

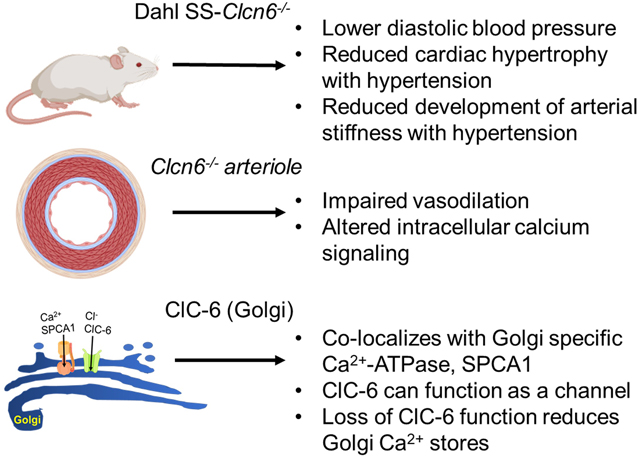
Abstract
Genome-wide association studies (GWAS) have found a number of potential genes involved in blood pressure regulation; however, the functional role of many of these candidates has yet to be established. One such candidate gene is CLCN6, which encodes the transmembrane protein, ClC-6. Although the CLCN6 locus has been widely associated with human blood pressure regulation, the mechanistic role of ClC-6 in blood pressure homeostasis at the molecular, cellular, and physiological levels is completely unknown. In this study, we demonstrate that rats with a functional knock out of ClC-6 on the Dahl Salt-Sensitive rat background (SS-Clcn6) have lower diastolic, but not systolic blood pressures. The effect of diastolic blood pressure attenuation was independent of dietary salt exposure in knockout animals. Moreover, SS-Clcn6 rats are protected from hypertension-induced cardiac hypertrophy and arterial stiffening; however, they have impaired vasodilation and dysregulated intracellular calcium handling. ClC-6 is highly expressed in vascular smooth muscle cells (vSMC) where it is targeted to the Golgi apparatus. Using bilayer electrophysiology, we provide evidence that recombinant human ClC-6 protein can function as a channel. Lastly, we demonstrate that loss of ClC-6 function reduces Golgi calcium stores, which may play a previously unidentified role in vascular contraction and relaxation signaling in vSMCs. Collectively, these data indicate that ClC-6 may modulate blood pressure by regulating Golgi calcium reserves which in turn contribute to vascular smooth muscle function.
Keywords: voltage-gated chloride channel, CLCN6, ClC-6, vasculature, hypertension, Dahl salt-sensitive rat, vascular smooth muscle
Graphical Abstract
Introduction
Hypertension is a worldwide epidemic that puts humans at increased risk of stroke, vascular damage, heart attack, heart failure, and kidney disease1. In the United States alone, nearly half the adult population is hypertensive2. Advances in genetic sequencing have begun to uncover chromosome regions and genes that are linked to variations in blood pressure (BP), potentially discovering new options for therapeutic targeting. Recent GWAS and whole exome sequencing in humans have linked many rare coding mutations and intronic single nucleotide polymorphisms in CLCN6 to lower BP and reduced hypertension and stroke risk3–7. An additional study in Dahl Salt-Sensitive (SS) rats found a similar genetic BP loci, and Clcn6 knockout rats have reduced BP, similar to humans8. Despite the mounting evidence that CLCN6 is likely a heritable modifier of hypertension risk, its mechanistic role in BP regulation at the molecular, cellular, and physiological levels remains completely unknown.
CLCN6 encodes the voltage-gated chloride channel 6 (ClC-6), which is one of 9 voltage-gated chloride channel (ClC) family members9. The ClCs have a range of physiological functions, including intracellular pH and chloride (Cl−) concentration control, cell volume homeostasis, and membrane excitability10,11. A unique attribute of the ClC family is that it is comprised of both channels and transporters, and at least two family members can function as either a transporter or a channel, depending on ionic concentrations or pH10,12–14. Studies characterizing CLCN6 localization with mRNA levels found that CLCN6 is broadly expressed and can be found in varying levels in colon, small intestine, ovaries, testes, lung, thymus, spleen, kidney, skeletal muscle, brain, heart, ocular trabecular network, and blood vessels9,15,16. Additional studies found that endogenous ClC-6 co-localizes with the late endo/lysosomal marker Lamp-1 in neurons, whereas over-expressing ClC-6 in a number of different cell lines results in mislocalization to early endosomes and recycling endosomes17–19. Localization in the endoplasmic reticulum (ER) with SERCA2 and overlap with GM130 in the Golgi in a heterologous expression system, has also been reported18,20,21. In neurons, ClC-6 has been proposed to function in proton (H+) exchange in acidified endosomes, with ClC-6 functioning as a Cl−/H+ antiporter19,22.
To date, no one has thoroughly examined the functional role of ClC-6 in BP regulation, despite the association with BP by GWAS3–5,8 and rare variant analysis7. Flister et al. reported that a Clcn6 knock out on the Dahl Salt-Sensitive (SS) rat background (SS-Clcn6) resulted in lowered diastolic and mean arterial blood pressure relative to controls after 10 days on a 4% NaCl diet8. However, this broad study only screened Clcn6 as one of six genes at the Agtrap-Plod1 locus with the ability to regulate BP and did not explore any mechanistic functions of ClC-6.
Here, we used the SS-Clcn6 rat to investigate the cardiac and vascular changes resulting from loss of ClC-6 protein function. We hypothesize that ClC-6 activity in vascular smooth muscle cells (vSMC) regulates luminal Golgi Ca2+ stores through an association with the Golgi-specific Ca2+-ATPase, SPCA1, and that Golgi Ca2+ may play an important role in vascular contraction-dilation signaling pathways and development of arterial stiffness. We demonstrate that the reduced diastolic BP in the SS-Clcn6 mutant attenuates BP independent of salt-sensitive effects, that these rats have reduced cardiac damage, and do not display an increase in arterial stiffness following development of hypertension. Furthermore, mesenteric resistance arteries in SS-Clcn6 rats have impaired vasodilation. We also demonstrate that ClC-6 is predominantly localized to the Golgi apparatus of vSMCs, and can function as a chloride channel, rather than a Cl−/H+ exchanger, as previously demonstrated22. Lastly, we demonstrate that loss of ClC-6 results in dysregulation of [Ca2+]i handling in vSMCs, and this is likely due to altered Golgi calcium stores. Collectively, these data provide novel mechanistic insight into the different effects on vasculature resulting from loss of ClC-6 function.
Methods
The data that support the findings of this study are available from the corresponding author upon reasonable request. The expanded Methods can be found in the Online Data Supplement.
Animals
All animal use and welfare adhered to the NIH Guide for the Care and Use of Laboratory Animals following a protocol reviewed and approved by the IACUC at the Medical College of Wisconsin. Animals were maintained in a standard 12/12 dark/light cycle with water and food provided ad libitum. The Dahl Salt-Sensitive (SS) rats (SS/JrHsdMcwi) have been inbred for more than 50 generations. All protocols used age-matched (8–12 week old male) SS-WT and SS-Clcn6 (SS-Clcn6em2Mcwi) rats maintained on a custom AIN-76 diet (Dyets, Inc.; 0.4% NaCl, #D113755). The SS-Clcn6 rats were generated using zinc finger nuclease (ZFN) mutagenesis resulting in a 15 base pair deletion in exon 13, which has previously been published8. BP measurements were obtained from telemeters as previously published23,24. After recovery from surgery, baseline measurements of arterial pressure were determined over 4–5 days with rats receiving the 0.4% NaCl diet before switching to 4% diet for 3 weeks (Dyets, Inc.; 0.4% NaCl, #D113756). Control groups (both SS-WT and SS-Clcn6 rats) were maintained on 0.4% NaCl diet for the same time frame. Rats were euthanized under isoflurane anesthesia. Plasma and urine electrolyte concentrations were determined by ABL800 FLEX blood gas analyzer (Radiometer America) as described25.
The online-only Data Supplement describes established methods, including immunohistochemical and immunofluorescence labeling26, echocardiography27,28, planar lipid bilayer electrophysiology29, pulse-wave velocity measurements30, pressure myography, and imaging of vSMC intracellular and Golgi calcium31.
Statistics
Results are presented as mean ± SEM, where N are independent animals and n are the number of replicates. Data were analyzed using GraphPad Prism 7 (GraphPad Software) and SigmaPlot 12.5 (SYSTAT Software). Data was compared using one-way ANOVA followed by Holm-Sidak post-hoc test or Two-way repeated measures ANOVA. Differences were considered statistically significant at p<0.05.
