

Abstract
Oligodendrocyte precursor cells (OPCs), also known as NG2 cells or polydendrocytes, are distributed widely throughout the developing and mature central nervous system. They remain proliferative throughout life and are an important source of myelinating cells in normal and demyelinating brain as well as a source of glioma, the most common type of primary brain tumor with a poor prognosis. OPC proliferation is dependent on signaling mediated by platelet-derived growth factor (PDGF) AA binding to its alpha receptor (PDGFRα). Here, we describe a group of structurally related compounds characterized by the presence of a basic guanidine group appended to an aromatic core that is effective in specifically repressing the transcription of Pdgfra but not the related beta receptor (Pdgfrb) in OPCs. These compounds specifically and dramatically reduced proliferation of OPCs but not that of astrocytes and did not affect signal transduction by PDGFRα. These findings suggest that the compounds could be further developed for potential use in combinatorial treatment strategies for neoplasms with dysregulated PDGFRα function.
Keywords: glioma, NG2, oligodendrocyte precursor, PDGF receptor, platelet-derived growth factor, proliferation
1 |. INTRODUCTION
Oligodendrocyte precursor cells (OPCs, also known as NG2 glia or polydendrocytes) are a glial cell population that is distributed widely in the central nervous system (CNS) and remains proliferative throughout life. They are identified by the expression of the alpha receptor for platelet-derived growth factor (PDGFRα) and NG2 proteoglycan, which are lost upon their terminal oligodendrocyte differentiation (Nishiyama, Komitova, Suzuki, & Zhu, 2009; Nishiyama, Lin, Giese, Heldin, & Stallcup, 1996a, 1996b). The production of the correct number of OPCs and oligodendrocytes is critically dependent on PDGF AA acting on PDGFRα (Calver et al., 1998; Fruttiger et al., 1999) throughout development as well as in the adult (Dang et al., 2019). Among other receptor tyrosine kinases that affect the oligodendrocyte lineage, epidermal growth factor receptor promotes transition from neural stem/progenitor cells into oligodendrocyte lineage cells (Galvez-Contreras, Quinones-Hinojosa, & Gonzalez-Perez,−2013), and fibroblast growth factor receptors (FGFRs) regulate various phases of oligodendrocyte lineage development, particularly their differentiation (Bansal, Kumar, Murray, Morrison, & Pfeiffer, 1996).
PDGFRα is a receptor tyrosine kinase, which is activated by dimeric ligands containing PDGF A, B, or C (Andrae, Gallini, & Betsholtz, 2008). Its close relative PDGFRβ is expressed by vascular mural cells (Ozerdem, Grako, Dahlin-Huppe, Monosov, & Stallcup, 2001; Zhu, Bergles, & Nishiyama, 2008) and neural stem cells in the subventricular zone (Ishii et al., 2008) but not by OPCs. The proliferative response of OPCs to PDGF AA is mediated through PDGFRα recruiting phosphatidylinositol-3-kinase (PI-3K) and subsequently activating Akt (Budde et al., 2010; McKinnon, Waldron, & Kiel, 2005). We previously showed that OPCs in the early postnatal white matter proliferate in response to PDGF AA through PI-3K-Akt and Wnt signaling (Hill, Patel, Medved, Reiss, & Nishiyama, 2013).
PDGFRα expression has emerged, along with other OPC gene products such as Olig2, as the molecular signature of the proneural subtype of highly malignant glioblastoma multiforme (The Cancer Genome Atlas Research Network, Nature, 2008; Verhaak et al., 2010). PDGFRA is also often amplified or mutated in pediatric high grade glioma (Jones & Baker, 2014; Paugh et al., 2010; Zarghooni et al., 2010). An autocrine/paracrine loop of aberrant PDGF signaling has been implicated in some gliomas (Hermanson et al., 1992; Nazarenko et al., 2012; Zheng et al., 2016). Lineage tracing studies have revealed OPCs as the cell of origin of some forms of experimental glioma (Galvao et al., 2014; Liu et al., 2011), suggesting that effective anti-glioma therapy could involve one that targets endogenous signaling pathways (Touat, Idbaih, Sanson, & Ligon, 2017). The limited anti-tumor effects of pharmaceutical reagents that block PDGFRα function could be attributed to multiple factors that include poor blood–brain barrier permeability and the complexity of the signaling pathways in these tumors (Alphandery, 2018; Phuphanich et al., 2017).
Here, we describe structurally related compounds with a basic guanidine moiety that specifically inhibited Pdgfra transcription and dramatically decreased OPC proliferation without causing significant cell death or differentiation. These guanidine compounds may function upstream of PDGFRα, which makes them unique from other anti-PDGFRA drugs that target the receptor or its downstream effectors (Nazarenko et al., 2012; Van Meir et al., 2010).
2 |. MATERIALS AND METHODS
2.1 |. Compounds
A diverse library of 1,520 small molecule compounds derived from commercial sources (ChemBridge Corporation, San Diego, CA) was obtained from the University of Connecticut High-Throughput Screening Facility in 96-well plate format containing 5 mM stocks of compounds dissolved in dimethyl sulfoxide (DMSO). Additional compounds were individually purchased from Hit2Lead (ChemBridge) and dissolved in DMSO at a stock concentration of 50 mM. For structure-activity relationship analysis, additional guanidine congeners were synthesized in house. In brief, aryl substituted primary or secondary amines were reacted with the guanidinylation reagent N,N-di-t-but-yloxylcarbonyl-S-methyl isothiourea. The resulting di-BOC substituted guanidines were deprotected by action of trifluoroacetic acid. The new guanidine compounds were purified to at least 95% purity and the structure confirmed by 1H NMR, 13C NMR, and MS analysis.
2.2 |. Plasmids
We generated Pdgfra-luciferase fragments that contained various lengths of the mouse Pdgfra 5′ flanking sequence by PCR using mouse genomic DNA as template (see Table 1 for primer sequences) and cloned them into pGL4.10 vector encoding Firefly luciferase 2 (Promega Corporation, Fitchburg, WI). The insertion site and sequence were confirmed by Sanger sequencing. Plasmids were purified with Plasmid Midi Kit (Qiagen, Germantown, MD).
TABLE 1.
Primers used to generate Pdgfra-luc constructs
| Plasmid name | Orientation | Primer sequence |
|---|---|---|
| Pdgfra-111 | Forward primer Reverse primer |
5′-CCGGTACCCAGAGAGCAAGGAGTCCTAGGG 5′-CCGAGCTCCTCTCCCTCAAGCTCCAACAG |
| Pdgfra-336 | Forward primer Reverse primer |
5′-CCGGTACCCACCCCCAAATTGGGAAGTC 5′-CCGAGCTCCTCTCCCTCAAGCTCCAACAG |
| Pdgfra-1000 | Forward primer Reverse primer |
5′-CCGGTACCTTGGTTCCTGGAGTGTCAGC 5′-CCGAGCTCCTCTCCCTCAAGCTCCAACAG |
| Pdgfra-1595 | Forward primer Reverse primer |
5′-CCGGTACCGTGCAAGCCTGTTCGCAGAC 5′-CCGAGCTCCTCTCCCTCAAGCTCCAACAG |
| Pdgfra-2229 | Forward primer Reverse primer |
5′-GACACCCTGGGTTGAGTGAC 5′-GGCTCGAGCTCTCCCTCAAGCTCCAACAG |
The plasmid pCDH-CMV-Pdgfra-EF1α-green fluorescent protein (GFP) encoding full-length mouse Pdgfra cDNA (CMV-Pdgfra-EF1a-GFP) (Huang et al., 2017) was obtained from Addgene (#99731). We generated control EF1α-GFP plasmid by digesting pCDH-CMV-PDGFRα-EF1α-GFP with XbaI and BsiWI to remove the Pdgfra coding sequence and religating the cut ends.
2.3 |. Cell culture
2.3.1 |. Cell lines
Oli-neu cells, a mouse OPC line generated by transformation with the neu oncogene (Jung et al., 1995; RRID:CVCL_IZ82), were plated on tissue culture dishes coated with 100 μg/ml poly-L-lysine (Sigma-Aldrich Corporation, St. Louis, MO) and maintained in growth medium consisting of Dulbecco’s Modified Eagle Medium/Ham’s F-12 (DMEM/F12) (Life Technologies, Carlsbad, CA), supplemented with N2 (Life Technologies) and 1% horse serum (Hyclone, Logan, UT) without PDGF AA. Mouse NIH3T3 cells were obtained from American Type Culture Collection (ATCC; Manassas, VA; RRID:CVCL_0594) and maintained in DMEM supplemented with 10% newborn calf serum (Sigma-Aldrich), 1% penicillin/streptomycin (Life Technologies), and 1% glutamine (Life Technologies). Cells were maintained at 37°C in 5% CO2.
2.3.2 |. Primary and secondary glial cell culture
OPCs from the cerebral cortex of perinatal CD rats (Charles River Laboratories, Wilmington, MA) or FVB or CD1 mice (Jackson Laboratory, Bar Harbor, ME; Charles River Laboratories, respectively) were prepared by the shake-off method (McCarthy & de Vellis, 1980; Yang, Watanabe, & Nishiyama, 2005) or directly by immunopanning (Dugas, Tai, Speed, Ngai, & Barres, 2006), with some modifications. For the shake-off method, mixed glial cultures were maintained in DMEM containing 10% fetal bovine serum (Life Technologies) for 6–8 days, after which the flasks were shaken overnight. Released cells were preplated on uncoated tissue culture dishes for 30 min to remove astrocytes and microglia, and the non-adherent cells were further enriched for OPCs by negative immunopanning with monoclonal antibodies O1 (RRID:AB_2314990) and O4 (RRID:AB_2314992) to remove oligodendrocytes (Sommer & Schachner, 1981; obtained from Dr S. Pfeiffer, University of Connecticut Health Center, Farmington, CT). To obtain rat OPCs directly from P4 rat cortex, dissociated cells were negatively immunopanned by O1 antibody, followed by positive selection with goat anti-mouse PDGFRα antibody (R&D Systems, Minneapolis, MN, 1 μg/ml; RRID:AB_2236897). Mouse OPCs were obtained directly from P1-P4 mouse cortex by immunopanning (Dugas et al., 2006) using positive selection with 2 μg/ml CD140a rat anti-mouse PDGFRα antibody (BD Biosciences, San Jose, CA, Cat# 558774, RRID:AB_397117). OPCs were maintained in DMEM supplemented with 100 μg/ml transferrin, 100 μg/ml bovine serum albumin, 16 μg/ml putrescine, 60 ng/ml progesterone, 40 ng/ml sodium selenite, 5 μg/ml N-acetyl cysteine, 10 ng/ml D-biotin, 5 μg/ml insulin, and 5 μM forskolin (from Sigma-Aldrich); 1 mM sodium pyruvate (Life Technologies), 1X trace elements B (Corning, Oneonta, NY); 10 ng/ml ciliary neurotrophic factor, 1 ng/ml neutrophin 3, and 20 ng/ml mouse PDGF AA (PeproTech, Rocky Hill, NJ).
To obtain enriched cultures of primary mouse astrocytes, mixed glial cultures were shaken off to remove loosely attached cells, and astrocytes that remained in the flask were trypsinized and collected. Astrocytes were maintained in DMEM supplemented with 10% fetal bovine serum (Life Technologies) 1% penicillin/streptomycin (Life Technologies), and 1% glutamine (Life Technologies). Cells were maintained at 37°C in 5% CO2.
Transfections of plasmids into P2 mouse OPCs or astrocytes was performed using Lipofectamine 2000 in 35 mm dishes, and transfected cells were incubated with the compounds starting on the following day for EdU incorporation or immunofluorescence assays.
2.3.3 |. Slice culture
Organotypic slice cultures were prepared from P8 mice that were double transgenic for NG2cre (Jackson Laboratory stock 008533) and the Cre reporter Z/EG (Jackson Laboratory stock 003920) as previously described (NG2cre:ZEG; Zhu et al., 2008; Hill et al., 2013; Sherafat, Hill, & Nishiyama, 2018). After 7 days, slices were treated with 10 μM compounds for 2 days in the presence or absence of 50 ng/ml human PDGF AA (R&D Systems). All animal procedures were approved by the University of Connecticut Institutional Animal Care and Use Committee.
2.4 |. Luciferase assays
Oli-neu cells were transfected with various Pdgfra-luciferase constructs using Lipofectamine 2000 (Invitrogen Corporation, Carlsbad, CA), along with the pGL4.73 (Promega) encoding Renilla luciferase at a ratio of 7 (pGL4.10) to 1 (pGL4.73). The day after transfection, cells were split into 96-well plates, and after 4 hr compounds and/or growth factors were added. Two days later, Firefly and Renilla luciferase activities were measured using the DualGlo Luciferase Assay System (Promega) and the Veritas Microplate Luminometer (Pomega). For positive control, we used pGL3-Promoter plasmid (Promega) that contained the SV40 promoter.
2.5 |. Primary screening
For screening by luciferase reporter assay, 50 μM of compounds dissolved in DMSO was added to Oli-neu cells transfected with Pdgfra-1595-luciferase plasmid (Pdgfra-luc), and luciferase activity was measured after 2 days. Wells with a drastic decrease in cell density suggesting compound cytotoxicity were marked and excluded from further analyses. For morphological and immunocytochemical assays, Oli-neu cells were treated with 25 μM compounds for 4 days without medium change, scored for changes in morphology and cell density by phase contrast microscopy, and then fixed and immunolabeled with the O1 antibody.
