

Abstract
Invasive lobular breast carcinoma (ILC), one of the major breast cancer histological subtypes, exhibits unique features compared to the well-studied ductal cancer subtype (IDC). The pathognomonic feature of ILC is loss of E-cadherin, mainly caused by inactivating mutations, but the contribution of this genetic alteration to ILC-specific molecular characteristics remains largely understudied. To profile these features transcriptionally, we conducted single-cell-RNA-sequencing on a panel of IDC and ILC cell lines, and an IDC cell line (T47D) with CRISPR-Cas9-mediated E-cadherin knock out (KO). Inspection of intra-cell line heterogeneity illustrated genetically and transcriptionally distinct subpopulations in multiple cell lines and highlighted rare populations of MCF7 cells highly expressing an apoptosis-related signature, positively correlated with a pre-adaptation signature to estrogen deprivation. Investigation of E-cadherin KO-induced alterations showed transcriptomic membranous systems remodeling, elevated resemblance to ILCs in regulon activation, and increased sensitivity to IFN-γ mediated growth inhibition via activation of IRF1. This study reveals single cell transcriptional heterogeneity in breast cancer cell lines and provides a resource to identify drivers of cancer progression and drug resistance.
Keywords: scRNA-seq, cell lines, heterogeneity, E-cadherin, lobular breast cancer
Introduction
Among subtyping systems of breast cancer, histological classification remains an essential criterion due to distinctive features of the major two subtypes—invasive lobular breast carcinoma (ILC) and invasive ductal breast carcinoma (IDC). ILC is the 6th most common cancer in women, with an estimated 40,000 new cases in 2019, despite accounting for a smaller proportion of breast cancer cases (~15%) compared to IDC (~75%)(1). ILC shows distinct signaling in pathways essential for breast cancer growth and proliferation compared to IDC – such as the WNT4 signaling in response to estrogen stimulus or blockade(2,3), increased PI3K/Akt signaling(4,5), enhanced IGF1-IGF1R activation(6), and dependency on ROS1(7), which suggest that ILC could benefit from unique treatment strategies. The most distinguishing molecular feature of ILC is loss of E-cadherin, largely arising from inactivating CDH1 mutations. E-cadherin loss disrupts adherens junctions(8) and leads to cells with a smaller and rounder morphology, a more scattered alignment within tumor stroma, and greater metastatic tropisms to ovaries, peritoneum or gastrointestinal (GI) tracts compared to IDC(9). Such loss often couples with other molecular features, including the aberrant cytosolic localization of p120(10). Meanwhile, E-cadherin-null tumor models also exhibit certain ILC resemblance: in vivo, the TP53 CDH1 dual KO mouse model showed elevated anoikis resistance and angiogenesis as well as GI tract or peritoneum dissemination similarly to human cases(11); while in vitro, hypersensitized PI3K/Akt signaling via GFR-dependent response was identified in both human and mouse ILC cell lines compared to their E-cadherin positive counterparts(4). Despite numerous clinical observations and biological models, it is currently unclear how E-cadherin loss leads to many of the lobular-specific features.
Intra-tumor heterogeneity (ITH) is a hallmark of treatment resistance and mortality in cancer(12). Multiple genetically distinct populations of cancer cells within the same tumor – typically arising from a series of mutational events—are dynamically selected by both intrinsic and external pressures and potentially preserve subclones with high invasiveness and/or drug resistance(13–16). In addition to genetic diversity, transcriptional heterogeneity is also a major driver of ITH in multiple cancer types(17–19). Such transcriptional variation, defined as cell states, appear transient and flexible in response to environmental stimuli while partially influenced by DNA alterations. Although ITH is frequently considered under in-vivo context, previous studies have shown there is considerable heterogeneity even for cell lines grown in culture. However, the extent of this intra-cell line heterogeneity in breast cancer models has not yet been comprehensively characterized(20,21).
To quantify ITH between cell lines, referred to as ICH (Inter-Cellular Heterogeneity), and investigate differences between IDC and ILC, we performed single cell RNA sequencing (scRNA-seq) on a panel of eight cell lines. We first investigated ICH in general: most cell lines consist of genetic subclones with unique copy number alterations (CNAs). Transcriptomic heterogeneity was shown for MCF7 cells specifically, revealing that it is dominated by cell cycle, in which cells dynamically transit through several well-defined phases. Despite the majority of cycling cells, a rare population exists distinctively outside the cell cycle with a non-transiting ‘dormant’ state. Characterization of such ‘outliers’ uncovered a unique apoptotic signature, which correlates with other functionally related signatures of dormancy in other cell lines and tumors.
We further inspected transcriptomic alterations induced by loss of E-cadherin, using CRISPR-mediated CDH1 KO in a commonly used IDC line, T47D. Simple deletion of CDH1 caused cells to cluster independently from wild type (WT) T47D cells in two-dimensional (2D) UMAP embedding. Given such distinctive and systemic differences are likely mediated by transcriptional factors (TFs)(22), we deduced regulon activation states from scRNA-seq data, which illustrated elevated resemblance towards two out of the three ILCs in CDH1 KO versus WT cells. Among the TFs identified, we found a regulon of IRF1 activated by CDH1 KO, which also show higher expression in luminal A ILC tumors than IDCs. While the mechanism whereby loss of E-cadherin activates IRF1 is not known, IRF1 regulon activation conforms to the less-proliferative, and potentially more immune-enriched(23) features of the lobular subtype.
Materials and Methods
This work doesn’t involve human subject and thus no written informed consent was required. Additional specific details of materials and methods are provided in Supplementary Materials
Generation of T47D and MCF7 CDH1 KO cells
Knockout of CDH1 was performed using CRISPR-Cas9 with the Gene Knockout Kit (V1) from Synthego (Redwood City, California). Four potential sgRNAs (1.CCGGTGTCCCTGGGCGGAGT, 2.CCTCTCTCCAGGTGGCGCCG, 3.GGCGTCAAAGCCAGGGTGGC, 4.CTCTTGGCTCTGCCAGGAGC) were selected based on sequence screening to target exons or introns of CDH1 and introduce a protein truncating indel. sgRNA 2 population resulted in the most complete protein depletion than other pools, and was thus chosen based on the sequencing results; demonstrating a 1bp deletion at exon 2 (c.321delC, in NM_004360.5), which caused a frameshift with a pre-mature stop codon at exon 16. To select pure KO clones, the sgRNA 2 cell population was single cell sorted. Clones were expanded, and knockout success was examined by Sanger sequencing and immunoblot. 8 clones with the least E-cadherin protein expression by immunoblot were then pooled in equal ratio and named T47D CDH1 KO. Images of KO and parental WT cells were obtained with 10X bright field using Olympus IX83 Inverted Microscope. MCF7 CDH1 KO cells were generated using the same method(sgRNA2) with a mixture of 8 single cell KO clones.
Cell line preparation
MCF7 (RRID: CVCL_0031), T47D (RRID: CVCL_0553), MDA-MB-134-VI (MM134, RRID: CVCL_0617), MCF10A (ATCC Cat# CRL-10317, RRID:CVCL_0598) and HEK293T (RRID: CVCL_0063) were all purchased from American Type Culture Collection (ATCC). SUM44-PE cells (SUM44, CVCL_3424) were purchased from Asterand and BCK4 cells were kindly provided by Britta Jacobsen, University of Colorado Anschutz, CO. MCF7 with ESR1-Y537S were generated previously(24). Cell identity was authenticated by by Short Tandem Repeat DNA profiling (University of Arizona), and cells were routinely tested to be mycoplasma free. Cells were maintained in continuous culture for less than 6 months with media indicated in supplementary materials.
Single-cell RNA sequencing
Nine groups of cells, each with viability > 90% based upon Trypan Blue staining and Invitrogen automated cell counting, were fixed separately at equal number (around 5–10 million cells per group) in 90% methanol at 4°C for 15 minutes and temporarily stored at −80°C, following the standard protocol from 10X website (https://assets.ctfassets.net/an68im79xiti/7rsw40AVqX3ZXwIl7MDj85/fb7ac4e1b324827f5b738ade5a02b650/CG000136_Demonstrated_Protocol_MethanolFixationCells_RevE.pdf). The cell suspension was rehydrated, mixed and processed following 10X Genomics 3’ Chromium v3.0 protocol at University of Pittsburgh Genomics Core. The library was sequenced with NovaSeq 6000 S1 flow cell at the UPMC Genome Center, getting around 400 million paired reads in total. 10,000 cells were targeted for sequencing from the rehydrated pool, and 8,953 cells were successfully demultiplexed after bioinformatics pre-processing in the unfiltered dataset from Cell Ranger html report.
scRNA-seq data pre-processing
Raw FASTQ data was aligned and quantified using GRCh38 reference with Cell Ranger (v3.0.2) (https://support.10xgenomics.com/single-cell-geneexpression/software/pipelines/latest/using/count) and velocyto CLI (v0.17.17)(25). The resulting loom files were loaded with scVelo (v0.1.25)(26) and processed with Scanpy (v1.4.4)(27). Doublet removal was performed using Srublet(28). Low quality cells and genes were filtered out by selecting cells expressing more than 2000 genes, having UMIs between 8000 and 10,000 with a mitochondrial gene percentage of less than 15%; and selecting genes with detectable expression in at least 2 cells, resulting in a final library of 4,614 cells and 21,888 genes.
From the filtered matrix, spliced and unspliced reads were normalized, converted to log scale, and imputed respectively by scVelo (v0.1.25)(26), using scvelo.pp.normalize_per_cell, scvelo.pp.log1p and scvelo.pp.moments. The top 30 principle components were calculated using the 3000 most variable genes. This was followed by dimensional reduction using UMAP (scanpy.tl.umap) and clustering with Louvain method (scanpy.tl.louvain, resolution=0.2), which demonstrated eight distinct clusters in 2D UMAP embedding.
Cell line identification
1,444 genes were present in all the three data sources (breast cell lines, HEK293T as reference cell lines; and scRNA-seq (3,000 genes by 4,614 cells) as query cells), which were selected for further analysis. Pearson correlation was calculated between each single cell in the 8 clusters and each reference cell line using log normalized counts. The reference with the most significantly right-skewed coefficient distribution was assigned to the query cluster. Every cluster was successfully assigned except cluster 2, which was by default BCK4. This was further confirmed by the high levels of MUC2 expression. WT and KO T47D cells were distinguished by CDH1 expression.
