

Abstract
The transdifferentiation of cardiac fibroblasts into myofibroblasts after cardiac injury has traditionally been defined by a unidirectional continuum from quiescent fibroblasts, through activated fibroblasts, and finally to fibrotic-matrix producing myofibroblasts. However, recent lineage tracing and single cell RNA sequencing experiments have demonstrated that fibroblast transdifferentiation is much more complex. Growing evidence suggests that fibroblasts are more heterogenous than previously thought, and many new cell states have recently been identified. This review reexamines conventional fibroblast transdifferentiation paradigms with a dynamic state space lens, which could enable a more complex understanding of how fibroblast state dynamics alters fibrotic remodeling of the heart. This review will define cellular state space, how it relates to fibroblast state transitions, and how the canonical and non-canonical fibrotic signaling pathways modulate fibroblast cell state and cardiac fibrosis. Finally, this review explores the therapeutic potential of fibroblast state space modulation by p38 inhibition, yes-associated protein (YAP) inhibition, and fibroblast reprogramming.
Keywords: Cellular State Space, Fibroblast Cell State, Cardiac Fibroblast, Cardiac Fibrosis, Myofibroblast, Matrifibrocyte
1. Fibroblast state space
1.1. The Landscape Model of Cellular State Space
Previously, fibroblast cell states were defined rigidly, as quiescent un-activated fibroblasts, activated proliferating fibroblasts, or matrix-secreting myofibroblasts [1]. Convention dictated that in response to a severe cardiac injury, cardiac fibroblasts would first proliferate and then transdifferentiate into myofibroblasts, a cell type that is critical for generating collagen-rich scar tissue and preventing cardiac rupture. As our understanding of heart disease and progressive cardiac remodeling has evolved, the cardiac fibroblast has emerged as a key target for heart failure therapeutics. Furthermore, recent genomic studies have greatly expanded the known complexity of cardiac fibroblast phenotypes, underscoring that the linear transdifferentiation model is insufficient to describe fibroblast fate [1–5]. To evolve the field’s understanding of injury-induced cardiac fibrosis, the complexity of the linear fibroblast transdifferentiation paradigm must be expanded. By improving the understanding of the dynamics of fibroblast state space, new therapeutic targets for treating maladaptive fibrotic remodeling may be identified.
What is cellular state space?
Cell state transitions such as development, growth, maturation, senescence, and transdifferentiation are critical in both normal physiology and disease. These cell transformations are driven by shifts in genetic profile, which can be described with a “state space” model. Classically, state space is a mathematical concept whereby dynamic systems can be described using a set of variables that comprise every possible state of the system. In the context of cell biology, this can be illustrated as a hilly landscape traversed by cells as they differentiate or transition between states [6,7]. A marble analogy is often used to think about cells moving dynamically through this landscape, where peaks and valleys represent unstable and stable cell states, respectively. The position on the landscape (x/y-dimension) is determined by a compressed set of multidimensional gene expression variables—essentially representing the relative similarity of cell states. The overall height (z-dimension) of the landscape represents the activation energy required to transition between stable cell states (Figure 1). Mathematical modeling has shown that this idea of state space landscape is grounded in mathematical theory [8]. In the heart, cardiac injuries deliver strong pro-fibrotic signals to cardiac fibroblasts. However, how signals modulate the cellular state of fibroblasts and the relationship of these state changes to matrix organization, quantity, and composition is still poorly understood.
Figure 1: Cardiac Fibroblast Movement in Cell State Space.
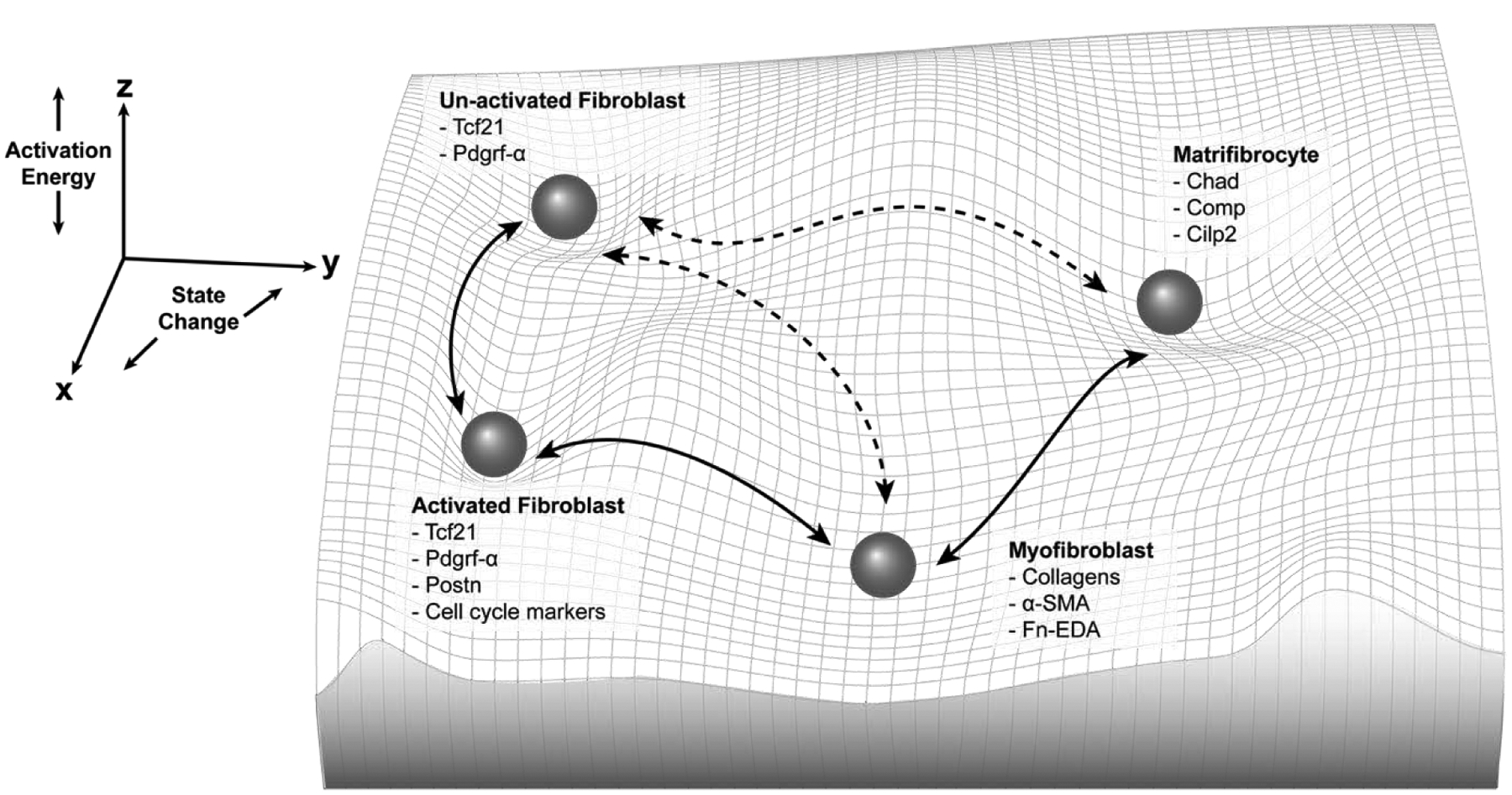
Cardiac fibroblasts have long been thought to exist on a linear continuum between an unactivated state and a fibrotic matrix secreting myofibroblast state. This figure depicts a more complete picture of cardiac fibroblast “state space.” State transition may be reversible indicated by the bidirectional arrow heads, new fibroblasts cell states may exist (e.g. matrifibrocytes), and transdifferentiation directly between these states might be possible indicated by dotted lines. Importantly the third dimension of state space “activation energy” is critical to understanding this concept. For example, the myofibroblast state could be unstable and not in a deep valley, indicating that a small amount of activation energy could induce a state change. In contrast, a more permanent cell state, like a cardiomyocyte, would have a much deeper valley to represent low likelihood of state transition. Known gene expression markers of the fibroblast states shown are listed, however it is important to note that a position in multidimensional state space is far more complex than a small number of marker genes.
Fibroblast cell state dynamics can inform overall matrix dynamics in the heart. Ideally, after cardiac injury, therapeutic targeting would be able to tune the heart’s fibrotic response to maintain structural integrity in the short term, preventing cardiac rupture, but reduce or abrogate cardiac fibrosis and maladaptive remodeling in the long term [9]. There is growing evidence to suggest that tactically controlling fibroblast state via their state space landscape effectively modulates the heart’s fibrotic response, and hence has therapeutic potential [10–13]. For example, if a therapeutic could reduce the activation energy for a myofibroblast to transition to a state with lower matrix secretion, pathologic fibrotic remodeling could be lessened. However, this goal requires identification of cardiac fibroblast state trajectories during injury, repair, and recovery, along with the regulators of those state transitions. Determining how modifying fibroblast state transitions, either through pathway inhibition, activation, or reprogramming, impacts fibroblast population dynamics will be vital towards understanding how therapeutic state modulation alters the physiological role of fibroblasts in the heart.
