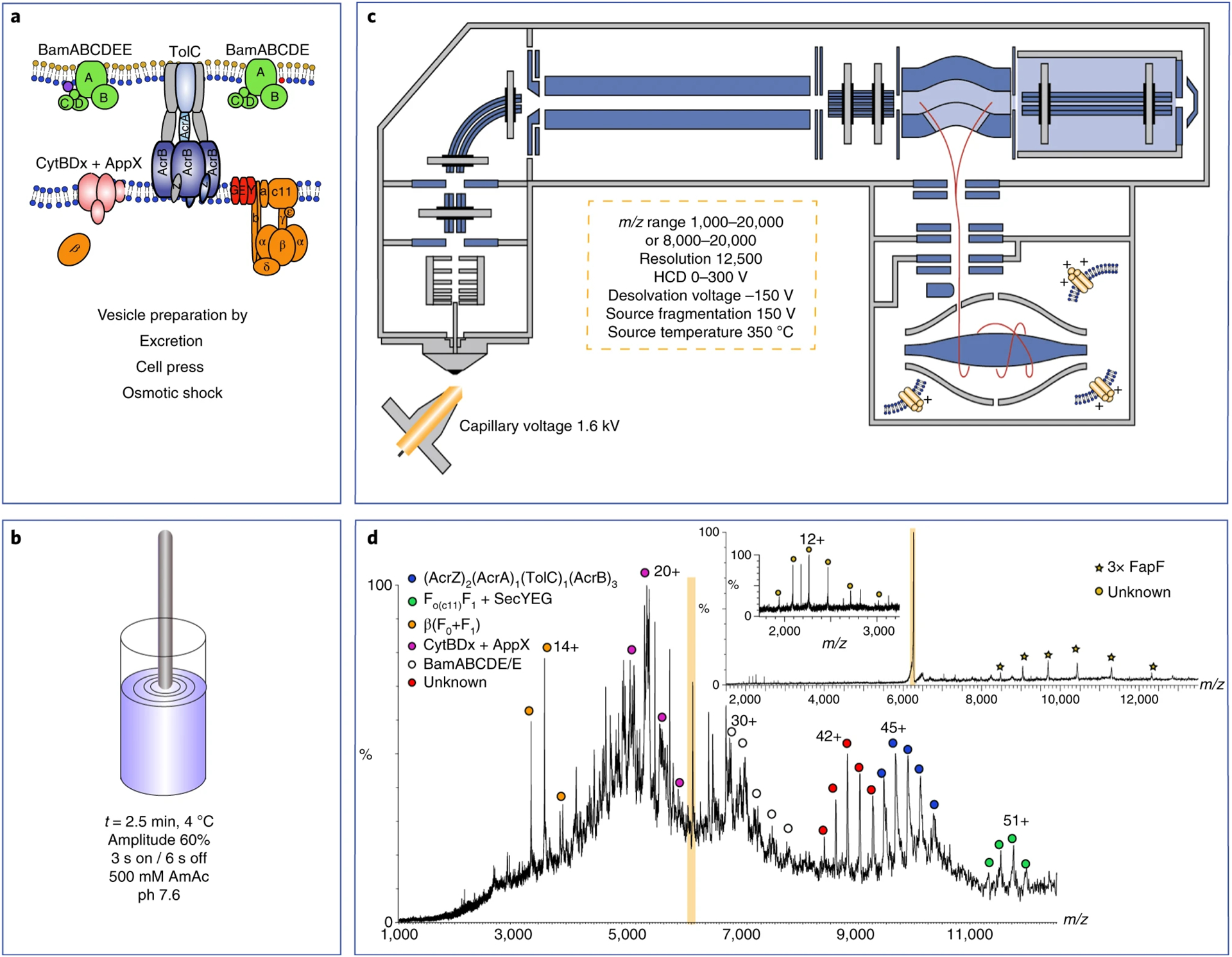
(A) Lipid vesicles are prepared directly from cellular membranes. (B) Lipid vesicles are then diluted into ammonium acetate and destabilized by sonication. (C) Instrument schematic of Q-Exactive UHMR Orbitrap used for analyzing membrane protein complexes ejected from vesicles. The instrumental parameters show the higher-energy conditions needed to generate mass spectra of sufficient quality. (D) Mass spectrum of protein complexes ejected from vesicles. Tandem MS (insets) reveals the FapF trimer (isolated peak highlighted in orange) bound to an unknown subunit. Reprinted by permission from Springer Nature. Nature Protocols, Chorev, D. S.; Tang, H.; Rouse, S. L.; Bolla, J. R.; von Kugelgen, A.; Baker, L. A.; Wu, D.; Gault, J.; Grunewald, K.; Bharat, T. A. M.; Matthews, S. J.; Robinson, C. V. The use of sonicated lipid vesicles for mass spectrometry of membrane protein complexes. Nat. Protoc. 2020, 15, 1690–1706 (ref 75). Copyright 2020.
Ionization: From Solution into the Gas Phase
Improving Buffer Tolerance with Submicron Emitters.
Native MS has relied heavily on static nano-ESI from pulled capillaries with 1–10 μm tip inner diameters and volatile ammonium acetate buffers.78–80 Nonvolatile salts and detergents can reduce the signal-to-noise ratio in native MS due to the formation of interfering high mass-to-charge ratio (m/z) clusters and adducts that broaden peaks. These challenges have limited the types of detergents and buffer components that are amenable for native MS.
Recently, Campuzano, Williams, and coworkers demonstrated that capillaries pulled to narrow submicron tip inner diameters show a much greater tolerance to nonvolatile salts and detergents for membrane proteins bacteriorhodopsin and AqpZ (Figure 4).81 Using these submicron emitters, the broad background signal from salt and detergent was significantly decreased compared to more common micron-scale emitters. The improved signal is due to the formation of smaller droplets with a lower cosolute-to-protein ratio, thereby reducing the chance of salt ions or micelles ending up in the same starting droplet as the protein analyte.82,83 Notably, this was the first reported use of ionic detergents for native MS of membrane proteins.
Figure 4.
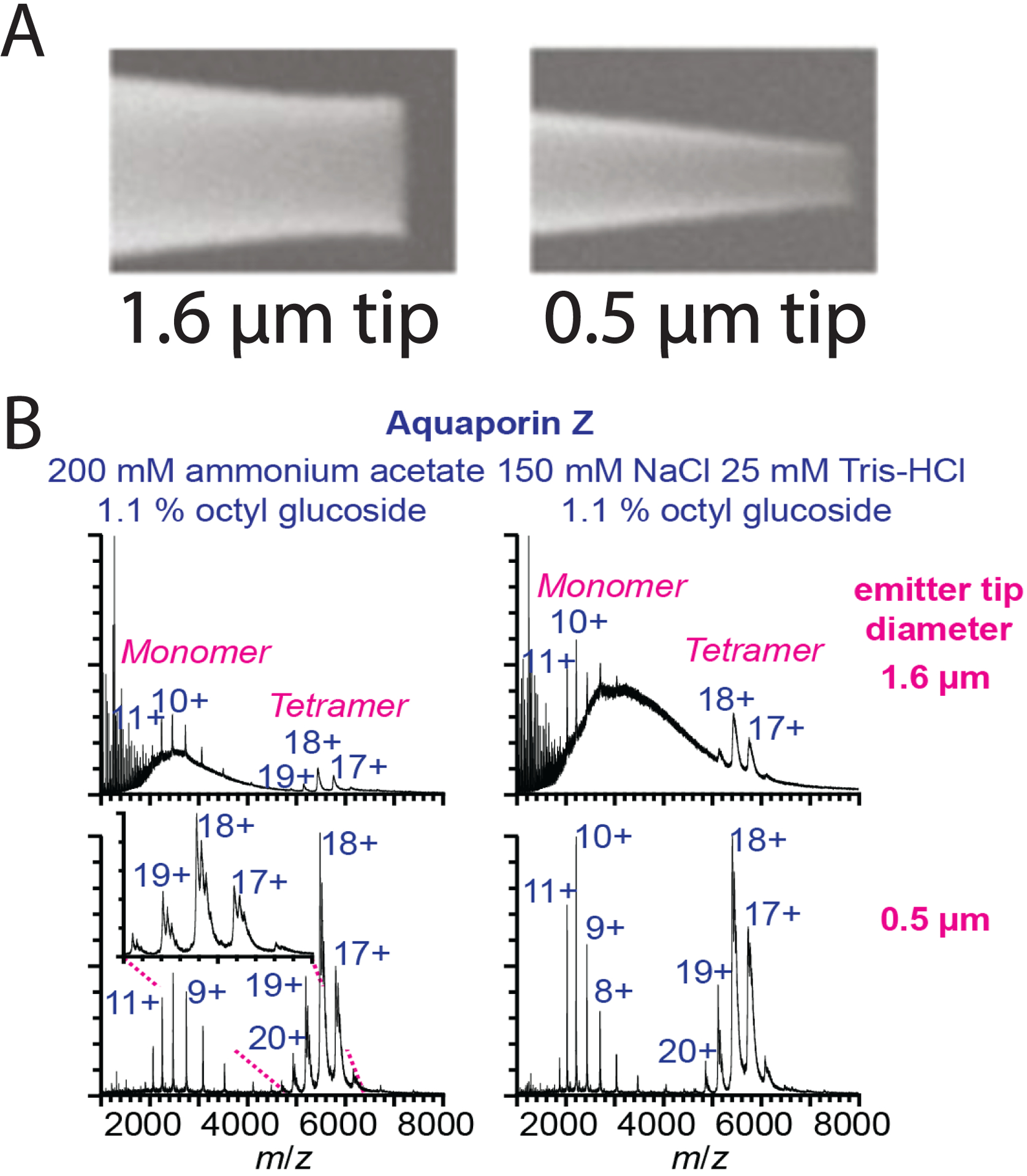
(A) Images of nano-ESI emitter tips at conventional 1.6 μm inner diameters and submicron 0.5 μm inner diameters. (B) Mass spectra of AqpZ using 1.6 μm or 0.5 μm tip diameter emitters in ammonium acetate or Tris-NaCl buffer showing greater tolerance to salt buffers with submicron emitters. Adapted from Susa, A. C.; Lippens, J. L.; Xia, Z.; Loo, J. A.; Campuzano, I. D. G.; Williams, E. R. Submicrometer Emitter ESI Tips for Native Mass Spectrometry of Membrane Proteins in Ionic and Nonionic Detergents. J. Am. Soc. Mass Spectrom. 2018, 29, 203–206 (ref 81). Copyright 2018 American Chemical Society.