CNA Inference
Two external datasets with scRNA-seq data of cell lines were integrated with our data for CNA inference - MCF7 and T47D cells from Hong et al.(29), and three MCF7 strains (WT-3,4,5) from Ben-David et al.(20) A raw count matrix from Hong et al.(29) was directly downloaded from GSE122743 while FASTQ files from Ben-David et al.(20) were reprocessed to derive the count matrix, following steps described in the scRNA-seq data pre-processing section. The average expression vector of the in-house MCF10A cells was utilized as reference.
Expression count matrix of each cell strain was first log normalized, sorted by genes’ chromosomal coordinates, and then merged with others using commonly detected genes. 300 cells were randomly selected from each strain. Only chromosome arms containing more than 100 genes were kept, and single cell expression was averaged with a moving 100 gene window. This averaged expression was used to fit a linear regression model against the MCF10A reference vector, and the residual value was assigned as the inferred CNA.
Identify cell subclusters from inferred CNA
We adopted a similar method as reported by Kinker et al.(30). For each cell line, the log normalized expression matrix was centered for each gene and moving average of 100 gene window was calculated along the genome. For each chromosome arm, a gaussian mixture model (GMM, implemented in scikit-learn, RRID:SCR_002577, https://scikit-learn.org/stable/) was fitted to the distribution. Selected chromosome arms should best fit with more than one component in GMM, each of which having more than 20 cells with confidence higher than 95%. These selected chromosome arms were then used for hierarchical clustering to identify subcluster of cells (Euclidean distance, Ward method). The top three hierarchical branching were extracted and differentially colored as the potential CNA subpopulation.
Identify cell subclusters from RNA
RNA counts of each cell line were extracted from raw data, filtered, log normalized and imputed, using the top 5000 variable genes following steps described in the scRNA-seq data pre-processing section. Cells were laid out in a force-directed graph drawing implemented in Scanpy(27). Louvain clustering was conducted in each cell line with resolution=1 individually as an empirical value; and with resolution = 0.4 for MCF7, as to generate 3 clusters in correspondence to the 3 clusters via other classification methods.
Cell cycle scoring
Five lists of phase marker genes (M/G1, G1/S, S, G2, G2/M) of the cell cycle were obtained(31). Log normalized expression of each cell line was acquired as described in the last section. For each cell line and phase, correlation coefficients were calculated between each gene and the average gene set expression. Genes with the highest (the upper 40% quantile) coefficients were selected as the refined cell cycle markers for that cell line. A score for each phase was calculated for each cell, as the average expression of cell cycle markers minus that of a randomly selected gene set of the same size (implemented in Scanpy(27)), which is standardized firstly in each cell then in each phase by z-score. The phase with the highest score was assigned to the cell.
RNA velocity analysis
For each cell line, the union of the refined cell cycle markers were selected to illustrate cell state transitioning dynamics, following the RNA velocity pipeline implemented in scVelo(26) using stochastic mode, with latent time calculated under the same setting. Stream arrows indicating transition dynamics were depicted on the force-directed graph drawing of individual cell line.
NMF clustering
NMF was performed as described by Puram, et al.(17) in MCF7 cells. The log normalized expression matrix of 5000 genes and 977 cells was centered for each gene to obtain the relative expression (Ei,j) for every gene i in each single cell j. Negative values was replaced by 0 and NMF was then conducted with k ranging from 5 to 15 (implemented in sklearn.decomposition.NMF). For each k, top 50 genes with highest D score(32) were defined as a gene set. Recurrent gene sets, which have Jaccard Index > 0.6 with at least one another gene set, were selected and merged as indicative features (101 genes). Ei,j of these 101 genes among MCF7 cells were standardized for each cell by cell-wise z-score and showed in heatmap, clustered for both genes and cells (Euclidean distance, Ward method). Clades from the top three hierarchical branching were selected as NMF clusters.
Differentially expressed genes
Differentially expressed genes (DEGs) for each cell line were derived by comparing this cell line versus all other cells using Wilcoxon test (BH adjustment, implemented in Scanpy(27)). To calculate DEGs in T47D CDH1 KO versus WT cells, the two groups were extracted from raw data, filtered, log normalized and imputed, using the top 3000 variable genes following steps described in the scRNA-seq data pre-processing section. DEGs were calculated comparing between each other (Wilcoxon test, BH adjustment). The top 100 genes with the smallest FDR were selected as marker genes for each group.
Gene set enrichment analysis
Gene lists of interest (marker genes of MCF7 NMF clusters; T47D WT and KO DEGs) were submitted to the gProfiler website(33) (RRID:SCR_006809, https://biit.cs.ut.ee/gprofiler/) for pathway enrichment analysis using over representation test with default parameters. Enriched terms of GO dataset (Biological Process and Cellular Component) were selected as input in Cytoscape (v3.7.1, RRID:SCR_003032)(34) Enrichment Map, in GeneMANIA Force Directed Layout (similarity_coefficient mode) colored by FDR.
GSVA scoring in TCGA and METABRIC
Expression count matrix of TCGA breast cancer dataset was obtained from cBioportal(35,36) (meta_RNA_Seq_v2_expression_median.txt) and log normalized as for sample i and gene j; corresponding breast cancer subtypes (PAM50, histology) were obtained from Ciriello et al.(37) METABRIC(38) RNA microarray data was downloaded from Synapse (RRID:SCR_006307) and log normalized. Gene set variation analysis (GSVA) of selected gene set was performed with GSVA R package(39), in ssgsea or gsva mode with default parameters. Comparisons between TCGA ILC versus IDC cases are all limited to LumA population unless otherwise specified.
For survival analysis, only estrogen receptor positive (ER+) and LumA patients were selected to avoid influence caused alone by PAM50 subtype. DormSig ssGSEA scores were stratified into low and high levels with optimized threshold using in surv_cutpoint and surv_categorize implemented in R survminer package (https://cran.r-project.org/web/packages/survminer/index.html). Kaplan-Meier curve were plotted for each group using disease free survival, with p value from log-rank test, and hazard ratio (HR) calculated using univariate Cox regression (coxph in R survival package, https://cran.r-project.org/web/packages/survival/index.html).
CDH1 exon and intron coverage
TCGA RNA BAM files were accessed from The Pittsburgh Genome Resource Repository (https://www.pgrr.pitt.edu/). Bulk RNA-seq BAM files of breast cancer cell lines were generated as described in Tasdemir et al.(40) RNA counts of each exon and intron of CDH1 was quantified with bedtools (v2.29.1, RRID:SCR_006646) counts mode and normalized by dividing the total number of counts within the sample. One representative ILC and IDC patient from TCGA(37), along with one cell sample from each cell line, were selected for visualization using Integrative Genome Viewer (https://software.broadinstitute.org/software/igv/), showing CDH1 coverage from intron 11 to exon16 with GRCh38 as reference genome.
Regulon activation profiles
Regulon activities were inferred from raw expression counts of each cell line following the default pySCENIC(41) pipeline (https://github.com/aertslab/pySCENIC). Each cell was eventually assigned with an AUC score for every regulon, indicating its activation status. These scores were then binarized into either “on” or “off” states, by setting an optimized threshold on the distribution of each regulon among all cells using the skimage.filters.threshold_minimum function. Target genes of each regulon were selected if it is commonly deduced under that TF in at least two of the following cell lines: T47D KO, MDA-MB-134-VI, SUM44-PE and BCK4 (Table S3).
Jaccard index
For every pair of cells, a Jaccard index was calculated using the two corresponding binarized regulon activation vector of the two cells, X and Y, as , a large value of which indicates higher similarity. For every two cell lines, the Jaccard index of all pairwise combinations between the two population was selected and plotted as the cumulative distribution.
Statistical analyses
All bioinformatic analyses were conducted in Python (v3.7, RRID:SCR_001658, RRID:SCR_002918) (http://www.python.org) or R (v3.6) (www.r-project.org). All statistical tests are two-sided, unless specified otherwise.
Code availability
Codes and important intermediate data will be available on github (https://github.com/leeoesterreich/scRNA-CellLines) upon publication.
Data availability
Processed files are deposited in Gene Expression Omnibus (RRID:SCR_005012) (GSE144320) and raw FASTQ files are deposited at SRP245420, which will be available upon publication.
Results
scRNA-seq of breast cancer cell lines
To investigate the effect of loss of E-cadherin in ILC, we generated T47D cells with CDH1 KO using CRISPR-Cas9, and ensured its depletion at the protein level. scRNA-seq was performed on this T47D KO strain and its parental WT strain, along with seven additional groups of cells: the IDC cell line MCF7, MCF7 with ESR1 Y537S mutation, referred to as MCF7-mut; three ILC cell lines: MDA-MB-134-VI, SUM44-PE, BCK4; as well as two immortalized but non-cancerous cell lines: the breast MCF10A, and human embryonic kidney 293 (HEK293T) cells. Each cell line was cultured separately in standardized conditions, mixed at similar number for standard 10X chemistry v3 library preparation, and sequenced with NovaSeq 6000 system (Fig. 1a).
Fig. 1. scRNA-seq of breast cancer and non-cancerous cell lines.