Results
CLC-6 Mutation Reduces Diastolic Blood Pressure
A Clcn6 mutant rat (SS-Clcn6em2Mcw, hereafter referred to as SS-Clcn6) was generated on the Dahl salt-sensitive background as part of a study to establish causality of GWAS candidates associated with BP8. We expanded on this study to compare daily BP values in male SS-WT and SS-Clcn6 rats over a 21 day 4.0% NaCl diet time course (Figure 1A). We found that genetic ablation of ClC-6 significantly reduced the diastolic pressure (DP), but did not significantly change the systolic pressure (SP). We determined that the DP was significantly lower on both the 0.4% NaCl diet and continuously (excluding day 2) throughout the 4% NaCl challenge (Figure 1, N≥10, p<0.05). Diurnal variations in blood pressure and heart rates were also not significantly different (Figure S1).
Figure 1. SS-Clcn6 cardiorenal phenotype.
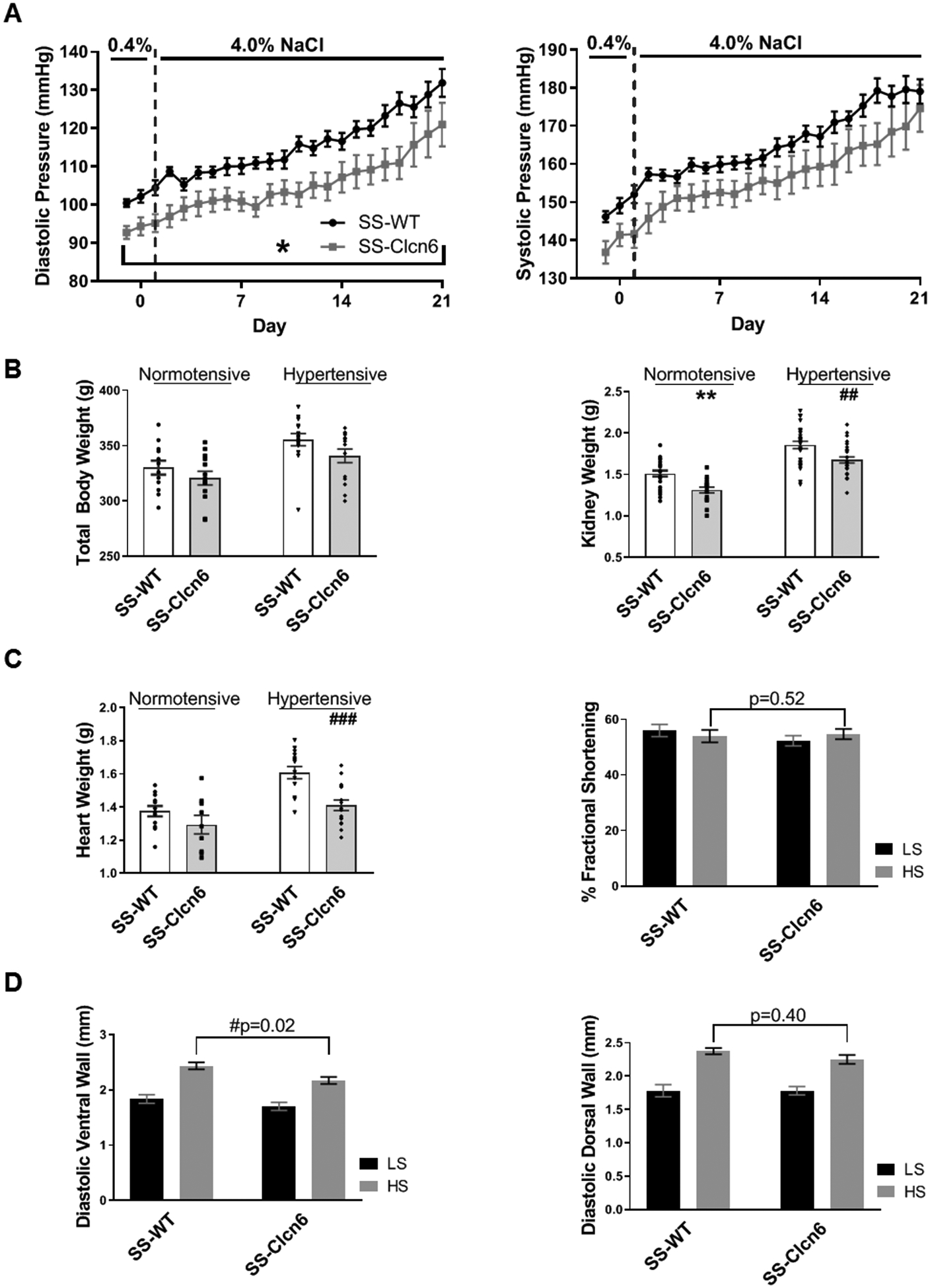
(A) Diastolic pressure (DP, left) and systolic pressure (SP, right). There are no significant differences between SS-WT (N=17) and SS-Clcn6 (N=12) in systolic pressure; however, the SS-Clcn6 rats have significantly lower DP on both the low (0.4% NaCl) and high (4% NaCl) salt diets (*p<0.05 for all days except day 2). (B) Total body weights of 11–12 weeks old normotensive or hypertensive SS-WT and SS-Clcn6 rats (N ≥ 12 rats). Kidney weights of normotensive or hypertensive SS-WT and SS-Clcn6 rats. SS-ClCn6 rats have significantly lower kidney weights in both conditions (N ≥ 18 kidneys, ** and ## p<0.01). (C) Loss of ClC-6 is cardio-protective. Heart weights of hypertensive SS-Clcn6 rats are significantly reduced compared to hypertensive SS-WT rats (left, N≥9, ### p<0.001). SS-WT and SS-Clcn6 rats underwent echocardiography before and after 3 weeks of a high salt diet to induce hypertension. Diastolic ventral wall thickness (D, left) was reduced in SS-Clcn6 rats on a high salt diet compared to SS-WT (N≥10, p<0.05). There was no significant difference in diastolic dorsal wall thickness (D, right; N≥10, p=0.61) or fractional shortening (C, right). Data were analyzed using a two-way ANOVA with repeated measures or using one-way ANOVA followed by Holm-Sidak post hoc test. Error bars are ± SEM.
There were no significant differences in total body weights between WT and mutant rats on a specific diet (LS or HS diet, Figure 1B, left), and body weights of both strains were increased on HS diet compared to LS. Since ClC-6 is expressed in the kidney with differing expression levels along the nephron we investigated what the effect of the ClC-6 mutation on renal size following a HS diet. Initially we compared kidney weights from age matched SS-WT and SS-Clcn6 rats on low and high salt diets (Figure 1B, right). Under both dietary conditions, the SS-Clcn6 rats had significantly lower kidney weights (LS, 1.51 ± 0.04 g vs 1.31 ± 0.03 g, N≥14 rats, **p<0.01; HS 1.90 ± 0.04 g vs 1.68 ± 0.04 g, N≥9 rats, ##p<0.01).
SS-Clcn6 Animals Have Reduced Cardiac Hypertrophy Compared to SS WT Rats
Next, we examined the cardiac phenotypes of SS-WT and SS-Clcn6 rats. For age-matched rats on LS diets, there were no significant differences between heart weights; however, on a HS diet, the SS-Clcn6 rats have reduced heart weights relative to controls (Figure 1C; 1.61 ± 0.04 g vs 1.41 ± 0.03 g, N≥14 rats, ###p<0.001). Additionally, there was no significant difference between heart weights of SS-Clcn6 rats on either LS or HS diets (Figure 1C; N≥9 rats, 1.30 ± 0.06 g vs 1.41 ± 0.03 g, p=0.15), whereas SS-WT rat heart weights were significantly increased (1.37 ± 0.03 g vs 1.61 ± 0.04 g, N≥12, p<0.001). To assess further functional differences and cardiac dimensions, we performed echocardiography on WT and mutant rats before and after 21 days of HS diet. Hypertrophy of the left ventricle wall was evaluated directly by echocardiography. We observed that when on the same high salt diet, SS-WT rats had significantly thicker ventral walls than SS-Clcn6 during diastole (Figure 1D, left; N≥10 rats, 2.43 ± 0.06 mm vs 2.17 ± 0.06 mm, #p<0.05). This difference was not observed in diastolic dorsal wall measurements (Figure 1D, right; N≥10 rats, 2.37 ± 0.05 vs 2.25 ± 0.07, p=0.39), or measurements from ventral or dorsal wall regions obtained during systole. There were no significant differences to the percentage of fractional shortening (% FS) either between WT and mutant or between LS and HS diets (Figure 1C, right).
