

Abstract
Animal studies have revealed gut microbial and metabolic pathways of blood pressure (BP) regulation, yet few epidemiological studies have collected microbiota and metabolomics data in the same individuals. In a population-based, Chinese cohort who did not report antihypertension medication use (30–69 years, 54% women), thus minimizing BP treatment effects, we examined multivariable-adjusted (e.g., diet, physical activity, smoking, kidney function), cross-sectional associations between measures of gut microbiota (16S rRNA, n=1003) and plasma metabolome (liquid chromatography-mass spectrometry, n=434) with systolic [SBP, mean (standard deviation)=126.0 (17.4) mmHg] and diastolic BP (DBP, [80.7 (10.7) mmHg]). We found that the overall microbial community assessed by principal coordinate analysis varied by SBP and DBP (permutational multivariate ANOVA p-value<0.05). To account for strong correlations across metabolites, we first examined metabolite patterns derived from principal component analysis and found that a lipid pattern was positively associated with SBP [linear regression coefficient (95% CI) per 1SD pattern score: 2.23 (0.72, 3.74) mmHg] and DBP [1.72 (0.81, 2.63) mmHg]. Among 1104 individual metabolites, 34 and 39 metabolites were positively associated with SBP and DBP (FDR-adjusted linear model p-value<0.05), respectively, including linoleate, palmitate, dihomolinolenate, eight sphingomyelins, four acyl-carnitines, and two phosphatidylinositols. Subsequent pathway analysis showed that metabolic pathways of long-chain saturated acyl-carnitine, phosphatidylinositol, and sphingomyelins were associated with SBP and DBP (FDR-adjusted Fisher’s exact test p-value<0.05). Our results suggest potential roles of microbiota and metabolites in BP regulation to be followed up in prospective and clinical studies.
Keywords: gut microbiota, circulating metabolome, blood pressure, population-based cohort, epidemiology, lipids
Graphical Abstract
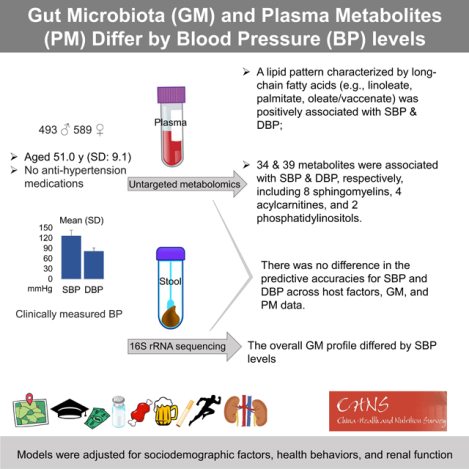
Summary
In a well-characterized Chinese adult cohort, we showed associations between gut microbiota and plasma metabolites with BP, indicating potential roles of microbial and metabolites groups, like lipids, in BP regulation.
INTRODUCTION
Hypertension is a leading modifiable risk factor for cardiovascular disease and mortality.1 Despite numerous clinical and public health efforts to curb the epidemic, the worldwide prevalence of hypertension has continued to increase over the past decade2 and the prevalence of controlled hypertension has remained low.3
The blood pressure (BP) regulatory system is multifactorial, involving interactions among host genetics,4 sociodemographic factors, and diet.5 The gut microbiota and host metabolome, which may reflect these complex interactions,6, 7 have been demonstrated to play fundamental roles in BP regulation in animal models8–11 and humans.12–14 In particular, the metabolome reflects a thorough snapshot of various metabolic processes, allowing the identification of novel biomarkers and pathogenic pathways of elevated BP.15 For example, the microbiota-mediated serum 4-hydroxyhippurate is positively associated with incident hypertension in blacks.16 Additionally, reductions in the overall gut microbial diversity and relative abundance of specific microbial groups, including Prevotella and Coprococcus, are associated with hypertension in animal models11, 17 and humans,13, 14, 18 albeit with small sample sizes. However, there is a lack of population-based studies that include microbial and metabolomic data along with phenotypic data, which is necessary to infer how microbiota influence host physiology through bioactive metabolites. Moreover, there is a need of studies conducted in populations with large burdens of hypertension but low rates of diagnosis and treatment for hypertension for the assessment of natural history of BP.
To this end, we used a well-characterized adult cohort from the 2015 China Health and Nutrition Survey (CHNS) to conduct two primary analyses: the association between (1) gut microbiota and (2) plasma metabolome with BP. We selected the CHNS because China has the greatest absolute burden of hypertension around the world19 coupled with a high rates of undiagnosed and untreated hypertension,20 making China an ideal context for studying BP while minimizing the medication effects.
METHODS
The data and code that support the findings of this study are available to researchers upon request. All phenotypic data can be accessed at the CHNS website (https://www.cpc.unc.edu/projects/china)
Study sample
We used data from the 2015 China Health and Nutrition Survey (CHNS). The CHNS is a prospective, household-based study across 12 provinces and three megacities, which vary substantially in geography, customs, economic development, and health indicators.21 Informed consent was obtained for all participants. The study met the standards for the ethical treatment of participants and was approved by the Institutional Review Boards of the University of North Carolina at Chapel Hill and the National Institute for Nutrition and Health, Chinese Center for Disease Control and Prevention. Participants of the 2015 survey aged 30–69 years from four southern provinces (Henan, Hunan, Guizhou, Guangxi) with BP data and gut microbiome or plasma metabolome data were eligible for analysis (n=1285, Figure S1). We excluded participants who were pregnant (n=1), self-reported use of antihypertension medication (n=99), or had missing covariates (n=86). For microbiota analysis, we additionally excluded 35 participants who currently used antibiotics, had diarrhea, inflammatory bowel disease, irritable bowel syndrome, or bowel removal. For metabolites analysis, we additionally excluded 16 participants who had detectable levels of four CVD dugs metabolites in plasma: metoprolol acid metabolite, alpha-hydroxymetoprolol, nifedipine, and valsartan. The total analysis sample had 1082 adults, with 1003 and 434 adults included in the microbiota and metabolomics analysis samples, respectively.
Blood pressure
Resting BP was measured by experienced physicians, who had completed a 7-day training session and passed a comprehensive reliability test. After a 5 min seated rest, systolic (SBP) and diastolic BP (DBP) were measured in triplicate (30-second interval between cuff inflation) using a standard mercury sphygmomanometer (measuring range: 0–300 mmHg) on the right arm (heart level in sitting position) rested on table with palm face up. The cuff size was selected according to standardized protocol.22 We used the average of the three readings as our measure of SBP and DBP. Hypertension was defined as SBP ≥140 mmHg, DBP ≥90 mmHg, or self-reported diagnosis.23
Gut microbiota
Participants collected stool samples at home using the QIAGEN collection kit (QIAGEN, Hilden, Germany) following standardized protocol. Samples were temporarily stored at foam boxes with frozen cold packs and brought to local community or village clinics immediately, where the samples were stored at −20°C. Then, samples were transported in cold-chain to laboratory and frozen at −80°C until processing. Samples were randomized for sequencing at Novogene Bioinformatics Technology Co., Ltd., Tianjin, China, so that batches were not related to specific collection centers. Bacterial DNA was extracted using TIANGEN DNA extraction kits (TIANGEN Biotech, Beijing, China). Sequencing for 16S rRNA targeting the V4 hypervariable region was performed using primers 515F/806R on the Illumina MiSeq PE250 platform. The raw sequencing reads were processed using the QIIME pipeline,24 with forward and reverse reads merged with fastq-join and filtered using a minimum quality score of 20. No sample was filtered out due to low quality. Operational Taxonomic Units (OTUs) were identified using open-reference method based on a threshold of 0.97, with chimeric OTUs detected by ChimeraSlayer being removed.25 Taxonomy was assigned based on the SILVA databases (Release 128). We rarefied the resulting taxonomic abundances of 1008 genera to 21,600 sequences/sample to correct for different sequencing depth (21,648–89,427 sequences/sample) before log10 transformation.
Plasma metabolomics
Fasting blood samples were collected within 3-days of fecal sample collection by clinicians following the same protocol for the collection, processing, and storage. Ethylenediamine tetraacetic acid was used as an anticoagulant and plasma was immediately separated through centrifugation and stored at −80°C. Detection and quantification of metabolites was performed by the partner campus of Metabolon Inc. in China using a nontargeted platform consisting of a Waters ACQUITY ultrahigh performance liquid chromatographer (Milford, MA) and a Thermo Scientific Q-Exactive high-resolution mass spectrometer (Waltham, MA).7 Methanol solvent was used to extract plasma samples, which were analyzed with several types of controls, including pooled experiment samples as technical replicate and extracted water samples as process blanks. Signals were extracted, peak identified, and processed using Metabolon’s software and hardware. Metabolites were identified by comparing to the mass-to-charge ratio, rendition time/index, and chromatographic data in the Metabolon reference library of purified standards and labeled according to Metabolomics Standards Initiative defined identification levels.26 Of the 1104 detected and quantified metabolites, we categorized 131 metabolites that were below detection limits (BDL) in 25%−50% samples to three groups (BDL, <median, ≥median) and 99 metabolites with >50% of BDL to binary variables (BDL, ≥detection limit). For 874 metabolites with ≤25% of BDL, we rescaled the raw area count of each metabolite to a median of one and imputed values BDL by the minimum value before log2 transformation.