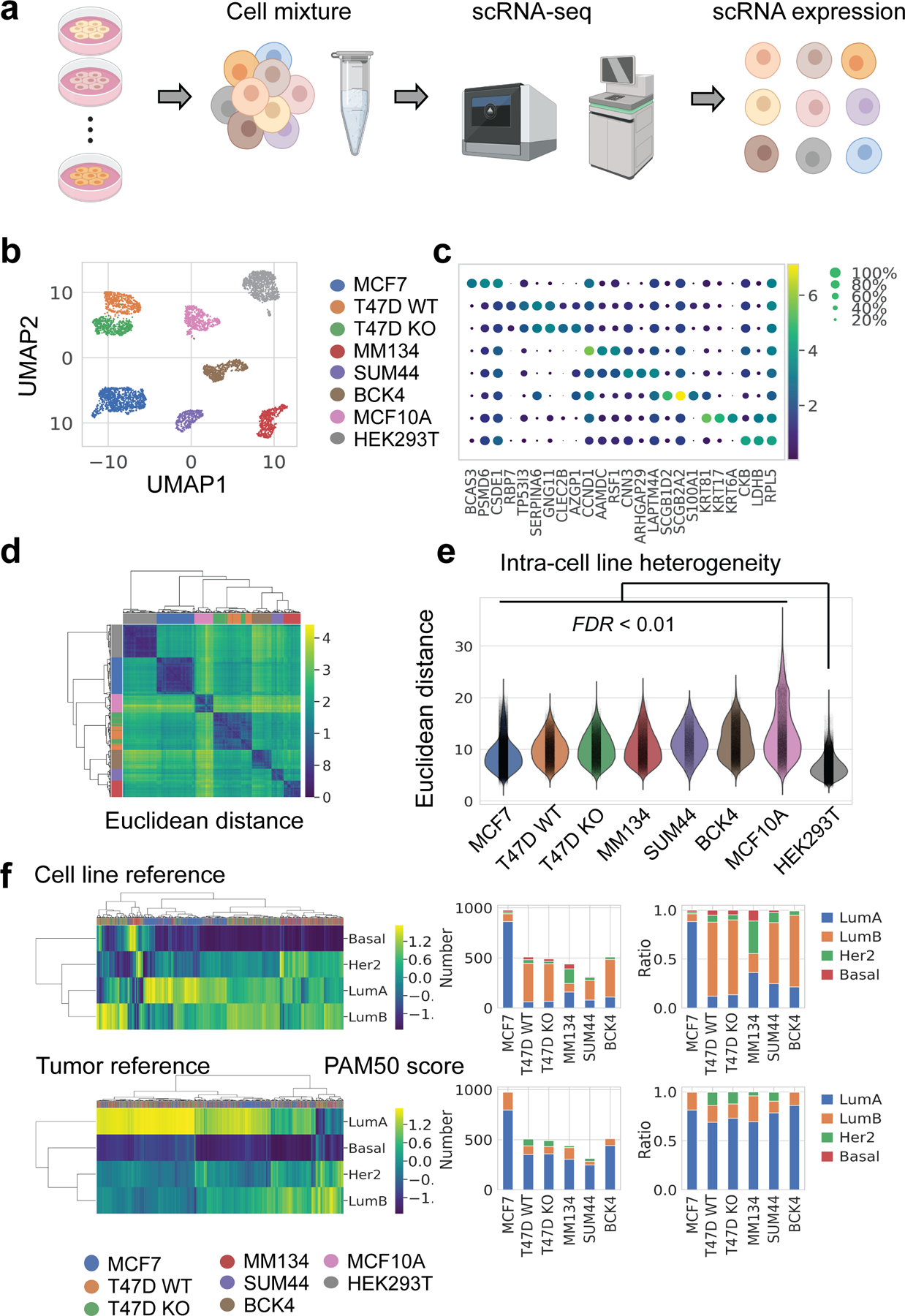
a. Schematic pipeline of scRNA-seq.
b. UMAP embeddings of 4,614 single cells in 8 clusters. Deconvoluted cell line identities are displayed in the same row as c. Number of cells: MCF7 (n=977), T47D WT (n=509), T47D KO (n=491), MM134 (n=439), SUM44 (n=314), BCK4 (n=512), MCF10A (n=491), HEK293T(n=881).
c. Marker gene expression of each cell line. Top three differentially expressed genes were plotted for each cell cluster which had the smallest FDR when compared with all other clusters (Wilcoxon test, Benjamini-Hochberg (BH) adjustment). Every dot is colored by average expression of the gene and sized by the fraction of cells expressing the gene within that cell line.
d. Hierarchical clustering (Euclidean distance, Ward’s method) of intercellular distances. xi,j in the matrix represents the Euclidean distance between cell i and cell j using the top 30 principle components from the original expression matrix. Corresponding cell lines are colored on side bars, with the same color label as in b, c.
e. Intercellular distances between every two single cells (calculated as Euclidean distance in d) within cell lines. Intra-cell line distances of all breast cell lines were higher than HEK293T (FDR < 0.01, Wilcoxon test, BH adjustment).
f. Prediction Analysis of Microarray 50 (PAM50) subtypes scores (left) and assignment (right) of every single cell, using typical cell lines (upper) or estrogen-positive tumors (lower) as reference. Corresponding cell lines are colored on top bar of heatmap. Bar plots showed both absolute number of cells or the ratio of each PAM50 subtypes. LumA: luminal A, LumB: luminal B, Her2: HER2-enriched, Basal: Basal-like.
Dimensional reduction in 2D UMAP revealed eight distinct clusters (Fig. 1b,c). We deconvoluted single cell identities by mapping transcriptomes of each cluster to six cell lines with available bulk-RNA reference. The scRNA-seq library showed good quality (Fig. S1a). Except for cluster 2, every cluster showed distinctive similarity to a specific bulk transcriptome, and thus identity was confidently assigned (Fig. S1b,c). Cluster 2 was by default assigned as BCK4, which has no bulk RNA-seq data, and this identity was further confirmed by the exclusively high mucin expression in this cell line (Fig. S1d). T47D CDH1 KO and WT cells were two proximal but discrete clusters and showed altered E-cadherin expression as expected (Fig. S1d). In contrast, the MCF7-mut and WT cells, despite being equally mixed, did not cluster separately, indicating limited transcriptomic differences when grown in standard media without estrogen deprivation. As these cells couldn’t be separated, we refer to them hereafter as MCF7 cells. After data pre-processing, the final single cell library consisted of 4,614 cells, approximately 500 cells for each cluster except MCF7 and HEK293T, both of which contain approximately 900 cells.
Expression of key breast cancer genes were examined (Fig. S1d), including hormone receptors (ESR1 for estrogen receptor, PGR for progesterone receptor, ERBB2 for human epidermal growth factor receptor 2), histology marker (CDH1 for E-cadherin) and proliferation indicator (MKI67 for Ki67). Consistent with the previous characterizations(42,43), ESR1 was expressed higher in the six breast cancer cell lines compared to MCF10A or HEK293T; PGR showed high expression in T47D and BCK4 cells and low to medium in others; and ERBB2 expression was higher in BCK4 than other cell lines. Consistent with histological classification, CDH1 was highly expressed in IDC cell lines (MCF7, T47D WT) and MCF10A, compared to ILC cell lines or HEK293T. All cell lines had abundant expression of MKI67. Despite cell-line specific expression, all markers showed a large variation in RNA abundance. Such heterogeneity is also reflected in PAM50 subtypes, calculated for each single cell with the subgroup-specific gene-centering method(44) (Fig. 1f) – each cell line exhibits several PAM50 calls in spite of the luminal subtype dominance.
To quantify inter and intra-cell line differences, we calculated Euclidean distance among all pairs of single cells (Fig. 1d). Cells from each cell line showed greater similarity to each other, and T47D WT and KO were highly similar to each other. This analysis highlighted the intrinsic inter-cell line distinctions. Intra-cell line distances were also selected and compared, revealing intra-cell line heterogeneity which was highest in MCF10A, relatively consistently among breast cancer cells, and lowest in HEK293T (Fig. 1e).
Inferred copy number aberrations (CNAs) reveal intra-cell line subpopulations that in part account for transcriptional heterogeneity
Cell lines are the most widely used laboratory model of cancer. However, studies have shown dissimilarities among breast cancer cell lines from different laboratories, potentially as a result of different culture conditions and/or intrinsic evolution of cells with genomic instability(20). Even within a single cell line, transcriptomic subpopulations exist – composing a small or median proportion of the whole population, and are only partially explained by CNA(21).
We examined genetic heterogeneity in cell lines using CNA inferred from scRNA-seq, a method described in multiple previous studies(17,29,45). Specifically, we followed calculation steps from Patel et. al. (45). To investigate the accuracy of this method, we extracted the real copy number of MCF7 and T47D cells from CCLE (46), the only two cell lines with available SNP array data. The inferred copy number from scRNA-seq recaptured the distinctive gain or losses of real copy number in general (Fig. 2a). We further tested the robustness of our data by integrating with public scRNA-seq data - T47D cells (29), and three MCF7 strains (20), each in standard media culture using Harmony(47). In both conditions, the external cell cluster correctly overlies with the in-house cluster of the same cell line identity (Fig. S2f).
Fig. 2. Intra-cell line subpopulations from inferred CNA.
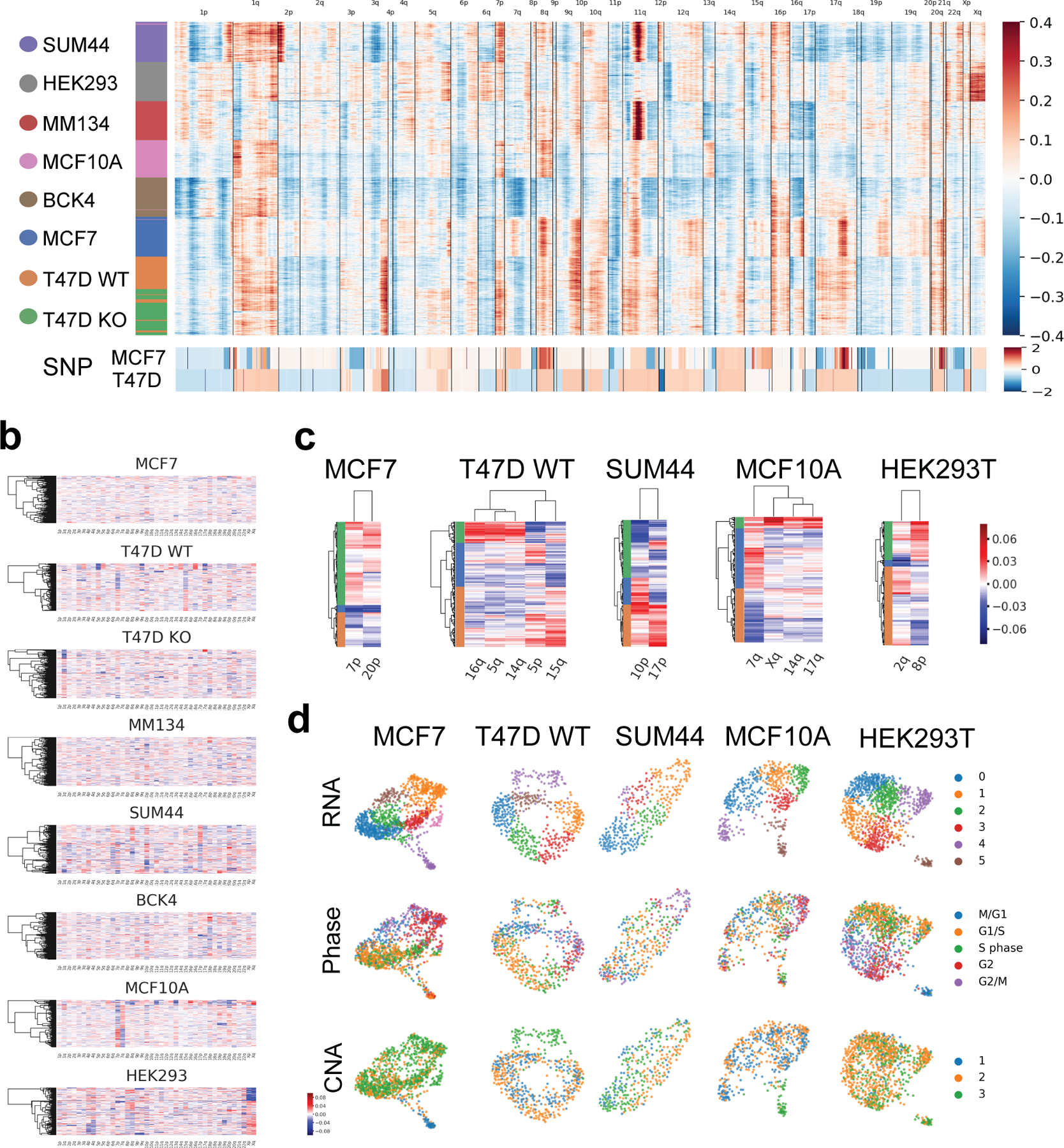
a. Inferred copy number (in log2 scale) of scRNA-seq (top), and Affymetrix SNP6.0 arrays (log2(CN/2)) of MCF7 and T47D cell lines from CCLE (46) (bottom).
b. Inferred CNA averaged for each chromosome arms with more than 100 genes expression.
c. Cell lines with identifiable intra-cell line CNA subpopulations based on selected chromosome arms, colored on heatmap side bars.
d. Intra-cell line RNA and CNA subpopulations, and cell cycle of cell lines in c. Clusters recurrently identified by both CNA and RNA are marked with square.