The Pulse-Wave Velocity of SS-Clcn6 Conduit Arteries Does Not Increase Following Development of Hypertension
To assess changes in arterial stiffness, SS-WT and SS-Clcn6 rat pulse-wave velocities (PWV) were measured before and after 3 weeks of high salt diet to induce hypertension using Doppler ultrasound. At baseline, SS-Clcn6 rats have faster PWVs (Figure 2A; N≥6, 3.62 ± 0.21 mm/msec vs 4.53 ± 0.30 mm/msec, *p<0.05); however, at the end of 3 weeks of HS diet, while the SS-WT rats have a significant increase in stiffness, the SS-Clcn6 rats do not, and there is no longer a significant difference in the final PWVs (Figure 2A; 4.58 ± 0.12 mm/msec vs 4.57 ± 0.12 mm/msec, p=0.99).
Figure 2. Arterial stiffness, dilation and remodeling.
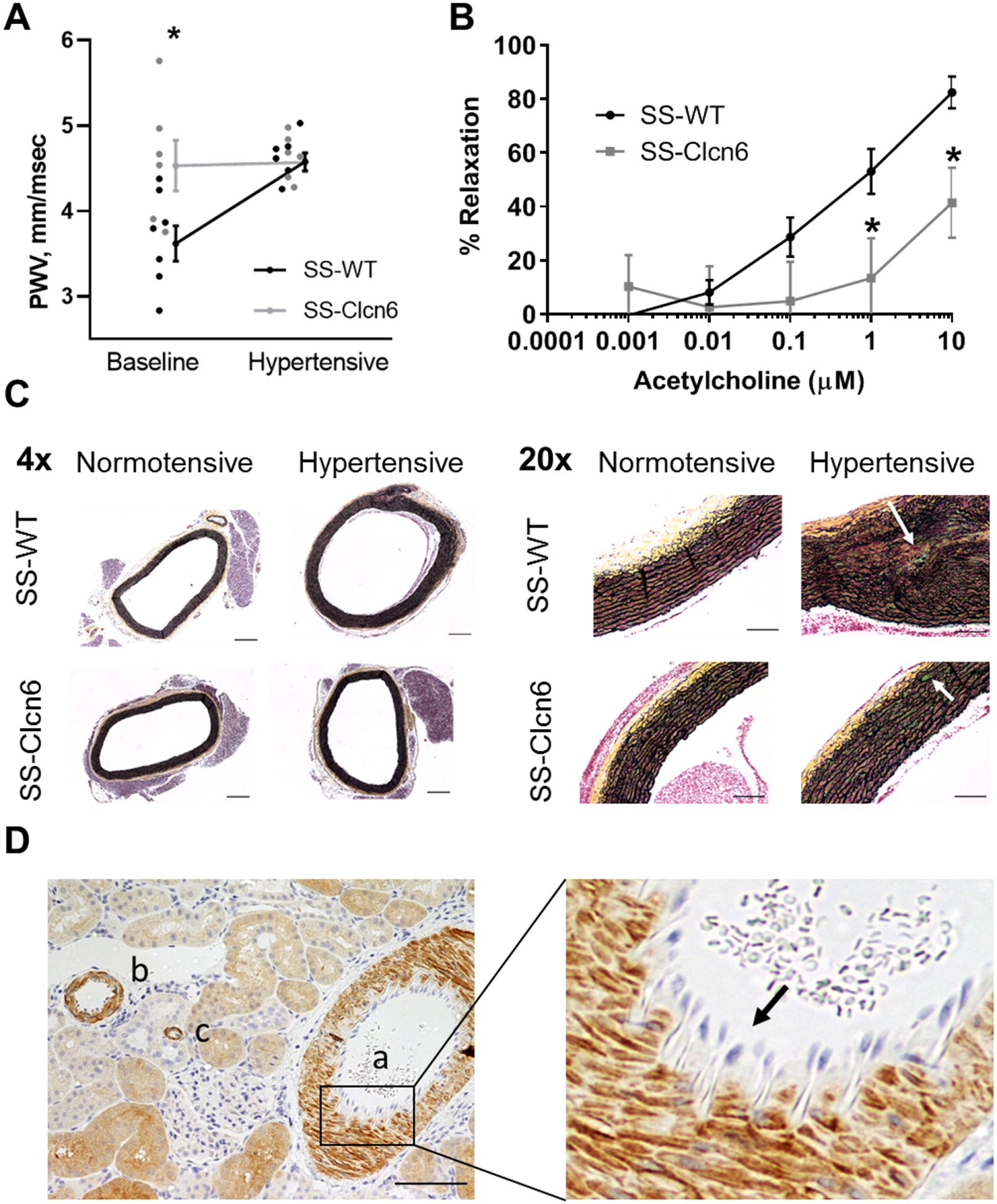
(A) Pulse-wave velocity (PWV) measurements were obtained for SS-WT and SS-Clcn6 rats at baseline and after 3 weeks of high salt diet to induce hypertension. SS-Clcn6 vessels have significantly increased PWVs at baseline (N=6 in both groups, *p<0.05). (B) Isolated arteries were maximally contracted with a thromboxane agonist and then exposed to increasing concentrations of acetylcholine (Ach). Arteries from SS-Clcn6 rats had significantly impaired vasodilation compared to SS-WT at 1 and 10 μM Ach (N=6 rats, n≥9 blood vessels, p<0.05). Error bars are ± SEM. (C) Movat pentachrome staining was used to examine collagen (yellow) and elastin fibers (black) in thoracic aorta sections from normotensive and hypertensive SS-WT or SS-Clcn6 rats. Cells are pink and calcification and glucosaminoglycans are aqua. The 4x representative images show medial thickening in the hypertensive SS-WT aorta (scale bars 200 μm). Additionally, the 20x images demonstrate elastin fragementation as well as the potential inititation some calcification (arrow) in the hypertensive SS-WT vessel wall, but not in the normotensive vessels. The hypertensive SS-Clcn6 tunica media does not display elastin fragmentation, but there appears to be some initiation of calcification (arrow) Scale bars are 100 μm. *20x images have been equally color saturated to provide better color contrast. (D) Immunohistochemistry labeling for ClC-6 (kidney section) demonstrating that ClC-6 is highly expressed in muscular (a) and large (b) arteries as well as arterioles (c). An enlarged area demonstrating the lack of ClC-6 expression in endothelial cells (arrow) in the intima is shown on the right.
Medial Thickening and Elastin Breakage in SS-WT Versus SS-Clcn6 Rats
Following the results of our PWV measurements, we formalin-fixed segments of the upper thoracic aortas from normotensive or hypertensive WT or knockout rats to look for structural changes using Movat Pentachrome, a histological stain that shows collagen in yellow and elastin in black. Cells can be observed in pink and calcification and glucosaminoglycans are aqua. We observed moderate medial thickening and areas with elastin breakage/disorganization in the hypertensive WT rats that was not present in the hypertensive SS-Clcn6 rat aortas, which are similar to the normotensive vessels. Representative images at 4x and 20x are shown in Figure 2C. Thus, while control Dahl SS rats develop salt sensitive hypertension, cardiac and vascular hypertrophy, as well as conduit vessel fibrosis and stiffening in response to chronic HS diets, the SS-Clcn6 rats are protected from SS hypertension and its related target organ damage.
Vessel Dilation Is Impaired in SS-Clcn6 Rats
We went on to examine the effect of ClC-6 knockout on the dilatory capability of resistance arteries. Third-order mesenteric resistance arteries were dissected out from SS-WT and SS-Clcn6 and cannulated and placed in a myography chamber with oxygenated in 95% O2/5% CO2 Krebs physiological salt solution at pH 7.4 and 37°C. After pressure equilibration, vessels were stimulated to maximal contraction with the thromboxane agonist, U46619. As incremental increases in acetylcholine (Ach) were added to the bath, the inner diameter of the vessel was measured with MyoView software, and percent dilation was calculated as the percent increase from the diameter at maximal dilation. We found that resistance arteries from the SS-Clcn6 rats had impaired dilation compared to SS-WT at 1 and 10 μM concentrations, as the percent relaxation was significantly less than WT (Figure 2B; 53 ± 8% vs 13 ± 15%, N=6 rats, n≥9 vessels, p<0.05 and 82 ± 6% vs 41 ± 13%, N=6 rats, n≥9 vessels, p<0.01, respectively).
SS-Clcn6 Rats Have Compromised Calcium Handling in vSMCs
The paradoxical vasodilation impairment is not likely to be a cause or contributor of the lower DBP in SS-Clcn6 rats, but rather a direct result of dysregulated smooth muscle calcium mobilization. To test this hypothesis, we examined ClC-6 tissue expression in multiple organ/systems. Initial studies characterizing ClC-6 mRNA and protein levels found that ClCN6 transcripts are broadly expressed at varying levels in a number of organs9,15–19. Using immunohistochemical analysis of rat tissue, we verified that ClC-6 is expressed in cardiomyocytes, lung smooth muscle, intestine smooth muscle and mucosal cells, neurons, and in different segments of the nephron (Figure S2). Importantly, we observed that ClC-6 is highly expressed in vSMCs, along the vascular tree from muscular and large arteries to arterioles, but absent from endothelial cells (Figure 2D).
