

Abstract
G protein-coupled receptors (GPCRs) have been exploited as primary targets for drug discovery, and GPCR dimerization offers opportunities for drug design and disease treatment. An important strategy for targeting putative GPCR dimers is the use of bivalent ligands, which are single molecules that contain two pharmacophores connected through a spacer. Here, we discuss the selection of pharmacophores, the optimal length and chemical composition of the spacer, and the choice of spacer attachment points to the pharmacophores. Furthermore, we review the most recent advances (from 2018 to the present) in the design, discovery and development of bivalent ligands. We aim to reveal the state-of-the-art design strategy for bivalent ligands and provide insights into future opportunities in this promising field of drug discovery.
Keywords: GPCR dimers, Bivalent ligands, Pharmacophores, Attachment points, Length and chemical composition
Introduction
G protein-coupled receptors (GPCRs) constitute the largest family of transmembrane (TM) receptors in humans, and they have been exploited as primary targets in drug discovery. For many years, GPCRs were considered to exist and function as monomeric entities, but in 1979, Hazum and colleagues reported that opioid receptors occurred in clusters on the surface of neuroblastoma cells [1]. To our knowledge, this was the first experimental suggestion that GPCRs form oligomers. GPCRs that have been reported to generate homomeric or heteromeric complexes include opioid receptors (ORs) [2,3], dopamine D1,2,3 receptors [4,5], metabotropic glutamate receptor 5 (mGluR5) [6,7], γ-aminobutyric acid receptors (GABAB1,B2) [8,9] and α2-adrenergic/M3 muscarinic receptors [10]. Most of the evidence supporting the existence of GPCR dimers also corroborates the existence of GPCR oligomers; such possibilities cannot be readily distinguished by currently available techniques. Therefore, the term dimer is often used, and will be adopted for the remainder of this article [11].
GPCR dimerization affords opportunities for drug design and the treatment of numerous diseases. An important strategy used in targeting putative GPCR dimers is the design of bivalent ligands, which are single molecules that contain two discrete pharmacophores linked by a spacer. They are grouped into two classes: homobivalent ligands (with two identical pharmacophores) and heterobivalent ligands (with two different pharmacophores) (Figure 1a). Targeting GPCR dimers with bivalent ligands that are designed to occupy dimeric binding sites can provide remarkable pharmacological benefits, such as higher binding affinity and selectivity, lower levels of drug–drug interactions (compared with co-administration of conventional therapeutics) and enhanced physiological responses [12]. Therefore, the bivalent-ligand approach represents a powerful strategy for overcoming obstacles associated with GPCR dimers.
Figure 1.

(a) A general schematic diagram for homobivalent and heterobivalent ligands. (b) The chemical structures of the bivalent ligand KDN21 and its two monomeric counterparts, NTI and GNTI. (c) The chemical structures of the bivalent ligand MCC22 and the two lead compounds oxymorphone and TAK-220.
The concept of bivalent ligands was first introduced in 1982 by the pioneer of the field, Philip Portoghese, in two reports [13,14]. Over the past four decades, increasing numbers of bivalent ligands have been synthesized to target GPCRs, and several reviews from the medicinal-chemistry perspective have summarized their development [12,15-21]. In this article, we focus on the most recent advances in the design, discovery and development of bivalent ligands targeting putative GPCR dimers from the perspectives of medicinal chemistry, structural biology and computational modeling. Specifically, we address two main factors: the selection of suitable monomeric pharmacophores and the choice of a spacer with an ideal length and chemical composition. Finally, we review the most recent advances in the field.
Selecting appropriate pharmacophores (lead compounds)
The selection of the pharmacophore (or pharmacophores) is crucial when targeting GPCR dimers. The ideal monomeric lead compound (or compounds) should selectively bind to the desired receptor and have several attachment sites that can be chemically functionalized to introduce the spacer. A general summary of common characteristics of lead compounds can be found in a review by Shonberg et al. [12]. The authors concluded that original lead compounds chosen for the synthesis of bivalent ligands are generally endowed with low to medium molecular weight (between 300 and 400 Da), and these lead compounds should have high specificity and generally high potency, with low nanomolar affinities.
Bivalent ligands targeting the δ-κ-opioid receptor heterodimer
To investigate the δ-κ-opioid receptor heterodimer in the spinal cord, a series of bivalent ligands were synthesized and evaluated by Portoghese and colleagues. In this study, the δ-selective antagonistic pharmacophore naltrindole (NTI, antagonist potency to [D-Ala2,D-Leu5]enkephalin Ke = 0.13 nM [22]) was tethered through variable-length spacers to the κ-selective antagonistic pharmacophore 5′-guanidino naltrindole (5′-GNTI, Ki = 0.18 nM [23]) to obtain the KDN series of bivalent compounds. In vivo antagonism of selective opioid receptor agonist-induced antinociceptive effects as well as in vitro binding data provided evidence, for the first time, of the bridging of the spinal δ-κ putative heterodimer by KDN-21 (Figure 1b) [24]. The in vivo study results suggested that selective targeting of the δ-κ heterodimer in the spinal cord but not in the brain could yield bivalent ligands as potential therapeutics as analgesics.
Bivalent ligands targeting the MOR–CCR5 heterodimer
In another study, a series of bivalent ligands that contained the μ-opioid receptor (MOR) agonist pharmacophore oxymorphone (Ki = 0.97 nM [25]) and the C-C chemokine receptor type 5 (CCR5) antagonist pharmacophore TAK-220 (IC50 = 3.5 nM [26]) linked through homologous spacers was synthesized (Figure 1c). These compounds were further evaluated to explore the possibility of MOR–CCR5 heterodimer involvement in the crosstalk between the two receptors; it is thought that chemokine release facilitates such crosstalk, which is unfavorable for the efficacy of morphine in the treatment of chronic pain. The results showed that the bivalent ligand MCC22 with a 22-atom spacer displayed remarkable antinociceptive activity (ED50 = 0.015 or 0.019 pmol/mouse) that was 2000 times or 700 times greater than that of morphine (ED50 = 35 or 15 pmol/mouse) when tested on lipopolysaccharide (LPS)- or complete Freund’s adjuvant (CFA)-inflamed mice [27]. These breathtaking results indicated that the MOR-CCR5 putative heterodimer could be a novel target for the treatment of chronic pain. In fact, a recent study further demonstrated that MCC22 has the potential to immensely improve the management of chronic pain related to chemotherapy-induced peripheral neuropathy given the profound potency of MCC22 and its lack of tolerance or rewarding properties [28]. Both studies corroborated the potential existence of the MOR–CCR5 heterodimer in vivo, and MCC22 offers a promising approach for the development of potent analgesics that are devoid of unwanted side effects.