There are several potential challenges with submicron emitters, including clogging, potential thermal destabilization, and increased interactions with the glass walls.84–87 However, Panczyk et al. observed no significant differences in soluble protein complex structure comparing micron and submicron capillaries.88 Because lipids are known to interact with glass surfaces,89 further research will be necessary to test how different membrane mimetics are affected and coating strategies that may prevent binding. Overall, submicron emitters present a promising approach to preserve membrane protein interactions during native MS while reducing nonspecific adducts.
Improving ESI with Charge Reduction.
The charge states of membrane protein ions influence their susceptibility to activation in the gas phase. Higher charge states increase Coulombic repulsion, potentially causing local unfolding and loss of noncovalent interactions. For example, DDM, one of the earliest detergents used for native MS, is no denaturing in solution but promotes higher charge states that can partially unfold membrane proteins in the gas phase.8,43 To combat this effect, charge-reducing methods can be used to better preserve noncovalent interactions and native membrane protein folding.90–93 Recently, several novel types of charge-reducing reagents have been shown to be effective for native MS of membrane proteins.
Petroff et al. used alkali metal acetate salts at low mill-molar concentrations for charge reduction of membrane protein ion channels.94 They discovered that charge reduction was dependent on the detergent, showing the highest reduction with polyethylene glycol (PEG)-based detergents and the least effect with LDAO. However, one limitation in alkali metal salt charge reduction is the peak broadening that accompanies higher additive concentrations due to adduction.
Following the observation that the chemical chaperone trimethylamine-N-oxide (TMAO) causes charge reduction,95 Patrick et al. reported potent charge reduction for three membrane proteins using TMAO.96 Native IM-MS analysis of membrane proteins with TMAO showed compact conformations, even under higher energy regimes normally associated with unfolding. Furthermore, TMAO charge reduction extended the spacing between adjacent charge states, allowing more bound lipids to be resolved than with typical conditions.
Inspired by the use of TMAO and imidazole91 for charge reduction of membrane proteins, Townsend et al. investigated the efficacy of several imidazole derivatives for soluble proteins, membrane proteins, and nanodisc complexes with antimicrobial peptides.97 Although TMAO provided the greatest charge reduction for membrane proteins in C8E4, imidazole derivatives with hydrophobic alkyl substituents in the 2-position provided more effective charge reduction than imidazole and showed narrower charge state distributions than TMAO. Furthermore, these imidazole derivatives were effective for charge reduction of nanodiscs, which could not be resolved with TMAO.
Lyu et al. recently investigated polyamines and cyclic amines for charge reduction by native IM-MS.98 Charge reduction was dependent on detergent, showing greater effect with C8E4 than DDM, and varied between several soluble proteins. Aliphatic polyamines such as spermine and spermidine provided significant charge reduction like TMAO but with several advantages. Similar charge reduction was achieved at concentrations 5 to 10-fold less than required for TMAO. Furthermore, these polyamines did not adduct to membrane proteins, minimizing broadening of mass spectral peaks.
Together, these studies show that small modifications to nano-ESI conditions can significantly improve native MS analysis of membrane proteins. We expect that further research into nondenaturing ionization conditions will continue to yield inexpensive and convenient strategies for preserving fragile membrane protein interactions and studying more complex systems.
Towards Native MS Imaging: DESI and LESA.
Mass spectrometry imaging (MSI) is used to profile the spatial distribution of molecules within biological substrates, such as thin tissue sections.99 Thus, an exciting goal is to extend native MSI to study the spatial distribution and interactions of membrane proteins in tissue. Recently, there has been progress in applications of desorption electrospray ionization (DESI) and liquid extraction surface analysis (LESA) for membrane protein analysis.
In DESI, a stream of solvent is electrosprayed towards an analyte-containing surface for desorption and delivery into the mass spectrometer.100 Ambrose et al. developed a native DESI-MS platform with the goal of enabling high-throughput membrane protein studies.101 This study demonstrated that soluble and membrane proteins could be liberated from clean surfaces while retaining native-like interactions. The binding of several ligands, including peptides, lipids, and antibiotics, to membrane proteins was observed after adding these ligands to the desorption spray. Although promising, native DESI-MS of membrane proteins has not been reported since this initial study, potentially owing to the sensitivity of DESI to geometric parameters and other difficulties in DESI of larger proteins, especially in the context of complex biological samples.102 However, a new Waters DESI source has shown initial promise for soluble protein complexes, and it may prove suitable for membrane proteins.103
LESA is a twist on ESI that allows for molecules to be analyzed from surface substrates.104 A robot arm lowers a solvent droplet onto a surface to form a liquid microjunction, allowing analytes to distribute between the substrate and the droplet. The arm then re-aspirates the analyte-containing droplet for direct infusion via ESI or nano-ESI. Cooper and coworkers demonstrated the feasibility of LESA-MS for membrane proteins, detecting intact trimeric membrane protein AmtB from a clean glass surface.105 Experiments on tissue samples showed that C8E4 detergent improved native protein extraction, but membrane protein complexes have not yet been detected in tissue to our knowledge, potentially due to their low natural abundance.106,107 Similar liquid sampling methods such as nano-DESI108 and Flowprobe sampling109 may also prove useful in native MSI of membrane proteins.
For both DESI and LESA, native membrane protein analysis has only been shown for clean glass substrates and further method development, including potentially ion mobility and top-down proteomics, may be necessary to achieve analysis of endogenous membrane proteins from tissue. However, these alternative ambient ionization techniques are uniquely equipped to advance high-throughput studies and directly analyze membrane proteins from biological substrates.
Laser-Based Ionization Approaches.
MS of intact proteins can also be performed using laser-based ionization techniques. Matrix-assisted laser desorption/ionization (MALDI) has been limited for native MS because it typically uses denaturing sample preparation conditions such as organic matrices and drying, requiring covalent crosslinking to stabilize noncovalent complexes for analysis.110 Recently, studies have demonstrated the capability for MALDI to preserve non-covalent interactions for proteins using liquid matrices.111,112 Other laser-based methods that have been shown to preserve native-like protein features include desorption by impulsive vibrational excitation (DIVE)113 and laser electrospray ionization (LEMS),114 but these techniques have not been applied to membrane proteins to our knowledge.
LILBID involves the generation of ions from aqueous droplets using an IR laser.115–117 Like MALDI, the ions generated by LILBID typically have less charge than in nano-ESI. While nano-ESI generally relies on collisional activation after ionization, LILBID uses the power of the IR laser to tune the degree of activation. Morgner and coworkers compared the performance of LILBID to nano-ESI for native MS of a range of membrane proteins.118 Compared to nano-ESI, LILBID was more tolerant to non-volatile buffers and salt and caused less unintended oligomer dissociation. The main disadvantage of LILBID is the low mass resolution when compared to nano-ESI, which is likely due to incomplete desolvation and the higher m/z values that result from only slightly charged analytes. Thus, improving instrumentation for LILBID-MS is an important focus for future membrane protein studies.
Ion Activation for Structural Elucidation
Following ionization, the next step in the MS experiment is ion activation, where several recent advances have been made in membrane protein analysis. Activation can be used to break noncovalent or covalent bonds, which we will refer to as dissociation and fragmentation, respectively. Ion activation methods in native MS have been covered in several excellent reviews,13,119,120 so we will focus here on new developments for membrane proteins.
Dissociating Membrane Protein Complexes by SID.
Subunits can be released from protein assemblies using controlled activation that disrupts noncovalent interactions without breaking covalent bonds. CID is the most common technique and is usually necessary to desolvate complexes and remove detergents. It can also be used to disrupt protein complexes to identify subunit composition and stoichiometry. Complementing CID, surface-induced dissociation (SID), pioneered for native MS by Wysocki and coworkers, involves a fast deposition of energy that breaks the weakest noncovalent interactions without global unfolding of protein subunits.121 Thus, SID can be used to study subunit architecture and connectivity. Harvey et al. reported the first use of SID with membrane proteins and showed that membrane proteins dissociate into subcomplexes that match their solution structure and can retain lipids after SID.122 Alternative dissociation methods to CID, including SID, IRMPD,123 and UVPD, which was recently shown to dissociate large protein complexes at low fluences,124 will certainly be useful to study the subunit architecture and structure of membrane protein complexes.
Top-Down: Fragmenting Membrane Proteins.
Top-down MS uses backbone fragmentation of proteins to reveal covalent and noncovalent modifications, complex organization, and primary sequence. Denaturing top-down and bottom-up proteomics are valuable for membrane proteins, and have been reviewed by Kar et al.125 Here, we focus on recent developments for top-down MS under native conditions, which according to recent standardized terminology, can fall under “complex-down” or “native top-down”.63
In complex-down MS, dissociated subunits are subjected to further activation for backbone fragmentation. Ro et al. used complex-down MS to identify post-translational modifications (PTMs) and the stoichiometry of copper binding to particulate methane monooxygenase (pMMO).126 Here, pMMO was ejected from either nanodiscs (described above) or micelles, dissociated into protein monomers, and then fragmented by CID to localize copper binding sites in the ejected monomers.
