

Abstract
Parkinson’s disease is characterized by the intracellular accumulation of α-synuclein (α-syn) amyloid fibrils, which are insoluble, β-sheet-rich protein aggregates. Raman spectroscopy is a powerful technique that reports on intrinsic molecular vibrations such as the coupled vibrational modes of the polypeptide backbone, yielding secondary structural information. However, in order to apply this method in cells, spectroscopically unique frequencies are necessary to resolve proteins of interest from the cellular proteome. Here, we report the use of 13C2H15N-labeled α-syn to study the localization of preformed fibrils fed to cells. Isotopic labeling shifts the amide-I (13C=O) band away from endogenous 12C=O vibrations, permitting secondary structural analysis of internalized α-syn fibrils. Similarly, 13C–2H stretches move to lower energies in the “cellular quiet” region, where there is negligible biological spectral interference. This combination of well-resolved, distinct vibrations allows Raman spectral imaging of α-syn fibrils across a cell, which provides conformational information with spatial context.
Keywords: Parkinson’s disease, amyloid, Raman spectroscopy, isotopic labeling, SK-MEL-28
1. Introduction
Parkinson’s disease (PD) afflicts millions of people worldwide and usually affects people over the age of 50. Clinical symptoms of resting tremor, slowness of movement, and rigidity are caused by the loss of dopaminergic cells in a midbrain region called the substantia nigra [1]. A cellular hallmark of the disease is the presence of cytoplasmic inclusions called Lewy bodies (LBs) [2], which are enriched in amyloid forms of α-synuclein (α-syn), a presynaptic protein [3]. α-Syn is genetically associated with PD, with mutations causing early-onset autosomal dominant PD, pointing to gain-of-toxicity mechanisms [4–9].
A vertebrate specific protein, α-syn is expressed primarily in neurons of the central nervous system (CNS) [10]. It is found at the highest concentrations (~20–100 μM [11, 12]) in the presynaptic spaces at the ends of axons. Lower levels of the protein are observed in other CNS cell types, and the gene is also transcribed in other tissues [10]. Within neurons, α-syn is mostly cytosolic and enriched in presynaptic nerve terminals with some fraction associated with synaptic vesicles [13]. Despite extensive study, pinpointing specific biological roles for α-syn has proven difficult. Mice with the α-syn gene (SNCA) knocked out present no obvious pathology [14]. There are two other α-syn-related proteins expressed in neurons, β-syn and γ-syn [13]. For mice with all three synuclein genes knocked out, age-dependent neuronal dysfunction was observed with altered synaptic structure and transmission [15]. Currently, it is thought that α-syn acts as a chaperone protein, aiding the rapid re-assembly of the SNARE complex of proteins in the exocytosis process of neurotransmitter release into the synapse [16–18].
In addition to the functional connection, there is cellular evidence supporting the involvement of α-syn-lipid interactions in cytotoxicity [19] including Golgi fragmentation [20, 21], mitochondrial fission [22], and lysosomal malfunction [23]. The relationship between membranes and α-syn pathology is further strengthened by observations that disease-related mutants exhibit different membrane-binding properties and that prefibrillar α-syn has been shown to permeabilize membranes [24–33]. The exact molecular mechanisms by which α-syn promotes membrane disruption remain poorly understood [34–36]. Most recently, cryoelectron microscopic images showed that LBs are crowded with lipid membranous materials along with amyloid fibrils [37].
The specific pathogenic roles of α-syn amyloid fibrils remain controversial. While prefibrillar aggregates had been thought to be the most cytotoxic [38], fibrils also can elicit cell death [39]. Numerous cellular compartments have been implicated and extracellular modes of action have been proposed [40, 41]. Emerging results indicate that fibril propagation may be an underlying mechanism for the spread of neurodegeneration in PD, in a manner similar to the infectivity of prion proteins in prion diseases [42, 43]. In vitro generated α-syn fibrils have been shown to seed and amplify endogenous α-syn aggregation leading to pathogenesis in cellular and animal models [44]. There is further evidence of cell-to-cell transfer of α-syn aggregates [40, 41]. Thus, there is still a need to structurally characterize amyloid fibrils in a cellular context to gain insights into how they contribute to disease progression.
A defining characteristic of all amyloids is a highly-ordered, unbranched filamentous morphology, where individual β-strands align perpendicularly to the filament axis. This structure is also known as a cross-β fold, owing to the X-ray diffraction patterns that filaments exhibit. Recent advances in cryoelectron microscopy have made fibril structures of α-syn [45–47] available at near-atomic resolution, adding to existing structures determined by solid-state NMR [48, 49]. However, due to their polymorphic nature, other α-syn structures are possible [50, 51] and remain to be determined from cellular and patient-derived materials [52]. While amyloids can be readily characterized in vitro at the ultrastructural level using atomic force or transmission electron microscopies and its β-sheet content analyzed by circular dichroism and Fourier transform infrared spectroscopies, direct observation of protein conformational changes and amyloid formation in cellular systems remains a challenge.
In this work, we use Raman spectral imaging to study α-syn fibrils in cellular environments by passively feeding human SK-MEL-28 cells with exogenous α-syn fibrils. We chose Raman spectroscopy for cellular studies because it has several advantages. First, protein amide bands, which arise from coupled vibrational modes of the polypeptide backbone, directly report on protein secondary structure; α-helix and β-sheet show characteristic Raman peaks [53]. Second, water is a weak Raman scatterer in the amide-I region in contrast to infrared spectroscopy, where water exhibits strong absorption [53]. Third, the simplicity of the experiment makes it easily adaptable to any inverted microscope which facilitates cellular imaging [54]. Finally, the use of visible light excitation allows for diffraction-limited spatial resolution.
To differentiate α-syn from the more abundant endogenous biomolecules, we prepared 13C2H15N-labeled α-syn in order to shift our signal of interest, the amide-I (predominantly C=O stretching) band and hence provide contrast against the overlapping vibrations from the cellular background. A particular advantage here is the availability of the aliphatic 13C‒2H stretching frequencies for facile identification of α-syn; these vibrations occur in a spectral region (2050–2300 cm−1) where there is little contribution from cytosolic biomolecules. This approach is analogous to the use of the unnatural amino acid homopropargylglycine (HPG), where the terminal alkyne (C≡C) bond yields a stretching frequency ~2112–2117 cm−1 [55]. To ensure that amyloid fibrils are internalized by cells, fibrils were co-stained with an emissive fluorophore (thioflavin-T [56]) and visualized by confocal fluorescence microscopy. Raman spectra of isotopically-labeled α-syn fibrils were measured in different spatial locations, where the amide-I (13C=O) and 13C–2H stretching bands confirmed the presence of β-sheet rich amyloid fibrils upon internalization. By generating maps using characteristic vibrational frequencies of endogenous biomolecules including nucleotides, lipids, and proteins, we find that the fibrils largely localized to the cellular periphery, with some incidence of colocalization with endogenous proteins and lipids in the perinuclear region. Interestingly, lipid accumulation in the cytosol was observed even in the absence of significant internalization of fibrils. Together, our results demonstrate the utility of isotopic labeling and Raman spectroscopy as an imaging approach in cellular investigations, directly reporting on both protein identity (13C–2H) and secondary structure (13C=O), information which is not easily obtained through other methods.
2. Results and Discussion
2.1. Isotopic labeling provides Raman spectral contrast against cellular background
Comparison of the Raman spectra of natural isotope abundance and 13C2H15N-labeled α-syn fibrils to a spectrum collected in the cytosol of cultured SK-MEL-28 cells reveals the advantages offered by isotopic labeling for studies of proteins in a cellular environment (Fig. 1A). The amide-I band (1664 cm−1) of natural isotope abundance α-syn fibrils (Fig. 1A, black curve) overlaps entirely with the much broader cellular background bands centered at 1656 cm−1, which likely are composed of contributions from endogenous protein amide-I bands and lipid C=C stretching modes (Fig. 1A, gray curve). 13C2H15N-labeled α-syn has an amide-I peak shifted by 44 cm−1 to 1620 cm−1 (Fig. 1A and 1B, blue curve), a slightly larger shift than a previous report using 13C-labeling alone [57]. This shift resolves the amide-I band of α-syn fibrils from cellular components, in principle permitting examination of α-syn secondary structure against a cellular background.
Fig 1. Raman spectroscopic comparison of α-syn fibrils with cellular background.