Covariates
Sociodemographic and behavioral information were collected using standard questionnaires administered by interviewers, including age, sex, education (yes/no completed high school), per-capita household income (household income/number of household member), ever smoking (yes/no), alcohol intake in the past year (yes/no), and total physical activity (METs/week). We assessed community-level urbanization using a validated urbanization index that encompasses 12 dimensions of urbanization,27 including population density, health infrastructure, sanitation, and transportation. We included two validated measures of diet, total energy intake28 and sodium intake,29 collected using three-consecutive 24-h diet recalls and household food inventories. We also included three clinically-measured health markers: (1) for kidney function, we used fasting serum creatinine concentration measured by picric acid method on Hitachi 7600 (Tokyo, Japan) to calculate estimated glomerular filtration rate (eGFR) based on the Chronic Kidney Disease Epidemiology Collaboration equation;30 (2) low-density lipoprotein cholesterol (LDL-C) was measured by the polyethylene glycol-modified enzyme method on Hitachi 7600; (3) we calculated body mass index (BMI) from weight over squared height (kg/m2) measured using calibrated beam scales and portable stadiometers, respectively.
Statistical analysis
Primary outcomes were SBP and DBP. In the microbiota analysis sample, we first analyzed the overall gut microbiota by examining the associations of genus-level within-person microbial diversity (α-diversity), measured by Shannon index and richness,31, 32 and between-person diversity (β-diversity), assessed by principal coordinate analysis (PCoA) based on Bray-Curtis dissimilarity matrix,33 with SBP and DBP using linear regression and permutational multivariate analysis of variance (PERMANOVA) with 999 permutations,34 respectively. PCoA axis score is a weighted sum of genera scores (Table S1). Then, we quantified the association between each of the first four PCoA axes, explaining 8.61%, 5.58%, 3.54%, and 3.16% of microbial variability, respectively, as well as 1008 specific genera with SBP and DBP using linear regression. We treated 110 genera detected in ≥25% of the sample as continuous variables and dichotomized the rest 898 rare genera to presence/absence. We adjusted all analyses for the following potential confounders in Model 1 based on a priori knowledge: age, sex, provinces, urbanization index (tertiles),35 education, per-capita household income (tertiles), total energy intake, animal-source food consumption,36 sodium consumption,37 total physical activity (tertiles), tobacco use, alcohol consumption, and eGFR.38 As BMI is a potential mediator for microbiota-BP relationship, we additionally adjusted for BMI in Model 2 as a sensitivity analysis to test whether the association was independent of BMI. Additionally, as lipid profile is correlated with BP and microbiota, we conducted a post-hoc analysis that additionally adjusted for the atherogenic LDL-C.
In the metabolomics analysis sample, we first analyzed the overall metabolome by separately grouping 874 metabolites (continuous variables, ≤25% BDL) into uncorrelated patterns to account for complex correlations across metabolites, using principal component analysis (PCA) followed by a varimax rotation to improve interpretation.39 Based on three criteria: eigenvalues >1, the point of inflection in scree plot, and interpretability,40 we selected three metabolite patterns (Table S2). Pattern score is a weighted sum of rotated and inverse factor loadings. Then, we assessed the association between each metabolite pattern, as well as 1104 individual metabolites with SBP and DBP, using the above-mentioned multivariable-adjusted linear models adjusting for batch. We used a Wald test to assess the statistical significance of 131 metabolites with three categories (BDL, <median, ≥median). Based on Model 1 results for individual metabolites, we calculated pathway enrichment score reflecting the degree to which a given pathway was associated with SBP or DBP, where k and n are numbers of BP-associated metabolites in the given pathway and all pathways, respectively, and m and N are numbers of tested metabolites in the given pathway and all pathways, respectively. We performed a Fisher’s exact test41 to evaluate whether the presence of BP-associated metabolites among identified compounds from a particular metabolic pathway was greater than expected by chance.
In a sub-sample of participants with microbiota and metabolite data (n=355), we examined the association between BP-associated microbiota features and BP-associated metabolites using linear models to understand the inter-correlation between microbiota and metabolites. Next, we conducted random forest regression (100 trees) which allows interaction across microbiota and metabolites,42 followed by a 5 iterations of 2-fold cross-validation (5×2cv) modified paired t-test of root mean squared errors (RMSE), a powerful test to compare the performance of learning algorithms with acceptable Type I error,43 to provide insight into which of the following data as a whole had the strongest association with BP: host factors (14 Model 1 covariates), microbiota (1008 genera), metabolites (1104 metabolites), microbiota + host factors, metabolites + host factors, microbiota + metabolites, and microbiota + metabolites + host factors.
We adjusted p-values for multiple comparisons using Benjamini-Hochberg method (false discovery rate, FDR)44 in comparisons across taxa, metabolites, and metabolic pathways for SBP and DBP separately. All statistical tests were two-sided with a significance level of 0.05. We used R 3.6.0 (http://www.r-project.org) and Python 3.5.1 (https://www.python.org) for data analysis.
RESULTS
Our sample had large variation in SBP [mean (SD): 126.0 (17.4) mmHg] and DBP [80.7 (10.7) mmHg], with 27.6% prevalence of hypertension (Table S3).
We first assessed the overall gut microbial measures. Within-person microbial diversity (Shannon index and richness) was not associated with SBP or DBP (Table S4, p-value=0.45–0.97). Between-person microbial diversity assessed by PCoA varied by SBP (Figure 1; PERMANOVA R2=0.20%, p-value=0.002) and DBP (Figure S2; PERMANOVA R2=0.14%, p-value<0.05). Only the fourth PCoA axis showed a clear separation of SBP (Figure 1), with higher axis score associated with higher SBP (Table S5). This axis was positively correlated with Rothia, Serratia, Enterobacteriaceae, Leuconostocaceae, and Fusobacterium, while negatively correlated with Coprococcus, Adlercreutzia, Eggerthella, and Raistonia. However, after correction for multiple hypothesis testing, none of the 1008 specific genera were associated with SBP or DBP at FDR-adjusted p-value<0.05 (Table S6). We observed similar results after additionally adjusted for LDL-C (Table S7–S9).
Figure 1. Microbial between-person diversity (β-diversity) assessed using principal coordinate analysis (PCoA) by systolic blood pressure (SBP).