These representative studies show that the selection of a suitable monomeric pharmacophore, even one with low to subnanomolar potency, can yield an optimal bivalent ligand for use as a pharmacological tool or even as a potential treatment. Interestingly, many lead compounds used in the design of GPCR bivalent ligands (e.g., NTI, GNTI and TAK-220) (see Figure 1b and 1c) have a molecular weight greater than 400 Da or even 500 Da. Thus, there seems to be no gold standard for assessing the molecular weight of selected lead compounds; the only requirements are that these small molecules should demonstrate potent and selective binding affinity for the desired GPCR, as well as favorable physiochemical properties.
Choosing attachment points on the two pharmacophores
The choice of appropriate attachment points on pharmacophores must be made before synthesizing bivalent ligands. Different choices would yield changes to the synthetic protocols and the generation of bivalent ligands with varied spatial arrangements of the pharmacophores, as well as distinct biological activities. Determining the best positions in which to attach the spacer on the two pharmacophore units can be achieved, in our opinion, through two approaches: traditional medicinal chemistry and structural biology paired with molecular modeling.
Traditional medicinal chemistry
An in-depth understanding of the known SARs of each monomeric counterpart and the feasibility of synthesizing them can aid the identification of the optimal attachment points. In this regard, SAR conclusions help to identify key functional groups on the chosen pharmacophores whose chemical modification would be deleterious to binding affinity and potency. The large size and intrinsic chemical nature of bivalent ligands can make their synthesis challenging. The following examples demonstrate rational decisions for determining attachment points for each pharmacophore.
Bivalent ligands targeting the ER homodimer
A recent study involved a series of bivalent ligands targeting the estrogen receptor (ER) homodimer. The selective ER downregulators 4-[1-(4-hydroxyphenyl)-2-phenyl-1-butenyl]cinnamic acid (GW7604) and cyclofenilacrylic acid were selected as lead compounds. Previous studies suggested that the rigidity of the acrylate moiety might be essential for retaining the crucial charge–charge repulsion interaction with D351 [29]. Treating the carboxylic acid moieties as the attachment points, bivalent ligands can be readily generated via amide bond formation maintaining the adjacent double bond. Thus, the monomeric lead compounds, bridged through diaminoalkane spacers (C2–C5), gave corresponding bivalent counterparts.
The success of this strategy is corroborated by results from in vitro assays showing that the cyclofenil-based bivalent compound AK-15b (Figure 2a) with a C4 spacer exhibited the highest binding affinity to ERα, with an ER downregulatory potency of 38% at 1 μM in MCF-7 cells. Furthermore, AK-15b completely blocked the recruitment of peroxisome proliferator-activated receptor-γ coactivator 1 (PGC1) and abolished the estradiol-induced transactivation effect in U2OS cells [30]. This study furnished ER modulators with a novel mode of action to prevent estrogen action.
Figure 2.

(a) The chemical structure of cyclofenilacrylic acid-based bivalent ligand AK-15b. (b) The chemical structures of SR141617A, PPHT, the initially designed CBR1-D2R bivalent ligands and SAR explorations derived from SR141617A. (c) The chemical structure of the MOR-CCR5 bivalent ligand VZMC001. (d) The chemical structures of eticlopride, NT(8–13) and designed D2R–NTS1R bivalent ligands, (e) The chemical structures of MTEP, DAPT, DAP, linkers and mGluR5–D2R bivalent ligand MQ-22a.
Bivalent ligands targeting the CB1R-D2R heterodimer
Grant et al. pursued a series of bivalent ligands targeting the cannabinoid-1 receptor (CB1R)–dopamine D2 receptor (D2R) heterodimer. The selective inverse agonist SR141617A, approved for the treatment of obesity in 2006, was chosen as the CB1R pharmacophore. For D2R, the selective agonist 2-(N-phenylethyl-N-propyl)-amino-5-hydroxytetralin (PPHT) was used. A series of bivalent ligands carrying 22–50-atom-length spacers with conjugation at the C3 position of the pyrazole ring of SR141617A and on the phenyl ring of PPHT (Figure 2b) was initially designed and synthesized. These bivalent compounds exhibited higher binding affinities (Ki = 0.84–4.2 nM) to hD2R than that of the parent agonist (±)-PPHT (Ki = 13.3 nM), but displayed poor affinities to hCB1R. Because there had been no systematic investigations of spacer attachment sites on SR141716A, the authors comprehensively surveyed alternative spacer conjugation sites using positions N1 and C5 (and C3 for comparison purposes) of the pyrazole core of SR141717A. This entailed the synthesis of series of SR141717A analogues through the incorporation of diverse fragments at the three positions of the pyrazole ring (Figure 2b), and subsequent evaluation of their binding affinities to hCB1R. Such SAR investigation efforts ultimately identified position C5 via an ester bond as a suitable conjugation site for the SR141716A pharmacophore [31].
Bivalent ligands targeting the MOR–CCR5 heterodimer
To better understand the crosstalk interaction between MOR and CCR5 while simultaneously exploring the relevance of this interaction for neuroAIDS, Arnatt el al. synthesized a series of bivalent ligands targeting the putative MOR-CCR5 heterodimer. The MOR antagonist naltrexone and the CCR5 antagonist maraviroc, an anti-HIV agent were chosen as the monomeric pharmacophores. The 6-carbonyl position of naltrexone was chosen as the attachment point on the *basis of examination of the MOR crystal structure. The 4′-position of the terminal phenyl ring in maraviroc was first selected as the attachment point, and the bivalent ligand VZMC001 was synthesized (Figure 2c) [32]. To investigate the influence of the spacer attachment position, another bivalent ligand with the same spacer was designed by switching the attachment position from the 4′-position to the 3′-position of the phenyl ring in maraviroc. VZMC001 was found to possess two-digit nanomolar binding affinity (Ki = 51.8 nM) and calcium mobilization antagonism (IC50 = 40.0 nM) to MOR, as well as moderate binding affinity (Ki = 239 nM) and calcium mobilization antagonism (IC50 = 126 nM) to CCR5. Furthermore, VZMC001 was 7- and 3.3-fold more potent than maraviroc in inhibiting viral entry in primary human astrocytes with and without morphine, respectively [33]. However, the 3 ′-position attachment led to much lower binding affinity for CCR5. This study demonstrated the importance of attachment points on the pharmacophore.