2.6 |. Cytotoxicity assays and flow cytometry
Oli-neu cells were treated for 16 hr with 50 μM compounds and labeled using Alexa Fluor 488 Annexin V/Dead Cell Apoptosis Kit and propidium iodide (Life Technologies). The cells were then washed once with PBS, and 10,000 cells were analyzed by flow cytometry (BD FACSCalibur, Becton Dickinson) in the Flow Cytometry Facility (Biotechnology-Bioservices Center, University of Connecticut, Storrs). Analysis was performed using FlowJo software.
2.7 |. Cell proliferation and differentiation assays
For proliferation experiments using EdU incorporation, cells were incubated in 10 μM 5-ethynyl-2′-deoxyuridine (EdU) (Life Technologies) during the last 12 hr (5 hr for slice cultures) and fixed with 4% paraformaldehyde (Electron Microscopy Sciences, Hatfield, PA). EdU was detected using the Click-iT EdU Alexa Fluor Imaging Kit (Life Technologies) or using the individual reagent components described previously (Zuo et al., 2018).
Cell proliferation was also assessed using the CellTiter-Glo 2.0 Cell viability assay (Promega) following the manufacturer’s guidelines. Cells plated in poly-lysine-coated 96-well white walled cell culture plates (Thermo Scientific Nunc, Rochester, NY) and grown for 2 days in the presence of compound with or without human PDGF AA (R&D Systems, 221-AA). On the day of the assay, an equal volume of CellTIter-Glo reagent was added to the cells for 10 min, and luminescence was measured using Gen5 2.07 (Biotek, Winooski, VT) on a Synergy HTX multi-mode microplate reader.
For differentiation experiments, rat OPCs were grown in medium containing 5 ng/ml PDGF AA and 10 μM compounds for 2 days. For a positive control for oligodendrocyte differentiation, we added 400 ng/ml thyroid hormone (T3, triiodothyronine) (Sigma-Aldrich). The cells were fixed and stained with the O1 antibody or processed for total RNA extraction.
2.8 |. Immunofluorescence labeling
For cell surface labeling, live cells were incubated with rabbit antibodies to PDGFRα (RRID:AB_2315173) and PDGFRβ (RRID: AB_2783647) (both obtained from Dr William Stallcup, Sanford Burnham Institute, La Jolla, CA; 1:1,000 dilution; Ozerdem et al., 2001) at room temperature for 30 min, fixed in cold ethanol or 4% paraformaldehyde, and subsequently blocked and permeabilized and incubated with other primary antibodies to intracellular antigens followed by secondary antibodies. For all other immunolabeling, cells and slice cultures were immunolabeled after 10 min of fixation in 4% paraformaldehyde as previously described (2006; Hill et al., 2013; Zuo et al., 2018) using mouse O1 (1:1 dilution) or chick anti-GFP (Aves Laboratories, Tigard, OR, RRID:AB_10000240), mouse anti-GFAP (Sigma, 1:500, RRID:AB_477010), rat anti-Ki67 (Invitrogen, 1:200, RRID:AB_10854564), mouse anti-Olig2 (Millipore, 1:500, RRID: AB_10807410), and mouse anti-Sox10 (Santa Cruz, 1:100, RRID: AB_10844002) antibodies. Following washes, cells were incubated in Alexa488-, Cy3- or Alexa647-conjugated secondary antibodies. To detect EdU, Click-iT reaction was performed after immunolabeling. The cells or slices were counterstained with Hoechst 33342 or DAPI and mounted Fluoro-Gel (Electron Microscopy Sciences) or Vectashield (Vector Laboratories, Burlingame, CA) and analyzed using a Zeiss Axiovert 200M microscope with ORCA EM camera or Leica SP8 confocal microscope housed in the University of Connecticut Advanced Light Microscope Facility.
2.9 |. Quantification of mRNA
Total RNA was collected from 500,000 Oli-neu cells or NIH3T3 cells or 250,000 primary OPCs using RNeasy Mini Kit (Qiagen). Reverse transcription was performed using 1 μg of total RNA, Superscript III (Invitrogen), and random primers. For quantitative PCR (qPCR) with SYBR Green PCR Master Mix (Life Technologies), we used 10 ng of cDNA and gene-specific primers listed in Table 2. The PCR primers were designed to amplify products spanning multiple exons to minimize amplifying genomic sequences. The PCR conditions were as follows: 2 min at 95°C followed by 39 cycles of denaturation (10 s at 95°C), annealing, and extension (30 s at 60°C). The specificity of PCR products was confirmed by the melting curve. Changes in transcript levels were determined using the relative quantification method and glyceraldehyde-3-phosphate-dehydrogenase (Gapdh) as the reference gene (Livak & Schmittgen, 2001). Each amplification reaction was performed on three independent RNA isolates.
TABLE 2.
PCR Primers used for qPCR
| Gene | Forward primer | Reverse primer |
|---|---|---|
| Mouse Gapdh | 5′-TGACAACTTTGGCATTGTGG | 5′-ATGCAGGGATGATGTTCTGG |
| Mouse Pdgfra | 5′-TCGAAGGCAGGCACATTTAC 5′-AGCTTGAGGGAGAGAAACAAACG |
5′-TTGAGTCTCCGGATCTGTGG 5′-CATAGCTCCTGAGACCTTCTCCTTC |
| Mouse Pdgfrb | 5′-CACCTTCTCCAGTGTGCTGAC | 5′-CGGAGTCCATAGGGAGGAAG |
| Mouse Fgfr1 | 5′-CTAACCGCAGAACTGGGATG | 5′-TGGACCAGGAGAGACTCCAC |
| Mouse Fgfr3 | 5′-TGCACAAGGTCTCTCGCTTC | 5′-TCAGCAGGCAGCTCAAGTTC |
| Mouse Cspg4 | 5′-GGCCTTGTTGGT CAGATCTACAG | 5′-GCAGCAGGTCTACTCTGGTCAGA |
| Mouse Olig2 | 5′-TTACAGACCGAGCCAACACC | 5′-GATGGGCGACTAGACACCAG |
| Mouse Sox10 | 5′-TACCCTCACCTCCACAATGC | 5′-AGTCCGGATGGTCCTTTTTG |
| Mouse Mbp | 5′-ACACACGAGAACTACCCATTATGG | 5′-TGTTCGAGGTGTCACAATGTTCTT |
| Mouse Gfap | 5′-GATCTATGAGGAGGAAGTTCGAGAA | 5′-CGTATTGAGTGCGAATCTCTCTCA |
| Mouse Cdkn1a | 5′-AACATCTCAGGGCCGAAAAC | 5′-CCTGACCCACAGCAGAAGAG |
| Mouse Cdkn1b | 5′-TGGGTCTCAGGCAAACTCTG | 5′-CCCTTTTGTTTTGCGAAGAAG |
2.10 |. Immunoblotting
Immunopurified OPCs from P1–4 mouse cortex were incubated overnight in 10 μM compounds without PDGF AA. On the following day, the cells were stimulated with 50 ng/ml PDGF-AA for 15 min at 37°C. Cells were then rinsed with chilled PBS, and proteins were extracted in chilled RIPA buffer (ThermoFisher), both containing 1x PhosSTOP (Sigma-Aldrich-Roche, 4906845001) and 1x Halt Protease Inhibitor Cocktail (ThermoFisher, 87786). The cleared lysate was frozen in liquid nitrogen and stored at −80°C until use. Protein concentration was measured using DC Protein Assay (Bio-Rad). 30 μg of reduced and denatured samples were electrophoresed through Bolt Bis-Tris Plus 8 or 12% polyacrylamide gels (ThermoFisher) in Bolt MOPS SDS running buffer (ThermoFisher) along with prestained molecular weight standards (LI-COR, 928–7000). Proteins were transferred to Immobilon FL membranes (EMD-Millipore) using a transfer buffer containing 1.25 mM bicine, 1.25 mM bis-Tris, pH 7.5, and 0.05 mM EDTA. Blots were immunolabeled according to the instructions specified by LI-COR, using Odyssey TBS (Tris-buffered saline) blocking buffer (LI-COR). The primary antibodies were rabbit anti-phospho-PDGFRα (3170, Cell Signaling), goat anti-mouse PDGFRα (AF1062, R&D Systems) and the rabbit antibodies to the following from Cell Signaling Technologies: phospho-PDGFRα (#3170, RRID: AB_2162348), AKT (#4685, RRID:AB_2225340), phospho-AKT (#4060, RRID:AB_2315049), ERK1/2 (#4695, RRID:AB_390779), phospho-ERK1/2 (#4370, RRID:AB_2315112), mTOR (#2983, RRID: AB_2105622), and phospho-mTOR (#5536, RRID:AB_10691552). Mouse antibody to GAPDH and rabbit antibody to heat shock protein Hsp90 (RRID:AB_2121214) were used for normalization. Secondary antibodies were donkey anti-goat or anti-rabbit antibodies conjugated with IRDye 800RD or IRDye 680RD (LI-COR). Blots were imaged on a LI-COR Odyssey Imager, and the density of protein bands was determined using Image Studio Lite.
2.11 |. Data analysis
Fluorescence intensity measurements were performed by capturing images under exposure conditions below pixel-saturation and using ImageJ to obtain mean integrated density. For PDGFRα immunofluorescence on OPCs, we obtained the mean integrated density of pixel gray-scale values in randomly selected fields and after background subtraction divided the value by the number of cells in the field. For PDGFRα immunofluorescence on CMV-Pdgfra-EF1a-GFP-transfected astrocytes, mean integrated density was obtained in regions of interest selected over GFP+ cells. The raw data for all the assays were imported into Excel, and statistical analyses were performed using Excel and GraphPad Prism 8.3. Values are expressed as means ± SDs. Images were assembled into figures using Adobe Photoshop.
3 |. RESULTS
3.1 |. Primary screening
To identify compounds that downregulated the transcription of Pdgfra in OPCs, we established a luciferase reporter assay using Oli-neu cells. We cloned various lengths of the mouse Pdgfra 5′ flanking sequences into pGL4.10 firefly luciferase reporter vector (Figure 1a) and transfected them into Oli-neu cells. Luciferase activity was assessed after 2 days in control growth medium or under differentiation conditions in the presence of 1 mM dibutyryl cAMP and 10 μM dexamethasone. Plasmid Pdgfra-1595, which contained 1595 nucleotides of the Pdgfra 5′ flanking sequence, showed the highest luciferase activity under control growth conditions. In differentiation media, luciferase activity from Pdgfra-1595 decreased by 35% compared to luciferase activity in growth media (Figure 1b). Of all the compounds tested, Pdgfra-1595 exhibited the most robust luciferase activity in growth media compared to differentiation media. Therefore, we used the Pdgfra-1595 promoter for the primary screening, and it will be referred to as Pdgfra-luc.
FIGURE 1.
