

Abstract
Extreme hyperbilirubinemia can cause bilirubin neurotoxicity. Infants with glucose-6-phosphate dehydrogenase (G6PD) deficiency can develop hemolysis and thus at high risk. We evaluated a device that quantitatively measures G6PD activity kinetically using digital microfluidics (DMF). Intra- and inter-instrument and -day imprecision (CVs) were first assessed. G6PD activity in 86 samples was then measured and compared between DMF and 2 reference methods. Overall DMF reproducibility was 3.8% over 5 days by 2 operators on 2 instruments. Mean intra- and inter-instrument variabilities were 3.6% and 3.9%, respectively (n=28), with a user variability of 4.3%. Mean G6PD activity was 6.40±4.62 and 6.37±4.62 U/g hemoglobin for DMF and reference method 1 (n=46) and 12.15±3.86 and 11.48±1.55 for DMF and method 2 (n=40), respectively, and strongly correlated (r=0.95 and 0.95) with mean biases of +0.04±2.90 and +0.67±1.55 for methods 1 and 2, respectively. The novel device could be used for early newborn G6PD screening.
Introduction
The deficiency of glucose-6-phosphate dehydrogenase (G6PD), the housekeeping enzyme involved in the pentose phosphate shunt in mature red blood cells (RBCs) (Figure 1), is an X-linked inherited disorder that results in an insufficient or absent NADPH-generating system, which affects over 400 million people worldwide, with the highest frequency in individuals of Mediterranean, Asian, or African ancestry.1–4 The coenzyme NADPH is essential for protection against oxidative damage of RBC membranes and insufficient production of NADPH can thus lead to hemolysis. G6PD deficiency is second to Rhesus (Rh) incompatibility in its severity and frequency. Based on the largest series, the best US estimate of G6PD deficiency in healthy adults is 3.5% (ranging from 0% to 12.2%). The severity of hemolysis varies among affected individuals.5–8 The extent of G6PD deficiency is often unpredictable or undiagnosed, and therefore can manifest as progressive extreme hyperbilirubinemia [EHB, total serum/plasma bilirubin (TB) > 25 mg/dL (428 μmol/L)] even if exposures to oxidative triggers (such as infection, moth balls, fava beans, certain drugs, etc) are avoided.9,10 Identification of G6PD deficiency in the newborn infant and education of parents to this condition, especially in regard to exposure of their infant to unsafe medications and/or dietary supplements, are critical. EHB, unrecognized or untreated, can progress to irreversible choreoathetoid cerebral palsy or acute kernicterus.9,10 Adverse outcomes, some milder, depend on biologic factors, choice of a screening test, and parental education.1,3,5,8 Whether screening for G6PD deficiency can be accomplished in a timely manner is uncertain. Decisions regarding universal or selective screening are made by the Secretary’s Advisory Committee on Heritable Disorders in Newborns and Children and included in the Recommended Uniform Screening Panel (RUSP) of the US Department of Human and Health Services and based on whether a test is feasible, accurate, practical, timely, and actionable.9,10 The types of screening vary by state and based on the Discretionary Advisory Committee on Heritable Disorders in Newborns and Children, which approves the Federal and State funding to 35 primary and 26 secondary conditions (80 total tests in CA) and do not include TB or G6PD testing.11
Fig. 1. Pentose phosphate pathway.
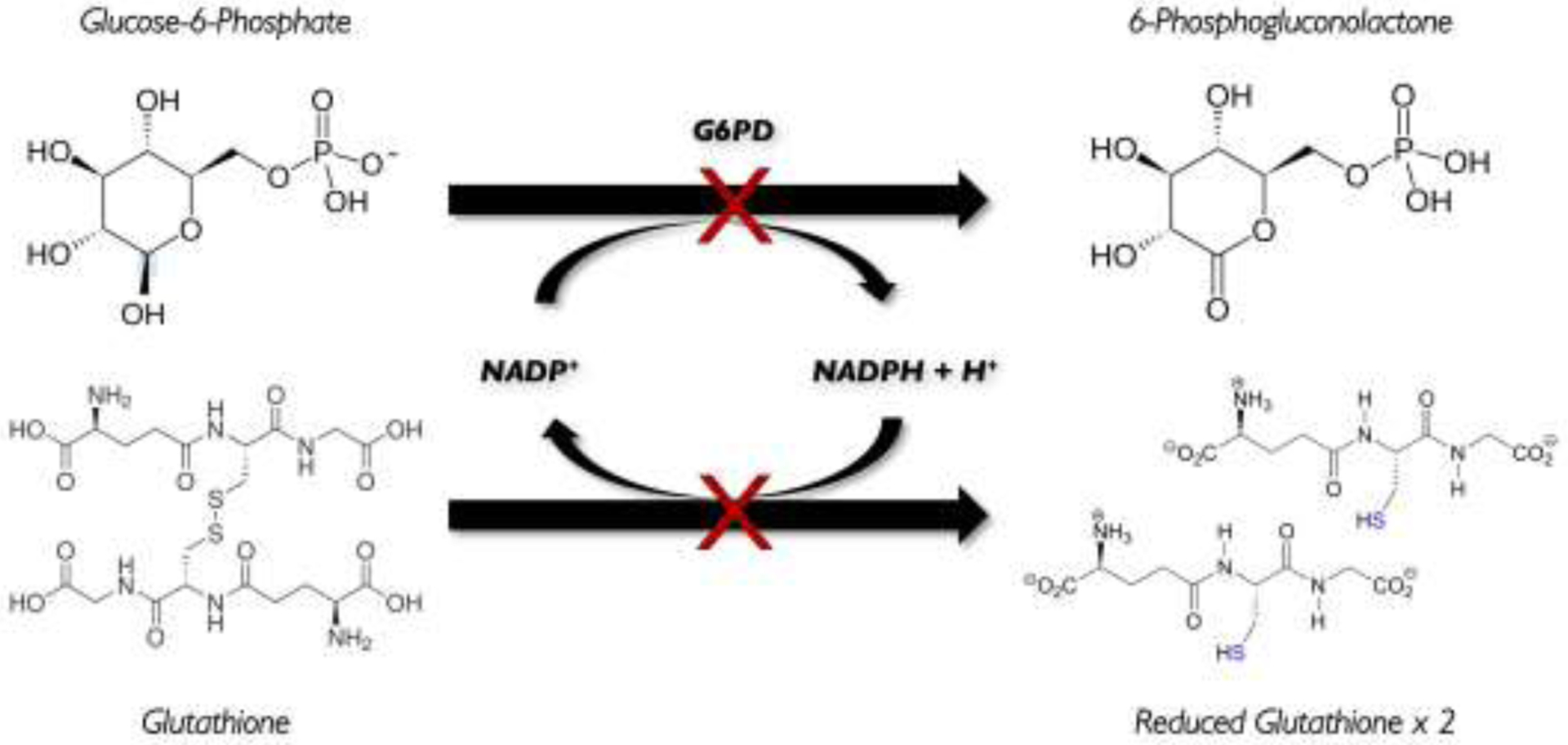
The enzyme glucose-6-phosphate dehydrogenase (G6PD) is involved in the pentose phosphate shunt that generates NADPH in mature red blood cells, which protects the cell membrane from oxidative damage; and if deficient, hemolysis can occur.