Native top-down involves gas-phase backbone cleavage without perturbing higher-order structure and interactions, which relies on fast electronic excitation without vibrational redistribution. Recently, Sipe et al. used a combination of native top-down and complex-down with ultraviolet photodissociation (UVPD) to investigate the effect of lipid binding on the conformational dynamics of the mechanosensitive channel of large conductance (MscL).127 Up to five bound lipids of phosphatidylcholine (PC), phosphatidylinositol (PI), and cardiolipin (CDL) lipids were observed by native MS. However, insufficient holo-fragment ion signal corresponding to MscL-phospholipid binding precluded localization of lipid binding, potentially due to photodissociation of lipids alongside the protein or difficulty finding low intensity fragment ions in complex spectra. Interestingly, the effect of lipid binding on the structural dynamics of MscL was probed using differences in fragment ion abundances (Figure 5). CDL and PI reduced backbone cleavages, suggesting increased stabilization or rigidity, whereas PC increased backbone cleavages, suggesting increased flexibility.
Figure 5.
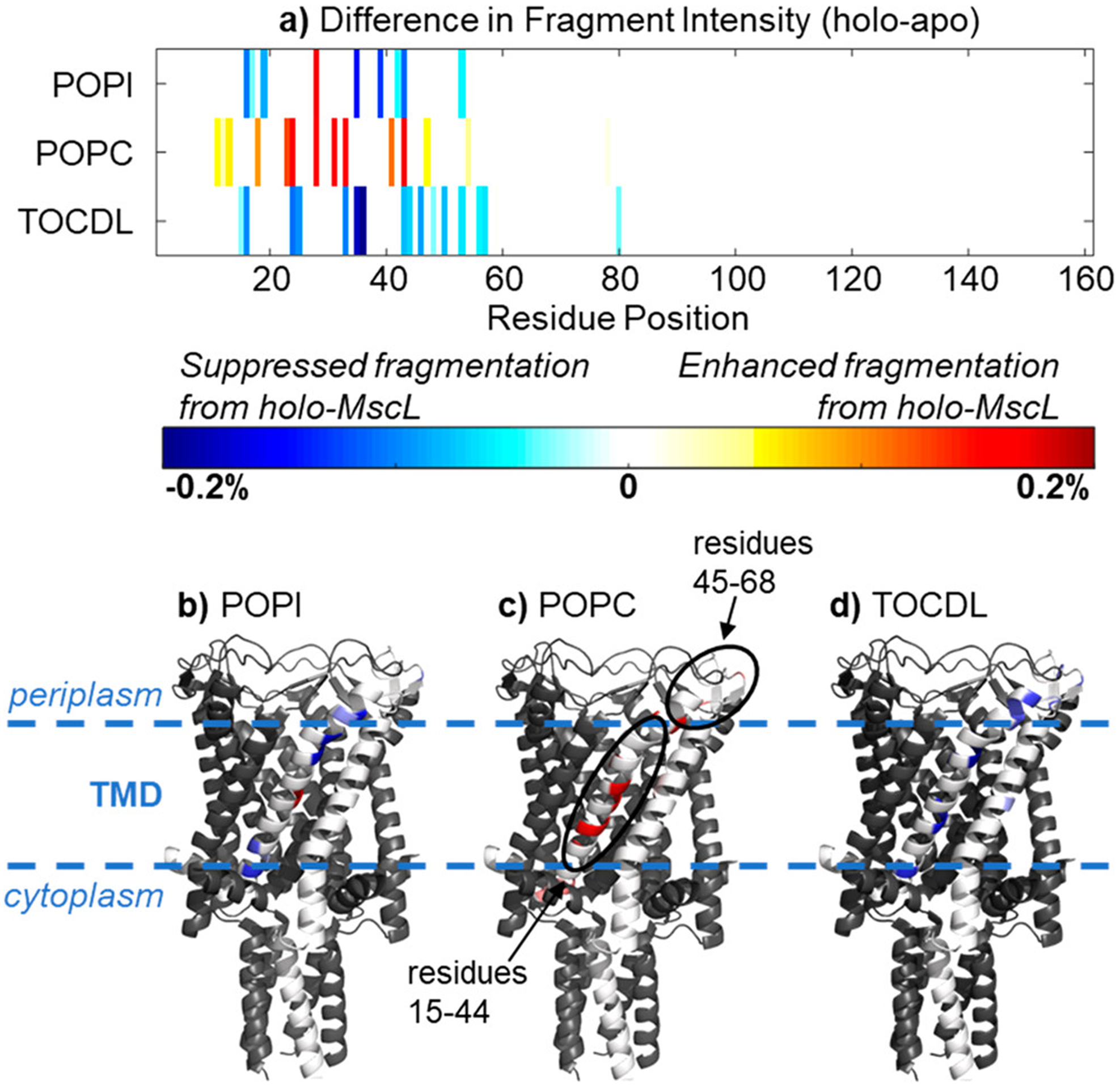
Differences in the UVPD fragment intensity between apo-MscL and holo-MscL bound to five lipids plotted by (A) residue position and (B-D) on a subunit of the crystal structure. The circled regions showed the most significant variations between apo-MscL and holo-MscL. Reprinted from Sipe, S. N.; Patrick, J. W.; Laganowsky, A.; Brodbelt, J. S. Enhanced Characterization of Membrane Protein Complexes by Ultraviolet Photodissociation Mass Spectrometry. Anal. Chem. 2020, 92, 899–907 (ref 127). Copyright 2020 American Chemical Society.
Together, these studies demonstrate the potential of native top-down and complex-down to study membrane protein structure and interactions. Because significant energy must be deposited to remove detergents or eject membrane proteins from membrane mimetics, ionization and sample preparation continue to be challenging for top-down analysis of membrane proteins. Furthermore, detergents and lipids may interfere with detecting peptide fragments. Advances in MS instrumentation, including new approaches to ion activation and post-fragmentation ion mobility, and membrane protein sample preparation will continue to drive improvements in top-down analysis of native membrane protein complexes.
Advances in Instrumentation
Ion Mobility: Probing Gas-Phase Conformations.
Ion mobility (IM) can be used to separate ions based on their gas-phase mobility through an inert gas.128,129 Thus, it can provide the collision cross section (CCS) for a membrane protein to show complex architecture and conformation.44 Tandem IM-MS (IM-MS/MS) can also separate species with the same m/z values, cleaning up spectra significantly. Finally, IM-MS collision-induced unfolding (CIU) experiments can be used to probe the stability and subunit organization of membrane proteins, exploring how lipid binding stabilizes membrane protein structure.8,130
Recently, Ruotolo and coworkers resolved translocator protein (TSPO)-ligand complexes and classified binding locations by CIU.131 They similarly used CIU to build a classification scheme for phenotype variants of a voltage-gated potassium channel voltage sensing domain.132 These methods are promising for classifying ligand binding and unknown variants for membrane proteins, but further research is necessary to understand the principles of gas-phase unfolding of membrane protein complexes, especially how membrane mimetics and detergents affect CIU.
Recently, Orbitrap mass analyzers have also been interfaced with ion mobility. Russell and coworkers developed a reverse-entry ion source (REIS)133 for high-mass Orbitrap instruments and interfaced this with a Fourier transform drift tube ion mobility mass spectrometer, which enabled IM analysis despite the relatively slow scan speed of the Orbitrap.134,135 Using the REIS instrument,133 mass spectra of AmtB with and without lipid binding showed comparable mass resolution to the unmodified instrument and improved mass resolving power over traditional time-of-flight (ToF) instruments, which was used to resolve three unique lipids bound to AmtB (Figure 6). These advances, along with recent work on interfacing field asymmetric ion mobility spectrometry (FAIMS) for native MS of soluble proteins,136 show that even slower mass analyzers like Orbitraps can benefit from IM-MS, which will ultimately improve analysis of membrane proteins.
Figure 6.
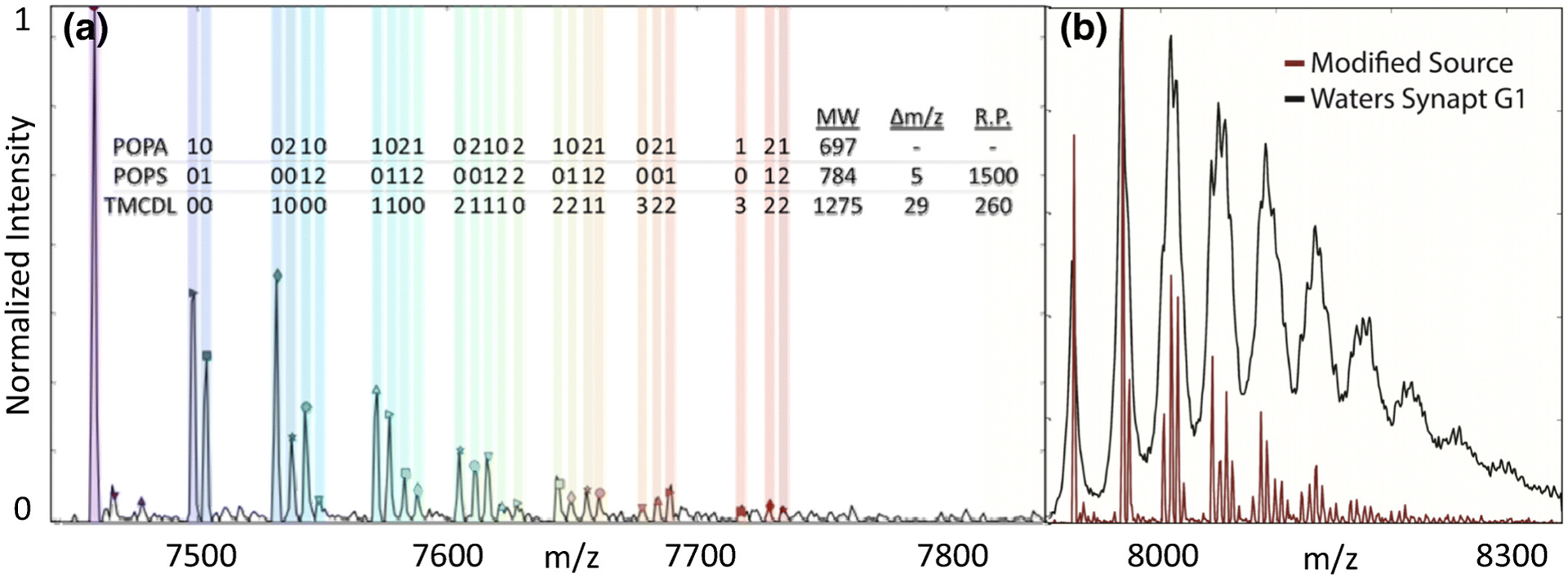
(A) Mass spectrum of AmtB bound to a mixture of CDL, PS, and phosphatidic acid (PA) lipids, which each show distinct peaks in the high-resolution spectrum. (B) ToF data (black) lacked resolution to distinguish the different types of bound lipids present in the higher-resolution Orbitrap data (red). Reprinted from Poltash, M. L.; McCabe, J. W.; Patrick, J. W.; Laganowsky, A.; Russell, D. H. Development and Evaluation of a Reverse-Entry Ion Source Orbitrap Mass Spectrometer. J. Am. Soc. Mass Spectrom. 2019, 30, 192–198 (ref 133). Copyright 2019 American Chemical Society.
