

Abstract
Ribonucleotide reductase (RNR), which is a heterodimeric tetramer composed of RRM1 and RRM2 subunits, is the rate-limiting enzyme in the synthesis of deoxyribonucleoside triphosphates (dNTPs) and essential for both DNA replication and the repair of DNA damage. The activity of RNR is coordinated with the cell cycle and regulated by fluctuations in the level of the RRM2 subunit. Multiple cancer types, including Ewing sarcoma tumors, are sensitive to inhibitors of RNR or a reduction in the levels of either the RRM1 or RRM2 subunits of RNR. Here, we show that the expression of the RRM2 protein is dependent on active protein synthesis and that 4E-BP1, a repressor of cap-dependent protein translation, specifically regulates the level of the RRM2 protein. Furthermore, inhibition of mTORC1/2, but not mTORC1, activates 4E-BP1, inhibits protein synthesis, and reduces the level of the RRM2 protein in multiple sarcoma cell lines. This effect of mTORC1/2 inhibitors on protein synthesis and RRM2 levels was rescued in cell lines with the CRISPR/Cas9-mediated knockout of 4E-BP1. In addition, the inducible expression of a mutant 4E-BP1 protein that cannot be phosphorylated by mTOR blocked protein synthesis and inhibited the growth of Ewing sarcoma cells in vitro and in vivo in a xenograft. Overall, these results provide insight into the multifaceted regulation of RRM2 protein levels and identify a regulatory link between protein translation and DNA replication.
INTRODUCTION
Ribonucleotide reductase (RNR) is the rate-limiting enzyme in the synthesis of deoxyribonucleosides (dNTPs) and the molecular target of multiple chemotherapy drugs, including gemcitabine, clofarabine, hydroxyurea, and iron chelators (1-3). RNR is a heterodimeric tetramer composed of RRM1 and RRM2 subunits and is essential for both DNA replication and the repair of DNA damage (3,4). Consequently, RNR inhibitors, which target either the active site of the RRM1 subunit or the dinuclear iron site of the RRM2 subunit, impair DNA replication in cancer cells and also synergize with a wide range of drugs that cause DNA damage (1). Notably, in previous work, we showed that Ewing sarcoma tumors are sensitive to inhibitors of RNR, or knockdown of RRM1 or RRM2 (5,6).
The activity of RNR is tightly regulated in cells and fluctuates during the cell cycle to coordinate dNTP production with DNA replication. The level of the RRM1 subunit of RNR is constant throughout the cell cycle, but the level of RRM2 fluctuates during the cell cycle and, thereby, coordinates cell-cycle progression with the balance between dNTP production and DNA replication (3,7). Although the RRM2 gene is a well-described target of the E2F1 transcription factor, additional transcriptional and non-transcriptional mechanisms are also reported to regulate the level of RRM2 protein in cells (7-11). For example, the CDK-mediated phosphorylation of RRM2 at Threonine-33 targets RRM2 for ubiquitination and degradation by the proteasome (12,13).
Inhibition of RNR, or a reduction in the level of the RRM2 protein, depletes nucleoside pools, causes DNA replication stress, and activates the Ataxia Telangiectasia and Rad3-Related Protein (ATR) and Checkpoint Kinase 1 (CHK1) pathway. ATR-CHK1 signaling, in turn, coordinates the stabilization of stalled replication forks, the repair of DNA damage, and, ultimately, the resumption of DNA replication (14-16). Notably, ATR and CHK1 inhibitors synergize, in vitro and in vivo, with RNR inhibitors and other drugs that cause DNA replication stress. For example, inhibition of the ATR-CHK1 pathway increases the toxicity of nucleoside analogues in a variety of cancers, including pancreatic, lung, and ovarian tumors (17-19). Similarly, we have shown that Ewing sarcoma tumors are sensitive to drug combinations that target RNR and CHK1 (5,13).
In the current study, we used a chemical-genetic approach to screen FDA-approved anticancer drugs and identify compounds that selectively target cancer cells with decreased RNR activity caused by a reduction in the level of the RRM2 protein. Notably, while investigating the mechanistic basis of a hit from the screen, we found that Ewing sarcoma cells, as well as multiple other cell types, are highly reliant on active protein synthesis to maintain a stable level of the RRM2 protein. In addition, we found that 4E-BP1, which represses cap-dependent protein translation by directly binding and sequestering the eukaryotic translation initiation factor 4E (eIF4E) that interacts with the m7G cap at the 5’ end of mRNA, regulates protein synthesis and decreases RRM2 protein levels in Ewing sarcoma cells. Furthermore, the inhibition of mTORC1/2, but not mTORC1, activated 4E-BP1 by blocking the inhibitory phosphorylation of 4E-BP1 at multiple sites, which disrupts the interaction of 4E-BP1 with eIF4E, and reduced protein synthesis and RRM2 protein levels. This effect of mTORC1/2 inhibitors on protein synthesis and RRM2 levels in Ewing sarcoma cells was rescued in cell lines with the CRISPR/Cas9-mediated knockout of 4E-BP1. Finally, we also found that the inducible expression of constitutively-active 4E-BP1 was sufficient to suppress tumor cell growth in vitro and in vivo in xenograft experiments. Overall, these results provide novel insight into the molecular mechanisms that link protein translation and 4E-BP1 activity to the regulation of DNA replication, as well as the cellular response to replication stress, in sarcomas.
MATERIALS AND METHODS
Cell lines and culture:
Cell lines were maintained at 37° C in a 5% CO2 atmosphere. The A673, TC32, TC71, SKNEP, and EW8 cell lines were provided by Dr. Kimberly Stegmaier (Dana-Farber Cancer Institute, Boston, MA). The HEK-293T, HT1080, and U2OS cell lines were obtained from ATCC. The RH30 and RD cell lines were provided by Dr. Munir Tanas (University of Iowa, Iowa City, IA). The ES6 and AGPN cell lines were kindly provided by the St. Jude Childhood Solid Tumor Network and the Childhood Cancer Repository (Children’s Oncology Group), respectively. The sarcoma cell lines were cultured as previously described (4, 5). The Jurkat, K562, Nalm6, REH, and SEM cell lines were obtained from ATCC or DSMZ. Cell lines were authenticated by DNA fingerprinting using the short tandem repeat (STR) method and used within 8-10 passages of thawing.
Chemical compounds:
Chemical compounds were purchased from Sigma (temsirolimus and MG132), Selleckchem (prexasertib, LY2603618, and TAK-228), APExBio (VE-822), Thermo Fisher Scientific (puromycin), and MedChemExpress (AZD1775, homoharringtonine, and AZD2014).
Doxycycline-inducible shRRM2 and shNT:
Cell lines with inducible shRNA targeting RRM2, or a non-targeting shRNA control (shNT), and fluorescent marker proteins were generated as described (13). In order to isolate cells with uniform and high expression of the shRNA and fluorescent protein, we used a brief doxycycline treatment (12-18 hours) to induce expression of the fluorescent protein followed by isolation of the cells with the highest 1-2% fluorescence by flow cytometry (Becton Dickinson FACS Aria).
Drug screen.
EW8 shRRM2 and shNT cells were plated in 384-well plates (Greiner Bio-One, 781091) at a density of 2,500 cells/well in the presence of doxycycline. Cells were allowed to attach overnight and then were treated with the FDA-approved oncology drug set (133 compounds; National Cancer Institute, Developmental Therapeutics Program). Each compound was added at a final concentration of 100 nM. Three CHK1 inhibitors and one ATR inhibitor were included in the screen as positive controls. DMSO was used as the negative control. Plates were then incubated at 37° C for 72 hr. Cell viability was quantified using Cell-Titer-Glo luminescence (Promega) and a FLUOstar Omega microplate reader (BMG Labtech) (5,20). The screen was performed in duplicate and data were analyzed using a normalized growth rate (GR) inhibition approach to account for the effects of the different growth rates of the shRRM2 and shNT cell lines on drug sensitivity metrics (21,22). GR values were calculated using the online GRcalculator tool (http://www.grcalculator.org/grtutorial/Home.html) (21).
Cell proliferation/viability and drug synergy:
Cell proliferation/viability was measured using the resazurin (AlamarBlue) fluorescence and Cell-Titer-Glo luminescence assays as previously described (5,20). Synergy data were analyzed using SynergyFinder (https://synergyfinder.fimm.fi) (23).
Immunoblotting:
Immunoblots were performed as previously described (4, 5). Antibodies to the following proteins were used in the immunoblots: phospho-Histone-139 H2A.X (Cell Signaling, #9718, 1:1000), phospho-Chk1-345 ( Cell Signaling, #2348, 1:1000), Chk1 (Cell Signaling, #2360, 1:1000), puromycin (Millipore, #AF488, 1:2000), 4E-BP1 (Cell Signaling, #9644, 1:1000), PARP (Cell Signaling, #9532, 1:1000), p-4E-BP1-37/46 (Cell Signaling, #2855, 1:1000), p-4E-BP1-65 (Cell Signaling, #9451, 1:1000), p-4E-BP1-70 (Cell Signaling, #9455, 1:1000), RRM1 (Cell Signaling, #8637, 1:1000), RRM2 (Santa Cruz, #398294, 1:500), FLAG (Sigma, F1804, 1:1000), actin (Proteintech, 60008-1, 1:5000), and tubulin (Proteintech, 66031-1, 1:2000). Protein loading for the immunoblots was normalized using cell number.