To further understand the vascular dysfunction observed in the pressure myography experiments, we isolated mesenteric resistance arteries and used 2-photon imaging with calcium sensitive dyes to interrogate intracellular calcium ([Ca2+]i) handing in vSMCs (Figure 3). Representative traces are shown in Figure 3B. Vessels at rest had basal [Ca2+]i activity, and the magnitude of the basal [Ca2+]i peaks was trending to be greater in SS-Clcn6 vSMCs (Figure 3C; N≥9 rats, n≥11 cells, 255 ± 38 a.u. vs 514 ± 132 a.u. p=0.06). There were no significant differences in peak [Ca2+]i magnitude between WT and SS-Clcn6 rats following contraction stimulation with 10 μM phenylephrine (PE) or relaxation stimulus with10 μM Ach. We also measured the length of time between calcium peaks to determine the periodicity of [Ca2+]i signaling in basal, contracted, and relaxed states. Under basal conditions and following 10 μM PE, the duration between [Ca2+]i peaks was not significantly different. When stimulated to relax, however, the SS-Clcn6 vascular cells continue to release [Ca2+]i more frequently (shorter length of time between peaks) than WT cells (Figure 3D; N≥4 rats, n≥9 cells, 86 ± 11 sec vs 23 ± 6 sec, p<0.001). Additionally, the time between peaks for SS-Clcn6 cells treated with 10 μM PE or 10 μM PE + 10 μM Ach were not significantly different (N≥4 rats, n≥9 cells, 34 ± 4 sec vs 23 ± 6 sec, p=0.836).
Figure 3. Intracellular Ca2+ handling in vascular smooth muscle cells.
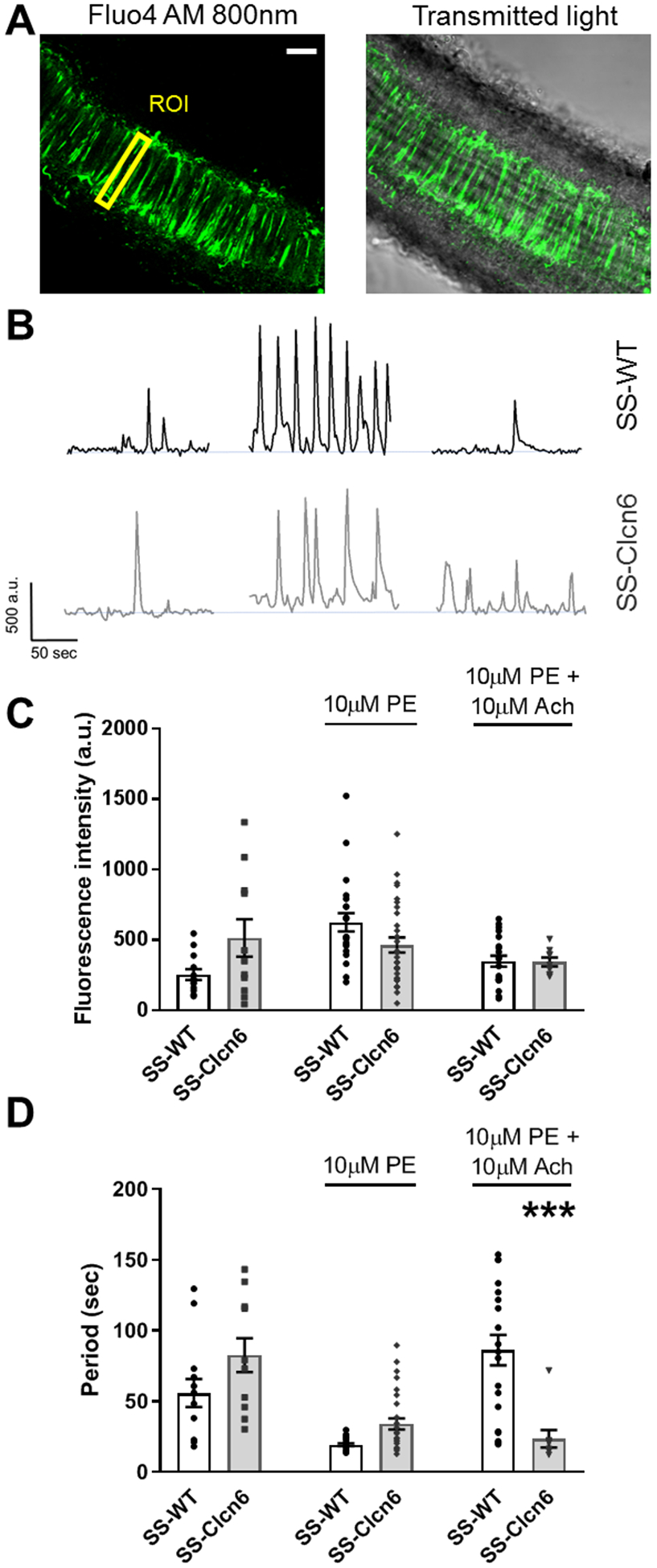
(A) Representative image of intracellular Ca2+ ([Ca2+]i) in vSMCs in a mesenteric artery imaged with 2-photon microscopy. Scale bar is 40 μm. Regions of interest (ROI) were defined for individual cells and changes in [Ca2+]i was measured as fluctuations in fluorescence intensity. (B) Representative Ca2+ traces from an individual cell before (basal) and after treatment with phenylephrine (PE) and acetylcholine (Ach) in SS-WT (black) and SS-Clcn6 (gray) resistance vessels. (C) Summarized data of [Ca2+]i spark intensity (N≥4 rats, n≥9 cells / condition). Under basal conditions, Ca2+ spike intensity is trending toward being significantly greater in the SS-Clcn6 vSMCs (p=0.06), whereas there are no differences in Ca2+ peak intensities between mutant and WT cells following contraction and relaxation stimuli. (D) Graph summarizing differences in Ca2+ release frequency. Under basal and contraction stimulus, the Ca2+ spark periodicity of WT and mutant cells are the same. Following relaxation stimulation (Ach), however, the SS-Clcn6 vSMCs maintain a higher rate of Ca2+ release, ie, the time between Ca2+ peaks is significantly reduced compared to SS-WT (N≥4 rats, n≥9 cells / condition, p<0.001) as determined by one way ANOVA. Error bars are ± SEM.
Subcellular Localization of ClC-6
Previous studies examining intracellular localization of ClC-6 have found that endogenous ClC-6 predominantly resides in the late endosomes/lysosomes17,18, in the endoplasmic reticulum (ER)20, or minimally in the Golgi apparatus18,21; however, the specific intracellular localization in vSMCs has not been previously assessed. We isolated vSMCs from rat aorta and assessed the co-localization of ClC-6 with multiple organelle markers. We found that endogenous ClC-6 in vSMCs is predominantly localized in the Golgi apparatus, with additional expression in intracellular punctae (Figure 4A). ClC-6 does not appear to strongly co-localize with Serca2 or Rab7 (Figure 4B–C), suggesting that ClC-6 is not targeted to the ER or late endosomes in vSMCs.
Figure 4. Intracellular localization of ClC-6.