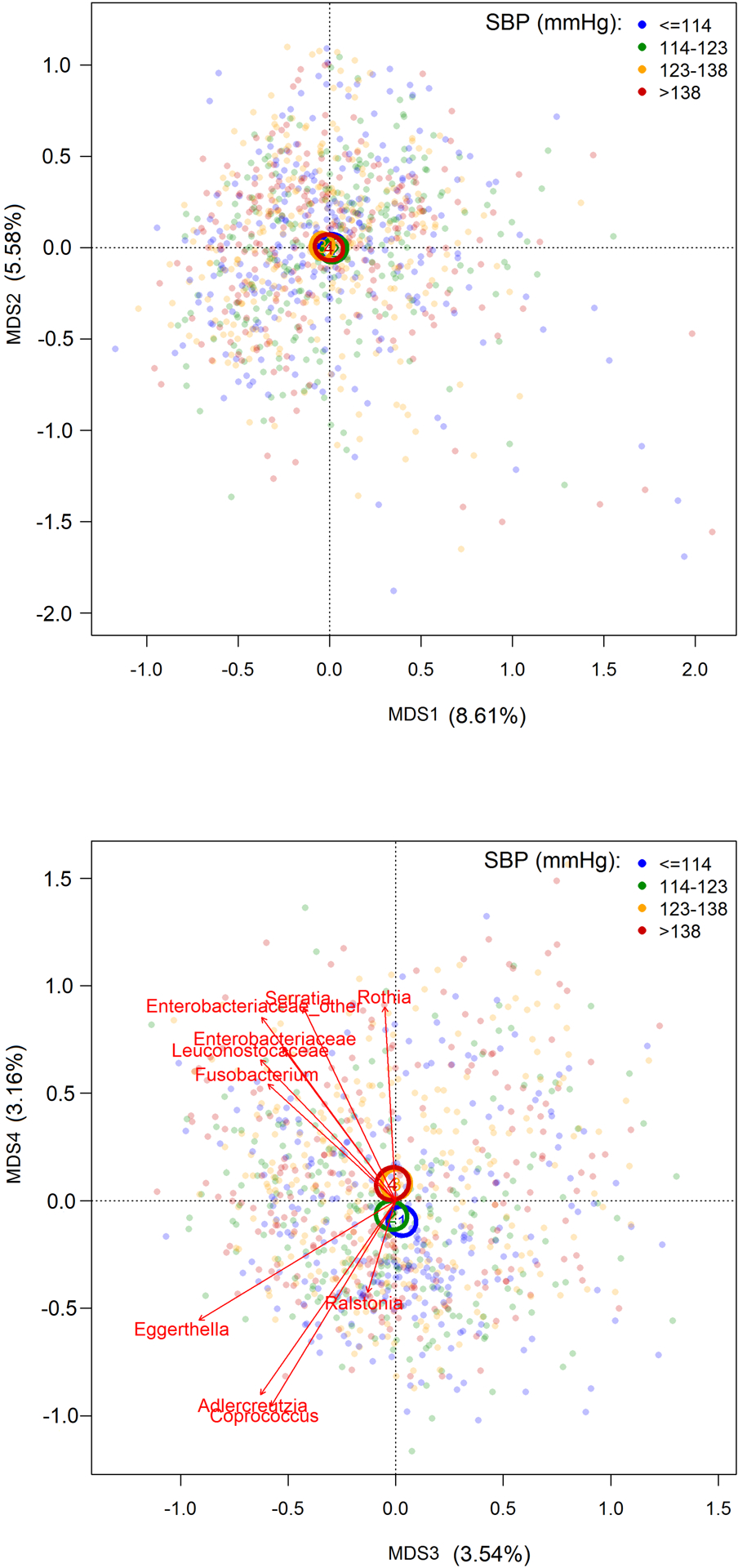
MDS, multidivisional scaling. Centroids illustrate the 95% CI for the mean location of each SBP (mmHg) quartile. Vectors for 10 taxa with the greatest contributions to MDS4 indicate the directions and strengths of their correlations with MDS4 (Table S1). In permutational multivariate analysis of variance (PERMANOVA, n=1003), SBP had R2 of 0.20% and p-value of 0.002, after adjusting for age, sex, provinces, urbanization index (≤64.2, 64.2–81.5, >81.5), per-capita household income (≤10, 10–21.6, >21.6), education, total energy intake, animal-source food, sodium, physical activity (≤57.4, 57.4–152, >152), smoking, alcohol, and estimated glomerular filtration rate (eGFR). Results remained the same after additional adjustment of BMI.
In plasma metabolite analysis, we identified three biologically possible patterns using PCA that each explained 9.63%, 4.79%, and 4.69% of variance (Table 1). The second pattern characterized by lipids, like linoleate, palmitate, and oleate/vaccinate, was positively associated with SBP [linear model coefficient (95% CI) per 1SD pattern score: 2.23 (0.72, 3.74) mmHg] and DBP [1.72 (0.81, 2.63) mmHg]. The results were slightly attenuated by adjustment of BMI [SBP: 1.88 (0.38, 3.38) mmHg; DBP: 1.45 (0.55, 2.35) mmHg].
Table 1.
Association between metabolite patterns with systolic and diastolic blood pressure (SBP and DBP, mmHg), Coefficient (95% confidence interval)
| Metabolite pattern | Metabolites contributing to each pattern | Eigenvalue | Variance explained | SBP | DBP | |||
|---|---|---|---|---|---|---|---|---|
| Model 1 | Model 2 | Model 1 | Model 2 | |||||
| Pattern 1 (nucleotide, amino acid, and peptide) | pseudouridine; 2,3-dihydroxy-5-methylthio-4-pentenoate (DMTPA); N-acetylthreonine; N,N-dimethyl-pro-pro; C-glycosyltryptophan; | orotidine; hydroxy-N6,N6,N6-trimethyllysine; 5,6-dihydrouridine; dimethylarginine (ADMA + SDMA); N6-acetyllysine | 84.19 | 9.63% | 1.81 (−0.24, 3.86) | 1.58 (−0.44, 3.60) | 0.24 (−1.01, 1.49) | 0.06 (−1.16, 1.28) |
| Pattern 2 (lipids, especially long-chain fatty acids) | linoleate (18:2n6); palmitate (16:0); oleate/vaccenate (18:1); 10-heptadecenoate (17:1n7); docosapentaenoate (DPA; 22:5n3); hexadecadienoate (16:2n6); | 10-nonadecenoate (19:1n9); margarate (17:0); dihomolinoleate (20:2n6); dihomolinolenate (20:3n3 or 3n6) | 41.83 | 4.79% | 2.23 (0.72, 3.74)** | 1.88 (0.38, 3.38)* | 1.72 (0.81, 2.63)** | 1.45 (0.55, 2.35)** |
| Pattern 3 (sphingomyelins, eicosanoid, short-chain fatty acids, and branched-chain amino acids) | sphingomyelin (d18:2/23:0, d18:1/23:1, d17:1/24:1); 3-methyl-2-oxobutyrate; leukotriene B4; 5-HETE; methionine sulfoxide; butyrate/isobutyrate (4:0); | 4-methyl-2-oxopentanoate; 1-(1-enyl-oleoyl)-GPE (P-18:1); 1-(1-enyl-palmitoyl)-GPE (P-16:0); 3-methyl-2-oxovalerate | 40.99 | 4.69% | 0.24 (−1.57, 2.05) | −0.02 (−1.80, 1.77) | 0.14 (−0.96, 1.24) | −0.05 (−1.13, 1.02) |
Patterns were derived from principal component analysis followed by a varimax rotation of 874 metabolites (n=434). Coefficient indicates BP associated with each 1SD of metabolites pattern score in linear regression. Contributing metabolites are metabolites with the highest absolute loadings for the respective pattern (Table S2). Model 1 was adjusted for age, sex, provinces, batch, urbanization index (≤64.2, 64.2–81.5, >81.5), per-capita household income (≤10, 10–21.6, >21.6), education, total energy intake, animal-source food, sodium, physical activity (≤57.4, 57.4–152, >152), smoking, alcohol, and estimated glomerular filtration rate (eGFR). Model 2 was additionally adjusted for BMI.
, p-value < 0.05;
, p-value<0.01.
To identify whether specific metabolites contributing to this lipid pattern drove the associations with SBP and DBP, we examined 1104 metabolites (Table S10–S12) and found that 34 and 39 metabolites were associated with SBP (Table 2) and DBP (Table 3) at Model 1 FDR-adjusted p-value<0.05, respectively, including eight sphingomyelins, four acyl-carnitines, and cholesterol. Among these SBP- and DBP-associated metabolites, 8 (23.5%) and 19 (48.7%) metabolites respectively had high loadings (>0.4) for the lipid pattern, including acyl-carnitines (C16, C26, C14, and C12), 1-palmitoleoylglycerol (16:1), and dihomolinolenate (20:3n3 or 3n6), which were positively associated with both BP measures. In contrast, we saw noticeably fewer SBP- and DBP-associated metabolites with high loadings for the other two metabolite patterns (0–11.8%). After adjusting for BMI, only nine and 17 metabolites remained statistically significantly associated with SBP and DBP (Model 2 FDR-adjusted p-value<0.05), respectively, including sphingomyelins (d18:1/23:0, d18:1/24:0, and d18:2/24:2) and acyl-carnitines (C16 and C14). After adjusting for LDL-C, results for metabolite patterns were similar to main model results (Table S13) and while only 19 metabolites remained statistically significantly associated with DBP (Table S14).
Table 2.
