

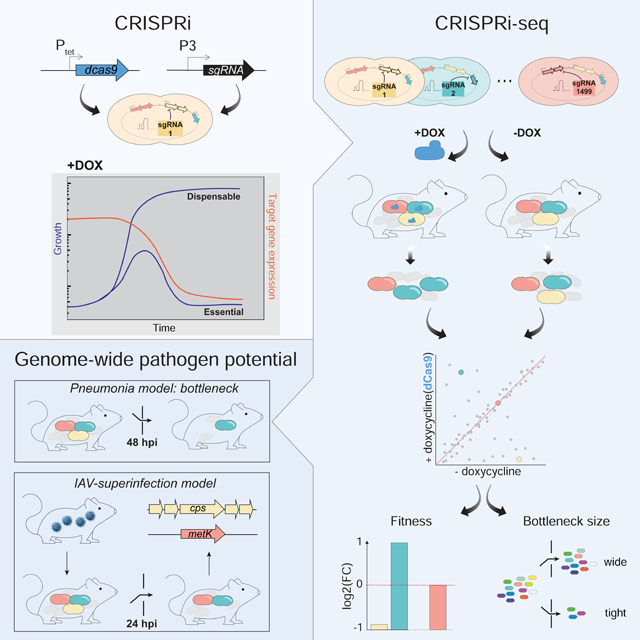
Summary
Streptococcus pneumoniae is an opportunistic human pathogen that causes invasive disease, including pneumonia, with greater health risks upon influenza A virus (IAV) co-infection. To facilitate pathogenesis studies in vivo, we developed an inducible CRISPR interference system (CRISPRi-seq) that enables genome-wide fitness testing in one sequencing step. We applied CRISPRi-seq to assess bottlenecks and identify pneumococcal genes important in a murine pneumonia model. A critical bottleneck occurs at 48 hours with few bacteria causing systemic infection. This bottleneck is not present during IAV superinfection, facilitating identification of pneumococcal pathogenesis-related genes. Top in vivo essential genes included purA, encoding adenylsuccinate synthetase, and the cps operon required for capsule production. Surprisingly, CRISPRi-seq indicated no fitness-related role for pneumolysin and metK, encoding S-adenosylmethionine synthetase, was essential in vitro but dispensable in vivo. This highlights advantages of CRISPRi-seq over transposon-based genetic screens as all genes, including essential genes, can be tested for pathogenesis potential.
Keywords: CRISPRi-seq, Streptococcus pneumoniae, bacterial pathogenesis, bottleneck, pneumonia, influenza A virus superinfection
Graphical Abstract
eTOC Blurb
Liu et al. developed CRISPRi-seq to enable in vivo genome-wide fitness testing of Streptococcus pneumoniae in one sequencing step. CRISPRi-seq revealed a bottleneck during murine pneumococcal infection not observed upon influenza virus co-infection, enabling identification of essential genes. By testing all genes, including essential genes, CRISPRi-seq has broad utility.
Introduction
Streptococcus pneumoniae is one of the most prevalent opportunistic human pathogens. The bacterium frequently colonizes the human nasopharynx but can cause severe diseases when it invades normally sterile sites. Invasive pneumococcal diseases, including pneumonia, sepsis, and meningitis, lead to millions of deaths per year (Weiser et al., 2018). S. pneumoniae is the leading agent of bacterial pneumonia worldwide (van der Poll and Opal, 2009). S. pneumoniae can pose even greater threats to global public health in combination with viral infections. An extreme example of this is the catastrophic influenza A virus (IAV) pandemic of 1918 where severe pneumococcal infections occurred in the aftermath of IAV infection and contributed significantly to excess morbidity and mortality (McCullers, 2014). Indeed, IAV can increase the susceptibility of the host to subsequent S. pneumoniae infection, which can render a mild influenza infection severe or even fatal (McCullers, 2006). While several key virulence factors are well studied, it remains unknown if or how the majority of the bacterium’s genome contributes to disease progression. Murine S. pneumoniae infection is commonly studied for modeling clinically relevant stages of disease including pneumonia and sepsis (Chiavolini et al., 2008) and the murine IAV coinfection model has been widely used to explore pneumococcal infection and transmission (Ivanov et al., 2013; Matarazzo et al., 2019; Siegel et al., 2014). High-throughput identification of important pneumococcal factors during the progression of bacterial pneumonia in the murine model can provide perspectives for understanding this leading human infectious disease.
Besides bacterial virulence factors, another important aspect influencing pathogenesis is the bottleneck imposed by the host during disease progression. The infection bottleneck is the combination of events such as immune responses and nutrient limitation that limit the size of the bacterial population causing infection. Quantification of bottleneck sizes in animal infection models can provide key insights for the host-pathogen interaction (Abel et al., 2015a). A small effective population of pneumococci during nasopharyngeal colonization, bacteremia and transmission has been observed (Li et al., 2013; Gerlini et al., 2014; Kono et al., 2016). Current studies in which pneumococcal pathogenesis bottlenecks were estimated used only three isogenic variants (Gerlini et al., 2014; Kono et al., 2016), and it remains unclear what the exact bottleneck sizes are in different pneumococcal disease models.
Large-scale identification of S. pneumoniae virulence determinants has been undertaken by signature-tagged mutagenesis (STM) and transposon-insertion sequencing (Tn-seq) studies (Chen et al., 2007; Hava and Camilli, 2002; Lau et al., 2001; Opijnen and Camilli, 2012; van Opijnen et al., 2009). However, these approaches have certain technical limitations, including the inability to investigate essential genes, and the fact that not all transposon insertions result in gene inactivation thus requiring large libraries to fully cover the genome (Cain et al., 2020). We previously harnessed an IPTG-inducible CRISPRi system for the functional study of essential genes in S. pneumoniae D39V in vitro with an arrayed library (Liu et al., 2017). This prior study showed the power of CRISPR interference (CRISPRi) for functional gene analysis. However, it was laborious to handle the arrayed library and the IPTG-inducible system is not ideal for in vivo studies due to limited pharmacokinetics information of IPTG in animal models. Recent work in Pseudomonas aeruginosa showed that an important virulence factor could be efficiently silenced in vivo using a constitutive promoter driving dCas9 (Qu et al., 2019). Here, we developed a doxycycline-inducible CRISPRi system for S. pneumoniae that is applicable to both in vitro and in vivo studies. In addition, we constructed a pooled CRISPRi library targeting nearly all operons of the prototypic S. pneumoniae strain D39V (Slager et al., 2018) that can readily be combined with Illumina sequencing (herein referred to as CRISPRi-seq). While pooled CRISPRi libraries have recently been reported for Escherichia coli, Staphylococcus aureus, Vibrio natriegens and Mycobacterium tuberculosis (Cui et al., 2018; Jiang et al., 2020; Lee et al., 2019; Wang et al., 2018; de Wet et al., 2018), they all used large pools of sgRNAs targeting each gene multiple times, often leading to off targeting (Cui et al., 2018). These libraries required deep sequencing to obtain enough statistical power on the abundance of each sgRNA in the population. In addition, for the conditions in which bottlenecks appear, for example during the infection process of pathogenic bacteria, a library with a large pool of mutants is not well suited for high throughput screening as it can lead to significant noise in calling gene fitness scores (Opijnen and Camilli, 2012). Here, we carefully selected a single sgRNA for every operon in S. pneumoniae D39V, thereby limiting off-target effects and reducing the pool of sgRNAs required to cover the entire genome.
In the present study, we first used CRISPRi-seq to measure bottleneck sizes of a commonly used murine pneumonia model. This identified an extreme bacterial bottleneck in progression from pneumonia to bacteremia. As our pooled CRISPRi library contains 1499 different genetic markers coded by the various sgRNAs, this allowed for precise measurement of bottlenecks demonstrating a large variation between hosts, and ultimately in disease progression – even in genetically identical inbred mice. We further applied CRISPRi-seq screening to an IAV pulmonary superinfection model to explore pneumococcal genes important for replication in the murine host. The top hits from this screen revealed many previously recognized virulence factors as well as additional factors that we experimentally confirmed. Together, we show that CRISPRi-seq is a robust method suitable for both bottleneck exploration and evaluation of gene fitness in vitro and in vivo.
Results
A doxycycline-inducible CRISPRi system in S. pneumoniae enables both in vitro and in vivo studies
To enable the study of S. pneumoniae genes in vivo, we designed a doxycycline-inducible CRISPRi system (Figure 1A and STAR methods). Doxycycline, a derivative of tetracycline, was used to induce TetR-controlled dCas9 expression because it has been extensively validated as inducer in rodent models due to its high potency and excellent tissue penetration (Redelsperger et al., 2016). To alleviate growth stress caused by doxycycline’s antimicrobial activity, TetM served as the antibiotic marker for dcas9 chromosome integration. TetM is a ribosome protection protein that confers tetracycline resistance by catalyzing the release of tetracycline from the ribosome (Dönhöfer et al., 2012).
Figure 1. A doxycycline-inducible CRISPRi system in S. pneumoniae.
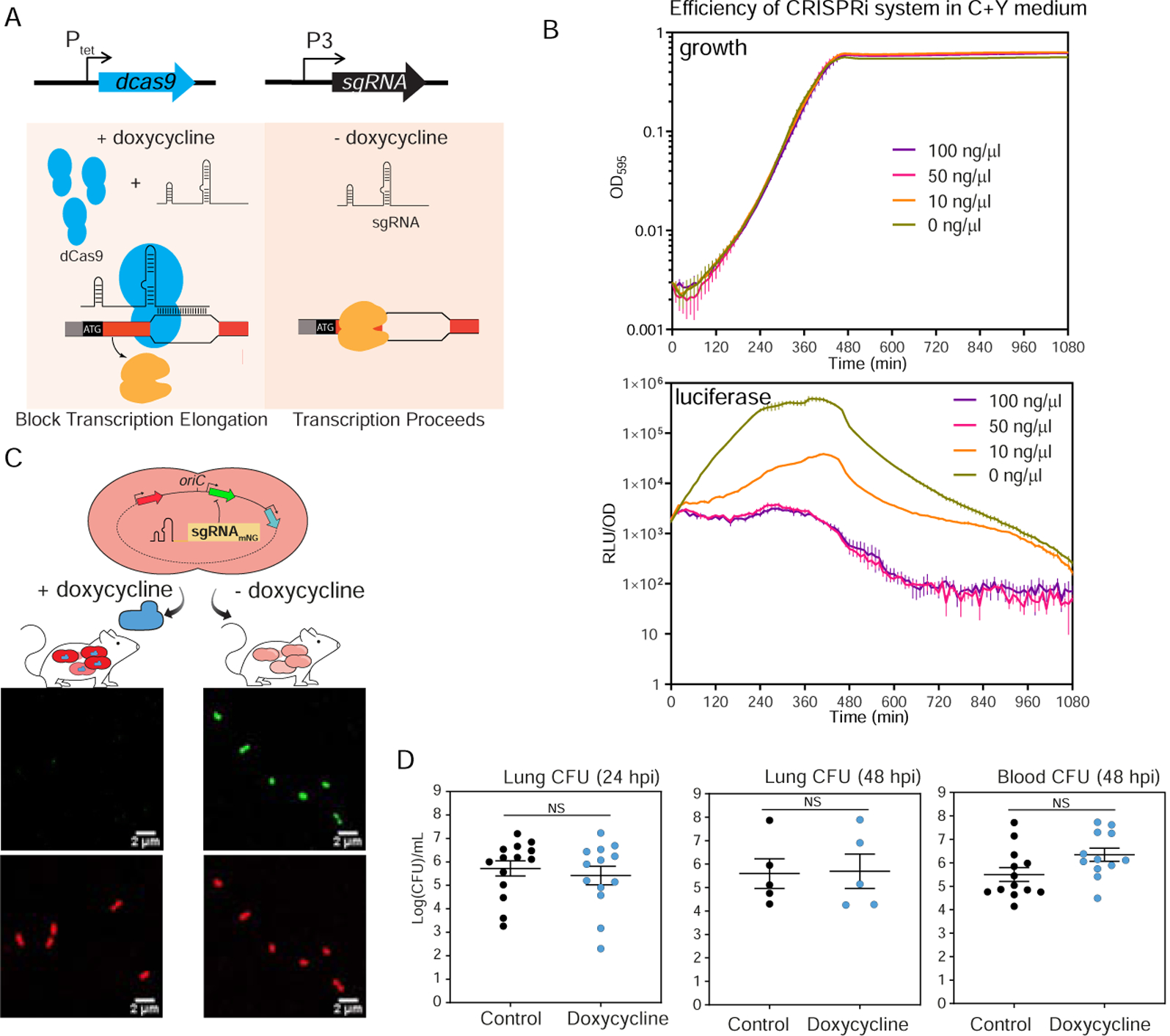
(A) The two key elements, dcas9 and sgRNA, were integrated into the chromosome of S. pneumoniae D39V and driven by a doxycycline-inducible promoter (Ptet) and a constitutive promoter (P3), respectively. With addition of doxycycline, dCas9 is expressed and binds to the target under the guidance of a constitutively expressed sgRNA. The specific dCas9-sgRNA binding to the target gene acts as a transcriptional roadblock. In the absence of the inducer, the target gene is transcribed. (B) The CRISPRi system was tested by targeting luc, which encodes firefly luciferase. The system was induced with doxycycline at different concentrations. Luciferase activity (RLU/OD) and cell density (OD595) were measured every 10 minutes. Top panel shows the growth and bottom panel shows the luciferase activity. The values represent averages of three replicates with SEM. (C) Reporter strain to assess in vivo activity of the doxycycline-inducible CRISPRi system. Strain VL2351 constitutively expresses mNeonGreen and mScarlet-I, and mNeonGreen is targeted by the sgRNA. Bacteria were collected from blood of mice on control or doxy-chow at 48 hpi and imaged with confocal microscopy in both the red and green channels. (D) Bacterial load at both lung and blood was quantified. Each dot represents a single mouse. Mean with SEM was plotted. There is no significant (NS) difference between the bacterial load in control- and doxycycline-treated mice (Mann Whitney U test).