Puromycin labeling:
Protein synthesis was assessed using puromycin labeling (SUnSET technique) as described (20,24).
Clonogenic assay:
Clonogenic assays were performed as described (5). The cells were continuously treated with doxycycline (2 μg/mL) or vehicle for 10-14 days. Colonies were then stained and counted using an inverted Olympus CKX41 microscope.
Quantitative reverse transcriptase PCR:
RT-qPCR was performed as previously described (20). The PCR primer sequences for RRM2 and GAPDH were 5’-CACGGAGCCGAAAACTAAAGC-3’ and 5’-CTGGGCTACACTGAGCACC-3’, respectively.
Xenograft:
The Institutional Animal Care and Usage Committee at the University of Iowa approved the animal studies and the studies were conducted in adherence with the NIH Guide for the Care and Use of Laboratory Animals. Approximately 1.0 × 106 doxycycline-inducible 4EBP1-Ala TC71 cells were mixed with 30% matrigel and injected subcutaneously into the flanks of 6-week old, female NCr mice. No statistical methods were used to determine the sample size. Mice were then randomized to either standard chow (n=8) or doxycycline-containing chow (Envigo Teklad, 625 mg/kg) (n=8). Tumor volumes were measured periodically, without blinding of the investigators, using calipers (volume = 0.5 x length x width2). Animals were sacrificed when a tumor reached 20 mm in any dimension. Differences in tumor growth rates were assessed by mixed-model two-way ANOVA (GraphPad Prism 8.01).
Cell cycle analysis:
Cell cycle analysis was performed in duplicate using the Click-iT EdU-488 kit for flow cytometry (ThermoFisher Scientific) as described (13).
Doxycycline-inducible 4E-BP1-Ala:
The full-length 4E-BP1 cDNA, with alanine substitutions at Thr37/46, Ser65, and Thr70 and a FLAG-tag, was obtained as a gene block (IDT; Coralville, IA) and inserted into the Lenti-X-Tet-One vector (Clontech). After verification by sequencing, the plasmid was used to make lentivirus, as described above.
4E-BP1 knockout cell lines:
CRISPR/Cas9-mediated knockout of 4E-BP1 was performed using the pLentiCRISPRv2 plasmid (Genscript) with a gRNA (GTGAGTTCCGACACTCCATC) targeting EIF4EBP1. Lentivirus was prepared as described above and cells were selected in 1 μg/mL puromycin 48 hours after transduction. The knockout cell lines were then used immediately after puromycin selection or single-cell cloned using flow cytometry (Becton Dickinson FACS Aria).
Reverse phase protein array (RPPA):
RPPA analysis of cell lines treated with HHT were performed by the RPPA Core Facility at the MD Anderson Cancer Center. Cells were provided to the core facility as frozen pellets. Protein extraction, RPPA analysis, and data normalization and analysis were performed according to the standard protocols of the facility (https://www.mdanderson.org/research/research-resources/core-facilities/functional-proteomics-rppa-core/rppa-process.html).
Statistical Analysis:
Student’s t-test two-tailed was used to calculate P-values for the comparison of two groups. Differences in the xenograft tumor growth rates were assessed by mixed-model two-way ANOVA. Statistical analyses were conducted using GraphPad Prism 8.01.
RESULTS
We developed a chemical-genetic screening approach to identify FDA-approved, anticancer drugs that selectively target cancer cells with a decreased level of the RRM2 protein. As reported in previous work, we used a doxycycline-inducible shRNA to modestly reduce the level of RRM2 in Ewing sarcoma cells and impair progression through S-phase (13). This partial reduction in the level of the RRM2 protein caused DNA replication stress, as assessed based on the phosphorylation of CHK1, but did not cause apoptosis or cell death (Figure 1A). Next, based on the known synergy between RNR and CHK1 inhibitors, as well as the activation of CHK1 by partial knockdown of RRM2, we tested whether RRM2 knockdown sensitizes the cells to inhibition of the CHK1 pathway (5,20). Figures 1B-C show that the treatment of the shRRM2 knockdown cells, but not the control (shNT) knockdown cells, with CHK1 inhibitors (prexasertib and LY2603618) decreased cell viability (Figure 1B) and increased DNA damage (γH2AX) and apoptosis (cleaved PARP) (Figure 1C). Consequently, CHK1 inhibitors were used as positive controls to optimize our screening approach.
Figure 1.
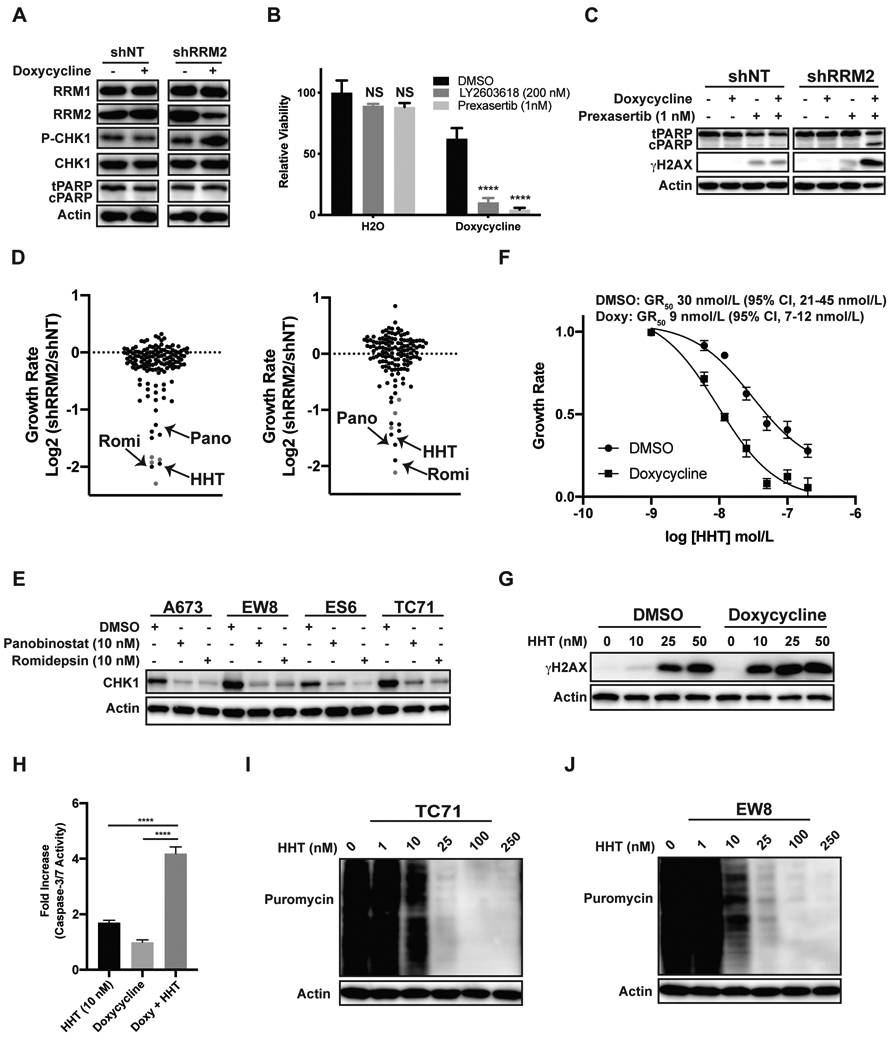
ATR-CHK1 inhibitors and homoharringtonine selectively target Ewing sarcoma cells with a partial reduction in the level of the RRM2 protein. (A) EW8 cell lines with doxycycline-inducible shRNA-mediated knockdown of RRM2, or a non-targeting shRNA (shNT), were treated with doxycycline or vehicle for 48 h. Cellular lysates were then collected for immunoblotting. (B) The shRRM2 cells, grown in the presence of doxycycline or vehicle, were treated with LY2603618 or prexasertib for 48 h. Cell viability was assessed 72 h after drug addition using the AlamarBlue assay. Error bars represent the mean ± SD of three technical replicates. The results are representative of two independent experiments. (C) The shRRM2 and shNT cells were treated with doxycycline, prexasertib, or vehicle for 24 h and then cellular lysates were collected for immunoblotting. (D) Results of the small molecule drug screen, which was performed in duplicate, for the shRRM2 and shNT cells. The log2 fold difference in the growth rate between the shRRM2 and shNT cell lines is shown for each drug. A normalized growth rate (GR) inhibition approach was used to analyze the data in order to account for the effects of the different growth rates of the shRRM2 and shNT cell lines. ATR and CHK1 inhibitors, which were included in the screen as positive controls, are shown in red. (E) Ewing sarcoma cell lines were treated with DMSO, panobinostat, or romidepsin for 24 h. Cellular lysates were then collected for immunoblotting. (F) Dose response curves for shRRM2 cells treated with different concentrations of HHT in the presence or absence of doxycycline. Cell viability was assessed 72 h after drug addition using the AlamarBlue assay. A normalized growth rate (GR) inhibition approach was used to analyze the data. Error bars represent the mean ± SD of three technical replicates. The results are representative of two independent experiments. (G) The shRRM2 cells, grown in the presence of DMSO or doxycycline, were treated with different concentrations of HHT for 24 h. Cellular lysates were then collected for immunoblotting. (H) The shRRM2 cells were treated with DMSO, doxycycline, HHT, or the combination of doxycycline and HHT for 24 h. Caspase-3/7 activation, relative to the cells treated with DMSO, was then quantified using a Casp-Glo 3/7 Luminescence assay. (I, J) TC71 and EW8 cells were treated with different concentrations of HHT for 24 h. Protein synthesis was then assessed using puromycin labeling. Protein loading for all the immunoblots was normalized using cell number.
