

Abstract
Rationale:
β1-adrenoceptors (β1ARs) exist at intracellular membranes and Organic Cation Transporter 3 (OCT3) mediates norepinephrine entry into cardiomyocytes. However, the functional role of intracellular β1AR in cardiac contractility remains to be elucidated.
Objective:
Test localization and function of intracellular β1AR on cardiac contractility.
Methods and Results:
Membrane fractionation, super-resolution imaging, proximity ligation, co-immunoprecipitation and single-molecule pulldown demonstrated a pool of β1ARs in mouse hearts that was associated with sarco/endoplasmic reticulum Ca2+-ATPase at the sarcoplasmic reticulum (SR). Local protein kinase A (PKA) activation was measured using a PKA biosensor targeted at either the plasma membrane (PM) or SR. Compared to wild type (WT), myocytes lacking OCT3 (OCT3KO) responded identically to the membrane-permeant βAR agonist isoproterenol in PKA activation at both PM and SR. The same was true at the PM for membrane-impermeant norepinephrine, but the SR response to norepinephrine was suppressed in OCT3KO myocytes. This differential effect was recapitulated in phosphorylation of the SR-pump regulator phospholamban. Similarly, OCT3KO selectively suppressed calcium transients and contraction responses to norepinephrine, but not isoproterenol. Furthermore, sotalol, a membrane-impermeant βAR-blocker suppressed isoproterenol-induced PKA activation at the PM, but permitted PKA activation at the SR, phospholamban phosphorylation and contractility. Moreover, pretreatment with sotatol in OCT3KO myocytes prevented norepinephrine induced PKA activation at both PM and the SR and contractility.
Conclusion:
Functional β1ARs exists at the SR and is critical for PKA-mediated phosphorylation of phospholamban and cardiac contractility upon catecholamine stimulation. Activation of these intracellular β1ARs requires catecholamine transport via OCT3.
Keywords: Beta-1 adrenergic receptor (β1AR), β-blockers, phospholamban (PLB), cardiac contractility and energetics, catecholamine, Organic Cation Transporter 3 (OCT3), adrenergic receptor
Subject Terms: Basic Science Research, Calcium Cycling/Excitation-Contraction Coupling, Cell Signaling/Signal Transduction, Contractile Function
Graphical Abstract
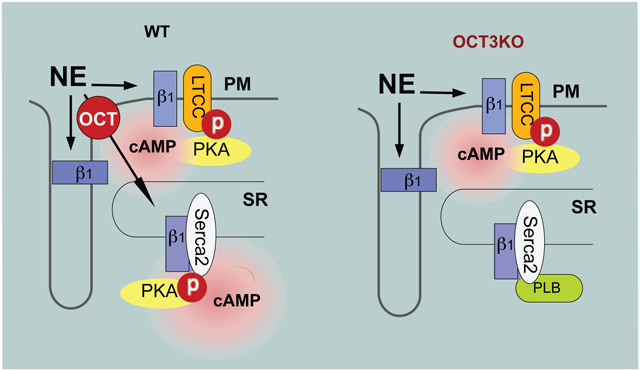
INTRODUCTION
Plasma membrane receptors are major mechanisms by which cells respond to extracellular stimuli, allowing cells to adapt to their surrounding environments and to modulate tissue/organ function in reaction to changes of neurohormones. Non-steroid hormones signal mainly through receptors at the plasma membrane (PM) to regulate cellular function. This includes sympathetic regulation of cardiac function via adrenergic receptors (ARs) on the PM of cardiomyocytes. Recently, a complementary thread of studies has found that neurotransmitters, peptide hormones, and growth factors also act on intracellular membrane receptors.1–4 Accumulating data indicate that intracellular G-protein coupled receptors (GPCRs) located at different organelles, e.g., mitochondria,5 endoplasmic reticulum membranes6 and Golgi,7 can be activated insides the cells and induce location-specific effects. The development of these observations helps expanding the current understanding of hormonal regulation of cardiac function.
β1ARs, a prototype GPCR, acts as a linchpin of sympathetic regulation in the heart and has been demonstrated to be localized and activated intracellularly in cardiomyocytes.8 However, the role of intracellular β1ARs in the regulation of cardiac excitation-contraction is still unknown. During the fight or flight response, activation of the sympathetic nerve and adrenal glands releases catecholamines (epinephrine, EPI and norepinephrine, NE) to activate βARs on myocytes, and thus enhance heart rate, cardiac contraction and relaxation.9, 10 These effects involve βARs stimulation-dependent cAMP and subsequent activation of protein kinase A (PKA) to enhance substrate phosphorylation. The PKA phosphorylation of different substrates occurs in different spatiotemporal nanodomains including L-type Ca2+ channel (LTCC) at the PM, phospholamban (PLB) and ryanodine receptor type 2 (RyR2) on the SR, and troponin I (TnI) and myosin binding protein C (MYBP-C) on the myofilaments.11, 12
Both NE and EPI are vital neurohormones in physiological stress responses. However, EPI and NE have extremely hydrophilic octanol/water partition coefficients (LogP =−1.37 and −1.24, respectively, https://pubchem.ncbi.nlm.nih.gov). After release from nerve termini, a small portion of catecholamine can enter myocytes, primarily mediated by corticosterone-sensitive organic cation transporter 3 (OCT3, also named extra-neuronal monoamine transporter, EMT).2, 13 OCT3-mediated NE uptake is necessary for activation of intracellular ARs in cardiomyocytes, including both nuclear α1ARs and Golgi-localized β1ARs.2, 8 Intriguingly, decreased NE uptake into myocardium and cardiac NE contents in heart failure patients are associated with significantly blunted inotropy response.14 In dilated cardiomyopathy patients, OCT2 expression is reduced and predicts the impairment of cardiac function.15 However, the functional role of NE uptake facilitated by OCT3 in adrenergic stimulation of cardiac contractile is unclear.
Activation of β1ARs induces spatial, temporal segregated cAMP-PKA signals in heart.12, 16 The application of Förster resonance energy transfer (FRET)-based biosensors have been instrumental in the investigation and dissection of compartmentalized cAMP/PKA signaling on the nanodomain scale.12, 17, 18 By using genetically encoded biosensors, previous studies from our group and others found that compartmentalized cAMP/PKA signal generated in response to βAR stimulation differs at the PM and SR local domains in both amplitude and kinetics.12, 17 The mechanism underlying this heterogeneity is still unclear. We hypothesize that a population of β1ARs is located near or at the SR and plays an important role in modulating calcium (Ca2+) cycling and cardiac contractility. In this study, we tested the expression, activation and functional effects of SR-localized β1AR in cardiac contractility. Our results provide novel insights into the localization and regulation of intracellular β1AR, therefore, offering potential and precise target for β1AR-associated therapeutic strategies.
METHODS
Data Availability.
The data, analytic methods, and study materials are made available to other researchers for purposes of reproducing the results or replicating the procedure. Please see the Major Resources Table and Expanded Materials & Methods in the Online Data Supplement.
All experiments including animal feeding, treatment, and tissue collection were approved by the Institutional Animal Care and Use Committees (protocol: 20234 and 20957) of the University of California at Davis and follow NIH guidelines. Male and female 2–4-month-old C57/BL6 and OCT3KO, newborn β1AR-KO, β2AR-KO and β1β2AR-KO mice were described previously.17, 19 Male Sprague Dawley outbred rats and New Zealand white rabbits were used in this study.
RESULTS
Existence and distribution of a distinct pool of β1AR at the SR in cardiomyocytes.
To examine the existence of different subcellular pools of β1ARs in adult mouse hearts, we fractionated heart tissues to separate the PM from intracellular membrane compartments. Sarcoplasmic endoplasmic reticulum Ca2+-ATPase 2 (SERCA2) and ryanodine receptor 2 (RyR2), which are SR resident proteins, were used as markers for the intracellular membrane fraction (non-PM), and insulin receptor (InsR) and lack of SERCA2/RyR2 were used to identify PM fraction. β1ARs were clearly detected in both PM and intracellular membrane fractions (non-PM) in WT mouse hearts, but not in β1AR-KO mouse hearts (Figure 1A and Online Figure IA), indicating that endogenous β1AR exists in at least two different pools in mouse hearts. In addition, quantitative radioligand-binding assay measured the functional β1ARs that bind to [I125] radio-labeled cyanopindolol in both PM and intracellular membrane fractions (Figure 1B), supporting the existence of an intracellular pool of functional β1ARs in hearts. We also applied a fluorescence dye BODIPY-tagged βAR antagonist propranolol (FL-Prop) to stain endogenous β1ARs in mouse adult ventricle myocytes (AVMs). Confocal images of FL-Prop fluorescent staining in WT mouse AVMs showed a distribution of βARs on both surface membrane and intracellular space (Online Figure IB), which was abrogated by addition of 1 μmol/L and 10 μmol/L propranolol (Online Figure IB). Moreover, compared with WT and β2AR-KO cardiomyocytes, the fluorescent dots and intensity of FL-Prop were greatly reduced in the β1AR-KO and β1β2AR-KO cardiomyocytes (Online Figure IC–D), indicating that Fl-Prop mainly stains endogenous β1AR.
Figure 1. Intracellular β1AR associates with SERCA2 but not RyR2 in hearts and isolated mouse adult ventricular cardiomyocytes (AVMs).