The efficiency of the doxycycline-inducible CRISPRi system was tested in vitro and in vivo. Efficiency of the CRISPRi system in vitro was tested in C+Y medium with different concentrations of doxycycline, by targeting luc, encoding luciferase (Figure 1B). As little as 10 ng/ml doxycycline was enough to strongly reduce (>20 fold) luciferase activity within 3 h, while 50 ng/ml doxycycline yielded a maximum repression efficiency without causing growth delay (Figure 1B). This demonstrates that the system is titratable in vitro. To test its functionality in vivo, the CRISPRi system targeting mNeonGreen was cloned into a dual-fluorescent reporter strain that constitutively expresses mNeonGreen and mScarlet-I. BALB/c mice were fed ad libitum with chow containing 200 ppm doxycycline hyclate or control chow for 2 days prior to infection, then infected with the reporter strain by intratracheal inoculation. At 48 h post infection (hpi), bacteria in blood samples were checked by confocal microscopy for both green (mNeonGreen) and red (mScarlet-I) fluorescence. As expected, both mNeonGreen and mScarlet-I signals were present in the samples harvested from mice fed with control chow, whereas the mNeonGreen signal was absent in mice receiving doxycycline (Figure 1C). Specific inhibition of S. pneumoniae mNeonGreen expression in mice fed with doxycycline confirmed functionality of the doxycycline-inducible CRISPRi system in vivo. Finally, we verified that this dose of doxycycline did not alter S. pneumoniae burden in blood or lungs at 24 hpi and 48 hpi (Figure 1D), giving us a tool to regulate dCas9 expression without interfering with bacterial survival.
A concise CRISPRi library targeting the entire genome of S. pneumoniae D39V
Due to the well-documented polar effects inherent to CRISPRi (Bikard et al., 2013; Liu et al., 2017; Peters et al., 2016; Qi et al., 2013), we adopted this technique to study gene function at the operon level. 1499 sgRNAs were selected to target 2111 out of 2146 genetic elements of S. pneumoniae D39V (Figure 2A). 35 elements are not included due to lack of a protospacer adjacent motif (PAM) or localization in repeat regions on the chromosome (Table S1). Potential (off-)targets of sgRNAs are listed in Table S2. This sgRNA library also covers core operons of other pneumococcal strains, like R6 (estimated 90.3% of annotated genetic elements covered), TIGR4 (72.2%), Hungary 19A-6 (66.6%), Taiwan 19F-14 (71.6%), 11A (66.8%), G54 (72.1%) (see supplementary text).
Figure 2. Workflow for construction of the pooled doxycycline-inducible CRISPRi library.
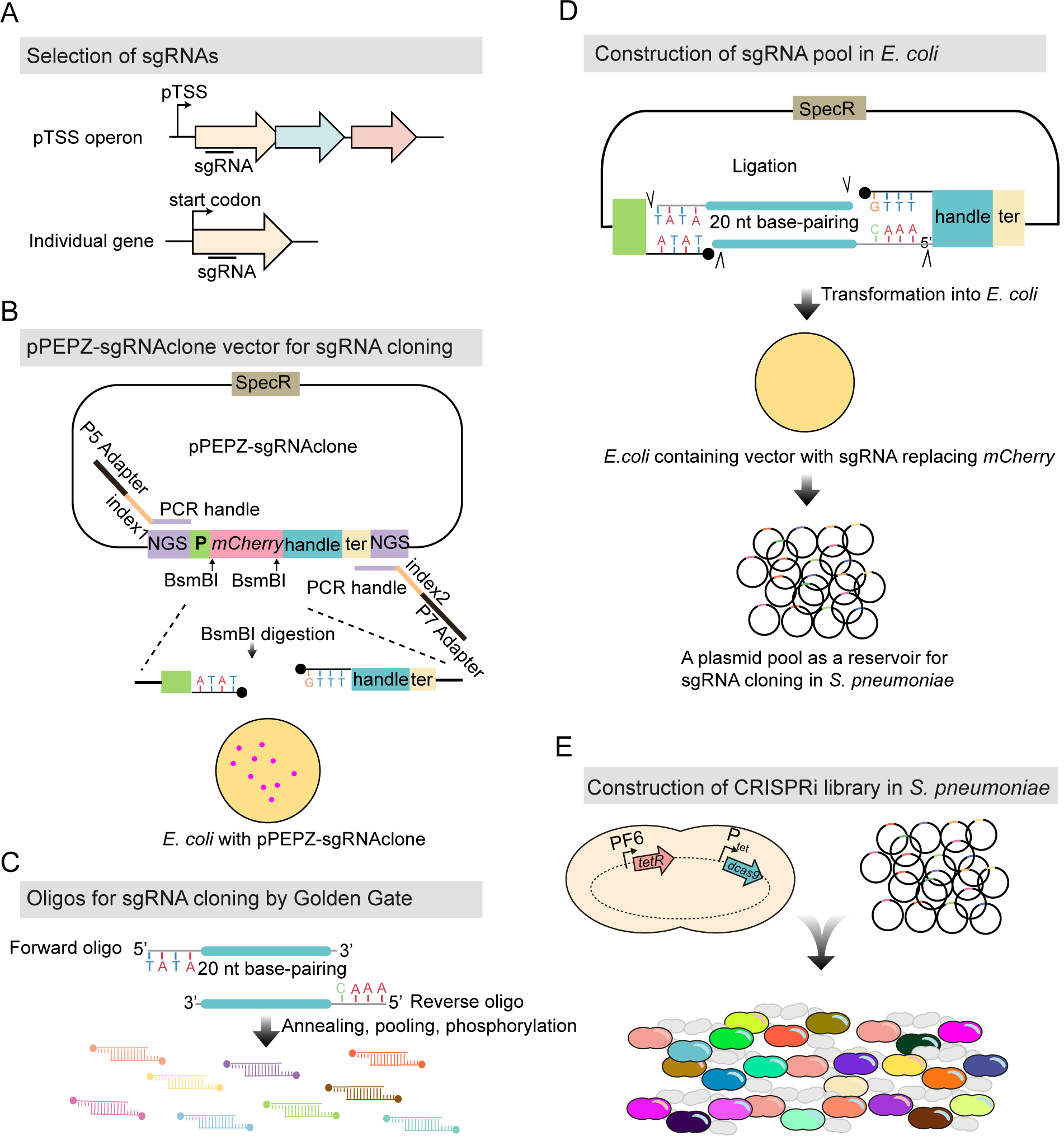
(A) 1499 sgRNAs were selected (see STAR methods), targeting 2111 genetic elements out of the 2146 in S. pneumoniae D39V. (B) The vector for sgRNA cloning, named pPEPZ-sgRNAclone, was designed to enable high efficiency Golden Gate cloning, monitoring false positive ratio, and construction of Illumina library in a one-step PCR. SpecR is the spectinomycin resistant marker; NGS indicates key elements which allow construction of an Illumina library by one-step PCR; P is the constitutive promoter which drives the expression of sgRNA; mCherry encodes a red fluorescent protein placed in the base-pairing region of sgRNA and flanked by a BsmBI site on each end; handle and ter represent the dCas9 handle binding region and terminator of the sgRNA. E. coli with the pPEPZ-sgRNAclone form red colonies resulting from the expression of mCherry. BsmBI digestion of the vector produces ends that are compatible with the sgRNA oligo annealing in (C). (C) Forward and reverse oligos were designed for each sgRNA containing 20 bp complementary to sgRNA and 4 nt overhangs compatible with the BsmBI digested vector. The oligos were annealed and pooled together followed by 5’ phosphorylation. (D) Ligation product of the digested vector (B) with the sgRNA annealing (C) was transformed into E. coli. E. coli transformed with the vector containing the sgRNA show white colonies due to replacement of mCherry with the sgRNA. 70,000 E. coli colonies were pooled together, and plasmids were purified and serve as an sgRNA reservoir. (E) Pooled plasmid library was transformed into a S. pneumoniae.
The sgRNA library was cloned into pPEPZ-sgRNAclone, a vector we engineered for efficient sgRNA cloning by Golden Gate Assembly (Figure 2 and STAR methods). To facilitate Illumina sequencing, key Illumina elements, including read 1, read 2, and adaptor sequences, were inserted flanking the sgRNA transcriptional unit. We transformed the cloned vector into E. coli first, to allow visual red/white screening of cloning efficiency: colonies containing the parental (mCherry) vector are red, while colonies containing the sgRNA construct are white. In our study, no red colony showed up among the 70,000 colonies, indicating this method was efficient in producing a high quality sgRNA pool. The E. coli library could also be used as a reservoir of sgRNAs for construction of reproducible CRISPRi libraries among S. pneumoniae D39V strains with different genetic backgrounds (e.g. mutant strains) or to transform other pneumococcal strains that contain the pPEPZ integration region, which is harbored by over 64% of all currently sequenced pneumococcal strains (Keller et al., 2019). The plasmid pool was then transformed into S. pneumoniae D39V with the above described doxycycline-inducible dcas9 (Figure 2E). To compare the doxycycline-inducible CRISPRi system to the IPTG-inducible CRISPRi system previously published by our group (Liu et al., 2017), the sgRNA library was also transformed into strain DCI23 (Liu et al., 2017).
Benchmarking CRISPRi-seq with both doxycycline- and IPTG-inducible libraries in vitro
CRISPRi screens with doxycycline- and IPTG-inducible libraries were performed in C+Y medium (Figure 3A). The sgRNAs were amplified by a one-step PCR and subsequently quantified by Illumina sequencing. Hence, this process was named CRISPRi-seq. Sequencing verified the presence of all 1499 sgRNAs in the uninduced samples of both doxycycline- and IPTG-inducible libraries (Figure 3B). In addition, the sgRNA abundance in the two libraries was very similar and only 1% of variance in sgRNA contents of induced samples is explained by difference in libraries (Figure S1A), confirming that our cloning strategy enabled repeatable transplantation of the sgRNA pool among parent strains with different genetic backgrounds. Induction of dCas9 (CRISPRi ON) by either doxycycline or IPTG resulted in a similar change in sgRNA profile (Figure 3B). The evaluated fitness, defined as the log2 fold change in sgRNA abundance upon induction, was highly consistent between the two CRISPRi libraries (Figure 3C); only five sgRNAs exhibited a statistically different abundance (log2FC>1, padj<0.05) (Figure S1B). The sgRNAs that were significantly less abundant upon dCas9 induction were categorized as targeting essential operons or genes. Likewise, sgRNAs that are more abundant upon induction were defined as costly, while sgRNAs that did not change were defined as neutral. Based on this definition, 339 sgRNAs were defined as targeting essential operons or genes, 1160 sgRNAs defined as neutral, and none defined as costly for S. pneumoniae growth in vitro (Table S3). Out of the 1499 sgRNAs, there were 1186 sgRNAs targeting individual genes, 162 sgRNAs targeting two-gene operons, and 151 sgRNAs targeting operons with three or more genes. Among these, 248 single-gene, 52 two-gene and 39 three-or-more-gene operons were found to be essential (Figure 3D). The majority of the essential genes defined by our CRISPRi-seq have been previously identified as essential or responsive by Tn-seq studies (Liu et al., 2017; Opijnen and Camilli, 2012; van Opijnen et al., 2009), indicating high consistency between the approaches (Figure S1CD and Table S4).
Figure 3. Fitness evaluation of CRISPRi targets under laboratory conditions.