A focused screen was then performed using a FDA-approved oncology drug set (133 compounds) to identify anticancer drugs that were more toxic to the shRRM2 cells than the shNT cells (Supplementary Figure 1A). All of the drugs were screened at a final concentration of 100 nM and three CHK1 inhibitors and one ATR inhibitor were included in the screen as positive controls (Z’-factor 0.73; Supplementary Figure 1B-C). The screen was performed in duplicate, on separate occasions, using different vials of thawed cells. In addition, in order to account for the effects of the different growth rates of the shRRM2 and shNT cell lines (Supplementary Figure 2) on drug sensitivity metrics, we used a normalized growth rate (GR) inhibition approach, as described by Hafner et al., to analyze the data (21,22). Briefly, the GR value is the ratio between the growth rates under treated and untreated conditions normalized to a single cell division. We observed strong correlation between the replicate screens (Supplementary Figure 1D-E) and the majority of the compounds, when tested at 100 nM, had a minimal effect on the viability of either the shRRM2 or shNT cells (Supplementary Tables 1-2). However, the drugs that decreased the viability of both the shRRM2 and shNT cells were compounds with reported toxicity toward Ewing sarcoma cell lines, including dactinomycin, anthracyclines, and alkaloids.
As a drug class, the ATR-CHK1 inhibitors (red data points) demonstrated the strongest selectivity for the shRRM2 cells relative to the shNT cells (Supplementary Tables 1-2; Figure 1D). Two histone deacetylase (HDAC) inhibitors, romidepsin and panobinostat, also showed similar selectivity toward the RRM2 knockdown cells. Homoharringtonine (HHT), which is FDA-approved for the treatment of chronic myeloid leukemia, was an additional hit with comparable selectivity to the ATR-CHK1 inhibitors in both screens (25). HDAC inhibitors are reported to regulate the levels of CHK1 and Figure 1E shows, in multiple Ewing sarcoma cell lines, that both romidepsin and panobinostat reduce levels of CHK1 (26-28). Consequently, we decided to focus our further efforts on HHT, which also showed strong selectivity for the shRRM2 cells. To validate the results of the screen we performed dose titration experiments with HHT in the presence and absence of doxycycline, or RRM2 knockdown. Figure 1F shows that the RRM2 knockdown cells are 2-3-fold more sensitive to HHT than the parental cells. Knockdown of RRM2 in the presence of low-dose HHT (10 nM) also increased DNA damage, as assessed using phosphorylation of H2AX, and apoptosis, based on Caspase-3/7 activation (Figure 1G and 1H). Similar results regarding enhanced sensitivity to HHT were obtained with two additional Ewing sarcoma cell lines, TC71 and AGPN, with doxycycline-inducible knockdown of RRM2 (Supplementary Figure 3).
HHT is a natural plant alkaloid and is reported to inhibit protein synthesis by binding in the A-site cleft of the ribosome and interfering with the docking of aminoacyl-tRNAs (29). Consequently, to investigate the mechanism of HHT, we used puromycin labeling to directly detect the synthesis of nascent proteins in Ewing sarcoma cells in the presence of HHT (24). Figures 1I-J show that HHT reduces protein synthesis in Ewing sarcoma cells.
HHT is a general inhibitor of protein translation and has been reported to cause apoptosis in cancer, but the critical proteomic targets of HHT are expected to be cell- and cancer-type dependent due to differences in protein stability and the unique dependencies of cancer cells (25,30,31). Consequently, we used reverse phase protein arrays (RPPA) to investigate the proteome of EW8 and TC71 cells treated with HHT, using a low concentration (10 nM) of the drug that sensitizes Ewing sarcoma cells to DNA replication stress (Figure 1G and Supplementary Tables 3-4). Notably, in both cell lines, RRM2 was one of the most highly down-regulated proteins (Figure 2A). This decrease in the level of the RRM2 protein caused by HHT was dose-dependent and occurred at drug concentrations used in the screen and subsequent experiments (Figures 2B-C). In previous work, we showed that the knockdown of RRM2 using siRNA, which is more potent than the shRNA used in the current study, caused DNA replication stress, apoptosis, and cell death as a single agent in Ewing sarcoma cells (6). Consequently, to determine whether the combination of HHT and shRRM2 caused toxicity by reducing the amount of RRM2 below a threshold level, we treated the shRRM2 knockdown cells with HHT (10 nM) and quantified RRM2 levels using immunoblotting. Figure 2D shows that HHT enhanced the loss of RRM2 in the shRRM2 knockdown cells.
Figure 2.
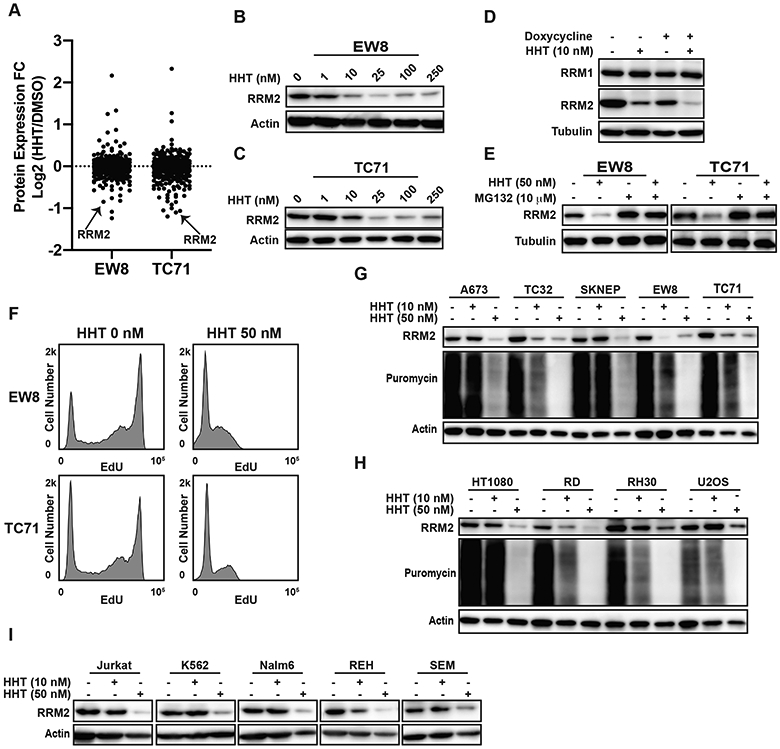
HHT inhibits protein synthesis and reduces the level of the RRM2 protein in Ewing sarcoma cell lines. (A) Comparison of protein expression (RPPA) in Ewing sarcoma cell lines treated with DMSO or HHT (10 nM) for 24 h. (B, C) EW8 and TC71 cell lines were treated with different concentrations of HHT for 24 h and then RRM2 levels were assessed by immunoblotting. (D) shRRM2 cells, grown in the presence or absence of doxycycline, were treated with HHT (10 nM) for 24 h. (E) EW8 and TC71 cells were treated with HHT (50 nM), MG132 (10 μM), or the drug combination for 6 h. Lysates were then collected for immunoblotting. (F) EW8 and TC71 were treated with HHT (50 nM) for 6 h and then DNA replication was quantified using EdU. (G) Ewing sarcoma cell lines were treated with HHT (10 nM or 50 nM) for 24 h. Cells were labeled with puromycin to quantify protein synthesis and then lysates were collected for immunoblotting. (H, I) Sarcoma and leukemia cell lines were treated with HHT (10 nM or 50 nM) for 24 h. Cellular lysates were then collected for immunoblotting. Protein loading for all of the immunoblots was normalized using cell number.
The RRM2 protein is degraded by the proteasome and Figure 2E shows that the proteasome inhibitor MG132 partially rescued the loss of RRM2 when protein synthesis was inhibited by HHT (12,13). In addition, as predicted for a drug that reduces RRM2 levels, Figure 2F shows that HHT also decreased DNA replication. We then tested the effect of HHT (10 nM and 50 nM) on RRM2 levels in additional Ewing sarcoma cell lines (A673, TC32, and SKNEP). The effect of the lower dose (10 nM) of HHT was variable between cell lines, but the higher dose (50 nM) of HHT depleted RRM2 in all of the Ewing sarcoma cell lines (Figure 2G). HHT (50 nM) also reduced the level of RRM2 in fibrosarcoma (HT1080), rhabdomyosarcoma (RD and Rh30), and osteosarcoma (U2OS) cell lines (Figure 2H). Similar results were obtained using a panel of leukemia cell lines (Figure 2I). Finally, we treated Ewing sarcoma cells with an additional protein synthesis inhibitor, nelfinavir, and quantified protein synthesis and RRM2 protein levels (32). Supplementary Figure 4 shows that nelfinavir reduced both protein synthesis and RRM2 levels in three Ewing sarcoma cell lines.