Structural biology and molecular modeling
Structural biology is fundamental for uncovering how macromolecules such as proteins generate their biological functions. An increasing number of GPCR crystal structures, either with or without ligand co-crystallization, have been resolved, including C-X-C chemokine receptor type 4 (CXCR4) [34], CB1R [35], CB2R [36], κ-opioid receptor (KOR) [37], δ-opioid receptor (DOR) [38] and jumping spider rhodopsin-1 [39]. GPCR homodimer constructs have been observed in some crystal structures, which provides evidence to support GPCR dimerization, as well as a template for designing bivalent ligands targeting putative GPCR dimers. Molecular modeling efforts based on structural biology observations afford insights into GPCR dimeric interactions and provide structure-based approaches for choosing appropriate attachment points on the pharmacophores.
Bivalent ligands targeting the D2R–NTS1R heterodimer
A study reported the development of bivalent ligands for the D2R–neurotensin NTS1 receptor (D2R-NTS1R) heterodimer. Similarly, the D2R-NTS1R heterodimer model was built on the basis of the D2R homology model (the D3R crystal structure [40] was used as a template) and the crystal structure of the NTS1R [41]. Based on the heterodimer model, it was concluded that spacers with a total length of ~55 Å were required to enable a bivalent-binding mode. The authors envisaged that at least two polyethylene glycol (PEG) units, in addition to a biphenyltriazole-based moiety, would be necessary to achieve that. Inspection of the ligand-bound crystal structures of D3R and NTS1R clearly revealed that the 4′-position on the pyrrole ring of eticlopride (a D3R antagonist) and the N terminus amino of NT(8–13) [neurotensin (8–13) peptide, an NTS1R agonist] are accessible from the extracellular side, and were therefore chosen as the attachment points for bridging the two lead compounds; the designed bivalent compound connected the two pharmacophores at these points (Figure 2d) [42]. These compounds showed high selectivity (up to three orders of magnitude) and binding affinities in the picomolar range for cells co-expressing D2R and NTS1R, compared with cells only expressing D2R. These examples present convincing evidence for applying structural biology and computational modeling to the successful design of bivalent ligands.
Choosing spacers with optimal length and chemical composition
It has been documented that the length of the spacer can have a profound influence on the binding affinity and functional activity of bivalent compounds. This optimal length has been determined for a number of different GPCR dimers, including the examples in this review [24,30,43-50].
A recent example is the report of bivalent ligands targeting the mGluR5–D2R heterodimer by Qian et al. in 2018 [47]. The negative allosteric modulator 3-[(2-methyl-4-thiazolyl)ethynyl]pyridine (MTEP) was chosen as a pharmacophore for mGluR5. The D2-likeR (D2R and D4R) agonist 5-hydroxy-2-(dipropylamino)tetralin (DPAT) and the D2R antagonist 1,4-disubstituted aromatic piperazine (DAP) were chosen as pharmacophores for D2R. Varying linker moieties were used in constructing the spacers to compare their influence on affinity. The bivalent ligand MQ-22a, bearing a 20-atom-length spacer, showed the highest potency; it exhibited a fourfold increase in binding affinity for cells co-expressing mGluR5 and D2R compared with cells solely expressing D2R, and a twofold increase compared with cells solely expressing mGluR5. In addition, MQ-22a had a fivefold higher affinity for mGluR5 than its monovalent counterpart in co-expressing cells (Figure 2e) [47].
On the basis of a systematic examination of the available literature, it seems there are no general rules that are applicable to all GPCRs when predicting the optimal spacer length for a bivalent ligand. However, the aforementioned examples afford a starting point for identifying the optimal spacer length.
The chemical composition of the spacers is also importance. Diverse chemical moieties, such as methylene units, PEG units, peptidic chains and cyclic core alkyl moieties, have been adopted as spacer components for bivalent ligands. Generally, the flexibility and hydrophilic–hydrophobic properties of the spacers are two main parameters that need to be considered. The use of constrained or rigid spacers often leads to decreased binding affinity and potency, whereas flexible spacers allow favorable positioning of each pharmacophore unit. Hydrophilic–hydrophobic properties of the spacers can not only influence the global solubility of bivalent ligands, but also determine the positioning of the spacers themselves. Hydrophobic spacers are likely to bridge the pharmacophores directly through the hydrophobic TM domains, whereas hydrophilic spacers might span the extracellular compartments of receptors. We have found that the PEG repeat (e.g., [44,47,51,52]) and peptidic chains (e.g.,[24,30,48,53]) seem to be the most popular spacers, because these allow for a gradual increase in spacer length with suitable intrinsic physiochemical properties [18]. In addition, spacers containing the 1,2,3-triazole moiety have been increasingly used in recent years, because the 1,2,3-triazole motif can be rapidly constructed through the efficient and prevalent CuAAC (copper(I)-catalyzed alkyne-azide cycloaddition) click reaction. This is exemplified by the diverse bivalent ligands targeting the D2R–NTS1R heterodimer [42], mGluR5–D2R heterodimer [47], serotonin-2A receptor (5-HT2AR) homodimer [54] and MOR–D2-likeR heterodimer [52].
With a combined structural biology and molecular modeling approach, medicinal chemists working with dimeric GPCR targets can not only identify the appropriate attachment points on each pharmacophore, but also estimate the distance between the two lead compounds in the binding pockets of each GPCR target after their designation. Thus, the length and chemical composition of spacers can be readily determined at the onset of drug design. In other words, a well-defined GPCR dimer model based on the available crystallography information for each target GPCR is the key to determining how long a spacer could be. SAR study results can then be used to determine the optimal length and chemical compositions of spacers to further optimize the bivalent ligand construct and pursue GPCR-dimerization investigations. A detailed summarization of the spacers used in the studies in this review can be found in Table 1.