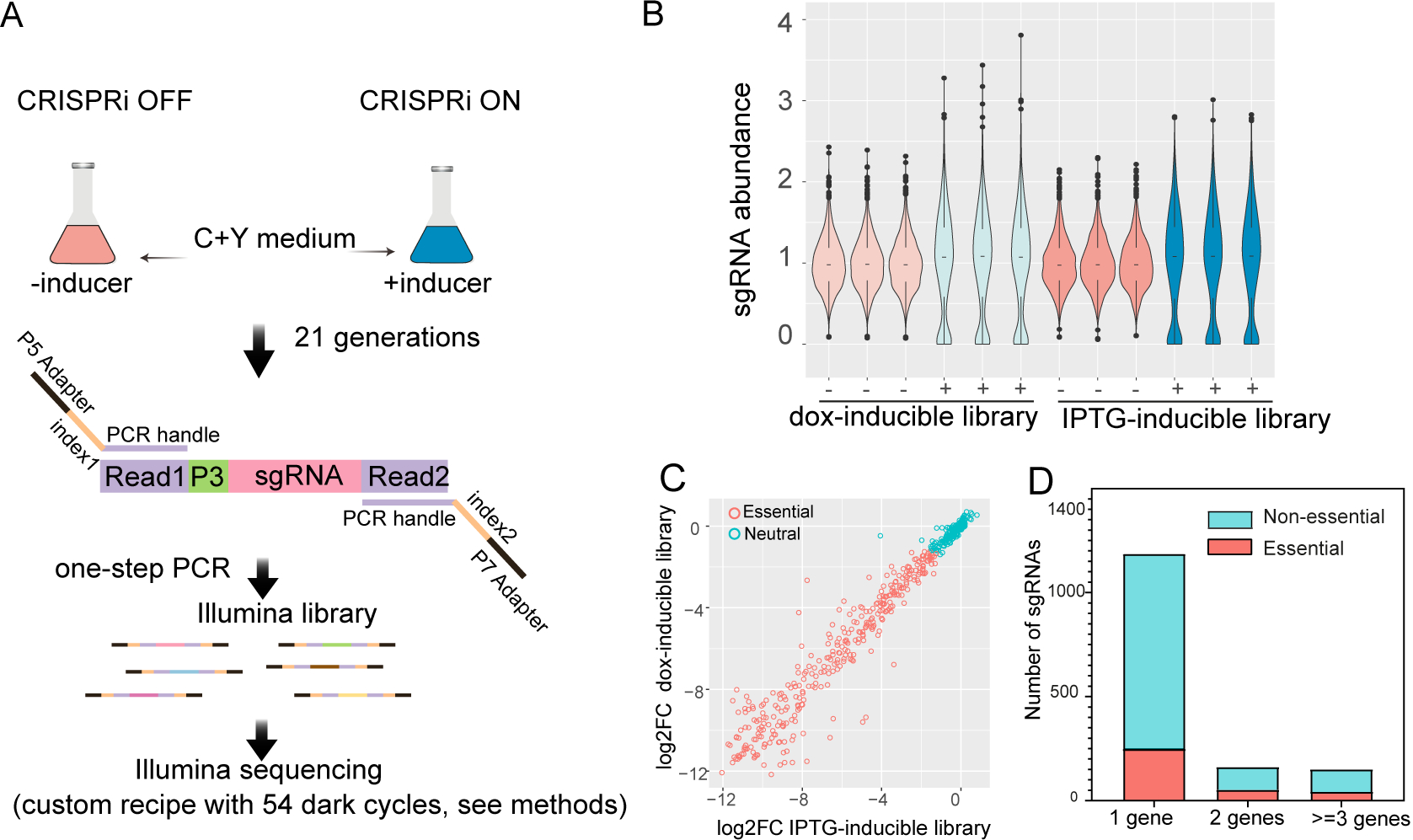
(A) Workflow of CRISPRi-seq. The CRISPRi libraries were cultured in C+Y medium in the absence (CRISPRi-OFF) or in the presence (CRISPRi-ON) of 10 ng/μl doxycycline or 1 mM IPTG. Bacteria were collected after approximately 21 generations of growth. Genomic DNA was isolated and used as a template for PCR. The forward oligo binds to Illumina amplicon element read 1 and contains the Illumina P5 adapter sequence; the reverse oligo binds to read 2 and contains the P7 adapter. Index 1 and index 2 were incorporated into the forward and reverse oligos respectively, for barcoding of different samples. (B) Violin plots showing the distribution of sgRNA abundance in each sample. ‘−’ represents control samples without inducer; ‘+’ represents induced samples. The abundance of sgRNA =1499*(counts of sgRNA)/(total counts of all sgRNAs). (C) Correlation of the fitness of targets evaluated by IPTG-inducible and doxycycline-inducible libraries. The log2FC, calculated with DEseq2, represents the fold change of sgRNA frequency between the control sample and induced sample. (D) Refinement of essential and non-essential genes of S. pneumoniae D39V by CRISPRi-seq. The sgRNAs were classified according to the number of their targets. 1 gene represents the sgRNAs targeting single gene operons; 2 represents two gene operons; >=3 s represents three or more gene operons. See also Figure S1.
Bottlenecks and heterogeneity of S. pneumoniae in mouse pneumonia
A random part of the bacterial population might die off during infection due to bottlenecks, which is caused by general stresses placed upon the bacterium within the host, such as nutrient restriction or innate immune system responses. The effective population size that gives rise to the final bacterial population that causes the infection is referred to as the bottleneck size. Bottlenecks have been reported for pneumococcal infection and transmission (Gerlini et al., 2014; Kono et al., 2016; Opijnen and Camilli, 2012), but precise estimations of their sizes are lacking due to prior ineffective methodologies. Any loss of sgRNAs of the CRISPRi library during infection can be attributed to a bottleneck effect, whose size can be estimated on the basis of allele frequencies (here: sgRNA frequencies) in the pool before and after infection (Abel et al., 2015b).
To examine pneumococcal bottlenecks during the commonly used murine model of pneumonia, standard inbred adult BALB/c mice were infected with the S. pneumoniae CRISPRi library via the intratracheal route and CRISPRi-seq was performed on bacteria isolated at 48 hpi (from both lung and blood samples) and 24 hpi (only from lung samples, as there are no detectable bacteria in the bloodstream at this time point) (Figure 4A). At 24 hpi, bottleneck sizes were relatively large (meaning most of the bacterial clones are still present), and covered more than 10-fold the library diversity in all samples except two (Figure 4B). Furthermore, estimated bottleneck sizes appeared to be smaller for CRISPRi-induced samples, which is likely due to the early drop-out of essential operons (Figure 4B).
Figure 4. Exploring bottleneck sizes during infection using CRISPRi-seq.
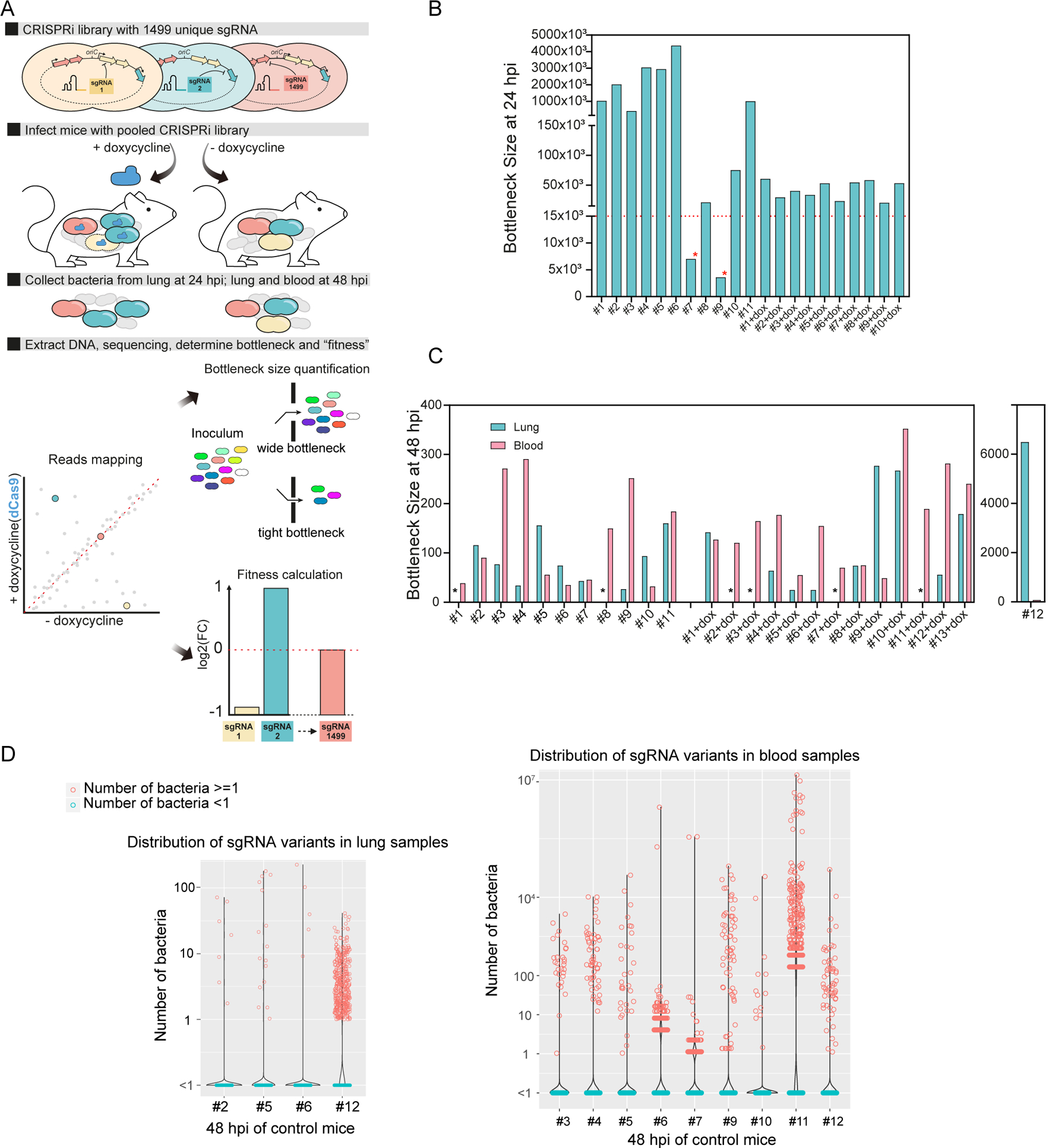
(A) Workflow of fitness cost and bottleneck evaluation in a mouse pneumonia model by CRISPRi-seq. (B) Bottleneck size of lung samples at 24 hpi. 11 mice were treated with control chow, and 10 mice were treated with doxycycline chow. The horizontal red dash line marks 14,990 bacterial cells, which is a 10-fold theoretical coverage of the CRISPRi library. The red asterisks point to mouse #7 and mouse #9 in the control group. The bottleneck size of these two mice is lower than 10-fold of the library diversity. (C) Bottleneck size in lung and blood at 48 hpi. The black asterisks point out the lung samples without successful collection of bacterial samples, which include mice without doxycycline treatment #1 and #8, mice treated with doxycycline #2-dox, #3-dox, #7-dox, and #11-dox. (D) The number of bacteria barcoded with different sgRNAs in the control group (no doxycycline treatment) was calculated according to the bacterial load and sgRNA abundance in the population. Violin plots show the distribution of bacteria number in the lung samples (left panel) and blood samples (right panel), each dot represents one bacterial variant. Notice that some mice were not shown here, because the total bacterial load was below the limit of detection and the bacterial numbers of each variant could not be calculated. See also Figure S2.
At 48 hpi, we observed a strong population size reduction in both lung and blood samples, and the bottleneck outcome was estimated to be as low as 25 bacterial cells responsible for causing disease (Figure 4C). dCas9 induction did not seem to have a substantial effect on bottleneck size estimations, suggesting that the bottleneck selection effect overshadows the CRISPRi selection effect (Figure 4C). Surprisingly, bottleneck sizes varied considerably between replicates and did not correlate between lung and blood samples of the same host (Figure 4C). Moreover, there was little to no overlap in the different surviving strains in blood and lung samples within mice, indicating independent bacterial survival in lung and blood invasion (Figure S2A). Taken together, these observations highlight the impact of bottlenecks on the outcomes of infection and strongly suggest that bacterial survival during infection in the mouse pneumonia model is highly heterogenous and bacterial survival is a stochastic event.
Quantification of the abundance of each mutant can provide information about bacterial replication and population expansion. To this end, we estimated the cell number of each mutant based on the abundance of each sgRNA in the library and the bacterial load in both lung and blood of the mice on control feed (Figure 4D). Dramatic stochastic changes in the genetic composition of the CRISPRi population were observed in all mice on control feed for both lung and blood samples at 48 hpi (Figure 4D). In addition, there was no correlation of bacterial genetic composition among samples from different mice, as individual mice have different dominant isogenic mutants (Table S7). Most strains have 0 sgRNA reads, indicating most bacteria were cleared from the lungs or failed to invade the bloodstream. Some lowly abundant strains appear to have managed to survive, but not to actively multiply in both host niches, with bacterial number estimates between 1–10. However, especially in the blood samples, some variants reached high cell numbers (up to 107), suggesting that invasion by a few clones was followed by rapid replication. High replication rates in blood were further supported by observed bacterial loads in blood that were much higher than the estimated bottleneck sizes (Figure S2B). Lastly, mouse number 12 seems to be less competent in clearing bacteria from the lung as clearly more variants survived, further stressing the importance of individual mouse effects despite being an inbred mouse strain (Figure 4C, D). Notice that here we used a published population level doubling time estimate for calculations of the bottleneck size (Opijnen and Camilli, 2012) (see Supplementary Information). However, as described in Figure 4D, we observed subpopulations with divergent behaviors, indicating high degrees of heterogeneity of bacterial growth in both lung and blood during infection. This brings challenges for accurate estimation of doubling time at the population level. Different destinies of pneumococcal cells in the mouse infection model may be explained by bacterial phenotypic diversity or host-response diversity (Kreibich and Hardt, 2015). It has been determined that individual bacteria may occupy different micro-environments and can thus be exposed to dramatically different stimuli (Davis et al., 2015), which may contribute to a level of randomness for certain pneumococcal clones to survive in the host. In addition, a single mouse passage can augment the virulence of some strains (Briles et al., 1981), such that within-host evolution for genetic adaptation may lead to the emergence of subpopulations with different fates.
CRISPRi-seq screen at 24 hpi identifies PurA as essential in a mouse pneumonia model
At 48 hpi, the effect of CRISPRi selection is overshadowed by a dramatic stochastic loss of mutants in the population while passing through the bottleneck, and thus this timepoint cannot be used to evaluate the fitness of targets by CRISPRi-seq (Figure 4C). However, earlier at 24 hpi, all mice except control mice numbers 7 and 9 exhibit a bottleneck size greater than 10-fold of the diversity in the CRISPRi library (Figure 4B). We thus analyzed the fitness of target genes during lung infection based on the sequencing data obtained at 24 hpi, excluding control mice numbers 7 and 9 (Table S5).
In vivo fitness was compared to growth in laboratory media to identify genes that became either more or less essential during infection (Figure 5A). There were 46 sgRNAs whose targets showed significantly differential fitness between in vivo and in vitro conditions (log2FC>1, padj<0.05), including 31 sgRNAs whose targets were more essential in vivo and 15 sgRNAs whose targets were less essential in vivo (Table S5). Seeking to identify virulence factors, we next focused on those genes that scored as more essential in vivo.