Association between individual metabolites and systolic blood pressure (SBP, mmHg)
| Metabolites | Pathway | Loading in lipid pattern* | Model 1 | Model 2 | |||
|---|---|---|---|---|---|---|---|
| Coefficient (95% CI) | q-value | Coefficient (95% CI) | q-value | ||||
| tricosanoyl sphingomyelin (d18:1/23:0)† | Sphingomyelins | ─ | 7.56 (4.52, 10.61) | 0.002 | 6.53 (3.43, 9.62) | 0.015 | |
| lignoceroyl sphingomyelin (d18:1/24:0)† | Sphingomyelins | ─ | 6.37 (3.59, 9.14) | 0.005 | 5.61 (2.83, 8.39) | 0.024 | |
| palmitoylcarnitine (C16)† | Fatty Acid Metabolism (Acyl Carnitine, Long Chain Saturated) | 0.50 | 7.26 (3.97, 10.56) | 0.007 | 6.95 (3.71, 10.2) | 0.015 | |
| 1-myristoyl-2-arachidonoyl-GPC (14:0/20:4)† | Phosphatidylcholine (PC) | ─ | 4.34 (2.32, 6.37) | 0.007 | 3.53 (1.45, 5.61) | 0.059 | |
| sphingomyelin (d18:2/24:2)† | Sphingomyelins | ─ | 4.53 (2.41, 6.64) | 0.007 | 4.46 (2.38, 6.54) | 0.015 | |
| 1-palmitoyl-2-linoleoyl-GPI (16:0/18:2)† | Phosphatidylinositol (PI) | ─ | 5.83 (3.02, 8.64) | 0.010 | 5.38 (2.59, 8.16) | 0.037 | |
| 1-palmitoyl-2-arachidonoyl-GPC (16:0/20:4n6)† | Phosphatidylcholine (PC) | ─ | 7.91 (4.01, 11.82) | 0.010 | 6.7 (2.77, 10.63) | 0.059 | |
| 1-palmitoyl-2-arachidonoyl-GPI (16:0/20:4)† | Phosphatidylinositol (PI) | ─ | 4.96 (2.51, 7.41) | 0.010 | 4.44 (2.01, 6.87) | 0.046 | |
| sphingomyelin (d18:1/21:0, d17:1/22:0, d16:1/23:0)† | Sphingomyelins | ─ | 6.38 (3.28, 9.49) | 0.010 | 5.03 (1.82, 8.25) | 0.088 | |
| behenoyl sphingomyelin (d18:1/22:0)† | Sphingomyelins | ─ | 7.35 (3.57, 11.14) | 0.014 | 5.91 (2.05, 9.77) | 0.092 | |
| sphingomyelin (d18:1/14:0, d16:1/16:0)† | Sphingomyelins | ─ | 7.21 (3.4, 11.01) | 0.019 | 5.65 (1.75, 9.55) | 0.118 | |
| cerotoylcarnitine (C26)† | Fatty Acid Metabolism (Acyl Carnitine, Long Chain Saturated) | 0.42 | 4.29 (2.02, 6.56) | 0.019 | 3.91 (1.66, 6.16) | 0.059 | |
| pantothenate (Vitamin B5) | Pantothenate and CoA Metabolism | ─ | 7.77 (3.63, 11.91) | 0.019 | 6.86 (2.74, 10.97) | 0.064 | |
| N2,N2-dimethylguanosine | Purine Metabolism, Guanine containing | ─ | 8.74 (3.99, 13.49) | 0.020 | 8.09 (3.4, 12.78) | 0.059 | |
| cholesterol† | Sterol | ─ | 8.48 (3.89, 13.07) | 0.020 | 7.67 (3.12, 12.22) | 0.062 | |
| sphingomyelin (d18:2/14:0, d18:1/14:1)† | Sphingomyelins | ─ | 5.75 (2.58, 8.91) | 0.022 | 4.57 (1.36, 7.79) | 0.121 | |
| adrenate (22:4n6) | Long Chain Polyunsaturated Fatty Acid (n3 and n6) | 0.71 | 3.15 (1.36, 4.93) | 0.028 | 2.9 (1.14, 4.67) | 0.066 | |
| 1-palmitoleoylglycerol (16:1)† | Monoacylglycerol | 0.57 | 2.26 (0.98, 3.53) | 0.028 | 1.75 (0.45, 3.05) | 0.143 | |
| 1-palmitoyl-2-palmitoleoyl-GPC (16:0/16:1) | Phosphatidylcholine (PC) | ─ | 3.57 (1.54, 5.59) | 0.028 | 3.01 (0.98, 5.04) | 0.107 | |
| myristoylcarnitine (C14)† | Fatty Acid Metabolism (Acyl Carnitine, Long Chain Saturated) | 0.53 | 2.96 (1.27, 4.64) | 0.029 | 3.01 (1.35, 4.66) | 0.046 | |
| branched-chain, straight-chain, or cyclopropyl 10:1 fatty acid (1) | Partially Characterized Molecules | ─ | 2.66 (1.13, 4.19) | 0.030 | 2.58 (1.07, 4.08) | 0.059 | |
| sphingomyelin (d18:2/16:0, d18:1/16:1)† | Sphingomyelins | ─ | 8.35 (3.52, 13.17) | 0.031 | 6.84 (1.99, 11.69) | 0.122 | |
| picolinoylglycine | Fatty Acid Metabolism (Acyl Glycine) | ─ | 3.34 (1.41, 5.28) | 0.031 | 2.75 (0.81, 4.69) | 0.122 | |
| N6-carbamoylthreonyladenosine | Purine Metabolism, Adenine containing | ─ | 6.16 (2.54, 9.78) | 0.032 | 5.57 (1.99, 9.16) | 0.088 | |
| dihomolinolenate (20:3n3 or 3n6)† | Long Chain Polyunsaturated Fatty Acid (n3 and n6) | 0.73 | 4.25 (1.75, 6.74) | 0.032 | 3.65 (1.16, 6.14) | 0.113 | |
| 1-stearoyl-2-arachidonoyl-GPC (18:0/20:4) | Phosphatidylcholine (PC) | ─ | 6.08 (2.52, 9.64) | 0.032 | 5.01 (1.44, 8.58) | 0.122 | |
| acetylcarnitine (C2) | Fatty Acid Metabolism (Acyl Carnitine, Short Chain) | 0.50 | 6.57 (2.63, 10.5) | 0.038 | 7 (3.13, 10.86) | 0.046 | |
| retinol (Vitamin A) | Vitamin A Metabolism | ─ | 5.16 (2.04, 8.28) | 0.040 | 4.48 (1.39, 7.58) | 0.118 | |
| argininate | Urea cycle; Arginine and Proline Metabolism | ─ | 3.93 (1.56, 6.3) | 0.040 | 3.19 (0.81, 5.57) | 0.148 | |
| 2,3-dihydroxy-5-methylthio-4-pentenoate (DMTPA) | Methionine, Cysteine, SAM and Taurine Metabolism | ─ | 7.88 (3.11, 12.66) | 0.040 | 6.51 (1.73, 11.29) | 0.135 | |
| laurylcarnitine (C12)† | Fatty Acid Metabolism (Acyl Carnitine, Medium Chain) | 0.49 | 2.25 (0.88, 3.62) | 0.042 | 2.36 (1.02, 3.71) | 0.059 | |
| Metabolites with 25–50% below detection limits (BDL): Reference=BDL | |||||||
| linoleoyl-linoleoyl-glycerol (18:2/18:2) [1] | Diacylglycerol | Below median | ─ | 2.29 (−1.6, 6.17) | 0.022 | 3.5 (−0.37, 7.37) | 0.046 |
| Above median | −4.56 (−8.39, −0.73) | −3.45 (−7.26, 0.36) | |||||
| oleoyl-linoleoyl-glycerol (18:1/18:2) [2] | Diacylglycerol | Below median | ─ | −3.07 (−6.99, 0.85) | 0.042 | −2.49 (−6.36, 1.38) | 0.088 |
| Above median | −7.31 (−11.39, −3.24) | −6.8 (−10.82, −2.78) | |||||
| Metabolites with >50% BDL: Reference=BDL | |||||||
| phenylalanylalanine | Dipeptide | Above limit of detection | ─ | −4.86 (−7.86, −1.85) | 0.047 | −4.94 (−7.89, −1.98) | 0.064 |
CI, confidence interval. Coefficient indicates SBP associated with a fold increase of the abundance or per category change of a metabolite in linear regression (n=434). The statistical significance of metabolites with 25–50% BDL was assessed using a Wald test. Model 1 was adjusted for age, sex, provinces, batch, urbanization index (≤64.2, 64.2–81.5, >81.5), per-capita household income (≤10, 10–21.6, >21.6), education, total energy intake, animal-source food, sodium, physical activity (≤57.4, 57.4–152, >152), smoking, alcohol, and estimated glomerular filtration rate (eGFR). Model 2 was additionally adjusted for BMI.
Pattern was derived from principal component analysis followed by a varimax rotation. Loadings>0.4 are listed.
Metabolites also associated with diastolic blood pressure.
Table 3.