We showed that Ewing sarcoma cells, as well other cell types, are reliant on active protein synthesis to maintain a stable level of the RRM2 protein. HHT inhibits protein synthesis by interfering with the docking of aminoacyl-tRNAs in the A-site of the ribosome, but we hypothesized that it may be possible to regulate the translation of the RRM2 protein using alternative approaches (29). In previous work, while studying the regulation of CHK1, we incidentally noted that the inhibition of mTORC1/2 reduces the level of RRM2 in Ewing sarcoma cell lines (20). Similarly, recent work by Muller et al. using polysome profiling showed that the mTORC1/2 inhibitor PP242 regulates the translation of RRM2 mRNA in pancreatic adenocarcinoma cells (33). Consequently, we next tested whether mTOR regulates the level of RRM2 using a panel of five Ewing sarcoma cell lines. Figure 3A shows that a 6-hour drug treatment with the mTORC1/2 inhibitor TAK-228, but not the mTORC1 inhibitor temsirolimus, blocked protein synthesis and reduced RRM2 protein levels.
Figure 3.
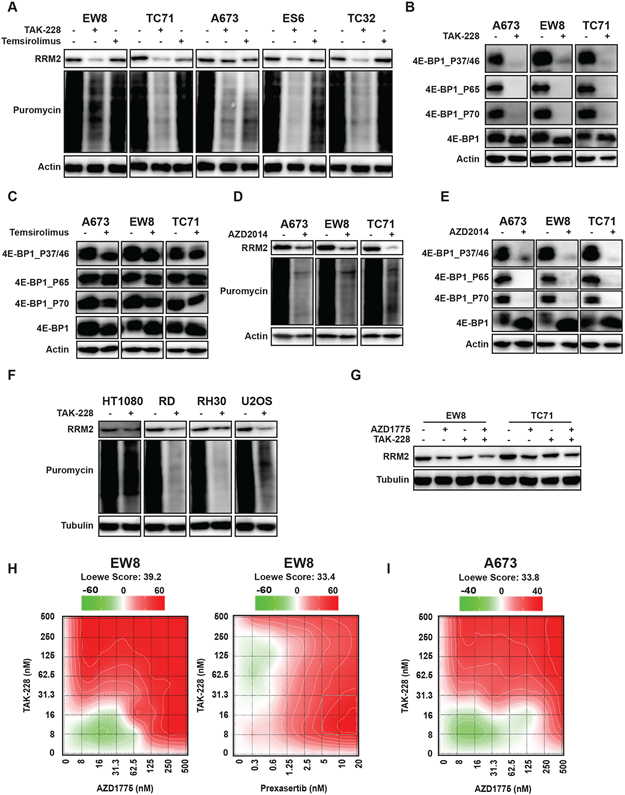
Inhibition of mTORC1/2 activates 4E-BP1, blocks protein synthesis, and reduces the level of the RRM2 protein. (A, B, C) Ewing sarcoma cell lines were treated with TAK-228 (1 μM) or temsirolimus (1 μM) for 6 h. Cells were labeled with puromycin to quantify protein synthesis and then lysates were collected for immunoblotting. (D, E) Ewing sarcoma cell lines were treated with AZD2014 (1 μM) for 6 h. Cells were labeled with puromycin to quantify protein synthesis and then lysates were collected for immunoblotting. (F) Sarcoma cell lines were treated with TAK-228 (1 μM) for 6 h. Cells were labeled with puromycin to quantify protein synthesis and then lysates were collected for immunoblotting. (G) EW8 and TC71 cells were treated with TAK-228 (100 nM), AZD1775 (100 nM), or the combination of the drugs for 6 h. (H, I) EW8 and A673 cells were treated for 72 h with a combination of TAK-228 and AZD1775, or TAK-228 in combination with prexasertib. Survival was assayed by Cell-Titer-Glo and each experiment was repeated 2 times. Loewe matrix plots for drug cooperativity are shown. Protein loading for all of the immunoblots was normalized using cell number.
The 4E-BP1 protein, which is regulated by the mTOR pathway and highly expressed in Ewing sarcoma tumors, represses protein translation by binding to the m7G cap-binding protein eIF4E and preventing the assembly of the eIF4F translation initiation complex (20,34,35). The phosphorylation of 4E-BP1, which is mediated by mTOR and other kinases, inactivates 4E-BP1 by disrupting the interaction between 4E-BP1 and eIF4E (36-38). Figure 3B shows that TAK-228 reduced the phosphorylation of 4E-BP1 at Thr37/46, Ser65, and Thr70. In contrast, temsirolimus had a minimal effect on 4E-BP1 phosphorylation and activation (Figure 3C). Similar results were obtained with an additional mTORC1/2 inhibitor, AZD2014 (Figure 3D-E). Next, we treated additional sarcoma cell lines, osteosarcoma (U2OS), rhabdomyosarcoma (RH30), and fibrosarcoma (HT1080), with TAK-228 and observed a decrease in protein synthesis and RRM2 protein levels, although the effect was reduced for HT1080 compared to the other cell lines. The effects of TAK-228 on protein synthesis in two non-transformed cell lines, BJ-tert and RPE-tert, were also reduced compared to the sarcoma cell lines (Supplementary Figure 5).
In previous work, we showed that the activation of CDK2 by inhibition of either CHK1 or WEE1 leads to the phosphorylation and proteasome-mediated degradation of RRM2 (13). Consequently, we next tested the hypothesis that blocking the translation of RRM2 mRNA while increasing the degradation of the RRM2 protein would more effectively target RRM2 levels. Figure 3G shows that the combination of a low dose of TAK-228 and a WEE1 inhibitor, AZD1775, enhanced the reduction in the level of the RRM2 protein. We and others previously showed that sarcoma cells are sensitive to TAK-228 (20,39). Figures 3H-I and Supplementary Figure 6 show that the combination of TAK-228 and AZD1775 or TAK-228 and prexasertib was synergistic with multiple Ewing sarcoma cell lines, as shown in the Loewe synergy matrix plots.
mTORC1/2 phosphorylates and regulates many downstream targets and critical pathways other than 4E-BP1 (38,40). Consequently, to determine whether 4E-BP1 regulates protein synthesis downstream of mTORC1/2, we used CRISPR/Cas9 to knockout 4E-BP1 in Ewing sarcoma cell lines (Figure 4A). We obtained efficient knockout of 4E-BP1, even prior to single cell cloning, so we used both the bulk cell population (prior to single cell cloning) (Figures 4) and single cell-derived clones (Supplementary Figure 7) in the subsequent experiments. Similar results were obtained with both cell populations, but a potential advantage of studying the pre-cloned cells, despite the incomplete knockout of 4E-BP1, is that it avoids the biases that can arise from single cell clones. Notably, in contrast to the parental cell lines, treatment of the 4E-BP1-KO cells with TAK-228 did not result in a reduction in protein synthesis (Figure 4B). Similarly, the knockout of 4E-BP1 partially rescued the effect of TAK-228 on the level of the RRM2 protein during a 6-hour drug treatment (Figure 4C). Similar results were obtained with an additional mTORC1/2 inhibitor, AZD2014 (Figure 4D). The TC71-4E-BP1-KO cell line showed a ~2-fold increase in resistance to TAK-228 in a dose-response assay (Figure 4E). However, the EW8-4E-BP1-KO cell line was minimally resistant to TAK-228 (Figure 4F). These results suggest that the toxicity of TAK-228 is caused by multiple mechanisms, consistent with the extensive cellular functions of the mTORC1 and mTORC2 pathways, and is not solely due to activation of 4E-BP1. Furthermore, based on the incomplete rescue of the level of the RRM2 protein in the 4E-BP1-KO cells treated with TAK-228, we tested whether inhibition of mTORC1/2 affects the level of RRM2 mRNA. We found that TAK-228 significantly down-regulated the level of RRM2 mRNA in Ewing sarcoma cells, which is consistent with reports that mTORC1 can regulate the transcription of RRM2 (Figures 4G) (41). Overall, these results with the 4E-BP1-KO cells suggest that the effects of inhibition of mTORC1/2 are multifaceted and that TAK-228 can target both RRM2 transcription and translation. In addition, we also recognize that mTORC1/2 regulates many downstream targets other than RRM2, which likely contribute to the effects of the drug.
Figure 4.
