

Abstract
Structure determination of membrane proteins is critical to the molecular understanding of many life processes, yet it has historically been a technically challenging endeavor. This past decade has given rise to a number of technological advancements, techniques, and reagents, which have facilitated membrane protein structural biology, resulting in an ever-growing number of membrane protein structures determined. To collate these advances, we have mined available literature to analyze the purification and structure determination specifics for all uniquely solved membrane protein structures from 2010-2019. Our analyses demonstrate the strong impact of single-particle cryo-electron microscopy on the field and illustrate how this technique has affected detergent and membrane mimetic usage. Furthermore, we detail how different structure determination methods, taxonomic domains and protein classes have unique detergent/membrane mimetic profiles, highlighting the importance of tailoring their selection. Our analyses provide a quantitative overview of where the field of membrane protein structural biology stands and how it has developed over time. We anticipate that these will serve as a useful tool to streamline future membrane protein structure determination by guiding the choice of detergent/membrane mimetic.
Keywords: Membrane proteins, structural biology, detergents, detergent mimetics, nanodiscs, amphipols, single-particle cryo-electron microscopy, X-ray crystallography
Graphical abstract
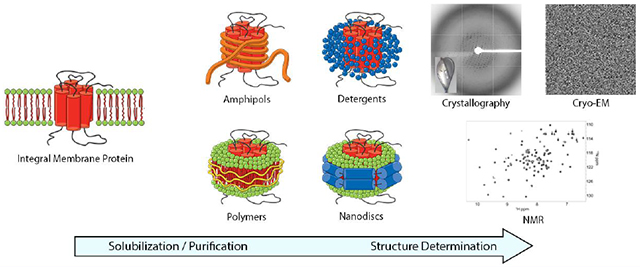
1. Introduction
Approximately a quarter of the human proteome encodes membrane proteins [1]. These proteins play an essential role in countless cellular functions and are integral in many processes of disease. Given this, it is unsurprising that membrane proteins are the molecular targets of over 40% of all FDA approved drugs [2, 3], and that the determination of their three-dimensional structures is of paramount importance. Structure determination of membrane proteins has historically been, and to a certain extent remains, a technically challenging endeavor – demonstrated by the fact that membrane proteins comprise less than 2% of all structures deposited in the Protein Data Bank (PDB) as of January 2020 [4]. These challenges can arise at any point in the workflow from gene to structure, for example: inadequate protein expression, poor solubilization efficiency from host lipid bilayers, limited long-term stability, as well as challenges at the structure determination stage per se, either by X-ray crystallography, single-particle cryo-electron microscopy (cryo-EM) or nuclear magnetic resonance (NMR) spectroscopy [5]. While these challenges have severely hindered the field, technological advances in expression systems, solubilization, purification techniques, and structure determination methods over the last decade have led to an ever-growing number of deposited membrane protein structures [6–8].
For membrane protein structure determination, identifying the best detergent or non-detergent alternative/membrane mimetic is critical for optimizing protein solubilization efficiency, yield, and stability [9–12]. Unfortunately, there are scarce standard guidelines regarding detergent usage for structural biology purposes. To address this, we have curated a dataset detailing the purification and structure determination specifics for all uniquely solved membrane protein structures – as classified by Dr. Stephen White’s database at UC Irvine (https://blanco.biomol.uci.edu/mpstruc/) – from 2010-2019 [13]. Here we analyze detergents and membrane mimetics (a.k.a. non-detergent alternatives) used for the solubilization and structure determination of membrane proteins, and draw correlations between these choices and the structure determining method of choice, protein taxonomic domain (i.e. prokaryotic or eukaryotic), and protein classification (i.e. ABC transporters, GPCRs, etc.). Our analyses demonstrate that over the last 5 years there has been a remarkable increase in the number of membrane protein cryo-EM structures determined, and an impressive improvement in their resolution. We exemplify how different structure determination methods, protein taxonomic domains and classes have unique detergent profiles, highlighting the importance of tailored detergent choice. Ultimately, we are hopeful that our work may serve as a useful tool to streamline membrane protein structure determination by guiding choices regarding detergent/membrane mimetic usage.
2. Methods
2.1. Dataset generation