Figure 5. CRISPRi-seq identified PurA as important for infection.
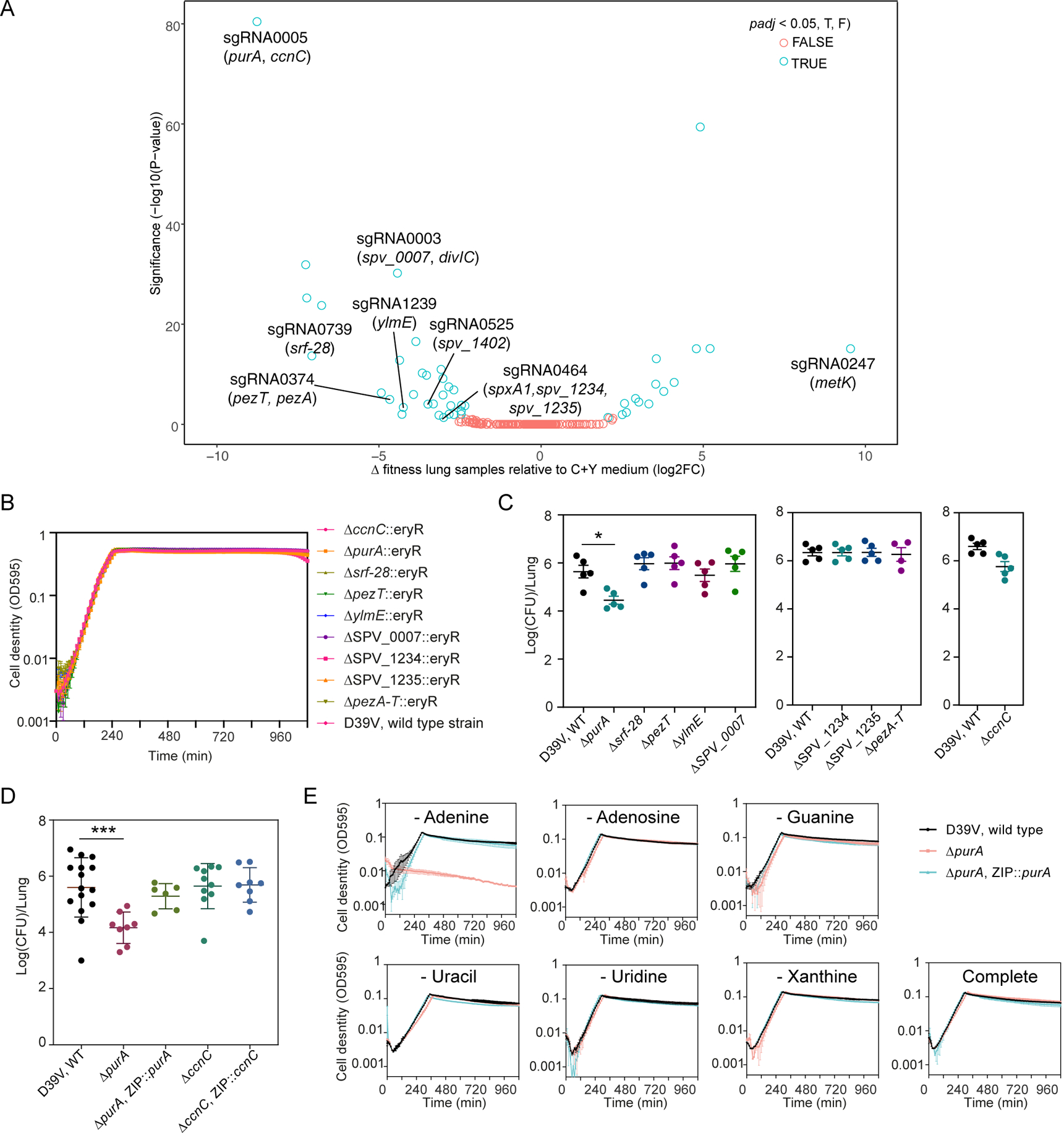
(A) Comparison of fitness cost of gene depletion by CRISPRi by different sgRNAs between the mouse lung infection model at 24 hpi and C+Y medium. The difference was shown as the log2 fold change between the two conditions by DEseq2 analysis and the P-values are adjusted by FDR. The highlighted sgRNAs were selected for follow-up studies. (B) Growth of the deletion mutants and the wild-type D39V strain in C+Y medium. Cell density was determined by measuring OD595nm every 10 minutes. The values represent averages of three technical replicates with SEM (same for panel E). (C-D) Mouse infection with individual mutants, compared to wild type D39V. Each dot represents a single mouse. Mean with SEM was plotted. (C) The mutants were tested in three batches of infection assays, for each assay the wild-type strain was tested in parallel. Significant difference between D39V and ΔpurA was tested by Sidak’s multiple comparisons test, and the adjusted p value is 0.0158. (D) Validation study of sgRNA0005 targets. The virulence of deletion mutants and complementation strains were tested and compared to wild type D39V. There was a significant difference between the wild-type and ΔpurA strain tested by Kruskal-Wallis test with Dunn’s post-analysis, and the adjusted p-value was 0.0007. Note that ectopic expression of purA complemented the phenotype of the purA deletion mutant. (E) Growth of ΔpurA in blood-like medium lacking adenine, adenosine, guanine, uracil, uridine, xanthine, and complete medium.
We selected 7 sgRNAs identified as targeting 13 neutral genes in C+Y laboratory growth media but predicted essential in vivo with an absolute log2FC>3 difference (Table S5). In line with the in vitro CRISPRi-seq data, most of the targeted genes (8) could be deleted without a detectable growth defect in C+Y medium (Figure 5B). Note that spv_2285 was not tested, as it is encodes a pseudo gene (Slager et al., 2018). However, divIC (targeted by sgRNA0003), spxA1 (sgRNA0464), and dpr (sgRNA0525) were identified as essential, since multiple attempts of deletion failed, corroborating the results of other studies (Liu et al., 2017; van Opijnen et al., 2009). Interestingly, for pezT and pezA, identified as an epsilon/zeta toxin-antitoxin system (Mutschler et al., 2011), single deletions of the toxin gene pezT or double deletion of pezA-T system were achieved and the resulting mutants showed no growth defect (Figure 5B). However, single deletion of the antitoxin gene pezA alone failed, indicating that the pezT-A toxin-antitoxin system is active in vitro.
Mice were then infected with the 8 viable knockout strains and the pezA-T double mutant individually by intratracheal challenge, and the bacterial load in the lung of each mutant was compared to wild-type strain D39V (Figure 5C). Among the 9 mutants, purA (targeted by sgRNA0005), which showed the biggest log2FC difference between infection and C+Y medium in the CRISPRi-seq screen, was confirmed to be attenuated. sgRNA0005 targets an operon consisting of ccnC and purA. Infection experiments with knockout and complementary strains of these two genes confirmed that deletion of purA, but not ccnC, led to strongly attenuated S. pneumoniae virulence (Figure 5D). PurA, an adenylosuccinate synthetase, was previously identified to have a virulence role in experimental pneumococcal meningitis (Molzen et al., 2011). As adenylsuccinate synthetase is important for purine biosynthesis, we suspect the attenuated virulence of the purA knockout mutant is caused by lack of purine availability in the corresponding in vivo niches. To probe this further, we used a synthetic blood-like medium (BLM) (Aprianto et al., 2018) to propagate the purA mutants. It showed that specific reduction of adenine in the media led to marked growth retardation (Figure 5E). Together, this shows that CRISPRi-seq, and by extension similar screens such as Tn-seq, is not very predictive in finding important virulence factors in a disease model with strong bottlenecks as only the most significant hit (purA) from the screen could be confirmed.
Identification of genes important for pneumococcal replication during influenza superinfection
In the above-described murine pneumonia model, S. pneumoniae was cleared rapidly from the lung as we used 108 CFU per lung as inoculum and the bacterial load at 24 hpi decreased to less than 104 CFU/ lung. Using CRISPRi-seq, we were able to track the bacterial composition in great detail for individual mice demonstrating that CRISPRi-seq is a useful tool for infection bottleneck studies. In order to obtain richer data identifying pneumococcal genes directly involved in virulence, we performed a CRISPRi-seq screen in a murine disease model that is known to promote intra-host bacterial replication, the IAV/ S. pneumoniae superinfection model (Figure 6A). Mice are first infected with 50 plaque forming units (PFU) of IAV followed seven days later by intranasal infection of 5×104 CFU of our S. pneumoniae D39V CRISPRi library (approximately 33-fold theoretical coverage of the library). The bacterial load at 24 hpi increased to about 108 CFU/lung in all animals (Figure 6B). After CRISPRi-seq analysis, we did not observe a bottleneck in the IAV superinfection model as the samples collected from the control mice clustered well with the samples from C+Y medium without the dCas9 inducer in a PCA, indicating similar sgRNA contents (Figure S3C). In addition, the sgRNA profile was similar when the CRISPRi system was uninduced either in vivo or in vitro (Figure S3D).
Figure 6. CRISPRi-seq in the influenza A virus pulmonary pneumococcal superinfection model.
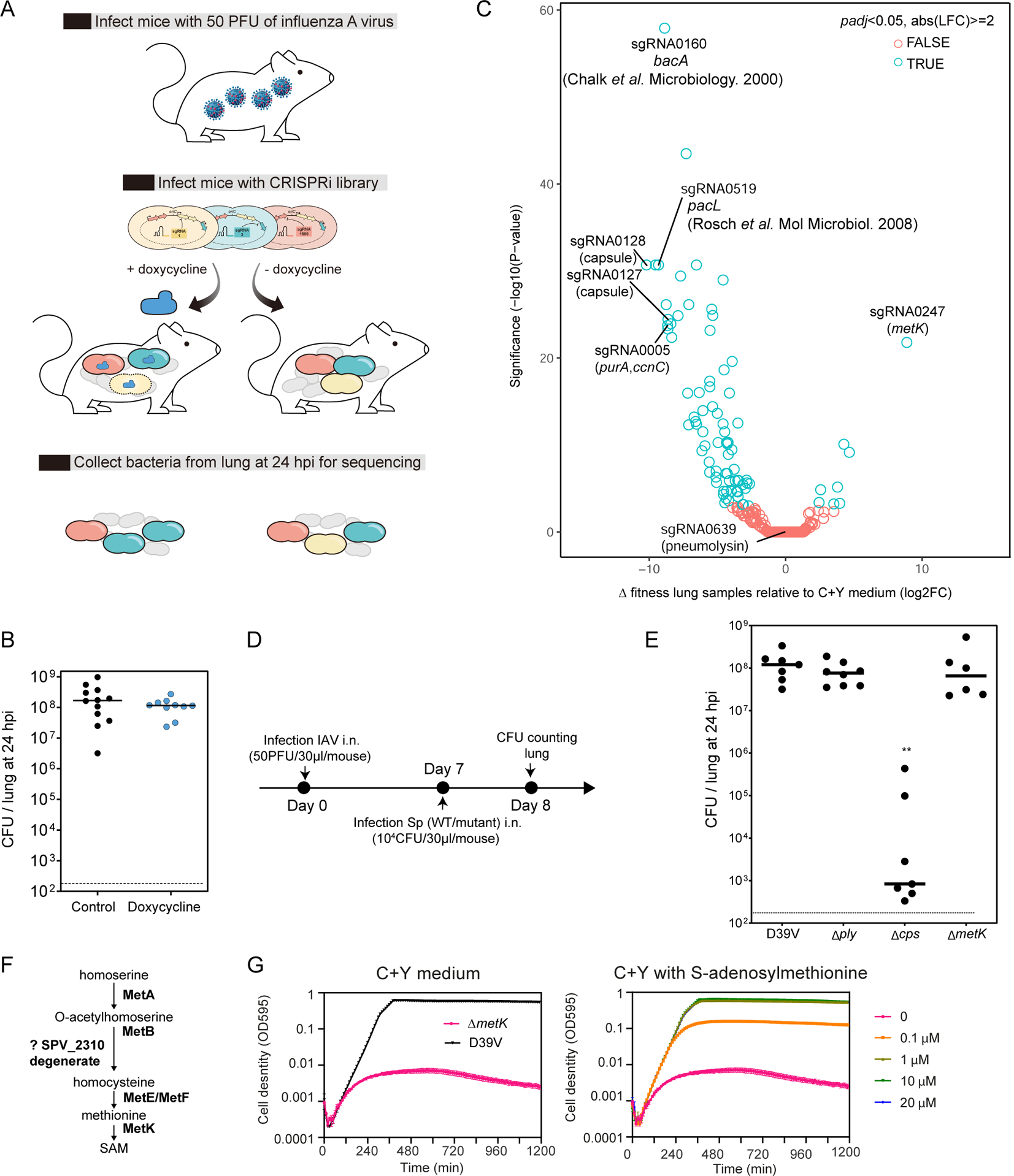
(A) Workflow of CRISPRi-seq screen. (B) The bacterial load in the lung at 24 hpi shows no impact of doxycycline. Horizontal bar indicates average. The inoculum used in this model is approximately 5×104 CFU intranasally (i.n.). (C) Comparison of fitness cost of gene between the IAV superinfection model at 24 hpi and C+Y medium. The difference was shown as the log2 fold change between the two conditions by DEseq2 analysis and the P-values are adjusted by FDR. Labelled circles represent sgRNAs targeting genes previously shown to be important for virulence or confirmed in the present study by mutational analysis. (D) Workflow for the confirmation study with individual strain in the IAV superinfection model. (E) IAV superinfection with pneumolysin deletion (Δply), capsule deletion (Δcps), and metK deletion (ΔmetK) mutant, compared to wild type D39V. Each dot represents a single mouse. ** indicate significantly different bacterial loads, p<0.05 Kruskal-Wallis one-way ANOVA. Horizontal bar indicates average. (F) The biosynthetic pathway of S-adenosylmethionine (SAM) synthesis. (G) Growth of the metK deletion mutant in C+Y medium supplemented with different concentrations of SAM. Mean and SEM of three replicates were shown. See also Figure S3.