Epigenetic and transcriptional analyses by high-throughput genome sequencing technologies like RNAseq, ATACseq, and CHIPseq may be used to molecularly define the state space occupied by cardiac fibroblasts. These new techniques enable unbiased and global snapshots of cellular state space transitions. Since transcript sequencing at the single cell level captures a snapshot of cells scattered along the differentiation or transdifferentiation trajectory; nuanced or ephemeral cell populations can be identified. This can be achieved using pseudotime analysis to understand how cells are moving through state space; however, pseudotime is best corroborated by serial sampling [14] and RNA labeling [15] to allow for more definitive analysis of how cells move through transcriptional space. This sort of analysis is exemplified in the field of hematopoietic progenitor differentiation [16,17], which showed that the differentiation of hemopoietic progenitors into either erythroid or myeloid lineages required destabilizing the progenitor state [17] and movement through metastable intermediate substates during the differentiation process [16]. Since state destabilization seems to be critical for state transition, cardiac fibroblasts may undergo similar cell state destabilizations to form fibrosis-producing myofibroblasts in the heart after injury. Approaching fibroblast transdifferentiation through the lens of multidimensional state space is critical to developing the next generation of therapeutic approaches for cardiac fibrosis.
1.2. Cardiac fibroblast states
While it was previously believed that cardiac myofibroblasts originated from several different sources, such as resident endothelial cells or circulating immune cells [18–23], genetic lineage tracing studies have shown that the majority (>95%) of activated myofibroblasts in the heart are derived from resident cardiac fibroblasts expressing Transcription Factor 21 (Tcf21) and Platelet-derived Growth Factor Receptor α (Pdgfrα) [24–28]. In the uninjured myocardium, quiescent Tcf21 and Pdgfrα expressing fibroblasts make up ~15% of the non-myocyte cardiac cell population, whereas activated fibroblasts and myofibroblasts are extremely rare or nonexistent [28]. This changes with injury or disease, as quiescent fibroblast progenitors begin transitioning to new states [1]. Fibroblast state transitions have been most characterized in the myocardial infarction (MI) model where within two days of injury, a subset of cardiac fibroblasts become proliferative and activated. These cells are marked by expression of the matricellular protein Periostin (Postn) [10,27,29] and minor to moderate expression of alpha smooth muscle actin (Acta2 or αSMA) [10,27,29], which gives these cells new contractile function. Importantly, Postn deletion blocks the formation of myofibroblasts [13], indicating that in addition to labeling the myofibroblast state it also regulates this transition. In the days following injury, these activated fibroblasts mature into fully activated myofibroblasts, which boost secretion of matrix proteins and alter the composition of the extracellular matrix (ECM) to form a rigid scar [13,30]. More mature myofibroblasts are identified by their strong expression of collagens I and III, αSMA, and a splice variant of fibronectin, Fibronectin Containing Extra Domain A (Fn-EDA) [10,26,27,29,31]. These cells also exhibit reduced expression of Tcf21 and Pdgfrα [10,32].
Recent studies demonstrate that fibroblast cell state is more complex than previously thought, suggesting that a dynamic state space model may more accurately depict fibroblast transdifferentiation then the traditional linear model (Figure 1). This is supported by recent findings regarding myofibroblast cell fate after injury resolution. Contrary to older studies, newer data has identified that most myofibroblasts don’t undergo apoptosis after injury resolution [33,34], but rather transition to a new state or regress back towards quiescence depending on the type of injury [2,10]. For example, around 14 days after myocardial infarction (MI) myofibroblasts appear to transition to a matrifibrocyte state that is molecularly defined by the expression of chondrocyte and osteogenic genes [2]. These matrifibrocytes are derived from myofibroblasts of the Postn lineage and are found in mature scars using the markers chondroadherin (Chad), cartilage oligomeric matrix protein (Comp), and CAP-Gly Domain Containing Linker Protein 2 (Clip2) (Figure 1). This subtype of cardiac fibroblast is postulated to chronically maintain the infarct scar. In contrast, Postn-traced myofibroblasts reverted to a less activated state after only two weeks of recovery from chronic stimulation of the heart via Angiotensin II (AngII) and phenylephrine (PE) [10]. These data raise questions about what is a “deactivated” myofibroblast, and how similar is this state to its original quiescent fibroblast progenitor. Another possible myofibroblast state transition is senescence. Research in cancer biology identified that senescent myofibroblasts often secrete enzymes that proteolytically degrade the extracellular matrix [35]. This state has also been identified in the heart and may be essential for reversing cardiac fibrosis during the heart’s recovery from injury [36]. The disappearance of myofibroblasts over time via transdifferentiation, deactivation, and senescence suggests that the myofibroblast state is transient and likely unstable [2,4]. Using a state space model, this could be described by positioning myofibroblasts in a very shallow “valley”, indicating that this state requires minimal activation energy to move into a more stable state (Figure 1). In the heart, myofibroblasts might transition back to the progenitor state or move towards a more mature fibroblast injury phenotype, such as the matrifibrocyte. Although this hypothesis has not been directly examined, it also opens up questions regarding where these cellular states reside in multidimensional molecular state space and how different fibroblast states can impact cardiac structure and function. The impact of redefining fibroblast transdifferentiation as a multidimensional state space model provides a more complex and comprehensive cellular and molecular road map that will be crucial for developing new targeted antifibrotic therapeutics.
1.3. Cardiac fibroblast & myofibroblast heterogeneity
Single cell RNA sequencing (scRNAseq) techniques have broadened the understanding of fibroblast cell state and provided evidence for a more nuanced fibroblast heterogeneity in both healthy and injured hearts. The general population of resident fibroblasts activate, proliferate, and increase their matrix production as described previously, but the molecular heterogeneity present in these populations suggests they consist of a diverse group of substates. However, these new clusters of molecularly distinct fibroblasts have not yet been functionally validated by other techniques. Three studies recently investigated the diversity of non-myocytes in murine hearts. Skelly et al. were one of the first groups to identify new cardiac fibroblast states by performing scRNAseq on cells isolated from uninjured hearts. A new fibrocyte population of cells was identified, expressing transcriptional markers of both fibroblasts (collagen type I alpha 1 chain (Col1a1), Pdgfrα, and Tcf21) and immune cells (high affinity immunoglobulin gamma Fc receptor I (Fcrg1), cluster of differentiation 14 (Cd14), and protein tyrosine phosphatase receptor type C (Ptprc)). However, the functional role of these cells in the heart at baseline or in injury was not investigated. Quiescent fibroblasts were also shown to produce growth factors for the maintenance of other cardiac cells, including neurons, endothelial cells, and mural cells, highlighting that cardiac fibroblasts play a larger role in cardiac homeostasis beyond matrix secretion [5]. These findings demonstrate cardiac fibroblast state is heterogenous and suggest these unique fibroblast clusters may have different functional roles.
Farbehi et al. expanded on this work, identifying new quiescent and activated fibroblast substates in response to MI. The quiescent fibroblast population segregated into two major groups marked by relative expression of the surface marker spinocerebellar ataxia 1 (Sca1). Fibroblasts with strong expression of Sca1 had higher expression of cell adhesion genes, whereas fibroblasts with low Sca1 expression had high expression of signal transduction genes. This would suggest that the quiescent fibroblasts have separate subpopulations involved in cell adhesion and signal transduction. Farbehi et al. also used lineage tracing to isolate and selectively sequence Pdgfrα expressing cells. By deep sequencing a fraction of the cells, they identified novel myofibroblast subtypes expressing both profibrotic and antifibrotic signatures. They also showed the canonical changes in the proportions of cells falling into each of these subtypes three to seven days post injury. In line with the established fibroblast state paradigm, activated and cycling fibroblasts peaked 3 days post infarction and myofibroblasts peaked 7 days post infarction [3], which is consistent with state change timelines established by other approaches [2]. Two novel fibroblast states were also identified. One was a transitory fibroblast state, which cell trajectory analysis placed as an intermediate substate between Sca1-low and the second novel state defined by Wnt inhibitory factor 1 (Wif1) expression. These cells expressed antifibrotic paracrine signaling and were present at all time points, persisting for at least 7 days after infarction. This fibrosis inhibition cell state opposes all the activating fibroblasts in the heart by expressing inhibitors of wingless-related integration site (WNT), cellular communication network factor 2 (Ccn2) and transforming growth factor beta (TGFβ) signaling and seems to be important for the timing of heart repair [3]. Farbehi et al. also reanalyzed the data from Skelly et al. and were able to find all of the same baseline cell populations identified in their work.