Dr. Stephen White’s database at UC Irvine (https://blanco.biomol.uci.edu/mpstruc/) was used to define unique Protein Data Bank (PDB) entries and provide basic information for each protein including year of release, class, species, and publication information [13]. To be defined as a unique membrane protein, the protein must be from a unique species in the dataset. For example, the structures of the lipopolysaccharide transporter LptB from E. coli, V. cholerae, E. cloacae, and S. flexneri are all included in the dataset. Point mutations and structures with substrate bound of proteins already in the dataset are not considered unique. For this set of 767 unique proteins, the literature was then manually curated to extract the detergent used for solubilization, purification, and structure determination for each protein. Furthermore, additional information, such as structure determination method and crystallization condition (if applicable) were obtained manually. For each PDB ID, a script was utilized to extract the PDB release date and the PDB structure weight from the Protein Data Bank. The final dataset used for analysis for this manuscript is available from the authors upon request.
2.2. Database Analysis
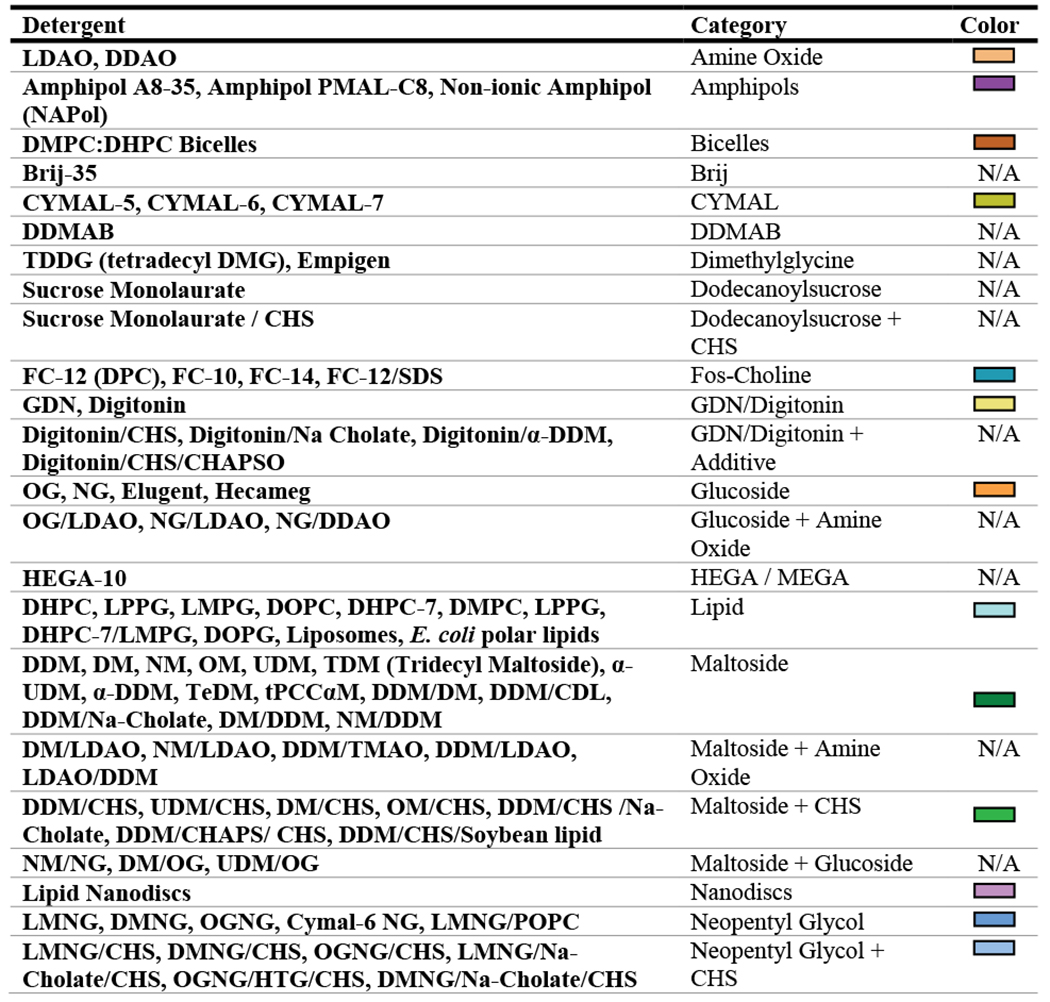
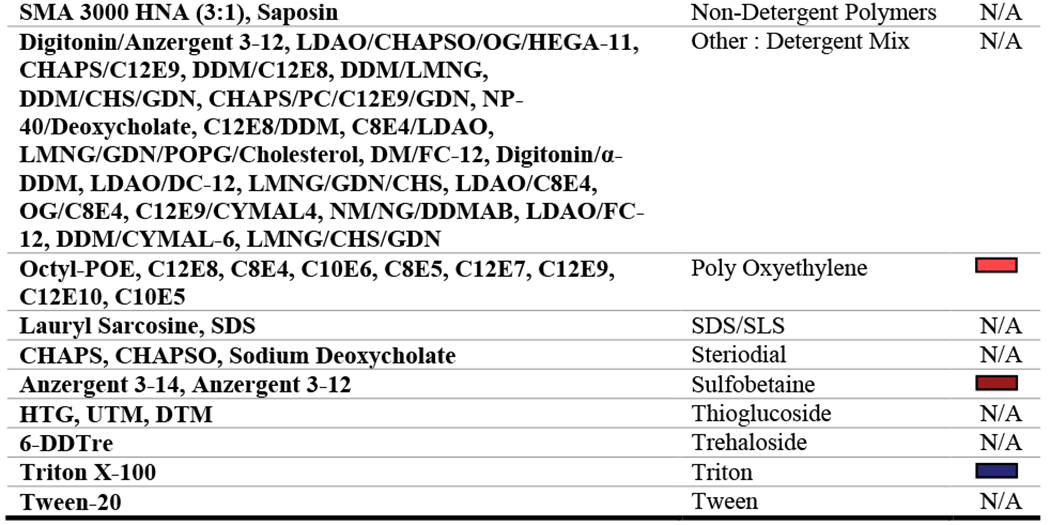
Data from 2010-2019 were analyzed. Of the 767 entries within this time period, 65 were excluded because they did not have transmembrane domains and did not utilize detergents. The variables that were analyzed include: year (PDB release date), protein molecular weight, protein taxonomic domain (prokaryotic or eukaryotic), protein type (alpha-helical or beta-barrel), protein class, structure determination method, solubilization detergent, structure determination detergent or non-detergent alternative, and use of detergent additives. Protein molecular weight was only analyzed for cryo-EM structures since only the molecular weight of the asymmetric unit was readily available from the PDB for those solved using X-ray crystallography. The ‘solubilization detergent’ was defined as the first detergent used (to extract the protein from the lipid bilayer) throughout the purification procedure, whereas the ‘structure determination condition’ was defined as the detergent or non-detergent alternative present during acquisition of structural data. Across the dataset, a total of 137 unique detergents, non-detergent alternatives, or detergent and additive combinations were used. Therefore, in an effort to simplify the analysis process, detergents were grouped into classes describing their chemical similarity (Table 1). For each analysis, the least commonly used detergents were collapsed into an “other” category which represented coverage of no more than 10% of the proteins analyzed. Excel 2016 PowerPivot functions were used to examine correlations between key variables listed above, and data were graphically represented using GraphPad Prism version 8.4.3 for Windows, GraphPad Software, San Diego, California USA, www.graphpad.com.
Table 1: Detergent classifications.
All detergents, detergent conditions, or non-detergent alternatives are grouped into chemically similar classes. N/A indicates that the condition was not used frequently enough to be plotted in our analyses and that any data for the condition would be collapsed into the ‘other’ category.
 |
 |
3. Results and Discussion
3.1. Trends in structure determination methods and the rise of cryo-EM
Our dataset contains 767 total entries from 2010-2019. Of these, 702 entries of transmembrane domain containing protein structures from 2010-2019 were analyzed (see exclusion criteria in Data Analysis). Within this subset, 187 structures were solved by cryo-EM, 479 by X-ray crystallography, and 36 by NMR (Figure 1A). In terms of general structural motifs, 578 proteins contained alpha-helical transmembrane domains, 103 proteins contained beta-sheet transmembrane domains, and 21 were monotopic proteins (Figure 1B).
Figure 1: Methods used in membrane protein structural biology and an overview of topology of transmembrane proteins within the dataset.
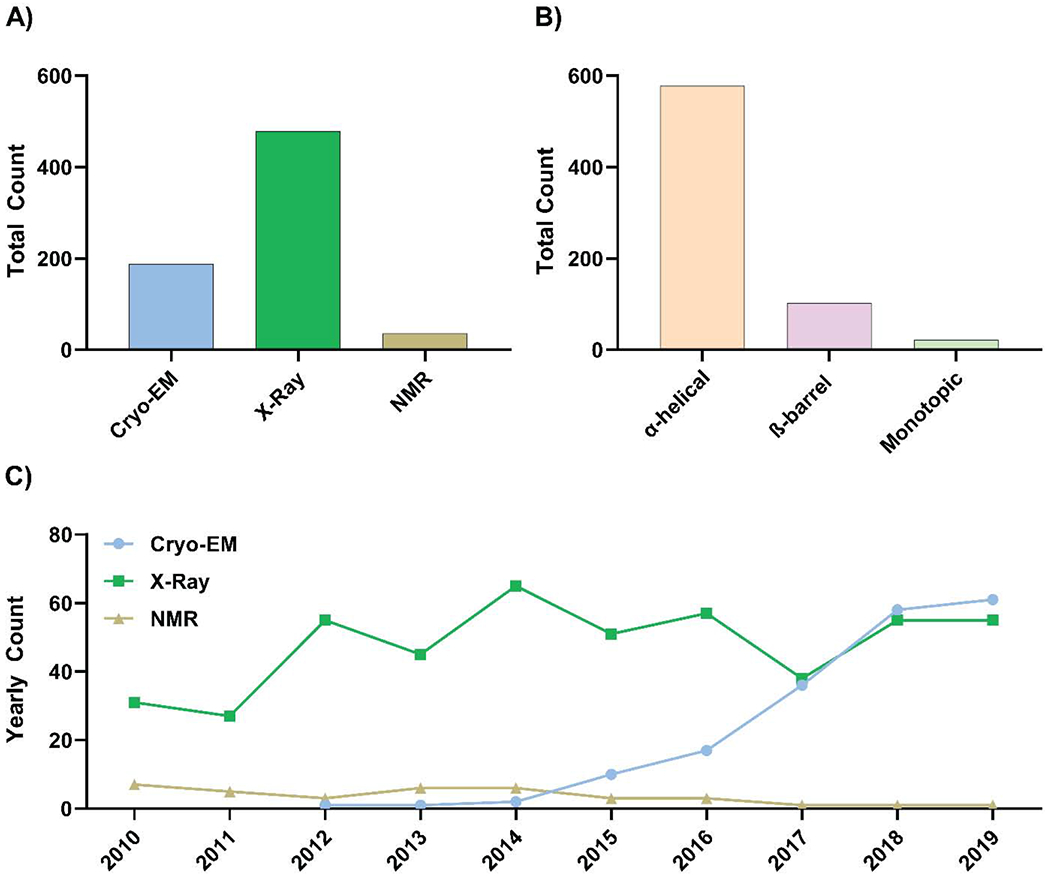
A) The total number of unique membrane proteins structures solved by cryo-EM, X-ray crystallography, or NMR from 2010-2019. B) Total counts of unique alpha-helical, beta-barrel, or monotopic transmembrane protein structures solved from 2010-2019. C) Yearly counts of unique membrane protein structures solved using either single-particle cryo-EM, X-ray crystallography, or NMR from 2010-2019. For (A) and (C), X-ray crystallographic structures include those solved using bicelle, dialysis, LCP, micro-batch, and vapor diffusion.