To characterize genetic ICH, we identified subpopulations from CNA using selected chromosome arms as described by Kinker et al.(21) (Fig. 2b,c; Fig. S2a). For cell lines exhibiting CNA subclones, we compared the genetic subclones with transcriptomic subclones (derived from Louvain clustering from normalized RNA expression, and annotated by phases deduced from cell cycle gene expression) (Fig. 2d). Most of the cell lines investigated (5 out of 8) showed distinct genetic subpopulations not attributed to the cell cycle or scRNA-seq library quality (Fig. 2d, Fig. S2b,c,d). Some but not all CNA clusters (CNA cluster 1 in MCF7, CNA cluster 3 in T47D WT and MCF10A), taking up a minority of the total population, corresponded to a transcriptomic subcluster based on both 2D layout and Louvain clustering (Fig. 2d). To further investigate relationship of the co-occurring CNA vs transcriptomic subclusters, we re-performed normalization and PCA on these three cell lines, but only with genes located on the aberrant chromosome arms in Fig. 2c. The same subpopulations were still distinguishable based on the top two PCs (Fig. S2e), which indicate the sufficiency of copy number aberrations in these subclusters to drive distinct transcriptomic profiles.
Signature derived from a dormant-like MCF7 subpopulation
To investigate transcriptomic ICH, we focused on MCF7 cells, which had sufficient cell numbers in favor of statistical analysis. To cluster cells and select indicative features simultaneously, we used non-negative matrix factorization (NMF), which generated a matrix highlighting three major blocks of cells with corresponding genes, referred to as NMF clusters/genes (Fig. 3a, Table S1).
Fig. 3. Transcriptomic heterogeneity in MCF7 cells.
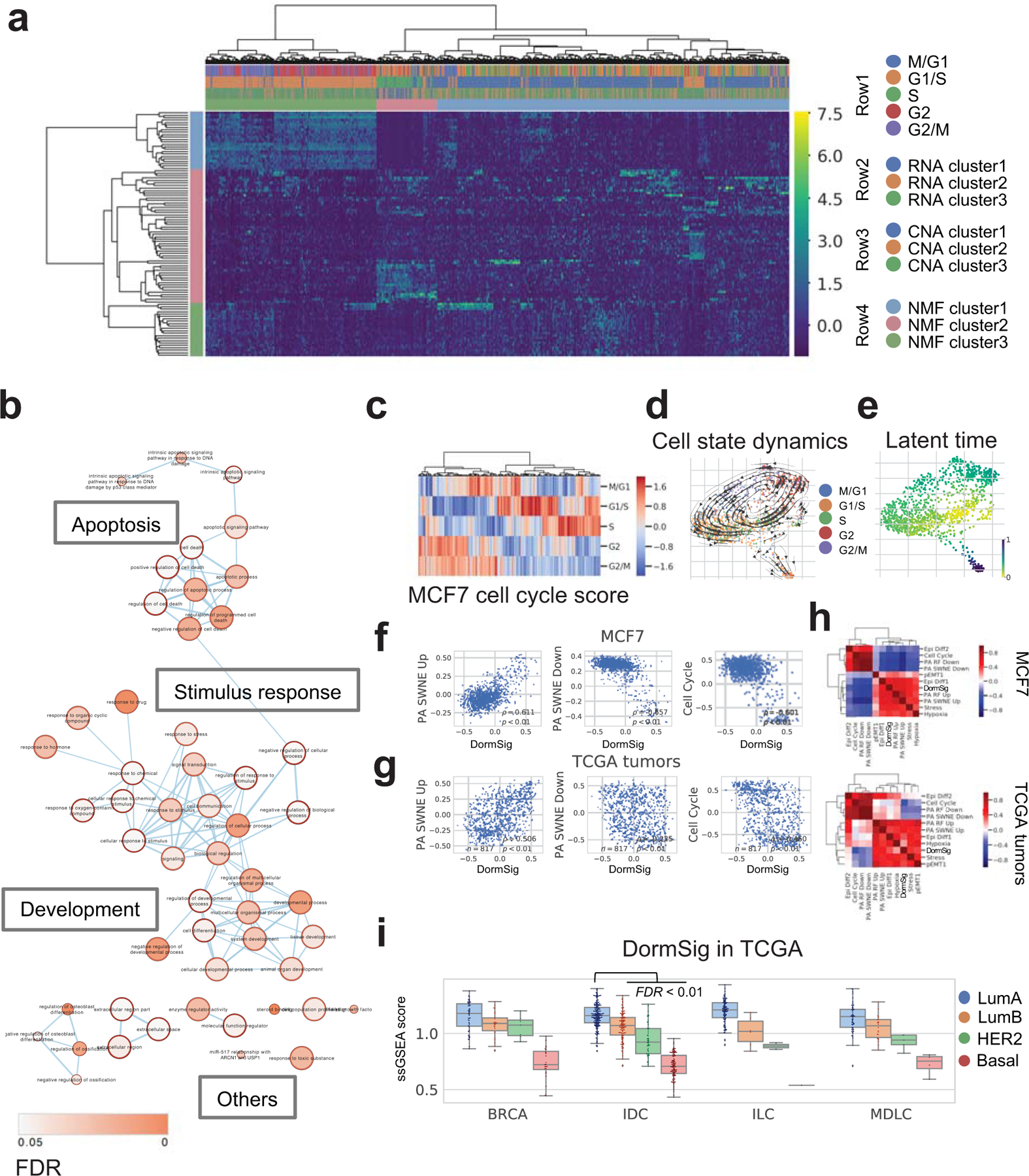
a. Clustering by Non-negative Matrix Factorization (NMF) in MCF7 cells (n=977). The first three rows of top bar showed respectively: cell cycle (row 1), RNA clusters (Louvain method, three clusters at resolution=0.4) (row 2) and CNA clusters (row 3). NMF clusters of cells and corresponding genes are shown in row 4 of top bar and the side bar.
b. GO enrichment of marker genes of NMF cluster 2 cells (pink side bar in Fig. 3a). Terms connections based on similarity; nodes colored by enrichment FDR (over-representation test, BH adjustment) in Cytoscape 3.7.1.
c. Cell cycle phase scores and assignments of MCF7.
d. Dynamical changes of cell states through the cell cycle. Cells are colored by the assigned phase in the force-directed graph drawing 2D layout. Arrows show directions of cell state transition from RNA velocity analysis.
e. Latent time among MCF7 cells from RNA velocity analysis, indicating developmental stages.
f. Co-expression of GSVA scores of DormSig with selected signatures in MCF7 cells (n=977). Correlation showed by Pearson ρ and p.
g. Co-expression of GSVA scores of DormSig with selected signatures (as in f) in TCGA breast tumors (n=817). Correlation showed by Pearson ρ and p.
h. Pearson correlation of GSVA scores of DormSig with selected signatures in MCF7 cells (n=977) and primary breast tumors from TCGA (n=817). Hierarchical clustering was performed using Euclidean distance and Ward’s method.
i. Single sample GSEA (ssGSEA) scores of DormSig in different subtypes of breast cancer from TCGA. (LumA: luminal A, LumB: luminal B, Her2: HER2-enriched, Basal: Basal-like; BRCA: breast cancer with unannotated histological subtype, IDC: invasive ductal carcinoma, ILC: invasive lobular carcinoma, MDLC: mixed ductal/lobular carcinoma). DormSig ssGSEA scores are higher in LumA IDCs (n=200) than each of the other subtypes in IDC tumors (LumB: n=122, Her2: n=51, Basal: n=107) (FDR < 0.01, Wilcoxon test, BH adjustment).
Most cells belonged to NMF cluster 1 or 3, which overlapped with the two major RNA clusters (1 and 2 in Fig. 3a) or CNA subclusters (2 and 3 in Fig. 3a). A comparison with cell cycle showed that NMF cluster 1 correspond to the mitotic phase (83% cells (237 out of 286)), while cluster 3 majorly consist of cells in non-mitotic phase (87% cells (513 out of 589)), further supported by Gene Ontology (GO) enrichment (Fig. 3a , Fig. S3a). The major effect of the cell cycle on transcriptional variation in MCF7 cells was demonstrated by highlighting the cell cycle phase of each cell (Fig. 3c), and the transition through different states predicted by RNA velocity analysis (Fig. 3d).
Despite the majority of cells apparently transiting through the cell cycle, there existed a minor population (NMF cluster 2) exhibiting a ‘dormant-like’ non-transiting state. This is consistently indicated by high latent time values from RNA velocity analysis (Fig. 3e). We thus refer to this cluster of cells as Dorm cells and their corresponding genes as DormSig (signature). An inspection of highly expressed genes in this cluster revealed an enrichment of apoptosis-related pathways (Fig. 3b). Interestingly, a recent report revealed that MCF7 cells contain a rare ‘pre-adapted endocrine resistant’ sub-population even when grown in regular media (DMEM, 10% fetal calf serum)(29). A pre-adaptation signature (highly expressed PA Up genes or lowly expressed PA Down genes), derived from these cells, revealed a negative correlation with cell cycle and was indicative of dormancy. It is hypothesized that these pre-adapted cells may endure growth inhibition by anti-estrogens via exhibiting the less-aggressive dormant-like features. Motivated by this discovery, we investigated the association of the DormSig with the pre-adapted signature. Despite a limited overlap in genes present in these two signatures (Fig. S3b), DormSig showed a significant correlation with both the PA Up (r=0.611, p<0.01) and PA Down signatures (r=−0.657, p<0.01) (Fig. 3f) in MCF7 cells. This correlation is similarly observed in TCGA breast tumors (Fig. 3g) or other breast cell lines (Fig. S3c,d,e). Expression correlation with other functionally-relevant tumor signatures (17) further illustrated a positive correlation of DormSig with partial EMT (epithelial–mesenchymal transition), stress and hypoxia (Fig. 3h, Fig. S3c). DormSig showed enrichment in Luminal A tumors, which has the best prognosis among all PAM50 subtypes (Fig. 3i). High expression of DormSig also indicates good prognosis, possibly due to its less aggressive manifestations, which holds true even when restricted to the estrogen receptor positive and LumA cohort, either for all patients or patients with hormone therapy (p = 0.022 by log rank test) (Fig. S3f).