Neonatal jaundice due to hyperbilirubinemia is observed in up to 80% of otherwise healthy, term and late-preterm newborns during the first week of life and is usually benign and transitional. However, in newborns who are undergoing hemolysis, EHB can occur and lead to bilirubin neurotoxicity if not treated in a timely manner. Infants who are at a particularly high risk are those with G6PD deficiency. Currently, testing of G6PD-deficiency is only reserved for infants with a family history or geographic or ethnic origin suggestive of this condition or in infants who have poor responses to phototherapy. G6PD status can be tested by quantitative biochemical methods (enzyme or cytochemical assays); semi-quantitative techniques; or molecular screening. But in most of the US, laboratory testing for G6PD enzyme activity is not routine nor part of universal newborn screening. Thus, newborns with undetected G6PD deficiency who are discharged from the hospital without appropriate parental education can later develop pathologic hyperbilirubinemia, and be at risk for developing bilirubin-induced neurologic dysfunction (BIND) if they are not re-admitted and treated in a timely fashion.12–14 Our objective was to evaluate a novel prototype device that quantitatively measures G6PD enzyme activity through an assessment of device performance and comparisons to known reference methods.
Materials and Methods
Devices
The prototype point-of-care device called FINDER® (Figure 2A) is developed by Baebies, Inc. (Durham, NC) and miniaturizes the digital microfluidics (DMF) technologies pioneered on SEEKER®,15 their FDA-cleared high-throughput laboratory-based instrument for screening newborn lysosomal storage disorders. DMF uses the rapid, coordinated manipulation of discrete droplets of blood and reagents by electrical control of surface tension, which is termed “electrowetting” (Figure 2B), in a disposable “chip” cartridge (Figure 2C). Here, blood and reagents droplets are sandwiched between an array of electrodes on a printed circuit board and a plastic top plate. In this system, droplets can be transferred between any 2 neighboring electrodes and transported anywhere within a network of contiguous electrodes. The cartridge also contains liquid reagents in a sealed reservoir, dried spots of assay reagents, lanes to mix samples and reagents, a detection zone, and a port for blood sample introduction. ~50 μL of whole blood is loaded and reagents are reconstituted once sample analysis is initiated using a tablet touchpad interface. Analysis takes approximately 15 min. The FINDER is designed for use in a hospital or clinic for rapid (< 15 min) point-of-care (POC) testing by medical personnel. G6PD enzyme activity was quantified using FINDER, which measures fluorescence kinetically on ~50 μL of whole blood.
Fig, 2. Digital microfluidics (DMF) method.