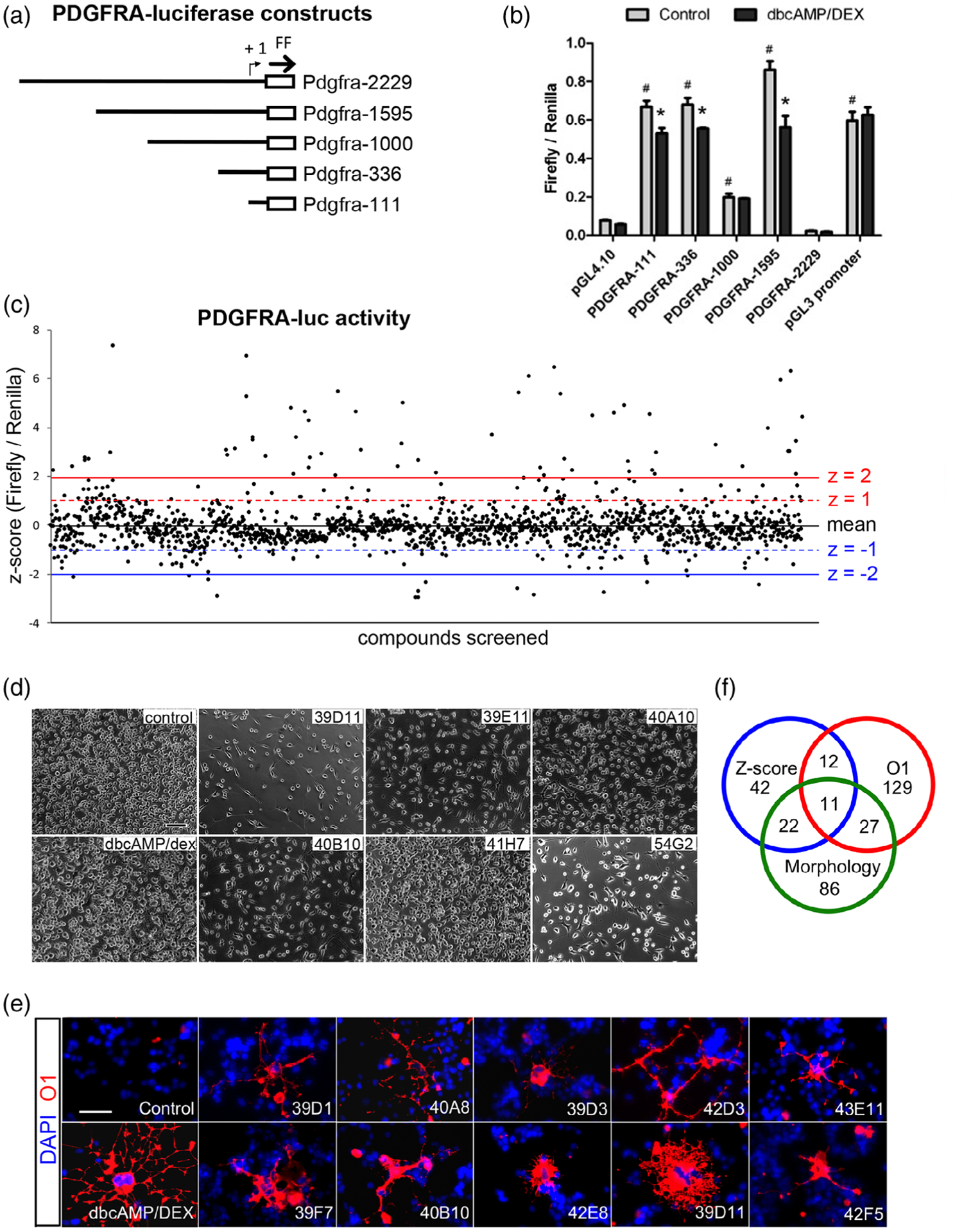
Primary screening for compounds that downregulated Pdgfra transcription (a) A schematic showing the different lengths of the mouse Pdgfra 5′-flanking/promoter regions used in luciferase assays. (b) Luciferase assays 2 days after transfecting the constructs into Oli-neu cells, showing firefly luciferase activity normalized to renilla luciferase activity. The cells were grown under control proliferative conditions (gray bars) or in the presence of dibutyryl cAMP/dexamethasone (dbcAMP/DEX) to promote oligodendrocyte differentiation (black bars). pGL4.10 is empty vector without Pdgfra promoter elements. pGL3 promoter is a positive control with SV40 promoter. # p < .001 relative to pGL4.10 control in proliferative conditions; * p < .001 relative to control in differentiation conditions. Two-way analysis of variance (ANOVA), Bonferroni posttest. n = 3. (c) Results of primary screening of 1,500 compounds. The y-axis represents the z-scores of normalized luciferase activities using Pdgfra-luc construct transfected into Oli-neu cells treated for 2 days with 50 μM compounds. (d) Phase contrast images of Oli-neu cells treated for 2 days with 50 μM compounds. Control, dimethyl sulfoxide (DMSO). Scale, 100 μm. (e) Staining of Oli-neu cells treated for 2 days with 50 μM compounds and stained with O1 antibody (red). Blue is DAPI labeling of nuclei. Scale, 50 μm. (f) Venn diagram showing the intersection of compounds that downregulated Pdgfra-luc with a z-score below 1 (blue; 87 compounds), altered Oli-neu cell morphology and/or reduced cell density (green; 146 compounds), and increased O1 immunoreactivity (red; 179 compounds)
Oli-neu cells were transfected with Pdgfra-luc and treated for 2 days in growth medium containing 50 μM compounds from the ChemBridge library, after which luciferase activity was measured. Out of 1,520 compounds evaluated in the assay, 931 downregulated Pdgfra-luc activity, and 87 of them downregulated Pdgfra-luc activity by more than 1 SD below the mean (Figure 1c). In parallel with the primary screen, we performed morphological assays on the entire compound set to identify compounds that affected oligodendrocyte differentiation. For these assays, Oli-neu cells were treated with the same compounds for 4 days in differentiation medium and were scored for cell density and cellular morphology by phase contrast microscopy (Figure 1d). The same cells were then assessed for oligodendrocyte differentiation by immunofluorescence labeling using the O1 antibody (Figure 1e), which detects a glycolipid antigen on newly differentiated oligodendrocytes (Sommer & Schachner, 1981). Out of 1,520 compounds tested, 146 caused a change in Oli-neu cell morphology to a more processed morphology and/or decreased their density (Figure 1d), and 179 compounds increased O1 immunofluorescence or the percentage of cells that were O1+ (Figure 1e). As shown in the summary Venn diagram (Figure 1f), 11 compounds reduced Pdgfra-luc and exhibited changes in cell density and O1 labeling.
3.2 |. Verification of Pdgfra downregulation by the hit compounds
We selected a total of 16 compounds for reevaluation for a full dose-response using the original luciferase assays. They included 7 of the 11 compounds that downregulated Pdgfra-luc with a Z score of <−1 and affected cell density and/or increased O1 expression as well as 9 additional compounds that downregulated Pdgfra-luc less prominently but had a pronounced effect on morphology/O1 expression. Seven of the compounds (39D11, 40A10, 40B10, 42E8, 54G2, 44C8, and 45C11) downregulated Pdgfra-luc activity in a dose-dependent manner (Figure 2a,b), whereas 39E11 failed to exhibit a dose-response effect. Compounds 39D11, 40A10, 40B10, and 39E11 had a strikingly similar chemical structure, derivatives of N-methyl-N-benzylguanidines (Figure 2a). We will refer to these as guanidine compounds. Three out of the four guanidine compounds (39D11, 40A10, and 40B11) caused a dose-dependent decrease in Pdgfra-luc with an IC50 of 51.8, 31.0, and 47.3 μM, respectively. The remaining four compounds of the seven that decreased Pdgfra-luc in a dose-dependent manner (42E8, 54G2, 44C8, and 45C11) were structurally unrelated to each other or to the guanidine compounds (Figure 2b).
FIGURE 2.
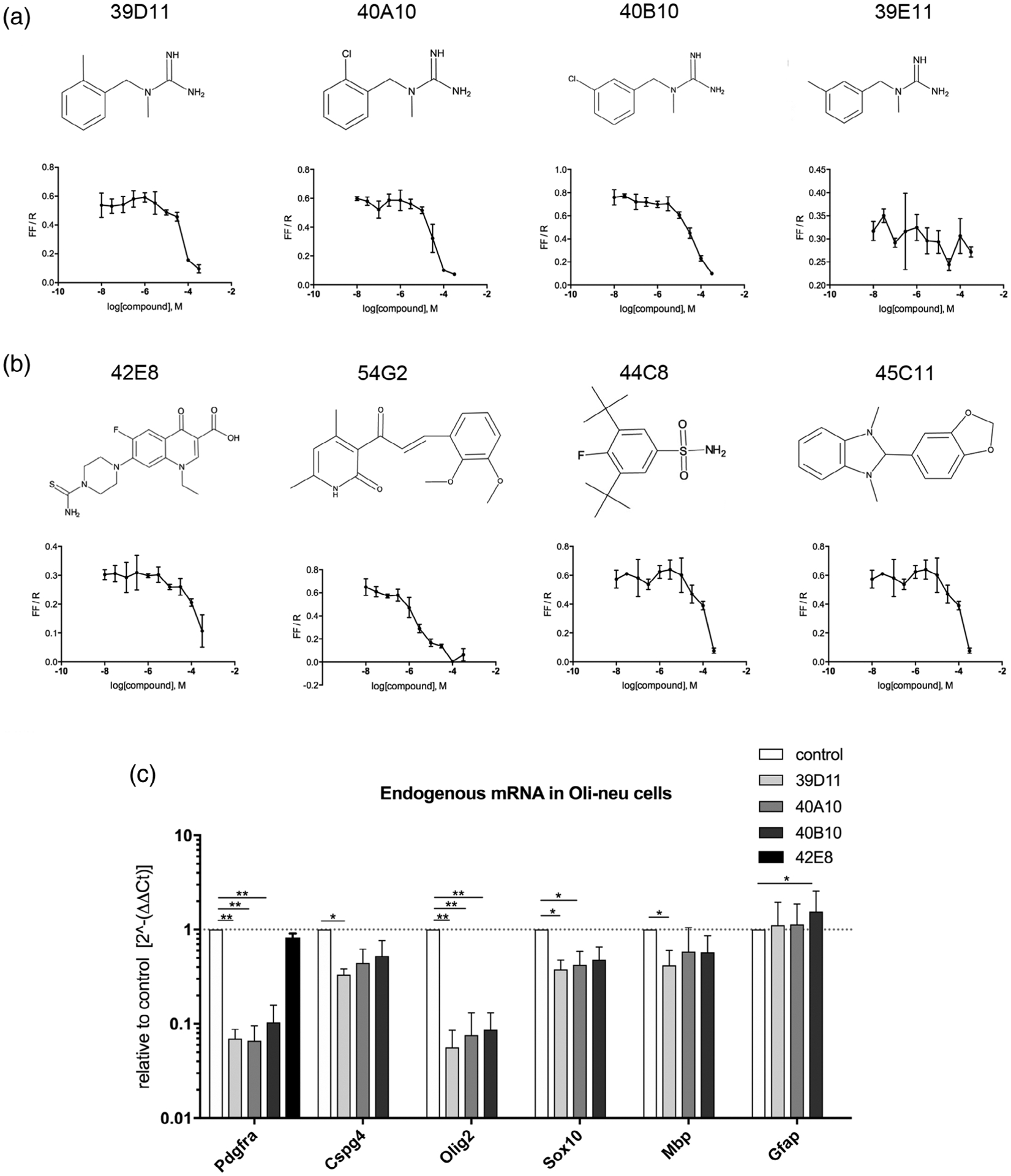
Effects of hits from the primary screen on Pdgfra transcription in Oli-neu cells. (a,b) Dose-response analyses of four guanidine compounds a) and four nonguanidine compounds (b) by luciferase assays using Oli-neu cells transfected with Pdgfra-luc. The y-axes represent firefly luciferase activity normalized to renilla luciferase in response to 2 days of incubation in 50 μM 3.3-fold serially diluted compounds. The structure of each compound is depicted above the graph. (c) Relative quantification of mRNA encoding Pdgfra, Cspg4, Olig2, Sox10, Mbp, and Gfap. Total RNA from Oli-neu cells treated with 50 μM compounds was used for quantitative PCR (qPCR), and the results are plotted relative to the levels in control cells treated with dimethyl sulfoxide (DMSO) (value of 1, dotted line). The y-axis is indicated on a log10 scale to reflect the magnitude of up- (above 1) and down (below 1)-regulation. * p < .05, ** p < .01, two-way analysis of variance (ANOVA), uncorrected Fisher’s LSD test. Dotted line represents y = 1.0
Since the Pdgfra-luc plasmid contained only 1,595 nucleotides of the promoter region of the mouse Pdgfra gene, downregulation of its activity does not necessarily reflect changes in transcription of the mouse Pdgfra gene. We used qPCR to examine the changes in endogenous Pdgfra mRNA levels after treating Oli-neu cells with the compounds (Figure 2c). The three guanidine leads, 39D11, 40A10, and 40B10 that elicited a dose-dependent decrease in Pdgfra-luc also prominently decreased endogenous Pdgfra transcripts by 14-, 15-, and 10-fold, respectively (Figure 2c), while their effects on Cspg4 mRNA encoding NG2 core protein were less robust, with 39D11 causing a threefold reduction. These three compounds also led to a >10-fold decrease in Olig2 mRNA but only marginally reduced transcripts encoding Sox10 or Mbp encoding myelin basic protein. The dichotomy of the effects on Olig2 and Sox10 is consistent with our previous observation that Sox10 expression becomes independent of Olig2 as OPCs become fully committed to the oligodendrocyte lineage during postnatal development (Zuo et al., 2018). Two of the three compounds did not significantly affect the levels of mRNA encoding the astrocyte-specific intermediate filament protein glial fibrillary acidic protein (Gfap), while one slightly increased it. These findings suggest that 39D11, 40A10, and 40B10 most prominently and selectively reduced Pdgfra and Olig2 transcription without altering the fate or differentiation state of Oli-neu cells. The compound 42E8, which did not have a guanidine moiety, reduced Pdgfra-luc activity but did not significantly affect endogenous Pdgfra or Olig2 mRNA levels (Figure 2c). Therefore, we focused our further profiling studies on the guanidine compounds.
3.3 |. Repression of Pdgfra transcription by structurally similar guanidine compounds
To further examine the structure-activity relationship of guanidine compounds, we synthesized 12 additional guanidine compounds, 001–012 (Figure 3a) and tested them for their effects on Pdgfra transcription in Oli-neu cells. The new compounds 001 and 007 were structurally identical to 40A10 and 40B10, respectively. Having established the critical role of the guanidine group in these compounds which can potentially engage in strong electrostatic interactions, we focused on determining key structure-activity relationships around the role of steric and electronic effects on the pendant aromatic ring. Altering the electronic properties of an aromatic ring such as the one in this lead series can have a significant impact on the ability of the molecule to engage in π-stacking and other hydrophobic interactions while alternative placement of these molecules can reveal additional opportunities for increasing target engagement. Analogs with strongly donating methoxy groups (002, 006, 112), mildly donating methyl groups, electron withdrawing chloro groups (001 and 007), and an unsubstituted analog (004) were prepared. In addition, the effect of an endocyclic nitrogen (010/011) was also examined in an attempt to enhance water solubility of the compounds. Two extended analogs (008 and 009) were also synthesized as well to investigate the importance of spacing between the aromatic moiety and the guanadino head group.
FIGURE 3.