Mass Analyzers.
As the demand for native MS has increased in recent years, so has the need for sensitive and high-resolution instrumentation. Early native MS of membrane proteins relied mostly on ToF analyzers, due in part to their wide mass range and fast scan speed that made them well suited for IM-MS. Over the last five years, new high-mass Orbitrap instruments, first the Q-Exactive EMR and later the UHMR, have been key in studying large macromolecular complexes because of their improved effective resolution, potentially due to improved desolvation.35,137,138 Membrane protein analysis from a variety of complex systems, including nanodiscs and lipid vesicles described above, has relied on Orbitrap instruments.57,76
Recently, Mallis et al. developed a higher-resolution extended mass range quadrupole (Q)-ToF platform with a modified static nano-ESI emitter.139 Other modifications included increasing the length of the drift tube, decreasing the pressure within the analyzer, and narrowing the pulse width for gating ions into the analyzer. Notably, the enhanced resolving power was sufficient to reveal the presence of several small molecule β-mercaptoethanol (β-ME) adducts bound to AmtB, demonstrating the potential of this platform for characterizing ligand and lipid binding to membrane proteins. Further development of higher-resolution ToF instruments, such as the Waters cyclic ion mobility instrument,140 show promise for high-resolution membrane protein IM-MS, but no other published examples with membrane proteins are yet available. Fourier transform ion cyclotron resonance (FT-ICR) mass analyzers also continue to be used for native MS of membrane proteins.68,141 Recently, Campuzano, Loo, and coworkers demonstrated the competitive performance of a commercial SolariX FT-ICR mass spectrometer for transmission and detection of membrane protein complexes, such as AqpZ, compared to ToF and Orbitrap instruments.76
Finally, there have been several recent advances in charge detection mass spectrometry (CDMS) for studying heterogeneous macromolecular assemblies. CDMS relies on detecting the charge of individual ions, avoiding the problem of peak overlap caused by insufficient resolving power and heterogeneous assemblies. Jarrold and coworkers have driven this work on home-built instruments over the last decade,142,143 applying CDMS to study large and polydisperse intact membrane protein systems like the nuclear pore complex,27 lipoprotein particles,144 and exosomes.145 Williams and coworkers have recently contributed to improving analytical methods on home-built CDMS instruments.146–149 Finally, CDMS on commercial Orbitrap instruments has been demonstrated for measuring complex proteoform mixtures and megadalton biomolecular assemblies.150,151 Together, these CDMS studies show promise for native MS of large, heterogenous membrane protein assemblies that could not be resolved with conventional approaches.
Advances in Experimental Design
Protein-Lipid Interactions.
Lipids surrounding membrane proteins can play critical structural and functional roles.152–154 Although detecting lipid binding to membrane proteins is challenging with conventional methods, native MS is particularly well suited for studying protein-lipid interactions because it is label free, can quantify binding of multiple species simultaneously, and can distinguish between lipids based on their masses. The use of native MS to study membrane protein-lipid interactions has been previously reviewed,16,155 so we will focus here on recent analytical developments, especially new experimental designs.
Laganowsky and coworkers have developed novel approaches to study the thermodynamics and allostery of lipid binding using native MS. First, they modified the ESI source to control the temperature of the analyte solution and the surrounding air.156 AmtB was incubated with phospholipids at different concentrations and temperatures, and the relative amounts of bound lipids were quantified by native MS. This approach enabled measurement of the thermodynamics and equilibria of individual lipid binding events. They then investigated how lipid binding allosterically affected AmtB binding to GlnK, a regulatory protein.157 Both lipid headgroups and chain lengths modulated AmtB-GlnK interactions. Laganowsky and coworkers similarly investigated allosteric regulation of protein-lipid interactions with AmtB by titrating binary lipid mixtures.158 They discovered that CDL and phosphatidylethanolamine (PE) displayed the strongest allosteric modulation, demonstrating that lipids can affect the binding of other lipids at remote sites. Finally, native MS suggested that rotation of the GIRK2 cytoplasmic domain allosterically modulates remote phosphatidyl-inositide-phosphate (PIP) binding sites.159 Overall, these methods provide novel experimental designs to study the allosteric regulation of protein-protein and protein-lipid interactions by lipids.
Bolla et al. recently developed a native MS approach for measuring the specificity of lipid binding based on competition with increasing amounts and exposure of detergents in solution.160 Following addition of E coli. polar lipids to the membrane protein presenilin homologue (PSH), increasing the concentration of nonyl-glucoside (NG) detergent gradually removed each lipid at comparable rates, consistent with predicted nonspecific interactions. In contrast, removal of CDL from leucine transporter (LeuT) required a higher NG concentration and longer incubation time than for removal of PG, suggesting that specifically-bound CDL is resistant to detergent exchange. Finally, only the second and third lipid-II adducts to MurJ could be removed by detergents, revealing a single specific lipid-II that could not be removed. Overall, this experimental strategy distinguishes the specificity of lipid interactions for a range of membrane proteins.
Another approach to identify lipid specificity involves combining native MS with MD simulations and mutagenesis. Politis and coworkers investigated the role of lipid binding in stabilizing the functional dimeric form of UapA, a eukaryotic purine transporter.161 Conventional LC-MS lipidomics and CID with native MS were used to identify co-purified PI and PE lipids that stabilized the UapA dimer. MD simulations predicted a PI binding site at the dimer interface, and disruption of the lipid binding site by mutagenesis resulted in loss of transport activity, demonstrating that specific UapA-lipid interactions are crucial for stabilizing the functional dimeric form. They similarly probed the role of phospholipids in either the oligomerization of BOR1p, a boron transporter,162 or the function of the SecA-SecYEG complex, a secretory translocase.163 Overall, these studies demonstrate the promise in coupling native MS with predictive MD, mutagenesis, and functional biochemical analysis to study the structure and functional effect of lipid binding to specific sites on membrane proteins.
Native-omics.
A key challenge in structural biology is determining the chemical identity of binding partners for membrane proteins. A significant number of data sets from crystallography and cryo-EM studies have poorly resolved ligand densities and thus unknown binding partners, especially from endogenous metabolites that are retained during purification. Although native MS can help detect the stoichiometry and mass of unknown bound ligands, the mass alone is often insufficient to unambiguously identify the molecule. Thus, native MS can be paired with conventional tandem MS to identify molecules extracted from the sample.36,152,164 For example, MS/MS-based lipidomics and native MS recently showed that PI was the predominant lipid bound to the Get1-Get2/WRB-CAML heterotetramer165 and identified lipids that co-purified with MurJ and FtsW.166 Although conventional MS/MS is useful for identification of small molecules co-purified with membrane proteins, information is lost when it is decoupled with the direct binding measurements of native MS through solution-phase extraction.
Recent work from Gault et al. sought to interface native MS with small molecule fragmentation to directly identify bound molecules ejected after native MS.167 This “native-omics” approach combines native MS with traditional “-omics” MS/MS (proteomics, lipidomics, and metabolomics) to better capture and define unknown ligands for membrane proteins. The main challenges in coupling these approaches is the instrumentation—the mass spectrometer must be capable of detecting both intact protein-ligand complexes of tens to hundreds of kilodaltons and fragmented ligands of tens to hundreds of daltons. Furthermore, the instrument needs the capability to perform multiple rounds of MS to progressively dissociate the protein-ligand assembly and yield ligand partners for fragmentation. This new integrative approach used a higher-mass modified tribrid Orbitrap mass spectrometer to first detect the intact protein-ligand complex, dissociate the complex to isolate the free ligand, and then fragment the ligand for identification with database matching (Figure 7). Using this approach, they determined the chemical identity of peptides, lipids, and drugs bound to membrane proteins. Native-omics was applied to TSPO to identify a series of homologous PE lipids that fit an ambiguous electron density from the X-ray structure. This novel experimental approach demonstrates that combining several separate experiments into a single instrumental workflow can solve critical challenges in identifying membrane protein interactions and complement conventional structural biology approaches.
Figure 7.