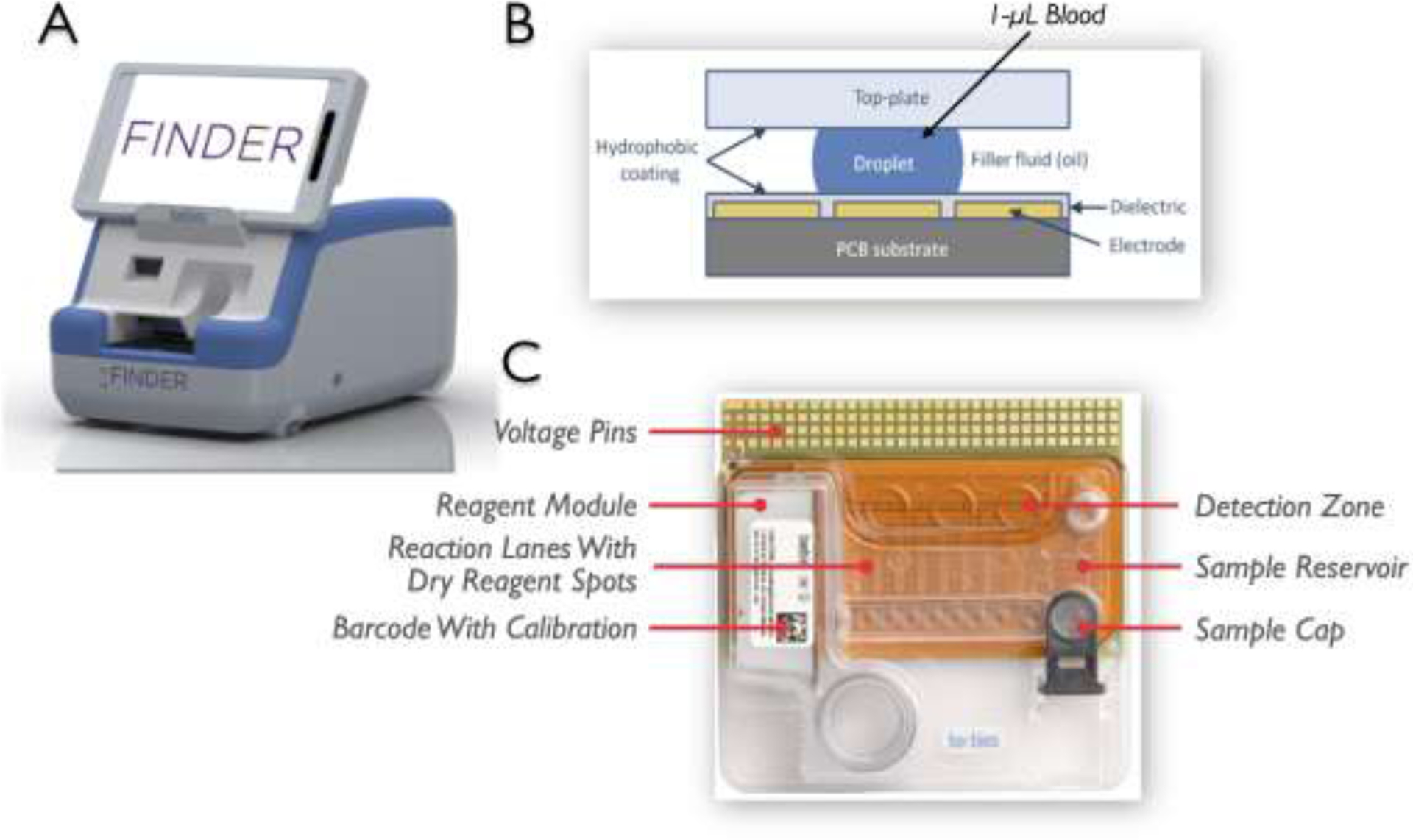
(A) The prototype point-of-care device is designed on a DMF platform; (B) DMF is based on electric fields to manipulate droplets of blood or “electrowetting”; and (C) Disposable “chip” cartridge is tightly regulated at 37°C and contains all the required reagents in dried form, which are then reconstituted once sample analysis is initiated. The assay requires ~50 μL of whole blood.
The methodology to quantitate G6PD activity used in the FINDER is based on the fluorescence of NADPH. In brief, droplets of whole blood are diluted at a ratio of 1:27 by mixing a sample droplet with 2 droplets of lysis solution, splitting off into 3 droplets, discarding 2 droplets, then continuing the serial dilutions. Absorbance of the diluted sample is measured at 540 nm to determine the concentration of hemoglobin (Hb). The remaining droplet is then mixed with substrate, plasma G6PD converts NADP to NADPH, and the fluorescence of NADPH is measured at 360-nm excitation and 460-nm emission at 8 time points and correlated directly to G6PD activity based on the rate of NADPH generation. G6PD activity is normalized to Hb concentrations.1,2,16
Reference Methods
The reference methods for measuring G6PD enzyme activity was performed by the Hillview RBC Special Studies Laboratory (Reference Method 1) as previously described15 and at Laboratory Corporation of America (LabCorp, Burlington, NC) (Reference Method 2).
Device Performance
For these studies, donated blood from a healthy, normal (non-G6PD-deficient) male was used and assayed daily for 5 consecutive days. All measurements were taken using 2 separate FINDER devices and by 2 separate users. We then evaluated intra- and inter-instrument and intra- and inter-day imprecision (CV).
Blood Samples for Method Comparison Studies
For the comparison studies with Reference Method 1, discarded whole blood samples were collected in EDTA-coated tubes and stored at 4°C prior to being transferred from the Hillview RBC Special Studies Laboratory (Palo Alto, CA) to Stanford. Based on previous experiences, RBC G6PD activity is stable for > 21 days when stored in EDTA and at 4°C.16 Sex, age, and collection date and time of the donor samples were recorded. Within 24 hrs of transfer, samples were analyzed on the FINDER devices.
For the comparison studies with Reference Method 2, 40 whole blood samples from adult males were obtained from BioIVT (Westbury, NY) across 4 days. Two mL of each whole blood sample was transferred into 2 EDTA Vacutainer® tubes and kept cold until transfer to LabCorp.
Comparison with a Lab-Developed Test at Stanford Hillview Lab: Reference Method 1
A method comparison study was performed by evaluating measurements using the DMF method and those performed with a lab-developed test at Stanford Hillview RBC Special Studies Laboratory. We compared the performance of the quantitative DMF G6PD assay with the gold standard fluorescence biochemical method on a convenience sample of discarded blood.
Comparison with FDA-Cleared Test at LabCorp: Reference Method 2
A similar method comparison study was performed by evaluating measurements using the DMF method and those performed with an FDA-cleared kit (Pointe Scientific G6PD Reagent Set and Test # 001917) run on a Roche Cobas® C501 module (6000 Series System) at LabCorp.
Statistical Analyses
Results were compared using paired t-tests, linear regression and Bland-Altman analyses. A weighted Deming fit was the fit of choice for the reference comparison with Reference Method 2 (LabCorp).