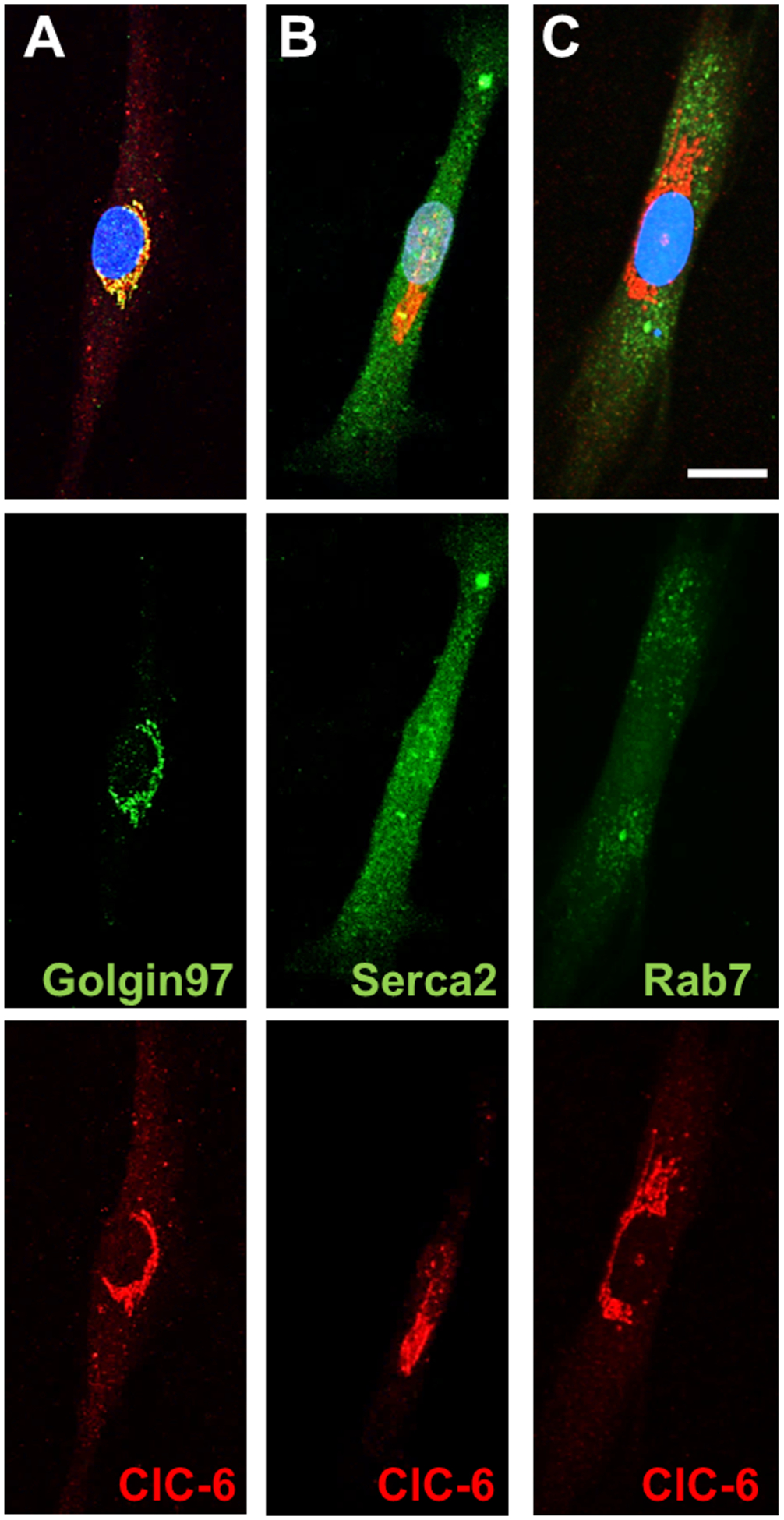
Representative images of ClC-6 expression with different organelle markers in isolated aorta vSMCs. Scale bar is 20 μm. The top images are the merged channels with nuclear staining. The middle images (green) show the different organelle markers, which are (A) Golgin-97, a resident trans-Golgi protein (B) Serca2, the ER localized Ca2+ ATPase, and (C) Rab7, a late endosomal marker. The bottom images (red) demonstrate the ClC-6 localization for each cell.
Electrophysiological Properties of ClC-6 in Lipid Bilayers
Previous research found that ClC-6 can function as a Cl−/H+ antiporter7,22. However, the electrochemical gradient and pH of the Golgi lumen are quite distinct from oocytes in which these studies were performed, which could result in different driving forces and activity of this protein. For example, another family member, ClC-3, behaves like a Cl−/H+ antiporter at the plasma membrane when expressed in HEK cells32, but becomes uncoupled at low pH (4.0) and its biophysical properties shift from exchanger to anion selective channel. To assess the functional characteristic of ClC-6 more fully, recombinant human ClC-6 protein was inserted into artificial lipid bilayers to measure electrophysiological activity (Figure 5). When inserted into phospholipid bilayers in asymmetrical salt concentration in cis- and trans-chambers (0.35/0.15 M KCl) to estimate anion-cation channel selectivity, ClC-6 displays step-like fluctuations typical of channel behavior (Figure 5A). The channel displays a large conductance (100 pS), which is independent of the transmembrane voltage, a feature typical of large protein pores in KCl solutions. The reversal potential of ClC-6 in our conditions was −8 mV, which corresponds to the weak anion selectivity (Figure 5C).
Figure 5. ClC-6 electrophysiological properties.
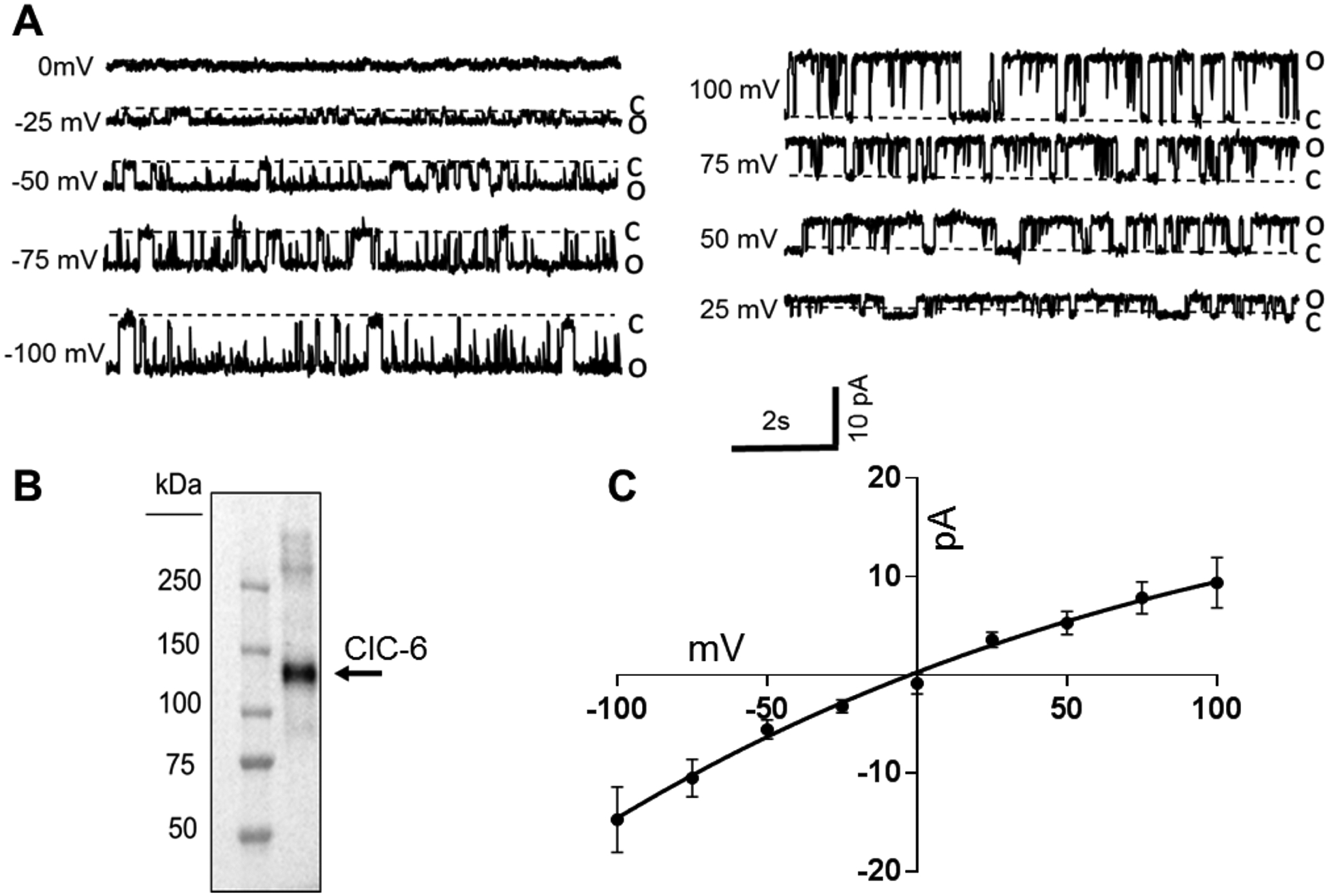
(A) Representative traces showing ClC-6 activity at different holding potentials, which were used to determine the single-channel current-voltage relationship. (B) Western blot of purified ClC-6 protein used for electrophysiological studies. Prediected molecular weight is 97 kDa plus a 28 kDa GST-tag. (C) Current-voltage (I-V) plot of voltage dependent ClC-6 conductance in bilayers composed of 80 mol% DPhPC and 20 mol% DPhPS in a 350:150 mM KCl, pH 7.4 bath solution gradient (n=15). Reversal potential is about −8 mV, which corresponds to non ideal anion selectivity. Error bars are ± SEM.