The mean resolution for membrane protein structures determined using single-particle cryo-EM has increased substantially from 4.2 ± 0.5 Å in 2015 to 3.6 ± 0.7 Å in 2019. X-ray crystallography, however, has solved membrane protein structures with a more consistent mean resolution of ~2.75 Å over the same time period (Table 2). The improvement in mean resolution for cryo-EM membrane protein structures is emblematic of general improvements beyond just membrane proteins, and is commonly referred to as the ‘resolution revolution’ [14]. This ‘revolution’ has been driven by technological advancements in transmission electron microscopes optics, direct detectors, image processing algorithms, and improved grid preparation tools [14–17]. Additionally, cryo-EM structures of membrane proteins have been specifically aided by the development of membrane mimetic systems such as amphipols and nanodiscs [18, 19] and the requirement for a substantially lower yield of protein compared to that required for X-ray crystallography [20]. These advancements have led to impressive growth in the number of single-particle cryo-EM membrane protein structures; indeed, cryo-EM now rivals X-ray crystallography as the favored method for membrane protein structure determination (Figure 1C). Furthermore, these developments have allowed for more structures of small membrane proteins (<200 kDa) to be solved using cryo-EM each year (Figure 2A), which fits into the larger narrative of the continuing decrease in lower size limits across the broader cryo-EM field [17, 21, 22].
Table 2: Mean nominal resolution of single-particle cryo-EM and X-ray crystallographic membrane protein structures.
The mean nominal resolutions of unique cryo-EM and X-ray crystallographic membrane protein structures from 2010-2019. Crystallographic structures include those solved by bicelle, dialysis, lipidic cubic phase (LCP), micro-batch, and vapor diffusion. Data represent mean +/− 1 S.D; n is indicated in parentheses where it is less than 3.
| Year | Cryo-EM (Å) | Crystallography (Å) |
|---|---|---|
| 2010 | N/A | 2.8 ± 0.7 |
| 2011 | N/A | 2.9 ± 0.5 |
| 2012 | 9.7 (n=1) | 2.8 ± 0.5 |
| 2013 | 3.3 (n=1) | 2.9 ± 0.4 |
| 2014 | 6.3 ± 1.3 (n=2) | 2.8 ± 0.5 |
| 2015 | 4.2 ± 0.5 | 2.8 ± 0.6 |
| 2016 | 4.3 ± 1.1 | 2.8 ± 0.6 |
| 2017 | 4.1 ± 1.3 | 2.8 ± 0.6 |
| 2018 | 3.7 ± 0.4 | 2.7 ± 0.6 |
| 2019 | 3.6 ± 0.7 | 2.7 ± 0.5 |
Figure 2: Cryo-EM, the resolution revolution, and its influence on the nature of structures solved.
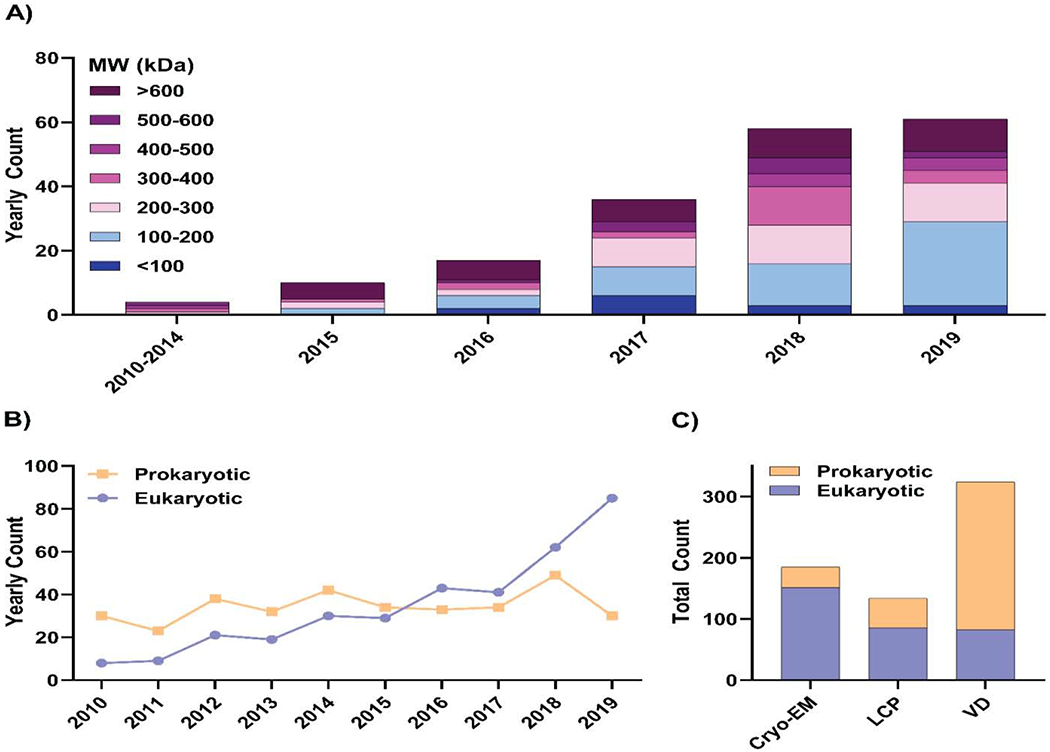
A) Yearly counts of unique single-particle cryo-EM membrane protein structures solved from 2010-2019 and a breakdown of their molecular weights in bins of 100 kDa. B) Yearly counts of unique prokaryotic and eukaryotic membrane protein structures solved from 2010-2019. C) Comparison of unique prokaryotic and eukaryotic membrane protein structures solved by either cryo-EM, LCP, or vapor diffusion (VD).
Issues surrounding protein stability and yield have classically been significant obstacles for solving eukaryotic membrane protein structure determination [8] because these proteins are typically less stable, and therefore more challenging to produce in large quantities compared to their prokaryotic counterparts. Despite this, there has been a continual increase in the number of eukaryotic membrane protein structures solved over the past decade (Figure 2B). This rise can in part be explained by advances in eukaryotic expression systems such as insect, mammalian, and yeast cell systems [23], in addition to improved sample preparation technologies such as nanodiscs, amphipols, and new detergents [10]. Interestingly, when comparing the number of prokaryotic and eukaryotic membrane protein structures solved using either cryo-EM, lipidic cubic phase (LCP), or vapor diffusion X-ray crystallography, it can be seen that the majority of cryo-EM and LCP structures are of eukaryotic proteins, whereas the majority of those solved using vapor diffusion are prokaryotic (Figure 2C). Evidently, the membrane-like environment provided by monoolein in LCP or nanodiscs in cryo-EM [12, 24, 25], alongside the lower protein yields required for cryo-EM [20], have been truly instrumental for this ~10-fold increase in eukaryotic membrane protein structures determined over the last 10 years, particularly as these techniques, once reserved for a select few labs, have become more accessible to the wider structural biology community.