CDH1 point mutation results in loss of spliced E-cadherin RNA and alters expression programs related to the ILC phenotype
CRISPR-Cas9-mediated CDH1 KO is induced by a single base pair deletion (C at 2753 of cDNA DQ090940.1) (Fig. S4a), which generated a premature stop codon, mimicking missense mutations found in ILC tumors and in genetically characterized ILC cell lines(48). Specifically, we used a mixture of eight single cell-derived clones, each with the aforementioned knock out, as to reduce potential off-target bias. This point mutation led to depletion of both E-cadherin RNA and protein (Fig. 4a, Fig. S4c), caused cells to lose cell-cell adhesion (Fig. 4a) and induced a profound effect on the transcriptome landscape, illustrated by distinct clustering of CDH1 KO and WT cells (Fig. S4b). When specifically examining spliced versus unspliced E-cadherin RNA abundance, the three ILC cell lines and T47D CDH1 KO showed depleted spliced RNA abundance but comparable unspliced RNA distribution compared to MCF7 and T47D WT cells (Fig. 4b). This suggests that CDH1 mutation does not affect the nascent RNA transcript but post-transcriptional events, e.g., causing insufficient splicing or rapid degradation(49). This result was mirrored from bulk RNA-seq in human tumors from TCGA(50) and cell lines, in which IDC vs ILC showed more difference in exon RNA sequencing coverage than intron regions of CDH1 (Fig. S4d,e).
Fig. 4. Differentially activated pathways in CDH1 KO vs WT T47D cells and ILC vs IDC tumors.
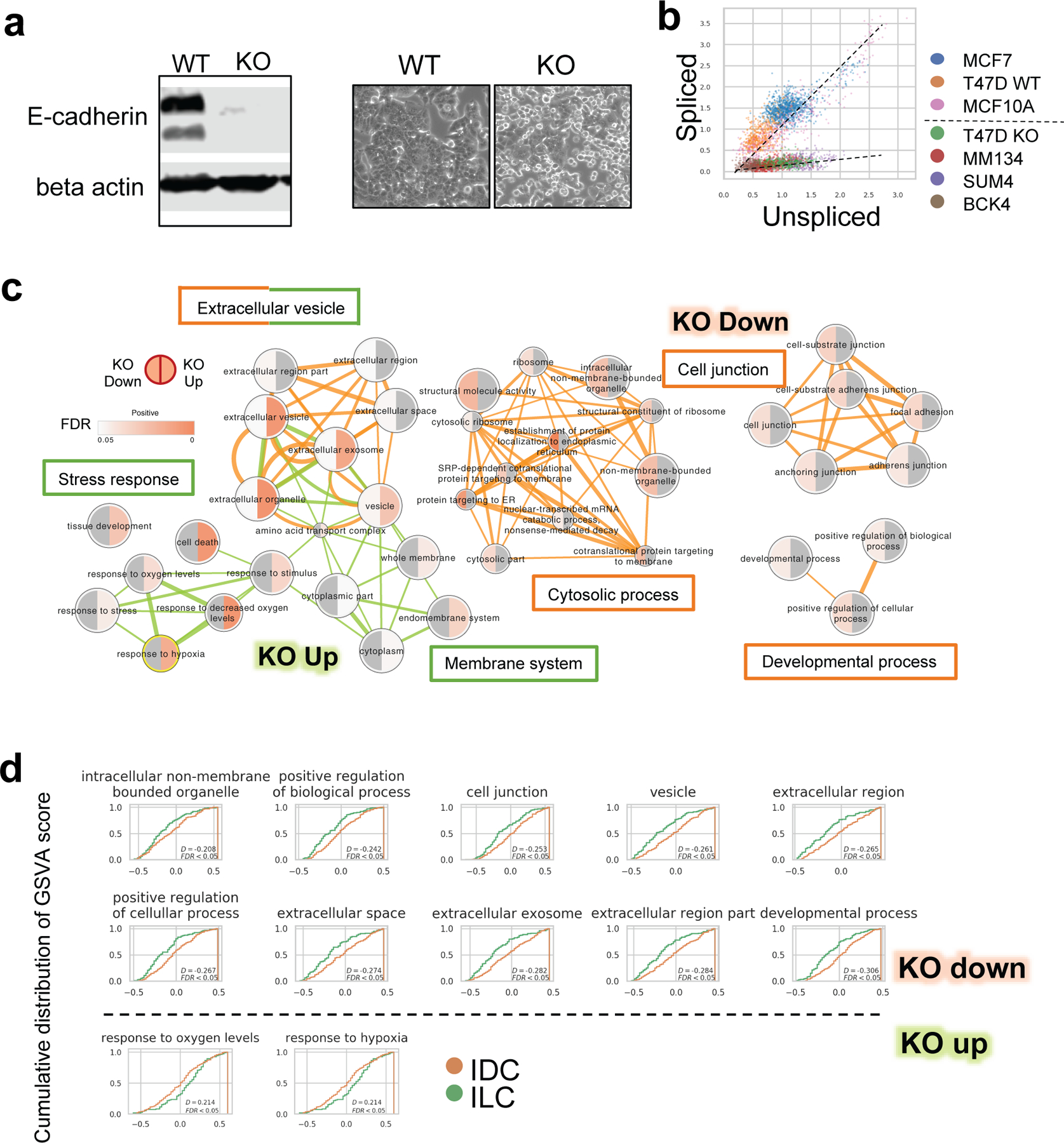
a T47D KO and WT cells. Left: E-cadherin expression by WB. Right: morphology under microscope
b. Normalized unspliced and spliced CDH1 RNA abundance among single cells.
c. Enriched Gene Ontology terms of down (red linked) and up (green linked) regulated genes after CDH1 KO in T47D cells. Terms connections based on similarity; nodes colored by enrichment FDR (over-representation test, BH adjustment) in Cytoscape 3.7.1.
d. Cumulative distribution of GSVA scores of selected signatures in TCGA LumA IDC (n=200) and ILC (n=106) tumors. Right shifted curve indicates distribution of higher score values.
We explored what genes and pathways were differentially expressed (DE) after CDH1 KO. As expected, cell junction-related components were down-regulated following loss of E-cadherin, along with some less specific pathways such as developmental or cytosolic processes (Fig. 4c). This is consistent with morphology changes in CDH1 KO cells, which were rounder with brighter margins, indicating decreased cell-cell contacts (Fig. 4a). Components in membranous system, endomembrane in particular, as well as stress response-related genes, were up-regulated in CDH1 KO cells (Fig. 4c). Interestingly, extracellular vesicle-related pathways were enriched in both up and down regulated genes (Fig. 4c).
To investigate whether the transcriptomic changes in CDH1 KO cells versus WT truly reflect ILC-IDC differences in tumors, we analyzed the expression of DE programs, refined by the overlap of DE genes with the original GO program (Table S2), among the IDC and ILC in TCGA LumA cases. The majority of down-regulated gene sets (10 out of the 16 deduplicated gene sets) and some up-regulated ones (2 out of the 13 deduplicated gene sets) showed significant differences between ILC and IDC tumors, consistent with the trend in CDH1 KO and WT models (Fig. 4d).
An IRF1 regulon is activated following loss of E-cadherin, with ILC and CDH1 KO cells showing increased growth inhibition by IFN-γ
Loss of E-cadherin in epithelial cells has been reported to induce expression of multiple transcript factors (TFs) and trigger profound downstream phenotypic changes, such as metastasis promotion through epithelial-mesenchymal transition (EMT)(51). While EMT does not seem to be a classical feature of ILCs(52,53), the vast transcriptomic changes in CDH1 KO cells strongly suggest involvement of downstream TFs. We therefore searched for TF regulatory modules (regulons) which are increased or decreased in activity following CDH1 deletion in T47D cells and investigated their expressions in ILC vs IDC tumors. Regulon activation profiles in each cell line was calculated using pySCENIC(41). Commonly deduced regulons were binarized with an optimized threshold on AUC distribution and merged for all cell lines, which were used for hierarchical clustering (Fig. 5a,b, Table S3). To more specifically quantify inter-cell line regulon activation differences, we measured the Jaccard Index between individual cells (Fig. 5c), where larger value indicates higher resemblance. Notably, T47D CDH1 KO cells showed higher similarity to two out of three ILC cells (MDA-MB-134-VI, BCK4) than the two IDC cell lines (MCF7, T47D WT), (Fig. 5c, FDRs < 0.01 based on two sample K-S test, BH adjustment). Of note, such increased similarity is claimed when restraining to activity of regulons showing in Fig. 5a (close clustering with ILCs), while from global transcriptomics the KO cells still most resemble its WT counterpart. This observation further supported that CDH1 KO in IDC cells initiates an ILC specific TF regulon activation.
Fig. 5. Regulon activation states in breast cell lines and TCGA tumors.