Most recently, McLellan et al. completed a scRNAseq study looking at the fibroblast populations present after an AngII infusion model. This study also found the Wif1 expressing cell population identified by Farbehi et al. Surprisingly, no αSMA expressing myofibroblast population was detected. Instead they identified two fibroblast subpopulations expressing the matricellular proteins Cilp and thrombospondin 4(Thbs4) [4]. Pseudotime analysis showed that Thbs4 expressing cells arose from Cilp cells. The Cilp and Thbs4 expressing fibroblasts also expressed markers similar to the matrifibrocyte (Comp, secreted frizzled related protein 2 (Sfrp2), and cellular communication network factor 5 (Ccn5)) indicating that these markers could be important for post-injury fibroblast states. Additionally, Cilp expressing fibroblasts expressed higher levels of matrix remodeling proteins. None of these groups expressed αSMA or had the molecular components of canonical myofibroblasts [1]. However, these cells did express Postn and seemed to be responsible for extracellular matrix production in this AngII infusion injury model. This suggests that different injury models may elicit a variety of fibroblast states in order to modulate the composition and quantity of extracellular matrix (Figure 2a). Notably, there are two types of fibrosis: replacement and reactive. Replacement fibrosis occurs when there is significant cardiomyocyte death, such as with MI, ischemia reperfusion (I/R), or cryoablation. The fibrotic response generated from these injuries is vital for preventing ventricular rupture after massive myocyte loss. In contrast, reactive fibrosis usually results in fibrosis that accumulates in the interstitial space. In animal models, reactive fibrosis is observed in models of pressure overload or treatment with a cocktail of AngII and PE [37]. To date it is unclear whether the same cellular and molecular mechanisms underlie these different types of fibrosis. Thus, the results of these scRNAseq studies begin to elucidate the different substates that fibroblasts might occupy in response to different injuries. Moreover, robust time course and genetic perturbation studies will be needed in order to ascertain if the substates identified represent novel cell states with discernable functions or are transition states and represent cells undergoing transdifferentiation.
Figure 2: Models of Cardiac Fibroblast State Transitions.
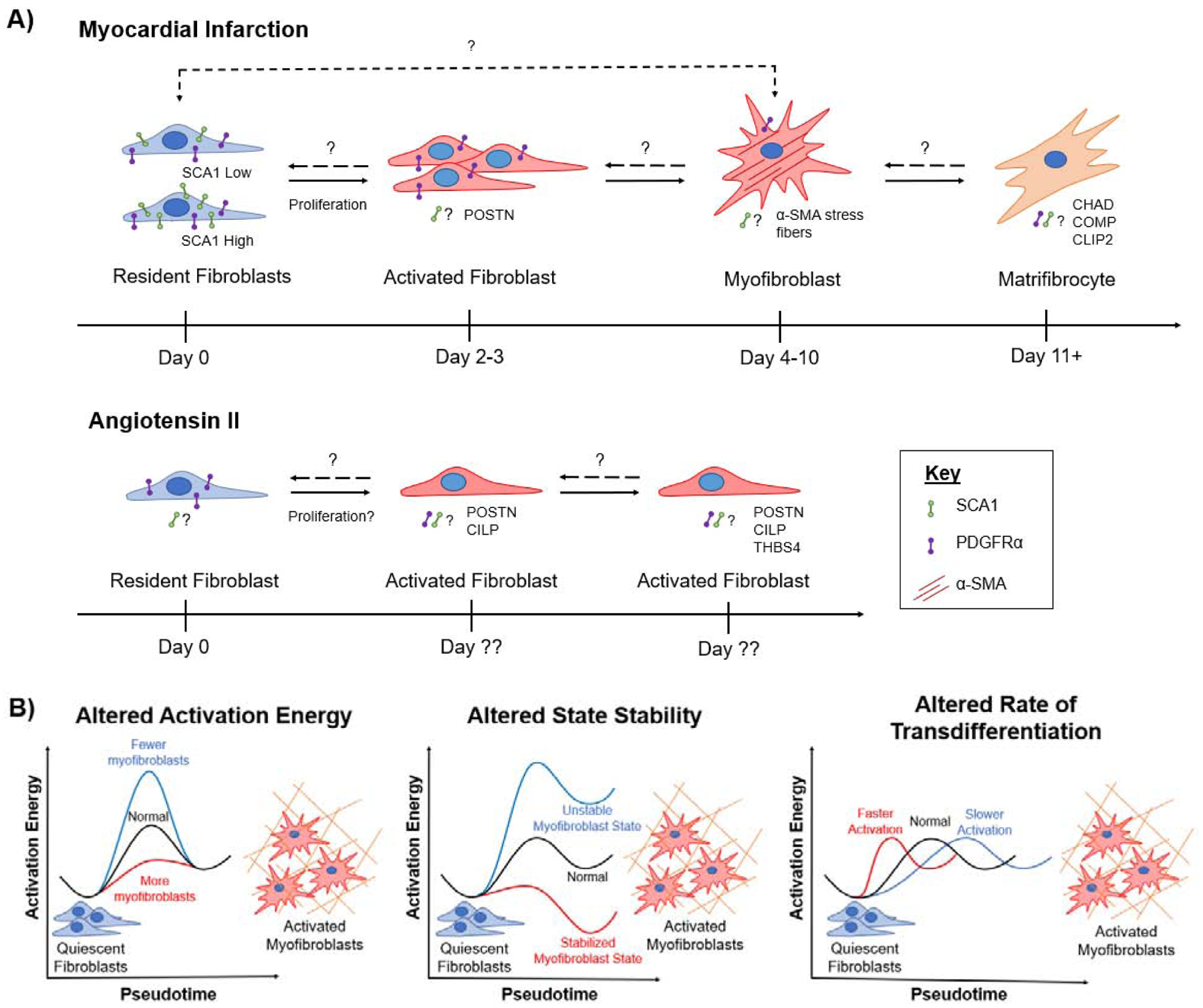
A) Differential Fibroblast Injury Response in Myocardial Infarction and Angiotensin-Infusion Injury Models. Recent RNAseq studies suggest that fibroblasts state transitions vary based on the injury stimulus. This figure summarizes what is known about fibroblast transdifferentiation in response to myocardial infarction and angiotensin II infusion, but also illustrates unknowns regarding: the proliferation phase in an Angiotensin model, the genotype (SCA1 high or low) of resident fibroblast population that transitions to a myofibroblast identity, and SCA1 expression as fibroblast undergo state transitions. B) State Space Modulation of Myofibroblast Identity. Perturbing genes in the fibrotic pathway can alter the fibroblast state space landscape, changing the number of α-SMA expressing myofibroblasts. Illustrations represent several hypothetical outcomes from fibroblast state space modulation.
These scRNAseq studies have also determined that intercellular communication between fibroblasts and other non-myocytes is critical for proper heart function [3–5]. By mapping expression of ligands and cognate receptors, these scRNAseq studies have started to elucidate these interactions. Skelly et al. found that fibroblasts are the most promiscuous cell type in the heart, with fibroblasts expressing ligands that target the other cardiac cell types and promote survival of specific cardiac cell populations. In vitro coculture experiments showed that fibroblast signaling is necessary but not sufficient for cardiac macrophage and endothelial cell growth [5]. McLellen et al. corroborated these results with their scRNAseq data set, showing that fibroblast signaling is crucial in the uninjured heart. Their analysis of ligand/receptor networks post AngII-induced hypertrophy showed an increase in communication signals between all cell types and an upregulation of signaling pathways associated with amino acid synthesis and collagen fibral organization. Downregulated signaling pathways in this study were enriched for inflammation and leukocyte/monocyte trafficking, indicating a suppression of the inflammatory response. In addition to direct signaling with other cell types, cardiac fibroblasts’ regulation of matrix structure and composition is an important signaling modality for other cell types in the heart. It has been shown that matrix composition—specifically the presence of the extracellular heparan sulfate proteoglycan agrin—can promote cardiomyocyte proliferation even in adult mammals [38]. Finally, fibroblasts can also interact with other cardiac cells through mechanotransduction. Coculture of macrophages and fibroblasts on a fibrillar collagen matrix showed that macrophages are attracted by the dynamic contraction of activated fibroblasts [39]. This could mean that αSMA positive contracting fibroblasts play a role in initiating the immune response post injury. Future work will need to continue elucidating the important role that fibroblasts play in intercellular signaling, both at baseline and in response to injury. The role cardiac fibroblasts play in signaling is at a nascent level of understanding. More in-depth reviews of communication between cardiac fibroblasts and other cell types can be found here [40,41].
Collectively, these scRNAseq studies demonstrate the limitations of the historic view of injury-induced fibroblast to myofibroblast transdifferentiation, underscoring the need for this paradigm to be expanded to include fibroblast and myofibroblast population heterogeneity, intermediate substates, and transient cell states. Each of these states have distinct patterns of gene expression and, presumably, different responsibilities within the injured myocardium, yet most of these studies lack true functional validation. This highlights a barrier in the field: integrating high dimensional data from transcriptome sequencing with protein expression and measurements of fibroblast structure and function. It is important to note that these scRNAseq experiments were carried out in mice—with the exception of McLellen et al., where findings were corroborated with human bulk RNAseq data [4]. Therefore, while it seems likely that the human fibroblast cell state landscape resembles that of the mouse fibroblast, the human fibroblast landscape still remains poorly characterized.