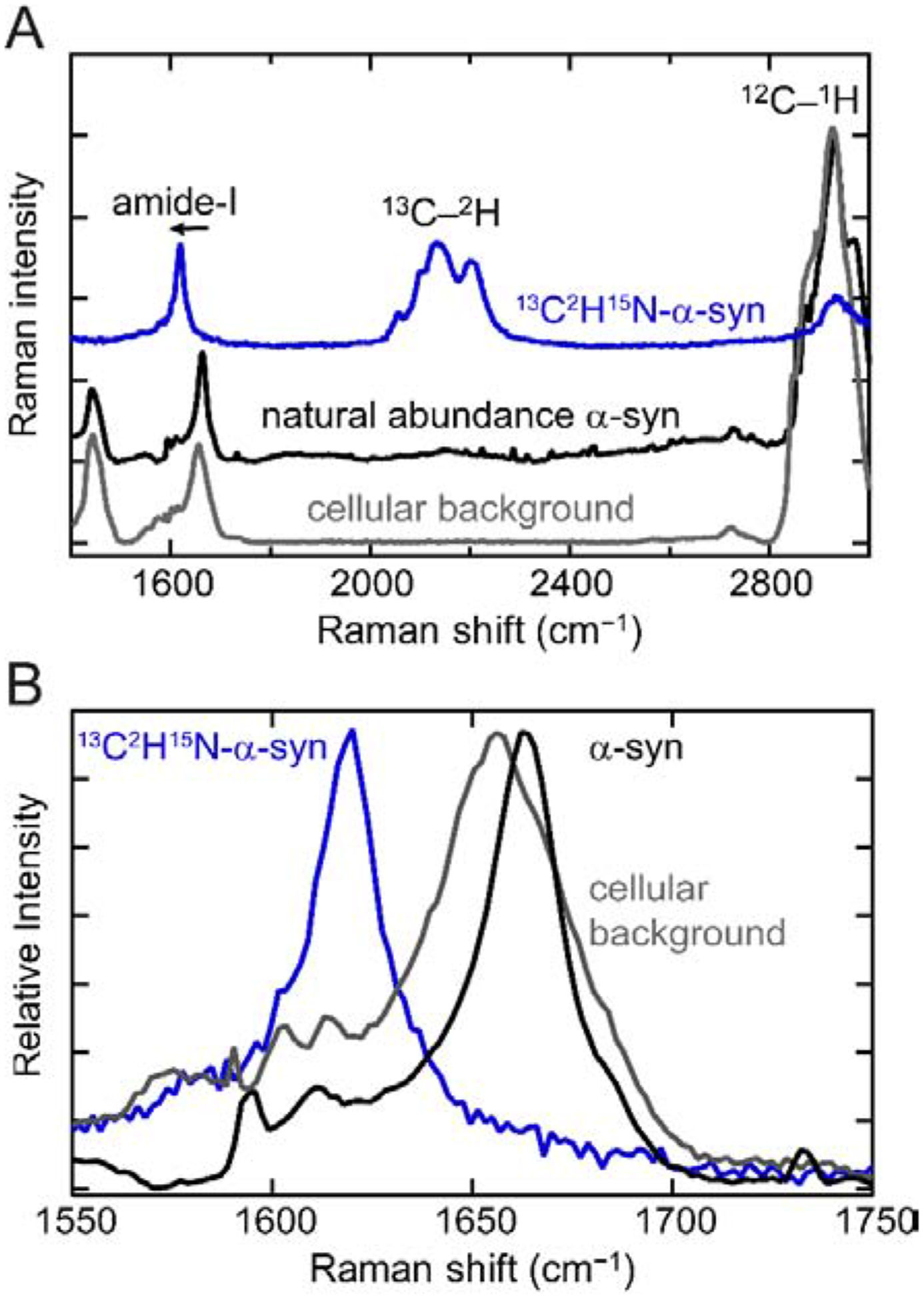
(A) The Raman spectrum of natural isotope abundance α-syn amyloid fibrils (black) overlaps with the cellular background (gray), whereas the Raman spectrum of 13C2H15N-labeled fibrils (blue) possesses several features that are distinctive against a cellular background. The 13C–2H stretching frequencies are red-shifted by approximately 800 cm−1 relative to the 12C–1H stretches, appearing in the “cellular quiet” region (1880–2700 cm−1). (B) An expanded view of the amide-I band. The characteristic β-sheet 13C=O amide-I peak (blue) is red-shifted by 44 cm−1 relative to the 12C=O amide-I band (black).
2.2. 13C–2H Stretching frequencies provide a unique cellular Raman signature
The effect of isotopic labeling on the C–H stretching modes in α-syn fibrils is much more dramatic, owing to the much larger change in reduced mass. The 13C–2H stretching bands are shifted by approximately 800 cm−1 relative to the 12C–1H stretching bands (Fig. 1A), placing the 13C–2H frequencies between ~2000 and 2300 cm−1 in the center of the “cellular quiet” region of the Raman spectrum from 1880 to 2700 cm−1. Because this region is devoid of Raman signals from any naturally occurring biomolecules, any vibrational modes in this range are not only readily detectable but also unique, thus allowing direct spectral identification of α-syn fibrils within a cell. There are multiple bands with comparable intensity to that of the amide-I band. For imaging purposes, this is advantageous as the total integrated area of 13C–2H stretches will offer enhanced signal-to-noise. Another key advantage of isotopic-labeling over other biosynthetic labeling techniques is that it is minimally perturbative to fibril formation, which we confirmed by thioflavin-T (ThT) monitored aggregation assays and transmission electron microscopy (TEM, Fig. 2). The duration of the lag and growth phases of 13C2H15N-labeled α-syn corresponds closely to those of the natural isotope abundance protein, and there are no ultrastructural differences apparent by TEM. Collectively, 13C2H15N-labeling provides two distinct spectroscopic handles to study α-syn fibrils against a cellular background, a structurally-sensitive amide-I 13C=O band and a readily detectable 13C–2H signal that uniquely reports on the presence of α-syn.
Fig. 2. Amyloid formation of 13C2H15N-α-syn.
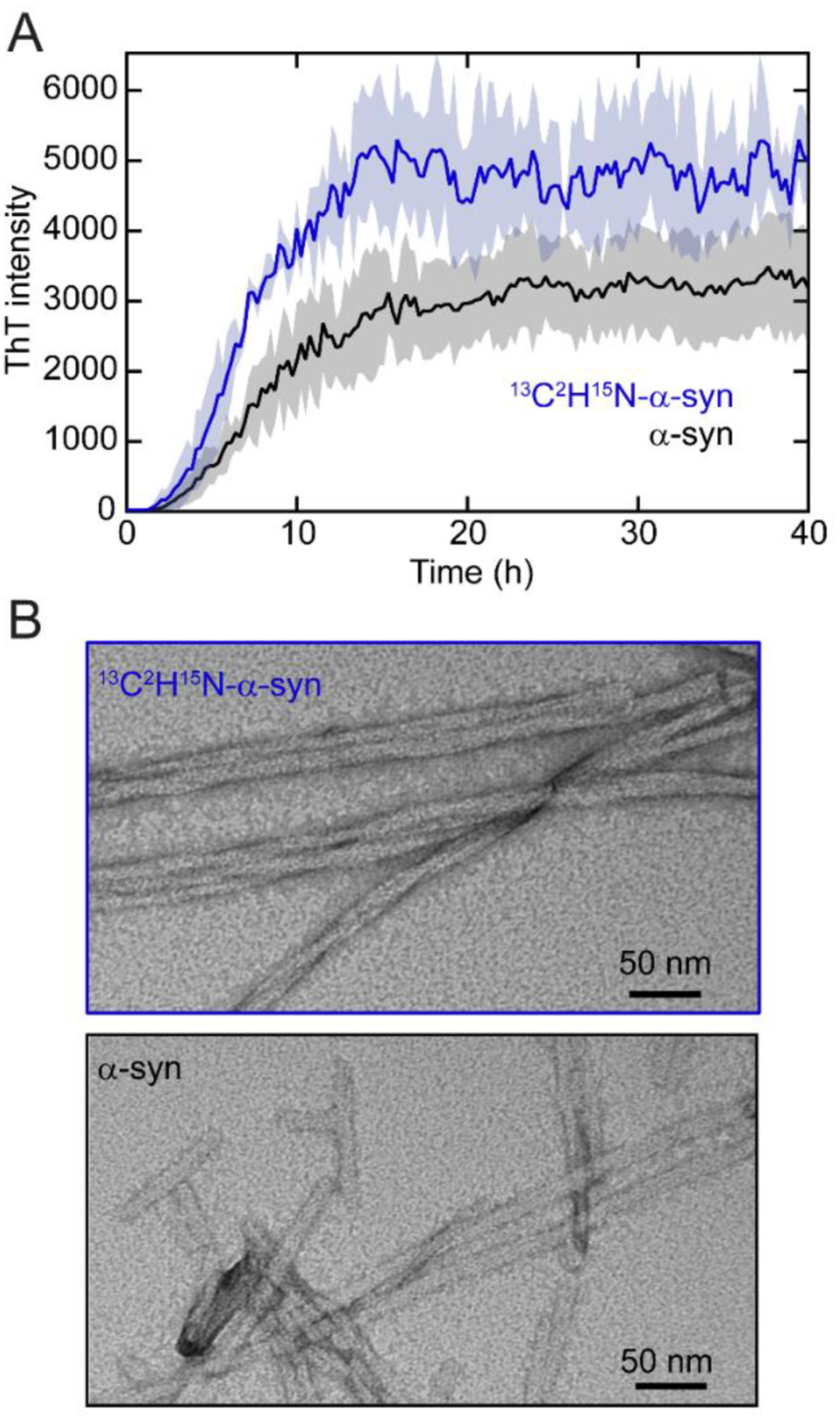
(A) ThT-monitored aggregation kinetics of natural abundance α-syn (black) and 13C2H15N-α-syn (blue). Averages (line) and standard deviations (shaded area) of three replicates are shown ([α-syn] = 50 μM and [ThT] = 5 μM in pH 5 buffer at 37 °C with shaking). (B) TEM images of 13C2H15N-α-syn (top) and α-syn (bottom) fibrils. Scale bars are as indicated.