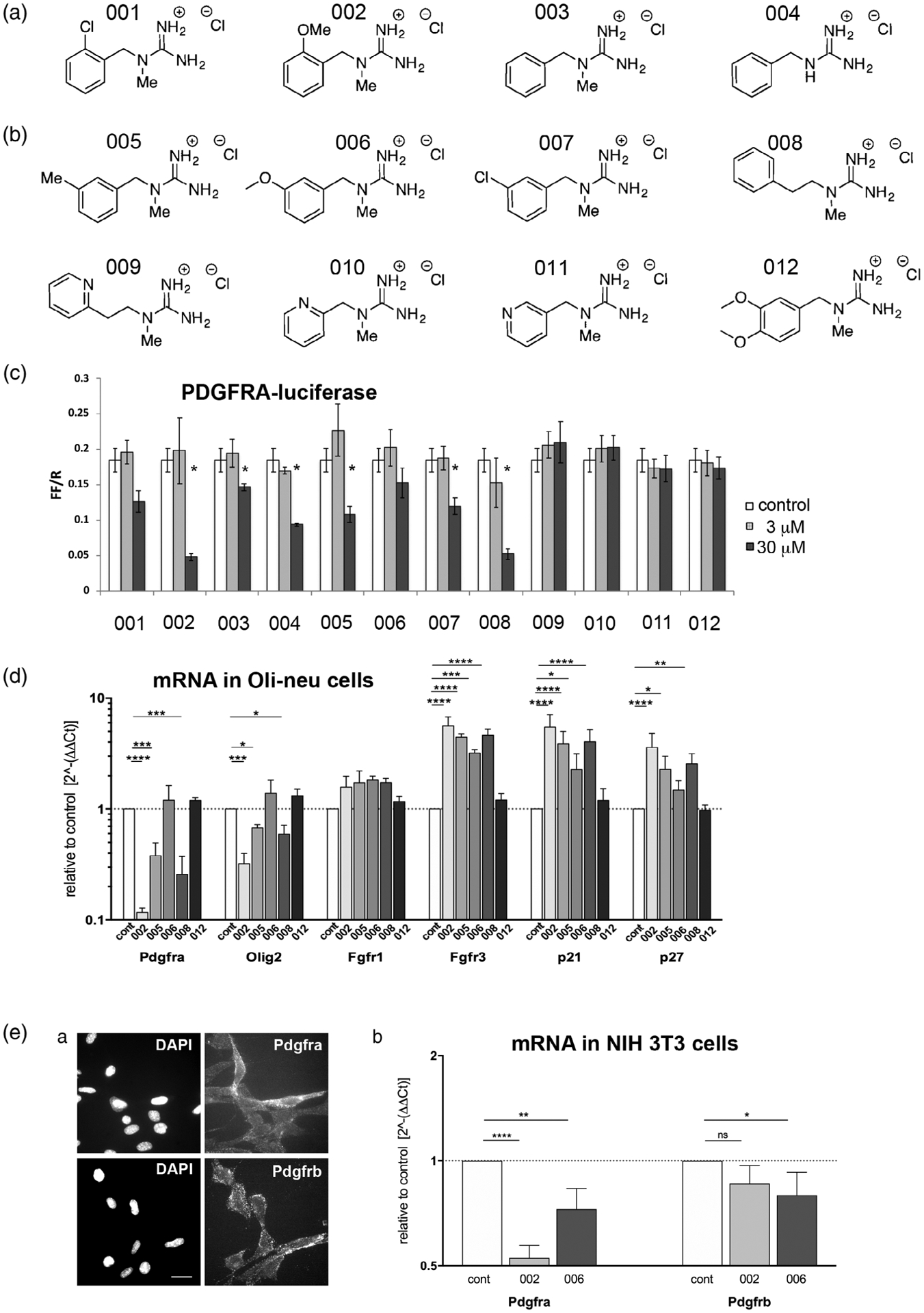
Newly synthesized guanidine compounds. (A) Chemical structure of the newly synthesized guanidine compounds. (B) Effects of the compounds on Pdgfra-luc activity in Oli-neu cells at 3 and 30 μM. White bars represent control dimethyl sulfoxide (DMSO)-treated cells. *p < .05. (C) Quantification of mRNA encoding Pdgfra, Olig2, Fgfr1, Fgfr3, p21Cip1 (Cdkn1a), and p27kip1 (Cdkn1b) after treatment of Oli-neu cells for 2 days with 50 μM guanidine compounds. The y-axis represents values relative to the levels in control DMSO-treated cells (dotted horizontal line) on a log10 scale. * p < .05, ** p < .01, *** p < .001, **** p < .0001, two-way analysis of variance (ANOVA), uncorrected Fisher’s LSD test. (D) Pdgfra and Pdgfrb expression on NIH3T3 mouse fibroblasts. (a) Immunofluorescence labeling of live unfixed NIH 3T3 cells using rabbit antibodies to Pdgfra (top) and Pdgfrb (bottom). Scale bar, 20 μm. (b) Effects of compounds 002 and 006 on Pdgfra and Pdgfrb mRNA levels examined by quantitative PCR (qPCR). Y-axis represents mRNA levels relative to control DMSO-treated cells (dotted horizontal line), plotted on log2 scale. * p < .05, ** p < .01, **** p < .0001, two-way analysis of variance (ANOVA), uncorrected Fisher’s LSD test
We first tested the new compounds in luciferase assays. At 30 μM, the compounds 002 and 008 most robustly downregulated Pdgfra-luc activity, while compounds 003, 004, 005, and 007 also significantly reduced Pdgfra-luc activity but less prominently (Figure 3b). Compounds 001 and 006 slightly reduced Pdgfra-luc activity, but the effects were not statistically significant. Compounds 009, 010, 011, and 012 did not alter Pdgfra-luc activity at all. Compounds 001 and 007 were less potent than their original structurally identical counterparts 40A10 and 40B10. We attributed this to several impurities observed by mass spectrometry in the commercial samples of compounds 40A10 and 40B10 (not shown). The two positional isomers 002 and 006, which differed only in the placement of the aromatic methoxy group, exhibited strikingly different activities, with the ortho-substituted analog 002 showing a robust down-regulation effect, whereas the meta-substituted 006 was largely ineffective. Compounds that contained a more hydrophobic chlorine (007) or methyl group (005) at the meta position of the phenyl ring showed increased potency relative to 006. An analog with a completely unsubstituted phenyl ring at C2 (003) showed a significant loss of potency. Interestingly, deletion of the guanidine N1-methyl group on the same unsubstituted system restored some activity (004) while elongation of the carbon chain between N1 and the benzyl ring (008) contributed to increased potency. Exchange of the phenyl ring for a pyridyl moiety (009–011) or addition of a second methoxy unit (012) abrogated the activity in the Pdgfra expression assay. The complete lack of effect of compounds 009–012 could be attributed to reduced permeability through the plasma membrane due to increased polarity as the logP of this later series ranges from 0.7 to 1 as compared to the active compounds with logP values ranging from 1.5 to 2.2.
We selected five representative compounds for further analyses. Compounds 002, 005, and 008 were chosen to represent those that decreased Pdgfra-luc, while compounds 006 and 012 were chosen to function as negative controls. When the compounds were added to Oli-neu cells and their effects on endogenous transcripts were assayed by qPCR, compounds 002, 005, and 008 caused a ninefold, threefold, and a fourfold decrease in Pdgfra mRNA, respectively, while compounds 006 and 012 had no effect (Figure 3c), consistent with their effects in luciferase assays. Compound 002 also decreased Olig2 mRNA, as did compounds 005 and 008 to a lesser degree.
FGF-2 increases Pdgfra expression on rat OPCs (McKinnon, Matsui, Dubois-Dalcq, & Aaronson, 1990; Nishiyama et al., 1996b), and when added to OPCs along with PDGF AA, it inhibits their oligodendrocyte differentiation and maintains their proliferative state (Bögler, Wren, Barnett, Land, & Noble, 1990). Of the high affinity receptors for FGF-2, Fgfr1, 2, and 3 are present on oligodendrocyte lineage cells. Fgfr2 is expressed only on terminally differentiated oligodendrocytes, while Fgfr1 and Fgfr3 are highly expressed on OPCs and are downregulated with terminal differentiation (Bansal et al., 1996; Zhang et al., 2014). None of the guanidine compounds significantly altered the level of Fgfr1 mRNA in Oli-neu cells (Figure 3c). However, compounds 002, 005, and 008 increased Fgfr3 mRNA more than 4.5-fold, and compound 006 also increased Fgfr3 mRNA by 3.9-fold, whereas compound 012 had no effect. Fgfr2 mRNA levels in Oli-neu cells were more than 10-fold lower than those of Fgfr1 and Fgfr3 and did not change with any of the guanidine compounds tested (not shown). Notably, the active compounds upregulated cell cycle inhibitors p21Cip1 (Cdkn1a) and p27Kip1 (Cdkn1b), suggesting cell cycle withdrawal (Casaccia-Bonnefil et al., 1997; Durand, Gao, & Raff, 1997; Ghiani et al., 1999).
PDGFRβ is another member of the PDGFR family, which is structurally closely related to PDGFRα but binds only PDGF BB and not PDGF AA (Andrae et al., 2008). PDGFRβ is not expressed by oligodendrocyte lineage cells but is present on pericytes and other mesenchymally derived cells (Stallcup, 2018). To examine whether the guanidine compounds also affected PDGFRβ expression, we performed qPCR on NIH3T3 mouse fibroblasts, which express both PDGFRα and PDGFRβ (Figure 3Da) (Grotendorst, Igarashi, Larson, Soma, & Charette, 1991). Addition of compound 002 caused a twofold reduction in Pdgfra but had no significant effect on Pdgfrb mRNA levels (Figure 3Db). Compound 006, which had no effect on Pdgfra mRNA levels in Oli-neu cells caused a small but significant decrease in Pdgfra mRNA NIH3T3 cells as well as a slight decrease in Pdgfrb mRNA. These results suggest that the guanidine compounds prominently downregulated Pdgfra mRNA on fibroblasts as well as on Oli-neu cells.
3.4 |. Guanidine compounds did not affect oligodendrocyte differentiation
Since downregulation of PDGFRα and upregulation of FGFR1/3, as well as cell cycle exit, are associated with oligodendrocyte differentiation from OPCs, we tested whether the guanidine compounds promoted oligodendrocyte differentiation using primary OPC cultures, since Oli-neu cells undergo limited differentiation. We incubated primary rat OPCs for 2 days with 10 μM guanidine compounds and obtained the percentage of cells that were positively labeled by the O1 antibody that detects glycolipids on differentiated oligodendrocytes (Figure 4a,b). When we treated the cells with 400 ng/ml T3, known to induce oligodendrocyte differentiation (Barres, Lazar, & Raff, 1994), there was a threefold increase in the proportion of O1+ cells, many of which had large cell bodies and expansive radial processes. However, none of the compounds caused a statistically significant increase in the percentage of O1+ cells, although some compounds including 39D11 and 40B10 resulted in a small increase over DMSO-treated controls. The poor cell survival in the differentiation media with reduced PDGF AA concentration (Baron, Decker, Colognato, & Ffrench-Constant, 2003; Barres et al., 1992) precluded us from increasing the compound concentration above 10 μM, which was below the IC50 for most and may not have been sufficient to elicit oligodendrocyte differentiation.
FIGURE 4.
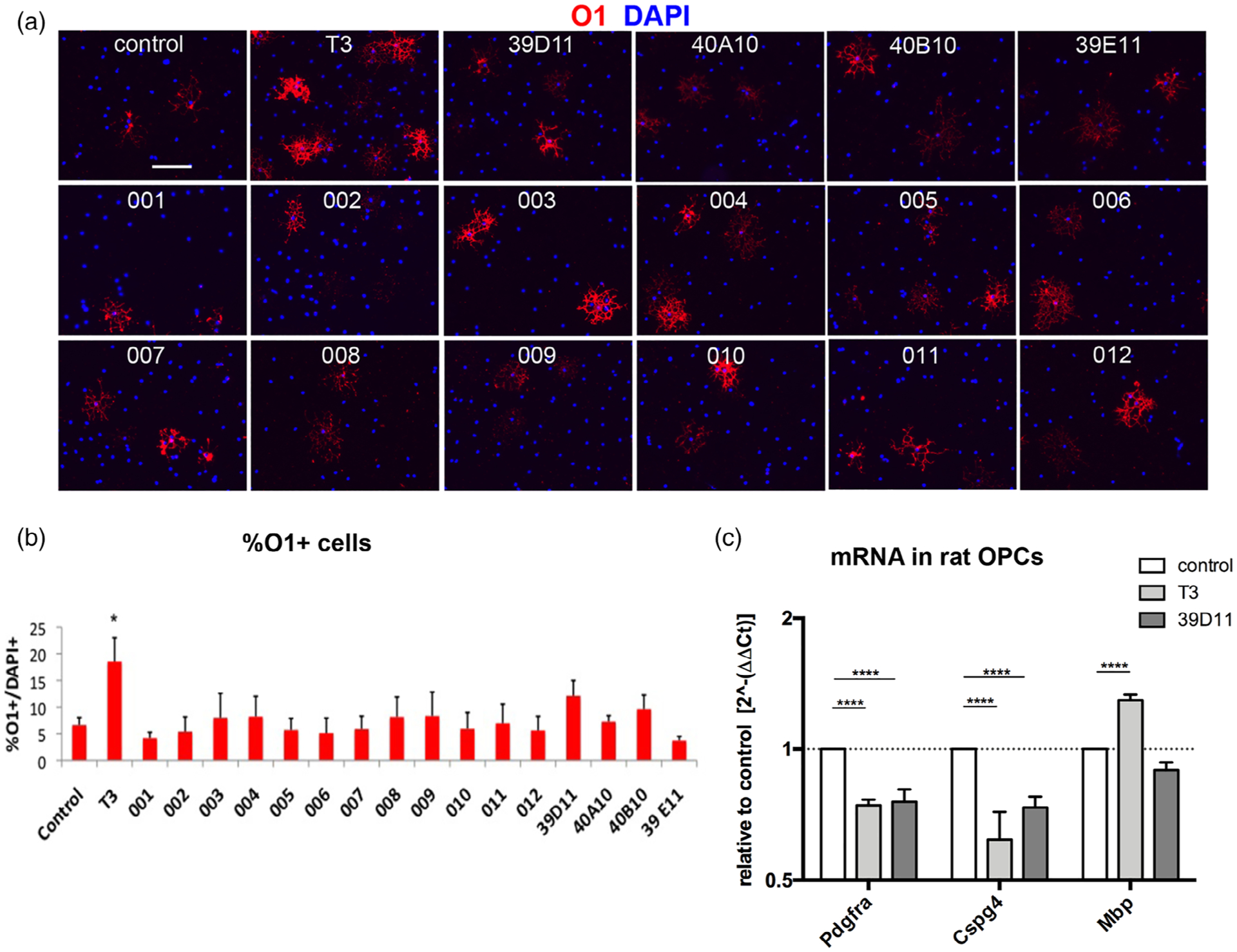
The effects of guanidine compounds on oligodendrocyte differentiation in primary oligodendrocyte precursor cells (OPCs). (a) Immunofluorescence labeling for the mature oligodendrocyte antigen O1 (red) in control rat primary OPCs grown in the presence of 5 ng/ml PDGF AA (control), in the presence of 5 ng/ml PDGF AA and 400 μM T3 (T3), or in the presence of the indicated guanidine compounds at 10 μ Blue represents DAPI. Scale, 100 μm. (b) The percentage of DAPI+ cells grown in the presence of the indicated compounds that were O1+. *, p < .05. (c) Reverse transcription-quantitative PCR (qPCR) for Pdgfra, NG2 (Cspg4), and Mbp after treatment with 5 ng/ml PDGF AA alone (control) or with 400 ng/ml T3 or 10 μM 39D11. mRNA levels are shown relative to dimethyl sulfoxide (DMSO)-treated control and plotted on a log2 scale. *, p < .05 compared to control, two-way analysis of variance (ANOVA), Tukey’s test
When we examined the levels of mRNA encoding Pdgfra, Cspg4, and Mbp in OPCs in the presence of 39D11, which marginally increased O1 expression, there was no increase in Mbp mRNA levels by 39D11, whereas T3 caused a 1.3-fold upregulation (Figure 4c). As in Oli-neu cells, 39D11 reduced Pdgfra mRNA in rat OPCs by 25%, as did T3 treatment. Both T3 and 39D11 also caused a 38 and 27% reduction in Cspg4 mRNA levels, respectively. Thus, while the guanidine compounds effectively downregulated Pdgfrα mRNA in rat OPCs, they did not cause overt oligodendrocyte differentiation.