Association between individual metabolites and diastolic blood pressure (DBP, mmHg)
| Metabolites | Pathway | Loading in lipid pattern* | Model 1 | Model 2 | ||
|---|---|---|---|---|---|---|
| Coefficient (95% CI) | q-value | Coefficient (95% CI) | q-value | |||
| lignoceroyl sphingomyelin (d18:1/24:0)† | Sphingomyelins | ─ | 4.85 (3.19, 6.52) | 2E-05 | 4.28 (2.62, 5.93) | 0.001 |
| behenoyl sphingomyelin (d18:1/22:0)† | Sphingomyelins | ─ | 6.16 (3.9, 8.43) | 1E-04 | 5.09 (2.8, 7.39) | 0.004 |
| tricosanoyl sphingomyelin (d18:1/23:0)† | Sphingomyelins | ─ | 4.95 (3.11, 6.8) | 1E-04 | 4.12 (2.26, 5.98) | 0.004 |
| cerotoylcarnitine (C26)† | Fatty Acid Metabolism (Acyl Carnitine, Long Chain Saturated) | 0.42 | 3.09 (1.71, 4.46) | 0.003 | 2.79 (1.45, 4.14) | 0.008 |
| Corticosterone | Corticosteroids | ─ | −1.39 (−2.02, −0.77) | 0.003 | −1.11 (−1.74, −0.49) | 0.033 |
| dihomolinolenate (20:3n3 or 3n6)† | Long Chain Polyunsaturated Fatty Acid (n3 and n6) | 0.73 | 3.28 (1.77, 4.79) | 0.004 | 2.83 (1.34, 4.31) | 0.020 |
| sphingomyelin (d18:2/24:2)† | Sphingomyelins | ─ | 2.8 (1.51, 4.09) | 0.004 | 2.75 (1.5, 4) | 0.004 |
| myristoylcarnitine (C14)† | Fatty Acid Metabolism (Acyl Carnitine, Long Chain Saturated) | 0.53 | 2.15 (1.13, 3.17) | 0.005 | 2.19 (1.2, 3.18) | 0.004 |
| cortolone glucuronide (1) | Corticosteroids | ─ | 2.34 (1.22, 3.45) | 0.005 | 1.7 (0.56, 2.83) | 0.112 |
| behenoyl dihydrosphingomyelin (d18:0/22:0) | Dihydrosphingomyelins | ─ | 1.84 (0.95, 2.72) | 0.006 | 1.2 (0.27, 2.14) | 0.182 |
| sphingomyelin (d18:1/21:0, d17:1/22:0, d16:1/23:0)† | Sphingomyelins | ─ | 3.85 (1.96, 5.74) | 0.007 | 2.73 (0.79, 4.67) | 0.133 |
| 1-palmitoyl-2-linoleoyl-GPI (16:0/18:2)† | Phosphatidylinositol (PI) | ─ | 3.46 (1.75, 5.18) | 0.007 | 3.11 (1.43, 4.79) | 0.024 |
| cis-4-decenoylcarnitine (C10:1) | Fatty Acid Metabolism (Acyl Carnitine, Monounsaturated) | 0.44 | 1.47 (0.73, 2.21) | 0.008 | 1.52 (0.81, 2.24) | 0.006 |
| laurylcarnitine (C12)† | Fatty Acid Metabolism (Acyl Carnitine, Medium Chain) | 0.49 | 1.63 (0.8, 2.46) | 0.009 | 1.72 (0.91, 2.52) | 0.006 |
| linoleate (18:2n6) | Long Chain Polyunsaturated Fatty Acid (n3 and n6) | 0.80 | 2.65 (1.26, 4.04) | 0.013 | 2.44 (1.08, 3.81) | 0.030 |
| decanoylcarnitine (C10) | Fatty Acid Metabolism (Acyl Carnitine, Medium Chain) | 0.48 | 1.43 (0.67, 2.19) | 0.014 | 1.46 (0.72, 2.2) | 0.013 |
| 1-palmitoleoylglycerol (16:1)† | Monoacylglycerol | 0.57 | 1.45 (0.68, 2.23) | 0.014 | 1.05 (0.27, 1.84) | 0.157 |
| palmitate (16:0) | Long Chain Saturated Fatty Acid | 0.80 | 3.51 (1.62, 5.41) | 0.014 | 3.11 (1.26, 4.97) | 0.054 |
| cis-4-decenoate | Medium Chain Fatty Acid | ─ | 2.06 (0.95, 3.17) | 0.014 | 2.03 (0.95, 3.11) | 0.021 |
| 1-myristoyl-2-arachidonoyl-GPC (14:0/20:4)† | Phosphatidylcholine (PC) | ─ | 2.29 (1.05, 3.53) | 0.014 | 1.59 (0.33, 2.85) | 0.188 |
| 5-dodecenoylcarnitine (C12:1) | Fatty Acid Metabolism (Acyl Carnitine, Monounsaturated) | 0.50 | 1.43 (0.66, 2.2) | 0.014 | 1.53 (0.78, 2.29) | 0.009 |
| 1-dihomo-linoleoylglycerol (20:2) | Monoacylglycerol | 0.64 | 1.39 (0.62, 2.15) | 0.018 | 1.06 (0.3, 1.83) | 0.141 |
| octanoylcarnitine (C8) | Fatty Acid Metabolism (Acyl Carnitine, Medium Chain) | 0.51 | 1.74 (0.78, 2.71) | 0.018 | 1.83 (0.9, 2.77) | 0.014 |
| 1-linoleoylglycerol (18:2) | Monoacylglycerol | 0.63 | 1.54 (0.68, 2.39) | 0.018 | 1.14 (0.28, 2) | 0.161 |
| 1-dihomo-linolenylglycerol (20:3) | Monoacylglycerol | 0.66 | 1.49 (0.65, 2.33) | 0.020 | 1.02 (0.17, 1.87) | 0.225 |
| palmitoylcarnitine (C16)† | Fatty Acid Metabolism (Acyl Carnitine, Long Chain Saturated) | 0.50 | 3.57 (1.55, 5.59) | 0.021 | 3.33 (1.36, 5.3) | 0.051 |
| tetrahydrocortisone glucuronide (5) | Corticosteroids | ─ | 1.65 (0.7, 2.6) | 0.024 | 1.22 (0.27, 2.17) | 0.182 |
| 1-palmitoyl-2-arachidonoyl-GPI (16:0/20:4)† | Phosphatidylinositol (PI) | ─ | 2.6 (1.11, 4.1) | 0.024 | 2.19 (0.72, 3.67) | 0.112 |
| sphingomyelin (d18:2/16:0, d18:1/16:1)† | Sphingomyelins | ─ | 5.09 (2.15, 8.03) | 0.024 | 3.9 (0.97, 6.82) | 0.161 |
| hexanoylcarnitine (C6) | Fatty Acid Metabolism (Acyl Carnitine, Medium Chain) | 0.55 | 1.79 (0.75, 2.83) | 0.025 | 1.83 (0.82, 2.84) | 0.027 |
| sphingomyelin (d18:2/14:0, d18:1/14:1)† | Sphingomyelins | ─ | 3.31 (1.38, 5.24) | 0.025 | 2.36 (0.42, 4.3) | 0.217 |
| 1-arachidonoyl-GPI (20:4) | Lysophospholipid | 0.55 | 3.53 (1.46, 5.6) | 0.026 | 2.99 (0.95, 5.02) | 0.118 |
| sphingomyelin (d18:1/14:0, d16:1/16:0)† | Sphingomyelins | ─ | 3.95 (1.63, 6.27) | 0.026 | 2.65 (0.29, 5.01) | 0.279 |
| cholesterol† | Sterol | ─ | 4.78 (1.98, 7.59) | 0.026 | 4.15 (1.4, 6.9) | 0.112 |
| 2-palmitoleoylglycerol (16:1) | Monoacylglycerol | 0.46 | 1.14 (0.46, 1.82) | 0.031 | 0.81 (0.13, 1.5) | 0.237 |
| hydantoin-5-propionate | Histidine Metabolism | ─ | 1.45 (0.58, 2.32) | 0.032 | 1.32 (0.47, 2.17) | 0.094 |
| 1-palmitoyl-2-arachidonoyl-GPC (16:0/20:4n6)† | Phosphatidylcholine (PC) | ─ | 3.94 (1.55, 6.33) | 0.035 | 2.94 (0.55, 5.32) | 0.206 |
| p-cresol sulfate | Benzoate Metabolism | ─ | −1.01 (−1.63, −0.4) | 0.036 | −0.88 (−1.48, −0.27) | 0.118 |
| palmitoleate (16:1n7) | Long Chain Monounsaturated Fatty Acid | 0.70 | 1.8 (0.69, 2.91) | 0.040 | 1.71 (0.63, 2.79) | 0.077 |
CI, confidence interval. Coefficient indicates DBP (mmHg) associated with a fold increase of the abundance of a given metabolite in linear regression (n=434). Model 1 was adjusted for age, sex, provinces, batch, urbanization index (≤64.2, 64.2–81.5, >81.5), per-capita household income (≤10, 10–21.6, >21.6), education, total energy intake, animal-source food, sodium, physical activity (≤57.4, 57.4–152, >152), smoking, alcohol, and estimated glomerular filtration rate (eGFR). Model 2 was additionally adjusted for BMI.
Pattern was derived from principal component analysis followed by a varix rotation. Loadings > 0.4 are listed.
Metabolites also associated with systolic blood pressure.
In pathway analysis that tested whether the number of positive or negative associations between BP and metabolites from a particular metabolic pathway was more than expected by chance (Table 4, Table S15), we found that diacylglycerol, acyl-carnitine (long chain saturated), phosphatidylcholine, phosphatidylinositol, sphingomyelins metabolic pathways were associated with SBP (FDR-adjusted p-value<0.05); and corticosteroids, acyl-carnitine (long chain saturated and median chain), monoacylglycerol, phosphatidylinositol, and sphingomyelins metabolic pathways were associated with DBP.