1.4. Reprograming and stem cell derivation of cardiac fibroblasts
The ability to reprogram fibroblasts to pluripotent stem cells with Yamanaka factors or to other cell types with myogenic transcription factors has also shed light on the stability and plasticity of differentiated fibroblasts and myofibroblasts [42,43]. The concept of reprogramming fibroblasts has been around since the 1980’s, when myogenic differentiation 1 (MyoD) was used to transdifferentiate fibroblasts into skeletal muscle cells [44]. Fibroblasts were successfully reprogrammed into cardiomyocytes using the three cardiac transcription factors Gata4, Mef2c, and Tbx5 (GMT) [45]. GMT has been used to reprogram fibroblasts both in vitro [45] and in vivo [46] with efficiencies of 10–15%. Later, reprograming efficiency both in vitro and in vivo was improved by the inclusion of the transcription factor Hand2 (GHMT) [47,48]. More recently in vitro reprogramming efficiencies of ~50–60% were achieved by blocking fibroblast activation pathways [49]. A major obstacle to reprogramming with transcription factors is that not all cells that express the reprogramming factors will undergo transdifferentiation [14,50]. The observation that inhibiting the myofibroblast phenotype dramatically improves reprograming drives home the idea that the cardiac fibroblast population is extremely heterogeneous and occupies an array of positions in state space at any given time [49]. The movement of reprogrammed cells through state space was recently observed using RNA velocity, reiterating the idea that these populations are dynamic [14]. Subpopulations of fibroblasts resistant to reprograming were identified, demonstrating that even cells thought to occupy similar positions in state space may have distinct activation energy requirements to move between states.
Reprogrammed cardiomyocytes are heterogenous, with some cells exhibiting highly organized sarcomeres and mature isoforms, while others have more disorganized sarcomeres and fetal isoform expression. To better understand the heterogeneity of these induced cardiomyocytes (iCM), single cell analysis was performed on fibroblasts converting to myocytes 3 days after GMT induction. Four cell states were identified: fibroblasts, intermediate fibroblasts, pre-iCM, and iCM. The pre-iCM state was identified as an unstable intermediary cell state expressing both fibroblast and cardiomyocyte markers. Proliferation was discovered to be a major delineating factor as to whether pre-iCM continued on to iCM or reverted back to a fibroblast state, with decreased proliferation enhancing reprogramming and vice versa. The splicing factor polypyrimidine tract-binding protein 1 (Ptpbp1) was also identified as an inhibitor to fibroblast transdifferentiation, with Ptpb1 inhibition greatly enhancing reprogramming efficiency [51]. Finally, since recent studies have carried out reprograming in both mouse and human cardiac fibroblasts [14], it seems likely that human and mouse cardiac fibroblast state space is similar. That being said, human fibroblasts have been more difficult to reprogram and need more reprogramming factors, suggesting that human fibroblasts are less plastic. Future research will need to elucidate exactly how the human cardiac fibroblast state space landscape differs from that of the mouse cardiac fibroblast.
The induced pluripotent stem cell (iPSC) field is also adding to our understanding of cardiac fibroblast biology, as protocols are developed for the directed differentiation of stem cells into a cardiac fibroblast lineage [52,53]. Metatranscriptomic analysis to place these iPSC derived fibroblasts in state space relative to the identified state spaces occupied by endogenous cardiac fibroblasts has not been done. This type of analysis, similar to what has been done for cardiac reprograming of fibroblasts [14], will elucidate how close these stem cell derived cells are to canonical cardiac fibroblasts. The complexity of the cardiac fibroblast lineage discussed above indicates that cardiac fibroblasts are more heterogeneous than once thought and hence could play varied roles in the heart. Therefore, iPSC derived fibroblasts will need to be defined in the complex state space of the endogenous cardiac fibroblast. Thus, modeling resident cardiac fibroblast cell states with iPSCs has proved challenging.
Robust lineage reporters for quiescent fibroblasts and myofibroblasts have dramatically moved forward the field of cardiac fibroblast biology. [10,24]. The next step forward is to use new single cell analysis tools to tease apart the genomic complexity behind any given lineage reporter. This will allow cardiac fibroblasts substates to be placed relative to each other in multidimensional state space (Figure 1). Lineage reporters permanently label a cell based on the expression of a single gene. While they are important for tracking a cells history, these cells can only be labeled once, limiting the identification of various molecular states a single cell might occupy throughout its differentiation trajectory. Therefore, development of a multi-lineage reporter may be useful for fully teasing apart fibroblast state dynamics. For now, scRNAseq experiments on isolated lineage-traced cells can begin to elucidate some of the complexities that underlie each cardiac fibroblast lineage [3,4].
2. Modulating Fibroblast Cell State to Alter Myocardial Fibrosis
Many factors must converge to drive cell state transitions. Injuries in the heart often involve several stimuli, such as ischemia, inflammation, or cell death, which overwhelmingly push cardiac fibroblasts towards activated states. Enhancing or inhibiting key genes can reshape the state space landscape, making it more or less likely to achieve a fibrotic fibroblast state. Therefore, these pathways can be leveraged to shift cardiac fibroblasts into more desirable cell states, mitigating fibrosis and halting maladaptive cardiac remodeling. The following section summarizes in vivo studies defining fundamental regulators of cardiac fibroblast state and how they have been therapeutically targeted to improve cardiac fibrosis and dysfunction.
2.1. TGFβ Pathway
TGFβ is a master regulator of the fibrotic response. The TGFβ receptor has two subunits: the type I and type II TGFβ receptors (Tgfbr1/2). In the canonical signaling pathway, binding of TGFβ to Tgfbr1/2 results in phosphorylation of the transcription factors Smad2 and Smad3. Once phosphorylated, these proteins complex with Smad4 in the cytoplasm, translocate to the nucleus, and promote a profibrotic genetic profile [54]. Modulating the expression of these proteins alters fibroblast cell state and, by extension, fibrosis in the heart due to injury.
In ischemic injuries, Smad3 expression is important for proper cardiac myofibroblast function, but does not appear to be a driver of the myofibroblast state. Knockdown of Smad3 increased αSMA expressing myofibroblast density and proliferation, but resulted in reduced [55] or disorganized collagen deposition [56]. Deletion in both the whole heart and Postn-expressing fibroblasts resulted in increased myofibroblast number [55,56], suggesting that Smad3 is not involved in initiation or maintenance of the myofibroblast state. However, myofibroblast morphology and alignment in the infarct area was impaired, and isolated fibroblasts exhibited decreases in collagen I and III [56], indicating that Smad3 is essential for mounting a proper fibrotic response to ischemic injuries. The mechanism behind the proliferative myofibroblast phenotype seen in these knockouts is unknown; it could be a direct effect of Smad3 deletion or secondary due to poor collagen deposition. In pressure overload models, Smad3 is also a critical regulator of myofibroblast function and state, with both global and fibroblast-specific Smad3 knockdown resulting in decreased interstitial fibrosis [54,57]. Smad2 knockout did not reduce fibrosis, suggesting Smad3 is the major driver of the fibrotic response in the canonical pathway. Deletion of Smad2 and Smad3 together (Smad2/3) decreased ECM gene expression, fibroblast proliferation, and αSMA positive myofibroblast number after transverse aortic constriction (TAC) [54]. These results suggest that Smad2/3 are important for fibroblast function, expansion, and induction of the αSMA positive myofibroblast state in pressure overload systems. The transcription factor Scleraxis has been identified as a key downstream effector of the Smad3-induced myofibroblast phenotype, with Scleraxis knockdown inhibiting the myofibroblast state [58,59]. While not studied in a disease context, decreased expression of Scleraxis may underlie the lack of myofibroblasts in Smad3 null mice. The opposing proliferative response of fibroblasts in ischemic and pressure overload models has not been studied. One possible explanation is the relative magnitudes of each injury: redundant fibrotic signaling pathways may take over in ischemic injuries to prevent ventricular rupture. This reiterates that fibroblasts likely have different cell states for responding to different injury stimuli. Notably, deletion of Smad2/3 did not confer any functional benefits. However, upstream knockdown of Tgfbr1/2 in fibroblasts decreased interstitial fibrosis, abrogated systolic and diastolic function, and improved cardiac hypertrophy [54]. While fibroblast specific effects were not investigated in this model, TGFβ signaling appears to affect heart function and hypertrophy through Smad2/3-independent pathways.
TGFβ signaling is not restricted to just fibroblasts. In pressure overloaded hearts, phosphorylated Smad3 has been found in the nuclei of cardiomyocytes and vascular cells, as well. Myocyte-specific deletion of Tgfbr2 reduced both pathological hypertrophic remodeling and interstitial fibrosis [60]. These data suggest that in pressure overload, myocyte TGFβ signaling also plays a role in defining fibroblast state, although exact fibroblast effects were not studied. In contrast, myocyte specific deletion of Smad2/3 in TAC mice did not reduce reactive fibrosis, indicating that the myocyte effect on fibroblast state may occur through secondary or non-canonical pathways [54]. For example, cardiomyocyte specific Smad4 knockout in uninjured mice changed sarcomere calcium sensitivity, causing these mice to develop a fibrotic, dilated cardiomyopathy (DCM) phenotype. This underscores how changes in myocyte health and function can alter fibroblast state.