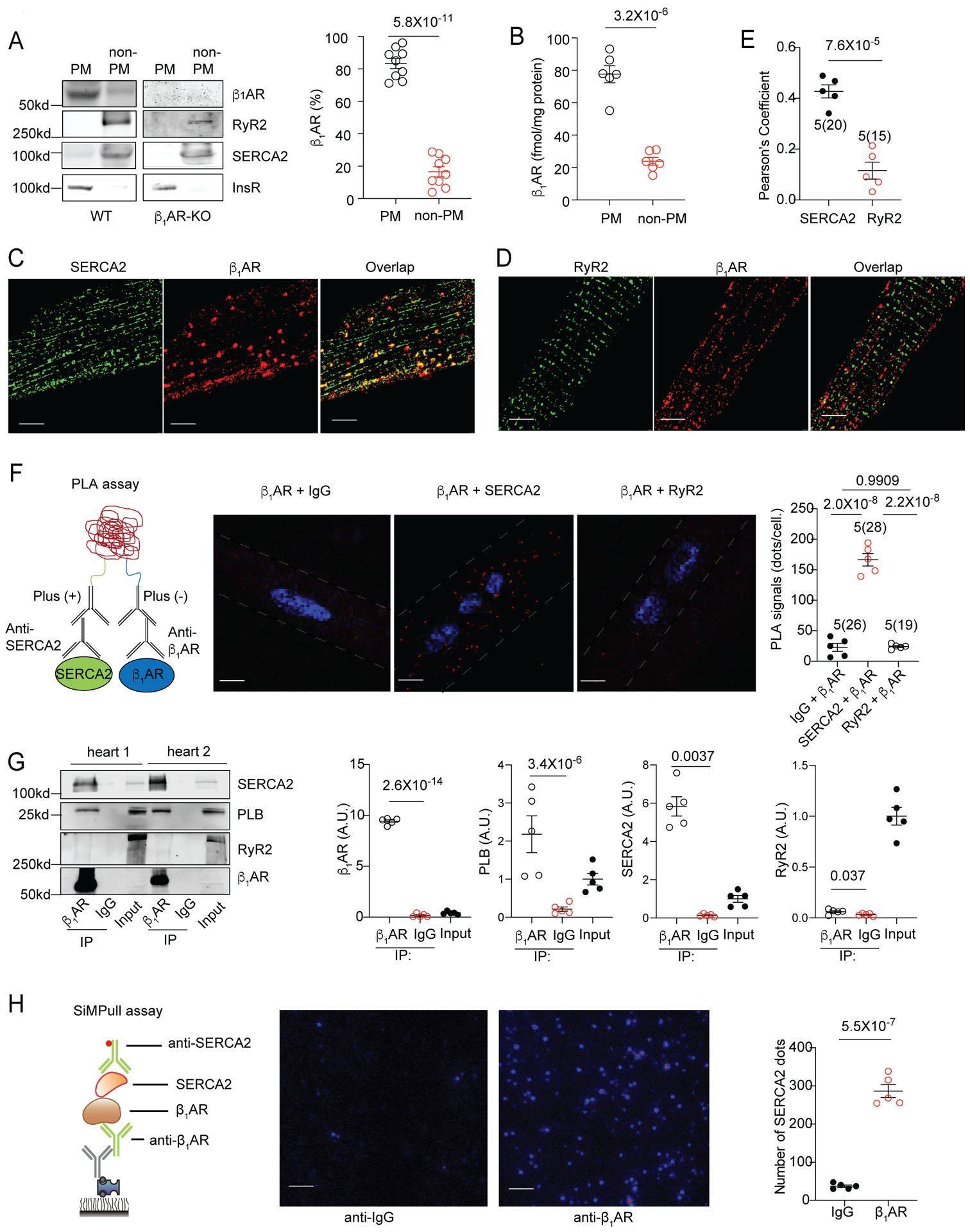
(A) WT and β1AR-KO mouse heart lysates were fractionated to assess cellular distribution of β1AR. Representative blots and the percentages of β1AR in the plasma membrane (PM) and intracellular membrane fractions (non-PM) over the total β1AR in WT and β1AR-KO mouse hearts. Data are shown in mean ± S.E.M. N = 9 mice. p values were obtained by student t-test. (B) β1AR densities in the PM and non-PM fractions of WT mouse hearts were determined by quantitative radioligand binding assay. Data are shown in mean ± S.E.M. N = 6 mice. p values were obtained by student t-test. (C-D) Representative confocal images showing distribution of β1AR, SERCA2, and RyR2 in isolated mouse AVMs. β1AR was overexpressed in AVMs by infection with recombinant adenovirus. Scale bar = 5 μm. (E) The overlap between staining from confocal images was evaluated by Pearson’s correlation coefficient using ImageJ. Data are shown in mean ± S.E.M; AVM (in the parenthesis) and mouse numbers are indicated in the figure. p value was obtained by t-test. (F) Schematic of in situ proximity ligation assay (PLA), exemplary fluorescence images (the whole cell images are in Online Figure II) and quantification of PLA signals after labeling with antibodies against β1AR and IgG, β1AR and SERCA2, and β1AR and RyR2 in AVMs, respectively. Positive PLA signal (red), DAPI (blue). Scale bar = 5 μm. Data are shown in mean ± S.E.M; AVM (in the parenthesis) and mouse numbers are indicated. p values were obtained by one-way ANOVA followed by Tukey multiple comparison test. (G) Representative images and quantitative assessment of PLB, SERCA2 and RyR2 coimmunoprecipitated with β1AR in WT mouse hearts. A.U. = arbitrary unit is defined as the ratio of intensity of proteins over inputs. Data are shown in mean ± S.E.M. (N = 5); p values were obtained by student t-test; (H) Schematic of SiMPull assay and representative images of SiMPull assay after endogenous β1AR, SERCA2 complex were pulled down with anti-β1AR or control IgG antibodies. The images were quantified using MATLAB. Scale bar = 5 μm. Data are shown in mean ± S.E.M. N = 5 mice. p values were obtained by t-test.
SR localized Ca2+ cycling proteins such as SERCA2 and RyR2 are crucial for the modulation of cardiac contractility in response to βAR activation.11 We hypothesize that a population of intracellular β1ARs are present at the SR. We examined a potential colocalization of overexpressed β1AR with SERCA2 or RyR2. β1AR displayed specific colocalization with SERCA2, but not RyR2, in mouse AVMs (Figure 1C–E and Online Figure IIA). As controls, overexpressed β2AR did not display overlap with either SERCA2 or RyR2 (Online Figure IIIA–B). Next, we applied proximity ligation assay (PLA) to further explore the proximal relationship between β1AR and SERCA2. We detected robust PLA signals in mouse AVMs co-stained for β1AR and SERCA2, suggesting that these β1ARs and SERCA2 are at 40 nm or less apart. Conversely, PLA signals were not detected after co-staining with antibodies against β1AR and IgG or against β1AR and RyR2 and only a small amount of PLA signals was detected after co-staining for β1AR and junctophilin 2 (JP2) (Figure 1F and Online Figure IIB–D). Moreover, endogenous β1AR was pulled down with β1AR antibody (Figure 1G). We found that both SERCA2 and its regulatory protein, phospholamban (PLB), were co-immunoprecipitated with the endogenous β1ARs. In contrast, RyR2 was not observed in the immunoprecipitation. Furthermore, we applied a super-sensitive single molecule pull down (SiMPull) assay20 to visualize individual β1AR-SERCA2 complex in heart lysate. Figure 1H shows that β1AR antibody was able to pull down a significant amount of SERCA2 molecules compared to control IgG antibody. Together, these data demonstrate that a population of β1ARs is localized at the SR and forms a complex with SERCA2 in AVMs.
Organic Cation Transporter 3 (OCT3) is required to activate the pool of β1ARs by catecholamine at the SR.
β-adrenoceptor stimulation results in activation of PKA, a key mediator for phosphorylating multiple Ca2+ handling proteins (e.g. PLB).21 To directly assess PKA activity induced by the SR-localized β1AR, we employed a genetically encoded Förster resonance energy transfer (FRET)-based A kinase activity reporter (AKAR) that is anchored at the intracellular SERCA2 complex on the SR (SR-AKAR3).17, 18 In comparison, the PKA activity induced by the PM-localized β1AR was assessed by an AKAR3 anchored on the PM (PM-AKAR3).
Previous studies showed that OCT3-mediated transportations of EPI or NE into cells are essential for activating intracellular adrenergic receptors in cardiomyocytes.2, 8 Here, we used mice with genetic deletion of OCT3 to examine the impacts of OCT3 on subcellular PKA activity induced by PM- or SR-localized β1AR (PM-β1AR or SR-β1AR). Unlike endogenous catecholamines, isoproterenol (ISO, LogP = 1.4) and dobutamine (LogP = 3.6), two membrane permeant β agonists, activate internal βAR via passive diffusion into myocytes (Figure 2A–B).7 Therefore, deleting OCT3 should selectively block the access of NE and EPI but not ISO and Dob to SR-localized β1AR (Figure 2A–B). NE, EPI, ISO and Dob all induced robust PKA activity at both the PM and SR in WT myocytes (Online Figure IVA–D). The kinetics of ligand induced PKA activity at the PM were comparable but were faster than those at the SR (Online Figure IVA–E). Among the SR-PKA activity, the Dob induced increases were faster than those by ISO, EPI and NE, consistent with its faster membrane permeability (Online Figure IV A–E). As predicted, the ISO-induced PKA activity at both the PM and SR was not affected by deletion of OCT3 (Figure 2C–D). However, deletion of OCT3 selectively attenuated NE-induced PKA activity at the SR without affecting PKA activity at the PM (Figure 2E–F). Moreover, inhibition of β1AR with CGP20712a completely abolished PKA activity induced by NE and ISO at both PM and the SR in AVMs whereas inhibition of β2AR with ICI118551 did not affect these PKA activities (Online Figure IV F–I). These observations were not due to change in β1AR expression in OCT3-KO hearts (Online Figure V). Additionally, we have examined PKA activity at the myofilaments (MF) with an AKAR3 anchored on the troponin complex (MF-AKAR318), which is adjacent to the SR. Our data show deletion of OCT3 selectively attenuated NE but not ISO induced PKA activity on the myofilaments (Online Figure VA). To explore specific PKA substrate affected by SR-localized β1AR activation, we then examined PKA phosphorylation of phospholemman (PLM, Ser63) and LTCC (Ser1928) at the PM, PLB (Ser16) and RyR2 (Ser2808) at the SR, TnI (Ser23/24) and MyBPC (Ser282, Ser273, Ser302) at myofilament. Deletion of OCT3 selectively attenuated NE-induced PKA phosphorylation of PLB and TnI without affecting PKA phosphorylation of PLM, LTCC, RyR2, and MyBPC (Figure 2G–H and Online Figure V B–H). In comparison, ISO-induced PKA phosphorylation of both PLM and PLB were not affected by OCT3 deficiency (Figure 2G–H). These data suggest that deletion of OCT3 blocks catecholamine-dependent activation of the β1ARs at the SR and the myofilament and downstream PKA-dependent PLB and TnI phosphorylation.
Figure 2. Differential local regulation of NE-induced β1AR/PKA activities in WT and OCT3KO AVMs.