Results
Device Performance
The limits of detection (LoD) of the DMF G6PD assay were determined to be 0.45 U/g Hb, and correlated well with the Hillview reference method with an LoD of 0.4 U/g Hb. Overall reproducibility for the assay across 5 days performed by 2 operators on 2 instruments was 3.8% with a mean G6PD enzyme activity of 4.90 ± 0.17 (4.33 to 5.36) and 5.18 ± 0.19 (4.75±5.57) U/g Hb, for the 2 devices for n = 28 measurements (Table 1). Mean interday and inter-instrument variabilities were 3.6% and 3.9 ± 1.2%, respectively, with a user variability of 4.3 ± 2.8% (Table 2), and were comparable to that reported by Baebies, Inc.
Table 1.
Device performance.
| Device | G6PD Activity, U/g Hb (range; n) |
|---|---|
| #1 | 4.90 ± 0.17 (4.33 to 5.36; 28) |
| #2 | 5.13 ± 0.19 (4.75 to 5.57; 28) |
Hb, hemoglobin
Table 2.
Device imprecision.
| Parameter | Mean ± SD % |
|---|---|
| Inter-Instrument | 3.9 ± 1.2 |
| Inter-Day | 3.6 |
| Intra-Day | 3.5 ± 1.0 |
| Inter-User | 4.3 ± 2.8 |
Method Comparison with Reference Method 1
We then performed a method comparison study between the DMF method and Reference Method 1 (Hillview). Samples were obtained from 46 discarded blood samples (20 females, 26 males) at Stanford ranging from 1-day to 71 yrs of age. In this convenience sample, mean (range) G6PD activity was 6.40 ± 4.62 (0.21 to 15.80) and 6.37 ± 4.62 (0.10 to 16.60) U/g Hb for the DMF and Reference Method 1, respectively (Table 3). DMF strongly correlated with Reference Method 1 (r = 0.95; slope = 0.95; y-intercept = 0.34). 57% (15/26) of males were G6PD-deficient (Figure 3A). Most G6PD-deficient samples (n = 17) were from males (n = 15 or 88.2%). A Bland-Altman plot showed that the bias and imprecision of the DMF method was only +0.04 with an imprecision of 2.90 U/g Hb of n = 46 subjects (Figure 4A).
Table 3. Comparison of the DMF and Reference Methods.
Glucose-6-phosphate dehydrogenase (G6PD) enzyme activity (U/g hemoglobin [Hb]) was measured in 46 subjects (age ranging from 2-days to 71-yrs-old; 20 females and 26 males).
| Reference Method | DMF Method | P-Value |
|---|---|---|
| ns | ||
| 6.37 ± 4.62 (0.10 to 16.60) n=46 |
6.40 ± 4.62 (0.21 to 15.80) n=46 |
|
| ns | ||
| 11.48 ± 1.55 (1.40 to 20.60) n=40 |
12.15 ± 3.86 (1.39 to 23.24) n=40 |
Fig. 3. Device performance comparison with Reference Methods: linear correlation.
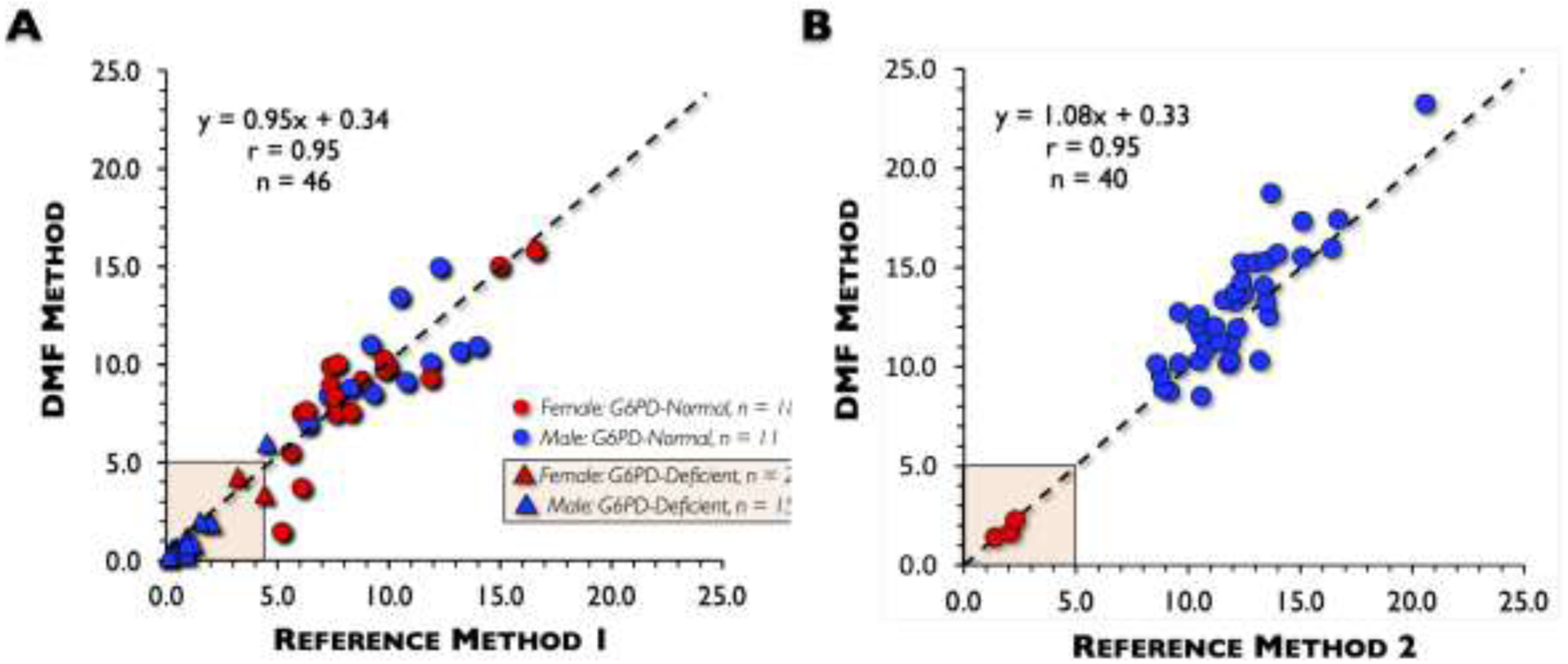
(A) When measurements using the digital microfluidics (DMF) method were compared to those of Reference Method 1, a correlation (r) of 0.95, with a slope of 0.95 and y-intercept of 0.34 was found. The majority of glucose-6-phosphate dehydrogenase (G6PD)-deficient samples (88.2%) were from males (15/17). (B) When measurements using the DMF were compared to those of Reference Method 2, a correlation (r) of 0.95, with a slope of 1.08 and y-intercept of −0.32 was found. The majority of samples had normal G6PD enzyme activity.