SS-Clcn6 vSMCs Have Reduced Golgi Ca2+ Stores
While the ER (or SR) is the largest Ca2+ sink in eukaryotic cells, the Golgi apparatus also has a high luminal Ca2+ concentration (~100 to 300 μM) and its own dedicated Ca2+-ATPase, the Secretory Pathway Ca2+-ATPase pump type 1 (SPCA1)33–37. SPCA1 is a Ca2+ (and Mn2+) pump that maintains luminal Golgi Ca2+ concentrations38–40 similar to the function of Serca in the ER; however, it is not inhibited by thapsigargin (TG), a classic Serca antagonist. Figure 6A demonstrates distinct co-localization of SPCA1 and ClC-6 in a vSMC. A recent study from Gallegos-Gomez et al, described a Ca2+ store released from the trans-Golgi by stimulation with emetine, by measuring changes in the organelle-specific Mg2+/Ca2+ specific dye, Mag-Fluo4 AM41. In order to determine whether loss of ClC-6 function affects Golgi Ca2+ reserves [Ca2+]golgi, we loaded primary cultured aorta vSMCs from SS-WT or SS-Clcn6 rats with Mag-Fluo4 and treated them with pharmacological agents to isolate the Golgi specific component. Cells were imaged in a 0 mM Ca2+ solution to eliminate extracellular calcium contributions. We applied 10 μM TG + 1 μM ATP to mobilize and empty ER/SR calcium stores, followed by application of 50 μM Emetine to deplete Golgi reserves. Representative images and summarized traces are shown in Figures 6B and 6C. Total [Ca2+]golgi was determined by area under the curve measurements. We establish that while there was no significant change in the calcium release stimulated by TG + ATP between groups, the total [Ca2+]golgi release from vSMCs from SS-Clcn6 was significantly reduced compared to controls (Figure 6D, (1.37 ± 0.93)∙104 a.u. vs (1.01 ± 0.57)∙ 104 a.u., n≥46 cells, **p<0.01).
Figure 6. Golgi Ca2+ stores are reduced in SS-Clcn6 vSMCs.
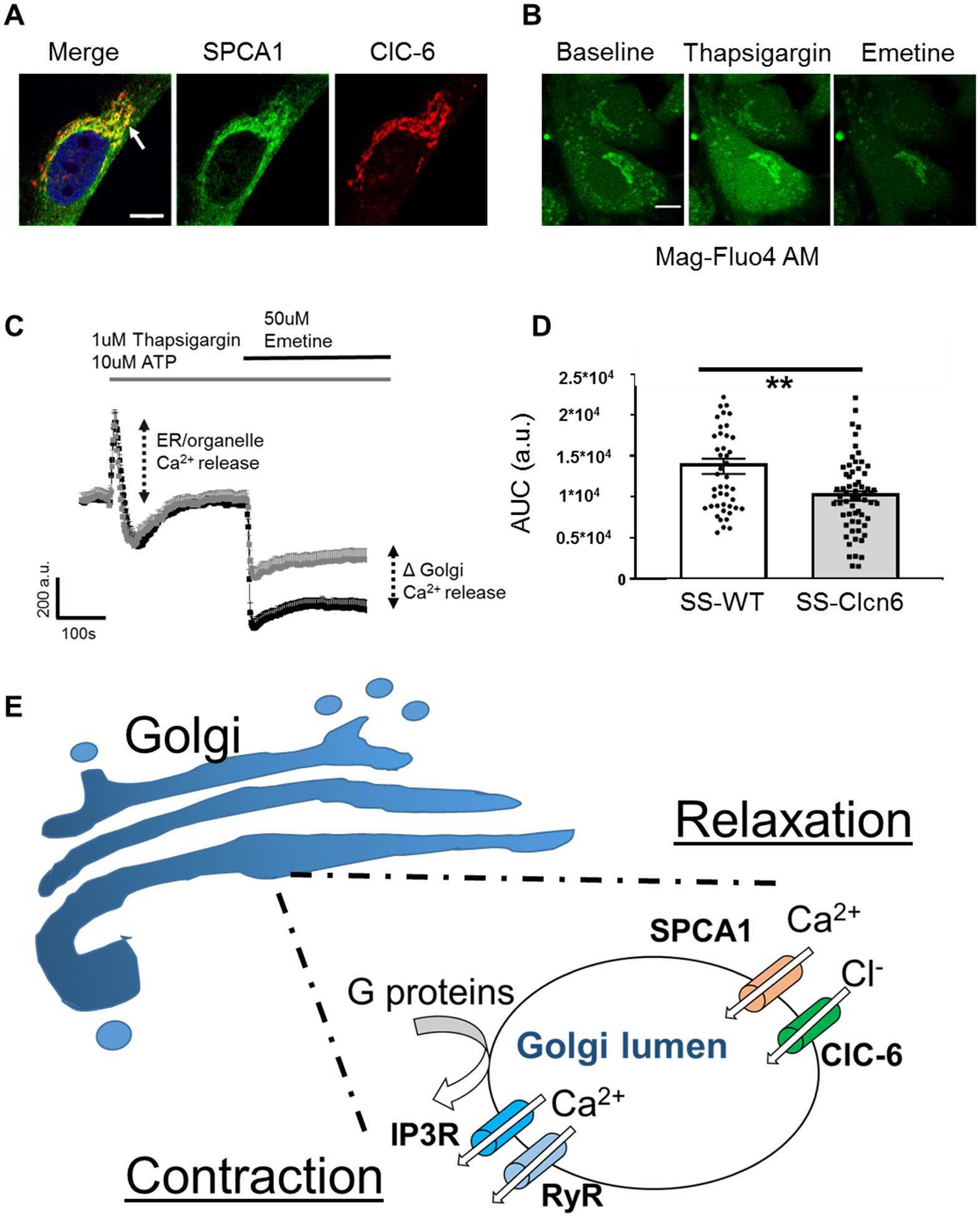
(A) Immunofluorescence labeling of SPCA1 (green) and ClC-6 (red) in a vSMC. Note the distinct area of co-localization (arrow). Scale bar is 10 μm. (B) Representative images of vSMCs loaded with Mag-Fluo4 at baseline (left), immediately after TG + ATP application (middle), and after Emetine application (right). Scale bar is 10 μm. First cells were stimulated with 1 μM Thapsigargin (TG) +10 μM ATP to release ER Ca2+, then stimulated with 50 μM Emetine to release Golgi Ca2+ stores. (C) Changes in Ca2+ over time were determined by ROIs drawn over the Golgi region. Average curve values ± SEM of SS-WT (black) or SS-Clcn6 (grey) vSMC Golgi Ca2+. There is no significant difference between the cells following 1 μM TG + 10 μM ATP; however, the [Ca2+]golgi release (as determined by the area under the curve (AUC) resulting from 50 μM Emetine) was significantly reduced in the SS-Clcn6 VSMCs. (D) Summary graph of AUC measurements from all experiments (N=4 experiments, n≥46 cells, **p<0.01). (E) Proposed molecular mechanism for ClC-6 in Golgi calcium handling. In response to a contractile stimulus such as phenylephrine, G-protein coupled signaling cascades result in release of lumenal [Ca2+]Golgi stores through IP3Rs and RyRs, which contribute to the [Ca2+]i pool. Upon relaxation, the secretory pathway calcium ATPase (SPCA1) removes cytosolic calcium back into the Golgi lumen, and this positive charge influx is balanced by concurrent chloride entry via ClC-6.
Discussion
Understanding the molecular and physiological factors that regulate BP are key to discovering new treatments for diseases like hypertension, which is a global health problem that causes heart disease, kidney failure, and stroke. Technological advances in genome sequencing have implicated a number of previously unknown genes in the regulation of BP, including CLCN6. In this work, we investigated how genetic ablation of CLCN6 regulates BP in the Dahl SS rat model, and determined that ClC-6 is highly expressed in vSMCs, where it is localized in the Golgi, and that loss of this protein’s function results in impaired vasodilation. Our data indicate that ClC-6 plays a role in [Ca2+]i handling in vSMCs, and suggest that ClC-6 regulates BP through alterations to vascular function. We speculate that due to its ability to function as an anion channel, chloride transport through ClC-6 functions as a charge balance to prevent changes to the electrochemical membrane potential of the Golgi apparatus during calcium reuptake via the secretory pathway calcium ATPase SPCA1 (Figure 6E). These data represent the first functional evidence of the role of ClC-6 protein in BP homeostasis.