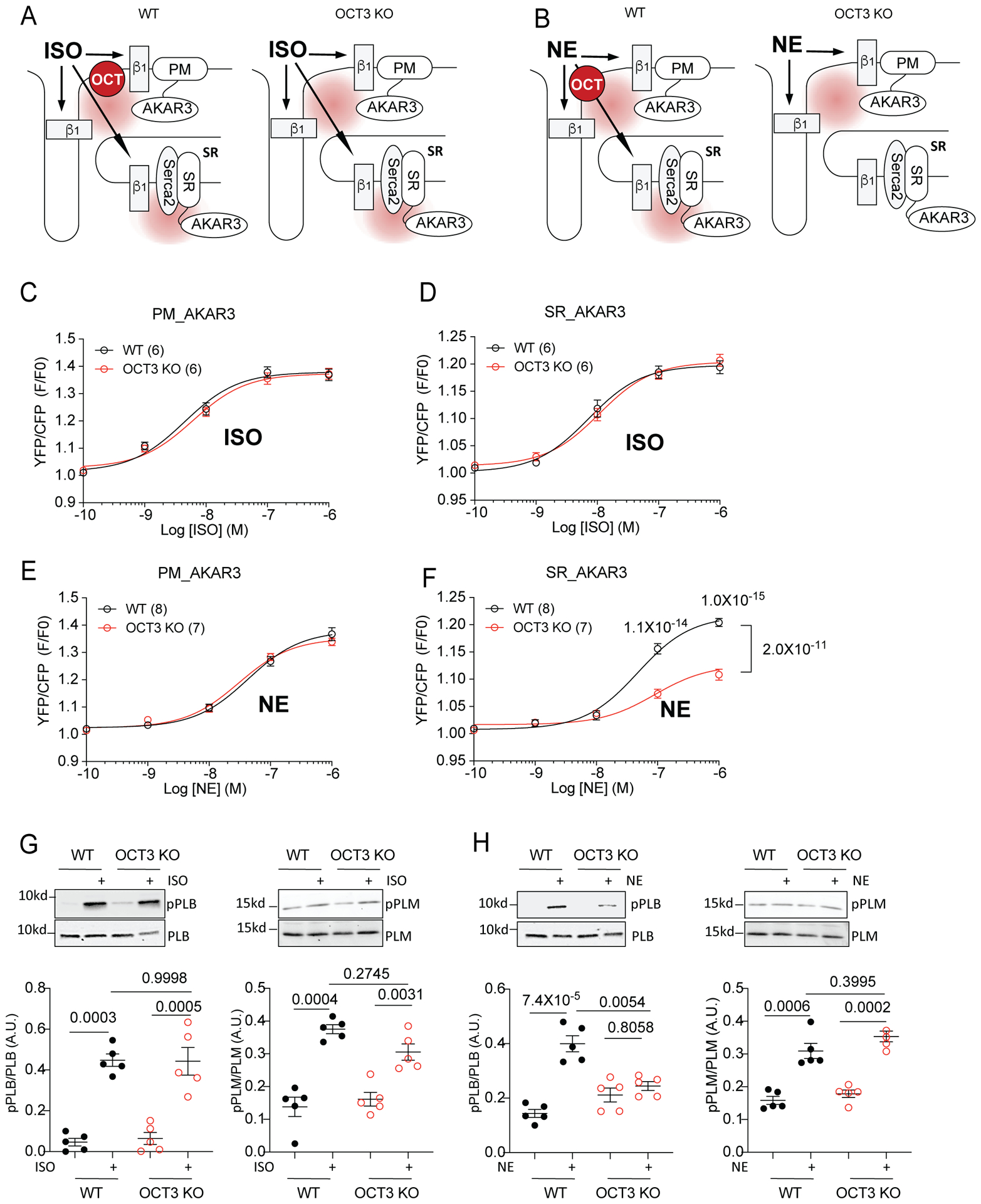
(A-B) Schematics of local activation of β1AR-induced PKA activity at subcellular locations and detection using FRET based biosensors (plasma membrane, PM-AKAR3 and sarcoplasmic reticulum, SR-AKAR3) in WT and OCT3KO AVMs. Isoproterenol (ISO), norepinephrine (NE). FRET assay was analyzed with F/F0 of YFP/CFP ratio. (C-D) Concentration-response curves of changes in YFP/CFP ratio after ISO stimulation in WT and OCT3KO AVMs expressing PM-AKAR3 or SR-AKAR3. Data were from 6 WT and 6 OCT3KO mice. (E-F) Concentration-response curves of changes in YFP/CFP ratio after NE stimulation in WT and OCT3KO AVMs expressing PM-AKAR3 or SR-AKAR3. Data were from 8 WT and 7 OCT3KO mice. Data are shown in mean ± S.E.M. p values were obtained by two-way ANOVA with Tukey’s multiple comparison test when comparing WT to OCT3-KO. (G-H) Detection of phosphorylation of phospholamban (PLB) at serine 16 and phosphorylation of phospholemman (PLM) at serine 63 in mouse AVMs after stimulation with 100 nmol/L ISO or 100 nmol/L NE. Data are shown in mean ± S.E.M. (N = 5). A.U. (arbitrary unit) is defined as the ratio of intensity of phosphorylated proteins over intensity of total proteins. p values were obtained by two-way ANOVA with Tukey’s multiple comparison test.
Functional consequences of the internal pool of β1ARs on adult ventricular myocyte.
We then assessed whether the deletion of OCT3 affects catecholamine induced Ca2+ handling and sarcomere shortening (SS) in isolated AVMs. Compared with basal conditions, stimulation with different βAR agonists (ISO, Dob, NE, and EPI) led to increased SS and Ca2+ transient amplitude, and decreased Ca2+ decay tau in both WT and OCT3KO AVMs (Figure 3A–H and Online Figure VIA–K). Dob induced a faster inotropic response than ISO and NE in WT AVMs. The increases in E-C coupling were blocked by β1AR antagonist CGP20712a, but not affected by β2AR inhibition with ICI118551 (Online Figure VII). Deletion of OCT3 did not affect ISO- or Dob-induced inotropic responses (Figure 3A–D and Online Figure VID–K). Conversely, the NE- and EPI-promoted responses in contraction, Ca2+ transients and [Ca2+]i decay tau were significantly attenuated in OCT3KO vs WT AVMs (Figure 3E–H and Online Figure VIJ–K). Deletion of OCT3 also shifted the NE dose-response curve of SS to the right without affecting ISO-induced dose-response curve (Figure 3I–J).
Figure 3. Deletion of Organic Catecholamine Transporter3 (OCT3) attenuates stimulation of myocyte contractility with norepinephrine and epinephrine.
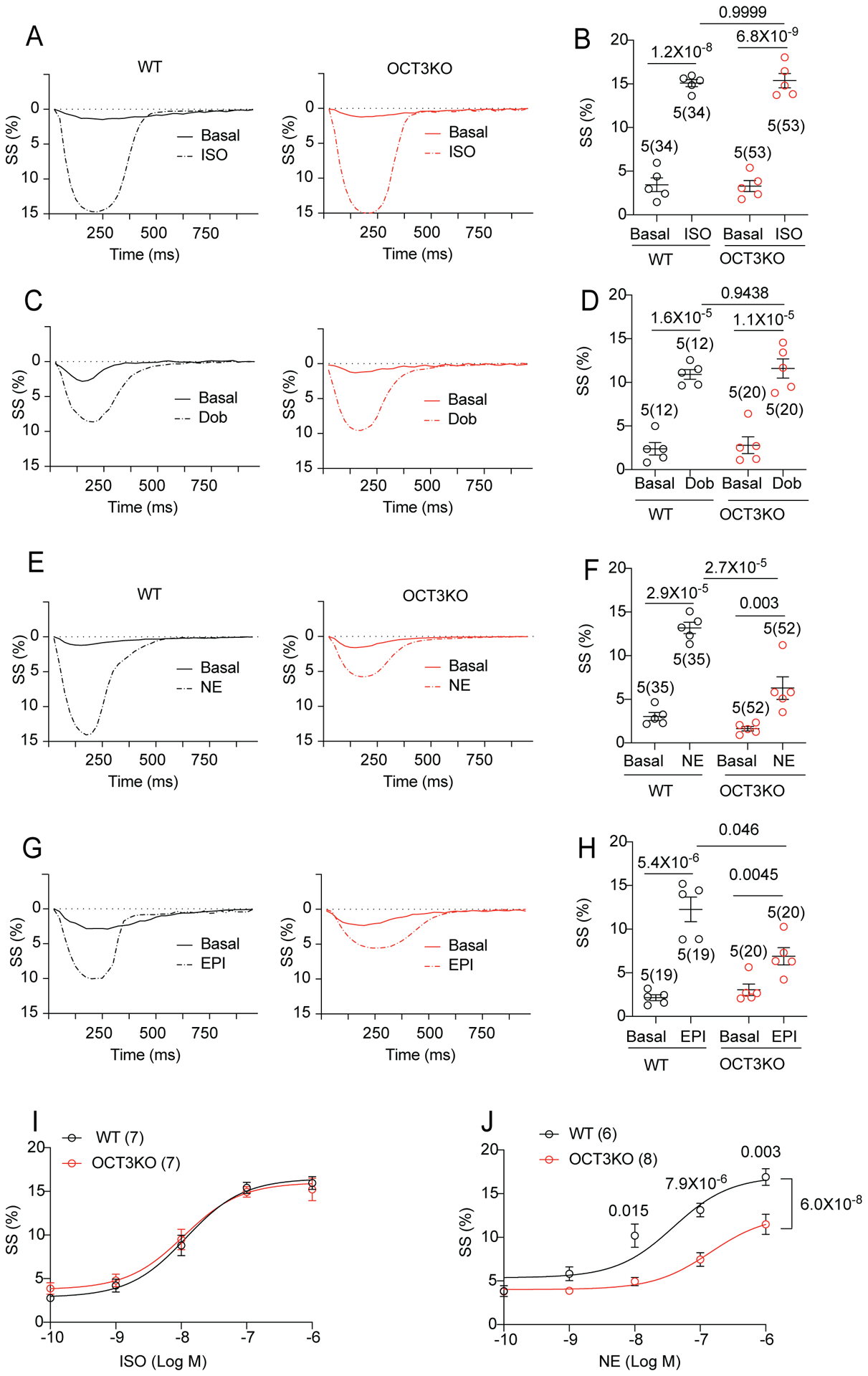
(A-B) Representative traces show sarcomere shortening (SS) before (close line) and after (dash line) stimulation with ISO (100 nmol/L) in the WT and OCT3KO mouse AVMs. The peak SS were quantified. (C-D) Representative traces show FS before (close line) and after (dash line) the application of dobutamine (Dob, 1μmol/L) in WT and OCT3KO AVMs. The peak SS were quantified. (E-F) Representative traces show SS before (close line) and after (dash line) the application of norepinephrine (NE, 100 nmol/L) in WT and OCT3KO AVMs. The peak SS were quantified. (G-H) Representative traces show SS before (close line) and after (dash line) the application of epinephrine (EPI, 100 nmol/L) in WT and OCT3KO AVMs. The peak SS were quantified. For panel A-H, data are shown in mean ± S.E.M. AVM (in the parenthesis) and animal numbers are indicated. p values were obtained by two-way ANOVA analysis followed with Tukey’s multiple comparison test. (I-J) Dose response curves of SS in AVMs after stimulation with ISO (7 WT and 7 OCT3-KO mice) or NE (6 WT and 8 OCT3-KO mice). p values were obtained by two-way ANOVA with Tukey’s multiple comparison test when comparing OCT3-KO to WT.