2.2. p38 Mitogen Activated Protein Kinase (MAPK)
TGFβ can activate both canonical and non-canonical fibrosis signaling pathways. When the TGFβ ligand binds to its receptors, the MAPK pathways are also activated, including the p38 mitogen-activated protein kinase (p38), extracellular signal-regulated kinase (ERK), and c-Jun-N-terminal kinase (JNK) [54]. While little work has been done in vivo with ERK and JNK in the context of cardiac fibrosis or fibroblasts, p38 has been identified as a major regulator of the fibrotic response in the heart [13]. In fact, increasing evidence is demonstrating that these non-canonical pathways may play a significant role in mediating fibroblast state and the fibrotic response to injury [13,61].
p38 is a crucial regulator of the myofibroblast state. In ischemic injuries, targeted genetic deletion of the p38α isoform from both Tcf21 and Postn-expressing fibroblasts drastically decreased reparative fibrosis, leading to massive mortality due to ventricular rupture [13]. In contrast to Smad3 deletion in ischemic injuries [55,56], knockdown of p38α in both fibroblast cell states grossly decreased the number of αSMA expressing myofibroblasts in the infarct and border zones [13]. These results were replicated in an AngII/PE reactive fibrosis model. In a gain of function experiment, p38 was activated by creating a transgenic mouse where the p38 regulator, mitogen activated kinase kinase 6 (MKK6), was made constitutively active. This model resulted in increased cardiac fibrosis and number of αSMA positive myofibroblasts at baseline and in response to I/R and AngII/PE-induced injuries. These data suggest that p38α is a potent activator of the myofibroblast state, essential for both mounting and sustaining the matrix secreting αSMA positive myofibroblast phenotype [13]. In the context of state space, it is possible that p38 deletion increases the activation energy required for myofibroblast transdifferentiation; whereas, p38 overexpression decreases this activation energy, accounting for the respective changes in myofibroblast number (Figure 2b).
In addition to TGFβ-driven signaling, recent work suggests that p38 incorporates spatial cues in its determination of fibroblast state [12]. Integration of fibroblast lineage tracing and matrix alignment analyses revealed that Postn and αSMA expressing myofibroblasts are most commonly found in the border zone of MI hearts, where the ECM is most aligned. The center of the scar, often characterized by disordered collagen organization [62] exhibited lower myofibroblast density. Myofibroblast morphology correlated with matrix alignment: myofibroblasts found in the aligned border zone were highly elongated, while those found in the disorganized scar were more circular. These results were recapitulated in vitro by culturing isolated adult cardiac fibroblasts on biomimetic nanofabricated devices with random or aligned patterns. Fibroblasts cultured on the aligned patterns showed increased myofibroblast transdifferentiation and expression of matrix proteins than those cultured on random patterns. TGFβ failed to exacerbate the myofibroblast phenotype on aligned patterns, potentially suggesting that chemical and mechanical cues converge onto a single pathway to induce myofibroblast transdifferentiation. Deletion of p38 abrogated pattern-induced αSMA positive myofibroblast transdifferentiation [12]. Moreover, alignment initiated p38-mediated stabilization of the yes-associated protein (YAP), a pathway that is highly studied in the context of cancer [63]. Transduction of a constitutively active nuclear YAP was sufficient to activate the myofibroblast cell state. These data suggest that p38 integrates both spatial and chemical cues to mediate fibroblast cell state [12].
2.3. Serum Response Factor (SRF), Transient Receptor Potential Channels (TRPC), and YAP
Multiple pathways have been found to converge or intersect with the p38 pathway, including the myocardin-related transcription factors-serum response factor (MRTF-SRF) axis, transient receptor potential cation channel subfamily C member 6 (TRPC6), and YAP. The transcription factor SRF is essential for vascular smooth muscle cell (SMC) differentiation [64] and has been implicated in inducing the contractile, SMC-like genetic profile in myofibroblasts. SRF binds to a promotor sequence known as the serum response element (also known as the CArG box) with the help of mechanosensitive transcription co-factors, such as MRTF-A and MRTF-B. The activity of these co-factors is highly dependent on actin dynamics, and is therefore regulated by Rho GTPases [65]. This signaling axis is known to activate due to injury, changes in mechanical forces and cell shape, or with TGFβ signaling (reviewed in [66]), highlighting its connection to the Smad and p38 pathways.
MRTF-A and TRPC6 are downstream effectors important for induction of the myofibroblast state. Global deletion of MRTF-A post-MI decreased fibrosis, non-myocyte proliferation, and overall number of αSMA positive fibroblasts, indicating that MRTF-A is necessary for the αSMA positive myofibroblast phenotype [65]. Transient receptor potential canonical family member 6 (TRPC6) has been identified as an essential target of SRF through the p38 pathway [61]. Previously linked to pathological hypertrophic remodeling, TRPC6 upregulation is known to activate calcineurin and nuclear factor of activated T cells (NFAT) [67]. Global knockout of TRPC6 in MI mice caused higher incidence of ventricular rupture, decreased function, and reduced collagen deposition. Mechanistic studies in vitro with isolated cardiac fibroblasts demonstrated that TRPC6 expression is directly upregulated by SRF and can be inhibited with p38 inhibitors. Moreover, TRPC6 overexpression was a potent activator of myofibroblasts, and TRPC6 knockout abrogated TGFβ-mediated myofibroblast transdifferentiation [61]. Together these data demonstrate that TRPC6 is a crucial downstream effector of p38-dependent transitions of fibroblasts into fibrosis-producing myofibroblasts.
YAP inhibition is essential for fibroblast specification during development and maintenance of the quiescent fibroblast cell state in adults. YAP is a transcriptional coactivator in the Hippo pathway that is negatively regulated by the kinases Lats1 and Lats 2 (Lats1/2) [63]. Deletion of Lats1/2 from the developing mouse heart prohibited epicardial differentiation into mature cardiac fibroblasts [11]. Deletion of Lats1/2 in adult cardiac fibroblasts destabilized the quiescent fibroblast state, causing spontaneous myofibroblast transdifferentiation (Figure 2b). Lats1/2 knockout mice had gross increases in fibrosis, both at baseline and with MI injury [63]. These data suggest that Lats1/2-dependent YAP inhibition is crucial for maintaining the quiescent fibroblast state. YAP signaling intersects with both p38 and SRF pathways. MRTF-A mRNA and protein levels have been shown to directly correlate with YAP expression. In MI, deletion of YAP from Tcf21 and Col1a1-expressing fibroblasts decreased collagen deposition, fibroblast proliferation, and αSMA positive fibroblast activation. Similar decreases in fibrosis were seen in AngII/PE-treated mice with loss of YAP function in Tcf21 fibroblasts. MRTF-A levels were also attenuated in the YAP knockouts, suggesting that YAP is required for MRTF-A activity [68]. These results demonstrate the importance of YAP/MRTF-A signaling in the myofibroblast state transition in response to ischemic and chronic injuries. In the p38 pathway, YAP appears to be a crucial component for transduction of mechanical signals via focal adhesions [12,69]. Notably, knockdown of YAP in fibroblasts did not increased mortality due to ischemic injury [68], indicating that other fibrotic signaling pathways may be responsible for the reparative fibrosis response, although this may also be an artifact of increased myocyte survival. In contrast, myocyte specific deletion of YAP resulted in dramatically increased fibrosis and mortality post-MI [70] and neonatal deletion of YAP in cardiomyocytes impaired regeneration [71]. The differential effects of YAP signaling in myocytes and fibroblasts again highlights how myocyte health can affect fibroblast cell state, but also underscores the level of specificity required to target this pathway therapeutically.
Collectively, these studies demonstrate that manipulating the expression of key proteins in the fibrotic cascade can alter fibroblast state space dynamics, resulting in altered global fibrosis. However, how state space is altered is still a major unknown for the field. This section reviewed how pathway manipulations can alter myofibroblast density in the myocardium. Based on this data, there appear to be at least three potential mechanisms for state space modulation: (1) a change in the activation energy required to reach a myofibroblast state, (2) a change in quiescent or activated fibroblast state stability, or (3) a change in the rate of myofibroblast transdifferentiation. With respect to the first mechanism, altered activation energy by p38 inhibition or enhanced activity likely alters the activation energy required to achieve a myofibroblast state. Because p38 inhibition results in dramatically fewer myofibroblasts in response to injury [13], it’s postulated that loss of p38 function increased the activation energy needed to achieve a myofibroblast state. In contrast, p38 activation by MKK6 resulted in spontaneous myofibroblast transdifferentiation without an injury stimulus [13], suggesting that this perturbation to p38 signaling lowered the activation energy, making the myofibroblast transition easier (Figure 2b). For the second mechanism (altered stability), we postulate that YAP regulates fibroblast and myofibroblast state stability because models with increased YAP activity (via Lats1/2 knockdown) caused a mass activation of myofibroblasts in both uninjured and infarcted mouse hearts [63]. Conversely, YAP inhibition decreased myofibroblast transdifferentiation [68]. Hence a reasonable hypothesis is that YAP inhibition stabilizes the quiescent fibroblast state while YAP activity stabilizes the myofibroblast state. (Figure 2b). With respect to the third mechanistic model (altered rates of transdifferentiation), there is minimal evidence to support this mechanism in the cardiac literature, but dermal wound closure which is mediated by myofibroblasts occurred much slower in TRPC6 knockout mice [61]. This suggest that modulations of some molecular regulators only slow down or speed up the transition to a myofibroblast state, without entirely inhibiting transdifferentiation (Figure 2b). However, the mechanisms for these changes in state space landscape are hypothetical and of course could include some combination of the mechanisms proposed here. Moreover, these mechanisms are simplified to only the conversion of resident fibroblasts to myofibroblasts, and a more robust analysis may need to examine the state transitions in between. For example, Scleraxis and Pdgfrα have been implicated in forming and maintaining resident cardiac fibroblasts [32,58]. This suggests that there are key regulators to every fibroblast state and demonstrates that the complexity of fibroblast state space transcends myofibroblast transdifferentiation. To truly begin elucidating how these regulators control state space, rigorous scRNAseq experimentation must be incorporated into pathway manipulation studies, although these experiments may be cost prohibitive. Adoption of analyses from the reprogramming field may be beneficial; for example, free energy calculations may be helpful for determining relative state stabilities [51]. RNA velocity and pseudotime calculations may be useful for characterizing state transitions and differentiation rates [3,4]. Other experiments, analyses, and calculations may yet need to be developed to be able to make robust state space determinations. For example, the field currently lacks methods for integrating multiomic transcriptional and proteomic changes. While scRNAseq will begin to uncover information about state space, these data must be validated with protein-based and physiologic assays. Ultimately, a better understanding of the regulators of the various fibroblast states, and how their modulation affects global cardiac fibrosis, will enable therapeutic targeting of specific states to inhibit maladaptive cardiac remodeling.