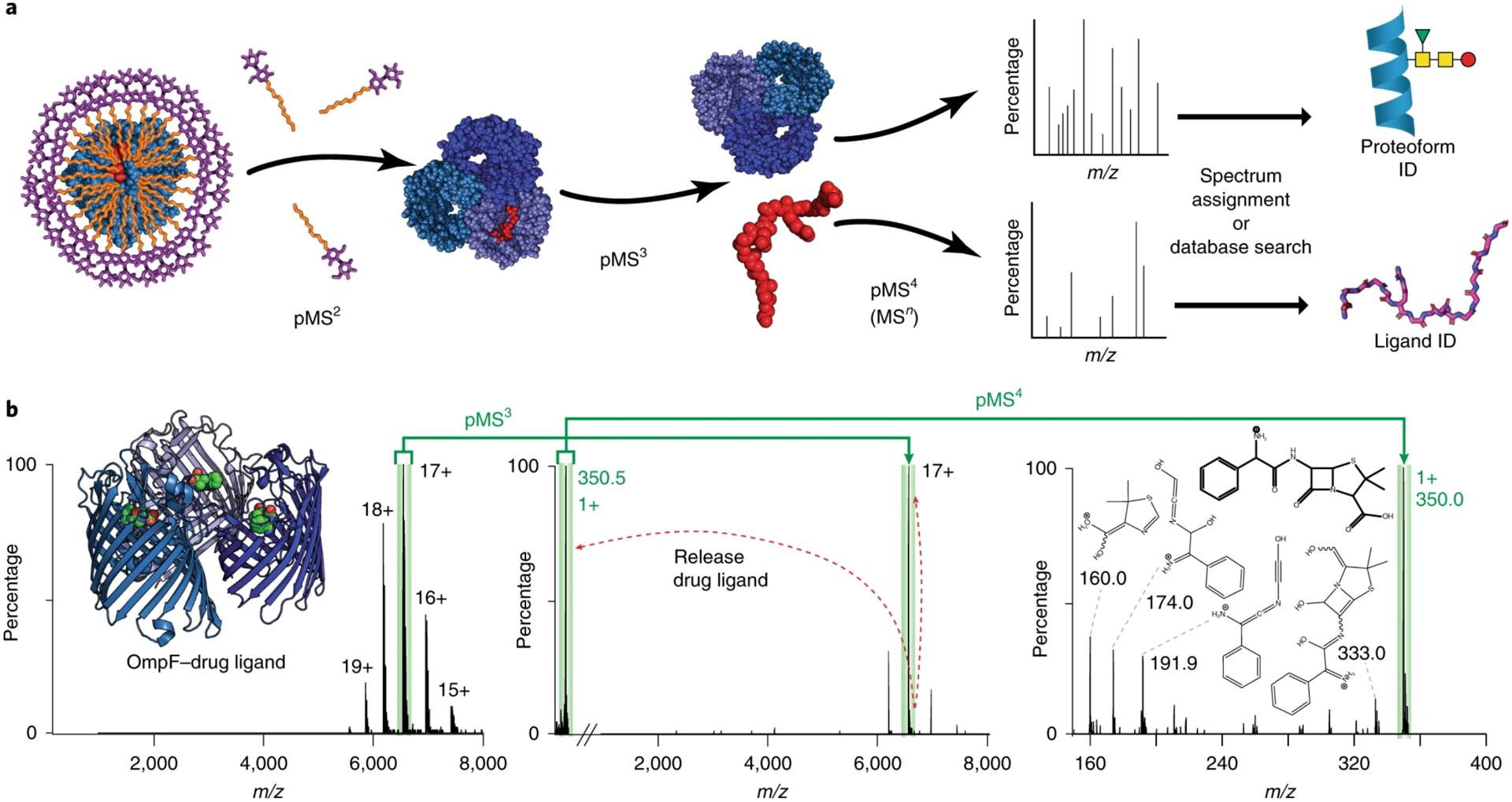
(A) Schematic of native-omics workflow showing ejection of membrane proteins from micelles, dissociation of bound ligands, and fragmentation of isolated ligands or protein complexes for identification. (B) Tandem MS process for identifying drug ligand bound to OmpF. Reprinted by permission from Springer Nature. Nature Methods, Gault, J.; Liko, I.; Landreh, M.; Shutin, D.; Bolla, J. R.; Jefferies, D.; Agasid, M.; Yen, H. Y.; Ladds, M.; Lane, D. P.; Khalid, S.; Mullen, C.; Remes, P. M.; Huguet, R.; McAlister, G.; Goodwin, M.; Viner, R.; Syka, J. E. P.; Robinson, C. V. Combining native and ‘omics’ mass spectrometry to identify endogenous ligands bound to membrane proteins. Nat. Methods 2020, 17, 505–508 (ref 167). Copyright 2020.
G-Protein Coupled Receptors.
GPCRs are critical for cellular processes and represent the largest fraction of membrane protein drug targets. However, the effects of small molecule binding on GPCR structure and signaling still remain poorly understood. Recently, Robinson and coworkers used native MS to investigate the selectivity and effect of lipid binding on three class A GPCRs: adenosine A2A receptor (A2AR), β1 adrenergic receptor (β1AR), and neurotensin receptor type 1 (NTSR1).49 Mini-G proteins, constructs that mimic the Gα subunit, were used to study the impact of lipids on G-protein coupling and selectivity.168 Comparing peak intensities for binding of different PI lipids showed that phosphatidylinositol-4,5-bisphosphate (PIP2) bound with the highest affinity to each GPCR. A larger abundance of phosphatidylserine (PS) and PI lipids was observed bound to A2AR after G-protein coupling, suggesting a stabilizing role of these lipids for active GPCR complexes.
Using mutagenesis to disrupt NTSR1 residues that bind PIP2, they developed a native MS strategy to localize lipid binding by incubating an equimolar solution of wild-type and mutant NTSR1 with PIP2. Preferential binding sites were identified by a decrease in the intensity of the PIP2bound peak for mutant NTSR1 relative to the PIP2-bound peak for wild-type NTSR1. Binding of PIP2 also enhanced coupling of mini-Gs to β1AR. In contrast, PS binding only slightly increased the coupling of mini-Gs to β1AR, and other mini-G proteins did not show strong PIP2 binding, suggesting specific interactions with PIP2 drive coupling of β1AR and mini-Gs. Overall, these data suggest that PIP2 is an allosteric modulator that enhances selectivity and stability of G-protein coupling. These studies highlight the power of native MS to interrogate the complex interactions of GPCRs.
Outlook and Future Perspectives
As shown above, there have been outstanding analytical advances in native MS of membrane proteins over the last few years that have built on broader advances in native MS and membrane protein biochemistry. Looking to the future, one key question is how to shift towards using native MS for studying eukaryotic membrane proteins. Many native MS structural studies have been performed using recombinant proteins from simple expression systems such as E. coli. Bacterial systems offer relatively fast expression and have limited PTMs to complicate the spectra. However, bacterial expression may be impossible or undesirable for eukaryotic membrane proteins, which often have extensive glycosylation. As research turns to eukaryotic membrane proteins with complex proteoforms, significant analytical developments will be needed to reduce heterogeneity in sample preparation and improve the instrumentation to handle complex samples. Furthermore, eukaryotic membrane proteins may be less stable, so new detergents and membrane mimetic strategies may be required to preserve the activity of these delicate proteins for native MS.
A key factor in these advances are instrumental developments in ionization, activation, ion mobility, and mass analyzers, especially in novel combinations of these methods. New instruments, such as the higher-mass tribrid Orbitrap Eclipse167 and Cyclic IMS,140 that allow multiple stages of activation, selection, and mass analysis will undoubtedly enable novel experiments. Online separation and flow injections systems are also promising for advancing native MS studies. An online buffer exchange protocol was recently developed for rapid screening of soluble proteins,169 and online size exclusion chromatography has been coupled to native MS for improved automation of CIU experiments.170 However, these strategies require further development for membrane proteins as high-flow methods may reduce sensitivity and may disrupt fragile membrane protein interactions.
Finally, we anticipate an increasing use of native MS to complement cryo-EM, X-ray crystallography, and NMR. Native MS is uniquely suited for identifying unknown interactions, characterizing polydisperse complexes, and quickly screening samples prior to structural analysis.171 The use of native MS to study membrane protein-lipid interactions is especially powerful and fills a critical gap in existing technologies. In addition, native MS will likely continue to be integrated with other structural MS techniques, such as hydrogen-deuterium exchange,172 crosslinking,173 fast photochemical oxidation of proteins,174 and recently-developed lipid exchange-mass spectrometry61 for unique membrane protein studies. Native MS is rapidly growing as applications, methodologies, and instrumentation expand, fostering a greater depth of information that can be gained for membrane protein analysis. We anticipate that native MS will continue to be an indispensable tool for solving challenging analytical problems surrounding membrane proteins and their interactions.
ACKNOWLEDGMENTS
The authors thank Frank Sobott for helpful suggestions on the manuscript. Funding was provided by the National Institute of General Medical Sciences and National Institutes of Health under Award Number R35 GM128624 to M.T.M. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
Biographies
James E. Keener earned his B.S. in chemistry from the University of California, Merced. He is currently a Ph.D. candidate in Prof. Michael Marty’s lab at the University of Arizona, where he received the Carl S. Marvel Memorial Fellowship. His research focuses on using native mass spectrometry and nanodiscs to study membrane protein-lipid interactions.
Guozhi Zhang earned her B.S. in chemistry from Kutztown University Pennsylvania. She is currently a Ph.D. candidate in Prof. Michael Marty’s lab at the University of Arizona, where her research focuses on lipid exchange in nanodiscs to study remodeling of lipids surrounding membrane proteins to find the optimal lipid environment.
Michael T. Marty earned his B.A. in chemistry and mathematics from St. Olaf College and his Ph.D. in chemistry from the University of Illinois Urbana-Champaign with Prof. Stephen Sligar. After postdoctoral research with Prof. Dame Carol Robinson at the University of Oxford, he joined the faculty at the University of Arizona, where he is currently an Assistant Professor in the Department of Chemistry and Biochemistry. His research focuses on interfacing nanodiscs with mass spectrometry to study interactions of membrane proteins, transmembrane peptides, and lipids.