Fig. 4. Device performance comparison with Reference Methods: bias and imprecision.
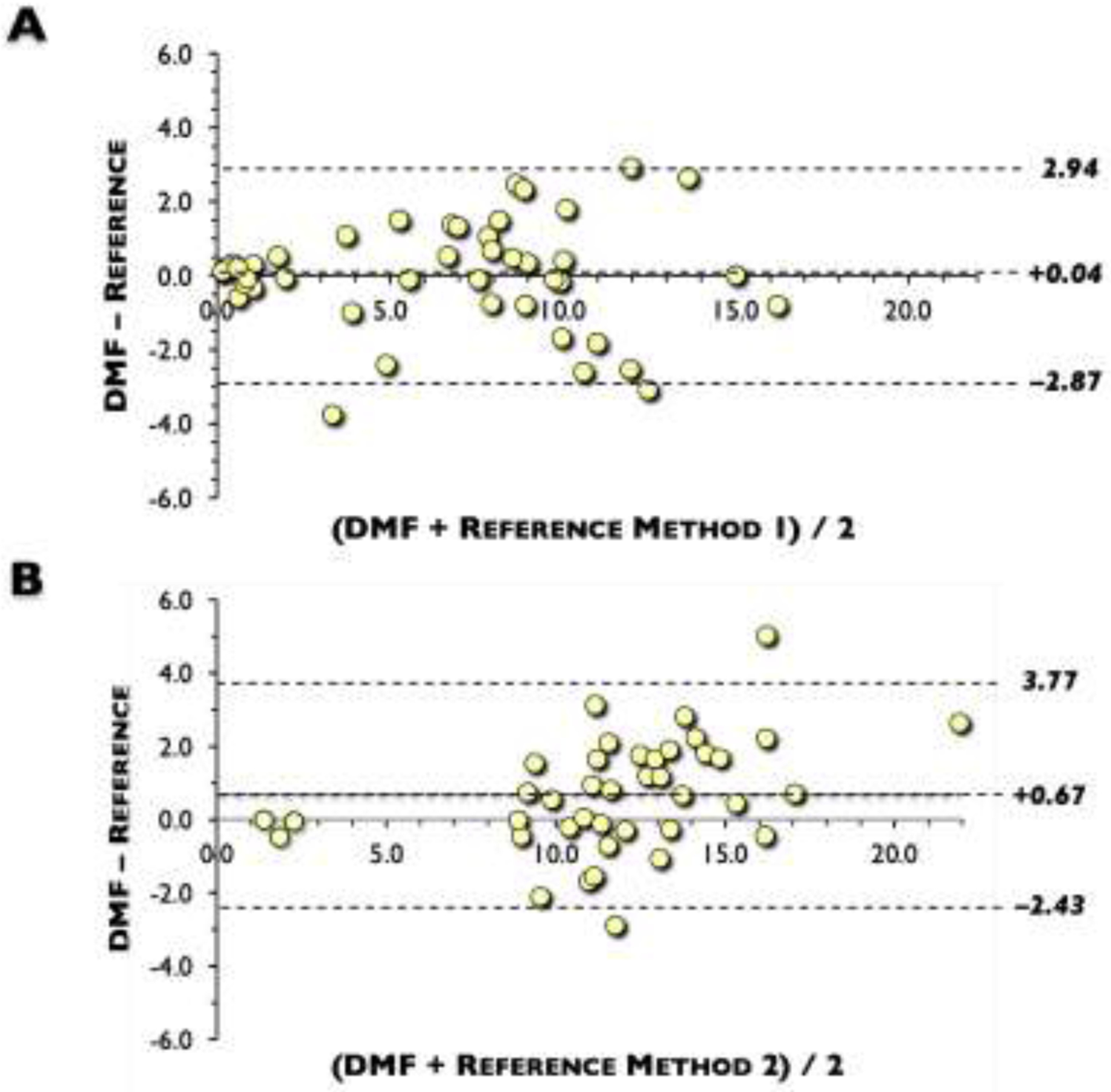
Bland-Altman plot showed that the bias and imprecision of the digital microfluidics (DMF) method to: (A) Reference Method 1, which showed only +0.04 with an imprecision of 2.90 U/g hemoglobin (Hb) of n = 46 subjects; (B). Reference Method 2, which showed that the bias of the DMF method was +0.67 U/g Hb and standard deviation of the difference was 1.55 U/g Hb of n = 40 subjects.
Method Comparison with Reference Method 2
We also performed a method comparison study between the DMF method and Reference Method 2 (LabCorp). 40 whole blood samples collected from adults with a mean age of 46.4 ± 14.3 yrs (ranging from 20 to 72) were run on both the FINDER and at LabCorp. 92.5% (37/40) of the samples were normal or above normal, 3 samples were deficient, and none were intermediate samples (3 to 8 U/g Hb). Consequently, the average (mean) G6PD activity was higher at 12.15 (1.39 to 23.2) and 11.48 (1.4 to 20.6) U/g Hb for DMF and Reference Method 2, respectively (Table 3). DMF also strongly correlated with Reference Method 2 (r = 0.95; slope = 1.08; y-intercept = −0.32) (Figure 3B). A Bland-Altman plot showed that the bias of the DMF method was only +0.67 U/g Hb and standard deviation of the difference was 1.55 U/g Hb of n = 40 subjects (Figure 4B).