3.5 |. Guanidine compounds inhibited OPC proliferation
3.5.1 |. Oli-neu cells
Since PDGF AA acting through PDGFRα constitutes the major mitogen for OPCs, we examined the effects of the structurally related guanidine compounds on OPC proliferation. When Oli-neu cells were treated with the guanidine compounds, 002, 005, and 008 reduced cell density, with 002 having the most prominent effect (Figure 5a). Compound 006 had a very small effect, and 012 had no effect. The magnitude of the antiproliferative effect correlated with the ability of these compounds to downregulate Pdgfra. Oli-neu cells treated with compounds 002 and 008 looked healthy and did not exhibit membrane blebbing or other features of dying cells. To determine whether the 002 reduced cell density by increasing cell death, we treated Oli-neu cells with 50 μM compounds for 16 hr and stained them for Annexin V, which binds to phosphatidylserine that is translocated to the outer leaflet and becomes accessible from the extracellular side during the early stages of apoptosis (Vermes, Haanen, Steffens-Nakken, & Reutelingsperger, 1995). Flow cytometry revealed that 9.43% of the cells treated with compound 002 were labeled with Annexin V, which was double that of DMSO-treated cells, but it was lower than cells treated with the inactive compound 006 (14.1%) (Figure 5b). Similarly, the proportion of the cells that had incorporated propidium iodide, which enters only into cells with compromised plasma membrane, after treatment with compound 002 (5.64%) was not different from control DMSO-treated cells (6.96%) and lower than the cells treated with 006 (10.4%). These findings suggest that the effective compounds reduced Oli-neu cell density by decreasing proliferation rather than increasing cell death.
FIGURE 5.
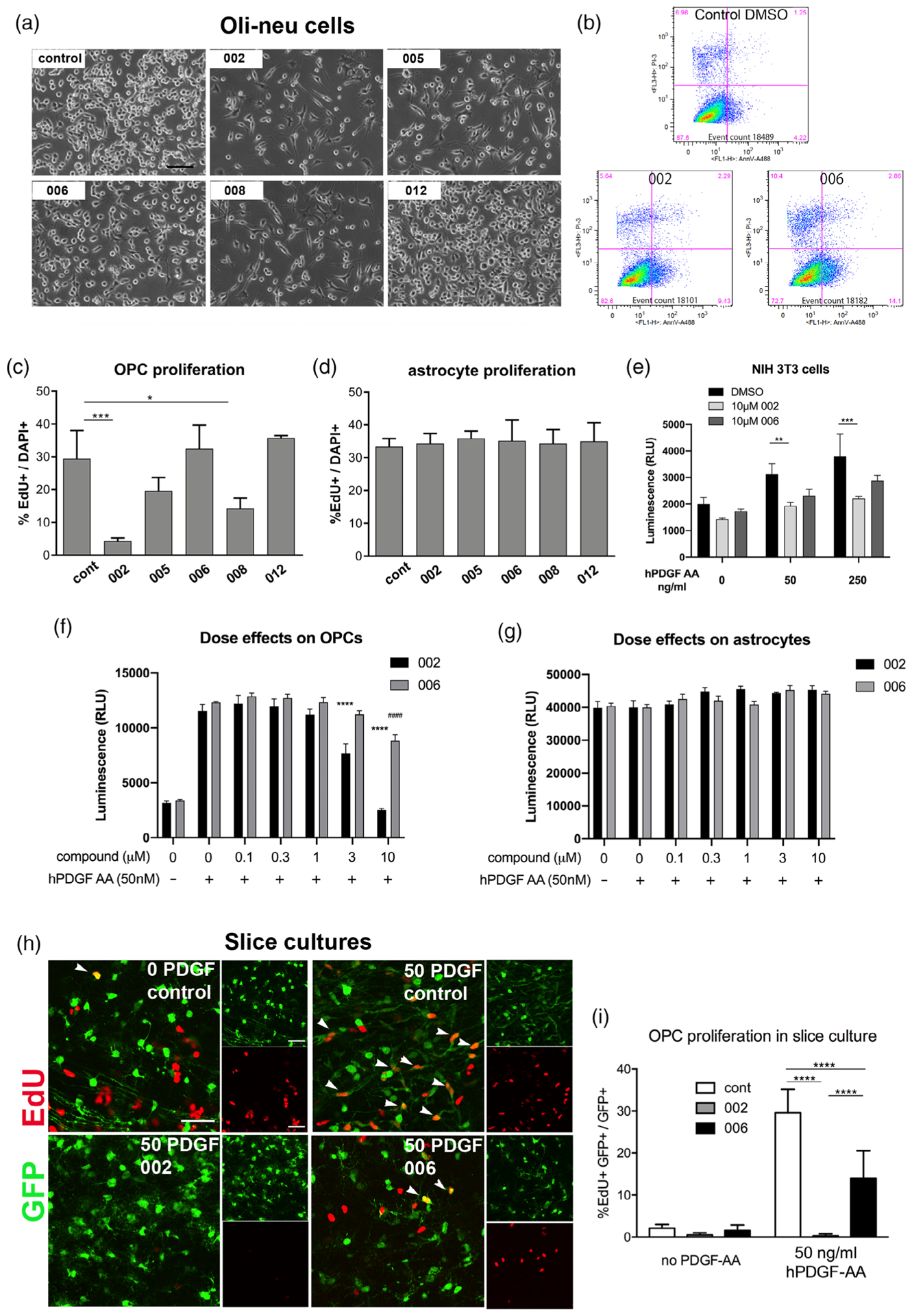
The effects of guanidine compounds on oligodendrocyte precursor cell (OPC) proliferation. (a) Phase contrast images of Oli-neu cells treated for 2 days with dimethyl sulfoxide (DMSO) (control) or 50 μM guanidine compounds 002, 005, 006, 008, or 012. Scale, 100 μm. (b) Flow cytometry of Oli-neu cells treated with DMSO (control) or 50 μM compounds 002 or 006 for 16 hr, stained with Annexin V (horizontal axis) and propidium iodide (vertical axis). (c,d) Quantification of EdU incorporation in OPCs (c) and primary astrocytes (d) after treatment with 50 μM compounds for 2 days. The graphs represent the percentage of EdU+ cells among all DAPI+ cells. * p < .05, *** p < .001. Dunnett’s multiple comparisons test. (e) Proliferation of NIH 3T3 cells measured by Cell Titer-Glo assay in 0, 50, and 250 ng/ml hPDGF AA in the presence of vehicle only or 10 μM 002 or 006. ** p = .0100, *** p = .0005. Compound 006 at 10 μM did not significantly reduce proliferation compared to DMSO alone (p = 0.1478 in 50 ng/ml PDGF AA and 0.0711 in 250 ng/ml PDGF AA). Tukey’s multiple comparisons test. (f,g) Dose–response effects of compounds 002 and 006 on OPC and astrocyte proliferation measured by Cell Titer-Glo assay. **** p < .0001 in 002-treated cells compared with DMSO only in the presence of PDGF AA. #### p < .0001 in 006-treated cells compared with DMSO only in the presence of PDGF AA. Sidak’s multiple comparisons test. (h) Detection of EdU in green fluorescent protein (GFP)+ OPCs in the white matter of slice cultures from the forebrain of P8 NG2cre:Z/EG double transgenic mice. Large panels indicate double labeling for GFP and EdU. Smaller panels are single channel images. Scale 50 μm. Arrowheads indicate EdU+ GFP+ cells. Top left: no PDGF AA. Other panels were incubated with 50 ng/ml human PDGF AA in the absence (control) or presence of 10 μM compound 002 or 006. (i) Quantification of EdU incorporation into OPCs in slice cultures treated with 002 or 006 in the absence or presence of 50 ng/ml human PDGF AA. **** p < .0001. Tukey’s multiple comparisons test
3.5.2 |. Primary glial cells and fibroblasts
To further determine whether the compounds inhibited cell proliferation, we treated primary OPC cultures for 2 days with 50 μM guanidine compounds and assayed for the incorporation of EdU given during the last 5 hr of incubation. Compounds 002 and 008 caused a sixfold and a twofold reduction in OPC proliferation, respectively, while the reduction by compound 005 did not reach significance (Figure 5c). When the compounds were added to primary cultures of astrocytes, there was no effect on EdU incorporation (Figure 5d), suggesting that the antiproliferative effects were specific to OPCs and likely due to the repression of Pdgfra, which is not expressed by astrocytes. Furthermore, compound 002, but not 006, reduced PDGF AA-mediated proliferation of NIH3T3 cells, as determined by the ATP-based luminescent assay CellTiter-Glo assay (Figure 5e). This suggests that the ability of compound 002 to reduce cell proliferation was not limited to OPCs but to other PDGFRα-expressing cells as well.
To examine whether antiproliferative effects of the compound 002 could be seen at lower concentrations, we performed a dose-response assay on primary mouse OPCs by treating them for 2 days with 0–10 μM guanidine compounds in the presence of PDGF AA using CellTiter-Glo assay. Compound 002 exhibited a dose-dependent antiproliferative effect. At 3 μM, 002 reduced OPC proliferation to 66.4% of control, while 10 μM 002 reduced OPC proliferation 4.6-fold, down to baseline levels of proliferation that occurs without exogenous PDGF-AA. By contrast, compound 006 reduced OPC proliferation only at 10 μM to 71.6% of control (Figure 5f). We did not observe any antiproliferative effects of 002 or 006 on astrocytes at the concentration range used (Figure 5g).
3.5.3 |. Slice cultures
We further verified the antiproliferative effects of the active guanidine compound 002 in a more physiological cellular context using organotypic slice cultures (Hill et al., 2013). We previously reported that in slice cultures, OPCs in white matter proliferate in response to exogenous PDGF AA. To test the effect of compounds 002 and 006 with very similar physicochemical properties but differential ability to downregulate Pdgfra, we incubated slice cultures from NG2cre: Z/EG mice, in which OPCs and their progeny expressed GFP, with 10 μM 002 or 006 for 2 days in the presence or absence of 50 ng/ml human PDGF AA, and assayed for EdU incorporation into GFP+ OPCs in the corpus callosum. In the absence of exogenous PDGF AA, the proportion of GFP+ cells that incorporated EdU was similarly low, below 3% (Figure 5h,i). Treatment with 002 reduced the basal proliferation fourfold to 0.53%, while 006 also caused a 24% reduction in basal proliferation. In response to PDGF AA, 30% of OPCs incorporated EdU in control slices. Addition of 002 almost completely suppressed OPC proliferation to 0.3%, while 006 caused a 50% reduction in OPC proliferation. The morphology of the GFP+ cells in the slices treated with 002 appeared similar to that of control slices without compounds, suggesting that 002 did not reduce EdU incorporation due to its cytotoxic effects. These observations indicate a potent antiproliferative effect of compound 002 on OPCs in the context of surrounding cells.
3.6 |. Guanidine compounds had minimum effects on activation of PDGFRα and its downstream effectors
To determine whether compound 002 inhibited PDGF-mediated OPC proliferation by acting on the cis-regulatory elements of the endogenous Pdgfra gene, we examined whether transfection of OPCs with CMV-Pdgfra-EF1a-GFP to expresses mouse Pdgfra under the constitutively active CMV promoter would render the transfected cells insensitive to the antiproliferative effects of 002. To test this, we first incubated OPCs for 2 days with 10 μM 002 or DMSO in the presence of 50 ng/ml PDGF AA and assayed for EdU incorporation in GFP+ cells. Compound 002 significantly decreased proliferation of GFP+ cells expressing CMV-Pdgfra-EF1a-GFP as well as control cells that had been transfected with EF1a-GFP (Figure 6a). However, since OPCs also expressed PDGFRα encoded by the endogenous gene, we could not accurately assess the contribution of PDGFRα transcribed and translated from CMV-Pdgfra relative to that from the endogenous gene.