REFERENCES
- (1).Krogh A; Larsson B; von Heijne G; Sonnhammer EL J. Mol. Biol 2001, 305, 567–580. [DOI] [PubMed] [Google Scholar]
- (2).Almen MS; Nordstrom KJ; Fredriksson R; Schioth HB BMC Biol 2009, 7, 50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (3).Overington J; Al-Lazikani B; Hopkins A Nat. Rev. Drug Discov 2006, 5, 993–996. [DOI] [PubMed] [Google Scholar]
- (4).Wagner S; Bader ML; Drew D; de Gier JW Trends Biotechnol. 2006, 24, 364–371. [DOI] [PubMed] [Google Scholar]
- (5).Wagner S; Baars L; Ytterberg AJ; Klussmeier A; Wagner CS; Nord O; Nygren PA; van Wijk KJ; de Gier JW Mol. Cell. Proteomics 2007, 6, 1527–1550. [DOI] [PubMed] [Google Scholar]
- (6).Gubellini F; Verdon G; Karpowich NK; Luff JD; Boel G; Gauthier N; Handelman SK; Ades SE; Hunt JF Mol. Cell. Proteomics 2011, 10, M111 007930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (7).Zweers JC; Wiegert T; van Dijl JM Appl. Environ. Microbiol 2009, 75, 7356–7364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (8).Laganowsky A; Reading E; Allison TM; Ulmschneider MB; Degiacomi MT; Baldwin AJ; Robinson CV Nature 2014, 510, 172–175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (9).Pliotas C; Dahl AC; Rasmussen T; Mahendran KR; Smith TK; Marius P; Gault J; Banda T; Rasmussen A; Miller S; Robinson CV; Bayley H; Sansom MS; Booth IR; Naismith JH Nat. Struct. Mol. Biol 2015, 22, 991–998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (10).Martfeld AN; Rajagopalan V; Greathouse DV; Koeppe RE, 2nd Biochim. Biophys. Acta, Biomembr 2015, 1848, 1849–1859. [DOI] [PubMed] [Google Scholar]
- (11).Hsia C-Y; Richards MJ; Daniel S Anal. Methods 2015, 7, 7076–7094. [Google Scholar]
- (12).Kaur U; Johnson DT; Chea EE; Deredge DJ; Espino JA; Jones LM Anal. Chem 2019, 91, 142–155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (13).Boeri Erba E; Signor L; Petosa CJ Proteomics 2020, 222, 103799. [DOI] [PubMed] [Google Scholar]
- (14).Sahin C; Reid DJ; Marty MT; Landreh M Biochem. Soc. Trans 2020, 48, 547–558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (15).Robinson CV Proc. Natl. Acad. Sci. U. S. A 2019, 116, 2814–2820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (16).Bolla JR; Agasid MT; Mehmood S; Robinson CV Annu. Rev. Biochem 2019, 88, 85–111. [DOI] [PubMed] [Google Scholar]
- (17).Bender J; Schmidt C Biol. Chem 2019, 400, 813–829. [DOI] [PubMed] [Google Scholar]
- (18).Calabrese AN; Radford SE Methods 2018, 147, 187–205. [DOI] [PubMed] [Google Scholar]
- (19).Kaur U; Meng H; Lui F; Ma R; Ogburn RN; Johnson JHR; Fitzgerald MC; Jones LM J. Proteome Res 2018, 17, 3614–3627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (20).Leney AC; Heck AJ J. Am. Soc. Mass Spectrom 2017, 28, 5–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (21).Azegami N; Saikusa K; Todokoro Y; Nagadoi A; Kurumizaka H; Nishimura Y; Akashi S Biochemistry 2013, 52, 5155–5157. [DOI] [PubMed] [Google Scholar]
- (22).Saikusa K; Osakabe A; Kato D; Fuchigami S; Nagadoi A; Nishimura Y; Kurumizaka H; Akashi S Anal. Chem 2018, 90, 8217–8226. [DOI] [PubMed] [Google Scholar]
- (23).Rostom AA; Fucini P; Benjamin DR; Juenemann R; Nierhaus KH; Hartl FU; Dobson CM; Robinson CV Proc. Natl. Acad. Sci. U. S. A 2000, 97, 5185–5190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (24).van de Waterbeemd M; Fort KL; Boll D; ReinhardtSzyba M; Routh A; Makarov A; Heck AJ Nat. Methods 2017, 14, 283–286. [DOI] [PubMed] [Google Scholar]
- (25).Walker LR; Marzluff EM; Townsend JA; Resager WC; Marty MT Anal. Chem 2019, 91, 9284–9291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (26).Walker LR; Marty MT Biochemistry 2020, 59, 2135–2142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (27).Kim SJ; Fernandez-Martinez J; Nudelman I; Shi Y; Zhang W; Raveh B; Herricks T; Slaughter BD; Hogan JA; Upla P; Chemmama IE; Pellarin R; Echeverria I; Shivaraju M; Chaudhury AS; Wang J; Williams R; Unruh JR; Greenberg CH; Jacobs EY, et al. Nature 2018, 555, 475–482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (28).Ziarek JJ; Baptista D; Wagner GJ Mol. Med 2018, 96, 18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (29).Ishchenko A; Gati C; Cherezov V Curr. Opin. Struct. Biol 2018, 51, 44–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (30).Cheng Y Cell 2015, 161, 450–457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (31).Loura LM; Prieto M Front. Physiol 2011, 2, 82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (32).Patching SG Biochim. Biophys. Acta, Biomembr 2014, 1838, 43–55. [DOI] [PubMed] [Google Scholar]
- (33).Sahu ID; Lorigan GA J. Phys. Chem. Biophys 2015, 5, 188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (34).Liko I; Allison TM; Hopper JT; Robinson CV Curr. Opin. Struct. Biol 2016, 40, 136–144. [DOI] [PubMed] [Google Scholar]
- (35).Gault J; Donlan JA; Liko I; Hopper JTS; Gupta K; Housden NG; Struwe WB; Marty MT; Mize T; Bechara C; Zhu Y; Wu B; Kleanthous C; Belov M; Damoc E; Makarov A; Robinson CV Nat. Methods 2016, 13, 333–336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (36).Gupta K; Donlan JAC; Hopper JTS; Uzdavinys P; Landreh M; Struwe WB; Drew D; Baldwin AJ; Stansfeld PJ; Robinson CV Nature 2017, 541, 421–424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (37).Laganowsky A; Reading E; Hopper JTS; Robinson CV Nat. Protoc 2013, 8, 639–651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (38).Stetsenko A; Guskov A Crystals 2017, 7, 197. [Google Scholar]
- (39).Kalipatnapu S; Chattopadhyay A IUBMB Life 2005, 57, 505–512. [DOI] [PubMed] [Google Scholar]
- (40).Cleary SP; Prell JS ChemPhysChem 2019, 20, 519–523. [DOI] [PubMed] [Google Scholar]
- (41).Wilson JW; Rolland AD; Klausen GM; Prell JS Anal. Chem 2019, 91, 10204–10211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (42).Wilson JW; Donor MT; Shepherd SO; Prell JS J. Am. Soc. Mass Spectrom 2020, 31, 1751–1754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (43).Reading E; Liko I; Allison TM; Benesch JLP; Laganowsky A; Robinson CV Angew. Chem., Int. Ed 2015, 54, 4577–4581. [DOI] [PubMed] [Google Scholar]
- (44).Konijnenberg A; Yilmaz D; Ingólfsson HI; Dimitrova A; Marrink SJ; Li Z; Vénien-Bryan C; Sobott F; Koçer A Proc. Natl. Acad. Sci. U. S. A 2014, 111, 17170–17175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (45).Urner LH; Maier YB; Haag R; Pagel KJ Am. Soc. Mass Spectrom 2019, 30, 174–180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (46).Urner LH; Liko I; Yen HY; Hoi KK; Bolla JR; Gault J; Almeida FG; Schweder MP; Shutin D; Ehrmann S; Haag R; Robinson CV; Pagel K Nat. Commun 2020, 11, 564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (47).Lee S; Mao A; Bhattacharya S; Robertson N; Grisshammer R; Tate CG; Vaidehi NJ Am. Chem. Soc 2016, 138, 15425–15433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (48).Urner LH; Schulze M; Maier YB; Hoffmann W; Warnke S; Liko I; Folmert K; Manz C; Robinson CV; Haag R; Pagel K Chem. Sci 2020, 11, 3538–3546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (49).Yen HY; Hoi KK; Liko I; Hedger G; Horrell MR; Song W; Wu D; Heine P; Warne T; Lee Y; Carpenter B; Pluckthun A; Tate CG; Sansom MSP; Robinson CV Nature 2018, 559, 423–427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (50).Yen HY; Hopper JTS; Liko I; Allison TM; Zhu Y; Wang D; Stegmann M; Mohammed S; Wu B; Robinson CV Sci. Adv 2017, 3, e1701016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (51).Guo Y Crystals 2020, 10, 86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (52).Hutchison JM; Shih KC; Scheidt HA; Fantin SM; Parson KF; Pantelopulos GA; Harrington HR; Mittendorf KF; Qian S; Stein RA; Collier SE; Chambers MG; Katsaras J; Voehler MW; Ruotolo BT; Huster D; McFeeters RL; Straub JE; Nieh MP; Sanders CR J. Am. Chem. Soc 2020, 142, 1271512729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (53).Marty MT; Hoi KK; Robinson CV Acc. Chem. Res 2016, 49, 2459–2467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (54).Bayburt TH; Grinkova YV; Sligar SG Nano Lett 2002, 2, 853–856. [Google Scholar]
- (55).Denisov IG; Grinkova YV; Lazarides AA; Sligar SG J. Am. Chem. Soc 2004, 126, 3477–3487. [DOI] [PubMed] [Google Scholar]