3. Therapeutics
Targeted genetic manipulation is essential for identifying the cell-specific role of a given signaling pathway, and these data can be leveraged to develop antifibrotic therapies for patients. In some cases, improvements in fibrosis do not always beget improvements in function. This could be because the myocytes are damaged to a point of no return, where lessened fibrosis cannot improve function. Alternatively, systems with less fibrosis could also have a fundamentally different type of fibrosis, with different biochemical composition, architecture, or organization resulting in an overall stiffer heart despite fibrotic attenuation. Hence alterations in the quantity of fibrosis may not be the proper index for evaluating the impact of a given therapeutic. To complicate matters further, human clinical trials don’t typically use fibrosis as an endpoint, despite advancements in non-invasive assessments of fibrosis by magnetic resonance imaging (MRI) with late gadolinium enhancement [72,73]. Instead, studies often rely on functional metrics as endpoints, even when testing antifibrotics. This makes it especially hard to determine how a given treatment is directly altering human cardiac fibrosis, let alone fibroblast state space. Therefore, the following section and Table 1 summarizes pre-clinical and clinical data from inhibiting two regulators, p38 and YAP, that show promise for altering global fibrosis and function through cardiac fibroblast state modulation. In addition, this section reviews fibroblast reprogramming approaches as a therapeutic.
Table 1: Summary of pre-clinical and clinical studies on P38 and YAP inhibitors.
The p38 inhibitors summarized here are all from studies within cardiac systems, whereas the YAP inhibitors are from a mix of renal, hepatic, and pulmonary systems.
| Paper/ Clinical Trial | Therapeutic(s) | Species | Injury Model | Result |
|---|---|---|---|---|
| Kyoi et al., 2006 [74] | SB203580* FR167653* |
Hamster | Dilated Cardiomyopathy | Decreased cardiac fibrosis, myocyte death, dilation, and hypertrophy. |
| See et al., 2004 [75] | RWJ-67657* | Rat | Myocardial Infarction | Improved systolic function; decreased dilation, αSMA protein expression, and collagen I deposition; enhanced myocyte hypertrophy. |
| Yamagami et al., 2015 [76] | Pirfenidone* | Mouse | Pressure Overload | Improved systolic function; decreased dilation and interstitial fibrosis. |
| Nguyen et al., 2010 [77] | Pirfenidone* | Rat | Myocardial Infarction | Improved systolic function; decreased fibrosis; decreased frequency of ventricular tachycardia; decreased fibrosis in the infarct zone, border zone, and non-infarct area of the heart. |
| Wang et al., 2013 [78] | Pirfenidone* | Mouse | Pressure Overload | Decreased mortality and cardiac fibrosis. |
| Yamazaki et al., 2012 [79] | Pirfenidone* | Mouse | Angiotensin II | Decreased hypertrophy and cardiac interstitial and perivascular fibrosis. |
| Newby et al., 2014 [80] NCT00910962 | Losmapimod* | Human Phase II | Myocardial Infarction | 535 patients: no increase in adverse cardiac reactions; decreased B-type natriuretic peptide concentrations after 12 weeks. |
| O’Donoghue et al., 2016 [81] NCT02145468 | Losmapimod* | Human Phase III | Myocardial Infarction | 3503 patients: no improvements in frequency of adverse cardiac events (e.g. sudden cardiac death, recurrent MI). |
| Muchir et al., 2012 [82] | ARRY-371797* | Mouse | Dilated Cardiomyopathy | Improved fractional shortening; decreased dilation and αSMA gene expression; no change in Col1a1 and Col1a2 gene expression. |
| NCT03439514 | ARRY-371797* | Human Phase III | Dilated Cardiomyopathy | Ongoing- set to be completed in 2024. |
| Liang et al., 2017 [84] | Verteporfin# | Mouse | Unilateral Ureteral Obstruction | Decreased renal interstitial fibrosis and number of αSMA expressing myofibroblasts. |
| Szeto et al., 2016 [85] | Verteporfin# | Mouse | Unilateral Ureteral Obstruction | Decreased renal interstitial fibrosis, number of αSMA expressing myofibroblasts, and Smad2/3 nuclearization. |
| Santos et al., 2020 [86] | Simvastatin# | Mouse | Bleomycin-induced Pulmonary Fibrosis | Decreased pulmonary interstitial fibrosis and collagen deposition. |
| Haak et al., 2019 [87] | Dihydrexidine# | Mouse | Bleomycin-induced Pulmonary Fibrosis Bile duct ligation |
Decreased pulmonary fibrosis and αSMA fluorescent intensity. Decreased hepatic fibrosis and αSMA fluorescent intensity. |
p38 inhibitor;
YAP inhibitor
3.1. p38 Inhibitors
p38 inhibitors are a promising class of drugs for reducing fibrosis and improving heart function. However, timing of p38-targeted therapeutics is crucial. In mouse models, early p38α deletion in Tcf21 expressing fibroblasts post-MI resulted in more dramatic decreases in fibrosis, improvements in function, and increases in mortality rates, compared to deletion from activating Postn positive fibroblasts [13]. This suggests p38 inhibition could be a double-edged sword: if administered too early, they may cause ventricular rupture, but if given too late, fibrosis and dysfunction may not be adequately attenuated. In a DCM hamster model, chronic administration of the p38 inhibitors SB203580 or FR167653 improved systolic function and decreased fibrosis [74]. Rats given the p38 inhibitor RWJ-67657 7 days after MI demonstrated decreased remote fibrosis and αSMA protein expression, but only moderate improvements in function [75]. Pirfenidone is a small molecule drug that has recently been approved to treat lung fibrosis. While pirfenidone has several proposed mechanisms of action, it has also been implicated in targeting TGFβ signaling and inhibiting p38 phosphorylation [76]. In rodents, pirfenidone has decreased cardiac fibrosis in response to MI [77], pressure overload [76,78], and AngII-induced injuries [79]. While pirfenidone was able to halt fibrosis progression in a chronic pressure overload model, it was not able to reverse existing fibrotic remodeling, highlighting the importance of preventative treatment [76]. In humans, a couple of anti-inflammatory p38 inhibitors have started clinical trials. Despite promising phase II clinical data (NCT00910962) [80], a large phase III clinical trial testing the effects of Losmapimod, a competitive inhibitor of p38, failed to improve adverse outcomes after MI (NCT02145468) [81]. Array BioPharma, a subsidiary of Pfizer, recently started a phase III clinical trial to investigate the effects of the anti-inflammatory and p38-inhibitor ARRY-371797 on DCM patients with mutations in the laminin A/C gene (NCT03439514), but this study will not be completed until 2024. This trial is supported by preclinical data in a mouse model with this laminin mutation, where treatment with ARRY-371797 decreased dilation and systolic dysfunction [82].
3.2. YAP Inhibitors
The YAP pathway has been identified as a key target for reducing cancer resistance to chemotherapies. Therefore, YAP inhibitors have been investigated thoroughly in this context (reviewed in [83]). However, given recent data proposing YAP as a potent activator of the fibrotic cascade [63,68], it may be helpful to review promising YAP inhibitors that could be applied to cardiac fibrosis. Verteporfin, a drug widely studied for treating macular degeneration, was recently found to have YAP inhibition properties. In the kidney, verteporfin treatment of mice with unilateral ureteral obstruction decreased interstitial fibrosis, αSMA expressing myofibroblast number, and Smad2/3 nuclearization [84,85]. In human isolated lung fibroblasts, a high-throughput screen of >13000 small molecules identified statins as potent YAP inhibitors, potentially explaining why patients with interstitial lung disease had better outcomes on statins. Simvastatin decreased established lung fibrosis in the bleomycin mouse model in a Lats1/2 independent manner [86]. Inhibition of the dopamine receptor D1 (DRD1), which couples to G-protein receptors, via dihydrexidine (DHX) disrupted YAP signaling in isolated fibroblasts and reduced lung and liver fibrosis in bleomycin and bile duct ligation models, respectively. DHX was also found to inhibit YAP nuclearization in isolated cardiac fibroblasts, although its use has not been tested in an in vivo cardiac fibrosis model [87]. Given that these YAP inhibitors have shown promise in other fibrotic systems, they may be useful for combatting cardiac fibrosis, as well.