Table 4.
Metabolic pathways associated with systolic (SBP) or diastolic blood pressure (DBP)
| Metabolic pathways | SBP | DBP | |||||||
|---|---|---|---|---|---|---|---|---|---|
| m* | k* | Enrichment score* | p-value† | q-value† | k* | Enrichment score* | p-value† | q-value† | |
| Corticosteroids | 6 | 0 | -- | -- | -- | 3 | 12.47 | 0.001 | 0.005 |
| Diacylglycerol | 3 | 2 | 18.77 | 0.004 | 0.019 | 0 | -- | -- | -- |
| Fatty Acid Metabolism (Acyl Carnitine, Long Chain Saturated) | 6 | 3 | 14.48 | 9E-04 | 0.009 | 3 | 12.47 | 0.001 | 0.005 |
| Fatty Acid Metabolism (Acyl Carnitine, Medium Chain) | 6 | 1 | 4.54 | 0.206 | 0.301 | 4 | 17.10 | 4E-05 | 3E-04 |
| Monoacylglycerol | 14 | 1 | 1.93 | 0.418 | 0.496 | 5 | 9.35 | 0.0002 | 0.001 |
| Phosphatidylcholine (PC) | 18 | 4 | 6.56 | 0.004 | 0.019 | 2 | 2.66 | 0.180 | 0.337 |
| Phosphatidylinositol (PI) | 5 | 2 | 11.24 | 0.013 | 0.049 | 2 | 9.72 | 0.017 | 0.048 |
| Sphingomyelins | 28 | 8 | 9.63 | 3E-06 | 6E-05 | 8 | 8.07 | 1E-05 | 2E-04 |
Enrichment score was calculated using (k/m)/[(n-k)/(N-m)], where k and n are numbers of BP-associated metabolites (Model 1 false discovery rate adjusted p-value<0.05) in a given pathway and all identified pathways (SBP: n=34; DBP: n=39), respectively; m and N are numbers of classified metabolites in a given pathway and all identified pathways(N=904), respectively.
P-value for each pathway was calculated using Fisher’s exact test and adjusted for false discovery rate (q-value) across pathways containing at least one BP-associated metabolite.
In a sub-sample of 355 participants with similar distributions of SBP [123.5 (16.7) mmHg] and DBP [79.8 (9.9)] to the full sample (Table S16), we conducted integrated microbiota and metabolite analysis to examine the inter-correlation between microbiota and metabolites and whether the microbiota and metabolite data had better BP predictive performance than host sociodemographic and behavioral risk factors. We observed no correlation between the BP-associated, fourth gut microbiota PCoA axis with any of the 54 BP-associated metabolites (Table S17, FDR-adjusted p-value≥0.27). Using random forest regression, we found comparable accuracies across host factors, microbiota, and metabolite data in predicting SBP and DBP (Figure S3, p-value>0.05).
DISCUSSION
In a population-based cohort of middle-aged Chinese adults, we found an association between the overall gut microbiota (between-person diversity) with SBP and DBP, after accounting for a wide range of sociodemographic factors, health behaviors, and kidney function. Using plasma metabolome data, we found that a lipid pattern and several individual metabolites like sphingomyelins, acyl-carnitines, and cholesterol, were positively associated with SBP and DBP. Our results suggest that in this population with high prevalence of untreated hypertension (27.6%), gut microbiota and plasma metabolites may play important roles in hypertension etiology.
Several studies have shown an association between the gut microbiota and BP.13, 14, 18 For example, a recent case-control study of 80 Brazilian adults14 showed lower microbial biodiversity along with lower proportions of butyrate-producing taxa like Roseburia, Coprococcus and Lachnospiraceae, but higher proportions of Enterobacteriaceae and Lactobacillus in individuals with high versus normal BP. The Sun et al. paper of 529 middle-aged US adults from Coronary Artery Risk Development in Young Adults (CARDIA) study found an inverse cross-sectional association between within-person microbial diversity with SBP and differences in the overall microbial community by SBP.18 Similarly, we observed differences in the overall gut microbial community by SBP and DBP in the current CHNS study. The US CARDIA cohort is quite different from the China population-based cohort (e.g., higher hypertension medication use, different diet and lifestyle in the US cohort). Furthermore, we excluded participants who used antihypertension medication from the current analysis, while 29.2% participants took antihypertension medications in the CARDIA analysis sample.18 Future prospective studies are needed to confirm the results of our study and previous research.
Metabolomics studies showing associations between microbial metabolites and BP further support the role of gut microbiota in BP regulation.16, 45 The International Population Study on Macronutrients and Blood Pressure (INTERMAP) study of 4630 middle-aged adults from USA, UK, Japan, and China, showed that urinary alanine and hippurate were positively and negatively associated with BP, respectively.45 The Atherosclerosis Risk in Communities (ARIC) study of 896 African Americans revealed that each one standard deviation increase in baseline serum 4-hydroxyhippurate was associated with 17% higher risk of incident hypertension.16 In our sample, we found that p-cresol sulphate from benzoate metabolism, a product of tyrosine and phenylalanine metabolism by anaerobic bacteria,46 was inversely associated with DBP. Additionally, we found comparable predictive accuracies between gut microbiota and plasma metabolome for BP, indicating that microbiota may play a role in metabolites-BP associations, as it has been shown that gut microbiota is involved in host lipid metabolism and modulates plasma metabolome in response to Angiotensin II.47, 48 Given that many microbiota-mediated metabolites were strongly associated with diet, for example, hippurate derived from dietary polyphenols,49 different dietary patterns across populations may relate to these different results across studies.
Host-derived metabolites like the ketone body β-hydroxybutyrate, acyl-carnitines, and long-chain fatty acids have also been suggested in mechanisms of BP regulation.15, 16, 50 For example, nutritional supplementation of a precursor of β-hydroxybutyrate attenuated hypertension in hypertensive rats fed a high-salt diet.50 In line with our findings, Menni et al. showed that in 3980 TwinsUK females, a few plasma carnitines, long chain fatty acids, and steroids were positively associated with BP, including hexadecanedioate, palmitate (16:0), octanoylcarnitine (C8), 10-heptadecenoate (17:1n7), and dihomolinoleate (20:2n6).15 In particular, hexadecanedioate, a dicarboxylic acid, consistently showed positive association with BP in two replication cohorts with both males and females.15 Subsequent analysis using rat model demonstrated that oral intake of hexadecanedioate increased BP, supporting a causal role of hexadecanedioate in BP regulation.15 In another study of 202 African and Caucasian men, serum long-chain and medium-chain acyl-carnitines (in Caucasians only) were positively associated with ambulatory BP.51 Similarly, we found positive associations between medium- and long-chain acyl-carnitines, long-chain fatty acids, and a lipid pattern driven by long-chain fatty acids with BP. Elevated levels of circulating acyl-carnitines and long-chain fatty acids may contribute to hypertension development, as acyl-carnitines are byproducts of incomplete β-oxidation and can accumulate in blood or urine when fatty acids are in excess for oxidation, thus stimulating proinflammatory pathways involving nuclear factor kappa B (NF-κB).52 Likewise, omega-6 fatty acid like linoleate may impair cardiovascular health as it can be metabolized to dihomolinoleate and then to arachidonic acid, a precursor for proinflammatory eicosanoids like leukotriene B4.53
In addition, we found that several sphingomyelins and the sphingomyelin metabolic pathway were each positively associated with BP. Ceramide as a precursor for sphingolipids is harmful to cardiovascular health, including impaired vasodilation.54 Excess sphingolipids occur when fatty acids exceed energy need or storage capacity of a cell.55 Several lipidomic studies have identified sphingolipids as candidate blood markers for cardiovascular diseases in humans.56–58 For example, Poss et al.58 found that 30 serum sphingolipids were elevated in subjects with coronary artery disease (CAD, n=462) than controls (n=212) and a sphingolipid risk score was more effective than conventional biomarkers like triglycerides and LDL-cholesterol in distinguishing CAD patients.
The strengths of our study include paired microbiota and metabolite data in a well-characterized cohort with clinically-measured BP. Moreover, the rich sociodemographic and behavioral data of the CHNS allowed us to account for a wide range of potential confounders. The low treatment rate for hypertension ensured sufficient sample size and large variation in BP, even after excluding people who took antihypertension medication to minimize medication effects. However, we cannot infer a causal relationship between gut microbiota, host metabolome, and BP due to the cross-sectional design, and our microbial 16S rRNA data did not provide functional information. Future studies are needed to confirm our findings, particularly, population-based studies with repeated measures paired with experimental studies to investigate the causal biological pathways modulating BP.