FIGURE 6.
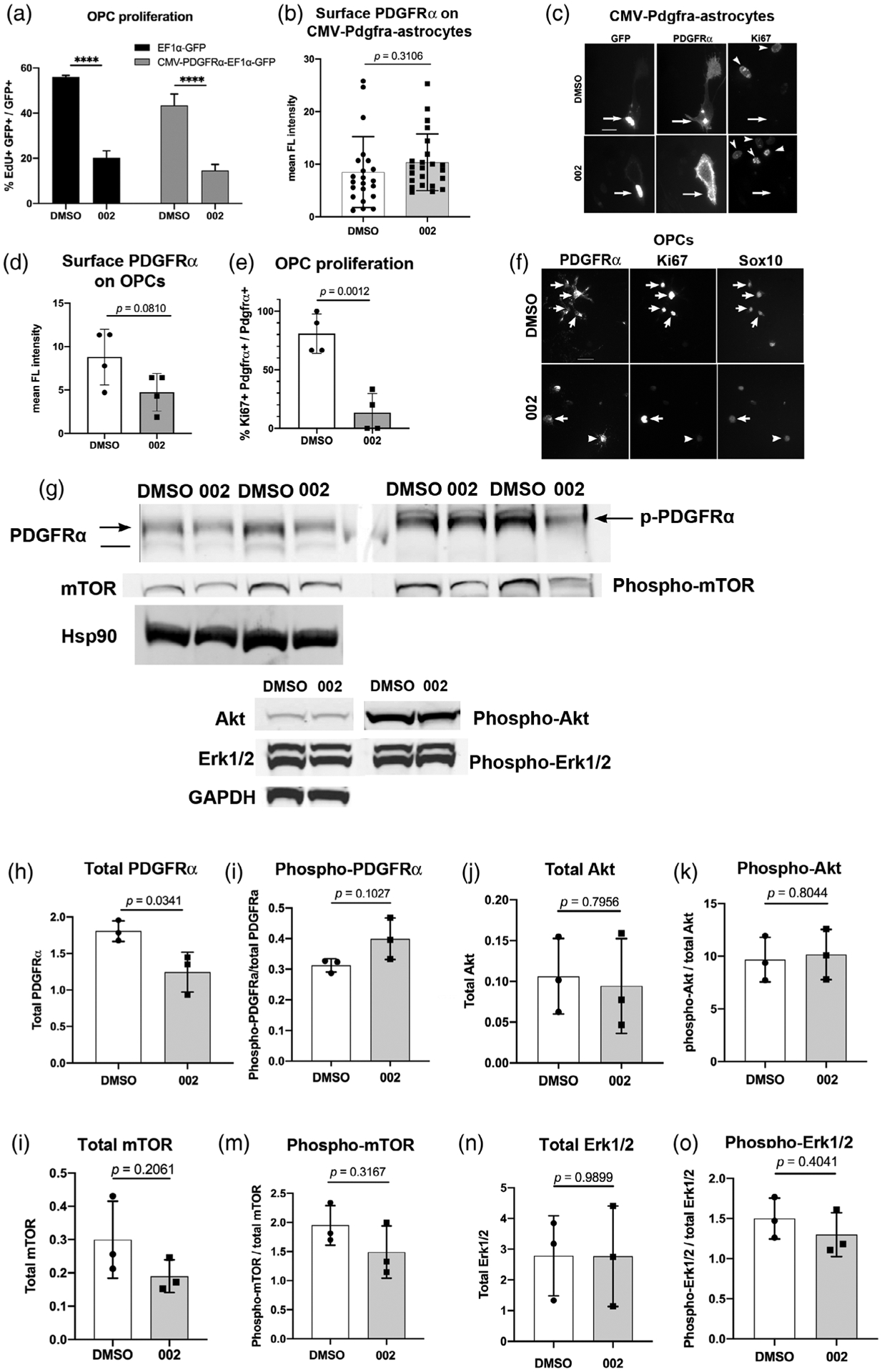
Effects of the guanidine compound 002 on PDGFRα and its downstream effectors. (a) Quantification of EdU incorporation by green fluorescent protein (GFP)+ primary mouse oligodendrocyte precursor cells (OPCs) that were transfected with EF1α-GFP (control) or CMV-Pdgfra-EF1a-GFP vectors and treated for 2 days with dimethyl sulfoxide (DMSO) or 002 in the presence of 50 ng/ml PDGF AA. **** p < .0001. Tukey’s multiple comparison test. (b) Quantification of cell surface PDGFRα immunofluorescence intensity on primary mouse astrocytes transfected with CMV-Pdgfra-EF1a-GFP and incubated for 2 days in DMSO or 002 in the presence of 50 ng/ml PDGF AA. The y-axis represents mean immunofluorescence intensity of surface PDGFRα immunolabeling in each field normalized by the number of GFP+ cells in the field. n = 22 (DMSO) and 23 (002). Two-tailed, unpaired Student’s t test. (c) Immunofluorescence for GFP, surface PDGFRα, and Ki67 in astrocytes transfected with CMV-Pdgfra-EF1a-GFP. Arrows indicate transfected cells, arrowheads indicate Ki67-immunoreactivity in untransfected cells. Scale = 25 μm. (d) Surface PDGFRα immunofluorescence intensity on primary mouse OPCs incubated for 2 days in DMSO or 002 in the presence of 50 ng/ml PDGF AA. A total of 4 fields with multiple PDGFRα+ cells (54 cells in DMSO and 34 cells in 002) were quantified. Two-tailed, unpaired Student’s t test. (e) The proportion of Ki67+ cells among PDGFRα+ OPCs in the same cells quantified in D after treatment with DMSO or 002. Two-tailed, unpaired Student’s t test. (f) Immunolabeling for surface PDGFRα, Ki67, and Sox10 in cells treated with DMSO or 002. Arrows indicate Ki67+ OPCs. Arrowheads indicate a cell that expresses PDGFRα but not Ki67. Scale = 25 μm. (g) Representative immunoblots of 30 μg of protein/lane from mouse OPCs that were treated overnight with DMSO or 002 and stimulated for 15 min with 50 ng/ml PDGFAA. The blots were stained for total and phosphorylated PDGFRα, AkT, mTOR, and Erk1/2. Blots for total PDGFRα and mTOR were normalized against Hsp90, and blots for total Akt and Erk1/2 were normalized against GAPDH. Arrows in the PDGFRα blots indicate 170-kDa upper PDGFRα bands that are also detected with anti-phospho-PDGFRα antibody, migrating slightly more slowly than the 160 kDa molecular weight marker (split between the two blots). The horizontal line on the left PDGFRα blot indicates the lower band corresponding to nonphosphorylated PDGFRα. (h–o) Quantification of the immunoblots for PDGFRα (h,i), Akt (j,k), mTOR (l,m), and the Erk1/2 p42/44 subunits of mitogen-activated protein kinase (MAPK) (n,o). The first graph for each set is total protein, normalized to Hsp90 (PDGFRα and mTOR) or GAPDH (Akt and Erk1/2). The second graph for each set is phosphorylated protein normalized to total protein. The p-values are from unpaired two-tailed Student’s t tests, n = 3
To better assess the effect of compound 002 on CMV promoter-driven PDGFRα, we transfected primary mouse astrocytes with CMV-Pdgfra-EF1a-GFP. Since astrocytes do not express PDGFRα, PDGFRα expression would be derived exclusively from the transfected vector and regulated by the CMV promoter. When we measured surface PDGFRα immunofluorescence intensity on live cells, we did not detect a difference in GFP+ astrocytes treated with 002 compared with those treated with DMSO (Figure 6b,c). Thus, PDGFRα expressed from CMV promoter in astrocytes was resistant to down-regulation by compound 002. Furthermore, even though the cells were cultured in the presence of PDGF AA, compound 002 did not reduce the number of GFP+ PDGFRα+ cells that expressed the proliferation antigen Ki67. Ki67 was detected in 12% of GFP+ PDGFRα+ astrocytes grown in DMSO and 19% among those grown in 002.
We next examined the effect of the compound 002 on the level of endogenous PDGFRα expressed by wild type OPCs. Immunofluorescence intensity of cell surface PDGFRα decreased by 50% on OPCs treated for 2 days with 002 compared to DMSO, but this did not reach statistical significance (Figure 6d,f). However, when we analyzed the data using paired t test to account for the differences in different batches of cell isolates, the difference was significant, with a p value of .0046. The reduction of surface PDGFRα in 002-treated OPCs was strikingly different from the case with CMV-Pdgfra-transfected astrocytes, on which surface PDGFRα was more robustly detected after 2 days in 002. Notably, the proportion of OPCs that expressed Ki67 was significantly lower after treatment with 002 (Figure 6e,f), consistent with the strong antiproliferative effects of 002 on OPCs shown in Figure 5. Under those conditions, we did not see any obvious changes in the level of Sox10 immunofluorescence in most cells, although a few cells appeared less brightly labeled (Figure 6f). There was no visible difference in the level of GFAP, or Olig2 immunofluorescence (not shown).
To more quantitatively investigate the effect of compound 002 on PDGFRα and its downstream signaling targets, we performed immunoblots of protein extracts from primary mouse OPCs that had been incubated overnight with DMSO or 002 and stimulated for 15 min with PDGF AA. Immunoblotting for total PDGFRα revealed two bands: a larger band at 170 kDa, which was also detected with anti-phospho-Y849-PDGFRα antibody (arrows in Figure 6g), and a less prominent lower band corresponding to nonphosphorylated receptor (horizontal line in Figure 6g). Treatment with 002 caused a 31% decrease in total PDGFRα (Figure 6h), while the level of phosphorylated PDGFRα, normalized to total PDGFRα, increased by 28%, though the latter did not reach statistical significance and likely reflects reduced total receptor levels (Figures 6i). Thus, compound 002 had no effect on PDGFRα activation, but significantly reduced the total level of PDGFRα, similar to the trend observed by immunofluorescence quantification.
Activated PDGFRα is known to recruit phosphatidyl inositol-3-kinase (PI-3K), which phosphorylates and activates Akt, which in turn activates mTOR. Immunoblotting for total and phosphorylated Akt did not reveal any effect of 002 (Figure 6g,j,k). Compound 002 caused a slight reduction in total and phosphorylated mTOR, but these changes did not reach significance (Figure 6g,l,m), although paired t test revealed a significance with a p value of .0042 (as opposed to 0.2061 by unpaired t test), which likely reflects slight variability among different batches of isolated cells, which could be influenced by the slight batch-to-batch differences in the degree of OPC maturity. We did not detect any change in total Erk1/2 mitogen-activated protein kinase (MAPK) or phospho-Erk1/2 after incubating the cells with compound 002 (Figure 6g,o,p), consistent with prior reports showing the lack of contribution of this pathway to PDGF-mediated OPC proliferation (Hill et al., 2013). Taken together, these results indicate that under the conditions that significantly reduced OPC proliferation, compound 002 only marginally reduced PDGFRα levels and did not affect signal transduction by PDGFRα.
4 |. DISCUSSION
An initial screening from a small commercial library using Pdgfra-luc assays in Oli-neu cells led to the identification of three structurally related guanidine compounds that repressed Pdgfra transcription. Limited synthetic expansion of the initial hits led to more potent aryl substituted guanidine derivatives such as compound 002, which most potently repressed Pdgfra transcription and OPC proliferation, while 008 and 005 were less potent. By contrast, a similar compound 006, which differed from compound 002 only by the placement of the aryl methoxy group, was significantly less effective. The active compounds also inhibited proliferation of other cells that expressed PDGFRα, such as NIH3T3 mouse fibroblasts, but not primary astrocytes that expressed PDGFRα under a heterologous constitutively active promoter.
4.1 |. Regulation of Pdgfra transcription by the guanidine compounds
PDGFRα is expressed by multiple cell types including OPCs, melanocytes, hematopoietic cells, and mesodermally derived cells such as fibroblasts, as well as on tumors arising from these cells (Demoulin & Essaghir, 2014; Indio et al., 2018; Zhang et al., 1998). A 2.2-kb 5′-flanking region of the human PDGFRA gene drives reporter expression faithfully in mesoderm and neural crest derived tissues (Zhang et al., 1998). There are three evolutionarily conserved enhancer elements C1–3 in the 5′-flanking region of the mouse Pdgfra gene, and during OPC specification the enhancer elements are bound by the high mobility group transcription factor Sox9 (Finzsch, Stolt, Lommes, & Wegner, 2008; Weider & Wegner, 2017). The Pdgfra-1595 promoter used in the luciferase assays contained the C2 and C3 but not C1 enhancer sequences. The longer Pdgfra-2229 construct encompassed the C1 sequence in addition to C2 and C3 but was significantly less active in Oli-neu cells than Pdgfra-1595.
In both Oli-neu cells and primary OPCs, the guanidine compounds that decreased Pdgfra transcription also downregulated NG2 (Cspg4) mRNA, which is not surprising, as NG2 is coordinately regulated with PDGFRα during development (McKinnon et al., 1990; Nishiyama et al., 1996b). We recently identified an evolutionarily conserved 1.45-kb enhancer in the first intron of the mouse Cspg4 gene, which is modulated by Sox10 and Olig2 and is sufficient to drive reporter expression in OPCs (Gotoh et al., 2018). NG2 is also highly expressed on proliferative progenitor cells of various lineages and is upregulated in tumors (Hidalgo & Logan, 2017; Stallcup, 2017; Stallcup & Huang, 2008; Yadavilli, Hwang, Packer, & Nazarian, 2016). It is not clear whether the decrease in Cspg4 mRNA is due to a direct effect of the compounds on its transcription or is secondary to reduced cell proliferation (Yadavilli et al., 2015).