Discussion
With limited evidence, the American Academy of Pediatrics (AAP) recommends testing for G6PD enzyme activity only in newborns with EHB who are to receive phototherapy, whose family history, ethnicity, or geographic origin are known risks for G6PD deficiency or in infants who do not respond to phototherapy well.8,17 The 2004 AAP Practice Guideline8 and its updated 2009 clarification17 guide use of phototherapy at lower TB thresholds; however, the evidence and process for operational directives are lacking. We proposed that applying 2 novel techniques: quantification of bilirubin load (concurrent G6PD and TB testing) to identify those most at risk; and a parental education approach to provide tailored instruction to parents based on their infant’s G6PD and TB status may lead to individualized (precision health) to prevent adverse outcomes in infants with G6PD deficiency. Altjhough the exact incidence of kernicterus in G6PD-deficient infants is not known, it has been estimated based only on neonatal mortality rates.18 According to the Canadian Paediatric Surveillance Program, of the 258 cases of infants with EHB (or having had an exchange transfusion), 20/93 (21.5%) were found to be G6PD-deficient.18,19 Similarly, Denmark, UK, and Ireland have reported that 108 newborns with EHB were all G6PD-deficient.18,20,21 In Utah, 28% (2/7 cases of kernicterus had G6PD deficiency.22 Independently, G6PD deficiency increases the risk of ABE with an odds ratio (OR) of 28.2 (95% cconfidence interval [CI]: 2.6, 307.7).18
The most frequent confounding biologic risk factors for EHB and kernicterus are sex, race, hemolysis, and late prematurity.23–28 Preterm infants have delayed heaptic maturation, and thus a lower expression of uridine 5’-diphosphoglucuronosyltransferase (UGT).29 Immature gastrointestinal function, and feeding difficulties that predispose preterm infants to dehydration, increased enterohepatic circulation of bilirubin, and decreased stool frequency add to diminished bilirubin elimination. Incremental genetic testing for other undiagnosed hemolytic diseases can enable the elucidation of causes of increased bilirubin production that may not be evident.30,31 Neonatal G6PD deficiency can setup a clinical “perfect storm” of hyperbilirubinemia with increased bilirubin production and concurrent UGT polymorphisms that overwhelms the immature bilirubin elimination process.32 On the other hand, in utero G6PD deficiency impacts embryonic epithelial-mesenchymal transition and embryonic development lethality needs a separate long-term exploration.33
Clinical manifestations of neonatal G6PD deficiency include early-onset, progressive, acute occurrence of EHB or a life-threatening “crisis”. In addition to race and sex, hemolysis, sepsis, prematurity, exposure to triggers/stressors, access to healthcare, and existence of comorbidities are key clinical risk factors. Of these, the AAP has guided the testing for G6PD deficiency in high-risk newborns based on their race and ethnicity.34,35 Identification of infants at risk for an acute hemolytic crisis has not been prospectively correlated to TB load and G6PD enzyme concentration.
G6PD genetic screening for 7 mutations is currently only performed in Pennsylvania and the District of Columbia using dried blood spots transported to a central laboratory with a turnaround time of several days.36,37 In Pennsylvania, where G6PD screening is routine, clinicians follow this general approach: after identifying an infant who is a presumptively deficient, the coordinator at the birthing hospital is contacted by the laboratory at about age 1 wk.37 This person is often a genetic counselor. All families of identified newborns are offered a single genetic counseling visit between the ages of 4 to 6 wks. They are also provided a list of medications and foods to avoid. Repeat testing of G6PD deficiency by a pediatric hematologist is recommended at age 3 to 6 mos. Finally, the information on the infant’s G6PD status is entered into their electronic medical record and also forwarded to the infant’s pediatrician. It has been reported that some parents do not come for their outpatient appointment at 2 to 3 wks after birth. In any case, this follow-up date is too late to identify and prevent most EHB cases.
Current practice tests for G6PD deficiency in infants when phototherapy prescription is being considered at time of readmission. The test results frequently only become available several days “after the crisis”. We propose that G6PD enzyme screening should be performed prior to discharge from a birthing facility. Thus, G6PD-deficient infants would be identified early, followed, and managed in an appropriate manner to achieve a lower TB burden. A specific threshold-based relationship between G6PD deficiency and TB levels has been elusive, and show that other critical factors also contribute to an intervention threshold. Clinical translation of biomarkers of hemolysis, developmental maturation, concurrent illnesses, or non-phototherapy interventions could alter the severity of progression to EHB. Avoidance of known triggers either through maternal milk or, direct exposure has been successful in malaria endemic regions of the world that have high incidence of G6PD deficiency.
Universal screening by biochemical methods without concurrent TB screening does not accurately detect all G6PD-deficient males (hemizygotes), and only detects some homozygote females. Even the use of intermediate G6PD activity thresholds can misclassify female heterozygotes as sufficient in > 50% of cases. Importantly, a heterozygote female reported as G6PD-normal may have a sizable contribution to the overall G6PD-deficient population. On the other hand, enzyme assays have been shown to discriminate G6PD deficiency in African-American males.