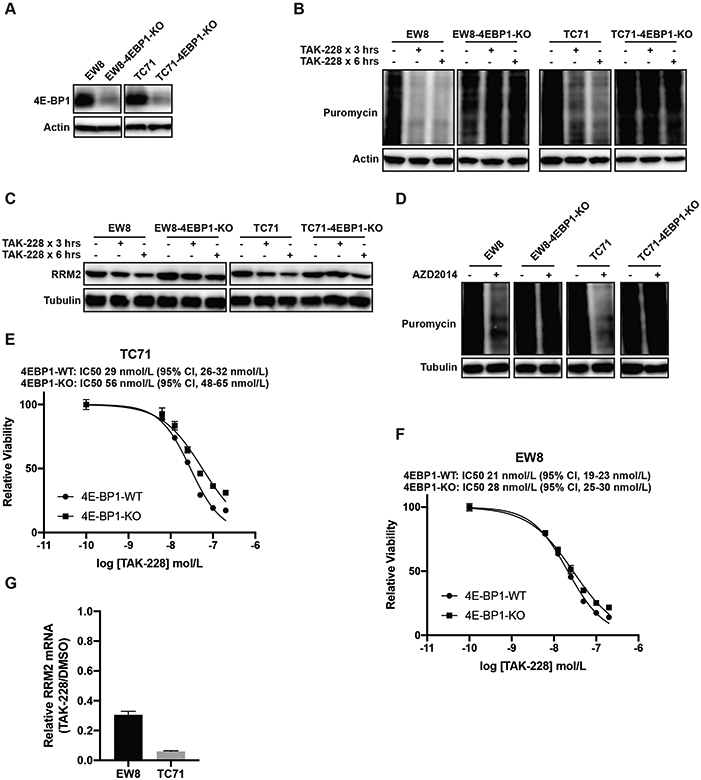
The CRISPR/Cas9-mediated knockout of 4E-BP1 in Ewing sarcoma cell lines rescues the effects of mTORC1/2 inhibitors on protein synthesis. (A) Immunoblot showing 4E-BP1 expression level after CRISPR/Cas9-mediated gene knockout. This immunoblot reflects 4E-BP1 levels in the bulk cell population prior to single cell cloning. (B, C) The 4E-BP1-KO and parental cells were treated with TAK-228 (1 μM) for 3 h or 6 h. Cells were labeled with puromycin to quantify protein synthesis and then lysates were collected for immunoblotting. (D) The 4E-BP1-KO and parental cells were treated with AZD2014 (1 μM) for 6 h. Cells were labeled with puromycin to quantify protein synthesis and then lysates were collected for immunoblotting. (E, F) Dose response curves for the 4E-BP1-KO and parental (4E-BP1-WT) cells. Cell viability was assessed 72 h after drug addition using the AlamarBlue assay. Error bars represent the mean ± SD of three technical replicates. The results are representative of two independent experiments. (G) RT-qPCR for RRM2 mRNA in EW8 and TC71 cells treated with TAK-228 (100 nM) for 24 h. Protein loading for all of the immunoblots was normalized using cell number.
Consequently, in order to investigate the specific role of 4E-BP1 in mediating toxicity in sarcoma cells, we generated a doxycycline-inducible, constitutively-active 4E-BP1 protein with alanine substitutions at the phosphorylation sites Thr37, Thr46, Ser65, and Thr70 (4E-BP1-Ala) (42). The inducible expression of 4E-BP1-Ala reduced the level of the RRM2 protein in Ewing sarcoma cell lines (Figure 5A). However, in contrast to TAK-228, the expression of 4E-BP1-Ala in Ewing sarcoma cells did not affect the level of RRM2 mRNA (Figure 5B). Expression of 4E-BP1-Ala also decreased overall protein synthesis, as assessed using puromycin labeling (Figure 5C). In addition, expression of 4E-BP1-Ala significantly decreased the growth of the cells (Figure 5D). Similarly, the inducible expression of 4E-BP1-Ala also significantly reduced colony formation in vitro (Figure 5E). Next, we implanted the TC71 4E-BP1-Ala cells in mice that were then fed either standard chow or doxycycline-containing chow. Figure 5F shows that that the inducible expression of 4E-BP1-Ala in vivo significantly reduced tumor growth in the xenograft experiment. Finally, Figure 5G shows that the inducible expression of 4E-BP1-Ala in additional sarcoma subtypes, fibrosarcoma, rhabdomyosarcoma, and osteosarcoma, also significantly reduced colony formation in vitro.
Figure 5.
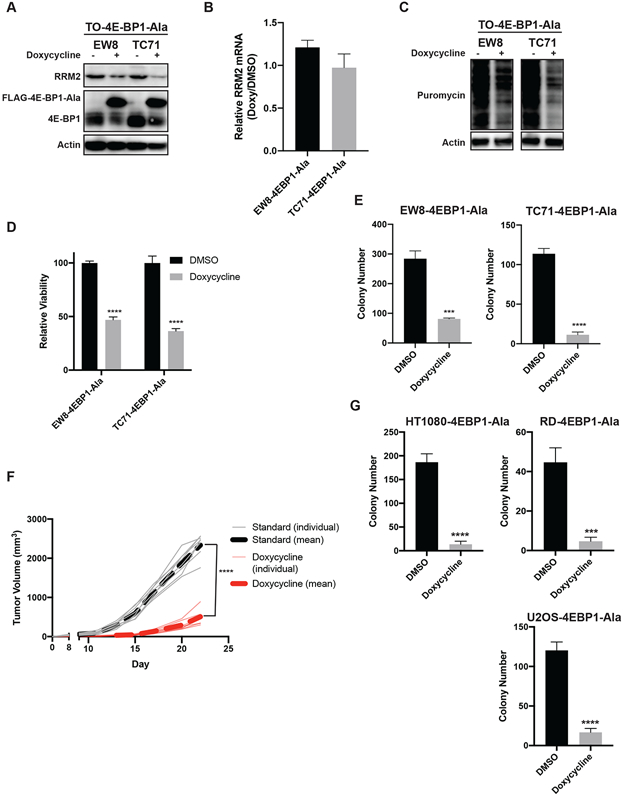
The inducible expression of constitutively-active 4E-BP1 reduces the level of the RRM2 protein and protein synthesis in sarcoma cells. (A) The doxycycline-inducible 4E-BP1-Ala cell lines were treated with doxycycline for 24 h. Cellular lysates were then collected for immunoblotting. (B) RT-qPCR for RRM2 mRNA in the 4E-BP1-Ala cell lines treated with doxycycline or DMSO for 24 h. (C) The doxycycline-inducible 4E-BP1-Ala cell lines were treated with doxycycline for 24 h. Cells were labeled with puromycin to quantify protein synthesis and then lysates were collected for immunoblotting. (D) The 4E-BP1-Ala cell lines were treated with doxycycline or DMSO for 72 h. Cell viability was then quantified using the AlamarBlue assay. Error bars represent the mean ± SD of three technical replicates. The results are representative of two independent experiments. (E) The 4E-BP1-Ala cell lines were treated with doxycycline or DMSO for 14 days and then the number of colonies were counted. Error bars represent the mean ± SD of three technical replicates. (F) TC71 cells with doxycycline-inducible expression of 4E-BP1-Ala were implanted subcutaneously (Day 0) in NCr mice. Mice were then fed standard chow (n=8) or doxycycline-containing chow (n=8). Tumor size was quantified every 2-3 days using caliper measurements. The plot shows the tumor growth for the individual mice, as well as the mean tumor volume (dotted line) for the mice receiving standard or doxycycline-containing chow. (G) HT1080, RD, and U2OS doxycycline-inducible 4E-BP1-Ala cell lines were treated with doxycycline for 24 h. Cells were labeled with puromycin to quantify protein synthesis and then lysates were collected for immunoblotting. Protein loading for all of the immunoblots was normalized using cell number.
DISCUSSION
Significant research has focused on understanding the pathologic mechanisms of the different oncogenes, such as EWS-FLI1, that drive the growth of sarcomas (43,44). However, the role of tumor suppressor genes (TSG) in sarcomas is less extensively studied than the function of driver oncogenes (45). In addition, tumor suppressors, such as TP53, are frequently lost in sarcomas due to deletions, nonsense mutations, frame-shift mutations, insertions, or missense mutations (46). Consequently, reactivating tumor suppressors is difficult and this challenge has limited drug discovery and the identification of novel therapeutic approaches for treating sarcomas (47,48).
In this work, we identified that the level of the RRM2 protein depends, in multiple cell types, on active protein synthesis and that 4E-BP1, a tumor suppressor that represses cap-dependent protein translation, specifically regulates the translation of the RRM2 protein. Notably, in previous work, we showed that the inhibition or knockdown of RRM2 in Ewing sarcoma cells inhibits cell growth and induces apoptosis in vitro and in vivo. The sensitivity of Ewing sarcoma cells to knockdown of RRM2 is due, in part, to the overexpression of the DNA restriction factor SLFN11, which is a direct transcriptional target of EWS-FLI1 (6,49-51). Consequently, we propose that 4E-BP1 regulates RRM2 levels in multiple cell types, but that the downstream effects of the loss of the RRM2 protein on cell viability and apoptosis may be more specific to Ewing sarcoma cells. However, other types of cancer are also sensitive to RNR inhibitors, or knockdown of RRM2, and we expect that the results obtained with Ewing sarcoma cells may also apply to additional tumor types.