To independently test the role of OCT3 in the SR-located β1AR/PKA signaling, we applied an OCT3/EMT inhibitor corticosterone (CORTI) to prevent NE transport into myocytes (Figure 4A–B). While CORTI had minimal effect on NE-induced PKA activity at the PM domain, it significantly attenuated NE-induced PKA activity at the SR domain (Figure 4C–D). CORTI did not change ISO-induced PKA activity at either the PM or SR domain in rat AVM (Figure 4C–D). In agreement, CORTI significantly reduced NE- but not ISO-induced PKA phosphorylation of PLB at Ser16 (Figure 4E–F). CORTI also significantly attenuated NE-induced increases in contractility, Ca2+ transient amplitude, and rate of Ca2+ transient decay (Figure 4G–I). However, these CORTI effects were absent in the responses to ISO stimulation (Figure 4J–L). Together, these data suggest that OCT3-dependent transport is required for stimulation of SR-localized β1AR/PKA/PLB signal cascade for optimal increases in Ca2+ handling and contractility.
Figure 4. Inhibition of organic catecholamine transporters reduces norepinephrine-promoted SR-localized β1AR signal and myocardial contractility.
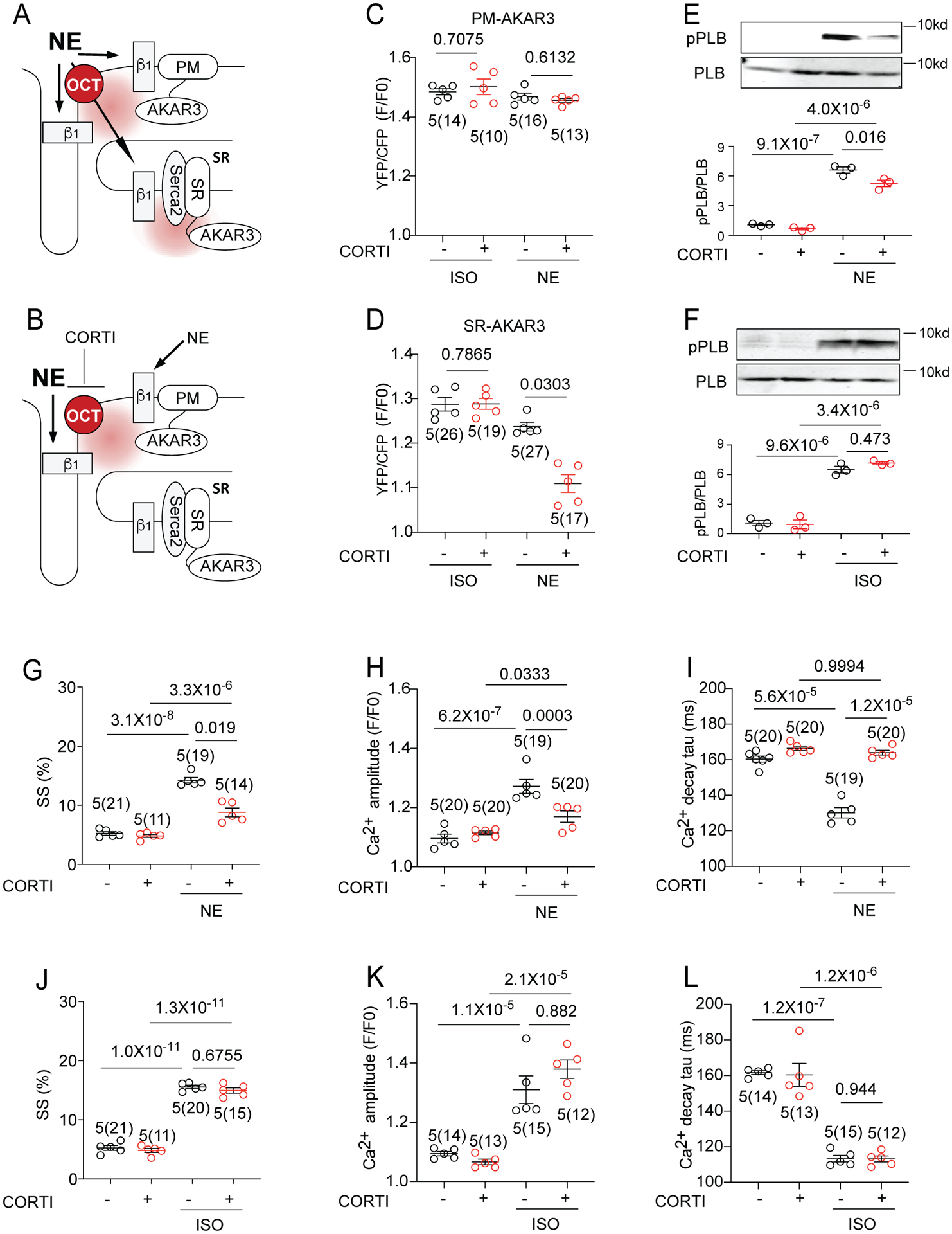
(A-B) Schematics show detection of β1AR-induced PKA activity at different subcellular locations with and without OCT3 inhibitor corticosterone (CORTI). (C-D) Rat AVMs expressing PM-AKAR3 or SR-AKAR3 FRET biosensor were pretreated with CORTI (1 μmol/L) before stimulation with NE (100 nmol/L) or ISO (100 nmol/L). FRET was analyzed as F/F0 of YFP/CFP ratio. Data show the maximal increases in YFP/CFP ratios in mean ± S.E.M. AVM (in the parenthesis) and rat numbers are indicated. p values were obtained by One-way ANOVA with Tukey’s multiple comparison test. (E-F) Representative immunoblot detection and quantification of phosphorylated serine 16 (pPLB) and total PLB in rat AVMs. Cells were treated with 5-minute incubation with ISO (100 nmol/L) or NE (100 nmol/L) in the absence and presence of CORTI pretreatment. A.U. (arbitrary units) is defined as the ratio of intensity of phosphorylated proteins over intensity of total proteins. Data are shown in mean ± S.E.M. (N = 3). p values were obtained by One-way ANOVA with Tukey’s multiple comparison test. (G-L) Rat AVMs were loaded with Ca2+ indicator, 5 μmol/L Fluo-4 AM and pretreated with CORTI (1 μmol/L) before stimulation with NE (100 nmol/L) or ISO (100 nmol/L). Sarcomere shortening (SS) and calcium transient amplitude and tau were recorded with 1Hz pacing. Data are shown in mean ± S.E.M. AVM (in the parenthesis) and animal numbers are indicated in figures. p values were obtained by One-way ANOVA with Tukey’s multiple comparison test.
Activation of the internal β1ARs at the SR was not blocked by membrane impermeant β-blockers.
Sotalol, a membrane impermeant β-blocker (LogP = 0.24), has limited access to intracellular β1AR.22 To further dissect the physiological function of the SR-β1AR, we applied sotalol to selectively block the PM-β1AR but permit access and activation of SR-β1AR by agonists. In contrast, propranolol, a membrane permeant β-blocker (LogP = 3.48), is expected to block βARs both on the PM and at intracellular membranes (Online Figure VIIIA). We tested the accessibilities of sotalol and propranolol to the β1ARs by examining their abilities to compete with fluorescent BODIPY-tagged propranolol (Fl-Prop) from binding to β1ARs. Confocal images of Fl-Prop fluorescent staining in WT mouse AVMs showed a distribution of βAR on both surface and intracellular membranes (Online Figure VIIIB). Sotalol pretreatment did not block the intracellular staining (Online Figure VIIIB); whereas addition of propranolol effectively competed against Fl-Prop from binding to β1AR in AVMs (Online Figure VIIIB). Addition of ISO and NE also effectively competed against Fl-Prop from binding to β1AR in AVMs. Thus, intracellular β1AR can be blocked by membrane permeant β-blockers but are less accessible to membrane impermeant β-blockers.
Sotalol was then applied to isolate PKA activity induced by local compartmentalized β1AR at the SR (Figure 5A–C). In WT, β1AR-KO and β2AR-KO mouse neonatal myocytes and WT mouse, rat, and rabbit AVMs, sotalol readily inhibited ISO induced and β1AR mediated PKA activity at the PM, but not those at the SR (Figure 5D–G, Online Figure IXA–D and Online Figure X). In rat AVMs, the IC50 of sotalol for inhibition of ISO-induced PKA activity at the SR was about 10-fold higher than that of PKA activity at the PM (Figure 5G, PM-AKAR3 IC50 = 2.22 ± 0.06 μmol/L; SR-AKAR3 IC50 = 21.5 ± 0.06 μmol/L). In contrast, the membrane permeant β-blocker propranolol inhibited ISO-induced PKA activity at both the PM and SR in a concentration-dependent manner (Figure 5F, PM-AKAR3 IC50 = 74.0 ± 0.05 nmol/L, SR-AKAR3 IC50 = 99.2 ± 0.06 nmol/L). Another poorly permeant β-blocker, atenolol (LogP = 0.6), acted similarly to sotalol by selectively inhibiting ISO-induced PKA activity only at the PM in rat AVMs (Online Figure IXE–F). In comparison, membrane permeant carvedilol (LogP = 4.19) inhibited ISO-induced PKA activity at both the PM and the SR (Online Figure IXE–F). Together, these data support that local activation of SR-β1AR-PKA signaling was minimally suppressed by membrane impermeant β-blockers, a conserved mechanism throughout different cardiac myocytes from different species. This also indicates that the β1AR at the SR are not activated at the PM and then translocated to the SR.
Figure 5. Activation of β1AR in the SR is essential for maximal stimulated contractility in AVMs.