Is heterozygosity in females a modifiable risk factor as assayed by quantitative biochemical assays of G6PD? If so, the identification of female heterozygosity represents a ratio of deficient and sufficient RBC pools and manifests as a normal or intermediate enzyme activity level that is falsely reassuring. Even applying intermediate G6PD enzyme thresholds may lead to the misclassification of female heterozygotes as being G6PD-sufficient. Molecular screening is the only means that can definitively identify a G6PD-deficient heterozygote female. Even with a normal G6PD result, newborn females in high-risk ethnic groups would still need to be closely monitored for jaundice, identification of heterozygosity, evidence of hemolysis, and EHB. Yet, the AAP guideline only relies on maternal self-reported ethnicity, race, and reliance on X-linked inheritance that oversimplifies and may misclassify racial ancestry and thus would lead to missed cases. It is crucial to know how many G6PD-deficient females might be unideintified if targeted screening is used. Steps needed to operationally and systematically establish G6PD levels is warranted and could be informed by data garnered from this study. In the meantime, these barriers to equities have to be minimized through reliance on medical signs of imminent medical emergencies that are mitigated by awareness, participation, and surveillance.
The FINDER can measure G6PD enzyme activity reproducibly in bench studies. In the clinical setting, it was found to highly correlate with the standard biochemical tests with minimal bias and imprecision. We conclude that the instrument could be used as an accurate POC screening tool for early newborn G6PD screening. Its clinical performance and diagnostic utility need to be further validated in a multicenter observational study. Combination of effective newborn screening and family empowerment could reduce EHB and spectrum of BIND, and therefore is highly significant.38,39 Currently, clinicians do not adhere to the AAP recommendations and manage TB and G6PD testing as two separate approaches that are not timely to prevent EHB. We suggest that a combined or near-simultaneous dual screening of these co-dependent conditions with concurrent education materials would substantially increase both provider and parental awareness of G6PD deficiency in the hospital and/or during home care.
Acknowledgements
We thank Professor Bertil Glader, who supervised the laboratory assays at Hillview, for his review of the data as well as his Laboratory Manager, Carolyn Wong. We acknowledge collaborative contributions and technical assistance from the Baebies team (Vamsee Pamula, Rama Sista, Susan Wilhelm, Ranier Ng, and Michael Bobadilla).
Supported by in part by Baebies, Inc., the David and Lucile Packard Foundation, and the National Institutes of Health: Eunice Kennedy Shriver National Institute of Child Health & Human Development (no. R44HD072853) and the National Heart, Lung, and Blood Institute (no. R44HL146016).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Disclosures
None of the authors have any financial interests to disclose.
References
- 1.WHO Working Group. Glucose-6-phosphate dehydrogenase deficiency. Bull World Health Organ. 1989;67:601–611. [PMC free article] [PubMed] [Google Scholar]
- 2.Beutler E Glucose-6-phosphate dehydrogenase deficiency. N Engl J Med. 1991;324:169–174. [DOI] [PubMed] [Google Scholar]
- 3.Cappellini MD, Fiorelli G. Glucose-6-phosphate dehydrogenase deficiency. Lancet. 2008;371:64–74. [DOI] [PubMed] [Google Scholar]
- 4.Valaes T, Karaklis A, Stravrakakis D, et al. Incidence and mechanism of neonatal jaundice related to glucose-6-phosphate dehydrogenase deficiency. Pediatr Res. 1969;3:448–458. [DOI] [PubMed] [Google Scholar]
- 5.Johnson L, Bhutani VK, Karp K, Sivieri E, Shapiro S. Clinical report from the pilot USA Kernicterus Registry (1992 to 2004). J Perinatol. 2009;29:S25–S45. [DOI] [PubMed] [Google Scholar]
- 6.Kaplan M, Beutler E, Vreman HJ, et al. Neonatal hyperbilirubinemia in glucose-6-phosphate dehydrogenase-deficient heterozygotes. Pediatrics. 1999;104:68–74. [DOI] [PubMed] [Google Scholar]
- 7.Watchko JF, Kaplan M, Stark AR, Stevenson DK, Bhutani VK. Should we screen newborns for glucose-6-phosphate dehydrogenase deficiency in the United States? J Perinatol. 2013;33:499–504. [DOI] [PubMed] [Google Scholar]
- 8.American Academy of Pediatrics Subcommittee on Hyperbilirubinemia. Management of hyperbilirubinemia in the newborn infant 35 or more weeks of gestation. Pediatrics. 2004;114:297–316. [DOI] [PubMed] [Google Scholar]
- 9.American Academy of Pediatrics Newborn Screening Authoring Committee. Newborn screening expands: recommendations for pediatricians and medical homes--implications for the system. Pediatrics. 2008;121:192–217. [DOI] [PubMed] [Google Scholar]
- 10.U.S. Health Resources and Services Administration. Recommended Uniform Screening Panel [updated December 2017; cited 2018 January 23rd]. Available from: https://www.hrsa.gov/advisory-committees/heritable-disorders/rusp/index.html.