Previous translational research on RNR has primarily focused on the identification and testing of small-molecule drugs that inhibit either the active site of the RRM1 subunit of RNR or the dinuclear iron site of the RRM2 subunit (1,52-55). We have identified a complementary approach to abrogate the activity of RNR by reducing the level of the RRM2 subunit, similar to the strategy used with proteolysis targeting chimera (PROTAC) drugs (56-60). Moreover, we showed that the mTORC1/2 inhibitor TAK-228 causes synergistic toxicity when combined with inhibitors of CHK1 and WEE1, which we previously showed activate CDK2 and target RRM2 for degradation by the proteasome. Consequently, we propose a model whereby the inhibition of CHK1-WEE1 signaling enhances the CDK-mediated degradation of RRM2 while the inhibition of mTORC1/2 blocks the synthesis of the RRM2 protein via the activation of 4E-BP1 (Figure 6). However, we also recognize that 4E-BP1 regulates the cap-dependent translation of many proteins other than RRM2. For example, we and others have shown that the mTOR pathway regulates the level of the CHK1 protein (20,61). In future work, we plan to use the doxycycline-inducible and constitutively-active 4E-BP1-Ala system, in combination with polysome profiling and proteomic approaches, to more extensively identify proteins and pathways regulated in sarcomas by active 4E-BP1.
Figure 6.
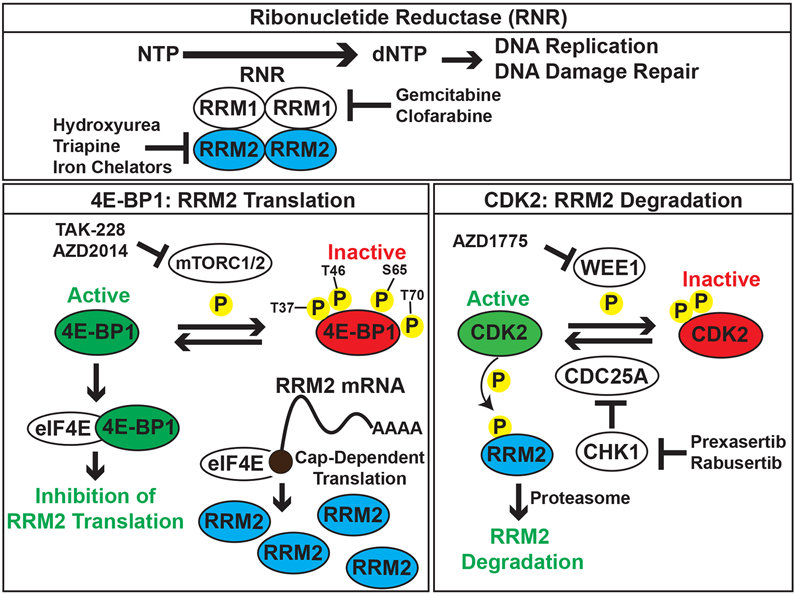
Integrated model for the regulation of RRM2 protein levels by the mTORC1/2, ATR-CHK1, and WEE1 pathways. In previous work, we found that the inhibition of ATR-CHK1-WEE1 signaling enhances the CDK-mediated degradation of RRM2 (13). In the current work, we showed that the inhibition of mTORC1/2 blocks the synthesis of the RRM2 protein via the activation of 4E-BP1.
Our data also provide further evidence for a critical link between protein synthesis and DNA replication, as well as the cellular response to DNA replication stress (33). Notably, DNA replication stress is a shared feature of many cancers because it is caused by both chemotherapy drugs, including inhibitors of RNR, and active oncogenes, such as c-Myc and Ras (62). Multiple sarcomas, including Ewing sarcoma, undifferentiated pleomorphic sarcoma (UPS), osteosarcoma, rhabdomyosarcoma, desmoplastic small round cell tumor, and synovial cell sarcoma, are sensitive to drugs that exacerbate DNA replication stress or impair the response to DNA replication stressors (63-70). Small molecule inhibitors of key components of the DNA replication stress pathway, including RNR, CHK1, ATR, and WEE1, are currently being evaluated in clinical trials (www.clinicaltrials.gov) (14,15,71-73). However, our data suggest that mTORC1/2 inhibitors, specifically via activation of 4E-BP1, may also target DNA replication. Notably, TAK-228 and AZD2014, as well as eIF4A inhibitors, have been shown to inhibit the growth of multiple sarcoma subtypes in vitro and in vivo in xenograft experiments (39,74,75). Furthermore, 4E-BP1 was recently identified to function as a tumor suppressor in malignant peripheral nerve sheath tumors and head and neck cancers (42,76). The combination of a WEE1 inhibitor and mTORC1/2 inhibitor has also been shown to inhibit the growth of KRAS-driven lung cancers (77).
We found that constitutively-active 4E-BP1, with alanine mutations at Thr37/46, Ser65, and Thr70, functions as a tumor suppressor in vitro and in vivo. However, the relative contributions of these different phosphorylation sites to the regulation of the activity of 4E-BP1 are poorly understood, in particular in sarcomas (36,37). For example, one model proposes that the phosphorylation of 4E-BP1 at Thr37/46 primes the protein for subsequent phosphorylation at Ser65 and Thr70, which then renders the hyperphosphorylated 4E-BP1 unable to bind eIF4E (36,37,61). Furthermore, different kinases, including mTORC1/2, p38 MAPK, ERK, PIM2, ATM, and CDK1, are reported to selectively phosphorylate different sites in 4E-BP1 in a cell-type dependent manner (38). For example, a recent publication reported that the mTORC1-mediated phosphorylation of 4E-BP1 at Thr37/46 facilitates the subsequent phosphorylation of 4E-BP1 at Ser65 and Thr70 by CDK12, which was recently identified as a therapeutic target in Ewing sarcoma tumors (61). We found in sarcoma cell lines that the inhibition of mTORC1/2, but not mTORC1, was sufficient to block the phosphorylation of 4E-BP1 at Thr37/46, Ser65, and Thr70. However, ongoing work in our laboratory is investigating whether inhibition of CDK12 enhances the effects of mTORC1/2 inhibitors on 4E-BP1 activation in sarcomas. In addition, we are also testing the relative contributions of the different phosphorylation sites in 4E-BP1 to the regulation of 4E-BP1 activity, as well as RRM2 levels and protein synthesis, in sarcomas.
In summary, our findings support a tumor-suppressive role for 4E-BP1 in sarcomas. We provide evidence that 4E-BP1 and mTORC1/2 regulate protein synthesis and, in particular, the level of the RRM2 protein. In addition, we also propose a model whereby the inhibition of CHK1-WEE1 signaling enhances the CDK-mediated degradation of RRM2 while the inhibition of mTORC1/2 blocks the synthesis of the RRM2 protein via the activation of 4E-BP1. Overall, our work provides novel mechanistic insight into the regulation of DNA replication, as well as the cellular response to DNA replication stress, by protein translation, 4E-BP1, and mTORC1/2.
Supplementary Material
ACKNOWLEDGMENTS
DJG is supported by a University of Iowa Dance Marathon Award, a Holden Comprehensive Cancer Center Sarcoma Multidisciplinary Oncology Group Seed Grant, a University of Iowa Oberley Seed Grant, St. Baldrick’s Research Foundation, Aiming for a Cure Foundation, The Matt Morrell and Natalie Sanchez Pediatric Cancer Research Foundation, and NIH Grant R37-CA217910. The authors would also like to acknowledge use of the University of Iowa Flow Cytometry and High Throughput Screening Core Facilities (NIH/NCI P30CA086862), as well as the RPPA Core Facility at MD Anderson Cancer Center (NCI # CA16672).
Footnotes
COMPETING INTERESTS
The authors declare that they do not have any competing interests.