3.3. Fibroblast Reprogramming
Reprogramming fibroblasts into cardiomyocytes is an enticing treatment for patients with ischemic heart disease who have lost a significant fraction of myocytes. The central hypothesis is that reprogrammed fibroblasts could replace the cells that were lost, improving function while also reducing fibrosis. In mice, injection of GMT factors after MI improved systolic function, reduced collagen deposition, and reduced fibrotic gene expression in the border zone. This method produces varying levels of maturity within iCM, with only half of the cells exhibiting organized sarcomeres and mitochondria [46].
However, translating these results to humans has proved difficult. It was quickly discovered that GMT alone was insufficient to reprogram human cardiac fibroblasts into cardiomyocytes, and that other factors and mRNAs were needed [88,89]. This suggests that there are species-related differences in cardiac fibroblast plasticity, with human fibroblasts having extra epigenetic regulation preventing reprograming. Although murine in vivo reprogramming has proven to be more robust than in vitro [47], in vivo reprogramming has not been demonstrated in large animal models or non-human primates. Moreover, no studies have investigated how removing a subset of fibroblasts affects long-term matrix homeostasis or response to future cardiac injuries. Given that mechanisms for sensing or compensating for low fibroblast numbers are unknown [32] and that fibroblasts provide additional roles in wound healing beyond matrix secretion [5], reprogramming these fibroblasts into cardiomyocytes may cripple the heart’s ability to maintain matrix homeostasis or offset future wound healing responses. These unknowns must be addressed before GMT can translate to humans.
4. Summary and Remaining Questions
4.1. Summary
Recent advances in lineage tracing reporters and scRNAseq has greatly expanded our understanding of fibroblast states and substates during injury-induced transdifferentiation. While fibroblasts were previously thought to have a unidirectional, linear transdifferentiation trajectory, these recent studies demonstrate that fibroblast state is more dynamic. scRNAseq studies have identified new fibroblast cell states and added complexity to the existing fibroblast transdifferentiation paradigm. Knowledge of myofibroblast fate has expanded, with studies demonstrating deactivation or progression into long-term, mature states (e.g. matrifibrocytes). Therefore, viewing fibroblast state changes according to the dynamic cellular state space model may allow for better visualization of the complexity of fibroblast cell states. This model may also allow for better understanding of how pathway perturbations can alter the fibroblast response to injury. By fully understanding how each pathway can modulate fibroblast state, therapeutics may be able to leverage cell state transitions to reduce pathological fibrosis in the heart.
4.2. Remaining Questions
There are many lingering gaps in knowledge surrounding fibroblast state and transdifferentiation trajectories. While scRNAseq studies have identified many new potential fibroblast cell states, the stability of these states and their physiological roles in cardiac homeostasis and scar formation are unknown. More time course scRNAseq studies are needed to determine the relative stability of these states and distinguish persistent cell states from transition states. These studies may also enable a better understanding of state space modulation, such as with the mechanisms proposed in Figure 2b. Moreover, these states need proteomic validation. The role of post-injury stable fibroblast states, such as the matrifibrocyte, in maintaining long-term matrix homeostasis is unknown. To answer this question, information about the scar microenvironment (e.g. stiffness, matrix content, and cellular composition) will need to be integrated with scRNAseq data to elucidate the function of homeostatic feedback loops between cardiac matrix proteins, cardiac fibroblasts, and other cardiac cell types. Once this is known, the field can begin to tease apart what role these fibroblast states play in chronic cardiac remodeling in heart failure. Satisfying these gaps in knowledge should enable targeted manipulation of the “activation energy” for fibroblast cell state transitions to prevent or slow the progression to heart failure. Along similar lines, it will be critical to connect the fibroblast state space model to the rest of the cell types in the heart. Crosstalk between cardiac cell types appears to be critical for regulation of wound healing and the inflammatory response. Ultimately, the therapeutics discussed in this review only halted progression of cardiac fibrosis, but never reversed it, thus highlighting that either 1) treatments must be administered prophylactically, or 2) novel therapeutics need to be developed to control fibroblast cell state transitions.
Highlights:
Cardiac fibroblast cell state is more dynamic and complex than previously thought.
Fibroblast state space can be altered by modulating TGFβ-related pathways.
Changes in fibroblast state modify the global fibrotic response to cardiac injury.
Fibroblast cell state may be a viable target for therapeutic intervention.
YAP and p38 inhibitors have targeted therapeutic potential for modulating fibroblast state space.
Acknowledgements
This work was supported by grants from the National Institutes of Health for JD (HL141187 & HL142624) and MR (HL128368 and P30AR074990), and a Graduate Research Fellowship from the National Science Foundation for IMR (DGE-1762114).
Abbreviations
- Acta2 or αSMA
Alpha smooth muscle actin
- AngII
Angiotensin II
- ATACseq
Assay for transposase-accessible chromatin using sequencing
- Ccn2
Cellular communication network factor 2
- Ccn5
Cellular communication network factor 5
- Cd14
Cluster of differentiation 14
- CHIPseq
Chromatin immunoprecipitation sequencing
- Col1a1
Collagen type I alpha 1 chain
- ECM
Extracellular matrix
- Fcrg1
High affinity immunoglobulin gamma Fc receptor I
- Fn-EDA
Fibronectin containing extra domain A
- GHMT
Gata4 (G) Hand2 (H) Mef2c (M) and Tbx5 (T)
- GMT
Gata4 (G) Mef2c (M) and Tbx5 (T)
- I/R
Ischemia reperfusion
- iCM
Induced cardiomyocyte
- MI
Myocardial infarction
- MRTF-SRF
Myocardin-related transcription factors-Serum response factor
- MyoD
Myogenic differentiation 1
- p38 MAPK
p38 mitogen-activated protein kinase
- Pdgfrα
Platelet-derived growth factor receptor α
- PE
Phenylephrine
- Postn
Periostin
- Ptpbp1
Polypyrimidine tract-binding protein 1
- Ptprc
Protein tyrosine phosphatase receptor type C
- Sca1
Spinocerebellar ataxia 1
- scRNAseq
Single cell RNA sequencing
- Sfrp2
secreted frizzled related protein 2
- TAC
transverse aortic constriction
- Tcf21
Transcription factor 21
- TGFβ
Transforming growth factor beta
- Thbs4
Thrombospondin 4
- TRPC6
Transient receptor potential cation channel subfamily C member 6
- Wif1
Wnt inhibitory factor 1
- WNT
Wingless-related integration site
- YAP
Yes-associated protein
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Declaration of Competing Interest
None
References
- [1].Tallquist MD, Molkentin JD, Redefining the identity of cardiac fibroblasts, Nat. Rev. Cardiol 14 (2017) 484–491. 10.1038/nrcardio.2017.57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Fu X, Khalil H, Kanisicak O, Boyer JG, Vagnozzi RJ, Maliken BD, Sargent MA, Prasad V, Valiente-Alandi I, Blaxall BC, Molkentin JD, Specialized fibroblast differentiated states underlie scar formation in the infarcted mouse heart, J. Clin. Invest 128 (2018) 2127–2143. 10.1172/JCI98215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Farbehi N, Patrick R, Dorison A, Xaymardan M, Janbandhu V, Wystub-Lis K, Ho JWK, Nordon RE, Harvey RP, Single-cell expression profiling reveals dynamic flux of cardiac stromal, vascular and immune cells in health and injury, Elife. 8 (2019) e43882 10.7554/eLife.43882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].McLellan MA, Skelly DA, Dona MSI, Squiers GT, Farrugia GE, Gaynor TL, Cohen CD, Pandey R, Diep H, Vinh A, Rosenthal NA, Pinto AR, High-Resolution Transcriptomic Profiling of the Heart During Chronic Stress Reveals Cellular Drivers of Cardiac Fibrosis and Hypertrophy, Circulation. (2020). 10.1161/circulationaha.119.045115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Skelly DA, Squiers GT, McLellan MA, Bolisetty MT, Robson P, Rosenthal NA, Pinto AR, Single-Cell Transcriptional Profiling Reveals Cellular Diversity and Intercommunication in the Mouse Heart, Cell Rep. 22 (2018) 600–610. 10.1016/j.celrep.2017.12.072. [DOI] [PubMed] [Google Scholar]