2.3. Cellular internalization of α-syn fibrils
Next, we verified the cellular uptake and internalization of α-syn fibrils in cultured SK-MEL-28 human melanoma cells. This fibril treatment mimics the cell-to-cell transmission mechanism believed to occur in PD progression and is widely considered to be a disease-related cellular model [44]. We chose SK-MEL-28 as a relevant model due to its high expression of endogenous α-syn [58], and because it shares many proteins with neuronal cells as it is derived from the neuronal crest [59], hence representing a native environment. Moreover, we are interested in studying SK-MEL-28 cells due to a connection between a higher incidence of melanoma in PD patients, which could involve α-syn [60].
Natural isotope abundance α-syn fibrils were first formed in the presence of ThT, then diluted into media, and added at a final concentration of 10 μM to SK-MEL-28 cells. After an hour of treatment, cells are visibly affected, adopting a less extended morphology with fewer processes compared to the untreated control cells (Fig. 3). Fibril internalization was examined by confocal fluorescence microscopy. ThT fluorescence was observed in some of the cells, where different localization patterns were seen, indicating unequal cellular uptake (Fig. 3B). There are different sized clusters of ThT emission with some isolated spots. In a few cells, it is also evident that there are larger masses of aggregates associated with cells. Internalization of the fibrils was confirmed by inspecting different focal planes and z-positions (Fig. 3C). Despite a lack of uniformity of uptake, SK-MEL-28 cells do internalize exogenously added fibrils.
Fig. 3. Cellular internalization of α-syn fibrils visualized by ThT emission.
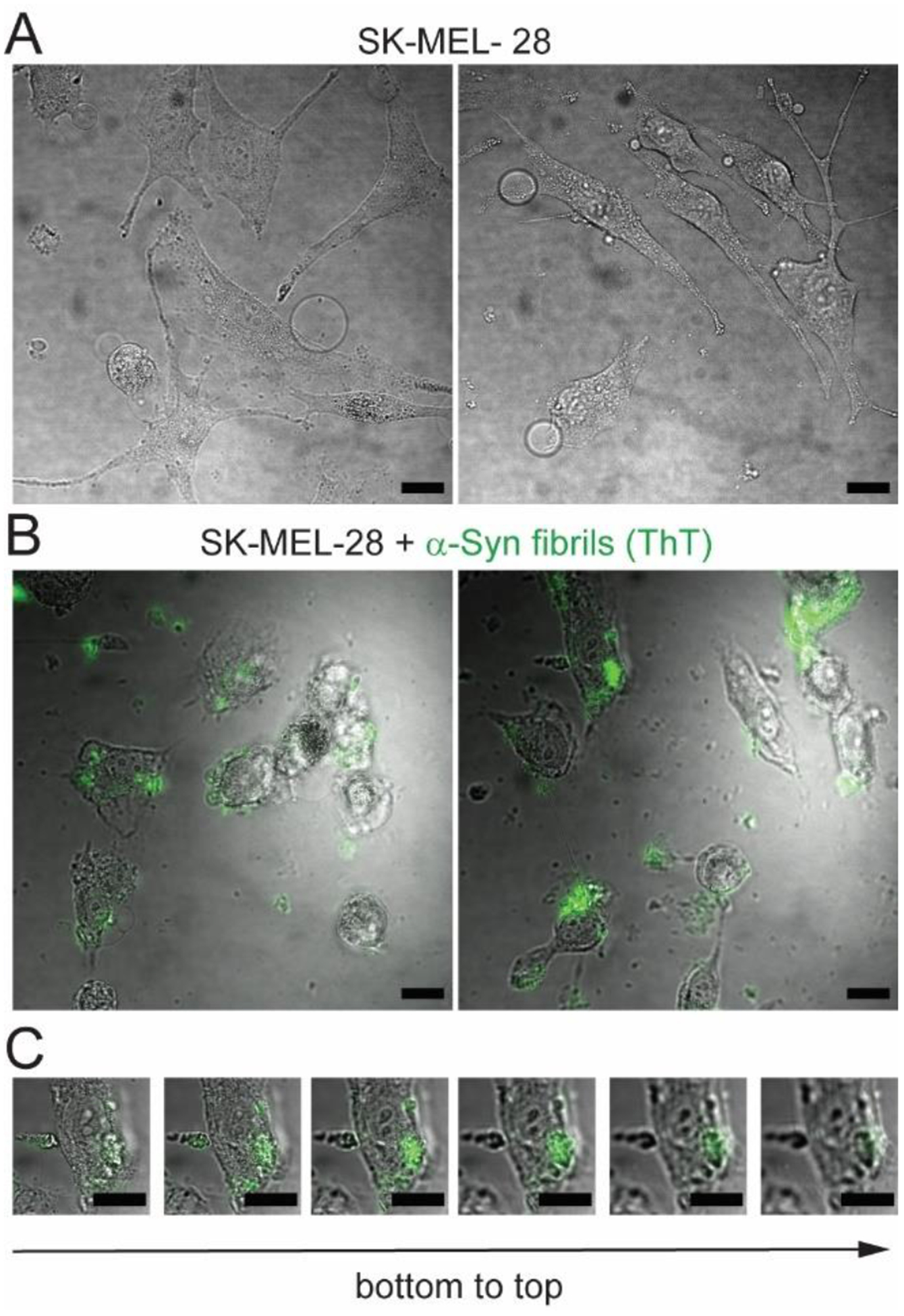
(A) Bright-field images of untreated control SK-MEL-28 cells. (B) Overlay of bright-field and confocal fluorescence images of SK-MEL-28 cells after 1 h treatment with ThT-stained α-syn amyloid fibrils (10 μM). (C) Confocal fluorescence z-stack images (z step-size = 5 μm) of an SK-MEL-28 cell after 1 h treatment with ThT-stained α-syn amyloid fibrils (10 μM). Scale bars are 20 μm.
2.4. Raman spectral imaging of α-syn fibrils in cells
SK-MEL-28 cells were treated with preformed α-syn fibrils without ThT, then washed, fixed, and imaged on a Raman microscope. Raman spectra at different spatial locations across the cell were measured with a spectral collection window from 1250–3000 cm−1, encompassing the fingerprint to the aliphatic C–H stretching regions. Spectra were corrected by selecting a spectrum from a location outside the cell containing the fewest spectral features and subtracting this spectrum from all spectra, then applying a second degree polynomial baseline. Specific spectral regions were integrated to generate Raman maps of various endogenous cellular components such as nucleotides (1550–1585 cm−1), proteins (2900–2950 cm−1), and lipids (2825–2855 cm−1) [61] as well as isotopically-labeled α-syn fibrils using both the 13C=O amide-I band (1590–1625 cm−1) and the distinctive 13C–2H stretching bands (2025–2265 cm−1). We show two Raman spectral images taken at a spatial resolution of 2 μm in Figure 4.
Fig. 4. Raman spectral imaging of SK-MEL-28 cells treated with 13C2H15N-α-syn fibrils.