The active guanidine compounds decreased Olig2 and Sox10 mRNA in Oli-neu cells, and there was a marginal reduction of Sox10 immunoreactivity but no noticeable reduction in Olig2 immunofluorescence on primary OPCs (not shown). The processes of Oli-neu cells and OPCs treated with compound 002 appeared less elongated, and in some cases, they seemed slightly more branched, but Mbp mRNA was not upregulated in either cell type. One of the original compounds marginally increased GFAP mRNA in Oli-neu cells, but in primary OPC cultures, we did not detect increased GFAP immunoreactivity among the treated group (not shown), suggesting that the compounds had not caused a fate switch to astrocytes. Furthermore, there was no evidence that the compounds caused OPCs to revert to a more stem cell-like phenotype, as judged by the lack of upregulation neural stem cell transcripts such as Nestin and Sox2 in Oli-neu cells (data not shown).
Several mechanisms are known to regulate Pdgfra transcription. During oligodendrocyte differentiation, microRNA species miR-219 and miR-338 are upregulated and directly repress Pdgfra mRNA (Dugas et al., 2010; Zhao et al., 2010). Interleukin-1β (IL-1β) reduces Pdgfra transcription by inducing the transcription factor C/EBPβ (CCAAT/enhancer-binding protein β), which binds to the Pdgfra promoter and modulates the proliferative response to PDGF-AA (Afink, Westermark, Lammerts, & Nister, 2004; Xie, Stroumza, & Graves, 1994). In OPCs, IL-1β inhibits OPC proliferation and PDGFRα expression by phosphorylating and activating the MAPK p38 (Vela, Molina-Holgado, Arevalo-Martin, Almazan, & Guaza, 2002). However, when we incubated Oli-neu cells with the p38 MAPK inhibitor SB202190 at 30 nM and 1 μM in the presence of compound 002, we did not observe reversal of the antiproliferative effect of 002 (data not shown). Nor did we observe any effects of SP600125, which inhibits the p38 MAPK target c-Jun kinase (data not shown).
Our immunofluorescence quantification showed that PDGFRα generated from endogenous Pdgfra gene on OPCs was more sensitive to the repressive effects of compound 002 than PDGFRα expressed from CMV promoter on astrocytes that do not express endogenous PDGFRα. This suggests that the compounds affect the endogenous Pdgfra promoter, directly or indirectly, by a mechanism that exists in OPCs. The magnitude of Pdgfra repression caused by the compounds was greater in Oli-neu cells than in primary OPCs, which could reflect an altered mechanism for Pdgfra mRNA regulation in Oli-neu cells that had been transformed with the neu oncogene (Jung et al., 1995).
4.2 |. Regulation of OPC proliferation by guanidine compounds
The guanidine compounds affected cell proliferation in a PDGFRα-dependent and cell type-specific manner. The antiproliferative effects were seen only in cells expressing PDGFRα from the endogenous promoter, and among those, more prominently in Oli-neu cells and OPCs than in NIH3T3 cells. However, the magnitude of inhibition of OPC proliferation by the compounds did not correlate with the magnitude of their effect on reducing PDGFRα protein levels or its downstream effectors. The short overnight exposure of the cells to the compound for the immunoblots to detect phosphorylation changes may not have been long enough relative to the half-life of these proteins (Rosenkranz et al., 2000). Alternatively, the guanidine compounds may be acting on another target that affects cell proliferation, which indirectly affects Pdgfra transcription.
We cannot rule out the possibility that the guanidine compounds reduce the viability of OPCs. The processes of primary OPCs appeared more retracted in the presence of compound 002 (Figure 6f), while those on Oli-neu cells were more elongated (Figure 5a,b). However, when plated on a more adhesive substrate, OPCs treated with 002 had slightly more numerous processes compared to the more bipolar morphology in DMSO (not shown), suggesting a more mature morphology. Furthermore, upregulation of mRNA encoding the cell cycle inhibitors p21cip1 and p27kip1 after treatment of Oli-neu cells indicates that the antiproliferative effect of the compounds could be attributed to cell cycle inhibition rather than a direct cytotoxic effect.
4.2.1 |. Possible targets of the guanidine compounds
The immunoblots and immunofluorescence intensity measurements consistently showed a marginal reduction of PDGFRα in OPCs treated with compound 002, and there was no reduction of phosphorylation of PDGFRα or their known targets Akt, mTOR, or Erk1/2. There is a self-regulatory autocrine loop, whereby Akt activation reduces Pdgfra transcription via mTOR, (Zhang et al., 2007). Furthermore, pathological activation of mTOR induced by deletion of Tuberous sclerosis complex-1 leads to postnatal reduction in OPC number and subsequent hypomyelination (Jiang et al., 2016). If the guanidine compounds affected a pathway that led to increased mTOR, this could repress Pdgfra transcription and trigger a compensatory or self-regulating fluctuation in mTOR via altered Akt signaling.
The mTOR-Pdgfra transcription loop can be influenced by several factors. For example, Src family kinases negatively regulate PDGFRα by promoting c-Cbl to ubiquitinate the receptor, increasing its degradation (Rosenkranz et al., 2000). Elevated levels of reactive oxygen species have been shown to reduce OPC proliferation by promoting c-Cbl activity via the Src family kinase Fyn leading to loss of PDGFRα (Li, Dong, Proschel, & Noble, 2007; Smith, Ladi, Mayer-Proschel, & Noble, 2000). The preservation of PDGFRα phosphorylation and its downstream signaling observed in this study in OPCs treated with compound 002 could reflect an equilibrium between repression of Pdgfra transcription by mTOR and compensatory restoration of Pdgfra transcription resulting from reduced PDGFRα, so that overall there is only a modest net change in PDGFRα and mTOR levels. We cannot rule out the possibility that the compounds exert a more direct effect on OPC cell cycle regulators, and that repression of Pdgfra transcription is a secondary phenomenon.
4.2.2 |. Therapeutic potential of the guanidine compounds
Although we have not identified the target of the guanidine compounds, the observed ability of the compounds to dramatically inhibit OPC proliferation and moderately repress Pdgfra transcription provide an attractive basis for further exploration of their potential in anti-glioma treatment. While previous studies on anti-PDGFR drugs have targeted the activation and downstream signaling of PDGFRA, the guanidine compounds described here are likely to act further upstream, repressing Pdgfra transcription, or by more directly inhibiting OPC proliferation. Thus, combinatorial use of the guanidine compounds and the traditional anti-PDGFRA agents or receptor tyrosine kinase inhibitors could provide an effective strategy.
Even with the limited set of analogs studied, the presence of clear structure-activity relationships suggests that these compounds were working through specific cellular targets. The nature and placement of the substituents on the aromatic ring significantly impacted the activity of the compounds. One important consequence of the incorporation of a guanidine group in the lead series is that this unit rendered the leads quite polar, and additional groups that further increase polarity such as pyridines may drastically limit cellular permeability. In future efforts, lead optimization will also prioritize the identification of modulators with the ability to penetrate the blood-brain barrier and allow more detailed in vivo studies in models of glioma or other CNS malignancies driven by PDGFRA. As these leads are still relatively small and hydrophilic, it should be possible to increase overall potency without overstepping key parameters associated with permeation of the blood-brain barrier. Overall, this initial structure-activity relationship profile indicated a variety of strategies that can be used to further enhance the activity of these leads.
In summary, we have described guanidine compounds that dramatically reduced OPC proliferation and selectively repressed Pdgfra transcription without affecting other receptor tyrosine kinases or activation of PDGFRα and its downstream targets. They are unique from the commonly used PDGFRA inhibitors in that they act upstream of PDGFRα, reducing its transcription rather than inhibiting the downstream signaling pathways. Thus, they could potentially be developed as an alternative or complementary approach to the currently available anti-PDGFR therapy.
ACKNOWLEDGMENTS
This work was conducted by grants from the NIH R21 NS 069960 (A. N. and D. W.), NS R01 NS07435 (A. N.), and S10OD016435 (A. N. for the purchase of SP8 confocal microscope for the University of Connecticut Advanced Light Microscopy Facility), and grants from the National Multiple Sclerosis Society RG4579 and RG-1612-26501 (A. N.). The authors thank Chris Fekete for many help-ful discussions on the interpretation of the data. The authors thank Dr William Stallcup (Sanford Burnham Institute) for antibodies to PDGFRα and PDGFRβ. The authors thank Drs Christopher O’Connell (Advanced Light Microscopy Facility) and Carol Norris (Flow Cytometry Facility) for their assistance and Youfen Sun for maintaining transgenic mice.
Funding information
National Institute of Neurological Disorders and Stroke, Grant/Award Numbers: R01 NS07435, R21 NS069960; National Institutes of Health, Grant/Award Number: S10OD016435; National Multiple Sclerosis Society, Grant/Award Numbers: RG-1612-26501, RG4579
Footnotes
CONFLICT OF INTEREST
The authors declare no conflict of interest.
DATA AVAILABILITY STATEMENT
The data that support the findings of this study are made available from the corresponding author upon reasonable request.
REFERENCES
- Afink G, Westermark UK, Lammerts E, & Nister M (2004). C/EBP is an essential component of PDGFRA transcription in MG-63 cells. Biochemical and Biophysical Research Communications, 315(2), 313–318. [DOI] [PubMed] [Google Scholar]
- Alphandery E (2018). Glioblastoma treatments: An account of recent industrial developments. Frontiers in Pharmacology, 9, 879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andrae J, Gallini R, & Betsholtz C (2008). Role of platelet-derived growth factors in physiology and medicine. Genes & Development, 22, 1276–1312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bansal R, Kumar M, Murray K, Morrison RS, & Pfeiffer SE (1996). Regulation of FGF receptors in the oligodendrocyte lineage. Molecular and Cellular Neurosciences, 7, 263–275. [DOI] [PubMed] [Google Scholar]
- Baron W, Decker L, Colognato H, & Ffrench-Constant C (2003). Regulation of integrin growth factor interactions in oligodendrocytes by lipid raft microdomains. Current Biology, 13, 151–155. [DOI] [PubMed] [Google Scholar]
- Barres BA, Hart IK, Coles HS, Burne JF, Voyvodic JT, Richardson WD, & Raff MC (1992). Cell death in the oligodendrocyte lineage. Journal of Neurobiology, 23, 1221–1230. [DOI] [PubMed] [Google Scholar]
- Barres BA, Lazar MA, & Raff MC (1994). A novel role for thyroid hormone, glucocorticoids and retinoic acid in timing oligodendrocyte development. Development, 120(5), 1097–1108. [DOI] [PubMed] [Google Scholar]
- Bögler O, Wren D, Barnett SC, Land H, & Noble M (1990). Cooperation between two growth factors promotes extended self-renewal and inhibits differentiaiton of oligodendrocyte-type-2 astrocyte (O-2A) progenitor cells. Proceedings of the National Academy of Sciences of the United States of America, 87, 6368–6372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Budde H, Schmitt S, Fitzner D, Opitz L, Salinas-Riester G, & Simons M (2010). Control of oligodendroglial cell number by the miR-17–92 cluster. Development, 137, 2127–2132. [DOI] [PubMed] [Google Scholar]
- Calver AR, Hall AC, Yu W-P, Walsh FS, Heath JK, Betsholtz C, & Richardson WD (1998). Oligodendrocyte population dynamics and the role of PDGF in vivo. Neuron, 20, 869–882. [DOI] [PubMed] [Google Scholar]
- Casaccia-Bonnefil A, Tikoo R, Kiyokawa H, Friedrich VJ, Chao MV, & Koff A (1997). Oligodendrocyte precursor differentiation is perturbed in the absence of the cyclin-dependent kinase inhibitor p27kip1. Genes and Development, 11, 2335–2346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dang TC, Ishii Y, Nguyen V, Yamamoto S, Hamashima T, Okuno N, … Sasahara M (2019). Powerful homeostatic control of oligodendroglial lineage by PDGFRalpha in adult brain. Cell Reports, 27, 1073–1089. [DOI] [PubMed] [Google Scholar]
- Demoulin JB, & Essaghir A (2014). PDGF receptor signaling networks in normal and cancer cells. Cytokine & Growth Factor Reviews, 25, 273–283. [DOI] [PubMed] [Google Scholar]
- Dugas JC, Cuellar TL, Scholze A, Ason B, Ibrahim A, Emery B, … Barres BA (2010). Dicer1 and miR-219 are required for normal oligodendrocyte differentiation and myelination. Neuron, 65, 597–611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dugas JC, Tai YC, Speed TP, Ngai J, & Barres BA (2006). Functional genomic analysis of oligodendrocyte differentiation. The Journal of Neuroscience, 26, 10967–10983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Durand B, Gao F-B, & Raff M (1997). Accumulation of the cyclin-dependent kinase inhibitor p27/kip1 and the timing of oligodendrocyte differentiation. EMBO Journal, 16, 306–317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Finzsch M, Stolt CC, Lommes P, & Wegner M (2008). Sox9 and Sox10 influence survival and migration of oligodendrocyte precursors in the spinal cord by regulating PDGF receptor alpha expression. Development, 135, 637–646. [DOI] [PubMed] [Google Scholar]