Table 1.
Summarization of the spacers utilized in the studies involved in this review
| Bivalent compound |
Spacer | Length of spacer (atoms) |
Target | In vitro activity | In vivo activity |
|---|---|---|---|---|---|
| KDN21 | 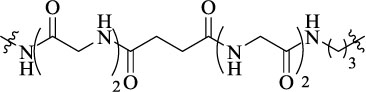 |
21 | δ–κ-opioid receptor heterodimer | Ki = 0.3 ± 0.0 nM (co-expressed δ–κ); Ki = 63 ± 6.3 nM (mixed δ + κ) | Agonist activity (ED50 =31 pmol) |
| MCC22 | 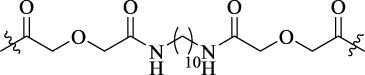 |
22 | MOR–CCR5a heterodimer | - | Antinociceptive activity (ED50 = 0.015 or 0.019 pmol/mouse) |
| AK-15b | 6 | ER homodimer | 38% ER downregulatory potency at 1 μM in MCF-7 cells | - | |
| VZMC001 | 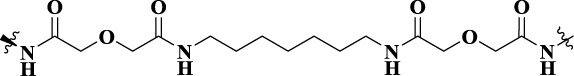 |
21 | MOR–CCR5a heterodimer | MOR (Ki = 51.8 nM, IC50 = 40.0 nM); CCR5 (Ki = 239 nM, IC50 = 126 nM) | - |
| - | 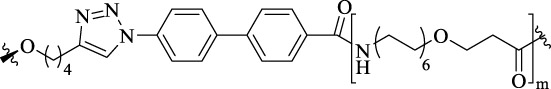 |
22–88 | D2R-NTS1R heterodimer | Binding affinities in the picomolar range for cells co-expressing D2R and NTS1R | - |
| MQ-22a | 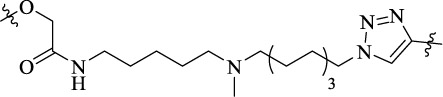 |
20 | mGluR5–D2R heterodimer | D2R (Ki = 50 ± 6.4 nM); D2R-mGluR5 (Ki = 13 ± 3.6 nM); mGluR5 (Ki = 2.3 ± 0.5 nM); D2R-mGluR5 (Ki= 1.1 ± 0.2 nM) | - |
| SG-15 | 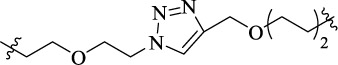 |
14 | 5-HT2AR homodimer | Inhibiting 5-HT (1 μM) induced calcium flux (IC50 = 36.5 nM) | - |
| CS-6c | 5-HT2AR homodimer | ERK1/2 activation (IC50 = 178 nM); 5-HT2AR (Ki = 5.8 nM) | Suppress hyperactivity | cocaine-evoked in a time-dependent manner (2 mg/kg) | |
| CJL-1-124 | PEDG20 | 20 | hMC4R homodimer | Stimulating the cAMP signaling pathway (EC50 = 4.7 nM) | - |
| DP-13 | 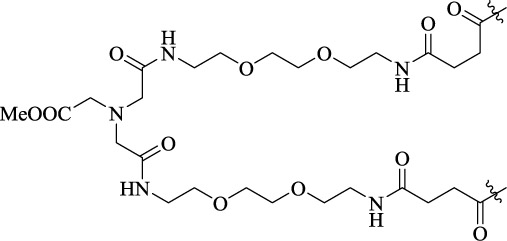 |
25 | D2R homodimer | KDB1 = 0.021 nM | - |
| SD-11 | 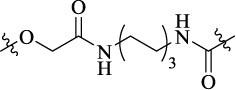 |
12 | MOR-CBR heterodimer | [3H]DAMGO (Ki = 18 nM); [3H]JWH-018 (Ki = 34 nM); Emax = 147%, EC50 = 215 nM | Antiallodynic effect (20 μg dose) |
| VZMX001 | 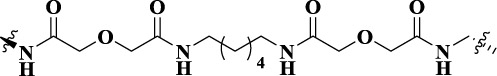 |
20 | MOR-CXCR4 heterodimer | MOR (Ki = 25.4 nM, IC50 = 61.9 nM); CXCR4 [IC50 = 17.2 μM (binding affinity), IC50 = 3.3 μM] | - |
| D24M | 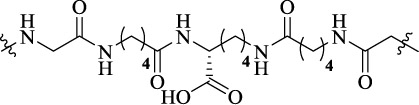 |
24 | MOR-DOR heterodimer | MDOR potency (0.85 nM), binding affinity (0.63 nM) | Reduced acute and chronic morphine withdrawal behaviors |
| MQ-12d | 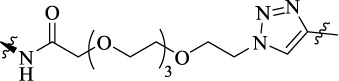 |
18 | MOR-D2-likeR heterodimer | MOR (Ki = 16 nM), D2R (Ki = 22 nM), MOR-D2R (Ki = 101 nM); MAPK phosphorylation (EC50 = 0.12 μM) | - |
5-HT2AR, serotonin-2A receptor; CBR, ; CCR5, C-C chemokine receptor type 5; CXCR4, C-X-C chemokine receptor type 4; D2R, dopamine D2 receptor; D2-likeR, D2R and D4R; DOR, δ-opioid receptor; ER, estrogen receptor; hMC4R, human melanocortin-4 receptor; MAPK, mitogen-activated protein kinase; MDOR, MOR-DOR heterodimer; mGluR5, metabotropic glutamate receptor 5; MOR, μ-opioid receptor; NTS1R, neurotensin NTS1 receptor.
Recent advances in the design and development of GPCR bivalent ligands (2018–present)
Here, we describe several representative examples of recent advances, with the aim of revealing the current trends in the design and development of GPCR bivalent ligands. Bivalent ligands targeting GPCR homodimers and heterodimers are discussed in turn.
Homobivalent ligands
Bivalent ligands targeting the 5-HT2AR homodimer
M100907 (volinanserin) is a highly selective 5-HT2AR antagonist that was first developed for the treatment of insomnia [55]. In 2018, M100907 was selected as a lead, and the homobivalent compound SG-15, containing a 1,2,3-triazole moiety, was synthesized and tested for its inhibitory effect on 5-HT-stimulated intracellular calcium release. SG-15 maintained its antagonistic property, inhibiting 5-HT (1 μM) induced calcium flux with an IC50 value of 36.5 nM, although its antagonism was inferior to that of M100907 (IC50 = 4.8 nM) (Figure 3a) [54].