3.2. The evolving landscape of detergents and membrane mimetics
The unique physical and chemical interactions between specific detergents and a membrane protein of interest are critical for efficient extraction of the protein from the cell membrane, and to maintain solubility and stability throughout the protein purification procedure [11, 12]. Inappropriate detergent choice can significantly reduce protein yields, cause aggregation, and interfere with structure determination strategies – for example by preventing the formation of crystal contacts (X-ray crystallography) [26, 27], or by affecting blotting and particle distribution on grids (cryo-EM) [10–12]. For these reasons, standard procedure commonly involves an initial small-scale screen of a series of detergents to identify which yield the largest quantity of soluble, active, homogeneous, and stable protein [5, 28–31].
Over the last decade, maltosides have been the detergent of choice for membrane protein solubilization, comprising 59.9% of conditions analyzed. Of this 59.9%, 17.7% combined maltosides with the lipidic additive cholesteryl hemisuccinate (CHS), whereas the remaining 42.2% did not. N-Dodecyl-P-D-Maltoside (DDM) is the most commonly used maltoside (84.2% of total maltoside use). DDM has a critical micelle concentration (CMC) of ~0.01%, an acyl chain length of 12 and a disaccharide headgroup [32]. These properties make DDM ideal for membrane protein solubilization at relatively low concentrations while maintaining protein stability – making it an excellent, cost-effective choice. Glucosides – such as octyl glucoside (OG) – on the other hand, are used less frequently for membrane protein solubilization – accounting for 5.8% of solubilization conditions analyzed in our dataset (Figure 3A). This is likely due to the fact that OG has a high CMC (0.5%), a shorter acyl chain length (8 carbons) and a monosaccharide headgroup – properties which reduce protein stability and call for large quantities of detergent for efficient solubilization and purification [32, 33].
Figure 3: Trends in detergents used for membrane protein solubilization.
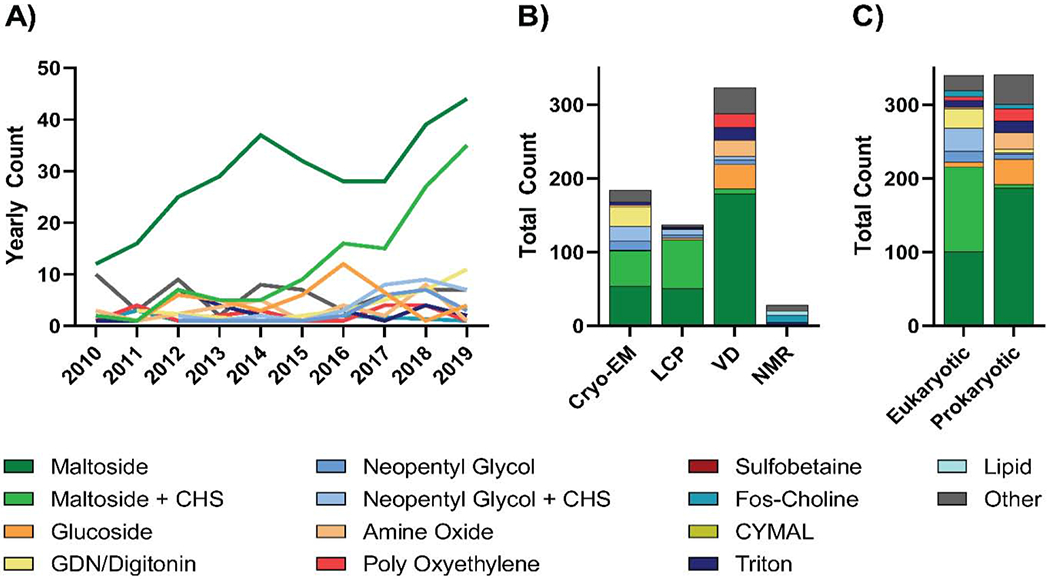
A) Year-by-year analysis of detergent classes used for solubilization of membrane proteins with unique structures solved between 2010 and 2019. B) Analysis of solubilization detergents used for unique membrane protein structures solved by either single-particle cryo-EM, LCP, vapor diffusion (VD), or NMR from 2010 to 2019. C) Comparison of detergents used to solubilize prokaryotic and eukaryotic membrane proteins with unique structures from 2010-2019. For each analysis, detergents were classified as outlined in Table 1 and the least commonly used detergents were collapsed into an “other” category which represented coverage of no more than 10% of proteins analyzed.
Distinct patterns in solubilization detergent choice emerge when proteins are separated on the basis of structure determination method (Figure 3B). For structures solved using cryo-EM and LCP, maltosides with and without CHS were the most commonly used solubilization conditions, with maltosides alone used 29.3% and 37.2% of the time for cryo-EM or LCP structures respectively, and maltosides with CHS used 26.1% and 48.2% of the time likewise respectively. (Figure 3B). For cryo-EM structures specifically, glyco-diosgenin (GDN) or digitonin (14.7%), neopentyl glycols with (10.9%) and without (6.5%) CHS were also commonly used solubilization conditions (Figure 3B). Alternatively, proteins with structures solved using vapor diffusion were predominantly solubilized by maltosides without CHS (55.4%), followed by glucosides (10.2%), amine oxides (6.8%), poly-oxyethylenes (5.9%), and triton (5.3%; Figure 3B). Finally, proteins with structures solved by NMR spectroscopy presented a unique solubilization condition profile, with fos-cholines and lipids being used 35.7% and 17.9% of the time respectively (Figure 3B).
Given that solubilization is one of the earliest steps in the purification procedure, these differences are most likely due to the varying protein types typically solved using each method rather than any aspect of the methods themselves. For example, when analyzing the relationship between solubilization detergent choices and taxonomic domain, it can be seen that CHS is used far more frequently for eukaryotic proteins than for prokaryotic proteins (43.8% vs 2.1%; Figure 3C). This is likely due to the fact that eukaryotic membranes typically contain cholesterol [34], and thus the addition of CHS is fitting and aids stabilization. Additionally, an increased use of GDN and digitonin can be seen for eukaryotic membrane protein solubilization compared to that of prokaryotic (7.9% vs 1.5 %; Figure 3C). This is likely attributed to the steroidal moiety present in GDN and digitonin mimicking the interaction of membrane proteins with cholesterol [35] and thus stabilizing these eukaryotic proteins by a similar mechanism.
While certain proteins are maintained in the same detergent from solubilization through to structure determination, in many instances it is beneficial to exchange these conditions to maintain protein stability and to optimize conditions for certain structure determination techniques [10]. For membrane proteins with structures solved by vapor diffusion and LCP crystallography, detergent conditions for solubilization were distinct from those used for structure determination 65.2% and 30.7% of the time respectively (Figure 4). This exchange often serves to place the protein in a shorter chain-length detergent to reduce the detergent micelle size and therefore increase the likelihood of crystal contact formation [26, 27] (Figure 4). While exchanges for vapor diffusion and LCP are limited to detergent, cryo-EM exchanges can encompass either detergent exchange, or detergent to non-detergent alternative exchange. The most commonly used non-detergent alternatives include amphipols and lipid nanodiscs.