When comparing doxycycline-induced sgRNA abundancy, we identified genes that were differentially essential during influenza superinfection compared to the in vitro C+Y medium (Figure 6C). We again found sgRNA0005 targeting purA among the top 10 hits. In addition, we identified two transcriptional units of the capsule locus, targeted by sgRNA0127 and sgRNA0128, in S. pneumoniae D39V to be important for survival during IAV superinfection (Figure 6C). Although the capsule is generally regarded as one of the most important virulence factors in S. pneumoniae, the essentiality of the capsule genes has not yet been implicated in the IAV superinfection model. We thus compared a capsule locus knockout mutant with the wild type D39V strain in the IAV superinfection model (Figure 6D). In line with the CRISPRi-seq screen, virulence of the capsule knockout mutant was significantly attenuated in the IAV superinfection model (Figure 6E). sgRNA0160, targeting bacA, was also among the top hits. Consistent with our screen, Chalker et al showed that a deletion mutant of bacA displayed no significant changes in growth rate or morphology in vitro, but was highly attenuated in a mouse model of infection (Chalker et al., 2000). Another top hit was sgRNA0519, targeting the operon consisting of pacL and SPV_1382. pacL, encoding an calcium-transporting ATPase, was previously shown to be essential for S. pneumoniae TIGR4 to survive in Balb/c mouse (Rosch et al., 2008). Among the top 10 hits, targets of 5 sgRNAs were proven by us or in previous studies to be important for pneumococcal virulence in different pneumococcal strains or animal models demonstrating that the data from CRISPRi-seq in the superinfection model is highly reliable.
Pneumolysin, encoded by ply, is the major pneumococcal toxin and previous studies showed that production of pneumolysin can increase pneumococcal adherence to the respiratory epithelium and facilitate S. pneumoniae colonization, invasion and dissemination (Nishimoto et al., 2020). Strikingly, the abundance of sgRNA0639, targeting ply, showed no difference between the in vivo and in vitro conditions in our IAV superinfection pneumonia model as determined by CRISPRi-seq (Figure 6C). To test whether this was not a false negative result, we performed infection assays in the superinfection model with a ply knockout mutant (Δply) and wild type D39V (Figure 6E). Indeed, we did not find any significant difference between wildtype D39V and Δply. Consistent with these findings, it was recently shown that pneumolysin promotes bacterial shedding allowing for transmission between hosts by induction of inflammation (Zafar et al., 2017), suggesting that pneumolysin might be more important for transmission rather than for survival in the host.
In vitro essential genes identified as non-essential in vivo highlight the power of CRISPRi-seq
One advantage of CRISPRi-seq over transposon-based genome-wide screens is the ability to evaluate the fitness of genes that are essential under laboratory conditions. From the performed CRISPRi-seq screens some sgRNAs were identified as essential in C+Y but neutral in lung infection. sgRNA0247 of this class of sgRNAs, targeting metK, showed the most significant fitness difference between the in vitro and in vivo condition in both the pneumonia and IAV superinfection model (Figure 5A, Figure 6C). The metK gene encodes S-adenosylmethionine synthetase, which catalyzes the formation of S-adenosylmethionine (SAM) from methionine and ATP (Figure 6F). By adding SAM in the growth plates, we were able to generate a metK deletion mutant. In line with its predicted function, the growth defect of the metK deletion mutant in C+Y medium could be completely rescued by addition of 1 μM SAM (Figure 6G). Infection of mice with the metK deletion mutant in the IAV superinfection model confirmed the observation of the CRISPRi-seq screen demonstrating a non-essential role of MetK during replication in the host (Figure 6E). The nonessentiality of MetK during lung infection might be explained by the presence of SAM in host tissue. Indeed, in human serum the SAM level was reported to be approximately 130 nM (Li et al., 2015).
Discussion
The principal contribution of this study is the development of a concise pooled CRISPR interference system, aided by the establishment of a sgRNA assessment algorithm, suitable for high-throughput quantitative genetic interaction screening on a genome-wide scale for the important human pathogen S. pneumoniae. Here, we adopted mouse feed containing doxycycline as in vivo inducer, demonstrating robust induction of the CRISPRi system engineered in S. pneumoniae. Future research should determine how titratable this system is in vivo and whether feeding by doxycycline-containing chow or direct injection is a preferred route for precise in vivo bacterial dCas9 induction. Fine-tuned induction of the system in vivo would also allow for the testing of virulence-related functions of otherwise essential genes, something currently not possible with Tn-seq approaches.
A main advantage of this concise doxycycline-inducible system is that it can be used for in vivo studies as the library size is small (1499 unique sgRNAs) so bacterial loads can be low and sequence depth does not need to be high. Using this system, we were able to map infection bottlenecks in a murine model of pneumococcal pneumonia and show that as little as 25 survived individual bacterial cells can finally cause systemic disease. In addition, CRISPRi-seq reveals that there is a large within-host and between-host variability in dealing with pneumococcal infection, strongly suggesting that future work would benefit from a single cell analytical approach to study pneumococcal infection. These findings are significant to our understanding of pneumococcal disease, given that in humans, the majority of S. pneumoniae exposures do not lead to severe disease, and disease manifestations can vary within a host over time. It would be interesting to see which host immune response are most successful at increasing the bottlenecks of bacterial pathogenesis (making the bottleneck tighter), and this information might inform on innovative therapies.
Previous Tn-seq studies demonstrated that due to severe bottlenecks, highly reduced library sizes were required making the subsequent data analysis challenging (Opijnen and Camilli, 2012). The beauty of the here described CRISPRi-seq platform is its efficient and precise quantification of the bottleneck during the infection as the pool of sgRNAs in the uninduced system can serve as a library of neutral barcodes. We note that CRISPRi-seq for bottleneck exploration is most effective if the system is tightly controllable. Although it cannot remove the bottleneck from the screening, as that is a property of the infection model, the data can directly inform on whether such bottlenecks exist and whether gene fitness analysis will be highly predictive. Using CRISPRi-seq, we found a strong bottleneck in the commonly used mouse pneumonia model making gene fitness predictions difficult. Indeed, out of the 7 sgRNAs that showed reduced in vivo fitness in the pooled CRISPRi-seq screen and were tested individually, only the top hit (sgRNA0003 targeting purA) could be confirmed (Figure 5). As CRISPRi-seq is a competitive assay, the fitness defect of mutants can be amplified. Determination of the competitive index (CI) is a highly sensitive method to detect differences in virulence between strains, however, it cannot determine whether a mutant is capable of causing progressive infection on its own (Cain et al., 2020). Therefore, we used individual infections (not competitions) to confirm hits from the CRISPRi-seq screens, which might have led to a reduced hit rate.
Regardless of their etiology, this study shows the existence of large bottlenecks in the commonly studied pneumococcal mouse pneumonia model. This finding begs the question of how useful this small animal system is for modeling human pneumococcal pneumonia. The presence of this strong bottleneck in this model limits the capability of CRISPRi-seq to identify virulence factors. Therefore, we performed CRISPRi-seq in the influenza superinfection model to explore S. pneumoniae genetic factors contributing to infection. We observed no bacterial bottleneck at 24 hours post infection in this model. Among the top 10 hits of potential virulence factors identified, so far, 5 of them were shown by us or in previous studies to be important for pneumococcal virulence. Current experiments in our laboratories are underway to test the remaining top hits for their roles in pneumococcal virulence and will be reported elsewhere.
Compared to traditional Tn-seq studies, the here-described CRISPRi-seq approach is easier to handle in terms of library construction as it just requires a single PCR step. In addition, Tn-seq does not readily allow for functional exploration of essential genes, and there are about 400 genes in S. pneumoniae that cannot be targeted by Tn-seq studies (Liu et al., 2017; Opijnen and Camilli, 2012; van Opijnen et al., 2009). By CRISPRi-seq we identified genes that are essential in laboratory medium but are non-essential in the host, which provides information to refine the list of therapeutic targets for S. pneumoniae. For example, MetK, the SAM synthetase involved in SAM and methionine pathways, was previously identified as potential drug target for M. tuberculosis (Berney et al., 2015). In contrast, our study shows that MetK is not a promising target for pneumococcal disease since it is not essential in vivo.
Here, we applied CRISPRi-seq on a murine pneumonia and IAV superinfection model as a proof-of-concept study. Future studies might apply the CRISPRi-seq approach to other established pneumococcal models of disease such as the zebrafish meningitis model and the Galleria mellanonela larvae invertebrate model (Cools et al., 2019; Jim et al., 2016; Rudd et al., 2016; Saralahti et al., 2014). It would also be interesting to perform a more detailed analysis of the infection models by querying different infection time points and different timing for induction of the CRISPRi system to obtain time-resolved and concentration-dependent fitness maps, which may be aided by different doxycycline-administration routes.
In summary, the here presented concise CRISPRi-seq setup can be used for studying pneumococcal pathogenesis. An additional benefit of CRISPRi-seq is that it can be used for bottleneck exploration. The library, its design rules and the underlying bioinformatic approaches developed here can now be expanded to study other infection-relevant conditions including testing of wild-type and knockout mouse strains and evaluation of antibiotics and other therapeutic interventions and may serve as an example for studies on other host-microbe interactions including human pathogens.
STAR*METHODS
RESOURCE AVAILABILITY
Lead Contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Jan-Willem Veening (jan-willem.veening@unil.ch).
Materials Availability
Key vector pPEPZ-sgRNAclone is available through Addgene (#141090). This study did not generate new unique reagents.
Data and Code Availability
The code produced in this study, and the raw files are available at GitHub (https://github.com/veeninglab/CRISPRi-seq). Results tables and analysis of the genomes tested in this study can be found on the Veeninglab website (https://www.veeninglab.com/crispri-seq). The fastq files generated from sequencing are uploaded to the Sequence Read Archive (SRA) on NCBI with accession number PRJNA611488.
EXPERIMENTAL MODELS AND SUBJECT DETAILS
Bacterial Strains and Growth Conditions
Streptococcus pneumoniae D39V (Slager et al., 2018) was used as the parent strain for this study. C+Y medium (pH=6.8), and Columbia agar or Tryptic soy agar plates supplied with 5% sheep blood were used to grow the strain and its derivatives. Working stock of the pneumococcal cells, named as “T2 cells”, were prepared by collecting cells at OD600 0.3 and then resuspending with fresh C+Y medium with 17% glycerol, and stored at −80°C. E. coli stbl3 (ThermoFisher) was used for subcloning of plasmids. LB agar with 100 μg/ml spectinomycin was used to select E. coli transformants. Strains and plasmids used in this study are listed Table S8. The oligos used for construction of mutants and strains used in this study are listed and described in Table S9.
Mice and Organ Collection
For the murine pneumonia model, the UCSD Institutional Animal Care and Use Committee approved all animal use and procedures (protocol number S00227M, V. Nizet). Two days prior to infection, 6–8 week old female BALB/c mice (Jackson Laboratories - 000651) were fed control feed or 200 ppm doxycycline feed ad libitum (Envigo TD.120769, with blue food coloring), allowing serum concentrations of doxycycline to stabilize prior to infection (Redelsperger et al., 2016). Bacterial libraries were grown in vitro in C+Y medium in the absence of selection (i.e. no doxycycline) to an OD600 of 0.4, sonicated for 3 seconds to break up chains of bacteria, and then resuspended in PBS at a concentration of 1 × 108 CFU per 30 μL. Mice were anesthetized with 100 mg/kg ketamine and 10 mg/kg xylazine (intraperitoneal administration), vocal cords were visualized with an otoscope and 30 μl bacteria was delivered into the lungs by pipetting. Mice were returned to the same cages after infection, containing doxycycline or control feed. At 24 or 48 hpi, mice were euthanized via CO2 asphyxiation, lungs were dissected and homogenized in 1 mL PBS, while blood was collected by cardiac puncture in the presence of EDTA to prevent clotting. Following tissue harvest, lung homogenate or blood was diluted in 15 ml C+Y medium (without selection), incubated at 37 °C with 5% CO2 until cultures reached an OD600 of 0.4. Samples were then pelleted and frozen before subsequent gDNA isolation and sequencing. For the comparison of fitness between murine pneumonia model and C+Y medium, see Table S5. The confirmation assays were performed with wild type and mutant strains in a similar way. Specifically, for confirmation assays, mice were all fed with control feed, and the bacterial load at lung were numerated by plating the lung homogenate or its dilutions onto Tryptic soy agar plate supplied with 5% sheep blood and incubation overnight.