Calcium signaling is unquestionably one of the most important and variable mechanisms controlling cellular function, as it can regulate excitation-contraction coupling, hormone secretion, apoptosis, growth, differentiation, and more. It has previously been demonstrated that roughly 55% of microsomal membrane Ca2+ uptake in vSMCs is TG insensitive, suggesting the presence of a large non-SR and non-mitochondrial Ca2+ sink38. The presence of such a large non-SR Ca2+ source suggests the Golgi and endosomes have functional roles in vSMC Ca2+ signal transduction pathways. Direct measurements of real-time changes in Golgi Ca2+ concentrations in intact vSMCs has never before been examined. The advantage of our approach compared to assays measuring Ca2+ reuptake in microsomal membranes is that we can perform the experiments without additional Ca2+ blockers and without disturbing the 3D integrity of organelle structures. We demonstrate that loss of ClC-6 reduces Golgi calcium stores, and suggest that Golgi calcium may have a hitherto unknown role in vascular tone.
Our data demonstrating impaired [Ca2+]i handling and relaxation is counter-intuitive to what might be predicted for a protective phenotype, and future studies will more thoroughly interrogate this phenotype in hypertensive vessels and in response to additional vasoconstriction and vasodilation stimuli. The elevated PWVs observed at baseline are also consistent with the impaired relaxation in the normotensive vessels, but the lack of increase following development of hypertension may be either a result of the overall reduced blood pressure or other changes resulting from Golgi dysfunction. The SS-Clcn6 rats had reduced heart weights and thinner ventral walls during diastole, but whether this was a secondary effect resulting from the reduced diastolic pressure or altered vascular compliance or a primary effect related to cardiac ClC-6 expression is still undetermined. The mechanism by which ClC-6 exerts this effect is currently unknown, but potentially results from impaired extracellular matrix protein trafficking, reduced vSMC proliferation, or increased apoptosis and remodeling during hypertension. In vSMCs, ClC-6 is predominantly located in the Golgi apparatus, which is the main director of protein trafficking in eukaryotic cells. Large extracellular molecules such as collagen rely on tightly regulated trafficking in order to form extracellular scaffolds. Alteration of intraluminal Golgi pH and calcium has the ability to disrupt proper protein trafficking, and impaired trafficking can diminish extracellular matrix proteins and focal adhesions, resulting in greater compliance. Additionally, disruption of Golgi lumen ionic balance has been shown to result in Golgi cisternae malformation, which causes increased cellular stress, leading to apoptosis42–46. Beyond this, during cell division, the Golgi apparatus undergoes disassembly before being divided and reassembled, and disruption of this process can halt cell division, thereby changing proliferation rates47. Lastly, as shown in Figure S2, ClC-6 is highly expressed in neurons and we have not yet interrogated potential changes to sympathetic drive resulting from loss of ClC-6.
Supplementary Material
Perspectives.
We have only just begun unravelling how alterations to ClC-6 function affect vascular function and convey protection against hypertension and stroke. We characterized the physiological changes resulting from ClC-6 genetic ablation to begin to elucidate the protective nature of rare variants in humans, and to develop a foundational understanding of the purpose of ClC-6 in arterial smooth muscle cells. We determined that ClC-6 loss reduces Golgi calcium stores, impairs vasodilation, and dysregulates intracellular calcium signaling. However, it also prevents or slows cardiac hypertrophy and development of vascular stiffness during development of hypertension. Additionally, while alterations to vascular relaxation, and stiffness are important factors involved in blood pressure regulation of the SS-Clcn6 rats, there may be additional changes, such as altered sympathetic signaling that contribute to the protective phenotype as well. Our results have elevated our understanding of how this understudied protein may be involved in blood pressure maintenance which will direct future investigations in the role of ClC-6 in Golgi calcium homeostasis and arterial tone. Furthermore, they suggest ClC-6 may be a novel therapeutic target for treatment of hypertension.
Novelty and Significance.
What is new?
These experiments present the first ever analysis of loss of ClC-6 function on arterial dilation, arterial stiffness, vascular remodeling, and intracellular calcium handling in vascular smooth muscle cells.
The role of Golgi calcium stores in vascular tone has not previously been investigated. Our studies suggest that disruption of these stores can impact vasodilation signaling.
What is relevant?
While many genetic studies have linked CLCN6 to blood pressure regulation, its functional role was previously unknown. These data indicate that it is involved in vascular smooth muscle cell calcium handling.
Knock out of ClC-6 on the Dahl Salt-Sensitive rat background has reduced diastolic blood pressure and is protective against cardiac hypertrophy and arterial stiffness resulting from hypertension, implicating ClC-6 block as a potential treatment strategy.
Summary
These results suggest ClC-6 may be a new potential therapeutic target in the treatment of hypertension, and additional studies are warranted to more thoroughly understand the role of this transporter in blood pressure homeostasis and vascular function.
Acknowledgments
We thank the Children’s Research Institute Histology Core for their assistance with immunohistochemistry. Thanks also to Dr. Curt D. Sigmund for equipment and additional support for the PWV measurements.
Sources of funding
This work was supported by the National Institute of Health grants: R35 HL135749, P01 HL116264 (AS), R56 DK121750 (OP), T32 HL134643 (CAK), Department of Veteran Affairs I01 BX004024 (AS).
Footnotes
Conflict of interest: The authors declare that they have no conflicts of interest with the contents of this article.
References
- 1.Bromfield S, Muntner P. High blood pressure: the leading global burden of disease risk factor and the need for worldwide prevention programs. Curr Hypertens Rep. 2013;15(3):134–136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Writing Group M, Mozaffarian D, Benjamin EJ, Go AS, Arnett DK, Blaha MJ, et al. Heart Disease and Stroke Statistics-2016 Update: A Report From the American Heart Association. Circulation. 2016;133(4):e38–360. [DOI] [PubMed] [Google Scholar]
- 3.Tomaszewski M, Debiec R, Braund PS, Nelson CP, Hardwick R, Christofidou P, et al. Genetic architecture of ambulatory blood pressure in the general population: insights from cardiovascular gene-centric array. Hypertension. 2010;56(6):1069–1076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Ji LD, Li JY, Yao BB, Cai XB, Shen QJ, Xu J. Are genetic polymorphisms in the renin-angiotensin-aldosterone system associated with essential hypertension? Evidence from genome-wide association studies. J Hum Hypertens. 2017;31(11):695–698. [DOI] [PubMed] [Google Scholar]
- 5.Zhang H, Mo XB, Xu T, Bu XQ, Lei SF, Zhang YH. Novel Genes Affecting Blood Pressure Detected Via Gene-Based Association Analysis. G3 (Bethesda). 2015;5(6):1035–1042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Pereira NL, Tosakulwong N, Scott CG, Jenkins GD, Prodduturi N, Chai Y, et al. Circulating atrial natriuretic peptide genetic association study identifies a novel gene cluster associated with stroke in whites. Circ Cardiovasc Genet. 2015;8(1):141–149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Yu B, Pulit SL, Hwang SJ, Brody JA, Amin N, Auer PL, et al. Rare Exome Sequence Variants in CLCN6 Reduce Blood Pressure Levels and Hypertension Risk. Circ Cardiovasc Genet. 2016;9(1):64–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Flister MJ, Tsaih SW, O’Meara CC, Endres B, Hoffman MJ, Geurts AM, et al. Identifying multiple causative genes at a single GWAS locus. Genome Res. 2013;23(12):1996–2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Brandt S, Jentsch TJ. ClC-6 and ClC-7 are two novel broadly expressed members of the CLC chloride channel family. FEBS Lett. 1995;377(1):15–20. [DOI] [PubMed] [Google Scholar]
- 10.Poroca DR, Pelis RM, Chappe VM. ClC Channels and Transporters: Structure, Physiological Functions, and Implications in Human Chloride Channelopathies. Front Pharmacol. 2017;8:151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Jentsch TJ, Pusch M. CLC Chloride Channels and Transporters: Structure, Function, Physiology, and Disease. Physiol Rev. 2018;98(3):1493–1590. [DOI] [PubMed] [Google Scholar]