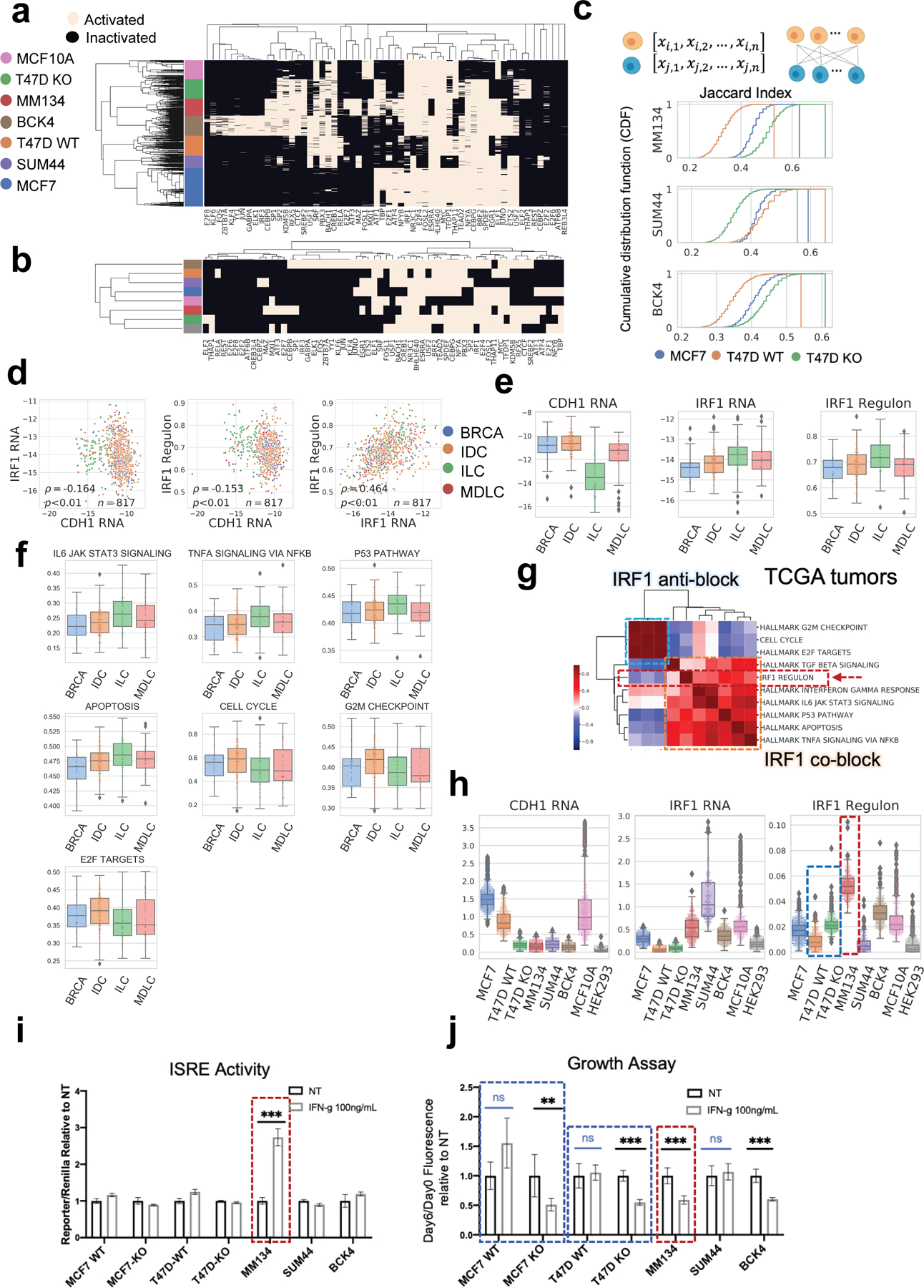
a. Binarized regulon activation profiles of each breast single cell deduced from scRNA-seq with pySCENIC.
b. Binarized TF activation profile for each breast cell line, based on the majority of single cell states in a. Hierarchical clustering by Jaccard distance, ward method.
c. Regulon activation similarity between each ILC cell line (reference) to MCF7, T47D WT and T47D KO (query), quantified by Jaccard Index. For each reference cell line (per row, labeled on y axis), Jaccard Index was calculated between individuals in the reference population and every single cell of the three query breast cell lines respectively, depicted in cumulative distribution. Larger Jaccard Index indicates higher similarity.
d. Log normalized expression of CDH1 RNA, IRF1 RNA, and AUC score of IRF1 regulon in breast cell lines. Difference between IDCs and ILCs are significant (FDR<0.05) in all the three cases.
e. Expression of CDH1, IRF1 (log normalized RNA abundance) and IRF1 regulon (ssGSEA score) in TCGA LumA cases. Difference between IDCs and ILCs are significant (FDR<0.05) in all the three cases.
f. ssGSEA scores of selected signatures (as in Fig. 5g) which showed significant difference between TCGA LumA IDC (n=200) and ILC (n=106) tumors. Signatures except for IRF1 Regulon or Cell Cycle are from MSigDB hallmark database. Difference between IDCs and ILCs are significant (FDR<0.05) in all cases.
g. Pearson correlation of ssGSEA scores of IRF1 regulon with relevant functional signatures in TCGA tumors (n=817). Signatures are divided to IRF1 co-block, which show positive correlation with IRF1 regulon; or IRF1 anti-block, which show negative correlation with IRF1 regulon.
h. Expression of CDH1, IRF1 (log normalized) and IRF1 regulon (AUC score from pySCENIC) in breast cell lines.
i. Relative ISRE transcriptional activity of IFN-γ (100ng/mL) stimulated cells than the non-treated group (six technical replicates for each group).
j. Relative growth at day 6 post IFN-γ (100ng/mL) stimulation compared to the non-treated group (six technical replicates for each group), by dsDNA Fluorescence quantification.
We next identified regulons specifically activated following CDH1 KO. Fourteen TFs were identified in this manner, which were further investigated regarding expression differences in LumA IDC and ILC in TCGA. Only IRF1 and CTCF showed significant differences (FDR<0.05), and only IRF1 exhibits higher expression in ILC (Fig. S5a,b). Intriguingly, IRF1 expression was also negatively correlated with CDH1 in tumors (Fig. 5d,e), which further supports its activation in a lobular specific and E-cadherin associated manner. Similar observations were obtained in cell lines where ILCs generally have lower CDH1 and higher IRF1 or IRF1 regulon activation levels while IDCs show the opposite (except IRF1 regulon score of SUM44-PE) (Fig. 5h).
IRF1 is a canonical target of IFNγ, and in a pathway known to affect cell survival and proliferation. We next therefore examined co-expression of IRF1 regulon (as deduced from pySCENIC results, Fig. S5c) activation with selected MSigDB hallmark signatures with relevant functions in tumors (Fig. 5g). Hierarchical clustering illustrated two distinct blocks, where IRF1 regulon positively correlates with IFNγ response, apoptosis, and signaling of TNFα, TGFβ and IL-6; while showing a negative association with cell cycle (Fig. 5g). Most pathways (4 out of 6) which were positively correlated showed enriched expression in ILC tumors given the difference is significant while all the three pathways with negative correlation showed the opposite (Fig. 5f). Similar trends were observed in IDC/ILC cell lines as well (Fig. S5h).
Based on the predicted IRF1 regulon activity difference between T47D WT vs KO, and WT IDCs vs ILCs, especially MM134 (Fig. 5h), we then investigated relevant phenotypes with the six breast cancer lines. To increase robustness of the study, we generated an additional IDC model with CDH1 knockout (MCF7-CDH1 KO cell line) (Fig. S5d) and included it into these analyses. RNA quantification by qRT-PCR shows higher baseline expression in ILCs than WT IDCs, and slight elevation in T47D KO compared to WT (Fig. S5e). As IRF1 is canonically activated by cytokines in TMEs, we used interferon-gamma (IFN-γ), one of the most common IRF1 stimulant, to investigate inducibility of the IRF1 regulon. Western blot shows significant elevation of IRF1 protein in all cell lines within 4 hours of IFN-γ (10ng/mL) stimulation, in which MM134 showed the highest induced expression (Fig. S5f,g). Similarly, MM134 was also most responsive to IFN-γ among all cell lines, shown as highest induced IRFs’ activity quantified by ISRE dual luciferase assay (Fig. 5i). Such high IRF1 inducibility of MM134 was consistent with computational results, in which MM134 scored top of IRF1 regulon activity (Fig. 5h). IFN-γ responsiveness was also measured in general phenotypes as cell growth, in which inhibition was observed in most cell lines on day 6 post treatment (Fig. 5j). Of note, both MCF7 and T47D KO were significantly inhibited by IFN-γ while their WT counterparts were barely influenced. The ILC cell lines MM134 and BCK4 also showed significant growth inhibition, while SUM44 was less responsive, but was again consistent with its lowest IRF1 regulon score from computation (Fig. 5h). Further knock down of IRF1 reverted the IFN-γ mediated growth inhibition in CDH1-KO MCF7 cells (Fig. S5i). In summary, these traits associate E-cadherin loss with IFN-γ responsiveness, as mediated by IRF regulon and influenced by other factors; while likely, IRF1 expression is required for IFN-γ induced growth inhibition in E-cadherin null cells.
Discussion
scRNA-seq allows for single cell resolution of the transcriptome and is fundamentally altering our understanding of normal development and cancer. In this report, we used scRNA-seq to investigate inter-cellular heterogeneity within each of the seven breast cancer cell lines. Of note, the generated scRNA-seq data of the three ILC cell lines and one CDH1 KO IDC model was novel, from which we uncovered and validated unique ILC features. scRNA-seq readily discerned differences between the cell lines, and genetic subclones were identified in most cell lines. Transcriptomic changes faithfully predicted the transition of cells through the cell cycle. However, in MCF7, a minor subpopulation of cells exist outside of the cell cycle, and these cells showed a dormancy related phenotype previously reported by other group(29). ILC cell lines were distinct from IDC cell lines, and genetic deletion of CDH1 caused transcriptional modeling in T47D as to be more similar to ILC than IDC cell lines. Specifically, we identify numerous novel pathways altered by E-cadherin loss including up regulation of the endomembranous system, as well as specific components of extracellular vesicle. These events are likely critical to membrane trafficking and signaling via membrane receptors, which we and others have reported(6,54), and suggest directions for future investigation. An investigation of activated regulons following loss of CDH1 identified IRF1, which was also activated in LumA ILC.
scRNA-seq of cell lines revealed genetic and transcriptomic subpopulations within cell lines. Although cell hashing would best delineate cell identity from a mixture seq result, our usage of stringent filtering, unsupervised clustering and marker genes is well capable of such deconvolution, as shown by correct clustering when integrated with two same cell lines (MCF7s and T47Ds) from public data. A previous report of scRNA-seq in cell lines identified genetic and transcriptomic subpopulations in many cell lines, but not MCF7(21). This inconsistency is unlikely due to strain artefacts, as our cell lines clustered correctly from both inferred CNA and scRNA with the same cell lines from two other independent datasets, including the dataset which didn’t identify subclones in MCF7(21). A possible reason is that we sequenced around five times the number of cells and thus had more power to find subpopulations. We found that MCF7 cells contained a subpopulation of non-cycling cells (“Dorm” cells) with a dormancy phenotype reported by others(29). Importantly, Dorm cells corresponded to a subpopulation with pre-adaptation (PA) to endocrine therapy – also identified through scRNA-seq. The PA signatures are reported to support cancer survival in acute hormone deprivation. This strengthens the concept of transcriptionally-distinct minor subpopulations, which are present at all times, but in case of a harsh environment (e.g. hormone starvation), use their dormant phenotypes to survive and ultimately cause endocrine resistance. Paradoxically, enrichment of DormSig in ER+ LumA tumors generally renders longer DFS for patients, likely due to the less invasive/proliferative nature of the disease, which postponed progression. On the other hand, it is also possible that late recurrences originating from Dorm cells could be harder to eliminate than less dormant/more proliferative tumor cells, a hypothesis that requires testing in future studies.
scRNA-seq showed that IDC and ILC cell lines have distinct transcriptional programs, similar to tumors in TCGA; and that genetic loss of CDH1 in an IDC cell line causes extensive transcriptional remodeling to make the resultant IDC CDH1 KO cell line to resemble ILC, in both morphology and pathways. E-cadherin deficiency in lobular breast cancer was shown to be functionally associated with other structural proteins, e.g., elevated reliance on p120 in cytokinesis regulation(7). From our data, we also observed structure-related transcriptomic changes after CDH1 KO, such as junctional disruption; along with other features as expression increasement in endomembrane system, stress response and certain exocytosis pathways. These phenotypes from cell models were similarly identified when comparing clinical IDC and ILC LumA tumors.