- (56).Bayburt TH; Sligar SG Protein Sci 2003, 12, 2476–2481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (57).Marty MT; Hoi KK; Gault J; Robinson CV Angew.Chem., Int. Ed 2016, 55, 550–554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (58).Keener JE; Zambrano DE; Zhang G; Zak CK; Reid DJ; Deodhar BS; Pemberton JE; Prell JS; Marty MT J. Am. Chem. Soc 2019, 141, 1054–1061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (59).Hoi KK; Robinson CV; Marty MT Anal. Chem 2016, 88, 6199–6204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (60).Kostelic MM; Ryan AM; Reid DJ; Noun JM; Marty MT J. Am. Soc. Mass Spectrom 2019, 30, 1416–1425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (61).Zhang G; Keener JE; Marty MT Anal. Chem 2020, 92, 5666–5669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (62).Debruycker V; Hutchin A; Masureel M; Ficici E; Martens C; Legrand P; Stein RA; McHaourab HS; Faraldo-Gomez JD; Remaut H; Govaerts C Nat. Struct. Mol. Biol 2020, 27, 829–835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (63).Ro SY; Schachner LF; Koo CW; Purohit R; Remis JP; Kenney GE; Liauw BW; Thomas PM; Patrie SM; Kelleher NL; Rosenzweig AC Nat. Commun 2019, 10, 2675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (64).Henrich E; Lohr F; Pawlik G; Peetz O; Dotsch V; Morgner N; de Kroon AI; Bernhard F Biochemistry 2018, 57, 5780–5784. [DOI] [PubMed] [Google Scholar]
- (65).Carlson ML; Young JW; Zhao Z; Fabre L; Jun D; Li J; Li J; Dhupar HS; Wason I; Mills AT; Beatty JT; Klassen JS; Rouiller I; Duong F eLife 2018, 7, e34085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (66).Li J; Han L; Li J; Kitova EN; Xiong ZJ; Prive GG; Klassen JS J. Am. Soc. Mass Spectrom 2018, 29, 1493–1504. [DOI] [PubMed] [Google Scholar]
- (67).Marty MT Int. J. Mass Spectrom 2020, 458, 116436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (68).Campuzano ID; Li H; Bagal D; Lippens JL; Svitel J; Kurzeja RJ; Xu H; Schnier PD; Loo JA Anal. Chem 2016, 88, 12427–12436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (69).Cleary SP; Li H; Bagal D; Loo JA; Campuzano IDG; Prell JS J. Am. Soc. Mass Spectrom 2018, 29, 2067–2080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (70).Han L; Kitov PI; Li J; Kitova EN; Klassen JS Anal. Chem 2020, 92, 3923–3931. [DOI] [PubMed] [Google Scholar]
- (71).Ravula T; Hardin NZ; Ramamoorthy A Chem. Phys. Lipids 2019, 219, 45–49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (72).Overduin M; Esmaili M Appl. Sci 2019, 9, 1230. [Google Scholar]
- (73).Hellwig N; Peetz O; Ahdash Z; Tascon I; Booth PJ; Mikusevic V; Diskowski M; Politis A; Hellmich Y; Hanelt I; Reading E; Morgner N Chem. Commun 2018, 54, 13702–13705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (74).Hesketh SJ; Klebl DP; Higgins AJ; Thomsen M; Pickles IB; Sobott F; Sivaprasadarao A; Postis VLG; Muench SP Biochim. Biophys. Acta, Biomembr 2020, 1862, 183192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (75).Chorev DS; Tang H; Rouse SL; Bolla JR; von Kugelgen A; Baker LA; Wu D; Gault J; Grunewald K; Bharat TAM; Matthews SJ; Robinson CV Nat. Protoc 2020, 15, 1690–1706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (76).Chorev DS; Baker LA; Wu D; Beilsten-Edmands V; Rouse SL; Zeev-Ben-Mordehai T; Jiko C; Samsudin F; Gerle C; Khalid S; Stewart AG; Matthews SJ; Grunewald K; Robinson CV Science 2018, 362, 829–834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (77).Hirst J; Kunji ERS; Walker JE Science 2019, 366. [DOI] [PubMed] [Google Scholar]
- (78).Fitzgerald MC; Chernushevich I; Standing KG; Whitman CP; Kent SB Proc. Natl. Acad. Sci. U. S. A 1996, 93, 6851–6856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (79).Rostom AA; Robinson CV Curr. Opin. Struct. Biol 1999, 9, 135–141. [DOI] [PubMed] [Google Scholar]
- (80).Benesch JLP; Sobott F; Robinson CV Anal. Chem 2003, 75, 2208–2214. [DOI] [PubMed] [Google Scholar]
- (81).Susa AC; Lippens JL; Xia Z; Loo JA; Campuzano IDG; Williams ER J. Am. Soc. Mass Spectrom 2018, 29, 203–206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (82).Susa AC; Xia Z; Williams ER Angew. Chem., Int. Ed 2017, 56, 7912–7915. [DOI] [PubMed] [Google Scholar]
- (83).Susa AC; Xia Z; Williams ER Anal. Chem 2017, 89, 3116–3122. [DOI] [PubMed] [Google Scholar]
- (84).Xia Z; Williams ER Analyst 2018, 144, 237–248. [DOI] [PubMed] [Google Scholar]
- (85).Mortensen DN; Williams ER Anal. Chem 2016, 88, 9662–9668. [DOI] [PubMed] [Google Scholar]
- (86).Mortensen DN; Williams ER Analyst 2016, 141, 5598–5606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (87).Xia Z; Williams ER J. Am. Soc. Mass Spectrom 2018, 29, 194–202. [DOI] [PubMed] [Google Scholar]
- (88).Panczyk EM; Gilbert JD; Jagdale GS; Stiving AQ; Baker LA; Wysocki VH Anal. Chem 2020, 92, 2460–2467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (89).Anderson TH; Min Y; Weirich KL; Zeng H; Fygenson D; Israelachvili JN Langmuir 2009, 25, 6997–7005. [DOI] [PubMed] [Google Scholar]
- (90).Hopper JTS; Sokratous K; Oldham NJ Anal. Biochem 2012, 421, 788–790. [DOI] [PubMed] [Google Scholar]
- (91).Mehmood S; Marcoux J; Hopper JTS; Allison TM; Liko I; Borysik AJ; Robinson CV J. Am. Chem. Soc 2014, 136, 17010–17012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (92).Pacholarz KJ; Barran PE EuPa Open Proteomics 2016, 11, 23–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (93).Sun N; Soya N; Kitova EN; Klassen JS J. Am. Soc. Mass Spectrom 2010, 21, 472–481. [DOI] [PubMed] [Google Scholar]
- (94).Petroff JT, 2nd Tong A; Chen LJ; Dekoster GT; Khan F; Abramson J; Frieden C; Cheng WWL Anal. Chem 2020, 92, 6622–6630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (95).Gault J; Lianoudaki D; Kaldmae M; Kronqvist N; Rising A; Johansson J; Lohkamp B; Lain S; Allison TM; Lane DP; Marklund EG; Landreh MJ Phys. Chem. Lett 2018, 9, 4082–4086. [DOI] [PubMed] [Google Scholar]
- (96).Patrick JW; Laganowsky AJ Am. Soc. Mass Spectrom 2019, 30, 886–892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (97).Townsend JA; Keener JE; Miller ZM; Prell JS; Marty MT Anal. Chem 2019, 91, 14765–14772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (98).Lyu J; Liu Y; McCabe JW; Schrecke S; Fang L; Russell DH; Laganowsky A Anal. Chem 2020, 92, 11242–11249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (99).Caprioli RM; Farmer TB; Gile J Anal. Chem 1997, 69, 4751–4760. [DOI] [PubMed] [Google Scholar]
- (100).Cooks RG; Ouyang Z; Takats Z; Wiseman JM Science 2006, 311, 1566–1570. [DOI] [PubMed] [Google Scholar]
- (101).Ambrose S; Housden NG; Gupta K; Fan J; White P; Yen HY; Marcoux J; Kleanthous C; Hopper JTS; Robinson CV Angew. Chem., Int. Ed 2017, 56, 14463–14468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (102).Sisley EK; Illes-Toth E; Cooper HJ TrAC, Trends Anal. Chem 2020, 124, 115534. [Google Scholar]
- (103).Bunch J; Yan B ChemRxiv 2020, DOI: 10.26434/chemrxiv.12925295.v12925291. [DOI] [Google Scholar]
- (104).Kertesz V; Van Berkel GJ J. Mass Spectrom 2010, 45, 252–260. [DOI] [PubMed] [Google Scholar]
- (105).Mikhailov VA; Griffiths RL; Cooper HJ Int. J. Mass Spectrom 2017, 420, 43–50. [Google Scholar]
- (106).Griffiths RL; Konijnenberg A; Viner R; Cooper HJ Anal Chem 2019, 91, 6962–6966. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (107).Hale OJ; Cooper HJ J. Am. Soc. Mass Spectrom 2020. [DOI] [PubMed] [Google Scholar]
- (108).Roach PJ; Laskin J; Laskin A Analyst 2010, 135, 2233–2236. [DOI] [PubMed] [Google Scholar]
- (109).Van Berkel GJ; Kertesz V; King RC Anal. Chem 2009, 81, 7096–7101. [DOI] [PubMed] [Google Scholar]
- (110).Kohler M; Neff C; Perez C; Brunner C; Pardon E; Steyaert J; Schneider G; Locher KP; Zenobi R Anal. Chem 2018, 90, 5306–5313. [DOI] [PubMed] [Google Scholar]
- (111).Beaufour M; Ginguene D; Le Meur R; Castaing B; Cadene MJ Am. Soc. Mass Spectrom 2018, 29, 1981–1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (112).Hale OJ; Cramer R Anal. Chem 2019, 91, 14192–14197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (113).Lu Y; Pieterse CL; Robertson WD; Miller RJD Anal. Chem 2018, 90, 4422–4428. [DOI] [PubMed] [Google Scholar]
- (114).Karki S; Shi F; Archer JJ; Sistani H; Levis RJ J. Am. Soc. Mass Spectrom 2018, 29, 1002–1011. [DOI] [PubMed] [Google Scholar]
- (115).Morgner N; Barth HD; Brutschy B Aust. J. Chem 2006, 59, 109–114. [Google Scholar]
- (116).Henrich E; Peetz O; Hein C; Laguerre A; Hoffmann B; Hoffmann J; Dotsch V; Bernhard F; Morgner N eLife 2017, 6, e20954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (117).Peetz O; Henrich E; Laguerre A; Lohr F; Hein C; Dotsch V; Bernhard F; Morgner N Anal. Chem 2017, 89, 12314–12318. [DOI] [PubMed] [Google Scholar]
- (118).Peetz O; Hellwig N; Henrich E; Mezhyrova J; Dotsch V; Bernhard F; Morgner NJ Am. Soc. Mass Spectrom 2019, 30, 181–191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (119).Chandler SA; Benesch JLP Curr. Opin. Chem. Biol 2018, 42, 130–137. [DOI] [PubMed] [Google Scholar]