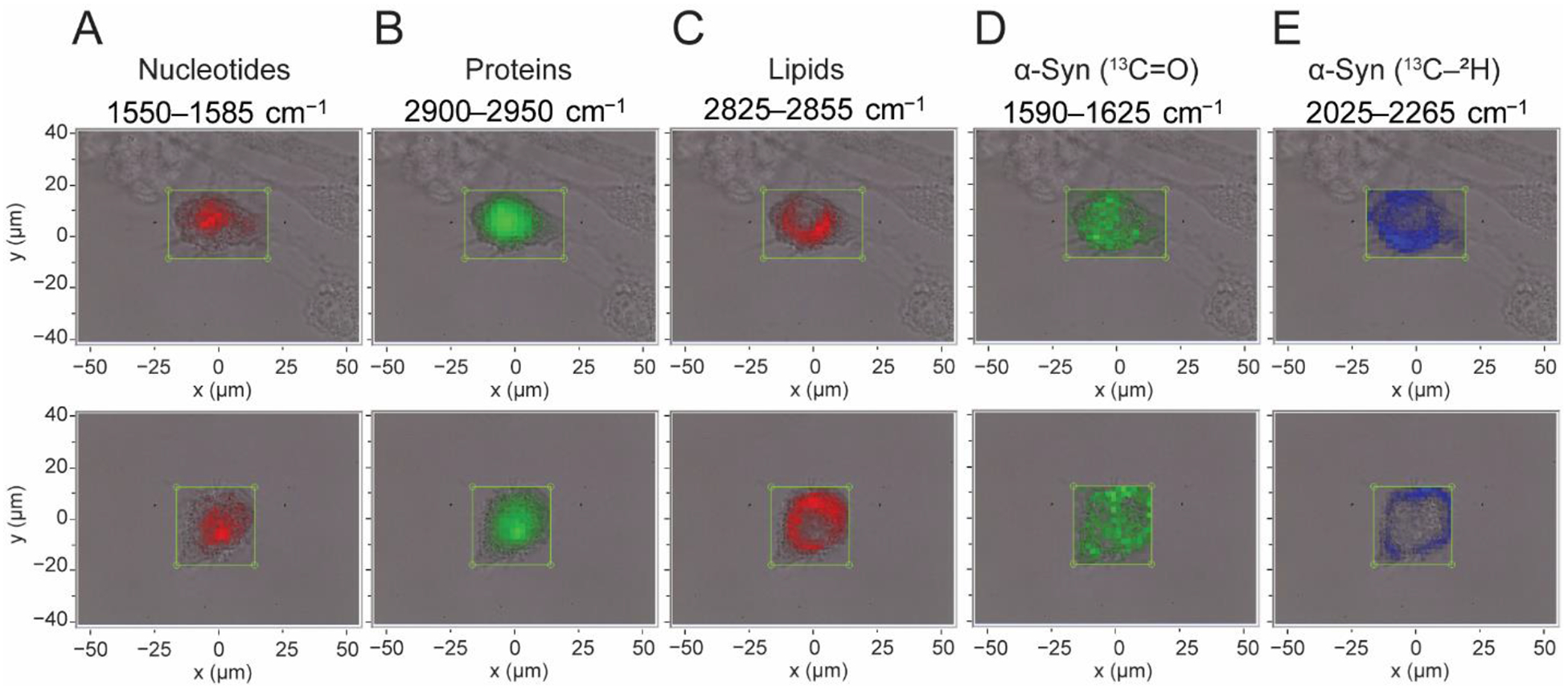
Maps of two individual cells indicating the locations of endogenous biomolecules including (A) nucleotides, (B) proteins, (C) lipids, along with the presence of 13C2H15N-α-syn fibrils as determined by (D) 13C=O amide-I and (E) 13C–2H stretching bands, generated by integrating regions of the Raman spectrum as indicated. Maps are overlaid with bright-field images, and boxes indicate the measurement area (2 μm steps).
Maps of the nucleotide spectral region clearly highlight the central location of nucleus with some diffuse intensity throughout the cytoplasm (Fig. 4A). Maps of the CH3 12C–1H stretching band which reports on endogenous proteins are more evenly diffused throughout the cell, but are again concentrated at the central portion of the cell (Fig. 4B). The distribution of lipids is indicated by the CH2 12C–1H stretching band, which is clearly excluded from the nucleus but is unexpectedly intense throughout the remainder of the cellular volume (Fig. 4C). This indicates an accumulation of lipids in the cytosol, which may be related to the membrane-disrupting properties of amyloid fibrils. Comparison of the maps generated using spectral regions constituting 13C=O amide-I and 13C–2H stretches show distinctive features. The amide-I peak appears to be more central and diffusive (Fig. 4D) whereas the 13C–2H peaks are more localized to the cellular periphery (Fig. 4E), with some fibril infiltration closer to the nucleus (Fig. 4E top panel). Although the fibril amide-I band is reasonably well-resolved from the congested fingerprint region, the relatively low intensity of this band resulted in comparatively noisy Raman maps. In contrast, the absence of interfering signals in the cellular quiet region yielded higher quality maps with superior signal-to-noise using the 13C–2H peaks.
2.5. Colocalization of α-syn fibrils with biomolecules in cells
Using these Raman maps, the colocalization of α-syn fibrils (13C–2H bands) and various cellular components can be examined (Fig. 5). α-Syn fibrils exhibited little to no colocalization with nucleotides (Fig. 5A) whereas there was partial colocalization with endogenous protein and lipid signatures, occurring largely at the interface of α-syn-rich and protein/lipid-rich regions, appearing as white pixels in Fig. 5B and 5C. In the cell where α-syn uptake appears to have been more significant (top panel), colocalization of α-syn with proteins and lipids was observed in the perinuclear region. These differences in colocalization are consistent with the variable uptake observed by ThT-staining and may represent different stages of fibril internalization and cell damage. Notably, although lipid bands are observed throughout much of the cell volume, they are not fully colocalized with the 12C–2H bands of the exogenous α-syn (Fig. 5C). This suggests that the cytosolic accumulation of lipids occurs without major infiltration by the fibrils themselves.
Fig. 5. Colocalization Raman maps.
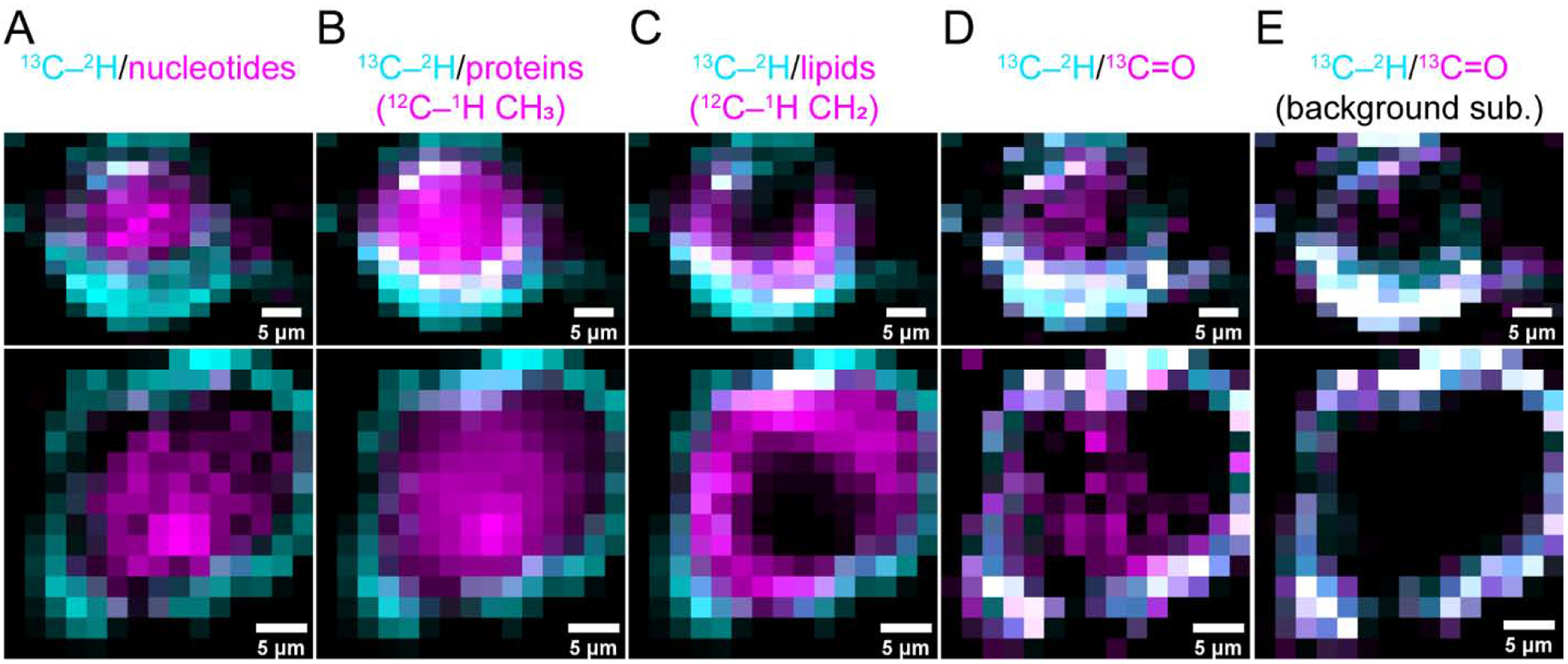
Composite Raman maps of 13C–2H stretching bands (cyan, 2025–2265 cm−1) with other biomolecules including (A) nucleotides (magenta, 1550–1585 cm−1), (B) endogenous proteins (magenta, 2900–2950 cm−1), and (C) lipids (magenta, 2825–2855 cm −1) as well as with the 13C=O amide-I peaks before (D) and after (E) cellular background subtraction (magenta, 1590–1625 and 1598–1626 cm−1, respectively). Colocalized areas appear as white. Two individual cells are shown in the upper and lower panels. Scale bars are as indicated.