The depletion of E-cadherin RNA and protein has been recognized in the majority of ILC tumors while promoter methylation is not associated with histological types(37). This on one hand, justifies our use of cell lines for modeling ILC tumors, where MDA-MB-134-VI, SUM44-PE and T47D all harbor little methylation at CDH1 promoter region (BCK4 had not been investigated)(55); and on the other hand, suggests post-transcriptional modifications as potential driver of E-cadherin depletion. Our observation of alterations in CDH1 spliced RNA, but not unspliced RNA in ILC from scRNA-seq data, provides evidence supporting this hypothesis. This was validated in TCGA bulk RNA-seq data via an approximation method of split exon/intron quantification, where we show more comparable intron RNA coverage in ILC as in IDC than exons. Notably, CDH1 in T47D KO and ILCs all bear a pre-mature termination codon (PTC) while not necessarily contain disruptive mutations at splicing site (BCK4 mutation is currently unknown). In this context, loss of spliced mRNA is likely to result from non-sense mediated decay (NMD), where the exon junction complex, including UPF2 and UPF3, were abnormally maintained on spliced RNA with premature stop codon, and eventually cause mRNA degradation (56). A similar finding of NMD-mediated E-cadherin loss has also been reported by Karam et al. in gastric cancer (57).
While E-cadherin is a membrane protein, its loss causes distinct transcriptional reprogramming, likely an indirect effect on TF activity, for example through inhibiting Kaiso’s TF activity as shown in mouse models(22). To investigate this further, we screened regulon activity computationally and identified IRF1, which showed elevated activity following CDH1 KO, meanwhile higher RNA expression in ILC cell lines or tumors. As a tumor suppressor, IRF1 inhibits proliferation and prompts cell death. In breast cancer, IRF1 depletion could well indicate endocrine resistance, while its induction by IFN-γ sensitize cancer cells to endocrine therapy(58). We then investigated IRF1 inducibility among cell lines under IFN-γ treatment, including two pairs of CDH1 KO IDCs (MCF7, T47D) and the same three ILC cell lines. IRF1 protein was produced 4 hours after stimulation for all cell lines, while ISRE activity was most significantly induced for MM134, consistent with its highest computational IRF1 regulon score. This indicates variable propensity of IRF regulon to IFN-γ stimulation, dependent on a cell-specific intrinsic regulon network, despite overall increased IRF1 expression. IFN-γ influence on cell growth was also investigated, where both KO IDCs (MCF7, T47D) and two ILCs with high computed IRF1 score (MM134, BCK4) exhibited significant inhibition, while both WT IDCs and one low-scoring ILC (SUM44) showed little response. Comparison between WT and KO signifies the role of E-cadherin loss in sensitizing cells to IFN-γ mediated growth-inhibition. While this may be only partially reflected by ISRE activity assay, IRF1 expression was likely required in such IFN-γ responsiveness shown by the KD experiment in MCF7 cells. Differential growth inhibition in ILCs, representing E-cadherin-null models representing lobular disease heterogeneity, indicates that such IFN-γ responsiveness is also affected by other genetic or epigenetic context. Our results hold promise for a subset of ILC patients, in which the tumors resemble MM134 and BCK4, in that these patients might benefit from IFN-γ treatment. Current IFN-γ combinatorial therapy in cancer includes an ongoing Phase I-II trial (NCT03112590(59)) in HER2+ breast cancers, but similar treatments could extend to patients with ILC with IFN-γ responsive markers. IFN-γ targets tumors in at least two ways – directly suppressing tumor proliferation, while at the same time increasing tumor antigenicity and augmenting active T cell function. We have showed the former in E-cadherin-null models, while validation of the latter needs to be performed in ILC and IDC immune-competent animal models in future investigations. Of note, statistical analysis from TCGA indicates immune signature enrichment in ER+ ILCs vs ER+ IDCs(23), which could well potentiate IFN-γ through the immune arm to provide ILC-specific benefits.
In summary, scRNA-seq of breast cancer cell lines has revealed significant intra-cell line genetic and transcriptomic heterogeneity, with identification of dormant cells likely primed for anti-estrogen resistance. Knockout of CDH1 in IDC mimics features of ILC and sensitizes ILC cells to IFN-γ mediated growth inhibition, which highlights the power of single cell sequencing to reveal unique features of breast cancer.
Supplementary Material
Statement of significance:
This study represents a key step towards understanding heterogeneity in cancer cell lines and the role of E-cadherin depletion in contributing to the molecular features of invasive lobular breast cancer.
Acknowledgments
This project benefited from resources provided by the University of Pittsburgh HSCRF Genomics Research Core, the UPMC Genome Center, the Pittsburgh Genomic Research Repository (dbGAP), and the University of Pittsburgh Center for Research Computing. The China Scholarship Council and Tsinghua University provided financial support for F.C. The authors would like to thank Jian Chen for outstanding technical support, and Dr. Fangping Mu and Uma Chandran for bioinformatics assistance.
Grant Support: This work was supported by the Breast Cancer Research Foundation [A.V.L. and S.O.]; Susan G. Komen Scholar awards [SAC110021 to A.V.L., SAC160073 to S.O.]; the National Cancer Institute [5F30CA203154 to K.M.L., P30CA047904]; Magee-Women’s Research Institute and Foundation, and the Shear Family Foundation. S.O. and A.V.L. are Hillman Fellows. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health or other Institutes and Foundations.
Footnotes
Conflicts of Interest / Competing Financial Interest Disclosure: The authors declare no potential conflicts of interest.
References
- 1.American Cancer Society. Breast Cancer Facts & Figures 2019–2020. Am Cancer Soc. 2019;1–44. [Google Scholar]
- 2.Sikora MJ, Cooper KL, Bahreini A, Luthra S, Wang G, Chandran UR, et al. Invasive Lobular Carcinoma Cell Lines Are Characterized by Unique Estrogen-Mediated Gene Expression Patterns and Altered Tamoxifen Response. Cancer Res. 2014;74:1463–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Sikora MJ, Jacobsen BM, Levine K, Chen J, Davidson NE, Lee AV., et al. WNT4 mediates estrogen receptor signaling and endocrine resistance in invasive lobular carcinoma cell lines. Breast Cancer Res. 2016;18:92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Teo K, Gómez-Cuadrado L, Tenhagen M, Byron A, Rätze M, van Amersfoort M, et al. E-cadherin loss induces targetable autocrine activation of growth factor signalling in lobular breast cancer. Sci Rep. 2018;8:15454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Ciriello G, Gatza ML, Beck AH, Wilkerson MD, Rhie SK, Pastore A, et al. Comprehensive Molecular Portraits of Invasive Lobular Breast Cancer. Cell. 2015;163:506–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Nagle AM, Levine KM, Tasdemir N, Scott JA, Burlbaugh K, Kehm J, et al. Loss of E-cadherin enhances IGF1-IGF1R pathway activation and sensitizes breast cancers to anti-IGF1R/InsR inhibitors. Clin Cancer Res. 2018;24:5165–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Bajrami I, Marlow R, van De Ven M, Brough R, Pemberton HN, Frankum J, et al. E-Cadherin/ROS1 inhibitor synthetic lethality in breast cancer. Cancer Discov. 2018;8:498–515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Canel M, Serrels A, Frame MC, Brunton VG. E-cadherin-integrin crosstalk in cancer invasion and metastasis. J Cell Sci. 2013;126:393–401. [DOI] [PubMed] [Google Scholar]
- 9.Dossus L, Benusiglio PR. Lobular breast cancer: incidence and genetic and non-genetic risk factors. Breast Cancer Res. 2015;17:37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Sarrió D, Pérez-Mies B, Hardisson D, Moreno-Bueno G, Suárez A, Cano A, et al. Cytoplasmic localization of p120ctn and E-cadherin loss characterize lobular breast carcinoma from preinvasive to metastatic lesions Oncogene. Nature Publishing Group; 2004;23:3272–83. [DOI] [PubMed] [Google Scholar]
- 11.Derksen PWB, Braumuller TM, Van Der Burg E, Hornsveld M, Mesman E, Wesseling J, et al. Mammary-specific inactivation of E-cadherin and p53 impairs functional gland development and leads to pleomorphic invasive lobular carcinoma in mice. DMM Dis Model Mech. 2011;4:347–58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Dagogo-Jack I, Shaw AT. Tumour heterogeneity and resistance to cancer therapies. Nat Rev Clin Oncol. 2018;15:81–94. [DOI] [PubMed] [Google Scholar]