- Fruttiger M, Karlsson L, Hall AC, Abramsson A, Calver AR, Bostrom H, … Richardson WD (1999). Defective oligodendrocyte development and severe hypomyelination in PDGF-A knockout mice. Development, 126, 457–467. [DOI] [PubMed] [Google Scholar]
- Galvao RP, Kasina A, McNeill RS, Harbin JE, Foreman O, Verhaak RG, … Zong H (2014). Transformation of quiescent adult oligodendrocyte precursor cells into malignant glioma through a multi-step reactivation process. Proceedings of the National Academy of Sciences of the United States of America, 111, E4214–E4223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Galvez-Contreras AY, Quinones-Hinojosa A, & Gonzalez-Perez O (2013). The role of EGFR and ErbB family related proteins in the oligodendrocyte specification in germinal niches of the adult mammalian brain. Frontiers in Cellular Neuroscience, 7, 258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghiani CA, Eisen AM, Yuan X, DePinho RA, McBain CJ, & Gallo V (1999). Neurotransmitter receptor activation triggers p27 (Kip1) and p21 (CIP1) accumulation and G1 cell cycle arrest in oligodendrocyte progenitors. Development, 126, 1077–1090. [DOI] [PubMed] [Google Scholar]
- Gotoh H, Wood WM, Patel KD, Factor DC, Boshans LL, Nomura T, … Nishiyama A (2018). NG2 expression in NG2 glia is regulated by binding of soxE and bHLH transcription factors to a Cspg4 intronic enhancer. Glia, 66, 2684–2699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grotendorst GR, Igarashi A, Larson R, Soma Y, & Charette M (1991). Differential binding, biological and biochemical actions of recombinant PDGF AA, AB, and BB molecules on connective tissue cells. Journal of Cellular Physiology, 149, 235–243. [DOI] [PubMed] [Google Scholar]
- Hermanson M, Funa K, Hartman M, Claesson-Welsh L, Heldin CH, Westermark B, & Nister M (1992). Platelet-derived growth factor and its receptors in human glioma tissue: Expression of messenger rna and protein suggests the presence of autocrine and paracrine loops. Cancer Research, 52, 3213–3219. [PubMed] [Google Scholar]
- Hidalgo A, & Logan A (2017). Go and stop signals for glial regeneration. Current Opinion in Neurobiology, 47, 182–187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hill RA, Patel KD, Medved J, Reiss AM, & Nishiyama A (2013). NG2 cells in white matter but not gray matter proliferate in response to PDGF. The Journal of Neuroscience, 33, 14558–14566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang H, Teng P, Mei R, Yang A, Zhang Z, Zhao X, & Qiu M (2017). Tmeff2 is expressed in differentiating oligodendrocytes but dispensable for their differentiation in vivo. Scientific Reports, 7, 337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Indio V, Astolfi A, Tarantino G, Urbini M, Patterson J, Nannini M, … Remondini D (2018). Integrated molecular characterization of gastro-intestinal stromal tumors (GIST) harboring the rare d842v mutation in PDGFRA gene. International Journal of Molecular Sciences, 19(3), 723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ishii Y, Matsumoto Y, Watanabe R, Elmi M, Fujimori T, Nissen J, … Funa K (2008). Characterization of neuroprogenitor cells expressing the PDGF beta-receptor within the subventricular zone of postnatal mice. Molecular and Cellular Neurosciences, 37(3), 507–518. [DOI] [PubMed] [Google Scholar]
- Jiang M, Liu L, He X, Wang H, Lin W, Wang H, … Lu QR (2016). Regulation of PERK-eIF2alpha signalling by tuberous sclerosis complex-1 controls homoeostasis and survival of myelinating oligodendrocytes. Nature Communications, 7, 12185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones C, & Baker SJ (2014). Unique genetic and epigenetic mechanisms driving paediatric diffuse high-grade glioma. Nature Reviews Cancer, 14 (10), 651–661. 10.1038/nrc3811 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jung M, Kramer E, Grzenkowski M, Tang K, Blakemore W, Aguzzi A, … Trotter J (1995). Lines of murine oligodendroglial precursor cells immortalized by an activated neu tyrosine kinase show distinct degrees of interaction with axons in vitro and in vivo. European Journal of Neuroscience, 7, 1245–1265. [DOI] [PubMed] [Google Scholar]
- Li Z, Dong T, Proschel C, & Noble M (2007). Chemically diverse toxicants converge on Fyn and c-Cbl to disrupt precursor cell function. PLoS Biology, 5, e35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu C, Sage JC, Miller MR, Verhaak RG, Hippenmeyer S, Vogel H, … Zong H (2011). Mosaic analysis with double markers reveals tumor cell of origin in glioma. Cell, 146, 209–221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Livak KJ, & Schmittgen TD (2001). Analysis of relative gene expression data using real-time quantitative pcr and the 2(−delta delta c [t]) method. Methods, 25, 402–408. [DOI] [PubMed] [Google Scholar]
- McCarthy KD, & de Vellis J (1980). Preparation of separate astroglial and oligodendroglial cell cultures from rat cerebral tissue. Journal of Cell Biology, 85, 890–902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McKinnon RD, Matsui T, Dubois-Dalcq M, & Aaronson SA (1990). FGF modulates the PDGF-driven pathway of oligodendrocyte development. Neuron, 5, 603–614. [DOI] [PubMed] [Google Scholar]
- McKinnon RD, Waldron S, & Kiel ME (2005). PDGF alpha-receptor signal strength controls an RTK rheostat that integrates phosphoinositol 3′-kinase and phospholipase C gamma pathways during oligodendrocyte maturation. The Journal of Neuroscience, 25, 3499–3508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nazarenko I, Hede SM, He X, Hedren A, Thompson J, Lindstrom MS, & Nister M (2012). PDGF and PDGF receptors in glioma. Upsala Journal of Medical Sciences, 117, 99–112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nishiyama A, Komitova M, Suzuki R, & Zhu X (2009). Polydendrocytes (NG2 cells): Multifunctional cells with lineage plasticity. Nature Reviews. Neuroscience, 10, 9–22. [DOI] [PubMed] [Google Scholar]
- Nishiyama A, Lin XH, Giese N, Heldin CH, & Stallcup WB (1996a). Co-localization of NG2 proteoglycan and pdgf alpha-receptor on O2A progenitor cells in the developing rat brain. Journal of Neuroscience Research, 43, 299–314. [DOI] [PubMed] [Google Scholar]
- Nishiyama A, Lin XH, Giese N, Heldin CH, & Stallcup WB (1996b). Interaction between NG2 proteoglycan and PDGF alpha-receptor on O2A progenitor cells is required for optimal response to pdgf. Journal of Neuroscience Research, 43, 315–330. [DOI] [PubMed] [Google Scholar]
- Ozerdem U, Grako KA, Dahlin-Huppe K, Monosov E, & Stallcup WB (2001). NG2 proteoglycan is expressed exclusively by mural cells during vascular morphogenesis. Developmental Dynamics, 222, 218–227. [DOI] [PubMed] [Google Scholar]
- Paugh BS, Qu C, Jones C, Liu Z, Adamowicz-Brice M, Zhang J, … Baker SJ (2010). Integrated molecular genetic profiling of pediatric high-grade gliomas reveals key differences with the adult disease. Journal of Clinical Oncology, 28, 3061–3068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Phuphanich S, Raizer J, Chamberlain M, Canelos P, Narwal R, Hong S, … Laubscher K (2017). Phase II study of MEDI-575, an anti-platelet-derived growth factor-alpha antibody, in patients with recurrent glioblastoma. Journal of Neuro-Oncology, 131, 185–191. [DOI] [PubMed] [Google Scholar]
- Rosenkranz S, Ikuno Y, Leong FL, Klinghoffer RA, Miyake S, Band H, & Kazlauskas A (2000). Src family kinases negatively regulate platelet-derived growth factor alpha receptor-dependent signaling and disease progression. The Journal of Biological Chemistry, 275, 9620–9627. [DOI] [PubMed] [Google Scholar]
- Sherafat A, Hill RA, & Nishiyama A (2018). Organotypic slice cultures to study oligodendrocyte proliferation, fate, and myelination. Methods in Molecular Biology, 1791, 145–156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith J, Ladi E, Mayer-Proschel M, & Noble M (2000). Redox state is a central modulator of the balance between self-renewal and differentiation in a dividing glial precursor cell. Proceedings of the National Academy of Sciences of the United States of America, 97, 10032–10037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sommer I, & Schachner M (1981). Monoclonal antibodies (O1 and O4) to oligodendrocyte surfaces: An immunocytological study in the central nervous system. Developmental Biology, 83, 311–327. [DOI] [PubMed] [Google Scholar]
- Stallcup WB (2017). NG2 proteoglycan enhances brain tumor progression by promoting beta-1 integrin activation in both cis and trans orientations. Cancers (Basel), 9, 31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stallcup WB (2018). The NG2 proteoglycan in pericyte biology. Advances in Experimental Medicine and Biology, 1109, 5–19. [DOI] [PubMed] [Google Scholar]
- Stallcup WB, & Huang FJ (2008). A role for the NG2 proteoglycan in glioma progression. Cell Adhesion & Migration, 2, 192–201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- The Cancer Genome Atlas Network. (2008). Comprehensive genomic characterization defines human glioblastoma genes and core pathways. Nature, 455, 1061–1068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Touat M, Idbaih A, Sanson M, & Ligon KL (2017). Glioblastoma targeted therapy: Updated approaches from recent biological insights. Annals of Oncology, 28, 1457–1472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Meir EG, Hadjipanayis CG, Norden AD, Shu HK, Wen PY, & Olson JJ (2010). Exciting new advances in neuro-oncology: The avenue to a cure for malignant glioma. CA: A Cancer Journal for Clinicians, 60, 166–193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vela JM, Molina-Holgado E, Arevalo-Martin A, Almazan G, & Guaza C (2002). Interleukin-1 regulates proliferation and differentiation of oligodendrocyte progenitor cells. Molecular and Cellular Neurosciences, 20, 489–502. [DOI] [PubMed] [Google Scholar]
- Verhaak RG, Hoadley KA, Purdom E, Wang V, Qi Y, Wilkerson MD, … Cancer Genome Atlas Research Network. (2010). Integrated genomic analysis identifies clinically relevant subtypes of glioblastoma characterized by abnormalities in PDGFRA, IDH1, EGFR, and NF1. Cancer Cell, 17, 98–110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vermes I, Haanen C, Steffens-Nakken H, & Reutelingsperger C (1995). A novel assay for apoptosis. Flow cytometric detection of phosphatidylserine expression on early apoptotic cells using fluorescein labelled annexin v. Journal of Immunological Methods, 184, 39–51. [DOI] [PubMed] [Google Scholar]
- Weider M, & Wegner M (2017). SoxE factors: Transcriptional regulators of neural differentiation and nervous system development. Seminars in Cell & Developmental Biology, 63, 35–42. [DOI] [PubMed] [Google Scholar]
- Xie JF, Stroumza J, & Graves DT (1994). IL-1 down-regulates platelet-derived growth factor-alpha receptor gene expression at the transcriptional level in human osteoblastic cells. Journal of Immunology, 153, 378–383. [PubMed] [Google Scholar]
- Yadavilli S, Hwang EI, Packer RJ, & Nazarian J (2016). The role of NG2 proteoglycan in glioma. Translational Oncology, 9, 57–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yadavilli S, Scafidi J, Becher OJ, Saratsis AM, Hiner RL, Kambhampati M, … Nazarian J (2015). The emerging role of NG2 in pediatric diffuse intrinsic pontine glioma. Oncotarget, 6, 12141–12155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang Z, Watanabe M, & Nishiyama A (2005). Optimization of oligodendrocyte progenitor cell culture method for enhanced survival. Journal of Neuroscience Methods, 149(1), 50–56. [DOI] [PubMed] [Google Scholar]
- Zarghooni M, Bartels U, Lee E, Buczkowicz P, Morrison A, Huang A, … Hawkins C (2010). Whole-genome profiling of pediatric diffuse intrinsic pontine gliomas highlights platelet-derived growth factor receptor alpha and poly (ADP-ribose) polymerase as potential therapeutic targets. Journal of Clinical Oncology, 28, 1337–1344. [DOI] [PubMed] [Google Scholar]
- Zhang H, Bajraszewski N, Wu E, Wang H, Moseman AP, Dabora SL, … Kwiatkowski DJ (2007). PDGFRs are critical for PI3K/Akt activation and negatively regulated by mTOR. The Journal of Clinical Investigation, 117, 730–738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang XQ, Afink GB, Svensson K, Jacobs JJ, Gunther T, Forsberg-Nilsson K, … Nister M (1998). Specific expression in mouse mesoderm- and neural crest-derived tissues of a human PDGFRA promoter/lacz transgene. Mechanisms of Development, 70, 167–180. [DOI] [PubMed] [Google Scholar]
- Zhang Y, Chen K, Sloan SA, Bennett ML, Scholze AR, O’Keeffe S, … Wu JQ (2014). An RNA-sequencing transcriptome and splicing database of glia, neurons, and vascular cells of the cerebral cortex. The Journal of Neuroscience, 34, 11929–11947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao X, He X, Han X, Yu Y, Ye F, Chen Y, … Lu QR (2010). Micro-RNA-mediated control of oligodendrocyte differentiation. Neuron, 65, 612–626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng Y, Yamamoto S, Ishii Y, Sang Y, Hamashima T, van De N, … Sasahara M (2016). Glioma-derived platelet-derived growth factor-BB recruits oligodendrocyte progenitor cells via platelet-derived growth factor receptor-alpha and remodels cancer stroma. The American Journal of Pathology, 186, 1081–1091. [DOI] [PubMed] [Google Scholar]
- Zhu X, Bergles DE, & Nishiyama A (2008). NG2 cells generate both oligodendrocytes and gray matter astrocytes. Development, 135, 145–157. [DOI] [PubMed] [Google Scholar]
- Zuo H, Wood WM, Sherafat A, Hill RA, Lu QR, & Nishiyama A (2018). Age-dependent decline in fate switch from NG2 cells to astrocytes after Olig2 deletion. The Journal of Neuroscience, 38, 2359–2371. [DOI] [PMC free article] [PubMed] [Google Scholar]