- 11.California Department of Public Health. Newborn Screening Program (NBS) [updated October 10, 2017; cited 2018 January 23rd]. Available from: https://www.cdph.ca.gov/Programs/CFH/DGDS/Pages/nbs/default.aspx. [Google Scholar]
- 12.Bhutani VK, Wong RJ. Bilirubin-induced neurologic dysfunction (BIND). Semin Fetal Neonatal Med. 2015;20:1. [DOI] [PubMed] [Google Scholar]
- 13.Bhutani VK, Wong RJ. Bilirubin neurotoxicity in preterm infants: risk and prevention. J Clin Neonatol. 2013;2:61–69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Johnson L, Brown AK, Bhutani VK. BIND – a clinical score for bilirubin-induced neurologic dysfunction in newborns. Pediatrics. 1999;104:746–747. [Google Scholar]
- 15.Bhutani VK, Kaplan M, Glader B, et al. Point-of-care quantitative measure of glucose-6-phosphate dehydrogenase enzyme deficiency. Pediatrics. 2015;136:e1268–e1275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Beutler E Red cell metabolism: a manual of biochemical methods. 3rd ed New York: Grune and Stratton, Inc; 1984. [Google Scholar]
- 17.Maisels MJ, Bhutani VK, Bogen D, et al. Hyperbilirubinemia in the newborn infant > or=35 weeks’ gestation: an update with clarifications. Pediatrics. 2009;124:1193–1198. [DOI] [PubMed] [Google Scholar]
- 18.Bhutani VK, Zipursky A, Blencowe H, et al. Neonatal hyperbilirubinemia and Rhesus disease of the newborn: incidence and impairment estimates for 2010 at regional and global levels. Pediatr Res. 2013;74:86–100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Sgro M, Campbell D, Shah V. Incidence and causes of severe neonatal hyperbilirubinemia in Canada. CMAJ. 2006;175:587–590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Ebbesen F, Andersson C, Verder H, et al. Extreme hyperbilirubinaemia in term and near-term infants in Denmark. Acta Paediatr. 2005;94:59–64. [DOI] [PubMed] [Google Scholar]
- 21.Manning D, Todd P, Maxwell M, Platt MJ. Prospective surveillance study of severe hyperbilirubinaemia in the newborn in the UK and Ireland. Arch Dis Child Fetal Neonatal Ed. 2007;92:F342–F346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Christensen RD, Agarwal AM, George TI, Bhutani VK, Yaish HM. Acute neonatal bilirubin encephalopathy in the State of Utah 2009–2018. Blood Cells Mol Dis. 2018;72:10–13. [DOI] [PubMed] [Google Scholar]
- 23.Missiou-Tsagaraki S Screening for glucose-6-phosphate dehydrogenase deficiency as a preventive measure: prevalence among 1,286,000 Greek newborn infants. J Pediatr. 1991;119:293–299. [DOI] [PubMed] [Google Scholar]
- 24.Bhutani VK, Vilms RJ, Hamerman-Johnson L. Universal bilirubin screening for severe neonatal hyperbilirubinemia. J Perinatol. 2010;30 Suppl:S6–S15. [DOI] [PubMed] [Google Scholar]
- 25.Chinevere TD, Murray CK, Grant E Jr., et al. Prevalence of glucose-6-phosphate dehydrogenase deficiency in U.S. Army personnel. Mil Med. 2006;171:905–907. [DOI] [PubMed] [Google Scholar]
- 26.Watchko JF. Hyperbilirubinemia in African American neonates: clinical issues and current challenges. Semin Fetal Neonatal Med. 2010;15:176–182. [DOI] [PubMed] [Google Scholar]
- 27.Herschel M, Beutler E. Low glucose-6-phosphate dehydrogenase enzyme activity level at the time of hemolysis in a male neonate with the African type of deficiency. Blood Cells Mol Dis. 2001;27:918–923. [DOI] [PubMed] [Google Scholar]
- 28.Kaplan M, Herschel M, Hammerman C, Hoyer JD, Stevenson DK. Hyperbilirubinemia among African American, glucose-6-phosphate dehydrogenase-deficient neonates. Pediatrics. 2004;114:e213–e219. [DOI] [PubMed] [Google Scholar]
- 29.Monaghan G, McLellan A, McGeehan A, et al. Gilbert’s syndrome is a contributory factor in prolonged unconjugated hyperbilirubinemia of the newborn. J Pediatr. 1999;134:441–446. [DOI] [PubMed] [Google Scholar]
- 30.Kaplan M, Muraca M, Hammerman C, et al. Imbalance between production and conjugation of bilirubin: a fundamental concept in the mechanism of neonatal jaundice. Pediatrics. 2002;110:e47. [DOI] [PubMed] [Google Scholar]
- 31.Ahlfors CE. The bilirubin binding panel: A Henderson-Hasselbalch approach to neonatal hyperbilirubinemia. Pediatrics. 2016;138:e20154378. [DOI] [PubMed] [Google Scholar]
- 32.Stevenson DK, Fanaroff AA, Maisels MJ, et al. Prediction of hyperbilirubinemia in near-term and term infants. Pediatrics. 2001;108:31–39. [DOI] [PubMed] [Google Scholar]
- 33.Wu YH, Lee YH, Shih HY, et al. Glucose-6-phosphate dehydrogenase is indispensable in embryonic development by modulation of epithelial-mesenchymal transition via the NOX/Smad3/miR-200b axis. Cell Death Dis. 2018;9:10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Beal AC, Chou SC, Palmer RH, et al. The changing face of race: risk factors for neonatal hyperbilirubinemia. Pediatrics. 2006;117:1618–1625. [DOI] [PubMed] [Google Scholar]
- 35.Padilla CD, Therrell BL. Newborn screening in the Asia Pacific region. J Inherit Metab Dis. 2007;30:490–506. [DOI] [PubMed] [Google Scholar]
- 36.Lin Z, Fontaine JM, Freer DE, Naylor EW. Alternative DNA-based newborn screening for glucose-6-phosphate dehydrogenase deficiency. Mol Genet Metab. 2005;86:212–219. [DOI] [PubMed] [Google Scholar]
- 37.Algur N, Avraham I, Hammerman C, Kaplan M. Quantitative neonatal glucose-6-phosphate dehydrogenase screening: distribution, reference values, and classification by phenotype. J Pediatr. 2012;161:197–200. [DOI] [PubMed] [Google Scholar]
- 38.G6PD Deficiency and Favism [cited 2018 January 23rd]. Available from: http://g6pddeficiency.org/wp/#.Wmf_GEtG1Z0.
- 39.Kaplan M, Hammerman C, Bhutani VK. Parental education and the WHO neonatal G-6-PD screening program: a quarter century later. J Perinatol. 2015;35:779–784. [DOI] [PubMed] [Google Scholar]