A composite image (Fig. 5D) generated from the 13C–2H and 13C=O amide-I bands highlights the unexpected incongruity between the two. Despite strong colocalization distal from the nucleus, there are 13C=O-positive pixels (magenta) devoid of 13C–2H signals in the middle of the cell. While intensity differences in the amide-I can be explained by fibril structural perturbations inside the cell, the 13C–2H bands should always persist because they are expected to be insensitive to structural changes. Upon inspection, it is apparent that the differences between these maps likely arise from cellular background interference, because the 13C=O amide-I band is not fully resolved from endogenous molecular vibrations. To correct for this, a background subtraction was applied. Briefly, an average cellular background spectrum was first generated from untreated SK-MEL-28 cells, then normalized to the area of the 12C=O amide-I band at each point on the maps of treated cells and then used for subtraction. Now, in the background subtracted maps (Fig. 5E), the cellular spectral interference is significantly reduced, dramatically improving colocalization between the 13C=O amide-I and 13C–2H signals, indicating the spatial locations of internalized α-syn fibrils.
2.6. Analysis of the 13C=O Amide-I Band in cellulo
The amide-I band is well known to be sensitive to protein secondary structure with peak shifts, broadening, and intensity changes indicating differences in conformational states. Although this feature has been widely used to examine protein conformational changes in vitro, it presents a challenge in cells. Amide-I shape changes are not only difficult to analyze against a cellular background, but also in the absence of an internal standard, their intensity changes can be misinterpreted as differences in local concentration where none may in fact exist. To address this, we employed the cellular background subtraction approach outlined previously and coupled it with normalization by the integrated area of 13C–2H bands in order to evaluate changes in the 13C=O amide-I band of α-syn fibrils within and across cells.
An additional Raman map made using 13C–2H bands is shown in Fig. 6A with the corresponding individual Raman spectra measured at different pixels shown in Fig. 6B and 6C. Intensity differences are observed in both the 13C–2H (Fig. 6B) and the 13C=O amide-I peaks (Fig. 6C). However, upon normalization to the areas of the 13C–2H bands, it is apparent that there is not a significant variation in the shape of the measured 13C–2H bands as a function of location (Fig. 6D). Similarly, the 13C=O amide-I bands of these normalized spectra are comparable in intensity, with consistent peak shapes across the map indicating that fibril structure is similar at all spatial locations (Fig. 6E). We do note that there are observable shifts in the peak position for a few of the spectra.
Fig. 6. Raman spectra of internalized fibrils.
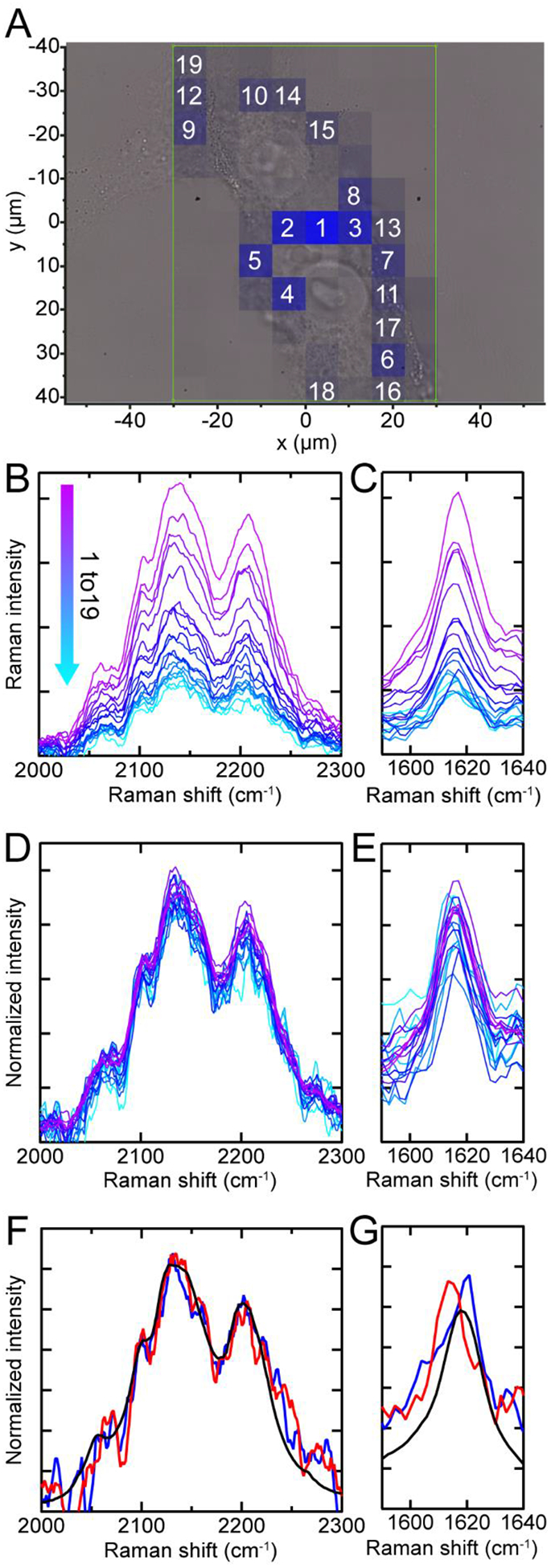
(A) Raman map of 13C–2H stretches (blue) overlaid with the bright-field image. Box indicates measurement area (7.5 μm steps). Raman spectra in the 13C–2H stretching and the amide-I regions with cellular background subtracted (B and C) and normalized by the area of the 13C–2H stretching bands (D and E). Their respective spatial locations are indicated by numbers in (A). The intervening spectral region was omitted for clarity. Comparison of additional Raman spectra (red and blue) to in vitro fibrils (black) in the 13C–2H (F) and 13C=O amide-I (G) regions. These are averaged and normalized spectra from two additional cells.
Consistent observations were also seen in other cells. Averaging spectra over an entire cell shows that there are no appreciable changes in the shape or frequency of the 13C–2H bands (Fig. 6F), while there are slight shifts in the 13C=O amide-I peak position of up to 7 cm−1 in the two extreme cases (Fig. 6G). In spite of these shifts, the relative intensities of the 13C=O amide-I and 13C–2H bands remain similar to that of in vitro fibrils (black curve) in all observed cases, suggesting that β-sheet-containing amyloid structure is retained upon cellular uptake.
These findings are in agreement with the results from a previous study that employed combined 13C- and HPG-labeling of α-syn [55], where cytosolic lipid accumulation was also observed in rat dopaminergic N27 cells treated with preformed fibrils. Here, however, we present mapping of whole cells by using the significantly more intense 13C–2H stretching bands compared to that of the C≡C stretching band of HPG. We note that a related approach was used in another recent study, in which poly-Q aggregates were metabolically labelled with deuterated glutamine amino acids and mapped in cells by stimulated Raman imaging [62]. While the use of metabolic labeling enables imaging of endogenously expressed proteins, it is restricted to proteins that are highly enriched in a specific amino acid with variable levels of background labeling. By comparison, our approach is entirely free of background signals and provides unambiguous localization of the internalized exogenously added proteins.
3. Conclusion
Isotopic labeling of α-syn amyloid fibrils combined with Raman spectroscopy offer a unique method for imaging exogenous proteins against the background of cellular components. 13C–2H stretching bands are intense, well-resolved from endogenous vibrational modes and insensitive to structural and environmental changes. This permits robust and reliable identification of α-syn distribution in a cellular environment and serves as an internal concentration standard relative to the amide-I band, which is sensitive to secondary structural changes. The 13C2H15N-labeled amide-I band is sufficiently separated from the amide-I band of endogenous proteins that changes in the peak frequency can be measured, allowing evaluation of structural integrity of the isotopically-labeled α-syn fibrils.
Raman mapping also affords simultaneous observation of endogenous cellular biomolecules, which revealed an accumulation of lipids in the cytosol, despite little α-syn buildup in this same region. Fibrils localize primarily to the cellular periphery, partially colocalizing with endogenous lipids and proteins. Critically, isotopic substitution is a uniquely nonperturbative labeling technique, which is an especially important concern in the study of amyloidogenic proteins, which have been demonstrated to be highly polymorphic, with fibril structure believed to be intricately linked to disease. The data presented here lay the groundwork for studying protein structural changes in the cellular environment, demonstrating the feasibility of mapping Raman spectra across entire cells and resolving the amide-I band from the cellular background, which has broad applicability to other amyloidogenic systems. Much remains to be explored, particularly development of spectroscopic approaches for direct observation of the monomer to fibril transition within a cell. A potential attractive methodology for future investigation is the incorporation of a genetically-encoded Raman probe, which would enable measurement of endogenously expressed proteins in a cellular environment.
4. Materials and Methods
4.1. Protein expression and purification
Natural isotope abundance α-syn was expressed in E. coli using the pRK172 plasmid [63] as described previously [64]. 13C2H15N-labeled α-syn was expressed in M9 media prepared with D-glucose (U-13C6, 1,2,3,4,5,6,6-D7), 15NH4Cl and D2O (Cambridge Isotopes Laboratories) according to previously published protocols [57, 65]. Proteins were purified by anion exchange chromatography as described previously [64]. Protein homogeneity was evaluated by SDS-PAGE. Protein molecular weights were confirmed by LC-ESI-MS (NHLBI Biochemistry Core Facility). Isotopic-labeling efficiency is estimated to be 96% based on the measured mass of 15,966.56 Da (expected mass: 16,026.95 Da). Purified protein was flash-frozen in liquid N2 and stored at −80 °C until use.