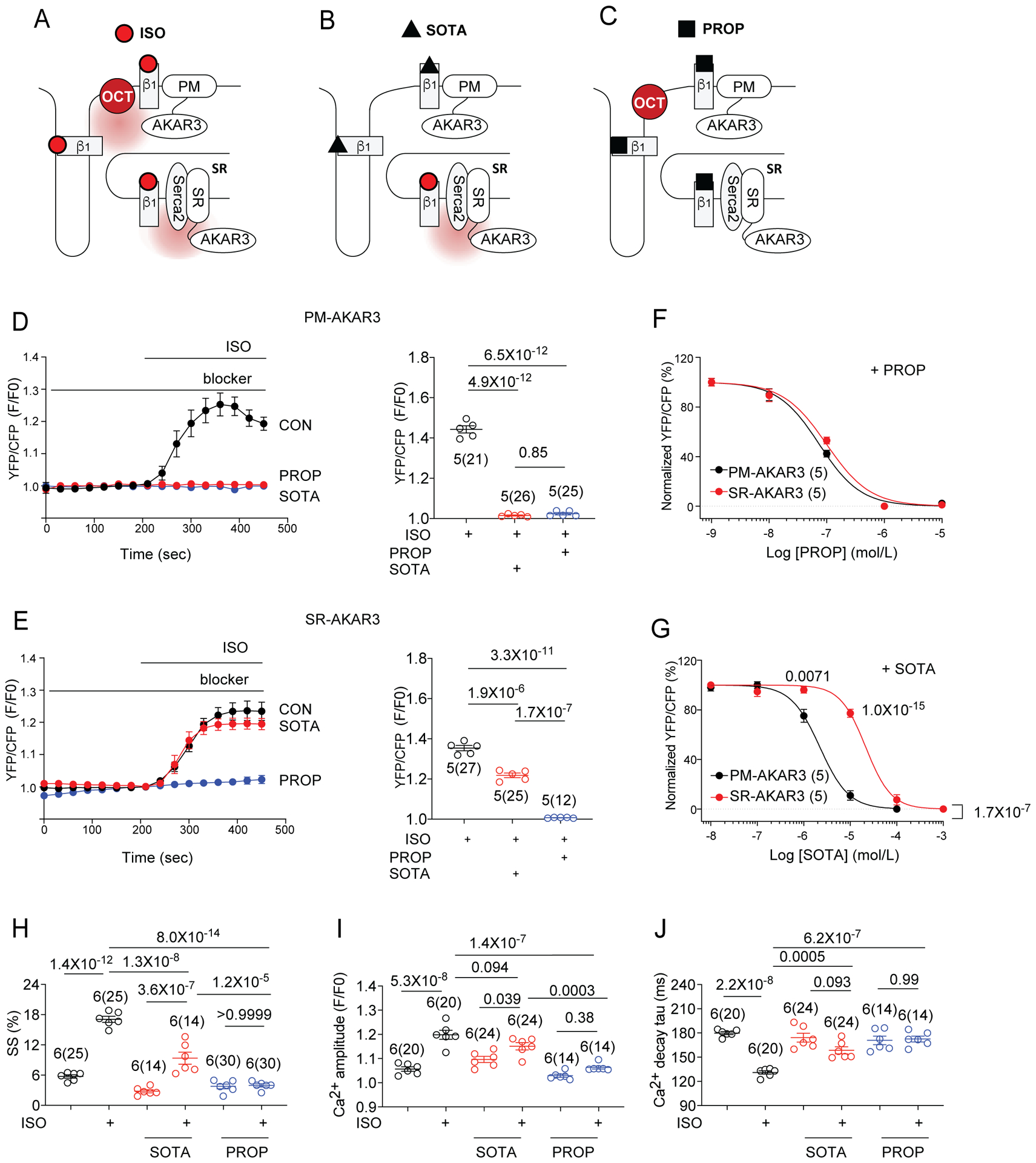
(A-C) Schematics show detection of β1AR-induced PKA activity at different subcellular locations (PM and SR) with FRET based biosensors in the presence of membrane impermeant β-blocker sotalol (SOTA) or the membrane permeant β-blocker propranolol (PROP). (D, E) Rat AVMs expressing PM-AKAR3 or SR-AKAR3 were stimulated with 100 nmol/L ISO with or without 5-mintue pretreatment of 25 μmol/L SOTA (blue) or 1 μmol/L PROP (red). Representative time courses show FRET response of PM-AKAR3 or SR-AKAR3 in AVMs. FRET was analyzed as F/F0 of YFP/CFP ratio. Data are shown in mean ± S.E.M. AVM (in the parenthesis) and rat numbers are indicated. p values were obtained by one-way ANOVA analysis with Tukey’s multiple comparison test. (F, G) Dose-dependent inhibition curves of PROP or SOTA on ISO-induced increases in FRET responses in AVMs expressing PM-AKAR3 or SR-AKAR3. Data show YFP/CFP ratios normalized against the maximal increases induced by ISO in the absence of β-blocker (N = 5 rats). p values were obtained by two-way ANOVA analysis followed by Tukey’s multiple comparison test when compared to OCT3-KO. (PM-AKAR3: PROP, IC50 = 7.399×10−8 mol/L, SR-AKAR3: PROP, IC50 = 9.92×10−8 mol/L; PM-AKAR3: SOTA, IC50 = 2.216×10−6 mol/L; SR-AKAR3, STOA, IC50 = 2.149×10−5 mol/L). (H-J) Rat AVMs were incubated with Ca2+ indicator (5 μM Fluo-4 AM) before 1Hz pacing. After pretreatment with SOTA (25 μmol/L) or PROP (1 μmol/L), sarcomere shortening (SS) and calcium transient were recorded before and after stimulation with 100 nmol/L ISO. The peak SS, amplitude of Ca2+ transient, and rate of Ca2+ decay (Tau) are shown as mean ± S.E.M. Animal numbers and cell numbers are indicated in figures. p values were obtained by one-way ANOVA analysis with Tukey’s multiple comparison test.
Activation of the β1AR at the SR promotes optimal cardiac contractility and Ca2+ handling in AVMs.
Consistent with the PKA activity detected at the SR, sotalol blocked ISO-induced PKA phosphorylation of LTCC at Ser1928 but did not affect ISO-induced PKA phosphorylation of PLB at Ser16 in mouse, rat or rabbit AVMs (Online Figure XI). On the other hand, propranolol abolished ISO-induced phosphorylation of PLB and LTCC (Online Figure XI). These data support that activation of the SR-β1AR signaling promotes SR-localized PKA phosphorylation of PLB in AVMs. Accordingly, ISO-induced increases in sarcomere shortening (SS), Ca2+ transient amplitude and deceases in tau, were partially reduced by sotalol but completely abolished by propranolol in rat AVMs (Figure 5H–J). These data suggest that phosphorylation of PLB by PKA is mediated by localized β1AR signaling at the SR and promotes myocyte contractility.
Similarly, sotalol selectively inhibited NE-induced PKA activity at the PM yet permitted increases in PKA activity in the SR local domain (Figure 6A–D and Online Figure XIIA–F). We then assessed whether OCT3 controls the NE-induced local PKA activity at the SR in the presence of sotalol. Deletion of OCT3 abolished PKA activity at the SR domain induced by NE in the presence of sotalol (Figure 6A–B and Online Figure XIIA–F). In contrast, ISO-induced PKA activity at the SR was not affected by OCT3KO and sotalol pretreatment (Figure 6C–D). In the presence of sotalol, deleting OCT3 completely blocked NE-induced increases in sarcomere shortening, Ca2+transient, and decreases in Ca2+ decay tau (Figure 6E–G) but did not affect the responses induced by ISO (Figure 6H–J). These data show that in the presence membrane impermeant β-blocker sotalol, NE can selectively stimulate SR-β1AR signaling to promote local PKA activity and enhance myocyte contractility. These activities can be blocked by deletion of OCT3 that is responsible for uptake of NE into AVMs.
Figure 6. Activation of β1AR at the SR promotes myocyte calcium cycling and contractility.
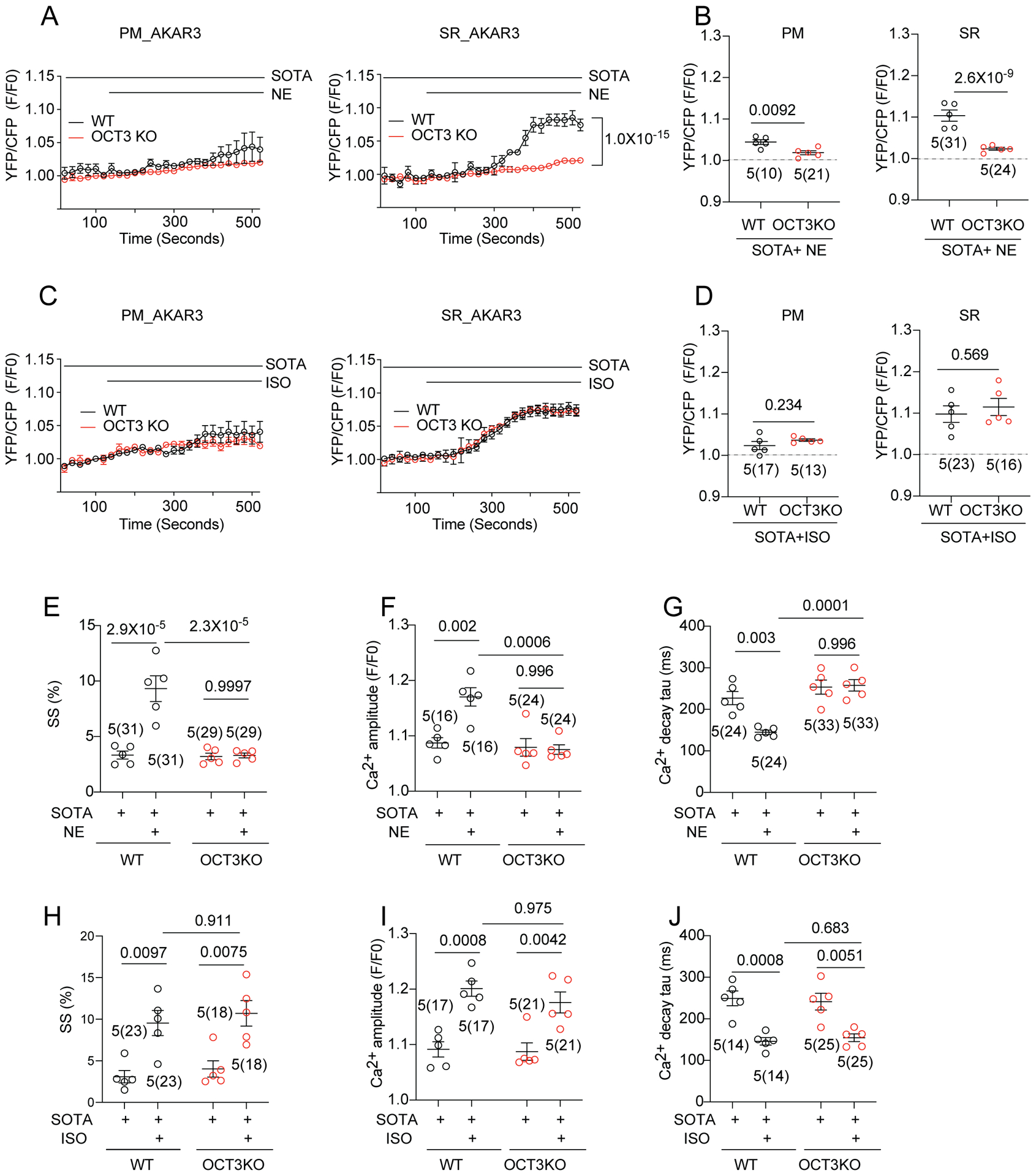
(A, B) WT and OCT3KO AVMs were used to express PM-AKAR3 or SR-AKAR3 biosensors. FRET was analyzed as F/F0 of YFP/CFP ratio. Representative curves and maximal changes show subcellular PKA FRET responses to NE (100 nmol/L) stimulation in AVMs pretreated with sotalol (SOTA, 25 μmol/L). Animal numbers and cell numbers (in the parenthesis) are indicated. p values were obtained by two-way ANOVA analysis followed with Tukey’s multiple comparison test when compared to OCT3-KO. (C, D) WT and OCT3KO mouse AVMs were used to express PM-AKAR3 or SR-AKAR3 biosensors. Representative curves and maximal changes show subcellular PKA FRET responses to ISO (100 nmol/L) stimulation in AVMs pretreated with sotalol (SOTA, 25 μmol/L). Animal numbers and cell numbers (in the parenthesis) are indicated. p values were obtained by two-way ANOVA analysis followed with Tukey’s multiple comparison test when compared to OCT3-KO. (E-G) AVMs from WT and OCT3 hearts were stimulated with NE (100 nmol/L) in the presence of SOTA (25 μmol/L) pretreatment. Data show maximal changes in sarcomere shortening (SS), Ca2+ transient amplitude, and rate of Ca2+ decay (Tau) as mean ± S.E.M. Animal numbers and cell numbers (in the parenthesis) are indicated in figures. p values were obtained by two-way ANOVA analysis with Tukey’s multiple comparison test. (H-J) AVMs from WT and OCT3 hearts were stimulated with ISO (100 nmol/L) in the presence of SOTA (25 μmol/L) pretreatment. Data show maximal changes in SS, Ca2+ transient amplitude, and Tau as mean ± S.E.M. Animal numbers and cell numbers (in the parenthesis) are indicated. p values were obtained by two-way ANOVA analysis with Tukey’s multiple comparison test.