- (120).Macias LA; Santos IC; Brodbelt JS Anal. Chem 2020, 92, 227–251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (121).Stiving AQ; VanAernum ZL; Busch F; Harvey SR; Sarni SH; Wysocki VH Anal. Chem 2019, 91, 190–209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (122).Harvey SR; Liu Y; Liu W; Wysocki VH; Laganowsky A Chem. Commun 2017, 53, 3106–3109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (123).Mikhailov VA; Liko I; Mize TH; Bush MF; Benesch JLP; Robinson CV Anal. Chem 2016, 88, 7060–7067. [DOI] [PubMed] [Google Scholar]
- (124).Greisch J-F; Tamara S; Scheltema RA; Maxwell HWR; Fagerlund RD; Fineran PC; Tetter S; Hilvert D; Heck AJR Chem. Sci 2019, 10, 7163–7171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (125).Kar UK; Simonian M; Whitelegge JP Expert Rev. Proteomics 2017, 14, 715–723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (126).Lermyte F; Tsybin YO; O’Connor PB; Loo JA J. Am. Soc. Mass Spectrom 2019, 30, 1149–1157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (127).Sipe SN; Patrick JW; Laganowsky A; Brodbelt JS Anal. Chem 2020, 92, 899–07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (128).Kalenius E; Groessl M; Rissanen K Nat. Rev. Chem 2018, 3, 4–14. [Google Scholar]
- (129).McCabe JW; Hebert MJ; Shirzadeh M; Mallis CS; Denton JK; Walker TE; Russell DH Mass Spectrom. Rev 2020, DOI: 10.1002/mas.21642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (130).Liu Y; Cong X; Liu W; Laganowsky AJ Am. Soc. Mass Spectrom 2017, 28, 579–586. [DOI] [PubMed] [Google Scholar]
- (131).Fantin SM; Parson KF; Niu S; Liu J; Polasky DA; Dixit SM; Ferguson-Miller SM; Ruotolo BT Anal. Chem 2019, 91, 15469–15476. [DOI] [PubMed] [Google Scholar]
- (132).Fantin SM; Huang H; Sanders CR; Ruotolo BT J. Am. Soc. Mass Spectrom 2020, DOI: 10.1021/jasms.1020c00288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (133).Poltash ML; McCabe JW; Patrick JW; Laganowsky A; Russell DH J. Am. Soc. Mass Spectrom 2019, 30, 192–198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (134).Poltash ML; McCabe JW; Shirzadeh M; Laganowsky A; Clowers BH; Russell DH Anal. Chem 2018, 90, 10472–10478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (135).Poltash ML; McCabe JW; Shirzadeh M; Laganowsky A; Russell DH TrAC, Trends Anal. Chem 2020, 124, 115533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (136).Hale OJ; Illes-Toth E; Mize TH; Cooper HJ Anal. Chem 2020, 92, 6811–6816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (137).Zubarev RA; Makarov A Anal. Chem 2013, 85, 5288–5296. [DOI] [PubMed] [Google Scholar]
- (138).Eliuk S; Makarov A Annu. Rev. Anal. Chem 2015, 8, 61–80. [DOI] [PubMed] [Google Scholar]
- (139).Mallis CS; Zheng X; Qiu X; McCabe JW; Shirzadeh M; Lyu J; Laganowsky A; Russell DH Int. J. Mass Spectrom 2020, DOI: 10.1016/j.ijms.2020.116451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (140).Giles K; Ujma J; Wildgoose J; Pringle S; Richardson K; Langridge D; Green M Anal. Chem 2019, 91, 8564–8573. [DOI] [PubMed] [Google Scholar]
- (141).Lippens JL; Nshanian M; Spahr C; Egea PF; Loo JA; Campuzano IDG J. Am. Soc. Mass Spectrom 2018, 29, 183–193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (142).Todd AR; Jarrold MF Anal. Chem 2019, 91, 14002–14008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (143).Keifer DZ; Pierson EE; Jarrold MF Analyst 2017, 142, 1654–1671. [DOI] [PubMed] [Google Scholar]
- (144).Lutomski CA; Gordon SM; Remaley AT; Jarrold MF Anal. Chem 2018, 90, 6353–6356. [DOI] [PubMed] [Google Scholar]
- (145).Brown BA; Zeng X; Todd AR; Barnes LF; Winstone JMA; Trinidad JC; Novotny MV; Jarrold MF; Clemmer DE Anal. Chem 2020, 92, 3285–3292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (146).Harper CC; Elliott AG; Lin HW; Williams ER J. Am. Soc. Mass Spectrom 2018, 29, 1861–1869. [DOI] [PubMed] [Google Scholar]
- (147).Elliott AG; Harper CC; Lin HW; Williams ER J. Am. Soc. Mass Spectrom 2019, 30, 946–955. [DOI] [PubMed] [Google Scholar]
- (148).Harper CC; Elliott AG; Oltrogge LM; Savage DF; Williams ER Anal. Chem 2019, 91, 7458–7465. [DOI] [PubMed] [Google Scholar]
- (149).Harper CC; Williams ER J. Am. Soc. Mass Spectrom 2019, 30, 2637–2645. [DOI] [PubMed] [Google Scholar]
- (150).Kafader JO; Melani RD; Durbin KR; Ikwuagwu B; Early BP; Fellers RT; Beu SC; Zabrouskov V; Makarov AA; Maze JT; Shinholt DL; Yip PF; Tullman-Ercek D; Senko MW; Compton PD; Kelleher NL Nat. Methods 2020, 17, 391–394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (151).Worner TP; Snijder J; Bennett A; Agbandje-McKenna M; Makarov AA; Heck AJR Nat. Methods 2020, 17, 395–398. [DOI] [PubMed] [Google Scholar]
- (152).Bechara C; Noll A; Morgner N; Degiacomi MT; Tampe R; Robinson CV Nat. Chem 2015, 7, 255–262. [DOI] [PubMed] [Google Scholar]
- (153).Lee AG Biochem. Soc. Trans 2011, 39, 761–766. [DOI] [PubMed] [Google Scholar]
- (154).Powl AM; East JM; Lee AG Biochemistry 2008, 47, 12175–12184. [DOI] [PubMed] [Google Scholar]
- (155).Frick M; Schmidt C Chem. Phys. Lipids 2019, 221, 145–157. [DOI] [PubMed] [Google Scholar]
- (156).Cong X; Liu Y; Liu W; Liang X; Russell DH; Laganowsky AJ Am. Chem. Soc 2016, 138, 4346–4349. [DOI] [PubMed] [Google Scholar]
- (157).Cong X; Liu Y; Liu W; Liang X; Laganowsky A Nat. Commun 2017, 8, 2203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (158).Patrick JW; Boone CD; Liu W; Conover GM; Liu Y; Cong X; Laganowsky A Proc. Natl. Acad. Sci. U. S. A 2018, 115, 2976–2981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (159).Liu Y; LoCaste CE; Liu W; Poltash ML; Russell DH; Laganowsky A Nat. Commun 2019, 10, 1352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (160).Bolla JR; Corey RA; Sahin C; Gault J; Hummer A; Hopper JTS; Lane DP; Drew D; Allison TM; Stansfeld PJ; Robinson CV; Landreh M Angew. Chem., Int. Ed 2020, 59, 3523–3528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (161).Pyle E; Kalli AC; Amillis S; Hall Z; Lau AM; Hanyaloglu AC; Diallinas G; Byrne B; Politis A Cell Chem. Biol 2018, 25, 840–848.e844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (162).Pyle E; Guo C; Hofmann T; Schmidt C; Ribiero O; Politis A; Byrne B Anal. Chem 2019, 91, 13071–13079. [DOI] [PubMed] [Google Scholar]
- (163).Corey RA; Pyle E; Allen WJ; Watkins DW; Casiraghi M; Miroux B; Arechaga I; Politis A; Collinson I Proc. Natl. Acad. Sci. U. S. A 2018, 115, 7967–7972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (164).Gupta K; Li J; Liko I; Gault J; Bechara C; Wu D; Hopper JTS; Giles K; Benesch JLP; Robinson CV Nat. Protoc 2018, 13, 1106–1120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (165).McDowell MA; Heimes M; Fiorentino F; Mehmood S; Farkas A; Coy-Vergara J; Wu D; Bolla JR; Schmid V; Heinze R; Wild K; Flemming D; Pfeffer S; Schwappach B; Robinson CV; Sinning I Mol. Cell 2020, 80, 72–86.e77. [DOI] [PubMed] [Google Scholar]
- (166).Bolla JR; Sauer JB; Wu D; Mehmood S; Allison TM; Robinson CV Nat. Chem 2018, 10, 363–371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (167).Gault J; Liko I; Landreh M; Shutin D; Bolla JR; Jefferies D; Agasid M; Yen HY; Ladds M; Lane DP; Khalid S; Mullen C; Remes PM; Huguet R; McAlister G; Goodwin M; Viner R; Syka JEP; Robinson CV Nat. Methods 2020, 17, 505–508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (168).Nehme R; Carpenter B; Singhal A; Strege A; Edwards PC; White CF; Du H; Grisshammer R; Tate CG PLoS One 2017, 12, e0175642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (169).VanAernum ZL; Busch F; Jones BJ; Jia M; Chen Z; Boyken SE; Sahasrabuddhe A; Baker D; Wysocki VH Nat. Protoc 2020, 15, 1132–1157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (170).Desligniere E; Ehkirch A; Botzanowski T; Beck A; Hernandez-Alba O; Cianferani S Anal. Chem 2020, 92, 12900–12908. [DOI] [PubMed] [Google Scholar]
- (171).Kieuvongngam V; Olinares PDB; Palillo A; Oldham ML; Chait BT; Chen J eLife 2020, 9, e51492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (172).Reading E; Hall Z; Martens C; Haghighi T; Findlay H; Ahdash Z; Politis A; Booth PJ Angew. Chem., Int. Ed 2017, 56, 15654–15657. [DOI] [PubMed] [Google Scholar]
- (173).Debelyy MO; Waridel P; Quadroni M; Schneiter R; Conzelmann A PLoS One 2017, 12, e0186840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (174).Lu Y; Zhang H; Niedzwiedzki DM; Jiang J; Blankenship RE; Gross ML Anal. Chem 2016, 88, 8827–8834. [DOI] [PMC free article] [PubMed] [Google Scholar]