- 12.Orhan G, Fahlke C, Alekov AK. Anion- and proton-dependent gating of ClC-4 anion/proton transporter under uncoupling conditions. Biophys J. 2011;100(5):1233–1241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Matsuda JJ, Filali MS, Collins MM, Volk KA, Lamb FS. The ClC-3 Cl−/H+ antiporter becomes uncoupled at low extracellular pH. J Biol Chem. 2010;285(4):2569–2579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Rohrbough J, Nguyen HN, Lamb FS. Modulation of ClC-3 gating and proton/anion exchange by internal and external protons and the anion selectivity filter. J Physiol. 2018;596(17):4091–4119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Lamb FS, Clayton GH, Liu BX, Smith RL, Barna TJ, Schutte BC. Expression of CLCN voltage-gated chloride channel genes in human blood vessels. J Mol Cell Cardiol. 1999;31(3):657–666. [DOI] [PubMed] [Google Scholar]
- 16.Comes N, Gasull X, Gual A, Borras T. Differential expression of the human chloride channel genes in the trabecular meshwork under stress conditions. Exp Eye Res. 2005;80(6):801–813. [DOI] [PubMed] [Google Scholar]
- 17.Poet M, Kornak U, Schweizer M, Zdebik AA, Scheel O, Hoelter S, et al. Lysosomal storage disease upon disruption of the neuronal chloride transport protein ClC-6. Proc Natl Acad Sci U S A. 2006;103(37):13854–13859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Ignoul S, Simaels J, Hermans D, Annaert W, Eggermont J. Human ClC-6 is a late endosomal glycoprotein that associates with detergent-resistant lipid domains. PLoS One. 2007;2(5):e474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Pressey SN, O’Donnell KJ, Stauber T, Fuhrmann JC, Tyynela J, Jentsch TJ, et al. Distinct neuropathologic phenotypes after disrupting the chloride transport proteins ClC-6 or ClC-7/Ostm1. J Neuropathol Exp Neurol. 2010;69(12):1228–1246. [DOI] [PubMed] [Google Scholar]
- 20.Buyse G, Trouet D, Voets T, Missiaen L, Droogmans G, Nilius B, et al. Evidence for the intracellular location of chloride channel (ClC)-type proteins: co-localization of ClC-6a and ClC-6c with the sarco/endoplasmic-reticulum Ca2+ pump SERCA2b. Biochem J. 1998;330(Pt 2):1015–1021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Stauber T, Jentsch TJ. Sorting motifs of the endosomal/lysosomal CLC chloride transporters. J Biol Chem. 2010;285(45):34537–34548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Neagoe I, Stauber T, Fidzinski P, Bergsdorf EY, Jentsch TJ. The late endosomal ClC-6 mediates proton/chloride countertransport in heterologous plasma membrane expression. J Biol Chem. 2010;285(28):21689–21697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Palygin O, Levchenko V, Ilatovskaya DV, Pavlov TS, Pochynyuk OM, Jacob HJ, et al. Essential role of Kir5.1 channels in renal salt handling and blood pressure control. JCI Insight. 2017;2(18):e92331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Endres BT, Priestley JR, Palygin O, Flister MJ, Hoffman MJ, Weinberg BD, et al. Mutation of Plekha7 attenuates salt-sensitive hypertension in the rat. Proc Natl Acad Sci U S A. 2014;111(35):12817–12822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Palygin O, Spires D, Levchenko V, Bohovyk R, Fedoriuk M, Klemens CA, et al. Progression of diabetic kidney disease in T2DN rats. Am J Physiol Renal Physiol. 2019;317(6):F1450–F1461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Pavlov TS, Levchenko V, O’Connor PM, Ilatovskaya DV, Palygin O, Mori T, et al. Deficiency of renal cortical EGF increases ENaC activity and contributes to salt-sensitive hypertension. J Am Soc Nephrol. 2013;24(7):1053–1062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Miller JJ, Aoki K, Mascari CA, Beltrame AK, Sokumbi O, North PE, et al. alpha-Galactosidase A-deficient rats accumulate glycosphingolipids and develop cardiorenal phenotypes of Fabry disease. FASEB J. 2019;33(1):418–429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Chuppa S, Liang M, Liu P, Liu Y, Casati MC, Cowley AW, et al. MicroRNA-21 regulates peroxisome proliferator-activated receptor alpha, a molecular mechanism of cardiac pathology in Cardiorenal Syndrome Type 4. Kidney Int. 2018;93(2):375–389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Montal M, Mueller P. Formation of bimolecular membranes from lipid monolayers and a study of their electrical properties. Proc Natl Acad Sci U S A. 1972;69(12):3561–3566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Wu J, Agbor LN, Fang S, Mukohda M, Nair AR, Nakagawa P, et al. Failure to Vasodilate in Response to Salt Loading Blunts Renal Blood Flow and Causes Salt-Sensitive Hypertension. Cardiovasc Res. 2020; doi: 10.1093/cvr/cvaa147). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Palygin O, Miller BS, Nishijima Y, Zhang DX, Staruschenko A, Sorokin A. Endothelin receptor A and p66Shc regulate spontaneous Ca(2+) oscillations in smooth muscle cells controlling renal arterial spontaneous motion. FASEB J. 2019;33:2636–2645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Matsuda JJ, Filali MS, Volk KA, Collins MM, Moreland JG, Lamb FS. Overexpression of CLC-3 in HEK293T cells yields novel currents that are pH dependent. Am J Physiol Cell Physiol. 2008;294(1):C251–262. [DOI] [PubMed] [Google Scholar]
- 33.Suzuki J, Kanemaru K, Iino M. Genetically Encoded Fluorescent Indicators for Organellar Calcium Imaging. Biophys J. 2016;111(6):1119–1131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Pizzo P, Lissandron V, Capitanio P, Pozzan T. Ca(2+) signalling in the Golgi apparatus. Cell Calcium. 2011;50(2):184–192. [DOI] [PubMed] [Google Scholar]
- 35.Van Baelen K, Dode L, Vanoevelen J, Callewaert G, De Smedt H, Missiaen L, et al. The Ca2+/Mn2+ pumps in the Golgi apparatus. Biochim Biophys Acta. 2004;1742(1–3):103–112. [DOI] [PubMed] [Google Scholar]
- 36.Wuytack F, Raeymaekers L, Missiaen L. PMR1/SPCA Ca2+ pumps and the role of the Golgi apparatus as a Ca2+ store. Pflugers Arch. 2003;446(2):148–153. [DOI] [PubMed] [Google Scholar]
- 37.Missiaen L, Raeymaekers L, Dode L, Vanoevelen J, Van Baelen K, Parys JB, et al. SPCA1 pumps and Hailey-Hailey disease. Biochem Biophys Res Commun. 2004;322(4):1204–1213. [DOI] [PubMed] [Google Scholar]
- 38.Wootton LL, Argent CC, Wheatley M, Michelangeli F. The expression, activity and localisation of the secretory pathway Ca2+-ATPase (SPCA1) in different mammalian tissues. Biochim Biophys Acta. 2004;1664(2):189–197. [DOI] [PubMed] [Google Scholar]
- 39.Vanoevelen J, Dode L, Van Baelen K, Fairclough RJ, Missiaen L, Raeymaekers L, et al. The secretory pathway Ca2+/Mn2+-ATPase 2 is a Golgi-localized pump with high affinity for Ca2+ ions. J Biol Chem. 2005;280(24):22800–22808. [DOI] [PubMed] [Google Scholar]
- 40.Vangheluwe P, Sepulveda MR, Missiaen L, Raeymaekers L, Wuytack F, Vanoevelen J. Intracellular Ca2+- and Mn2+-transport ATPases. Chem Rev. 2009;109(10):4733–4759. [DOI] [PubMed] [Google Scholar]
- 41.Gallegos-Gomez ML, Greotti E, Lopez-Mendez MC, Sanchez-Vazquez VH, Arias JM, Guerrero-Hernandez A. The Trans Golgi Region is a Labile Intracellular Ca2+ Store Sensitive to Emetine. Sci Rep. 2018;8(1):17143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Micaroni M, Perinetti G, Berrie CP, Mironov AA. The SPCA1 Ca2+ pump and intracellular membrane trafficking. Traffic. 2010;11(10):1315–1333. [DOI] [PubMed] [Google Scholar]
- 43.van Raam BJ, Lacina T, Lindemann RK, Reiling JH. Secretory stressors induce intracellular death receptor accumulation to control apoptosis. Cell Death Dis. 2017;8(10):e3069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Okunade GW, Miller ML, Azhar M, Andringa A, Sanford LP, Doetschman T, et al. Loss of the Atp2c1 secretory pathway Ca2+-ATPase (SPCA1) in mice causes Golgi stress, apoptosis, and midgestational death in homozygous embryos and squamous cell tumors in adult heterozygotes. J Biol Chem. 2007;282(36):26517–26527. [DOI] [PubMed] [Google Scholar]
- 45.Sepulveda MR, Vanoevelen J, Raeymaekers L, Mata AM, Wuytack F. Silencing the SPCA1 (secretory pathway Ca2+-ATPase isoform 1) impairs Ca2+ homeostasis in the Golgi and disturbs neural polarity. J Neurosci. 2009;29(39):12174–12182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Machamer CE. The Golgi complex in stress and death. Front Neurosci. 2015;9:421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Carlton JG, Jones H, Eggert US. Membrane and organelle dynamics during cell division. Nat Rev Mol Cell Biol. 2020;21(3):151–166. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.