Figure 3.

(a) The chemical structures of the lead compound M100907 and the bivalent compound SG-15. (b) The chemical structure of the bivalent compound CS-6c. (c) The chemical structures of the hMC4R antagonist, hMC4R agonist and bivalent compounds CJL-1-124 and CJL-5-74. (d) The chemical structures of the lead compound NAPS and the bivalent compounds DP-12 and DP-13.
Using M100907 as the lead compound, the same research groups then reported another series of homobivalent compounds bearing spacers with different chemical compositions. All bivalent compounds exhibited inhibitory effects on 5-HT-mediated phosphorylation of extracellular regulated kinases 1/2 (ERK1/2) in h5-HT2AR-Chinese hamster ovary (CHO) cells, and they showed binding affinity to 5-HT2AR at a nanomolar level. Furthermore, one of these bivalent compounds, CS-6c (2 mg/kg), demonstrated in vivo efficacy in suppressing cocaine-evoked hyperactivity in a time-dependent manner (Figure 3b) [44].
Bivalent ligands targeting the hMC4R homodimer
In 2019, Lensing and coworkers reported first-in-class biased unmatched bivalent ligands (BUmBLs) targeting the human melanocortin-4 receptor (hMC4R) homodimer [56]. These compounds contained one tetrapeptide antagonist pharmacophore and one tetrapeptide agonist pharmacophore. The BUmBL CJL-1-124 (EC50 = 4.7 nM), with one 20-atom polyethylene diamine diglycolyic acid (PEDG20) spacer, exhibited a more potent biased agonism profile than the bivalent ligand CJL-5-74 (EC50 = 5.9 nM), with two PEDG20 units, in stimulating the cyclic adenosine monophosphate (cAMP) signaling pathway (Figure 3c). Additionally, all synthesized BUmBLs only partially activated the β-arrestin 2 recruitment pathway at concentrations up to 10 μM in in vitro assays. This rigorous strategy has the potential to be readily applied to other GPCR targets to study putative GPCR-biased signaling pathways.
Bivalent ligands targeting the D2R homodimer
Taking N-(p-aminophenethyl)spiperone (NAPS), derived from the marketed D2R antagonist spiperone as the pharmacophore unit, Pulido and colleagues designed and synthesized two bivalent ligands, DP-12 and DP-13, with different length spacers targeting the D2R homodimer [51]. DP-13, bearing a 25-atom spacer, had a 3.3-fold higher affinity (KDB1 = 0.021 nM) than the 35-atom-spacer-containing DP-12 (KDB1 = 0.07 nM) (Figure 3d). Moreover, the fused peptide TAT–TM6, synthesized by fusing the cell-penetrating HIV-1 transactivator of transcription (TAT) peptide with a synthetic peptide containing the amino acid sequence of TM6, decreased the binding of the bivalent compound DP-13 [(KDB1 (DP-13 + TM6) =1.1 nM versus KDB1 (DP-13) = 0.021 nM)]; however, it did not show any influence on the monovalent compound in native tissues. This confirmed that the D2R homodimer forms through the TM6 interface. This versatile bivalent chemical platform generated ligands that simultaneously target both orthosteric binding sites of the D2R homodimer.
Heterobivalent ligands
Bivalent ligands targeting the MOR–CBR heterodimer
A series of bivalent compounds targeting the putative MOR–CBR heterodimer was designed and synthesized in 2019. In this study, the selective agonist oxycodone and an encephalin-related tetrapeptide, Tyr-D-Ala-Gly-Phe, were employed as the lead compounds for MOR activation. The agonist JWH-018 was used as the CBR pharmacophore. The bivalent derivatives SD-11 and SD-19 exhibited the highest binding affinity in each sub-series and demonstrated agonist profiles in [35S]-GTPγS binding assays in rat brain membrane homogenates. Furthermore, SD-11 and SD-19 exhibited antinociceptive effects similar to their lead compounds at a 20 μg/kg dose after spinal administration in a chronic osteoarthritis pain model (Figure 4a) [48]. These in vivo study results support the strategy of targeting GPCR dimers to develop analgesics with new mechanisms of action.
Figure 4.

(a) The chemical structures of the lead compounds oxycodone, tetrapeptide and JWH-018, and the bivalent compounds SD-11 and SD-19. (b) The chemical structure of the MOR–CXCR4 bivalent ligand VZMX001 (with attachment points shown). (c) The chemical structures of the DOR antagonist (Tyr-Tic-OH), MOR antagonist (H-Tyr-Pro-Phe-D1Nal-NH2), and the bivalent compound D24M. (d) The chemical structures of the MOR agonist hydromorphone, the D2-likeR agonist DPAT and antagonist DAP, and the bivalent compound MQ-12d.
Bivalent ligands targeting the MOR–CXCR4 heterodimer
Recently, Reinecke and colleagues reported VZMX001, the first bivalent ligand targeting the putative MOR-CXCR4 heterodimer (Figure 4b). The MOR antagonist naltrexone was selected as the MOR pharmacophore, with an attachment point at the C6 position. The antagonist IT1t was chosen to target CXCR4 owing to its co-crystallization with CXCR4. In the CXCR4-IT1t crystal structure complex (Protein Data Bank code 3ODU), it was observed that the cyclohexyl moieties of IT1t pointed toward the extracellular region. Given the rotational C2 symmetry of IT1t, a cyclohexyl group was selected as the attachment point. The bivalent ligand VZMX001 exhibited two-digit nanomolar binding affinity to the MOR with a Ki value of 25.4 nM. In calcium mobilization assays with MOR-CHO cells, VZMX001 inhibited [D-Ala2-MePhe4-Gly(ol)5]encephalin (DAMGO)-induced Ca2+ flux with an IC50 value of 61.9 nM. Furthermore, in antibody binding assays with CXCR4-CHO cells, VZMX001 retained binding affinity to CXCR4 (IC50 = 17.2 μM). In calcium mobilization assays, VZMX001 inhibited stromal cell-derived factor 1 (SDF1)-induced calcium flux with an IC50 value of 3.3 μM [57]. This structure-based ligand could be employed as a hit for developing chemical probes to help reveal the mechanism of the putative MOR–CXCR4 heterodimer formation in opioid-use-accelerated HIV-1 replication.