REFERENCES
- 1.Aye Y, Li M, Long MJC, Weiss RS. Ribonucleotide reductase and cancer: biological mechanisms and targeted therapies. Oncogene. 2015. April 16;34(16):2011–21. [DOI] [PubMed] [Google Scholar]
- 2.Cerqueira NM, Fernandes PA, Ramos MJ. Ribonucleotide reductase: a critical enzyme for cancer chemotherapy and antiviral agents. Recent Pat Anticancer Drug Discov. 2007;2(1):11–29. [DOI] [PubMed] [Google Scholar]
- 3.Nordlund P, Reichard P. Ribonucleotide reductases. Annu Rev Biochem. 2006;75:681–706. [DOI] [PubMed] [Google Scholar]
- 4.Larsson K-M, Jordan A, Eliasson R, Reichard P, Logan DT, Nordlund P. Structural mechanism of allosteric substrate specificity regulation in a ribonucleotide reductase. Nat Struct Mol Biol. 2004. November;11(11):1142–9. [DOI] [PubMed] [Google Scholar]
- 5.Goss KL, Koppenhafer SL, Harmoney KM, Terry WW, Gordon DJ. Inhibition of CHK1 sensitizes Ewing sarcoma cells to the ribonucleotide reductase inhibitor gemcitabine. Oncotarget. 2017. October 20;8(50):87016–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Goss KL, Gordon DJ. Gene expression signature based screening identifies ribonucleotide reductase as a candidate therapeutic target in Ewing sarcoma. Oncotarget. 2016. September 27;7(39):63003–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Chabes AL, Björklund S, Thelander L. S Phase-specific transcription of the mouse ribonucleotide reductase R2 gene requires both a proximal repressive E2F-binding site and an upstream promoter activating region. J Biol Chem. 2004. March 12;279(11):10796–807. [DOI] [PubMed] [Google Scholar]
- 8.Zhang Y-W, Jones TL, Martin SE, Caplen NJ, Pommier Y. Implication of checkpoint kinase-dependent up-regulation of ribonucleotide reductase R2 in DNA damage response. J Biol Chem. 2009. July 3;284(27):18085–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Kotova I, Chabes AL, Lobov S, Thelander L, Björklund S. Sequences downstream of the transcription initiation site are important for proper initiation and regulation of mouse ribonucleotide reductase R2 gene transcription. Eur J Biochem. 2003. April;270(8):1791–801. [DOI] [PubMed] [Google Scholar]
- 10.DeGregori J, Kowalik T, Nevins JR. Cellular targets for activation by the E2F1 transcription factor include DNA synthesis- and G1/S-regulatory genes. Mol Cell Biol. 1995. August;15(8):4215–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Truman AW, Kristjansdottir K, Wolfgeher D, Ricco N, Mayampurath A, Volchenboum SL, et al. Quantitative proteomics of the yeast Hsp70/Hsp90 interactomes during DNA damage reveal chaperone-dependent regulation of ribonucleotide reductase. J Proteomics. 2015. January 1;112:285–300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Pfister SX, Markkanen E, Jiang Y, Sarkar S, Woodcock M, Orlando G, et al. Inhibiting WEE1 Selectively Kills Histone H3K36me3-Deficient Cancers by dNTP Starvation. Cancer Cell. 2015. November 9;28(5):557–68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Koppenhafer SL, Goss KL, Terry WW, Gordon DJ. Inhibition of the ATR-CHK1 Pathway in Ewing Sarcoma Cells Causes DNA Damage and Apoptosis via the CDK2-Mediated Degradation of RRM2. Mol Cancer Res. 2020. January;18(1):91–104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Rundle S, Bradbury A, Drew Y, Curtin NJ. Targeting the ATR-CHK1 Axis in Cancer Therapy. Cancers (Basel). 2017. April 27;9(5). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Lecona E, Fernández-Capetillo O. Replication stress and cancer: it takes two to tango. Exp Cell Res. 2014. November 15;329(1):26–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Iyer DR, Rhind N. The Intra-S Checkpoint Responses to DNA Damage. Genes (Basel). 2017. February 17;8(2). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Hill SJ, Decker B, Roberts EA, Horowitz NS, Muto MG, Worley MJ, et al. Prediction of DNA Repair Inhibitor Response in Short-Term Patient-Derived Ovarian Cancer Organoids. Cancer Discov. 2018. September 13;8(11):1404–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Liu Y, Li Y, Wang X, Liu F, Gao P, Quinn MM, et al. Gemcitabine and Chk1 Inhibitor AZD7762 Synergistically Suppress the Growth of Lkb1-Deficient Lung Adenocarcinoma. Cancer Res. 2017. September 15;77(18):5068–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Wallez Y, Dunlop CR, Johnson TI, Koh S-B, Fornari C, Yates JWT, et al. The ATR Inhibitor AZD6738 Synergizes with Gemcitabine In Vitro and In Vivo to Induce Pancreatic Ductal Adenocarcinoma Regression. Mol Cancer Ther. 2018. June 11;17(8):1670–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Koppenhafer SL, Goss KL, Terry WW, Gordon DJ. mTORC1/2 and Protein Translation Regulate Levels of CHK1 and the Sensitivity to CHK1 Inhibitors in Ewing Sarcoma Cells. Mol Cancer Ther. 2018. October 3;17(12):2676–88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Clark NA, Hafner M, Kouril M, Williams EH, Muhlich JL, Pilarczyk M, et al. GRcalculator: an online tool for calculating and mining dose-response data. BMC Cancer. 2017. October 24;17(1):698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Hafner M, Niepel M, Chung M, Sorger PK. Growth rate inhibition metrics correct for confounders in measuring sensitivity to cancer drugs. Nat Methods. 2016. May 2;13(6):521–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Ianevski A, He L, Aittokallio T, Tang J. SynergyFinder: a web application for analyzing drug combination dose-response matrix data. Bioinformatics. 2017. August 1;33(15):2413–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Schmidt EK, Clavarino G, Ceppi M, Pierre P. SUnSET, a nonradioactive method to monitor protein synthesis. Nat Methods. 2009. April;6(4):275–7. [DOI] [PubMed] [Google Scholar]
- 25.Lam SSY, Ho ESK, He B-L, Wong W-W, Cher C-Y, Ng NKL, et al. Homoharringtonine (omacetaxine mepesuccinate) as an adjunct for FLT3-ITD acute myeloid leukemia. Sci Transl Med. 2016. October 5;8(359):359ra129. [DOI] [PubMed] [Google Scholar]
- 26.Li X, Su Y, Madlambayan G, Edwards H, Polin L, Kushner J, et al. Antileukemic activity and mechanism of action of the novel PI3K and histone deacetylase dual inhibitor CUDC-907 in acute myeloid leukemia. Haematologica. 2019. February 28; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Zhao J, Xie C, Edwards H, Wang G, Taub JW, Ge Y. Histone deacetylases 1 and 2 cooperate in regulating BRCA1, CHK1, and RAD51 expression in acute myeloid leukemia cells. Oncotarget. 2017. January 24;8(4):6319–29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Li X, Su Y, Hege K, Madlambayan G, Edwards H, Knight T, et al. The HDAC and PI3K dual inhibitor CUDC-907 synergistically enhances the antileukemic activity of venetoclax in preclinical models of acute myeloid leukemia. Haematologica. 2020. March 12; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Gürel G, Blaha G, Moore PB, Steitz TA. U2504 determines the species specificity of the A-site cleft antibiotics: the structures of tiamulin, homoharringtonine, and bruceantin bound to the ribosome. J Mol Biol. 2009. May 29;389(1):146–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Tang R, Faussat A-M, Majdak P, Marzac C, Dubrulle S, Marjanovic Z, et al. Semisynthetic homoharringtonine induces apoptosis via inhibition of protein synthesis and triggers rapid myeloid cell leukemia-1 down-regulation in myeloid leukemia cells. Mol Cancer Ther. 2006. March;5(3):723–31. [DOI] [PubMed] [Google Scholar]
- 31.Chen R, Guo L, Chen Y, Jiang Y, Wierda WG, Plunkett W. Homoharringtonine reduced Mcl-1 expression and induced apoptosis in chronic lymphocytic leukemia. Blood. 2011. January 6;117(1):156–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.De Gassart A, Demaria O, Panes R, Zaffalon L, Ryazanov AG, Gilliet M, et al. Pharmacological eEF2K activation promotes cell death and inhibits cancer progression. EMBO Rep. 2016. August 29;17(10):1471–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Müller D, Shin S, Goullet de Rugy T, Samain R, Baer R, Strehaiano M, et al. eIF4A inhibition circumvents uncontrolled DNA replication mediated by 4E-BP1 loss in pancreatic cancer. JCI Insight. 2019. November 1;4(21). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Sonenberg N, Hinnebusch AG. Regulation of translation initiation in eukaryotes: mechanisms and biological targets. Cell. 2009. February 20;136(4):731–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Hinnebusch AG, Ivanov IP, Sonenberg N. Translational control by 5’-untranslated regions of eukaryotic mRNAs. Science. 2016. June 17;352(6292):1413–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Gingras AC, Gygi SP, Raught B, Polakiewicz RD, Abraham RT, Hoekstra MF, et al. Regulation of 4E-BP1 phosphorylation: a novel two-step mechanism. Genes Dev. 1999. June 1;13(11):1422–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Ayuso MI, Hernández-Jiménez M, Martín ME, Salinas M, Alcázar A. New hierarchical phosphorylation pathway of the translational repressor eIF4E-binding protein 1 (4E-BP1) in ischemia-reperfusion stress. J Biol Chem. 2010. November 5;285(45):34355–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Qin X, Jiang B, Zhang Y. 4E-BP1, a multifactor regulated multifunctional protein. Cell Cycle. 2016;15(6):781–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Slotkin EK, Patwardhan PP, Vasudeva SD, de Stanchina E, Tap WD, Schwartz GK. MLN0128, an ATP-competitive mTOR kinase inhibitor with potent in vitro and in vivo antitumor activity, as potential therapy for bone and soft-tissue sarcoma. Mol Cancer Ther. 2015. February;14(2):395–406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Musa J, Orth MF, Dallmayer M, Baldauf M, Pardo C, Rotblat B, et al. Eukaryotic initiation factor 4E-binding protein 1 (4E-BP1): a master regulator of mRNA translation involved in tumorigenesis. Oncogene. 2016. September 8;35(36):4675–88. [DOI] [PubMed] [Google Scholar]