OCT3 deficiency impairs catecholamine transport in heart and attenuates epinephrine induced inotropy and heart rate in vivo.
Finally, we sought to understand the impacts of intracellular β1AR activation on adrenergic regulation of cardiac function in vivo. We observed no differences in heart weight/body weight ratios of OCT3KO and WT mice (Figure 7A), indicating a grossly normal cardiac structure in OCT3KO hearts. However, deletion of OCT3 reduced endogenous NE concentration in hearts compared to WT controls, whereas OCT3KO and WT mice had comparable NE concentrations in the plasma (Figure 7B–C). Consequently, OCT3KO mice show lower cAMP concentrations in hearts relative to WT controls (Figure 7D). These data indicate disrupted catecholamine transport and potential intracellular adrenergic receptor activation by OCT3 deletion. Compared to WT, OCT3KO mice showed no difference in cardiac function, including ejection fraction (EF) and heart rate (HR) at baseline conditions (Figure 7E–I). These observations obtained by echocardiography are probably relevant to adaptive gene expressions in myocardium due to OCT3 deletion. The expression of PLB and PLB/SERCA2 ratio were decreased whereas expression of RyR2 was increased in OCT3KO hearts (Online Figure XIIIA–B). Furthermore, we detected cardiac β-adrenergic response with 10 μg/kg EPI or ISO injection in vivo. Compared to WT, OCT3KO mice showed no difference in cardiac function after 10 μg/kg ISO administration (Figure 7G–I). However, OCT3KO displayed significantly blunted EF and HR responses to EPI injection relative to WT (Figure 7G–I). These data show that OCT3 is essential for enhancing myocardial contractility induced by epinephrine in vivo.
Figure 7. Deletion of OCT3 reduces catecholamine uptake, cAMP signal and inotropic response in response to adrenergic stimulation of mouse hearts.
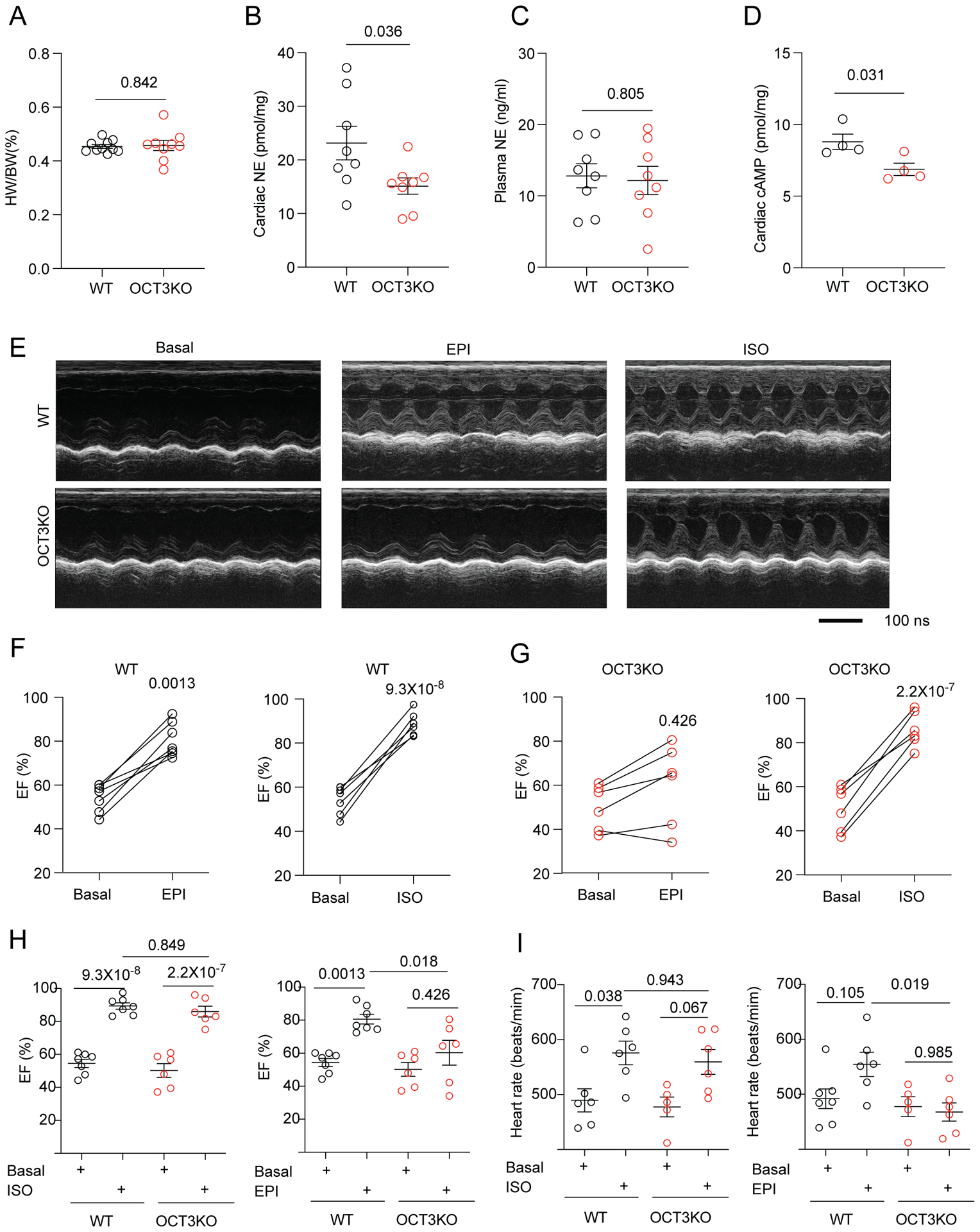
(A) Heart weight/body weight ratio (HW/BW %) in WT and OCT3-KO mice. Data are shown as mean ± S.E.M. (N = 8). p values were obtained by student t-test. (B, C) Quantitative determination of endogenous NE in cardiac tissues and the plasma. Data are shown as mean ± S.E.M. (N = 8). p values were obtained by student t-test. (D) Quantitative determination of cAMP levels in cardiac tissues. Data are shown as mean ± S.E.M. (N = 4). p values were obtained by student t-test. (E) Representative echocardiographic images of WT (N = 7) and OCT3KO (N = 6) mice at baseline and after intraperitoneal injection of 10 μg/kg isoproterenol (ISO) or epinephrine (EPI). (F-I) Cardiac ejection fraction (EF) of individual mice before and after intraperitoneal injection of ISO or EPI in WT (N = 7) and OCT3KO (N = 6). Quantification of EF and heart rate (HR) in WT and OCT3KO mice before and after injection with ISO or EPI. Data are shown as mean ± S.E.M. p values were obtained by two-way ANOVA analysis with Tukey’s multiple comparison test.
DISCUSSION
Adrenergic signaling is one of the most important mechanisms for regulating cardiac function and is typically blunted/disturbed in cardiac diseases including HF.23 In this study, we provide evidence to support the presence of a pool of β1AR at the SR that is essential for enhancing contractility in cardiomyocytes. We also show that monoamine transporter OCT3/EMT mediated catecholamine uptake is essential for activation of the SR-β1AR and for promoting phosphorylation of PLB by PKA and cardiac contractility. Furthermore, our data suggest that different clinically used β-blockers differ significantly in their ability to suppress internal β1AR-PKA signaling due to different membrane permeability. While membrane-permeant β blockers such as propranolol and carvedilol are able to block activation of intracellular β1ARs, membrane impermeant β-blockers such as sotalol and atenolol cannot readily access intracellular β1AR. Therefore, this study opens a novel avenue to optimize therapeutic strategies for targeting cardiac β1 adrenergic signaling in different clinical settings.
Intracellular adrenergic receptor signaling at the SR.