Supplementary Material
PERSPECTIVES.
Our study provides substantial observational evidence for the associations between gut microbiota and plasma metabolites with BP in a population-based cohort of middle-aged Chinese adults. The overall microbial community varied by BP. Several individual metabolites (e.g., lignoceroyl sphingomyelin, cerotoylcarnitine, and dihomolinolenate) and an overall lipid metabolite pattern characterized by long-chain fatty acids were positively associated with BP, suggesting a role of circulating lipids in hypertension. Further analyses with longitudinal data and refined microbial composition data in larger samples are needed to fully elucidate the causal relationship between gut microbiota, host metabolites, and BP, thereby informing effective early interventions and treatments for hypertension.
NOVELTY AND SIGNIFICANCE.
What Is New?
Our study fills the gap of lacking population-based studies investigating both gut microbiota and circulating metabolomics in association with blood pressure (BP).
Our sample is unique in that may participants with hypertension were untreated, allowing us to minimize the medication effects.
What Is Relevant?
Our findings support a difference in the overall gut microbiota by BP.
We identified a novel lipid pattern and several lipid metabolites (e.g., sphingomyelins, acyl-carnitines) positively associated with BP.
ACKNOWLEDGEMENTS
We thank Dr. Misa Graff and Dr. Kari North for assistance with methodology, and Dr. Shufa Du, Ms. Guifeng Jin, and Dr. Hsiao-Chuan Tien for database assistance.
SOURCES OF FUNDING:
This work was supported by the NIH and National Institute of Diabetes and Digestive and Kidney Diseases (R01- DK104371). We are grateful to the Eunice Kennedy Shriver National Institute of Child Health and Human Development (NICHD) for R01 HD30880 and the NIH Fogarty grant D43 TW009077 for financial support for the CHNS data collection and analysis files from 1989 to 2015 and future surveys. We are also grateful for funding from the NICHD to Carolina Population Center (CPC) at the UNC-CH (NIH grant P2C HD050924). We thank the National Institute for Nutrition and Health (NINH) and China Center for Disease Control and Prevention (CCDC). Y.W is grateful to the Sanofi funding award 29230-50347-466001 for educational support. M.C.B.T is supported by Ruth L. Kirschstein National Research Service Awards (NRSA) Genetic Epidemiology of Heart, Lung, and Blood (HLB) Traits Training Grant (GenHLB, T32HL129982) funded by National Heart, Lung, and Blood Institute (NHLBI). We are grateful to the National Center for Advancing Translational Sciences (NCATS), National Institutes of Health, for pilot funding through Grant Award Number UL1TR002489.
Footnotes
DISCLOSURES
None.
REFERENCES
- 1.Benjamin EJ, Muntner P and Bittencourt MS. Heart disease and stroke statistics-2019 update: a report from the American Heart Association. Circulation. 2019;139:e56–e528. [DOI] [PubMed] [Google Scholar]
- 2.Mills KT, Bundy JD, Kelly TN, Reed JE, Kearney PM, Reynolds K, Chen J and He J. Global burden of hypertension: analysis of population-based studies from 89 countries. Journal of Hypertension. 2015;33:e2. [Google Scholar]
- 3.Fryar CD, Ostchega Y, Hales CM, Zhang G and Kruszon-Moran D. Hypertension prevalence and control among adults: United States, 2015–2016. 2017. [PubMed]
- 4.Lu X, Wang L, Lin X, Huang J, Charles Gu C, He M, Shen H, He J, Zhu J and Li H. Genome-wide association study in Chinese identifies novel loci for blood pressure and hypertension. Human molecular genetics. 2014;24:865–874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.He J, Gu D, Chen J, Jaquish CE, Rao DC, Hixson JE, Chen J-c, Duan X, Huang J-f and Chen C-S. Gender difference in blood pressure responses to dietary sodium intervention in the GenSalt study. Journal of hypertension. 2009;27:48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Rothschild D, Weissbrod O, Barkan E, Kurilshikov A, Korem T, Zeevi D, Costea PI, Godneva A, Kalka IN and Bar N. Environment dominates over host genetics in shaping human gut microbiota. Nature. 2018;555:210–215. [DOI] [PubMed] [Google Scholar]
- 7.Long T, Hicks M, Yu H-C, Biggs WH, Kirkness EF, Menni C, Zierer J, Small KS, Mangino M and Messier H. Whole-genome sequencing identifies common-to-rare variants associated with human blood metabolites. Nature genetics. 2017;49:568. [DOI] [PubMed] [Google Scholar]
- 8.Yang T, Richards EM, Pepine CJ and Raizada MK. The gut microbiota and the brain–gut–kidney axis in hypertension and chronic kidney disease. Nature Reviews Nephrology. 2018;14:442–456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Pluznick J A novel SCFA receptor, the microbiota, and blood pressure regulation. Gut microbes. 2014;5:202–207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Mell B, Jala VR, Mathew AV, Byun J, Waghulde H, Zhang Y, Haribabu B, Vijay-Kumar M, Pennathur S and Joe B. Evidence for a link between gut microbiota and hypertension in the Dahl rat. Physiological genomics. 2015;47:187–197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Yang T, Santisteban MM, Rodriguez V, Li E, Ahmari N, Carvajal JM, Zadeh M, Gong M, Qi Y and Zubcevic J. Gut dysbiosis is linked to hypertension. Hypertension. 2015;65:1331–1340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Morris D, Latif S, Hardy M and Brem A. Endogenous inhibitors (GALFs) of 11β-hydroxysteroid dehydrogenase isoforms 1 and 2: Derivatives of adrenally produced corticosterone and cortisol. The Journal of steroid biochemistry and molecular biology. 2007;104:161–168. [DOI] [PubMed] [Google Scholar]
- 13.Li J, Zhao F, Wang Y, Chen J, Tao J, Tian G, Wu S, Liu W, Cui Q and Geng B. Gut microbiota dysbiosis contributes to the development of hypertension. Microbiome. 2017;5:14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Silveira-Nunes G, Durso DF, Cunha EHM, Maioli TU, Vieira AT, Speziali E, Corrêa-Oliveira R, Martins-Filho OA, Teixeira-Carvalho A and Franceschi C. Hypertension Is Associated With Intestinal Microbiota Dysbiosis and Inflammation in a Brazilian Population. Frontiers in pharmacology. 2020;11:258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Menni C, Graham D, Kastenmüller G, Alharbi NH, Alsanosi SM, McBride M, Mangino M, Titcombe P, Shin S-Y and Psatha M. Metabolomic identification of a novel pathway of blood pressure regulation involving hexadecanedioate. Hypertension. 2015;66:422–429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Zheng Y, Yu B, Alexander D, Mosley TH, Heiss G, Nettleton JA and Boerwinkle E. Metabolomics and incident hypertension among blacks: the atherosclerosis risk in communities study. Hypertension. 2013:HYPERTENSIONAHA. 113.01166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Adnan S, Nelson JW, Ajami NJ, Venna VR, Petrosino JF, Bryan RM Jr and Durgan DJ. Alterations in the gut microbiota can elicit hypertension in rats. Physiological genomics. 2016;49:96–104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Sun S, Lulla A, Sioda M, Winglee K, Wu MC, Jacobs DR Jr, Shikany JM, Lloyd-Jones DM, Launer LJ and Fodor AA. Gut Microbiota Composition and Blood Pressure: The CARDIA Study. Hypertension. 2019;73:998–1006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Wang Z, Chen Z, Zhang L, Wang X, Hao G, Zhang Z, Shao L, Tian Y, Dong Y and Zheng C. Status of hypertension in China: results from the China Hypertension Survey, 2012–2015. Circulation. 2018;137:2344–2356. [DOI] [PubMed] [Google Scholar]
- 20.Xi B, Liang Y, Reilly KH, Wang Q, Hu Y and Tang W. Trends in prevalence, awareness, treatment, and control of hypertension among Chinese adults 1991–2009. International journal of cardiology. 2012;158:326–329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Popkin BM, Du S, Zhai F and Zhang B. Cohort Profile: The China Health and Nutrition Survey—monitoring and understanding socio-economic and health change in China, 1989–2011. International journal of epidemiology. 2009;39:1435–1440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Chobanian AV, Bakris GL, Black HR, Cushman WC, Green LA, Izzo JL Jr, Jones DW, Materson BJ, Oparil S and Wright JT Jr. Seventh report of the joint national committee on prevention, detection, evaluation, and treatment of high blood pressure. hypertension. 2003;42:1206–1252. [DOI] [PubMed] [Google Scholar]
- 23.Unger T, Borghi C, Charchar F, Khan NA, Poulter NR, Prabhakaran D, Ramirez A, Schlaich M, Stergiou GS and Tomaszewski M. 2020 International Society of Hypertension global hypertension practice guidelines. Hypertension. 2020;75:1334–1357. [DOI] [PubMed] [Google Scholar]