Figure 4: Changes in conditions between solubilization and structure determination for different structure determination methods.
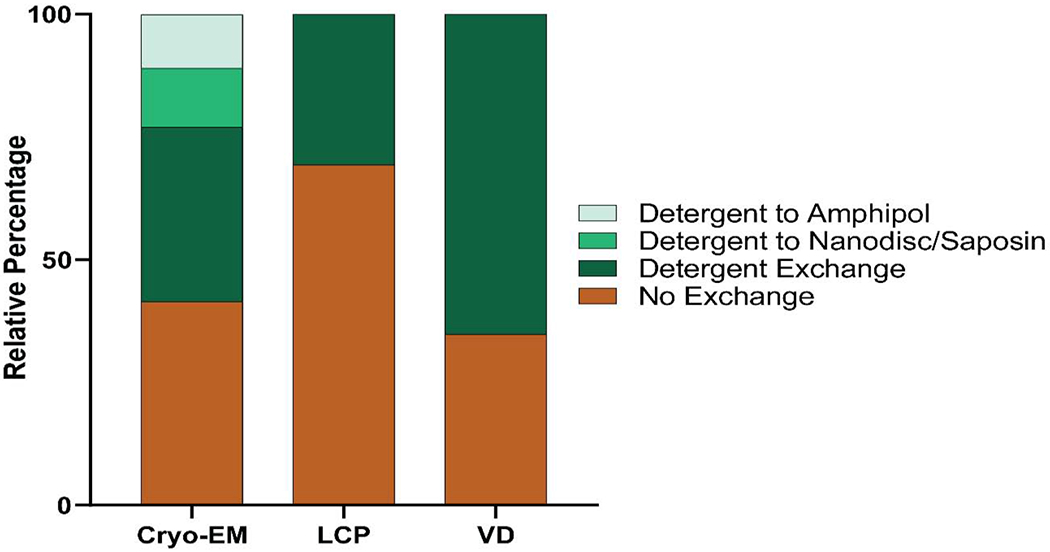
No exchange signifies that the protein was maintained in the same detergent condition throughout the entire protein purification and structure determination process. Detergent exchange signifies that detergent was exchanged into a different detergent. For single-particle cryo-EM specifically, proteins could also be exchanged into lipid nanodiscs/saposin or amphipols for structure determination.
Amphipols are amphipathic polymers that wrap around hydrophobic transmembrane domains to keep membrane proteins soluble and stable in solution [19, 36], whereas lipid nanodiscs utilize amphipathic membrane scaffold proteins to wrap transmembrane proteins in a discoidal lipid bilayer [18, 37, 38]. Similarly, saposin-derived lipid nanoparticles, in which saposin A serves to reconstitute membrane proteins into a lipid environment, have also been utilized [39]. For membrane proteins with structures solved using cryo-EM, conditions changed between solubilization and structure determination 58.5% of the time, either into a different detergent (35.5%), into nanodisc/saposin (12.0%), or into amphipol (10.9%; Figure 4).
Whether a condition exchange occurred or not, maltosides without and with CHS remained the favored conditions of choice for structure determination – accounting for 28.0% and 10.3% of all structure determining conditions respectively – followed by poly-oxyethylenes (10.3%), glucosides (9.3%), and GDN/Digitonin (8.4%; Figure 5A). Despite still being the favored choice, there is a notable net reduction in the use of maltosides at the structure determination stage compared to that seen at the solubilization stage, and this decrease is observed for all structure determination methods (Figure 4 and Figure 5).
Figure 5: Trends in structure determination conditions for membrane protein structures.
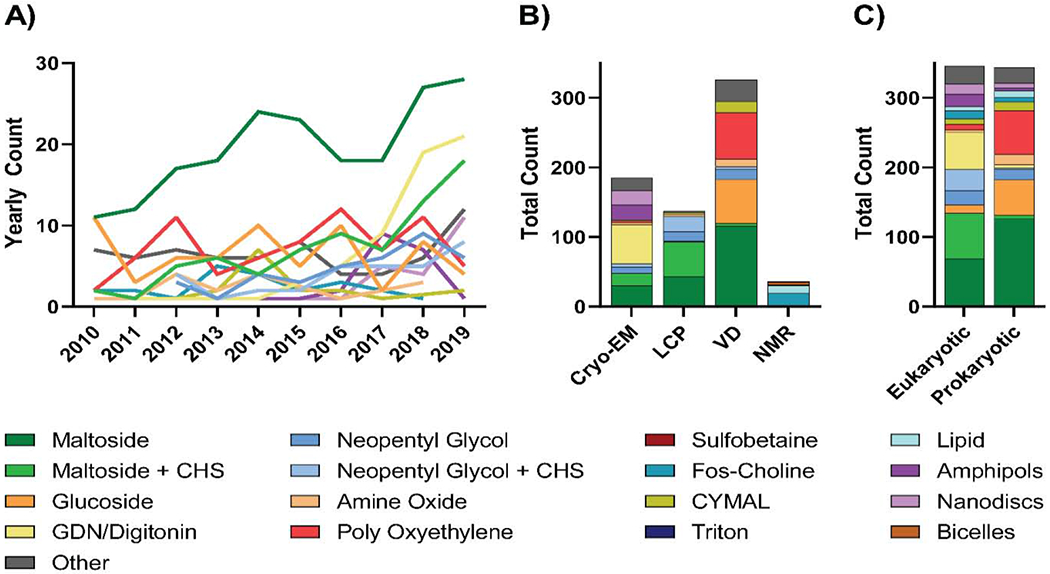
A) Year-by-year analysis of structure determination conditions used for unique membrane protein structures solved between 2010 and 2019. B) Analysis of structure determination conditions used for unique membrane protein structures solved using either single-particle cryo-EM, LCP, vapor diffusion (VD), NMR from 2010 to 2019. C) Comparison of structure determination conditions used for prokaryotic and eukaryotic membrane proteins with unique structures from 2010-2019. For each analysis, detergents were classified as outlined in Table 1 and the least commonly used detergents were collapsed into an “other” category which represented coverage of no more than 10% of proteins analyzed.
Unique patterns for structure determination conditions evolved when proteins were separated on the basis of structure determination method (Figure 5B). For cryo-EM, GDN/digitonin is the most commonly used condition – accounting for 30.3% of conditions, followed in order by maltosides without CHS (16.2%), amphipols (11.4%), nanodiscs (11.4%), and maltosides with CHS (9.7%; Figure 5B). This is in stark contrast with vapor diffusion and LCP. For LCP, maltosides with or without CHS were used for most proteins (36.5% and 31.4% respectively) followed by neopentyl glycols with CHS (16.1%) and without CHS (10.2%). Alternatively, maltosides without CHS (35.6%), poly-oxyethylenes (20.6%), and glucosides (19.3%) were the predominant conditions used for proteins with structures solved using vapor diffusion. Similar to what was observed when analyzing the choice of solubilization detergent, these method-specific differences in structure determination condition can, in part, be due differences in the type of protein (i.e. prokaryotic or eukaryotic) most commonly solved by each of the methods (Figure 5C). However, given that the detergent and non-detergent alternative choices can directly impact, or be impacted by, structure determination methods [10, 27], direct comparisons can be made between conditions used for distinct structure determination techniques. For example, fos-choline detergents were used 47.2% of the time for protein structures solved by NMR (Figure 5B) as they provide a small micelle size which allows for fast isotropic protein-detergent complex tumbling; however, concerns have been raised regarding the propensity of fos-cholines to partially denature membrane proteins [40, 41]. Alternatively, amphipols and lipid nanodiscs – while not compatible with crystallographic analysis – carry several benefits for structure determination using cryo-EM and, coincidently, both were utilized in 11.4% of cryo-EM structures. Amphipols are suitable for cryo-EM analysis because they don’t form micelles, thus minimizing background and avoiding contrast issues caused by free detergent micelles [10, 12]. Furthermore, the reconstitution of membrane proteins into amphipols can restrict their conformational flexibility, which can improve the homogeneity of the particles analyzed for cryo-EM [12]. However, it has been suggested that conformations induced by reconstitution into amphipols may in some instances have the potential to differ from physiologically relevant states [42]. On the other hand, lipid nanodiscs carry the same benefits as amphipols – assuming empty nanodiscs have been adequately removed – while providing the additional advantage of imaging the protein in a near-native lipid environment [18, 37, 38]. This has resulted in an increase in popularity since their original development [43–45], and we anticipate that use of these non-detergent alternatives, alongside others such as styrene maleic acid (SMA) [46], diisobutylene/maleic acid (DIBMA) copolymer [47], saposin [39], and peptidiscs [48], will continue to grow. Finally, cryo-EM has recently been used to solve the structures of membrane proteins reconstituted into small, uniformly sized proteoliposomes [49, 50]. This technique will likely aid structure determination of membrane transporters and channels in energetically unfavorable conformations by creating gradients across the liposomal membrane.