Superinfection experiments complied with national, institutional and European regulations and ethical guidelines, were approved by our Institutional Animal Care and Use Committee (animal facility agreement C59–350009, Institut Pasteur de Lille; reference: APAFIS#5164, protocol 2015121722429127_v4, J.C. Sirard) and were conducted by qualified, accredited personnel. Male C57BL/6JRj mice (6–8 weeks old) (Janvier Laboratories, Saint Berthevin, France, or Envigo, Huntingdon, UK) were maintained in individually ventilated cages (Innorack® IVC Mouse 3.5) and handled in a vertical laminar flow biosafety cabinet (Class II Biohazard, Tecniplast). Prior to infections by intranasal (i.n.) route, each mouse was anesthetized by intraperitoneal injection of 1.25 mg of ketamine plus 0.25 mg of xylazine in 250 μl of PBS. On day 1, flu infection is performed intranasally (i.n.) with 50 plaque-forming units (PFU) of the pathogenic, murine-adapted H3N2 influenza A virus strain Scotland/20/74 in 30 μl of PBS (Ivanov et al., 2013; Matarazzo et al., 2019). On day 7, S. pneumoniae infection is done i.n with frozen working stocks of S. pneumoniae at 5 × 104 CFU (single strain) or 5 × 105 CFU (CRISPRi library) in 30 μl of PBS as described previously (Muñoz et al., 2010). In CRISPRi experiments, mice remained on control diet or were provided a diet supplemented with doxycycline (200 mg/kg, Ssniff Spezialdiäten GmbH) on day 3 post-flu infection (4 days prior to pneumococcal infection). At 24 h post-pneumococcal infection, mice were euthanized by intraperitoneal injection of 5.47 mg of sodium pentobarbital in 100 μl of PBS and lungs were sampled in 1 mL PBS. Lung homogenates were plated to evaluate CFU counts or mixed with deoxyribonuclease I (10 μg/ml, Sigma-Aldrich) and filtered through 100 μm and 40 μm meshes, centrifuged at 16,000 g and bacterial genomic DNA was extracted using NucleoSpin Microbial DNA (Macherey-Nagel) for further analysis. The high bacterial load at 24 hpi enabled us to compare two ways of sample preparation: 1) culturing bacteria isolated from tissue in medium before collection and genomic DNA isolation; 2) direct isolation of genomic DNA of bacteria from the lung tissue. The sequencing data showed that there was no significant difference between the two methods, as the samples clustered according to the treatment but not to the way of sample collection in the PCA analysis (Figure S3A), and the sgRNA distribution was very similar between the two approaches (Figure S3B).
METHOD DETAILS
Construction of A Doxycycline Inducible CRISPRi System in S. pneumoniae D39V
The doxycycline inducible CRISPRi system was constructed based on our previously published IPTG-inducible CRISPRi system in S. pneumoniae (Liu et al., 2017) and a newly developed pneumococcal tet-inducible platform (Sorg et al., 2020). First, a constitutively expressed pneumococcal codon-optimized tetR driven by promoter PF6 was amplified from D-T-PEP9Ptet (Sorg et al., 2020) and integrated into the chromosome at the prs1 locus in D39V strain, resulting in strain VL2210. Three fragments were assembled to make the Ptet-dcas9 construct for integration at the bgaA locus. Fragment 1 containing upstream of bgaA and tetM was amplified from DCI23 (Liu et al., 2017) and digested with XbaI; fragment 2 containing tet-inducible promoter PT4–1, here named Ptet, was amplified from plasmid pPEP8T4–1 (Sorg et al., 2020), and digested with XbaI/NotI; fragment 3 containing the coding region of dcas9 and downstream of bgaA locus was amplified from strain DCI23 and digested with NotI. The three fragments were then ligated followed by transformation into VL2210 by selecting with 1 μg/ml tetracycline, resulting in strain VL2212.
Construction of the Dual Fluorescent Reporter Strain and Confocal Microscopy
The codon optimized mNeonGreen was digested from pASR110 (pPEPZ-Plac-mNeonGreen) with BglII and XhoI and cloned into pPEPY-Plac (Keller et al., 2019), followed by transformation into strain VL2212, resulting in strain VL2339. The DNA fragment for insertion of hlpA-mScarlet-I was amplified from strain VL1780 (Kurushima et al., 2020) and transformed into VL2339, resulting in the final dual fluorescent reporter strain VL2351. To verify doxycycline levels were sufficient in vivo to induce inhibition via CRISPRi, mice were switched to feed containing doxycycline (or control feed) two days prior to infection, and then infected with the reporter strain via intra-tracheal infection, during which time mice remained on doxycycline feed (or control feed). At 48 hours post infection, whole blood was collected via cardiac puncture followed by hypotonic lysis of red blood cells and subsequent resuspension of remaining cells in PBS. Samples were placed on a glass slide, heat fixed and mounted in Cytoseal. Slides were imaged using a Leica TCS SPE Confocal microscope with a 63X objective, LAS X acquisition software, and processed using FIJI (Schindelin et al., 2012).
Construction of Knockout and Complementary Mutants in S. pneumoniae
The erythromycin resistant marker, encoded by eryR, was used as selection marker for the knockout mutants. Three fragments were assembled by Golden Gate cloning with either BsaI or BsmBI for each knockout mutant: Fragment 1 containing upstream of the target gene including its promoter sequence; fragment 2 containing eryR coding region with RBS; fragment 3 containing downstream of the target gene. The assembled DNA fragment was then transformed into D39V with 0.5 μg/ml erythromycin for selection. Notice that for making the ΔmetK strain, 10 μM SAM was supplemented in the agar plate. To make the complementary strains, the target gene with its native promoter was amplified from genomic DNA of D39V and ligated with upstream and downstream homologous fragments of the ZIP locus (Keller et al., 2019) followed by transformation into the knockout mutant with 100 μg/ml spectinomycin for selection. Primers used here are listed in Table S9.
Construction of the Pooled CRISPRi Library
Construction of vector pPEPZ-sgRNAclone
Integration vector pPEPZ (Keller et al., 2019) was used as backbone. A gBlock containing Illumina read 1 sequence, P3 promoter, mCherry flanked by BsmBI sites, dCas9 handle binding and terminator region of sgRNA, Illumina read 2 sequence, 8 bp Illumina index sequence and P7 adaptor sequence in order, was synthesized by Integrated DNA Technologies (IDT). In this design, mCherry provides the sgRNA base-pairing cloning sites and will be replaced with 20 bp specific sequences for targeting different genes. The Illumina sequences across the sgRNA cloning sites work as primer binding handles for one-step PCR amplification of the sgRNA sequence, in order to prepare amplicon library for Illumina sequencing. For the Sequence (5’−3’) of the gBlock, see below. GATCTAGCAGATCTGAGAGGATCCCCATTCTACAGTTTATTCTTGACATTGCACTGTCCCCCTGGTATAATAACTATATGAGACGAGGAGGAAAATTAATGAGCAAAGGAGAAGAAGATAACATGGCAATCATCAAAGAATTTATGCGTTTCAAAGTTCACATGGAAGGTTCTGTAAACGGACACGAATTTGAAATTGAAGGTGAAGGTGAAGGCCGTCCTTATGAAGGAACACAAACGGCAAAGCTGAAAGTAACAAAAGGCGGACCGCTTCCGTTTGCATGGGATATCCTTTCTCCGCAATTCATGTACGGTTCAAAAGCATACGTGAAGCATCCGGCTGATATTCCTGATTATTTGAAGCTGTCATTCCCTGAAGGCTTCAAATGGGAGCGTGTGATGAACTTTGAAGATGGCGGTGTTGTTACTGTTACTCAAGATTCAAGCCTTCAAGACGGTGAATTTATTTACAAAGTGAAGCTGCGCGGAACAAACTTCCCATCTGACGGACCTGTCATGCAAAAGAAAACAATGGGCTGGGAAGCAAGCTCTGAACGCATGTATCCAGAGGACGGTGCTTTAAAAGGAGAAATCAAACAGCGTTTGAAGCTGAAAGACGGCGGACACTATGACGCTGAAGTGAAAACAACTTACAAAGCGAAAAAGCCGGTTCAGCTTCCAGGTGCTTACAACGTAAACATCAAACTTGATATTACAAGCCACAATGAAGATTATACGATTGTTGAACAATATGAACGCGCTGAAGGCCGTCATTCAACTGGCGGAATGGATGAGCTTTACAAATAACGTCTCGGTTTAAGAGCTATGCTGGAAACAGCATAGCAAGTTTAAATAAGGCTAGTCCGTTATCAACTTGAAAAAGTGGCACCGAGTCGGTGCTTTTTTTCTGTCTCTTATACACATCTCCGAGCCCACGAGACTAAGGCGAATCTCGTATGCCGTCTTCTGCTTGCTCGAGGCGTATCTAGACGAGATC
The gBlock was digested with BamHI/XholI, and then ligated with BamHI/XholI digested pPEPZ, followed by transformation into E. coli stbl3 selected with 100 μg/ml spectinomycin. The E. coli strain with this vector forms bright red colonies. The vector pPEPZ-sgRNAclone was deposited at Addgene (catalog #141090). Selection of sgRNAs
Primary operons (pTSS operons) were annotated in S. pneumoniae D39V strain in a previous study (Slager et al., 2018). First, for all the identified pTSS operons, one sgRNA targeting the non-template strand with high specificity and close proximity to the pTSS was designed for each operon. However, the pTSS operons cover only about 65% of the genetic elements of S. pneumoniae D39V. For genes that are not covered by pTSS operons, one sgRNA was selected for each gene. S. pneumoniae has multiple types of repeat regions, such as BOX elements, Repeat Units of the Pneumococcus (RUP), SPRITEs and IS elements (Slager et al., 2018). There are some sgRNAs targeting genes located in repeat regions, and as such these sgRNAs have multiple targeting sites. The sgRNAs and targets are listed in Table S1. Post-hoc target identification, including off-target sites, was performed with a custom R script (https://github.com/veeninglab/CRISPRi-seq), of which the results are shown in Table S2 and analyzed separately (https://www.veeninglab.com/crispri-seq, “Pneumococcal sgRNA library efficiency exploration”).
Cloning of sgRNAs by Golden Gate Assembly
Two oligos were designed for each sgRNA (Figure 2). The two oligos were then annealed in TEN buffer (10 mM Tris, 1 mM EDTA, 100 mM NaCl, pH 8) in a thermocycler, 95°C for 5 minutes followed by slowly cooling down to room temperature. The annealed oligos were then pooled together at equimolar concentration, followed by phosphorylation with T4 polynucleotide kinase (New England Biolabs). The vector pPEPZ-sgRNAclone was digested with BsmBI and carefully purified by gel extraction to ensure removal of the mCherry fragment. The annealed oligos and digested pPEPZ-sgRNAclone were then ligated with T4 ligase, followed by transformation into E. coli stbl3 and selected with 100 μg/ml spectinomycin on LB agar plates. In total, more than 70,000 individual transformant colonies were obtained and collected, providing about a 50-fold theoretical coverage of the 1499 sgRNAs. No red colonies were visually present, indicating a very low false positive rate of the cloning. The oligos for cloning of sgRNAs are listed in Table S9.
Construction of the pooled CRISPRi library in S. pneumoniae D39V
Plasmids from the E. coli library with the sgRNA pool were isolated, and transformed into CSP-induced competent S. pneumoniae VL2212 (this study) and DCI23 (Liu et al., 2017) to construct doxycycline-inducible and IPTG-inducible CRISPRi libraries, respectively. More than 107 individual transformant colonies were obtained and collected for both of the strains.
sgRNA Library Target and Efficiency Evaluation
All potential sgRNA binding sites on the S. pneumoniae D39V genome were identified using the R package CRISPRseek (Zhu et al., 2014), taking into account PAM presence and allowing up to eight mismatches between spacers and genome. We set the maximum number of allowed mismatches to eight, because of (1) the exponential growth of computation time with this parameter and (2) any potential effect on a site with >8 mismatches was assumed to be negligible. The S. pneumoniae D39V genome (Slager et al., 2018) was downloaded from NCBI (CP027540.1) and read into R using the biomartr package (Drost and Paszkowski, 2017). All identified binding sites can be found in Table S2.
In addition to the standard CRISPRseek output, we assessed for each binding site if it overlapped with any genetic element annotated with a locus_tag key in the GFF file on the non-template (NT) strand. If any, the locus tag was added to the table (“NTgene”), as well as which part of the sgRNA corresponding to that binding site was overlapping (“coverPart”: complete, 5`- or 3`-end) and with how many base pairs (“coverSize”) including the PAM. In case one binding site overlapped multiple annotated elements, both were inserted as a row in the table, with matching “site” numbers.
Furthermore, we estimated the relative retained repression activity (“reprAct”) of each sgRNA binding site compared to a hypothetical zero-mismatch binding site on the same locus, based on the mismatches with the sgRNA spacer. Retained repression activity depends on both the number and the within-spacer position of mismatches (Qi et al., 2013). Furthermore, the retained activity for an sgRNA with two adjacent mismatches appears to be the product of their individual retained scores, relative to a zero-mismatch silencing effect (Qi et al., 2013). We assumed this multiplication principle also holds for >2 and non-adjacent mismatches. Therefore, we computed per sgRNA, per binding site, the expected repression activity as the product of the nucleotide-specific retained activity scores as reported by Qi et al. (2013), estimated from their Figure 5D and averaged over the three intra-spacer regions they defined (Qi et al., 2013). The resulting score represents the estimated retained repression activity of the sgRNA on the binding site, relative to a hypothetical zero-mismatch binding site for the same sgRNA on the same chromosomal locus, on the [0,1] interval.
According to this method, the maximum repression effect of any site with >8 mismatches would be 0.77% of the hypothetical zero-mismatch effect. This was indeed considered negligible, supporting our decision to only consider sgRNA binding sites with eight mismatches or less.
Lastly, we added to the table the relative distance of the binding site to the start codon of the genetic element it binds to, if any (“dist2SC”). This distance is normalized to the [0,1] interval using feature scaling, where a distance of 0 means binding on the start codon or partially overlap with the 5’-end of the element, and a distance of 1 means binding at or partial overlap with the far 3’-end of the element. Smaller distances are associated with more efficient transcription repression (Qi et al., 2013).