Bivalent ligands targeting the MOR–DOR heterodimer
In 2018, Olson et al. reported the design and evaluation of a series of synthetic peptides as selective bivalent antagonists with variable-length spacers of 15–41 atoms for the MOR–DOR heterodimer (MDOR) [53]. A low-affinity MOR antagonist (H-Tyr-Pro-Phe-D1Nal-NH2) and a moderate-affinity DOR antagonist (Tyr-Tic-OH) were selected as pharmacophore units. D24M, with a 24-atom-length spacer, possessed the highest MDOR potency, affinity and selectivity (0.85 nM, 0.63 nM and ≥ 89-fold, respectively). It was also found that D24M dose-dependently antagonized tail flick antinociception generated by the known MDOR agonist CYM51010 in mice [58]. Moreover, D24M significantly reduced acute and chronic morphine withdrawal behaviors. These in vivo study results suggest that D24M could be a novel drug candidate for treating opioid dependence (Figure 4c).
Bivalent ligands targeting the MOR–D2-likeR heterodimer
Another example of the influence of spacers with different lengths was reported by Qian et al. They designed and evaluated a series of MOR agonist/antagonist-D2-likeR agonist/antagonist heterobivalent ligands that targeted MOR-D2-likeR heterodimers. The MOR agonist hydromorphone and antagonist naltrexone, as well as the D2-likeR agonist DPAT and antagonist DAP, were used as pharmacophores. PEG units with varying length (18–24 atoms) were employed as spacers. The results indicated that MQ-12d (bearing an 18-atom spacer) demonstrated excellent potency with high efficacy both in β-arrestin 2 recruitment for MOR and mitogen-activated protein kinase phosphorylation (MAPK-P) for D4R. MQ-12d was further characterized by a biphasic competition binding curve for the MOR–D4R heterodimer (Figure 4d) [52]. This study provided more pharmacological tools for investigating the putative MOR–D4R heterodimer.
Concluding remarks and prospects
Accumulating evidence has corroborated the natural existence of GPCR dimers. However, the pharmacology and biology of GPCR dimerization at the cellular and tissue levels remains unclear. Bivalent ligands can bridge two pharmacophore units as potential chemical probes and pharmacological tools for investigating the GPCR-dimerization process. With the growing number of GPCR crystal structures and the development of molecular modeling techniques, dimeric GPCRs have been defined as potential targets for drug discovery. The design of bivalent ligands targeting putative GPCR dimers on the basis of the available information shows promise, and is a requisite from a medicinal chemistry perspective.
For the rational design of bivalent ligands, the primary considerations are the selection of potent and selective monomeric ligands, appropriate attachment points on pharmacophore units, and spacers with optimal length and chemical compositions. This necessitates diligent effort and considerable deliberation. The successful combination of these aspects would result in dramatic improvement in potency as well as affinity of the bivalent ligands. As shown by the studies presented here, diverse bivalent ligands targeting different putative GPCR homodimers and heterodimers have been designed and synthesized in past decades; in some cases, the bivalent compounds have demonstrated vast superiority over their monomeric lead counterparts, indicating promise for clinical applications.
There are still challenges in the development of bivalent ligands that need to be addressed and resolved. First, bivalent ligands tend to be larger, more lipophilic and more structurally complicated than their monomeric counterparts. Most of the bivalent ligands discussed in this review have molecular weights above 700 Da, or even 1000 Da. These unfavorable physicochemical properties often lead to unsatisfactory cell penetration ability and a poor pharmacokinetic profile, which means that it could be difficult to yield a ‘drug-like’ molecule. Second, some of the synthesized bivalent compounds might possess decreased binding affinities and/or less favorable selectivity profiles toward the target GPCRs when compared with the lead compounds. It is therefore thought that such bivalent ligands are not capable of stimulating the desired pharmacological activities, and some unwanted off-target effects might occur. Third, the dimerization from the crystal packaging might not reflect the real situation in vivo, because the conditions used for crystallography are usually too artificial or even harsh physiologically. Consequently, some potent bivalent ligands designed on the basis of crystal complexes might be inactive when applied to in vivo studies. Last, most of the experimental evidence for GPCR dimerization is from in vitro studies. Although the results might indicate a close distance between individual receptors, the studies do not directly support a physical association in endogenous systems, which will hinder further development of the bivalent ligands as potential therapeutics.
As shown in this review, apart from the opioid receptor field, the in vivo potential of bivalent ligands has not been extensively explored. Furthermore, several studies have questioned the existence of dimerization of some GPCRs by showing that some bivalent ligands turned out to act on separate monomers rather than dimers. For example, Wang et al. demonstrated that MOR and DOR segregated in dorsal horn interneurons, and in amygdalar and cortical neurons as well [59]. Another study showed that the two pharmacophores of the synthesized bivalent ligands MMG22 and MMG10 target MOR and mGluR5 as separate receptor monomers instead of targeting their heterodimer in the spared nerve injury-induced model of neuropathic pain [60].
To date, none of bivalent compounds targeting putative GPCR dimers has been approved for clinical applications. However, with the continuous breakthroughs in structural biology and computational techniques, as well as with more and more robust and comprehensive in vitro and in vivo biological evaluation methods, we think that it is only a matter of time before such a drug emerges.
Highlights.
Highly potent and selective pharmacophores are essential for targeting G protein-coupled receptor (GPCR) dimers.
The choice of appropriate attachment points on pharmacophores is pivotal.
Spacers with optimal length and chemical composition are needed.
Recent advances in the design and development of GPCR bivalent ligands are summarized.
Acknowledgments
This work was partially supported by NIH/NIDA Grants DA024022, DA044855 and DA050311 (Y.Z.).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Conflicts of interest
The authors declare no competing financial interests.