3.3. Class-specific trends in structure determination methods and detergent usage
To assist in optimal future detergent and structure determination technique selection, we have analyzed the conditions used to solve the top ten classes of membrane proteins represented within the dataset (Figure 6). These are classified by Dr. Stephen White’s database [13] and listed in alphabetical order include: ATP Binding Cassette (ABC) transporters; beta-barrel proteins: monomeric/dimeric; beta-barrel proteins: porins and relatives; ‘other’ ion channels (these include: glutamate receptors, otopetrin protein channels, CALHM2, among others); Na+/K+/H+ ion channels; Transient Receptor Potential (TRP) channels; G-Protein-Coupled Receptors (GPCRs); Major Facilitator Superfamily (MFS) transporters; Sec and Translocase proteins; and Solute Carrier (SLC) transporters.
Figure 6: Conditions used to solve structures of the ten most-commonly solved membrane proteins classes.
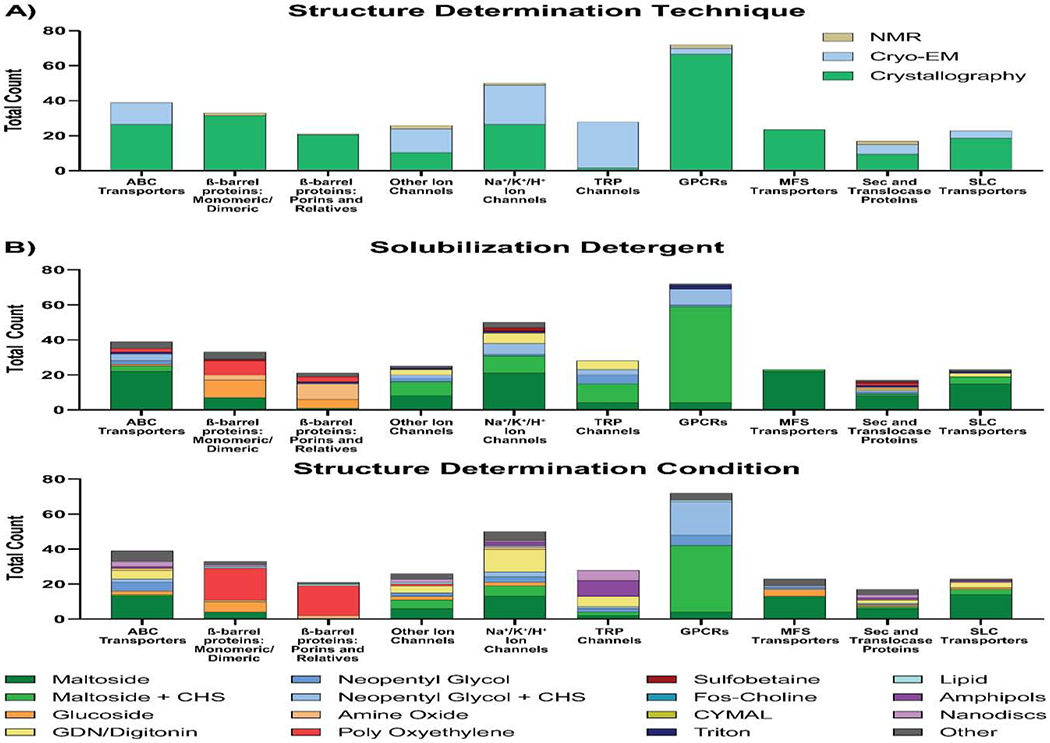
A) Structure determination techniques used to solve unique structures of the ten most commonly solved classes of membrane proteins, as classified by Dr. Stephen White’s Database at UC Irvine (https://blanco.biomol.uci.edu/mpstruc/) [13]. B) Analysis of the solubilization detergents (top) and the structure determination conditions (bottom) used for each protein class. For each analysis, detergents were classified as outlined in Table 1 and the least commonly used detergents were collapsed into an “other” category which represented coverage of no more than 10% of proteins analyzed.
Across the ten different classes, GPCRs, MFS transporters, and beta-barrel proteins were predominantly solved by crystallography, TRP channels were almost exclusively solved by cryo-EM, and the remaining five classes by a mixed representation of the three techniques (Figure 6A). This analysis is in some way skewed because cryo-EM only became a technique of choice for membrane protein structure determination in more recent years (Figure 1C and Table 2) [10]. However, it also reflects that certain protein classes are more suitable for specific structure determination techniques due to properties such as size and flexibility [22, 51].
Different protein classes favored unique subsets of detergents for both protein solubilization and structure determination because shared structural motifs are likely stabilized by certain detergents more favorably than by others. For eight of these ten classes, at least half of the proteins were solubilized using maltosides (with or without CHS; Figure 6B, top panel). Maltosides without CHS were used more often than maltosides with CHS for ABC transporters (56.4% and 7.7% respectively), Na+/K+/H+ ion selective channels (42.0% and 20.0% respectively), MFS transporters (95.7% and 4.4% respectively), Sec and Translocase Proteins (47.1% and 5.9% respectively), and SLC transporters (65.2% and 17.4% respectively; Figure 6B, top panel). Alternatively, maltosides with CHS were used more often than without CHS for TRP channels (39.3% and 14.3% respectively) and GPCRs (76.4% and 5.6% respectively; Figure 6B, top panel). Other’ ion channels were solubilized by maltosides with and without CHS equally at 32.0% each. In addition to maltosides, ABC transporters were commonly solubilized using neopentyl glycols with and without CHS (10.3% and 5.1% respectively), poly-oxyethylenes (5.1%), triton (2.6%), and glucosides (2.6%); GPCRs by neopentyl glycols with and without CHS (12.5% and 1.39% respectively), and triton (2.8%); SLC transporters by GDN/Digitonin (8.7%) and triton (4.4%); and Sec-Trans proteins used a variety of different detergents (Figure 6B, top panel). For TRP, Na+/K+/H+, and ‘other’ ion channels, the most common solubilization conditions used included neopentyl glycols with CHS (10.7%, 12.0%, and 8.0% respectively), neopentyl glycols without CHS (17.9%, 2.0%, and 8.0% respectively), and GDN/Digitonin (17.9%, 12.0%, and 12.0% respectively). In contrast to the eight classes of alpha-helical proteins, the two classes of beta-barrel proteins have remarkably distinct solubilization condition profiles (Figure 6B, top panel). Monomeric/dimeric beta-barrel proteins were most often solubilized by glucosides (30.3%), followed by poly-oxyethylenes (24.2%), and maltosides without CHS (21.2%; Figure 6B, top panel). Beta-barrel porins and relatives were predominately solubilized by amine oxides (42.9%), glucosides (23.8%), and poly-oxyethylenes (14.3%; Figure 6B, top panel). These unique solubilization detergent profiles for beta-barrel proteins is exemplary of how secondary/tertiary structure of transmembrane proteins can influence the detergents that they favorably interact with.