4.2. Amyloid formation and kinetics
Protein solutions were exchanged into pH 5 buffer (10 mM NaOAc and 100 mM NaCl) using a PD-10 desalting column (GE Healthcare) and then filtered (YM100 membranes; Millipore) immediately prior to aggregation to remove any preformed aggregates. Protein solutions were then incubated in 1.5 mL Eppendorf tubes at 37 °C and shaken at 600 rpm (Mini-Micro 980140 shaker, VWR) for 3–5 days to form fibrils. For ThT-monitored kinetics, α-syn and ThT were diluted into acetate buffer to final concentrations of 50 μM and 5 μM, respectively. Samples were aliquoted into a 384-well polypropylene microplate (Greiner Bio-One cat. 781209) with a 2.0 mm glass bead in each well and sealed with an optically clear adhesive film (Applied Biosystems cat. 4311971). ThT fluorescence was recorded at 30 min intervals on a Tecan Infinite M200 microplate reader (37 °C with 3-mm amplitude linear shaking) using excitation and emission wavelengths of 415 nm and 480 nm, respectively.
4.3. Raman Spectral Imaging
Raman spectra were recorded on a home-built Raman microscope [54] as previously described [55]. Briefly, the 514-nm line of an argon-ion laser (CVI Melles Griot, 35-MAP-431–200) was passed through a clean-up filter (Semrock, LL01-514-25) and directed to the sample using a dichroic mirror (Semrock, LPD01–514RU-25×36–1.1) and a 60× water-immersion objective (Olympus, UPLSAPO60XW) on an inverted microscope (Olympus IX71). Spontaneous Raman Stokes scattering was collected by the same objective, then directed through a filter (Semrock, LP02–514RE-25) into an imaging spectrometer (Horiba Scientific, iHR 320, 1200 gr/mm). A pinhole (400-μm, Thorlabs) at the imaging plane was used before the 50-μm entrance slit. Individual spectra were collected with 25 accumulations (10-s integration time) from 500–3700 cm−1 for in vitro fibril data or 2–3 accumulations (5–20-s integration time) from 1250–3000 cm−1 for cellular data on a liquid nitrogen cooled, back illuminated deep-depletion CCD array (Horiba Scientific, Symphony II, 1024×256 px, 26.6 mm × 6.6 mm, 1 MHz repetition rate, high-gain enabled, pixels 120–146 in the y-direction were binned). Bright-field images were collected using a USB 2.0 camera (iDS, UI-1220-C). Raman mapping was achieved using a Märzhäuser Wetzler motorized XYZ-stage (SCAN IM 120×80, Tango controller). Daily calibration of imaging spectrometer was performed using neat cyclohexane (20 μL in a sealed capillary tube). Bandpass and accuracy were found to be <12 cm−1 and ± 1 cm−1, respectively.
4.4. Transmission electron microscopy
α-Syn fibrils were deposited on 400 mesh formvar-coated copper grids (Electron Microscopy Sciences) for 1 min and excess sample was wicked away with filter paper. The grids were washed by quickly applying and wicking away a drop of deionized water. Grids were then stained with 1% uranyl acetate for 1 min and excess liquid was wicked away with filter paper. Grids were imaged on a JEOL EM-1200 EXII electron microscope (accelerating voltage 80 keV) equipped with an AMT XR-60 digital camera (NHLBI EM Core Facility).
4.5. Cell culture and fibril treatment
SK-MEL-28 (ATCC HTB-72) cells were maintained in phenol-free minimum essential medium (MEM, Thermo Fisher 41061307) supplemented with 10% fetal bovine serum (FBS, ATCC 30–2020) and 2% penicillin/streptomycin at 37 °C in 5% CO2. When cells reached 70–80% confluency, they were trypsinized by addition of 0.05% trypsin-EDTA (Gibco, Cat. No. 2520056) for 5 min at RT.
For confocal fluorescence imaging of ThT-stained fibrils, resuspended cells were added to poly-D-lysine coated coverslips at a 1:10 dilution and allowed to grow for 24 h. Fibrils stained with ThT were pelleted by centrifugation (16,000 ×g, 20 °C, 30 min), resuspended in fresh PBS (KD Medical, Cat. No. RGF-3210), diluted into growth media and added to cells. An identical volume of PBS was added to the control cells to mirror the fibril treatment condition. After 1 h incubation, cells were washed 3× with PBS and fixed in a 2.5% paraformaldehyde solution (Electron Microscopy Sciences, Cat. No. 15710) for 15 min, washed 3× with PBS, and stored at 4 °C until imaging.
For Raman imaging experiments, resuspended cells were added to Nunc Lab-Tek II chambered slides (Thermo Fisher, Cat. No. 155411) at a 1:10 dilution and allowed to grow for 24–48 h. Fibrils were pelleted by centrifugation (16,000 ×g, 20 °C, 30 min), resuspended into PBS and probe-tip sonicated for 30-s on a 50% duty cycle before diluting to a final concentration of 25 μM into growth media, and then added to cells. After 24 h incubation, cells were washed, fixed and stored as before.
4.6. Confocal fluorescence microscopy
Samples were imaged using a PlanApo N 60×/1.42 NA oil objective (Olympus, Tokyo, Japan) on an Olympus IX73 inverted microscope fitted with a Thorlabs Confocal Laser Scanner (CLS-SL) fiber coupled to a multichannel CMLS-E laser source. ThT was excited using a 405-nm laser and fluorescence was collected using a 482/18 nm Brightline bandpass filter (Semrock) by a high-sensitivity GaAsP photomultiplier tube. A 50-μm pinhole was used and the scale was 0.202 μm/pixel.
4.7. Raman spectral imaging analysis
Raman spectra were analyzed in the LabSpec 6 software (Horiba Scientific). For in vitro spectra, separate buffer background spectra were acquired and subtracted from fibril spectra before applying a second degree baseline polynomial fit. For Raman maps, a spectrum from the periphery with the fewest spectral features was selected as a background and subtracted from all spectra. A second degree polynomial baseline fit was then applied to all spectra. Maps of cellular features were prepared by integrating with a linear baseline subtraction over a defined spectral region at all map points. To further remove background contributions, Raman spectra from a map of an untreated control SK-MEL-28 cell were averaged together, normalized to the area of the 12C=O amide-I band at each point on a map of fibril-treated cells and subtracted from the spectrum at each point. Cellular data were smoothed using a Savitzky-Golay algorithm.
Colocalization images were prepared in the FIJI software. A gaussian blur was applied to the Raman maps and subtracted from the raw image to remove background signal. Colocalized pixels were identified by a Boolean algebra intersection function, and composite images were generated by merging the colocalized pixels with the original Raman maps as three color channels.