References
- 1.Hazum E et al. (1979) Opiate (enkephalin) receptors of neuroblastoma cells: occurrence in clusters on the cell surface. Science 206,1077–1079 [DOI] [PubMed] [Google Scholar]
- 2.Cvejic S and Devi LA (1997) Dimerization of the δ opioid receptor: implication for a role in receptor internalization. Journal of Biological Chemistry 272, 26959–26964 [DOI] [PubMed] [Google Scholar]
- 3.Portoghese PS (2001) From models to molecules: opioid receptor dimers, bivalent ligands, and selective opioid receptor probes. Journal of Medicinal Chemistry 44, 2259–2269 [DOI] [PubMed] [Google Scholar]
- 4.Ng GYK et al. (1996) Dopamine D2 receptor dimers and receptor-blocking peptides. Biochemical and Biophysical Research Communications 227, 200–204 [DOI] [PubMed] [Google Scholar]
- 5.Fiorentini C et al. (2010) Dimerization of dopamine D1 and D3 receptors in the regulation of striatal function. Current Opinion in Pharmacology 10, 87–92 [DOI] [PubMed] [Google Scholar]
- 6.Kunishima N et al. (2000) Structural basis of glutamate recognition by a dimeric metabotropic glutamate receptor. Nature 407, 971–977 [DOI] [PubMed] [Google Scholar]
- 7.Moreno Delgado D et al. (2017) Pharmacological evidence for a metabotropic glutamate receptor heterodimer in neuronal cells. eLife 6, e25233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Kniazeff J et al. (2004) Locking the dimeric GABAB G-protein-coupled receptor in its active state. The Journal of Neuroscience 24, 370–377 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Rondard P et al. (2008) Functioning of the dimeric GABAB receptor extracellular domain revealed by glycan wedge scanning. The EMBO Journal 27, 1321–1332 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Maggio R et al. (1993) Coexpression studies with mutant muscarinic/adrenergic receptors provide evidence for intermolecular ‘cross-talk’; between G-protein-linked receptors. Proceedings of the National Academy of Sciences 90, 3103–3107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Bouvier M (2001) Oligomerization of G-protein-coupled transmitter receptors. Nature Reviews Neuroscience 2, 274–286 [DOI] [PubMed] [Google Scholar]
- 12.Shonberg J et al. (2011) Design strategies for bivalent ligands targeting GPCRs. ChemMedChem 6, 963–974 [DOI] [PubMed] [Google Scholar]
- 13.Erez M et al. (1982) Narcotic antagonistic potency of bivalent ligands which contain β-naltrexamine. Evidence for simultaneous occupation of proximal recognition sites. Journal of Medicinal Chemistry 25, 847–849 [DOI] [PubMed] [Google Scholar]
- 14.Portoghese PS et al. (1982) Opioid agonist and antagonist bivalent ligands as receptor probes. Life Sciences 31,1283–1286 [DOI] [PubMed] [Google Scholar]
- 15.Halazy S (1999) G-protein coupled receptors bivalent ligands and drug design. Expert Opinion on Therapeutic Patents 9, 431–446 [Google Scholar]
- 16.William SM Jr, (2004) Bivalent ligands for G protein-coupled receptors. Current Pharmaceutical Design 10, 2015–2020 [DOI] [PubMed] [Google Scholar]
- 17.Ao Z et al. (2007) Receptor dimerization – rationale for the design of bivalent ligands. Current Topics in Medicinal Chemistry 7, 343–345 [DOI] [PubMed] [Google Scholar]
- 18.Isabelle B-B et al. (2008) Bivalent ligands as specific pharmacological tools for G protein-coupled receptor dimers. Current Drug Discovery Technologies 5, 312–318 [DOI] [PubMed] [Google Scholar]
- 19.Hiller C et al. (2013) Class A G-protein-coupled receptor (GPCR) dimers and bivalent ligands. Journal of Medicinal Chemistry 56, 6542–6559 [DOI] [PubMed] [Google Scholar]
- 20.Arnatt CK and Zhang Y (2014) Bivalent ligands targeting chemokine receptor dimerization: molecular design and functional studies. Current Topics in Medicinal Chemistry 14, 1606–1618 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Newman AH et al. (2020) 2016 Philip S. Portoghese Medicinal Chemistry Lectureship: Designing bivalent or bitopic molecules for G-protein coupled receptors. The whole is greater than the sum of its parts. Journal of Medicinal Chemistry 63, 1779–1797 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Portoghese PS et al. (1988) Naltrindole, a highly selective and potent non-peptide δ opioid receptor antagonist. European Journal of Pharmacology 146, 185–186 [DOI] [PubMed] [Google Scholar]
- 23.Jones RM and Portoghese PS (2000) 5 ′ -Guanidinonaltrindole, a highly selective and potent κ -opioid receptor antagonist. European Journal of Pharmacology 396, 49–52 [DOI] [PubMed] [Google Scholar]
- 24.Bhushan RG et al. (2004) A bivalent ligand (KDN-21) reveals spinal δ and κ opioid receptors are organized as heterodimers that give rise to δ1 and κ2 phenotypes. Selective targeting of δ-κ heterodimers. Journal of Medicinal Chemistry 47, 2969–2972 [DOI] [PubMed] [Google Scholar]
- 25.Haddou TB et al. (2014) Pharmacological investigations of N-substituent variation in morphine and oxymorphone: opioid receptor binding, signaling and antinociceptive activity. PLOS ONE 9, e99231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Imamura S et al. (2006) Discovery of a piperidine-4-carboxamide CCR5 antagonist (TAK-220) with highly potent anti-HIV-1 activity. Journal of Medicinal Chemistry 49, 2784–2793 [DOI] [PubMed] [Google Scholar]
- 27.Akgün E et al. (2015) Inhibition of Inflammatory and neuropathic pain by targeting a mu opioid receptor/chemokine receptor5 heteromer (MOR-CCR5). Journal of Medicinal Chemistry 58, 8647–8657 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Cataldo G et al. (2019) The bivalent ligand MCC22 potently attenuates hyperalgesia in a mouse model of cisplatin-evoked neuropathic pain without tolerance or reward. Neuropharmacology 158,107598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kieser KJ et al. (2010) Characterization of the pharmacophore properties of novel selective estrogen receptor downregulators (SERDs). Journal of Medicinal Chemistry 53, 3320–3329 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Knox A et al. (2020) Development of bivalent triarylalkene- and cyclofenil-derived dual estrogen receptor antagonists and downregulators. European Journal of Medicinal Chemistry 192,112191. [DOI] [PubMed] [Google Scholar]
- 31.Grant PS et al. (2019) Divalent cannabinoid-1 receptor ligands: a linker attachment point survey of SR141716A for development of high-affinity CB1R molecular probes. Bioorganic & Medicinal Chemistry Letters 29, 126644. [DOI] [PubMed] [Google Scholar]