In terms of conditions used for structure determination, there is substantially greater diversity across all protein classes compared to those used for solubilization (Figure 6B). Maltosides either with and without CHS remained the predominant condition for GPCRs (52.8% and 5.6% respectively), MFS transporters (0% and 56.5% respectively), SLC transporters (13.0% and 60.9% respectively), ABC transporters (2.6% and 33.3% respectively), Na+/K+/H+ channels (12.0% and 26.0% respectively), ‘other’ ion channels (19.2% and 23.1% respectively), and Sec and Translocase proteins (5.9% and 35.3% respectively; Figure 6B, bottom panel). Amongst these classes, in addition to maltosides, GPCRs were commonly solved in neopentyl glycols with or without CHS (26.4% and 8.3% respectively); MFS transporters commonly in glucosides (17.4%) or neopentyl glycols with or without CHS (4.4% and 4.4% respectively); SLC transporters in GDN/Digitonin (13.0%); ABC transporters in GDN/Digitonin (12.8%), neopentyl glycols with or without CHS (5.1% and 12.8%), nanodiscs (7.7%) or glucosides (5.1%); Na /K+/H+ channels in GDN/Digitonin (26.0%); ‘other’ ion channels in GDN/Digitonin (15.4%), neopentyl glycols without CHS (7.7%), glucosides (7.7%), or nanodiscs (7.7%); and Sec and Translocase proteins in nanodiscs (11.8%), GDN/Digitonin (11.8%), or amphipols (5.9%; Figure 6B, bottom panel). Interestingly, the majority of TRP channel structures were solved in either amphipols (32.1%), nanodiscs (21.4%), or GDN/Digitonin (21.4%; Figure 6B, bottom panel). The prevalent use of amphipols and nanodiscs for TRP channels has also been observed when examining all TRP channel structures, i.e. including non-unique structures that are not part of our dataset [52–55]. In stark contrast with these alpha-helical classes, the structures of two beta-barrel protein classes, monomeric/dimeric and porins and relatives, were most commonly solved in poly-oxyethylenes (54.6% and 81.0% respectively; Figure 6B, bottom panel). These poly-oxyethylenes were rarely used for structure determination of alpha-helical proteins, suggesting a unique synergy between them and the secondary/tertiary structural arrangements of beta-barrel proteins. For beta-barrel monomeric/dimeric proteins, glucosides (18.2%) and maltosides without CHS (12.1%) were also commonly used, and for beta-barrel porins and relatives, amine oxides (9.5%) were also commonly used.
Whilst these analyses are, in our opinion, informative, it should be noted that for these data, there may likely be a degree of selection bias in detergent choice since researchers may be inclined to employ the same detergent conditions used previously for related proteins. As the detergent and membrane mimetic toolkit continues to expand, this selection bias might possibly cause a lag in the use of new detergent types as more conventional choices such as maltosides will continue to dominate. In some cases, adhering to conventional choices may be adequate and can provide useful guidance for detergent selection, for example GPCRs are most commonly solubilized in maltosides with CHS and have had their structures solved using either maltosides with CHS or neopentyl glycols with CHS (Figure 6B). However, for the majority of protein classes, there is a wider variety of detergents used which highlights the importance of wider detergent screening. We hope that our analysis can provide a good start point for detergents to select for screening and may assist in guiding research where resources for screening are not as readily available.
4. Conclusions
The past decade has been an exceptionally transformative period for membrane protein structural biology. This dataset and our analyses summarize the key changes that have occurred in these ten years, particularly through the lens of detergent and non-detergent alternative use for distinct structure determination methods, taxonomic domains, and membrane protein classes. This work offers structural biologists a roadmap for detergent/membrane mimetic choice, to streamline membrane protein structure determination.
Highlights.
Tailored detergent selection is crucial for membrane protein structural biology
Generation of a detergent and membrane mimetic dataset covering 2010-2019
Protein type and structure determination method impact detergent choice
Nanodiscs and amphipols have increased in popularity over the last decade
Cryo-EM has significantly impacted membrane protein structural biology
5. Acknowledgments
We thank Brianna Young and David Weber for having provided a sample NMR spectrum for the graphical abstract. This work was supported by NIH grants (R35 GM132120 to FM). BCC was supported by the Rabi Scholars Program of the Columbia University in the City of New York, and RJC by the Simons Society of Fellows (Award Number: 578646). Funding sources had no involvement in the preparation and submission of this article for publication.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Declaration of competing interests
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
E.E.P. Jr. was formerly employed by Anatrace Products, LLC, a manufacturer and vendor of detergents and lipids.
6. References
- 1.Fagerberg L, et al. Prediction of the human membrane proteome. Proteomics, 2010. 10(6): p. 1141–9. [DOI] [PubMed] [Google Scholar]
- 2.Drews J, Drug discovery: a historical perspective. Science, 2000. 287(5460): p. 1960–4. [DOI] [PubMed] [Google Scholar]
- 3.Overington JP, Al-Lazikani B, and Hopkins AL, How many drug targets are there? Nat Rev Drug Discov, 2006. 5(12): p. 993–6. [DOI] [PubMed] [Google Scholar]
- 4.Berman HM, et al. The Protein Data Bank. Nucleic Acids Res, 2000. 28(1): p. 235–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Wiener MC, A pedestrian guide to membrane protein crystallization. Methods, 2004. 34(3): p. 364–72. [DOI] [PubMed] [Google Scholar]
- 6.Shimizu K, et al. Comparative analysis of membrane protein structure databases. Biochim Biophys Acta Biomembr, 2018. 1860(5): p. 1077–1091. [DOI] [PubMed] [Google Scholar]
- 7.Allen JP, Recent innovations in membrane-protein structural biology. F1000Res, 2019. 8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.He Y, Wang K, and Yan N, The recombinant expression systems for structure determination of eukaryotic membrane proteins. Protein Cell, 2014. 5(9): p. 658–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Seddon AM, Curnow P, and Booth PJ, Membrane proteins, lipids and detergents: not just a soap opera. Biochim Biophys Acta, 2004. 1666(1-2): p. 105–17. [DOI] [PubMed] [Google Scholar]
- 10.Thonghin N, et al. Cryo-electron microscopy of membrane proteins. Methods, 2018. 147: p. 176–186. [DOI] [PubMed] [Google Scholar]
- 11.Arachea BT, et al. Detergent selection for enhanced extraction of membrane proteins. Protein Expr Purif, 2012. 86(1): p. 12–20. [DOI] [PubMed] [Google Scholar]
- 12.Mio K and Sato C, Lipid environment of membrane proteins in cryo-EM based structural analysis. Biophys Rev, 2018. 10(2): p. 307–316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.White SH, Biophysical dissection of membrane proteins. Nature, 2009. 459(7245): p. 344–6. [DOI] [PubMed] [Google Scholar]
- 14.Kuhlbrandt W, Biochemistry. The resolution revolution. Science, 2014. 343(6178): p. 1443–4. [DOI] [PubMed] [Google Scholar]