Accumulating evidence suggests the presence of GPCRs and G proteins inside mammalian cells.24 Increasing number of intracellular GPCRs have been identified in animal hearts, including angiotensin3 and adrenergic receptors.2, 8 In cardiomyocytes, previous studies have characterized both β1 and α1ARs, but not β2AR on the nuclear envelope.1, 2 β1ARs were also found to reside in the Golgi apparatus and initiate an internal Gs-cAMP signal from the Golgi apparatus, and this internal signal is independent of receptors at the PM.7, 8 However, whether ARs are localized at other subcellular locations and their functional implications in cardiac contractility remain to be elucidated. Here, we used a combination of sophisticated molecular probes, genetically modified mice and pharmacological inhibitors to uncover important details of the intracellular β1AR-cAMP-PKA pathway at the SR. We provide solid evidence of the presence of intracellular β1ARs at the SR in cardiomyocytes, which is associated with SERCA2 and PLB, consistent with a recent report that exogenously expressed β1ARs bind to SERCA2 in fibroblasts.25 Meanwhile, our data do not support the presence of a cardiac β2AR-SERCA2 complex at the SR membrane. This identification of SR-localized β1ARs provides a novel possibility to explain precisely compartmentalized β1AR signaling in cardiomyocytes. Upon βAR activation the locally anchored PKA is activated and promotes phosphorylation of targets within a nanodomain, including LTCC at the PM, PLB on the SR, and troponin I and myosin binding protein C on the myofilaments.12, 18, 26 The present work shows that activation of the intracellular β1ARs at the SR is necessary for promoting local PKA activity to phosphorylate PLB and TnI in cardiomyocytes, and probably plays a more critical role in rate-limiting cardiac outputs during the physiological fight-or-flight responses. The intracellular β1AR and PKA activity was essential to optimize enhancement of Ca2+ transients and cardiac contractility. Yet, much remains to be learnt about the composition, regulation, and function of this emerging β1AR signaling machinery at the SR in hearts.
Requirement of catecholamine-uptake for intracellular β1ARs activation.
Based on lipophilicity, different βARs ligands access intracellular β1AR either by passive diffusion or transporter-mediated uptake. For instance, the current and prior studies show that hydrophilic catecholamine NE and epinephrine require OCT3/EMT for cellular uptake,2 whereas hydrophobic ligands (e.g. ISO) can passively cross the PM.7, 8 During sympathetic stimulation, catecholamine is released from nerve termini to stimulate adrenergic receptors on cardiomyocytes. Extracellular catecholamines are quickly reduced mainly by re-uptake at nerve termini.27 However, cardiomyocytes can also take up a portion of catecholamines via OCT3/EMT.28 A key question is raised by these observations: what is the function of the catecholamine uptake into cardiomyocytes? Traditionally, it is considered as a mechanism to clear excess catecholamines to prevent overstimulation of cardiac adrenergic signaling.14 However, recent research revealed that intracellular NE activates Golgi-localized β1ARs to promote PI4P hydrolysis.8 In this work, for the first time, we show that inhibition of NE uptake by OCT3/EMT reduces intracellular PKA activity at the SR as well as contractility in cardiomyocytes. Accordingly, OCT3/EMT dependent-catecholamine uptake is essential for optimal βAR-induced enhancement of contractility in cardiac muscles. Deletion of OCT3 attenuated EPI-induced increases in cardiac EF. Meanwhile, deletion of OCT3 also blunted EPI-induced increases in cardiac HR, supporting a notion that SR Ca2+ content contributes to sinoatrial nodal pacemaker cells for physiological heart rate increases.29 Further studies may be merited to address impacts of intracellular β1ARs activation on SR Ca2+ content and Ca2+ handling in myocytes. It also remains to be examined whether other transporters such as OCT2 may affect activation of intracellular β1ARs in hearts. These observations highlight the transporter-dependent uptake of catecholamines as a potential drug target for clinical applications in a variety of cardiovascular conditions.
Similarly, clinically used β-blockers differ significantly in their membrane permeability. Carvedilol is a commonly used β-blocker in HF therapy, and it can permeate the PM and inhibit β1AR at both the PM and intracellular membrane compartments. Thus, it may potentially inhibit detrimental effects of chronic activation of the intracellular β1AR such as nuclear PKA activity for maladaptive gene expression. In comparison, membrane impermeant β-blockers such as sotalol and atenolol, which should mainly block β1AR only at the PM,7, 8 are not widely used for HF therapy. Nonetheless, these β-blockers are effective as anti-arrhythmic drugs,30 probably in part due to their ability to inhibit β1AR on the PM. Our study thus provides a conceptual platform to evaluate and assess individual β-blockers for more efficacious therapy in different clinical applications.
Integrity of β1ARs at the cell surface and subcellular membrane.
Recent and previous studies indicate that the activation of intracellular βAR/PKA signal generates localization-specific and substrate specific effects.7, 8 Membrane localization of βARs and their associated complexes precisely fine-tune βAR activation, which is essential to maintain the cellular function and appropriate responses to extracellular stimuli, such as catecholamines.16, 26 More importantly, we found that the integral regulation of β1AR at both cell surface and the SR is required to achieve maximal enhancement of cardiac contraction via optimally coordinating PKA-dependent phosphorylation of substrates in multiple loci. Impaired integrity of β1AR/cAMP/PKA signaling and blunted cardiac contractile response to β-adrenergic stimulation have been implicated in pathological stress conditions.26 It has been reported that in HF βAR activation induced PKA activity is significantly inhibited or abolished at the SR and at the myofilaments, but well-preserved at the PM,12, 18 suggesting the dysregulation of intracellular βAR/PKA signal is involved in the pathogenesis of HF. Besides, a large body of studies revealed that in human HF, NE reuptake was impaired.31 Although cardiac NE release by sympathetic nerves was increased in HF patients, cardiac NE uptake and cardiac NE contents were significantly reduced. Accordingly, OCT transporters expression is reduced in dilated cardiomyopathy patients.15 Those observations are consistent with our findings in OCT3KO mice that disrupted catecholamine uptake leads to decreased NE concentrations and cAMP in the myocardium. Meanwhile, OCT3KO mice show attenuated inotropic response to catecholamines. Taken together, these findings suggest a decreased activation of intracellular β1AR at the SR due to dysregulated OCT3 may contribute to the decreased PKA activity and phosphorylation of PLB associated with cardiac dysfunction. The integrity β1ARs at intracellular membranes and their role in development of HF remains to be further explored.
In summary, this study uncovers a novel signaling paradigm in which a SR-localized intracellular β1AR is functionally crucial in adrenergic stimulation of cardiac contractility. Moreover, the data not only help to broaden our current understanding of the general role of β1AR signaling in cardiac regulation, but also open up a novel platform to evaluate and optimize β-blockers in clinical applications based on their “biased” properties in selective inhibition of cardiac β1AR at distinct subcellular locations.
Supplementary Material
NOVELTY AND SIGNIFICANCE.
What Is Known?
β1-adrenoceptors (β1ARs) are the major subtype driving cardiac contractility in response to catecholamine stimulation.
β1-adrenoceptors (β1ARs) exist in intracellular membranes.
Organic Cation Transporter 3 (OCT3) mediates norepinephrine entry into cardiomyocytes.
What New Information Does This Article Contribute?
A pool of β1AR are associated with the sarco-endoplasmic reticulum calcium ATPase (SERCA2) complex at the sarcoplasmic reticulum (SR).
Activation of the SR localized β1AR is essential to promote phosphorylation of phospholamban.
Organic Cation transporter 3 is required for catecholamines (norepinephrine and epinephrine) to enter cells and stimulate the β1AR at the SR.
Activation of intracellular SR β1AR is essential to promote cardiac contractility.
Functional β1ARs exists at the SR and is critical for PKA-mediated phosphorylation of phospholamban and cardiac contractility upon catecholamine stimulation. Activation of these intracellular β1ARs requires catecholamine transport via OCT3.
For decades, catecholamines have been shown to signal via βARs on the plasma membrane to regulate heart contractile functions. Little is known about the presence and function of intracellular βARs in cardiomyocytes. We show the presence of of intracellular β1AR associated with SERCA2 on the SR. Activation of this SR-localized β1AR is essential for promoting PKA-dependent phosphorylation of PLB and optimizing the cardiac contraction response. Intracellular β1AR activation is regulated by catecholamine uptake via the OCT3 transporter or membrane permeant β-blockers in cardiomyocytes. These findings provide new mechanistic insight into the composition, localization and regulation of β1AR signaling. Targeting intracellular β1AR and its regulators could have potential therapeutic utility in diseases related to cardiac contractile function.
ACKNOWLEDGEMENTS
We thank Johanna Borst and Logan Bailey for isolation of rabbit adult cardiomyocytes, Toni West for manuscript editing, and Mohammad Sahtout for statistical analysis.
SOURCE OF FUNDING
This work was supported by National Institutes of Health grants R01-HL127764 and R01-HL147263 (YKX), R01-HL133832 and P01-HL141084 (DMB), R01 NS078792, R01-AG055357, and R01-MH097887 (JWH), a VA Merit grant 01BX002900 (YKX), and National Natural Science Foundation of China grant 81700252 (ML). QS is a recipient of American Heart Association postdoctoral fellowship. YKX is an established American Heart Association investigator.
Nonstandard Abbreviations and Acronyms:
- AR
Adrenergic receptors
- AVMs
Adult ventricular cardiomyocytes
- AKARs
A kinase activity reporters
- FL-Prop
BODIPY-tagged propranolol
- Ca2+
Calcium
- CORTI
Corticosterone
- Dob
Dobutamine
- EF
Ejection fraction
- EPI
Epinephrine
- FRET
Förster resonance energy transfer
- FS
Fractional shortening
- ISO
Isoproterenol
- NE
Norepinephrine
- OCT3/EMT
Organic Cation Transporter 3/extraneuronal monoamine transporter
- PLB
Phospholamban
- PLM
Phospholemman
- PM
Plasma membrane
- PKA
Protein kinase A
- PLA
Proximity ligation assay
- RyR2
Ryanodine receptor 2
- JP2
Junctophilin 2
- SS
Sarcomere Shortening
- SERCA2
Sarcoplasmic endoplasmic reticulum Ca2+-ATPase 2
- SR
Sarcoplasmic reticulum
- WT
Wild type
- cAMP
Cyclic AMP
- OCT3KO
OCT3 knockout
- LTCC
L-type calcium channel
Footnotes
Publisher's Disclaimer: This article is published in its accepted form. It has not been copyedited and has not appeared in an issue of the journal. Preparation for inclusion in an issue of Circulation Research involves copyediting, typesetting, proofreading, and author review, which may lead to differences between this accepted version of the manuscript and the final, published version.
DISCLOSURES
None.