- 32.Yuan Y et al. (2012) Design and synthesis of a bivalent ligand to explore the putative heterodimerization of the mu opioid receptor and the chemokine receptor CCR5. Organic & Biomolecular Chemistry 10, 2633–2646 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Arnatt CK et al. (2016) Exploration of bivalent ligands targeting putative mu opioid receptor and chemokine receptor CCR5 dimerization. Bioorganic & Medicinal Chemistry 24, 5969–5987 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Qin L et al. (2015) Crystal structure of the chemokine receptor CXCR4 in complex with a viral chemokine. Science 347, 1117–1122 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Hua T et al. (2017) Crystal structures of agonist-bound human cannabinoid receptor CB1. Nature 547, 468–471 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Li X et al. (2019) Crystal structure of the human cannabinoid receptor CB2. Cell 176, 459–467.e413 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Che T et al. (2018) Structure of the nanobody-stabilized active state of the kappa opioid receptor. Cell 172, 55–67.e15 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Claff T et al. (2019) Elucidating the active δ-opioid receptor crystal structure with peptide and small-molecule agonists. Science Advances 5, eaax9115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Varma N et al. (2019) Crystal structure of jumping spider rhodopsin-1 as a light sensitive GPCR. Proceedings of the National Academy of Sciences 116, 14547–14556 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Chien EYT et al. (2010) Structure of the human dopamine D3 receptor in complex with a D2/D3 selective antagonist. Science 330, 1091–1095 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.White JF et al. (2012) Structure of the agonist-bound neurotensin receptor. Nature 490, 508–513 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Hübner H et al. (2016) Structure-guided development of heterodimer-selective GPCR ligands. Nature Communications 7, 12298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Daniels DJ et al. (2005) A bivalent ligand (KDAN-18) containing δ-antagonist and κ-agonist pharmacophores bridges δ2 and κ1 opioid receptor phenotypes. Journal of Medicinal Chemistry 48, 1713–1716 [DOI] [PubMed] [Google Scholar]
- 44.Soto CA et al. (2018) Novel bivalent 5-HT2A receptor antagonists exhibit high affinity and potency in vitro and efficacy in vivo. ACS Chemical Neuroscience 9, 514–521 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Soulier J-L et al. (2005) Design and synthesis of specific probes for human 5-HT4 receptor dimerization studies. Journal of Medicinal Chemistry 48, 6220–6228 [DOI] [PubMed] [Google Scholar]
- 46.Russo O et al. (2007) Synthesis of specific bivalent probes that functionally interact with 5-HT4 receptor dimers. Journal of Medicinal Chemistry 50, 4482–4492 [DOI] [PubMed] [Google Scholar]
- 47.Qian M et al. (2018) Synthesis toward bivalent ligands for the dopamine D2 and metabotropic glutamate 5 receptors. Journal of Medicinal Chemistry 61, 8212–8225 [DOI] [PubMed] [Google Scholar]
- 48.Dvorácskó S et al. (2019) Preparation of bivalent agonists for targeting the mu opioid and cannabinoid receptors. European Journal of Medicinal Chemistry 178, 571–588 [DOI] [PubMed] [Google Scholar]
- 49.Daniels DJ et al. (2005) Opioid-induced tolerance and dependence in mice is modulated by the distance between pharmacophores in a bivalent ligand series. Proceedings of the National Academy of Sciences of the United States of America 102, 19208–19213 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Soriano A et al. (2009) Adenosine A2A receptor-antagonist/dopamine D2 receptor-agonist bivalent ligands as pharmacological tools to detect A2A-D2 receptor heteromers. Journal of Medicinal Chemistry 52, 5590–5602 [DOI] [PubMed] [Google Scholar]
- 51.Pulido D et al. (2018) Design of a true bivalent ligand with picomolar binding affinity for a G protein-coupled receptor homodimer. Journal of Medicinal Chemistry 61, 9335–9346 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Qian M et al. (2018) Design, synthesis, and biological evaluation of bivalent ligands targeting dopamine D2-like receptors and the μ-opioid receptor. ChemMedChem 13, 944–956 [DOI] [PubMed] [Google Scholar]
- 53.Olson KM et al. (2018) Synthesis and evaluation of a novel bivalent selective antagonist for the mu-delta opioid receptor heterodimer that reduces morphine withdrawal in mice. Journal of Medicinal Chemistry 61, 6075–6086 [DOI] [PubMed] [Google Scholar]
- 54.Gilbertson SR et al. (2018) Synthesis and activity of functionalizable derivatives of the serotonin (5-HT) 5-HT2A receptor (5-HT2AR) antagonist M100907. Bioorganic & Medicinal Chemistry Letters 28, 1381–1385 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Bradley RT et al. (2008) 5-HT2A inverse-agonists for the treatment of insomnia. Current Topics in Medicinal Chemistry 8, 969–976 [DOI] [PubMed] [Google Scholar]
- 56.Lensing CJ et al. (2019) Developing a biased unmatched bivalent ligand (BUmBL) design strategy to target the GPCR homodimer allosteric signaling (cAMP over β-arrestin 2 recruitment) within the melanocortin receptors. Journal of Medicinal Chemistry 62, 144–158 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Reinecke BA et al. (2020) Design and synthesis of a bivalent probe targeting the putative mu opioid receptor and chemokine receptor CXCR4 heterodimer. RSC Medicinal Chemistry 11, 125–131 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Gomes I et al. (2013) Identification of a μ-δ opioid receptor heteromer-biased agonist with antinociceptive activity. Proceedings of the National Academy of Sciences 110 (29), 12072–12077 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Wang D et al. (2018) Functional divergence of delta and mu opioid receptor organization in CNS pain circuits. Neuron 98, 90–108.e105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Peterson CD et al. (2017) Bivalent ligand that activates mu opioid receptor and antagonizes mGluR5 receptor reduces neuropathic pain in mice. Pain 158, 2431–2441 [DOI] [PMC free article] [PubMed] [Google Scholar]