- 13.Navin N, Kendall J, Troge J, Andrews P, Rodgers L, McIndoo J, et al. Tumour evolution inferred by single-cell sequencing Nature. Nature Publishing Group; 2011;472:90–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Wang Y, Waters J, Leung ML, Unruh A, Roh W, Shi X, et al. Clonal evolution in breast cancer revealed by single nucleus genome sequencing Nature. Nature Publishing Group; 2014;512:155–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kim C, Gao R, Sei E, Brandt R, Hartman J, Hatschek T, et al. Chemoresistance Evolution in Triple-Negative Breast Cancer Delineated by Single-Cell Sequencing Cell. Cell Press; 2018;173:879–893.e13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Gao R, Davis A, McDonald TO, Sei E, Shi X, Wang Y, et al. Punctuated copy number evolution and clonal stasis in triple-negative breast cancer. Nat Genet. 2016;48:1119–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Puram SV, Tirosh I, Parikh AS, Patel AP, Yizhak K, Gillespie S, et al. Single-Cell Transcriptomic Analysis of Primary and Metastatic Tumor Ecosystems in Head and Neck Cancer. Cell. 2017;171:1611–1624.e24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Chung W, Eum HH, Lee HBHOH-BH-O, Lee K-MM, Lee HBHOH-BH-O, Kim K-TT, et al. Single-cell RNA-seq enables comprehensive tumour and immune cell profiling in primary breast cancer Nat Commun. Nature Publishing Group; 2017;8:15081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Neftel C, Laffy J, Filbin MG, Hara T, Shore ME, Rahme GJ, et al. An Integrative Model of Cellular States, Plasticity, and Genetics for Glioblastoma. Cell. 2019;178:835–849.e21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Ben-David U, Siranosian B, Ha G, Tang H, Oren Y, Hinohara K, et al. Genetic and transcriptional evolution alters cancer cell line drug response. Nature. 2018;560:325–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kinker GS, Greenwald AC, Tal R, Regev A, Tirosh I. Pan-cancer single cell RNA-seq uncovers recurring programs of cellular heterogeneity. bioRxiv. 2019; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Van De Ven RAH, Tenhagen M, Meuleman W, Van Riel JJG, Schackmann RCJ, Derksen PWB. Nuclear p120-catenin regulates the anoikis resistance of mouse lobular breast cancer cells through Kaiso-dependent Wnt11 expression. DMM Dis Model Mech. 2015; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Du T, Zhu L, Levine KM, Tasdemir N, Lee AV., Vignali DAA, et al. Invasive lobular and ductal breast carcinoma differ in immune response, protein translation efficiency and metabolism Sci Rep. Springer US; 2018;8:1–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Bahreini A, Li Z, Wang P, Levine KM, Tasdemir N, Cao L, et al. Mutation site and context dependent effects of ESR1 mutation in genome-edited breast cancer cell models. Breast Cancer Res. 2017;19:60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.La Manno G, Soldatov R, Zeisel A, Braun E, Hochgerner H, Petukhov V, et al. RNA velocity of single cells Nature. Nature Publishing Group; 2018;560:494–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Bergen V, Lange M, Peidli S, Wolf FA, Theis FJ. Generalizing RNA velocity to transient cell states through dynamical modeling. bioRxiv. 2019; [DOI] [PubMed] [Google Scholar]
- 27.Wolf FA, Angerer P, Theis FJ. SCANPY: large-scale single-cell gene expression data analysis. Genome Biol. 2018;19:15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Wolock SL, Lopez R, Klein AM. Scrublet: Computational Identification of Cell Doublets in Single-Cell Transcriptomic Data Cell Syst. Elsevier Inc; 2019;8:281–291.e9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Hong SP, Chan TE, Lombardo Y, Corleone G, Rotmensz N, Bravaccini S, et al. Single-cell transcriptomics reveals multi-step adaptations to endocrine therapy Nat Commun. Springer Science and Business Media LLC; 2019;10:3840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Kinker GS, Greenwald AC, Tal R, Regev A, Tirosh I, Orlova Z, et al. Pan-cancer single cell RNA-seq uncovers recurring programs of cellular heterogeneity. bioRxiv. 2019;807552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Whitfield ML, Sherlock G, Saldanha AJ, Murray JI, Ball CA, Alexander KE, et al. Identification of Genes Periodically Expressed in the Human Cell Cycle and Their Expression in Tumors Solomon MJ, editor. Mol Biol Cell. American Society for Cell Biology (ASCB); 2002;13:1977–2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Zhu X, Ching T, Pan X, Weissman SM, Garmire L. Detecting heterogeneity in single-cell RNA-Seq data by non-negative matrix factorization. PeerJ. 2017;5:e2888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Raudvere U, Kolberg L, Kuzmin I, Arak T, Adler P, Peterson H, et al. g:Profiler: a web server for functional enrichment analysis and conversions of gene lists (2019 update) Nucleic Acids Res. Oxford University Press; 2019;47:W191–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Shannon Paul 1, Markiel Andrew 1, Ozier Owen, 2 Baliga Nitin S., 1 Wang Jonathan T., 2 Ramage Daniel 2, Amin Nada 2, Schwikowski Benno, 1, 5 and Ideker Trey 2, 3, 4 5, 山本隆久, et al. Cytoscape: A Software Environment for Integrated Models. Genome Res. 1971;13:426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Cerami E, Gao J, Dogrusoz U, Gross BE, Sumer SO, Aksoy BA, et al. The cBio Cancer Genomics Portal: An open platform for exploring multidimensional cancer genomics data. Cancer Discov. 2012;2:401–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Gao J, Aksoy BA, Dogrusoz U, Dresdner G, Gross B, Sumer SO, et al. Integrative Analysis of Complex Cancer Genomics and Clinical Profiles Using the cBioPortal. Sci Signal. 2013;6:pl1–pl1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Ciriello G, Gatza ML, Beck AH, Wilkerson MD, Rhie SK, Pastore A, et al. Comprehensive Molecular Portraits of Invasive Lobular Breast Cancer Cell. Cell Press; 2015;163:506–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Curtis C, Shah SP, Chin S-F, Turashvili G, Rueda OM, Dunning MJ, et al. The genomic and transcriptomic architecture of 2,000 breast tumours reveals novel subgroups. Nature. 2012;486:346–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Hänzelmann S, Castelo R, Guinney J. GSVA: gene set variation analysis for microarray and RNA-Seq data. BMC Bioinformatics. 2013;14:7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Tasdemir N, Bossart EA, Li Z, Zhu L, Sikora MJ, Levine KM, et al. Comprehensive phenotypic characterization human invasive lobular carcinoma cell lines in 2D and 3D cultures. Cancer Res. 2018;78:6209–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Aibar S, González-Blas CB, Moerman T, Huynh-Thu VA, Imrichova H, Hulselmans G, et al. SCENIC: Single-cell regulatory network inference and clustering Nat Methods. Nature Publishing Group; 2017;14:1083–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Dai X, Cheng H, Bai Z, Li J. Breast cancer cell line classification and Its relevance with breast tumor subtyping. J Cancer. 2017;8:3131–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Jacobsen B, Badtke M, Jambal P, Horwitz K. Abstract #287: <strong>BCK4 cells are a new model of mucinous human breast cancer</strong> Cancer Res. 2009;69:287 LP – 287. [Google Scholar]
- 44.Zhao X, Rødland EA, Tibshirani R, Plevritis S. Molecular subtyping for clinically defined breast cancer subgroups. Breast Cancer Res. 2015;17:1–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Patel AP, Tirosh I, Trombetta JJ, Shalek AK, Gillespie SM, Wakimoto H, et al. Single-cell RNA-seq highlights intratumoral heterogeneity in primary glioblastoma. Science (80- ). 2014;344:1396–401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Ghandi M, Huang FW, Jané-Valbuena J, Kryukov GV., Lo CC, McDonald ER, et al. Next-generation characterization of the Cancer Cell Line Encyclopedia. Nature. 2019;569:503–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Korsunsky I, Millard N, Fan J, Slowikowski K, Zhang F, Wei K, et al. Fast, sensitive and accurate integration of single-cell data with Harmony Nat Methods. Nature Research; 2019;16:1289–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Christgen M, Derksen PWB. Lobular breast cancer: Molecular basis, mouse and cellular models. Breast Cancer Res. 2015;17:1–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Alberts B, Johnson A, Lewis J, Raff M, Roberts K, Walter P. Molecular Biology of the Cell. Fifth ed. Garland Science; 2007. [Google Scholar]
- 50.Ciriello G, Gatza ML, Beck AH, Wilkerson MD, Rhie SK, Pastore A, et al. Comprehensive Molecular Portraits of Invasive Lobular Breast Cancer. [DOI] [PMC free article] [PubMed]
- 51.Onder TT, Gupta PB, Mani SA, Yang J, Lander ES, Weinberg RA. Loss of E-cadherin promotes metastasis via multiple downstream transcriptional pathways. Cancer Res. 2008;68:3645–54. [DOI] [PubMed] [Google Scholar]
- 52.McCart Reed AE, Kutasovic JR, Vargas AC, Jayanthan J, Al-Murrani A, Reid LE, et al. An epithelial to mesenchymal transition programme does not usually drive the phenotype of invasive lobular carcinomas. J Pathol. 2016; [DOI] [PubMed] [Google Scholar]
- 53.Bossart EA, Tasdemir N, Sikora MJ, Bahreini A, Levine KM, Chen J, et al. SNAIL is induced by tamoxifen and leads to growth inhibition in invasive lobular breast carcinoma. Breast Cancer Res Treat. 2019; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Qian X, Karpova T, Sheppard AM, McNally J, Lowy DR. E-cadherin-mediated adhesion inhibits ligand-dependent activation of diverse receptor tyrosine kinases EMBO J. Oxford University Press; 2004;23:1739–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Lombaerts M, Van Wezel T, Philippo K, Dierssen JWF, Zimmerman RME, Oosting J, et al. E-cadherin transcriptional downregulation by promoter methylation but not mutation is related to epithelial-tomesenchymal transition in breast cancer cell lines. Br J Cancer. 2006;94:661–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Baker KE, Parker R. Nonsense-mediated mRNA decay: terminating erroneous gene expression. Curr Opin Cell Biol. 2004;16:293–9. [DOI] [PubMed] [Google Scholar]
- 57.Karam R, Carvalho J, Bruno I, Graziadio C, Senz J, Huntsman D, et al. The NMD mRNA surveillance pathway downregulates aberrant E-cadherin transcripts in gastric cancer cells and in CDH1 mutation carriers. Oncogene. 2008;27:4255–60. [DOI] [PubMed] [Google Scholar]
- 58.Schwartz JL, Shajahan AN, Clarke R. The Role of Interferon Regulatory Factor-1 (IRF1) in Overcoming Antiestrogen Resistance in the Treatment of Breast Cancer. Int J Breast Cancer. 2011;2011:1–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Han H, Khong H, Costa R, Loftus L, Goodridge D, Henry T, et al. Abstract P2-09-15: A phase I study of interferon-gamma (γ)plus weekly paclitaxel, trastuzumab and pertuzumab in patients with HER-2 positive breast cancer Cancer Res. American Association for Cancer Research (AACR); 2019. page P2-09-15-P2-09–15. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Processed files are deposited in Gene Expression Omnibus (RRID:SCR_005012) (GSE144320) and raw FASTQ files are deposited at SRP245420, which will be available upon publication.