- 15.Cheng Y, Single-Particle Cryo-EM at Crystallographic Resolution. Cell, 2015. 161(3): p. 450–457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Sgro GG and Costa TRD, Cryo-EM Grid Preparation of Membrane Protein Samples for Single Particle Analysis. Front Mol Biosci, 2018. 5: p. 74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Merk A, et al. Breaking Cryo-EM Resolution Barriers to Facilitate Drug Discovery. Cell, 2016. 165(7): p. 1698–1707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Bayburt TH and Sligar SG, Membrane protein assembly into Nanodiscs. FEBS Lett, 2010. 584(9): p. 1721–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Popot JL, et al. Amphipols from A to Z. Annu Rev Biophys, 2011. 40: p. 379–408. [DOI] [PubMed] [Google Scholar]
- 20.Wang HW and Wang JW, How cryo-electron microscopy and X-ray crystallography complement each other. Protein Sci, 2017. 26(1): p. 32–39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Lyumkis D, Challenges and opportunities in cryo-EM single-particle analysis. J Biol Chem, 2019. 294(13): p. 5181–5197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Nygaard R, Kim J, and Mancia F, Cryo-electron microscopy analysis of small membrane proteins. Curr Opin Struct Biol, 2020. 64: p. 26–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kesidis A, et al. Expression of eukaryotic membrane proteins in eukaryotic and prokaryotic hosts. Methods, 2020. [DOI] [PubMed] [Google Scholar]
- 24.Cherezov V, Lipidic cubic phase technologies for membrane protein structural studies. Curr Opin Struct Biol, 2011. 21(4): p. 559–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Liu W, et al. LCP-Tm: an assay to measure and understand stability of membrane proteins in a membrane environment. Biophys J, 2010. 98(8): p. 1539–48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Birch J, et al. The fine art of integral membrane protein crystallisation. Methods, 2018. 147: p. 150–162. [DOI] [PubMed] [Google Scholar]
- 27.Loll PJ, Membrane proteins, detergents and crystals: what is the state of the art? Acta Crystallogr F Struct Biol Commun, 2014. 70(Pt 12): p. 1576–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kawate T and Gouaux E, Fluorescence-detection size-exclusion chromatography for precrystallization screening of integral membrane proteins. Structure, 2006. 14(4): p. 673–81. [DOI] [PubMed] [Google Scholar]
- 29.Kotov V, et al. High-throughput stability screening for detergent-solubilized membrane proteins. Sci Rep, 2019. 9(1): p. 10379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Vergis JM, Purdy MD, and Wiener MC, A high-throughput differential filtration assay to screen and select detergents for membrane proteins. Anal Biochem, 2010. 407(1): p. 1–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.White MA, et al. Characteristics affecting expression and solubilization of yeast membrane proteins. J Mol Biol, 2007. 365(3): p. 621–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.VanAken T, et al. Alkyl glycoside detergents: synthesis and applications to the study of membrane proteins. Methods Enzymol, 1986. 125: p. 27–35. [DOI] [PubMed] [Google Scholar]
- 33.Rosevear P, et al. Alkyl glycoside detergents: a simpler synthesis and their effects on kinetic and physical properties of cytochrome c oxidase. Biochemistry, 1980. 19(17): p. 4108–15. [DOI] [PubMed] [Google Scholar]
- 34.Fantini J and Barrantes FJ, How cholesterol interacts with membrane proteins: an exploration of cholesterol-binding sites including CRAC, CARC, and tilted domains. Front Physiol, 2013. 4: p. 31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Chae PS, et al. A new class of amphiphiles bearing rigid hydrophobic groups for solubilization and stabilization of membrane proteins. Chemistry, 2012. 18(31): p. 9485–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Autzen HE, Julius D, and Cheng Y, Membrane mimetic systems in CryoEM: keeping membrane proteins in their native environment. Curr Opin Struct Biol, 2019. 58: p. 259–268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Denisov IG and Sligar SG, Nanodiscs for structural and functional studies of membrane proteins. Nat Struct Mol Biol, 2016. 23(6): p. 481–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Efremov RG, Gatsogiannis C, and Raunser S, Lipid Nanodiscs as a Tool for High-Resolution Structure Determination of Membrane Proteins by Single-Particle Cryo-EM. Methods Enzymol, 2017. 594: p. 1–30. [DOI] [PubMed] [Google Scholar]
- 39.Frauenfeld J, et al. A saposin-lipoprotein nanoparticle system for membrane proteins. Nat Methods, 2016. 13(4): p. 345–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Warschawski DE, et al. Choosing membrane mimetics for NMR structural studies of transmembrane proteins. Biochim Biophys Acta, 2011. 1808(8): p. 1957–74. [DOI] [PubMed] [Google Scholar]
- 41.Chipot C, et al. Perturbations of Native Membrane Protein Structure in Alkyl Phosphocholine Detergents: A Critical Assessment of NMR and Biophysical Studies. Chem Rev, 2018. 118(7): p. 3559–3607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Earl LA, et al. Cryo-EM: beyond the microscope. Curr Opin Struct Biol, 2017. 46: p. 71–78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Gorzelle BM, et al. Amphipols can support the activity of a membrane enzyme. J Am Chem Soc, 2002. 124(39): p. 11594–5. [DOI] [PubMed] [Google Scholar]
- 44.Nath A, Atkins WM, and Sligar SG, Applications of phospholipid bilayer nanodiscs in the study of membranes and membrane proteins. Biochemistry, 2007. 46(8): p. 2059–69. [DOI] [PubMed] [Google Scholar]
- 45.Tribet C, Audebert R, and Popot JL, Amphipols: polymers that keep membrane proteins soluble in aqueous solutions. Proc Natl Acad Sci U S A, 1996. 93(26): p. 15047–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Parmar M, et al. Using a SMALPplatform to determine a sub-nm single particle cryo-EM membrane protein structure. Biochim Biophys Acta Biomembr, 2018. 1860(2): p. 378–383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Oluwole AO, et al. Solubilization of Membrane Proteins into Functional Lipid-Bilayer Nanodiscs Using a Diisobutylene/Maleic Acid Copolymer. Angew Chem Int Ed Engl, 2017. 56(7): p. 1919–1924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Carlson ML, et al. The Peptidisc, a simple method for stabilizing membrane proteins in detergent-free solution. Elife, 2018. 7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Tonggu L and Wang L, Cryo-EM sample preparation method for extremely low concentration liposomes. Ultramicroscopy, 2020. 208: p. 112849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Yao X, Fan X, and Yan N, Cryo-EM analysis of a membrane protein embedded in the liposome. Proc Natl Acad Sci U S A, 2020. 117(31): p. 18497–18503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Shoemaker SC and Ando N, X-rays in the Cryo-Electron Microscopy Era: Structural Biology’s Dynamic Future. Biochemistry, 2018. 57(3): p. 277–285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Liao M, et al. Structure of the TRPV1 ion channel determined by electron cryo-microscopy. Nature, 2013. 504(7478): p. 107–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Paulsen CE, et al. Structure of the TRPA1 ion channel suggests regulatory mechanisms. Nature, 2015. 520(7548): p. 511–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Pumroy RA, et al. Structural insights into the gating mechanisms of TRPV channels. Cell Calcium, 2020. 87: p. 102168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Vinayagam D, et al. Electron cryo-microscopy structure of the canonical TRPC4 ion channel. Elife, 2018. 7. [DOI] [PMC free article] [PubMed] [Google Scholar]