Acknowledgment
This work is supported by the Intramural Research Program at the National Institutes of Health, National Heart, Lung, and Blood Institute. Parts of this research were performed on instruments maintained by the NHLBI Biochemistry (LC-MS) and Electron Microscopy (TEM) core. We thank Michael Goedert (MRC Laboratory of Molecular Biology) for the pRK172 plasmid. The content of this work is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Declaration of interests
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
References
- [1].Lees AJ, Hardy J, Revesz T, Parkinson’s disease, Lancet, 373 (2009) 2055–2066. [DOI] [PubMed] [Google Scholar]
- [2].Baba M, Nakajo S, Tu PH, Tomita T, Nakaya K, Lee VMY, Trojanowski JQ, Iwatsubo T, Aggregation of α-synuclein in Lewy bodies of sporadic Parkinson’s disease and dementia with Lewy bodies, Am J Pathol, 152 (1998) 879–884. [PMC free article] [PubMed] [Google Scholar]
- [3].Spillantini MG, Schmidt ML, Lee VM, Trojanowski JQ, Jakes R, Goedert M, α-Synuclein in Lewy bodies, Nature, 388 (1997) 839–840. [DOI] [PubMed] [Google Scholar]
- [4].Polymeropoulos MH, Lavedan C, Leroy E, Ide SE, Dehejia A, Dutra A, Pike B, Root H, Rubenstein J, Boyer R, Stenroos ES, Chandrasekharappa S, Athanassiadou A, Papapetropoulos T, Johnson WG, Lazzarini AM, Duvoisin RC, DiIorio G, Golbe LI, Nussbaum RL, Mutation in the α-synuclein gene identified in families with Parkinson’s disease, Science, 276 (1997) 2045–2047. [DOI] [PubMed] [Google Scholar]
- [5].Kruger R, Kuhn W, Muller T, Woitalla D, Graeber M, Kosel S, Przuntek H, Epplen JT, Schols L, Riess O, Ala30Pro mutation in the gene encoding α-synuclein in Parkinson’s disease, Nat Genet, 18 (1998) 106–108. [DOI] [PubMed] [Google Scholar]
- [6].Zarranz JJ, Alegre J, Gomez-Esteban JC, Lezcano E, Ros R, Ampuero I, Vidal L, Hoenicka J, Rodriguez O, Atares B, Llorens V, Gomez Tortosa E, del Ser T, Munoz DG, de Yebenes JG, The new mutation, E46K, of α-synuclein causes Parkinson and Lewy body dementia, Ann Neurol, 55 (2004) 164–173. [DOI] [PubMed] [Google Scholar]
- [7].Lesage S, Anheim M, Letournel F, Bousset L, Honore A, Rozas N, Pieri L, Madiona K, Durr A, Melki R, Verny C, Brice A, French G Parkinson’s Disease Genetics Study, G51D α-synuclein mutation causes a novel Parkinsonian-pyramidal syndrome, Ann Neurol, 73 (2013) 459–471. [DOI] [PubMed] [Google Scholar]
- [8].Appel-Cresswell S, Vilarino-Guell C, Encarnacion M, Sherman H, Yu I, Shah B, Weir D, Thompson C, Szu-Tu C, Trinh J, Aasly JO, Rajput A, Rajput AH, Jon Stoessl A, Farrer MJ, α-Synuclein p.H50Q, a novel pathogenic mutation for Parkinson’s disease, Mov Disord, 28 (2013) 811–813. [DOI] [PubMed] [Google Scholar]
- [9].Pasanen P, Myllykangas L, Siitonen M, Raunio A, Kaakkola S, Lyytinen J, Tienari PJ, Poyhonen M, Paetau A, Novel α-synuclein mutation A53E associated with atypical multiple system atrophy and Parkinson’s disease-type pathology, Neurobiol Aging, 35 (2014) 2180 e2181–2185. [DOI] [PubMed] [Google Scholar]
- [10].Ueda K, Fukushima H, Masliah E, Xia Y, Iwai A, Yoshimoto M, Otero DA, Kondo J, Ihara Y, Saitoh T, Molecular cloning of cDNA encoding an unrecognized component of amyloid in Alzheimer disease, Proc Natl Acad Sci USA, 90 (1993) 11282–11286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Wilhelm BG, Mandad S, Truckenbrodt S, Krohnert K, Schafer C, Rammner B, Koo SJ, Classen GA, Krauss M, Haucke V, Urlaub H, Rizzoli SO, Composition of isolated synaptic boutons reveals the amounts of vesicle trafficking proteins, Science, 344 (2014) 1023–1028. [DOI] [PubMed] [Google Scholar]
- [12].Iwai A, Masliah E, Yoshimoto M, Ge NF, Flanagan L, Desilva HAR, Kittel A, Saitoh T, The precursor protein of non-A-beta component of Alzheimers-disease amyloid is a presynaptic protein of the central-nervous-system, Neuron, 14 (1995) 467–475. [DOI] [PubMed] [Google Scholar]
- [13].Clayton DF, George JM, The synucleins: A family of proteins involved in synaptic function, plasticity, neurodegeneration and disease, Trends Neurosci, 21 (1998) 249–254. [DOI] [PubMed] [Google Scholar]
- [14].Abeliovich A, Schmitz Y, Farinas I, Choi-Lundberg D, Ho WH, Castillo PE, Shinsky N, Verdugo JM, Armanini M, Ryan A, Hynes M, Phillips H, Sulzer D, Rosenthal A, Mice lacking α-synuclein display functional deficits in the nigrostriatal dopamine system, Neuron, 25 (2000) 239–252. [DOI] [PubMed] [Google Scholar]
- [15].Greten-Harrison B, Polydoro M, Morimoto-Tomita M, Diao L, Williams AM, Nie EH, Makani S, Tian N, Castillo PE, Buchman VL, Chandra SS, αβγ-Synuclein triple knockout mice reveal age-dependent neuronal dysfunction, Proc Natl Acad Sci USA, 107 (2010) 19573–19578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Burre J, Sharma M, Tsetsenis T, Buchman V, Etherton MR, Südhof TC, α-Synuclein promotes SNARE-complex assembly in vivo and in vitro, Science, 329 (2010) 1663–1667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Burre J, Sharma M, Südhof TC, α-Synuclein assembles into higher-order multimers upon membrane binding to promote SNARE complex formation, Proc Natl Acad Sci USA, 111 (2014) E4274–4283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Kaur U, Lee JC, Unroofing site-specific α-synuclein lipid interactions at the plasma membrane, Proc Natl Acad Sci USA, 117 (2020) 18977–18983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Meredith GE, Totterdell S, Petroske E, Santa Cruz K, Callison RC Jr., Lau YS, Lysosomal malfunction accompanies α-synuclein aggregation in a progressive mouse model of Parkinson’s disease, Brain Res, 956 (2002) 156–165. [DOI] [PubMed] [Google Scholar]
- [20].Gosavi N, Lee HJ, Lee JS, Patel S, Lee SJ, Golgi fragmentation occurs in the cells with prefibrillar α-synuclein aggregates and precedes the formation of fibrillar inclusion, J Biol Chem, 277 (2002) 48984–48992. [DOI] [PubMed] [Google Scholar]
- [21].Fujita Y, Ohama E, Takatama M, Al-Sarraj S, Okamoto K, Fragmentation of Golgi apparatus of nigral neurons with α-synuclein-positive inclusions in patients with Parkinson’s disease, Acta Neuropathol, 112 (2006) 261–265. [DOI] [PubMed] [Google Scholar]
- [22].Nakamura K, Nemani VM, Azarbal F, Skibinski G, Levy JM, Egami K, Munishkina L, Zhang J, Gardner B, Wakabayashi J, Sesaki H, Cheng Y, Finkbeiner S, Nussbaum RL, Masliah E, Edwards RH, Direct membrane association drives mitochondrial fission by the Parkinson disease-associated protein α-synuclein, J Biol Chem, 286 (2011) 20710–20726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Freeman D, Cedillos R, Choyke S, Lukic Z, McGuire K, Marvin S, Burrage AM, Sudholt S, Rana A, O’Connor C, Wiethoff CM, Campbell EM, α-Synuclein induces lysosomal rupture and cathepsin dependent reactive oxygen species following endocytosis, PLoS One, 8 (2013) e62143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Conway KA, Lee SJ, Rochet JC, Ding TT, Williamson RE, Lansbury PT, Acceleration of oligomerization, not fibrillization, is a shared property of both α-synuclein mutations linked to early-onset Parkinson’s disease: Implications for pathogenesis and therapy, Proc Natl Acad Sci USA, 97 (2000) 571–576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Conway KA, Harper JD, Lansbury PT, Accelerated in vitro fibril formation by a mutant α-synuclein linked to early-onset Parkinson disease, Nat Med, 4 (1998) 1318–1320. [DOI] [PubMed] [Google Scholar]
- [26].Lashuel HA, Petre BM, Wall J, Simon M, Nowak RJ, Walz T, Lansbury PT, α-Synuclein, especially the Parkinson’s disease-associated mutants, forms pore-like annular and tubular protofibrils, J Mol Biol, 322 (2002) 1089–1102. [DOI] [PubMed] [Google Scholar]
- [27].Choi W, Zibaee S, Jakes R, Serpell LC, Davletov B, Crowther RA, Goedert M, Mutation E46K increases phospholipid binding and assembly into filaments of human α-synuclein, FEBS Lett, 576 (2004) 363–368. [DOI] [PubMed] [Google Scholar]
- [28].Fares MB, Ait-Bouziad N, Dikiy I, Mbefo MK, Jovicic A, Kiely A, Holton JL, Lee SJ, Gitler AD, Eliezer D, Lashuel HA, The novel Parkinson’s disease linked mutation G51D attenuates in vitro aggregation and membrane binding of α-synuclein, and enhances its secretion and nuclear localization in cells, Hum Mol Genet, 23 (2014) 4491–4509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Bussell R, Eliezer D, Residual structure and dynamics in Parkinson’s disease-associated mutants of α-synuclein, J Biol Chem, 276 (2001) 45996–46003. [DOI] [PubMed] [Google Scholar]