The custom R script used to produce this table can be found on GitHub (https://github.com/veeninglab/CRISPRi-seq). The script is written in a generic way, allowing to run the complete pipeline described above for any given NCBI genome, as we did for S. pneumoniae strains TIGR4 (AE005672.3), R6 (AE007317.1), Hungary19A-6 (CP000936.1), Taiwan19F-14 (CP000921.1), 11A (CP018838.1) and G54 (CP001015.1). Results tables and analysis of these genomes can be found on the Veeninglab website (https://www.veeninglab.com/crispri-seq).
CRISPRi-seq Screen in Laboratory Medium
The screen was done over approximately 21 generations of growth in triplicates. The pooled libraries were grown in C+Y medium at 37°C to OD595=0.3 as preculture. Then, the precultures were diluted 1:100 into C+Y medium with or without inducer, 1 mM IPTG or 10 ng/ml doxycycline. When OD595 reached 0.3, cultures were diluted into fresh medium by 1:100 again. Another 1:100 dilution was done in the same fashion, so in total three times of 1:100 dilution, ensuring about 21 generations of induction and competition (doubling time of approximately 26 min). The multiple dilutions with fresh medium were performed to make sure that bacteria stay in exponential phase during the treatment to prevent stationary phase-induced autolysis. Bacteria were collected when OD600=0.3 after the third dilution, and the pellets were used for gDNA isolation with the Wizard Genomic DNA Purification Kit (Promega) as described previously (Liu et al., 2017). The fitness evaluated by IPTG- or doxycycline- inducible library is listed in Table S3.
Library Preparation, Sequencing and Data Analysis
The Illumina libraries were prepared by one-step PCR with oligos listed in Table S9. The isolated gDNAs of S. pneumoniae were used as template for PCR. The index 1, index 2 and adapter sequence were introduced by this one-step PCR. N701-N712 were used as index 1, and N501-N508 were used as index 2 (illumine barcodes). For the sequence of the amplicon, see below. AATGATACGGCGACCACCGAGATCTACACTAGATCGC(N501)TCGTCGGCAGCGTCAGATGTGTATAAGAGACAGCCATTCTACAGTTTATTCTTGACATTGCACTGTCCCCCTGGTATAATAACTATANNNNNNNNNNNNNNNNNNNNGTTTAAGAGCTATGCTGGAAACAGCATAGCAAGTTTAAATAAGGCTAGTCCGTTATCAACTTGAAAAAGTGGCACCGAGTCGGTGCTTTTTTTCTGTCTCTTATACACATCTCCGAGCCCACGAGACTAAGGCGA(N701)ATCTCGTATGCCGTCTTCTGCTTG Note that we used i5 (N501) and i7 (N701) as example barcodes to show the amplicon sequence. The highlighted 20N represents the base-pairing region of sgRNAs. For full annotation of the amplicon, please refer to “CRISPRiseq amplicon.dna” file on https://www.veeninglab.com/crispri-seq.
For each 50 μl of PCR reaction, 4 μg of gDNA was used as input template, which enables us to obtain sufficient PCR products with as little as 8 cycles of PCR. In addition, we have tested 10 cycles, 20 cycles and 30 cycles of PCR reaction, and no significant difference was observed (data not shown), indicating no detectable bias introduced by PCR. The amplicons (304 bp) were then purified from a 2% agarose gel. Concentrations of amplicons were then determined by a Qubit assay (Q32854, ThermoFisher Scientific). Purified amplicons were sequenced on a MiniSeq (Illumina) with a custom sequencing protocol. The first 54 cycles for sequencing of common sequence of amplicons were set as dark cycles, and the following 20 cycles were used for sequencing of the diversified base-pairing region of sgRNA. The fastq files generated from sequencing are uploaded to the Sequence Read Archive (SRA) on NCBI with accession number PRJNA611488.
The 20 bp base-pairing sequences were trimmed out from read 1 according to their position with Trimmomatic Version 0.36 (Bolger et al., 2014). To map the sgRNA sequences, a pseudogenome containing all the sgRNA sequences was prepared, and the sgRNA sequences on the pseudogenome were annotated with sgRNA numbers, 1 to 1499. Then the trimmed reads were mapped to the pseudogenome with Bowtie 2 (Langmead and Salzberg, 2012). The sgRNAs were counted with featureCounts (Liao et al., 2014), all the resulting raw counts are shown in Table S7. The count data of sgRNAs were then analyzed with the DESeq2 package in R (Love et al., 2014) for evaluation of fitness cost of each sgRNA. We tested against a log2FC of 1, with an alpha of 0.05. Whenever log2FC are visualized or reported, these are shrunk with the apeglm method (Zhu et al., 2019). The R script used for analysis is available at https://github.com/veeninglab/CRISPRi-seq. The size of infection bottlenecks was calculated as reported previously (Abel et al., 2015b). The in vivo doubling time of S. pneumoniae used in the calculation was based on a previous Tn-seq study (Opijnen and Camilli, 2012) as 108 minutes.
Growth and Luciferase Assays
For Figures 1B, 6A and 6F, the working stock of each mutant, T2 cells, were thawed and diluted 1:100 into fresh C+Y medium, or C+Y medium with doxycycline at different final concentrations, or with different concentrations of S-(5’-Adenosyl)-L-methionine (A7007, Sigma Aldrich), as the initial cell culture. For Figure 6D, the T2 cells were thawed and diluted 1:100 into fresh Blood Like Medium (BLM) without nucleobases solution, or supplemented with individual nucleobases component (adenine, adenosine, guanine, uracil, uridine and xanthine), or with all the components (Aprianto et al., 2018). Then 300 μl of the initial culture was aliquoted into each well of 96-well plates with 3 replicates. Cell density were monitored by measuring OD595 every 10 minutes with a Tecan Spark microtiter plate reader at 37°C. Luciferase assay (Figure 1B) was performed as previously described (Liu et al., 2017). Luciferin (D-Luciferin sodium salt, SYNCHEM OHG) was added into C+Y medium at final concentration of 450 μg/ml as substrate of the luciferase. Luminescence signals were measured every 10 minutes with a Tecan Spark microtiter plate. Growth and luciferase activity curves were plotted with Prism 8 as described previously (Sorg and Veening, 2015).
Bottleneck Population Size Estimation
The size of infection bottlenecks was calculated as reported previously (Abel et al., 2015b). The doubling time of S. pneumoniae used in the calculation was based on a previous Tn-seq study (Opijnen and Camilli, 2012) as 108 minutes. To calculate the bottleneck size (Nb), we assume that the changes in sgRNA frequencies in the mice without doxycycline treatment are introduced by random survival of S. pneumoniae that pass through a population bottleneck. The equations we used for calculation of bottleneck size are described in a previous study (Abel et al., 2015b).
And
where k is the total number of distinct alleles, here is the number of sgRNAs in the library; fi,0 the frequency of sgRNAi at time 0, fi,s the frequency of sgRNAi at sampling, g the number of generations during infection, and S0 and Ss the number of sgRNA reads at time 0 or at sampling, respectively. The data used for this calculation are in Table S7.
QUANTIFICATION AND STATISTICAL ANALYSIS
Data analyses were performed with GraphPad Prism (v8.0) and R(v3.6.1). Data shown in plots are averages of at least 3 replicates with SEM. For animal infection assays, at least 5 mice were used for each group, and differences were determined using the Mann-Whitney U test for comparing two groups, Kruskal-Wallis test with Dunn’s post-analysis for comparing multiple groups. P values were stated in the figure legends. Sample size and statistical tests are also reported in the figure legends.
ADDITIONAL RESOURCES
Code, output and analyses for sgRNA library evaluation on any given genome: https://veeninglab.com/crispri-seq
Supplementary Material
Table S1. List of sgRNA and targets.xlsx, related to STAR methods and Figure 2.
Table S2. All binding sites sgRNA library in D39V.xlsx, related to STAR methods.
Table S3. Fitness evaluated by IPTG- and Tet-inducible libraries, related to Figure 3.
Table S4. Comparison of Tn-seq with CRISPRi-seq, related to Figure 3.
Table S5. Comparison of fitness between murine pneumonia model and C+Y medium, related to Figure 4 and 5.
Table S6. Comparison of fitness between IAV coinfection model and C+Y medium, related to Figure 6.
Table S9. Oligos used in this study, related to STAR methods.
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Bacterial and Virus Strains | ||
| Bacterial strains are listed in Table S8 | This paper | N/A |
| Murine-adapted H3N2 influenza A virus | Dr Mustapha Si-Tahar (University of Tours) | strain Scotland/20/74 |
| Chemicals, Peptides, and Recombinant Proteins | ||
| D-luciferine | Synchem | CAS:115144-35-9 |
| Wizard Genomic DNA Purification Kit | Promega | Cat#A1120 |
| NucleoSpin Microbial DNA | Macherey-Nagel | Cat#740235.50 |
| Deposited Data | ||
| Sequencing output (Fastq files) | This paper | SRA: PRJNA611488 |
| Code for sgRNA binding site identification and efficiency evaluation | This paper | https://github.com/veeninglab/CRISPRi-seq |
| Binding sites for sgRNA library | This paper | https://veeninglab.com/crispri-seq |
| Experimental Models: Organisms/Strains | ||
| 6–8 week old female BALB/c mice | Jackson Laboratories | 000651 |
| 7-week old male C57BL/6JRj | Janvier Laboratories | C57BL/6JRj |
| Oligonucleotides | ||
| gBlock for pPEPZ-sgRNAclone construction (See SI for sequence) | Integrated DNA Technologies | N/A |
| Oligos for sgRNA pool (See Table S9) | Synbio Technologies | N/A |
| Other oligos for PCR (See Table S9) | Sigma | N/A |
| Recombinant DNA | ||
| pPEPZ-sgRNAclone | This paper | Addgene #141090 |
| Software and Algorithms | ||
| Prism v8.0 | GraphPad Software | https://www.graphpad.com/ |
| Bowtie2 | Langmead and Salzberg, 2012 | http://bowtie-bio.sourceforge.net/bowtie2/index.shtml |
| featureCounts | Liao et al., 2014 | N/A |
| ImageJ v2.0 | National Institutes of Health | https://imagej.nih.gov/ij/ |
| R v3.6.1 | The R Foundation for Statistical Computing | https://www.r-project.org/ |
| Illustrator CC | Adobe | https://www.adobe.com |
Highlights.
CRISPRi-seq enables in vivo genome-wide fitness testing in one sequencing step
Identification of a strong bottleneck in a S. pneumoniae murine pneumonia model
CRISPRi-seq reveals pneumococcal genes critical during influenza virus superinfection
CRISPRi-seq can test gene fitness of all genes, including essential genes
ACKNOWLEGEMENTS
We appreciate all members of the Veening lab for stimulating discussions and thank Lance Keller for feedback on the manuscript. We thank Indiana Castro and Charlotte Costa for technical help on the IAV co-infection model. This work was supported by the Swiss National Science Foundation (SNSF) (project grant 31003A_172861 to J.W.V.), SNSF JPIAMR grant (40AR40_185533 to J.W.V.) and SNSF NCCR ‘AntiResist’ (51NF40_180541 to J.W.V.). Work in the Nizet lab is supported by NIH grant AI145325. J.M.K. was supported by the University of California President’s Postdoctoral Fellowship Program (UC PPFP). J.C.S. received funding from Inserm, University of Lille, Institut Pasteur de Lille and the European Union’s Horizon 2020 research and innovation programme under grant agreement No 847786.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
DECLARATION OF INTERESTS
The authors declare no conflicting interests.