- 24.Caporaso JG, Kuczynski J, Stombaugh J, Bittinger K, Bushman FD, Costello EK, Fierer N, Pena AG, Goodrich JK and Gordon JI. QIIME allows analysis of high-throughput community sequencing data. Nature methods. 2010;7:335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Haas BJ, Gevers D, Earl AM, Feldgarden M, Ward DV, Giannoukos G, Ciulla D, Tabbaa D, Highlander SK and Sodergren E. Chimeric 16S rRNA sequence formation and detection in Sanger and 454-pyrosequenced PCR amplicons. Genome research. 2011;21:494–504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Sumner LW, Amberg A, Barrett D, Beale MH, Beger R, Daykin CA, Fan TW-M, Fiehn O, Goodacre R and Griffin JL. Proposed minimum reporting standards for chemical analysis. Metabolomics. 2007;3:211–221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Jones-Smith JC and Popkin BM. Understanding community context and adult health changes in China: development of an urbanicity scale. Social science & medicine. 2010;71:1436–1446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Yao M, Lichtenstein A, Roberts S, Ma G, Gao S, Tucker K and McCrory M. Relative influence of diet and physical activity on cardiovascular risk factors in urban Chinese adults. International journal of obesity. 2003;27:920. [DOI] [PubMed] [Google Scholar]
- 29.Batis C, Gordon-Larsen P, Cole SR, Du S, Zhang B and Popkin B. Sodium intake from various time frames and incident hypertension among Chinese adults. Epidemiology (Cambridge, Mass). 2013;24:410–418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Levey AS, Stevens LA, Schmid CH, Zhang YL, Castro AF, Feldman HI, Kusek JW, Eggers P, Van Lente F and Greene T. A new equation to estimate glomerular filtration rate. Annals of internal medicine. 2009;150:604–612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Oksanen J, Kindt R, Legendre P, O’Hara B, Stevens M, Oksanen M and Suggests M. The vegan package: community ecology package. R package version. 2007;1:15–11. [Google Scholar]
- 32.Peet RK. The measurement of species diversity. Annual review of ecology and systematics. 1974;5:285–307. [Google Scholar]
- 33.Faith DP, Minchin PR and Belbin L. Compositional dissimilarity as a robust measure of ecological distance. Vegetatio. 1987;69:57–68. [Google Scholar]
- 34.Anderson MJ. Permutational multivariate analysis of variance (PERMANOVA). Wiley StatsRef: Statistics Reference Online. 2014:1–15. [Google Scholar]
- 35.Winglee K, Howard AG, Sha W, Gharaibeh RZ, Liu J, Jin D, Fodor AA and Gordon-Larsen P. Recent urbanization in China is correlated with a Westernized microbiome encoding increased virulence and antibiotic resistance genes. Microbiome. 2017;5:121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Popkin BM and Du S. Dynamics of the nutrition transition toward the animal foods sector in China and its implications: a worried perspective. The Journal of nutrition. 2003;133:3898S–3906S. [DOI] [PubMed] [Google Scholar]
- 37.Du S, Neiman A, Batis C, Wang H, Zhang B, Zhang J and Popkin BM. Understanding the patterns and trends of sodium intake, potassium intake, and sodium to potassium ratio and their effect on hypertension in China–. The American journal of clinical nutrition. 2013;99:334–343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Sekula P, Goek O-N, Quaye L, Barrios C, Levey AS, Römisch-Margl W, Menni C, Yet I, Gieger C and Inker LA. A metabolome-wide association study of kidney function and disease in the general population. Journal of the American Society of Nephrology. 2016;27:1175–1188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Antonelli J, Claggett BL, Henglin M, Kim A, Ovsak G, Kim N, Deng K, Rao K, Tyagi O and Watrous JD. Statistical workflow for feature selection in human metabolomics data. Metabolites. 2019;9:143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Conway JM and Huffcutt AI. A review and evaluation of exploratory factor analysis practices in organizational research. Organizational research methods. 2003;6:147–168. [Google Scholar]
- 41.McDonald JH. Handbook of biological statistics: sparky house publishing; Baltimore, MD; 2009. [Google Scholar]
- 42.Pedregosa F, Varoquaux G, Gramfort A, Michel V, Thirion B, Grisel O, Blondel M, Prettenhofer P, Weiss R and Dubourg V. Scikit-learn: Machine learning in Python. Journal of machine learning research. 2011;12:2825–2830. [Google Scholar]
- 43.Dietterich TG. Approximate statistical tests for comparing supervised classification learning algorithms. Neural computation. 1998;10:1895–1923. [DOI] [PubMed] [Google Scholar]
- 44.Benjamini Y and Hochberg Y. Controlling the false discovery rate: a practical and powerful approach to multiple testing. Journal of the Royal statistical society: series B (Methodological). 1995;57:289–300. [Google Scholar]
- 45.Holmes E, Loo RL, Stamler J, Bictash M, Yap IK, Chan Q, Ebbels T, De Iorio M, Brown IJ and Veselkov KA. Human metabolic phenotype diversity and its association with diet and blood pressure. Nature. 2008;453:396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Chen Y-Y, Chen D-Q, Chen L, Liu J-R, Vaziri ND, Guo Y and Zhao Y-Y. Microbiome–metabolome reveals the contribution of gut–kidney axis on kidney disease. Journal of translational medicine. 2019;17:1–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Kindt A, Liebisch G, Clavel T, Haller D, Hörmannsperger G, Yoon H, Kolmeder D, Sigruener A, Krautbauer S and Seeliger C. The gut microbiota promotes hepatic fatty acid desaturation and elongation in mice. Nature communications. 2018;9:1–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Cheema MU and Pluznick JL. Gut microbiota plays a central role to modulate the plasma and fecal metabolomes in response to angiotensin II. Hypertension. 2019;74:184–193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Pallister T, Jackson MA, Martin TC, Zierer J, Jennings A, Mohney RP, MacGregor A, Steves CJ, Cassidy A and Spector TD. Hippurate as a metabolomic marker of gut microbiome diversity: Modulation by diet and relationship to metabolic syndrome. Scientific reports. 2017;7:1–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Chakraborty S, Galla S, Cheng X, Yeo J-Y, Mell B, Singh V, Yeoh B, Saha P, Mathew AV and Vijay-Kumar M. Salt-responsive metabolite, β-hydroxybutyrate, attenuates hypertension. Cell reports. 2018;25:677–689. e4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Mels CM, Schutte AE, Erasmus E, Huisman HW, Schutte R, Fourie CM, Kruger R, Van Rooyen JM, Smith W and Malan NT. L-carnitine and long-chain acylcarnitines are positively correlated with ambulatory blood pressure in humans: the SABPA study. Lipids. 2013;48:63–73. [DOI] [PubMed] [Google Scholar]
- 52.Adams SH, Hoppel CL, Lok KH, Zhao L, Wong SW, Minkler PE, Hwang DH, Newman JW and Garvey WT. Plasma acylcarnitine profiles suggest incomplete long-chain fatty acid β-oxidation and altered tricarboxylic acid cycle activity in type 2 diabetic African-American women. The Journal of nutrition. 2009;139:1073–1081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Harris WS and Shearer GC. Omega-6 fatty acids and cardiovascular disease: friend, not foe? 2014. [DOI] [PubMed]
- 54.Chaurasia B and Summers SA. Ceramides–lipotoxic inducers of metabolic disorders. Trends in Endocrinology & Metabolism. 2015;26:538–550. [DOI] [PubMed] [Google Scholar]
- 55.Summers SA, Chaurasia B and Holland WL. Metabolic Messengers: ceramides. Nature Metabolism. 2019:1–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Havulinna AS, Sysi-Aho M, Hilvo M, Kauhanen D, Hurme R, Ekroos K, Salomaa V and Laaksonen R. Circulating ceramides predict cardiovascular outcomes in the population-based FINRISK 2002 cohort. Arteriosclerosis, thrombosis, and vascular biology. 2016;36:2424–2430. [DOI] [PubMed] [Google Scholar]
- 57.Laaksonen R, Ekroos K, Sysi-Aho M, Hilvo M, Vihervaara T, Kauhanen D, Suoniemi M, Hurme R, März W and Scharnagl H. Plasma ceramides predict cardiovascular death in patients with stable coronary artery disease and acute coronary syndromes beyond LDL-cholesterol. European heart journal. 2016;37:1967–1976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Poss AM, Maschek JA, Cox JE, Hauner BJ, Hopkins PN, Hunt SC, Holland WL, Summers SA and Playdon MC. Machine learning reveals serum sphingolipids as cholesterol-independent biomarkers of coronary artery disease. The Journal of Clinical Investigation. 2020;130. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.