- [30].Jensen PH, Nielsen MS, Jakes R, Dotti G, Goedert M, Binding of α-synuclein to brain vesicles is abolished by familial Parkinson’s disease mutation, J Biol Chem, 273 (1998) 26292–26294. [DOI] [PubMed] [Google Scholar]
- [31].Jo E, Fuller N, Rand RP, St George-Hyslop P, Fraser PE, Defective membrane interactions of familial Parkinson’s disease mutant A30P α-synuclein, J Mol Biol, 315 (2002) 799–807. [DOI] [PubMed] [Google Scholar]
- [32].Khalaf O, Fauvet B, Oueslati A, Dikiy I, Mahul-Mellier AL, Ruggeri FS, Mbefo MK, Vercruysse F, Dietler G, Lee SJ, Eliezer D, Lashuel HA, The H50Q mutation enhances α-synuclein aggregation, secretion, and toxicity, J Biol Chem, 289 (2014) 21856–21876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Ding TT, Lee SJ, Rochet JC, Lansbury PT Jr., Annular α-synuclein protofibrils are produced when spherical protofibrils are incubated in solution or bound to brain-derived membranes, Biochemistry, 41 (2002) 10209–10217. [DOI] [PubMed] [Google Scholar]
- [34].Butterfield SM, Lashuel HA, Amyloidogenic protein-membrane interactions: Mechanistic insight from model systems, Angew Chem Int Ed Engl, 49 (2010) 5628–5654. [DOI] [PubMed] [Google Scholar]
- [35].Beyer K, Mechanistic aspects of parkinson’s disease: Alpha-synuclein and the biomembrane, Cell Biochem Biophys, 47 (2007) 285–299. [DOI] [PubMed] [Google Scholar]
- [36].Dikiy I, Eliezer D, Folding and misfolding of α-synuclein on membranes, Biochim Biophys Acta, 1818 (2012) 1013–1018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Shahmoradian SH, Lewis AJ, Genoud C, Hench J, Moors TE, Navarro PP, Castano-Diez D, Schweighauser G, Graff-Meyer A, Goldie KN, Sutterlin R, Huisman E, Ingrassia A, Gier Y, Rozemuller AJM, Wang J, Paepe A, Erny J, Staempfli A, Hoernschemeyer J, Grosseruschkamp F, Niedieker D, El-Mashtoly SF, Quadri M, Van IWFJ, Bonifati V, Gerwert K, Bohrmann B, Frank S, Britschgi M, Stahlberg H, Van de Berg WDJ, Lauer ME, Lewy pathology in Parkinson’s disease consists of crowded organelles and lipid membranes, Nat Neurosci, 22 (2019) 1099–1109. [DOI] [PubMed] [Google Scholar]
- [38].Caughey B, Lansbury PT, Protofibrils, pores, fibrils, and neurodegeneration: Separating the responsible protein aggregates from the innocent bystanders, Annu Rev Neurosci, 26 (2003) 267–298. [DOI] [PubMed] [Google Scholar]
- [39].Bousset L, Pieri L, Ruiz-Arlandis G, Gath J, Jensen PH, Habenstein B, Madiona K, Olieric V, Bockmann A, Meier BH, Melki R, Structural and functional characterization of two α-synuclein strains, Nat Commun, 4 (2013) 2575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Emmanouilidou E, Melachroinou K, Roumeliotis T, Garbis SD, Ntzouni M, Margaritis LH, Stefanis L, Vekrellis K, Cell-produced α-synuclein is secreted in a calcium-dependent manner by exosomes and impacts neuronal survival, J Neurosci, 30 (2010) 6838–6851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Alvarez-Erviti L, Seow Y, Schapira AH, Gardiner C, Sargent IL, Wood MJ, Cooper JM, Lysosomal dysfunction increases exosome-mediated α-synuclein release and transmission, Neurobiol Dis, 42 (2011) 360–367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Guo JL, Lee VM, Cell-to-cell transmission of pathogenic proteins in neurodegenerative diseases, Nat Med, 20 (2014) 130–138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].Goedert M, Falcon B, Clavaguera F, Tolnay M, Prion-like mechanisms in the pathogenesis of tauopathies and synucleinopathies, Curr Neurol Neurosci Rep, 14 (2014) 495. [DOI] [PubMed] [Google Scholar]
- [44].Volpicelli-Daley LA, Luk KC, Lee VM, Addition of exogenous α-synuclein preformed fibrils to primary neuronal cultures to seed recruitment of endogenous α-synuclein to Lewy body and Lewy neurite-like aggregates, Nat Protoc, 9 (2014) 2135–2146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [45].Guerrero-Ferreira R, Taylor NM, Mona D, Ringler P, Lauer ME, Riek R, Britschgi M, Stahlberg H, Cryo-em structure of α-synuclein fibrils, eLife, 7 (2018) 18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [46].Li Y, Zhao C, Luo F, Liu Z, Gui X, Luo Z, Zhang X, Li D, Liu C, Li X, Amyloid fibril structure of α-synuclein determined by cryo-electron microscopy, Cell Res, 28 (2018) 897–903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [47].Ni X, McGlinchey RP, Jiang J, Lee JC, Structural insights into α-synuclein fibril polymorphism: Effects of Parkinson’s disease-related C-terminal truncations, J Mol Biol, 431 (2019) 3913–3919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [48].Tuttle MD, Comellas G, Nieuwkoop AJ, Covell DJ, Berthold DA, Kloepper KD, Courtney JM, Kim JK, Barclay AM, Kendall A, Wan W, Stubbs G, Schwieters CD, Lee VM, George JM, Rienstra CM, Solid-state NMR structure of a pathogenic fibril of full-length human α-synuclein, Nat Struct Mol Biol, 23 (2016) 409–415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [49].Vilar M, Chou HT, Luhrs T, Maji SK, Riek-Loher D, Verel R, Manning G, Stahlberg H, Riek R, The fold of α-synuclein fibrils, Proc Natl Acad Sci USA, 105 (2008) 8637–8642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [50].Boyer DR, Li B, Sun C, Fan W, Sawaya MR, Jiang L, Eisenberg DS, Structures of fibrils formed by α-synuclein hereditary disease mutant H50Q reveal new polymorphs, Nat Struct Mol Biol, 26 (2019) 1044–1052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [51].Zhao K, Li Y, Liu Z, Long H, Zhao C, Luo F, Sun Y, Tao Y, Su XD, Li D, Li X, Liu C, Parkinson’s disease associated mutation E46K of α-synuclein triggers the formation of a distinct fibril structure, Nat Commun, 11 (2020) 2643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [52].Schweighauser M, Shi Y, Tarutani A, Kametani F, Murzin AG, Ghetti B, Matsubara T, Tomita T, Ando T, Hasegawa K, Murayama S, Yoshida M, Hasegawa M, Scheres SHW, Goedert M, Structures of α-synuclein filaments from multiple system atrophy, Nature, 585 (2020) 464–469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [53].Pelton JT, McLean LR, Spectroscopic methods for analysis of protein secondary structure, Anal Biochem, 277 (2000) 167–176. [DOI] [PubMed] [Google Scholar]
- [54].Flynn JD, McGlinchey RP, Walker RL 3rd, Lee JC, Structural features of α-synuclein amyloid fibrils revealed by Raman spectroscopy, J Biol Chem, 293 (2018) 767–776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [55].Flynn JD, Gimmen MY, Dean DN, Lacy SM, Lee JC, Terminal alkynes as Raman probes of α-synuclein in solution and in cells, ChemBioChem, 21 (2020) 1582–1586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [56].LeVine H 3rd, Quantification of β-sheet amyloid fibril structures with Thioflavin T, Methods Enzymol, 309 (1999) 274–284. [DOI] [PubMed] [Google Scholar]
- [57].Flynn JD, Jiang Z, Lee JC, Segmental 13C-labeling and Raman microspectroscopy of α-synuclein amyloid formation, Angew Chem Int Ed Engl, 57 (2018) 17069–17072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [58].Pan T, Zhu J, Hwu WJ, Jankovic J, The role of α-synuclein in melanin synthesis in melanoma and dopaminergic neuronal cells, PLoS One, 7 (2012) e45183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [59].Scott G, Zhao Q, Rab3a and SNARE proteins: Potential regulators of melanosome movement, J Invest Dermatol, 116 (2001) 296–304. [DOI] [PubMed] [Google Scholar]
- [60].Dean DN, Lee JC, Defining an amyloid link between Parkinson’s disease and melanoma, Proc Natl Acad Sci USA, 117 (2020) 22671–22673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [61].Schie IW, Kiselev R, Krafft C, Popp J, Rapid acquisition of mean Raman spectra of eukaryotic cells for a robust single cell classification, Analyst, 141 (2016) 6387–6395. [DOI] [PubMed] [Google Scholar]
- [62].Miao K, Wei L, Live-cell imaging and quantification of polyQ aggregates by stimulated raman scattering of selective deuterium labeling, ACS Cent Sci, 6 (2020) 478–486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [63].Jakes R, Spillantini MG, Goedert M, Identification of two distinct synucleins from human brain, FEBS Lett, 345 (1994) 27–32. [DOI] [PubMed] [Google Scholar]
- [64].Pfefferkorn CM, Lee JC, Tryptophan probes at the α-synuclein and membrane interface, J Phys Chem B, 114 (2010) 4615–4622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [65].Jiang Z, Heinrich F, McGlinchey RP, Gruschus JM, Lee JC, Segmental deuteration of α-synuclein for neutron reflectometry on tethered bilayers, J Phys Chem Lett, 8 (2017) 29–34. [DOI] [PMC free article] [PubMed] [Google Scholar]