References
- Abel S, Wiesch P.A. zur, Davis BM, and Waldor MK. (2015a). Analysis of Bottlenecks in Experimental Models of Infection. PLOS Pathog. 11, e1004823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Abel S, Abel zur Wiesch P, Chang H-H, Davis BM, Lipsitch M, and Waldor MK (2015b). Sequence tag–based analysis of microbial population dynamics. Nat. Methods 12, 223–226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aprianto R, Slager J, Holsappel S, and Veening J-W (2018). High-resolution analysis of the pneumococcal transcriptome under a wide range of infection-relevant conditions. Nucleic Acids Res. 46, 9990–10006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berney M, Berney-Meyer L, Wong K-W, Chen B, Chen M, Kim J, Wang J, Harris D, Parkhill J, Chan J, et al. (2015). Essential roles of methionine and S-adenosylmethionine in the autarkic lifestyle of Mycobacterium tuberculosis. Proc. Natl. Acad. Sci. U. S. A 112, 10008–10013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bikard D, Jiang W, Samai P, Hochschild A, Zhang F, and Marraffini LA (2013). Programmable repression and activation of bacterial gene expression using an engineered CRISPR-Cas system. Nucleic Acids Res. 41, 7429–7437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bolger AM, Lohse M, and Usadel B (2014). Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinforma. Oxf. Engl 30, 2114–2120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Briles DE, Nahm M, Schroer K, Davie J, Baker P, Kearney J, and Barletta R (1981). Antiphosphocholine antibodies found in normal mouse serum are protective against intravenous infection with type 3 Streptococcus pneumoniae. J. Exp. Med 153, 694–705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cain AK, Barquist L, Goodman AL, Paulsen IT, Parkhill J, and van Opijnen T (2020). A decade of advances in transposon-insertion sequencing. Nat. Rev. Genet 1–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chalker AF, Ingraham KA, Lunsford RD, Bryant AP, Bryant J, Wallis NG, Broskey JP, Pearson SC, and Holmes DJ (2000). The bacA gene, which determines bacitracin susceptibility in Streptococcus pneumoniae and Staphylococcus aureus, is also required for virulence. Microbiol. Read. Engl 146 (Pt 7), 1547–1553. [DOI] [PubMed] [Google Scholar]
- Chen H, Ma Y, Yang J, O’Brien CJ, Lee SL, Mazurkiewicz JE, Haataja S, Yan J-H, Gao GF, and Zhang J-R (2007). Genetic requirement for pneumococcal ear infection. PloS One 3, e2950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chiavolini D, Pozzi G, and Ricci S (2008). Animal Models of Streptococcus pneumoniae Disease. Clin. Microbiol. Rev 21, 666–685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cools F, Torfs E, Aizawa J, Vanhoutte B, Maes L, Caljon G, Delputte P, Cappoen D, and Cos P (2019). Optimization and Characterization of a Galleria mellonella Larval Infection Model for Virulence Studies and the Evaluation of Therapeutics Against Streptococcus pneumoniae. Front. Microbiol 10, 311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cui L, Vigouroux A, Rousset F, Varet H, Khanna V, and Bikard D (2018). A CRISPRi screen in E. coli reveals sequence-specific toxicity of dCas9. Nat. Commun 9, 1912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davis KM, Mohammadi S, and Isberg RR (2015). Community behavior and spatial regulation within a bacterial microcolony in deep tissue sites serves to protect against host attack. Cell Host Microbe 17, 21–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dönhöfer A, Franckenberg S, Wickles S, Berninghausen O, Beckmann R, and Wilson DN (2012). Structural basis for TetM-mediated tetracycline resistance. Proc. Natl. Acad. Sci 109, 16900–16905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Drost HG, and Paszkowski J (2017). Biomartr: Genomic data retrieval with R. Bioinformatics 33, 1216–1217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gerlini A, Colomba L, Furi L, Braccini T, Manso AS, Pammolli A, Wang B, Vivi A, Tassini M, Rooijen N. van , et al. (2014). The Role of Host and Microbial Factors in the Pathogenesis of Pneumococcal Bacteraemia Arising from a Single Bacterial Cell Bottleneck. PLOS Pathog. 10, e1004026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hava DL, and Camilli A (2002). Large-scale identification of serotype 4 Streptococcus pneumoniae virulence factors: Pneumococcal virulence factors. Mol. Microbiol 45, 1389–1406. [PMC free article] [PubMed] [Google Scholar]
- Ivanov S, Renneson J, Fontaine J, Barthelemy A, Paget C, Fernandez EM, Blanc F, De Trez C, Van Maele L, Dumoutier L, et al. (2013). Interleukin-22 reduces lung inflammation during influenza A virus infection and protects against secondary bacterial infection. J. Virol 87, 6911–6924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang W, Oikonomou P, and Tavazoie S (2020). Comprehensive Genome-wide Perturbations via CRISPR Adaptation Reveal Complex Genetics of Antibiotic Sensitivity. Cell 180, 1002–1017.e31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jim KK, Engelen-Lee J, van der Sar AM, Bitter W, Brouwer MC, van der Ende A, Veening J-W, van de Beek D, and Vandenbroucke-Grauls CMJE (2016). Infection of zebrafish embryos with live fluorescent Streptococcus pneumoniae as a real-time pneumococcal meningitis model. J. Neuroinflammation 13, 188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keller LE, Rueff A-S, Kurushima J, and Veening J-W (2019). Three New Integration Vectors and Fluorescent Proteins for Use in the Opportunistic Human Pathogen Streptococcus pneumoniae. Genes 10, 394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kono M, Zafar MA, Zuniga M, Roche AM, Hamaguchi S, and Weiser JN (2016). Single Cell Bottlenecks in the Pathogenesis of Streptococcus pneumoniae. PLOS Pathog. 12, e1005887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kreibich S, and Hardt W-D (2015). Experimental approaches to phenotypic diversity in infection. Curr. Opin. Microbiol 27, 25–36. [DOI] [PubMed] [Google Scholar]
- Kurushima J, Campo N, van Raaphorst R, Cerckel G, Polard P, and Veening J-W (2020). Unbiased homeologous recombination during pneumococcal transformation allows for multiple chromosomal integration events. ELife 9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Langmead B, and Salzberg SL (2012). Fast gapped-read alignment with Bowtie 2. Nat. Methods 9, 357–359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lau GW, Haataja S, Lonetto M, Kensit SE, Marra A, Bryant AP, McDevitt D, Morrison DA, and Holden DW (2001). A functional genomic analysis of type 3 Streptococcus pneumoniae virulence: Functional genomics of Streptococcus pneumoniae. Mol. Microbiol 40, 555–571. [DOI] [PubMed] [Google Scholar]
- Lee HH, Ostrov N, Wong BG, Gold MA, Khalil AS, and Church GM (2019). Functional genomics of the rapidly replicating bacterium Vibrio natriegens by CRISPRi. Nat. Microbiol 1. [DOI] [PubMed] [Google Scholar]
- Li T, Yu G, Guo T, Qi H, Bing Y, Xiao Y, Li C, Liu W, Yuan Y, He Y, et al. (2015). The Plasma S-adenosylmethionine Level is Associated With the Severity of Hepatitis B-Related Liver Disease. Medicine (Baltimore) 94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Y, Thompson CM, Trzciński K, and Lipsitch M (2013). Within-Host Selection Is Limited by an Effective Population of Streptococcus pneumoniae during Nasopharyngeal Colonization. Infect. Immun 81, 4534–4543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liao Y, Smyth GK, and Shi W (2014). featureCounts: an efficient general purpose program for assigning sequence reads to genomic features. Bioinforma. Oxf. Engl 30, 923–930. [DOI] [PubMed] [Google Scholar]
- Liu X, Gallay C, Kjos M, Domenech A, Slager J, Kessel S.P. van, Knoops K, Sorg RA, Zhang J-R, and Veening J-W. (2017). High-throughput CRISPRi phenotyping identifies new essential genes in Streptococcus pneumoniae. Mol. Syst. Biol 13, 931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Love MI, Huber W, and Anders S (2014). Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 15, 550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matarazzo L, Casilag F, Porte R, Wallet F, Cayet D, Faveeuw C, Carnoy C, and Sirard J-C (2019). Therapeutic Synergy Between Antibiotics and Pulmonary Toll-Like Receptor 5 Stimulation in Antibiotic-Sensitive or -Resistant Pneumonia. Front. Immunol 10, 723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCullers JA (2006). Insights into the interaction between influenza virus and pneumococcus. Clin. Microbiol. Rev 19, 571–582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCullers JA (2014). The co-pathogenesis of influenza viruses with bacteria in the lung. Nat. Rev. Microbiol 12, 252–262. [DOI] [PubMed] [Google Scholar]
- Molzen TE, Burghout P, Bootsma HJ, Brandt CT, van der Gaast-de Jongh CE, Eleveld MJ, Verbeek MM, Frimodt-Møller N, Østergaard C, and Hermans PWM. (2011). Genome-Wide Identification of Streptococcus pneumoniae Genes Essential for Bacterial Replication during Experimental Meningitis. Infect. Immun 79, 288–297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muñoz N, Van Maele L, Marqués JM, Rial A, Sirard J-C, and Chabalgoity JA (2010). Mucosal administration of flagellin protects mice from Streptococcus pneumoniae lung infection. Infect. Immun 78, 4226–4233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mutschler H, Gebhardt M, Shoeman RL, and Meinhart A (2011). A Novel Mechanism of Programmed Cell Death in Bacteria by Toxin–Antitoxin Systems Corrupts Peptidoglycan Synthesis. PLoS Biol. 9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nishimoto AT, Rosch JW, and Tuomanen EI (2020). Pneumolysin: Pathogenesis and Therapeutic Target. Front. Microbiol 11, 1543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Opijnen T, and Camilli A (2012). A fine scale phenotype–genotype virulence map of a bacterial pathogen. Genome Res. 22, 2541–2551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Opijnen T, Bodi KL, and Camilli A (2009). Tn-seq: high-throughput parallel sequencing for fitness and genetic interaction studies in microorganisms. Nat. Methods 6, 767–772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peters JM, Colavin A, Shi H, Czarny TL, Larson MH, Wong S, Hawkins JS, Lu CHS, Koo B-M, Marta E, et al. (2016). A Comprehensive, CRISPR-based Functional Analysis of Essential Genes in Bacteria. Cell 165, 1493–1506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van der Poll T, and Opal SM (2009). Pathogenesis, treatment, and prevention of pneumococcal pneumonia. The Lancet 374, 1543–1556. [DOI] [PubMed] [Google Scholar]
- Qi LS, Larson MH, Gilbert LA, Doudna JA, Weissman JS, Arkin AP, and Lim WA (2013). Repurposing CRISPR as an RNA-guided platform for sequence-specific control of gene expression. Cell 152, 1173–1183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qu J, Prasad NK, Yu MA, Chen S, Lyden A, Herrera N, Silvis MR, Crawford E, Looney MR, Peters JM, et al. (2019). Modulating Pathogenesis with Mobile-CRISPRi. J. Bacteriol 201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Redelsperger IM, Taldone T, Riedel ER, Lepherd ML, Lipman NS, and Wolf FR (2016). Stability of Doxycycline in Feed and Water and Minimal Effective Doses in Tetracycline-Inducible Systems. J. Am. Assoc. Lab. Anim. Sci. JAALAS 55, 467–474. [PMC free article] [PubMed] [Google Scholar]
- Rosch JW, Sublett J, Gao G, Wang Y-D, and Tuomanen EI (2008). Calcium efflux is essential for bacterial survival in the eukaryotic host. Mol. Microbiol 70, 435–444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rudd JM, Ashar HK, Chow VT, and Teluguakula N (2016). Lethal Synergism between Influenza and Streptococcus pneumoniae. J. Infect. Pulm. Dis 2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saralahti A, Piippo H, Parikka M, Henriques-Normark B, Rämet M, and Rounioja S (2014). Adult zebrafish model for pneumococcal pathogenesis. Dev. Comp. Immunol 42, 345–353. [DOI] [PubMed] [Google Scholar]
- Schindelin J, Arganda-Carreras I, Frise E, Kaynig V, Longair M, Pietzsch T, Preibisch S, Rueden C, Saalfeld S, Schmid B, et al. (2012). Fiji: an open-source platform for biological-image analysis. Nat. Methods 9, 676–682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Siegel SJ, Roche AM, and Weiser JN (2014). Influenza Promotes Pneumococcal Growth during Coinfection by Providing Host Sialylated Substrates as a Nutrient Source. Cell Host Microbe 16, 55–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Slager J, Aprianto R, and Veening JW (2018). Deep genome annotation of the opportunistic human pathogen Streptococcus pneumoniae D39. Nucleic Acids Res. 46, 9971–9989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sorg RA, and Veening J-W (2015). Microscale insights into pneumococcal antibiotic mutant selection windows. Nat. Commun 6, 8773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sorg RA, Gallay C, and Veening J-W (2020). Synthetic gene regulatory networks in the opportunistic human pathogen Streptococcus pneumoniae. B Proc. Natl. Acad. Sci in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Maele L, Carnoy C, Cayet D, Ivanov S, Porte R, Deruy E, Chabalgoity JA, Renauld J-C, Eberl G, Benecke AG, et al. (2014). Activation of Type 3 innate lymphoid cells and interleukin 22 secretion in the lungs during Streptococcus pneumoniae infection. J. Infect. Dis 210, 493–503. [DOI] [PubMed] [Google Scholar]
- Wang T, Guan C, Guo J, Liu B, Wu Y, Xie Z, Zhang C, and Xing X-H (2018). Pooled CRISPR interference screening enables genome-scale functional genomics study in bacteria with superior performance. Nat. Commun 9, 2475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weiser JN, Ferreira DM, and Paton JC (2018). Streptococcus pneumoniae : transmission, colonization and invasion. Nat. Rev. Microbiol 1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Wet TJ, Gobe I, Mhlanga MM, and Warner DF (2018). CRISPRi-Seq for the Identification and Characterisation of Essential Mycobacterial Genes and Transcriptional Units. BioRxiv, 10.1101/358275 [DOI] [Google Scholar]
- Zafar MA, Wang Y, Hamaguchi S, and Weiser JN (2017). Host-to-Host Transmission of Streptococcus pneumoniae Is Driven by Its Inflammatory Toxin, Pneumolysin. Cell Host Microbe 21, 73–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu A, Ibrahim JG, and Love MI (2019). Heavy-tailed prior distributions for sequence count data: removing the noise and preserving large differences. Bioinforma. Oxf. Engl 35, 2084–2092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu LJ, Holmes BR, Aronin N, and Brodsky MH (2014). CRISPRseek: A Bioconductor package to identify target-specific guide RNAs for CRISPR-Cas9 genome-editing systems. PLoS ONE 9. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1. List of sgRNA and targets.xlsx, related to STAR methods and Figure 2.
Table S2. All binding sites sgRNA library in D39V.xlsx, related to STAR methods.
Table S3. Fitness evaluated by IPTG- and Tet-inducible libraries, related to Figure 3.
Table S4. Comparison of Tn-seq with CRISPRi-seq, related to Figure 3.
Table S5. Comparison of fitness between murine pneumonia model and C+Y medium, related to Figure 4 and 5.
Table S6. Comparison of fitness between IAV coinfection model and C+Y medium, related to Figure 6.
Table S9. Oligos used in this study, related to STAR methods.
Data Availability Statement
The code produced in this study, and the raw files are available at GitHub (https://github.com/veeninglab/CRISPRi-seq). Results tables and analysis of the genomes tested in this study can be found on the Veeninglab website (https://www.veeninglab.com/crispri-seq). The fastq files generated from sequencing are uploaded to the Sequence Read Archive (SRA) on NCBI with accession number PRJNA611488.