- 41.He Z, Hu X, Liu W, Dorrance A, Garzon R, Houghton PJ, et al. P53 suppresses ribonucleotide reductase via inhibiting mTORC1. Oncotarget. 2017. June 20;8(25):41422–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Wang Z, Feng X, Molinolo AA, Martin D, Vitale-Cross L, Nohata N, et al. 4E-BP1 Is a Tumor Suppressor Protein Reactivated by mTOR Inhibition in Head and Neck Cancer. Cancer Res. 2019. April 1;79(7):1438–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Knott MML, Hölting TLB, Ohmura S, Kirchner T, Cidre-Aranaz F, Grünewald TGP. Targeting the undruggable: exploiting neomorphic features of fusion oncoproteins in childhood sarcomas for innovative therapies. Cancer Metastasis Rev. 2019;38(4):625–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Okimoto RA, Wu W, Nanjo S, Olivas V, Lin YK, Ponce RK, et al. CIC-DUX4 oncoprotein drives sarcoma metastasis and tumorigenesis via distinct regulatory programs. J Clin Invest. 2019. July 22;129(8):3401–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Lee EYHP, Muller WJ. Oncogenes and tumor suppressor genes. Cold Spring Harb Perspect Biol. 2010. October;2(10):a003236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Kim J, Kim JH, Kang HG, Park SY, Yu JY, Lee EY, et al. Integrated molecular characterization of adult soft tissue sarcoma for therapeutic targets. BMC Med Genet. 2018. December 31;19(Suppl 1):216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Guo XE, Ngo B, Modrek AS, Lee W-H. Targeting tumor suppressor networks for cancer therapeutics. Curr Drug Targets. 2014. January;15(1):2–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Wang H, Han H, Mousses S, Von Hoff DD. Targeting loss-of-function mutations in tumor-suppressor genes as a strategy for development of cancer therapeutic agents. Semin Oncol. 2006. August;33(4):513–20. [DOI] [PubMed] [Google Scholar]
- 49.Tang S-W, Bilke S, Cao L, Murai J, Sousa FG, Yamade M, et al. SLFN11 Is a Transcriptional Target of EWS-FLI1 and a Determinant of Drug Response in Ewing Sarcoma. Clin Cancer Res. 2015. September 15;21(18):4184–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Murai J, Thomas A, Miettinen M, Pommier Y. Schlafen 11 (SLFN11), a restriction factor for replicative stress induced by DNA-targeting anti-cancer therapies. Pharmacol Ther. 2019. May 23; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Zoppoli G, Regairaz M, Leo E, Reinhold WC, Varma S, Ballestrero A, et al. Putative DNA/RNA helicase Schlafen-11 (SLFN11) sensitizes cancer cells to DNA-damaging agents. Proc Natl Acad Sci USA. 2012. September 11;109(37):15030–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Moorthy NS, Cerqueira NM, Ramos MJ, Fernandes PA. Development of ribonucleotide reductase inhibitors: a review on structure activity relationships. Mini Rev Med Chem. 2013;13(13):1862–72. [DOI] [PubMed] [Google Scholar]
- 53.Zhou B, Su L, Hu S, Hu W, Yip MLR, Wu J, et al. A small-molecule blocking ribonucleotide reductase holoenzyme formation inhibits cancer cell growth and overcomes drug resistance. Cancer Res. 2013. November 1;73(21):6484–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.van der Donk WA, Yu G, Pérez L, Sanchez RJ, Stubbe J, Samano V, et al. Detection of a new substrate-derived radical during inactivation of ribonucleotide reductase from Escherichia coli by gemcitabine 5’-diphosphate. Biochemistry. 1998. May 5;37(18):6419–26. [DOI] [PubMed] [Google Scholar]
- 55.Cerqueira NM, Pereira S, Fernandes PA, Ramos MJ. Overview of ribonucleotide reductase inhibitors: an appealing target in anti-tumour therapy. Curr Med Chem. 2005;12(11):1283–94. [DOI] [PubMed] [Google Scholar]
- 56.Burslem GM, Smith BE, Lai AC, Jaime-Figueroa S, McQuaid DC, Bondeson DP, et al. The advantages of targeted protein degradation over inhibition: an RTK case study. Cell Chem Biol. 2018. January 18;25(1):67–77.e3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Paiva S-L, Crews CM. Targeted protein degradation: elements of PROTAC design. Curr Opin Chem Biol. 2019. April 17;50:111–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Pettersson M, Crews CM. PROteolysis TArgeting Chimeras (PROTACs) - Past, present and future. Drug Discov Today Technol. 2019. April;31:15–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Lai AC, Crews CM. Induced protein degradation: an emerging drug discovery paradigm. Nat Rev Drug Discov. 2017;16(2):101–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Neklesa TK, Winkler JD, Crews CM. Targeted protein degradation by PROTACs. Pharmacol Ther. 2017. June;174:138–44. [DOI] [PubMed] [Google Scholar]
- 61.Choi SH, Martinez TF, Kim S, Donaldson C, Shokhirev MN, Saghatelian A, et al. CDK12 phosphorylates 4E-BP1 to enable mTORC1-dependent translation and mitotic genome stability. Genes Dev. 2019. April 1;33(7–8):418–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Gaillard H, García-Muse T, Aguilera A. Replication stress and cancer. Nat Rev Cancer. 2015. May;15(5):276–89. [DOI] [PubMed] [Google Scholar]
- 63.Jones SE, Fleuren EDG, Frankum J, Konde A, Williamson CT, Krastev DB, et al. ATR is a therapeutic target in synovial sarcoma. Cancer Res. 2017. December 15;77(24):7014–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Lee JA, Jeon D-G, Cho WH, Song WS, Yoon HS, Park HJ, et al. Higher Gemcitabine Dose Was Associated With Better Outcome of Osteosarcoma Patients Receiving Gemcitabine-Docetaxel Chemotherapy. Pediatr Blood Cancer. 2016. May 16;63(9):1552–6. [DOI] [PubMed] [Google Scholar]
- 65.Saini P, Li Y, Dobbelstein M. Wee1 is required to sustain ATR/Chk1 signaling upon replicative stress. Oncotarget. 2015. May 30;6(15):13072–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Lowery CD, VanWye AB, Dowless M, Blosser W, Falcon BL, Stewart J, et al. The checkpoint kinase 1 inhibitor prexasertib induces regression of preclinical models of human neuroblastoma. Clin Cancer Res. 2017. August 1;23(15):4354–63. [DOI] [PubMed] [Google Scholar]
- 67.Stewart E, Federico SM, Chen X, Shelat AA, Bradley C, Gordon B, et al. Orthotopic patient-derived xenografts of paediatric solid tumours. Nature. 2017. September 7;549(7670):96–100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Stewart E, McEvoy J, Wang H, Chen X, Honnell V, Ocarz M, et al. Identification of Therapeutic Targets in Rhabdomyosarcoma through Integrated Genomic, Epigenomic, and Proteomic Analyses. Cancer Cell. 2018. September 10;34(3):411–426.e19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Lowery CD, Dowless M, Renschler M, Blosser W, VanWye AB, Stephens JR, et al. Broad spectrum activity of the checkpoint kinase 1 inhibitor prexasertib as a single agent or chemopotentiator across a range of preclinical pediatric tumor models. Clin Cancer Res. 2019. April 1;25(7):2278–89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Laroche-Clary A, Lucchesi C, Rey C, Verbeke S, Bourdon A, Chaire V, et al. CHK1 inhibition in soft-tissue sarcomas: biological and clinical implications. Ann Oncol. 2018. April 1;29(4):1023–9. [DOI] [PubMed] [Google Scholar]
- 71.Zhao H, Piwnica-Worms H. ATR-mediated checkpoint pathways regulate phosphorylation and activation of human Chk1. Mol Cell Biol. 2001. July;21(13):4129–39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Do K, Wilsker D, Ji J, Zlott J, Freshwater T, Kinders RJ, et al. Phase I Study of Single-Agent AZD1775 (MK-1775), a Wee1 Kinase Inhibitor, in Patients With Refractory Solid Tumors. J Clin Oncol. 2015. October 20;33(30):3409–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Leijen S, van Geel RMJM, Pavlick AC, Tibes R, Rosen L, Razak ARA, et al. Phase I study evaluating WEE1 inhibitor AZD1775 as monotherapy and in combination with gemcitabine, cisplatin, or carboplatin in patients with advanced solid tumors. J Clin Oncol. 2016. December 20;34(36):4371–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Wang X, Lai P, Zhang Z, Huang M, Wang L, Yin M, et al. Targeted inhibition of mTORC2 prevents osteosarcoma cell migration and promotes apoptosis. Oncol Rep. 2014. July;32(1):382–8. [DOI] [PubMed] [Google Scholar]
- 75.Chang L-S, Oblinger JL, Burns SS, Huang J, Anderson LW, Hollingshead MG, et al. Targeting protein translation by rocaglamide and didesmethylrocaglamide to treat MPNST and other sarcomas. Mol Cancer Ther. 2020. March;19(3):731–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Ki DH, Oppel F, Durbin AD, Look AT. Mechanisms underlying synergy between DNA topoisomerase I-targeted drugs and mTOR kinase inhibitors in NF1-associated malignant peripheral nerve sheath tumors. Oncogene. 2019. August 23;38(39):6585–98. [DOI] [PubMed] [Google Scholar]
- 77.Hai J, Liu S, Bufe L, Do K, Chen T, Wang X, et al. Synergy of WEE1 and mTOR Inhibition in Mutant KRAS-Driven Lung Cancers. Clin Cancer Res. 2017. November 15;23(22):6993–7005. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.