REFERENCES
- 1.Boivin B, Lavoie C, Vaniotis G, Baragli A, Villeneuve LR, Ethier N, Trieu P, Allen BG and Hebert TE. Functional beta-adrenergic receptor signalling on nuclear membranes in adult rat and mouse ventricular cardiomyocytes. Cardiovasc Res. 2006;71:69–78. [DOI] [PubMed] [Google Scholar]
- 2.Wright CD, Chen Q, Baye NL, Huang Y, Healy CL, Kasinathan S and O’Connell TD. Nuclear alpha1-adrenergic receptors signal activated ERK localization to caveolae in adult cardiac myocytes. Circ Res. 2008;103:992–1000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Escobales N, Nunez RE and Javadov S. Mitochondrial angiotensin receptors and cardioprotective pathways. Am J Physiol Heart Circ Physiol. 2019;316:H1426–H1438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Ibarra C, Vicencio JM, Estrada M, Lin Y, Rocco P, Rebellato P, Munoz JP, Garcia-Prieto J, Quest AF, Chiong M, Davidson SM, Bulatovic I, Grinnemo KH, Larsson O, Szabadkai G, Uhlen P, Jaimovich E and Lavandero S. Local control of nuclear calcium signaling in cardiac myocytes by perinuclear microdomains of sarcolemmal insulin-like growth factor 1 receptors. Circ Res. 2013;112:236–45. [DOI] [PubMed] [Google Scholar]
- 5.Benard G, Massa F, Puente N, Lourenco J, Bellocchio L, Soria-Gomez E, Matias I, Delamarre A, Metna-Laurent M, Cannich A, Hebert-Chatelain E, Mulle C, Ortega-Gutierrez S, Martin-Fontecha M, Klugmann M, Guggenhuber S, Lutz B, Gertsch J, Chaouloff F, Lopez-Rodriguez ML, Grandes P, Rossignol R and Marsicano G. Mitochondrial CB(1) receptors regulate neuronal energy metabolism. Nature neuroscience. 2012;15:558–64. [DOI] [PubMed] [Google Scholar]
- 6.Revankar CM, Cimino DF, Sklar LA, Arterburn JB and Prossnitz ER. A transmembrane intracellular estrogen receptor mediates rapid cell signaling. Science. 2005;307:1625–30. [DOI] [PubMed] [Google Scholar]
- 7.Irannejad R, Pessino V, Mika D, Huang B, Wedegaertner PB, Conti M and von Zastrow M. Functional selectivity of GPCR-directed drug action through location bias. Nature chemical biology. 2017;13:799–806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Nash CA, Wei W, Irannejad R and Smrcka AV. Golgi localized beta1-adrenergic receptors stimulate Golgi PI4P hydrolysis by PLCepsilon to regulate cardiac hypertrophy. Elife. 2019;8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Lefkowitz RJ, Rockman HA and Koch WJ. Catecholamines, cardiac beta-adrenergic receptors, and heart failure. Circulation. 2000;101:1634–7. [DOI] [PubMed] [Google Scholar]
- 10.Xiang Y and Kobilka BK. Myocyte adrenoceptor signaling pathways. Science. 2003;300:1530–2. [DOI] [PubMed] [Google Scholar]
- 11.Bers DM. Cardiac excitation-contraction coupling. Nature. 2002;415:198–205. [DOI] [PubMed] [Google Scholar]
- 12.Surdo NC, Berrera M, Koschinski A, Brescia M, Machado MR, Carr C, Wright P, Gorelik J, Morotti S, Grandi E, Bers DM, Pantano S and Zaccolo M. FRET biosensor uncovers cAMP nano-domains at beta-adrenergic targets that dictate precise tuning of cardiac contractility. Nature communications. 2017;8:15031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kawai H, Fan TH, Dong E, Siddiqui RA, Yatani A, Stevens SY and Liang CS. ACE inhibition improves cardiac NE uptake and attenuates sympathetic nerve terminal abnormalities in heart failure. Am J Physiol. 1999;277:H1609–17. [DOI] [PubMed] [Google Scholar]
- 14.Eisenhofer G The role of neuronal and extraneuronal plasma membrane transporters in the inactivation of peripheral catecholamines. Pharmacology & therapeutics. 2001;91:35–62. [DOI] [PubMed] [Google Scholar]
- 15.Grube M, Ameling S, Noutsias M, Kock K, Triebel I, Bonitz K, Meissner K, Jedlitschky G, Herda LR, Reinthaler M, Rohde M, Hoffmann W, Kuhl U, Schultheiss HP, Volker U, Felix SB, Klingel K, Kandolf R and Kroemer HK. Selective regulation of cardiac organic cation transporter novel type 2 (OCTN2) in dilated cardiomyopathy. Am J Pathol. 2011;178:2547–59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Xiang YK. Compartmentalization of beta-adrenergic signals in cardiomyocytes. Circ Res. 2011;109:231–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Liu S, Li Y, Kim S, Fu Q, Parikh D, Sridhar B, Shi Q, Zhang X, Guan Y, Chen X and Xiang YK. Phosphodiesterases coordinate cAMP propagation induced by two stimulatory G protein-coupled receptors in hearts. Proc Natl Acad Sci U S A. 2012;109:6578–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Barbagallo F, Xu B, Reddy GR, West T, Wang Q, Fu Q, Li M, Shi Q, Ginsburg KS, Ferrier W, Isidori AM, Naro F, Patel HH, Bossuyt J, Bers D and Xiang YK. Genetically Encoded Biosensors Reveal PKA Hyperphosphorylation on the Myofilaments in Rabbit Heart Failure. Circ Res. 2016;119:931–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Cui M, Aras R, Christian WV, Rappold PM, Hatwar M, Panza J, Jackson-Lewis V, Javitch JA, Ballatori N, Przedborski S and Tieu K. The organic cation transporter-3 is a pivotal modulator of neurodegeneration in the nigrostriatal dopaminergic pathway. Proc Natl Acad Sci U S A. 2009;106:8043–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Jain A, Liu R, Ramani B, Arauz E, Ishitsuka Y, Ragunathan K, Park J, Chen J, Xiang K and Ha J. Probing Cellular Protein Complexes via Single Molecule Pull-down. Nature. 2011;473:484–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Brittsan AG and Kranias EG. Phospholamban and cardiac contractile function. J Mol Cell Cardiol. 2000;32:2131–9. [DOI] [PubMed] [Google Scholar]
- 22.Detroyer A, Vander Heyden Y, Carda-Broch S, Garcia-Alvarez-Coque MC and Massart DL. Quantitative structure-retention and retention-activity relationships of beta-blocking agents by micellar liquid chromatography. J Chromatogr A. 2001;912:211–21. [DOI] [PubMed] [Google Scholar]
- 23.Lohse MJ, Engelhardt S and Eschenhagen T. What is the role of beta-adrenergic signaling in heart failure? Circ Res. 2003;93:896–906. [DOI] [PubMed] [Google Scholar]
- 24.Ortiz Zacarias NV, Lenselink EB, AP IJ, Handel TM and Heitman LH. Intracellular Receptor Modulation: Novel Approach to Target GPCRs. Trends in pharmacological sciences. 2018;39:547–559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Yang Z, Kirton HM, MacDougall DA, Boyle JP, Deuchars J, Frater B, Ponnambalam S, Hardy ME, White E, Calaghan SC, Peers C and Steele DS. The Golgi apparatus is a functionally distinct Ca2+ store regulated by the PKA and Epac branches of the beta1-adrenergic signaling pathway. Sci Signal. 2015;8:ra101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Bers DM, Xiang YK and Zaccolo M. Whole-Cell cAMP and PKA Activity are Epiphenomena, Nanodomain Signaling Matters. Physiology (Bethesda). 2019;34:240–249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kvetnansky R, Sabban EL and Palkovits M. Catecholaminergic systems in stress: structural and molecular genetic approaches. Physiol Rev. 2009;89:535–606. [DOI] [PubMed] [Google Scholar]
- 28.Obst OO, Rose H and Kammermeier H. Characterization of catecholamine uptake2 in isolated cardiac myocytes. Mol Cell Biochem. 1996;163–164:181–3. [DOI] [PubMed] [Google Scholar]
- 29.Wu Y, Valdivia HH, Wehrens XH and Anderson ME. A Single Protein Kinase A or Calmodulin Kinase II Site Does Not Control the Cardiac Pacemaker Ca2+ Clock. Circ Arrhythm Electrophysiol. 2016;9:e003180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Anderson JL and Prystowsky EN. Sotalol: An important new antiarrhythmic. Am Heart J. 1999;137:388–409. [DOI] [PubMed] [Google Scholar]
- 31.Rose CP, Burgess JH and Cousineau D. Tracer norepinephrine kinetics in coronary circulation of patients with heart failure secondary to chronic pressure and volume overload. J Clin Invest. 1985;76:1740–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Liu SB, Zhang J and Xiang YK. FRET-based direct detection of dynamic protein kinase A activity on the sarcoplasmic reticulum in cardiomyocytes. BBRC. 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.West TM, Wang Q, Deng B, Zhang Y, Barbagallo F, Reddy GR, Chen D, Phan KS, Xu B, Isidori A and Xiang YK. Phosphodiesterase 5 Associates With beta2 Adrenergic Receptor to Modulate Cardiac Function in Type 2 Diabetic Hearts. J Am Heart Assoc. 2019;8:e012273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Foulon P and De Backer D. The hemodynamic effects of norepinephrine: far more than an increase in blood pressure! Ann Transl Med. 2018;6:S25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Wang Q, Liu Y, Fu Q, Xu B, Zhang Y, Kim S, Tan R, Barbagallo F, West T, Anderson E, Wei W, Abel ED and Xiang YK. Inhibiting Insulin-Mediated beta2-Adrenergic Receptor Activation Prevents Diabetes-Associated Cardiac Dysfunction. Circulation. 2017;135:73–88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Reddy GR, West TM, Jian Z, Jaradeh M, Shi Q, Wang Y, Chen-Izu Y and Xiang YK. Illuminating cell signaling with genetically encoded FRET biosensors in adult mouse cardiomyocytes. J Gen Physiol. 2018;150:1567–1582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Shi Q, Li M, Mika D, Fu Q, Kim S, Phan J, Shen A, Vandecasteele G and Xiang YK. Heterologous desensitization of cardiac beta-adrenergic signal via hormone-induced betaAR/arrestin/PDE4 complexes. Cardiovasc Res. 2017;113:656–670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Pogwizd SM. Nonreentrant mechanisms underlying spontaneous ventricular arrhythmias in a model of nonischemic heart failure in rabbits. Circulation. 1995;92:1034–48. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The data, analytic methods, and study materials are made available to other researchers for purposes of reproducing the results or replicating the procedure. Please see the Major Resources Table and Expanded Materials & Methods in the Online Data Supplement.
All experiments including animal feeding, treatment, and tissue collection were approved by the Institutional Animal Care and Use Committees (protocol: 20234 and 20957) of the University of California at Davis and follow NIH guidelines. Male and female 2–4-month-old C57/BL6 and OCT3KO, newborn β1AR-KO, β2AR-KO and β1β2AR-KO mice were described previously.17, 19 Male Sprague Dawley outbred rats and New Zealand white rabbits were used in this study.