- [6].Waddington CH, The Principles of Embryology, 1st Editio, Routledge, 2017. https://www.amazon.com/Principles-Embryology-Routledge-Revivals-Waddingtonebook/dp/B0744P5QYD/ref=sr_1_1?dchild=1&keywords=Waddington%2C+CH.+Principles+of+embryology.&qid=1600057668&s=books&sr=1-1. [Google Scholar]
- [7].Fagan MB, Waddington redux: Models and explanation in stem cell and systems biology, Biol. Philos 27 (2012) 179–213. 10.1007/s10539-011-9294-y. [DOI] [Google Scholar]
- [8].Marr C, Zhou JX, Huang S, Single-cell gene expression profiling and cell state dynamics: Collecting data, correlating data points and connecting the dots, Curr. Opin. Biotechnol 39 (2016) 207–214. 10.1016/j.copbio.2016.04.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Bretherton R, Bugg D, Olszewski E, Davis J, Regulators of cardiac fibroblast cell state, Matrix Biol. (2020). 10.1016/j.matbio.2020.04.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Kanisicak O, Khalil H, Ivey MJ, Karch J, Maliken BD, Correll RN, Brody MJ, Lin S-CJ, Aronow BJ, Tallquist MD, Molkentin JD, Genetic lineage tracing defines myofibroblast origin and function in the injured heart, Nat. Commun 7 (2016) 12260 10.1038/ncomms12260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Xiao Y, Hill MC, Zhang M, Martin TJ, Morikawa Y, Moise AR, Wythe JD, Martin JF, Hippo signaling plays an essential role in cell state transitions during cardiac fibroblast development, Dev. Cell 45 (2019) 153–169. 10.1016/j.devcel.2018.03.019.Hippo. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Bugg D, Bretherton RC, Kim P, Olszewski E, Nagle A, Schumacher AE, Chu N, Gunaje J, DeForest CA, Stevens K, Kim D-H, Davis JM, Infarct Collagen Topography Regulates Fibroblast Fate Via p38-Yap-TEAD Signals, Circ. Res (2020) 1306–1322. 10.1161/circresaha.119.316162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Molkentin JD, Bugg D, Ghearing N, Dorn LE, Kim P, Sargent MA, Gunaje J, Otsu K, Davis J, Fibroblast-specific genetic manipulation of p38 MAPK in vivo reveals its central regulatory role in fibrosis, Circulation. 136 (2018) 549–561. 10.1161/CIRCULATIONAHA.116.026238.Fibroblast-specific. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Zhou Y, Liu Z, Welch JD, Gao X, Wang L, Garbutt T, Keepers B, Ma H, Prins JF, Shen W, Liu J, Qian L, Single-Cell Transcriptomic Analyses of Cell Fate Transitions during Human Cardiac Reprogramming, Cell Stem Cell. 25 (2019) 149–164.e9. 10.1016/j.stem.2019.05.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Cao J, Zhou W, Steemers F, Trapnell C, Shendure J, Sci-fate characterizes the dynamics of gene expression in single cells, Nat. Biotechnol 38 (2020) 980–988. 10.1038/s41587-020-0480-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Olsson A, Venkatasubramanian M, Chaudhri VK, Aronow BJ, Salomonis N, Singh H, Grimes HL, Single-cell analysis of mixed-lineage states leading to a binary cell fate choice, Nature. 537 (2016) 698–702. 10.1038/nature19348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Mojtahedi M, Skupin A, Zhou J, Castaño IG, Leong-Quong RYY, Chang H, Trachana K, Giuliani A, Huang S, Cell Fate Decision as High-Dimensional Critical State Transition, PLoS Biol. 14 (2016) 1–28. 10.1371/journal.pbio.2000640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Hinz B, Phan SH, Thannickal VJ, Galli A, Bochaton-Piallat ML, Gabbiani G, The myofibroblast: One function, multiple origins, Am. J. Pathol 170 (2007) 1807–1816. 10.2353/ajpath.2007.070112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Crawford JR, Haudek SB, Cieslik KA, Trial J, Entman ML, Origin of developmental precursors dictates the pathophysiologic role of cardiac fibroblasts, J. Cardiovasc. Transl. Res 5 (2012) 749–759. 10.1007/s12265-012-9402-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Haudek SB, Cheng J, Du J, Wang Y, Hermosillo-Rodriguez J, Trial JA, Taffet GE, Entman ML, Monocytic fibroblast precursors mediate fibrosis in angiotensin-II-induced cardiac hypertrophy, J. Mol. Cell. Cardiol 49 (2010) 499–507. 10.1016/j.yjmcc.2010.05.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Haudek SB, Trial JA, Xia Y, Gupta D, Pilling D, Entman ML, Fc receptor engagement mediates differentiation of cardiac fibroblast precursor cells, Proc. Natl. Acad. Sci. U. S. A 105 (2008) 10179–10184. 10.1073/pnas.0804910105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Haudek SB, Xia Y, Huebener P, Lee JM, Carlson S, Crawford JR, Pilling D, Gomer RH, Trial JA, Frangogiannis NG, Entman ML, Bone marrow-derived fibroblast precursors mediate ischemic cardiomyopathy in mice, Proc. Natl. Acad. Sci. U. S. A 103 (2006) 18284–18289. 10.1073/pnas.0608799103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Moeller A, Gilpin SE, Ask K, Cox G, Cook D, Gauldie J, Margetts PJ, Farkas L, Dobranowski J, Boylan C, O’Byrne PM, Strieter RM, Kolb M, Circulating fibrocytes are an indicator of poor prognosis in idiopathic pulmonary fibrosis, Am. J. Respir. Crit. Care Med 179 (2009) 588–594. 10.1164/rccm.200810-1534OC. [DOI] [PubMed] [Google Scholar]
- [24].Acharya A, Baek ST, Huang G, Eskiocak B, Goetsch S, Sung CY, Banfi S, Sauer MF, Olsen GS, Duffield JS, Olson EN, Tallquist MD, The bHLH transcription factor Tcf21 is required for lineage-specific EMT of cardiac fibroblast progenitors, Development. 139 (2012) 2139–2149. 10.1242/dev.079970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Moore-Morris T, Guimarães-Camboa N, Banerjee I, Zambon AC, Kisseleva T, Velayoudon A, Stallcup WB, Gu Y, Dalton ND, Cedenilla M, Gomez-Amaro R, Zhou B, Brenner DA, Peterson KL, Chen J, Evans SM, Resident fibroblast lineages mediate pressure overload-induced cardiac fibrosis, J. Clin. Invest 124 (2014) 2921–2934. 10.1172/JCI74783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Furtado MB, Costa MW, Pranoto EA, Salimova E, Pinto AR, Lam NT, Park A, Snider P, Chandran A, Harvey RP, Boyd R, Conway SJ, Pearson J, Kaye DM, Rosenthal NA, Cardiogenic genes expressed in cardiac fibroblasts contribute to heart development and repair, Circ. Res 114 (2014) 1422–1434. 10.1161/CIRCRESAHA.114.302530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Ali SR, Ranjbarvaziri S, Talkhabi M, Zhao P, Subat A, Hojjat A, Kamran P, Müller AMS, Volz KS, Tang Z, Red-Horse K, Ardehali R, Developmental heterogeneity of cardiac fibroblasts does not predict pathological proliferation and activation, Circ. Res 115 (2014) 625–635. 10.1161/CIRCRESAHA.115.303794. [DOI] [PubMed] [Google Scholar]
- [28].Pinto AR, Ilinykh A, Ivey MJ, Kuwabara JT, D’Antoni ML, Debuque R, Chandran A, Wang L, Arora K, Rosenthal NA, Tallquist MD, Revisiting Cardiac Cellular Composition, Circ. Res 118 (2016) 400–409. 10.1161/CIRCRESAHA.115.307778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Kaur H, Takefuji M, Ngai CY, Carvalho J, Bayer J, Wietelmann A, Poetsch A, Hoelper S, Conway SJ, Möllmann H, Looso M, Troidl C, Offermanns S, Wettschureck N, Targeted Ablation of Periostin-Expressing Activated Fibroblasts Prevents Adverse Cardiac Remodeling in Mice, Circ. Res 118 (2016) 1906–1917. 10.1161/CIRCRESAHA.116.308643. [DOI] [PubMed] [Google Scholar]
- [30].Tomasek JJ, Gabbiani G, Hinz B, Chaponnier C, Brown RA, Myofibroblasts and mechano-regulation of connective tissue remodelling, Nat. Rev. Mol. Cell Biol 3 (2002) 349–363. 10.1038/nrm809. [DOI] [PubMed] [Google Scholar]
- [31].Dhanesha N, Chorawala MR, Jain M, Bhalla A, Thedens D, Nayak M, Doddapattar P, Chauhan AK, Fn-EDA (Fibronectin Containing Extra Domain A) in the Plasma, but Not Endothelial Cells, Exacerbates Stroke Outcome by Promoting Thrombo-Inflammation, Stroke. 50 (2019) 1201–1209. 10.1161/STROKEAHA.118.023697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Ivey MJ, Kuwabara JT, Riggsbee KL, Tallquist MD, Platelet derived growth factor receptor alpha is essential for cardiac fibroblast survival, Am. J. Physiol. Circ. Physiol (2019). 10.1152/ajpheart.00054.2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Takemura G, Ohno M, Hayakawa Y, Misao J, Kanoh M, Ohno A, Uno Y, Minatoguchi S, Fujiwara T, Fujiwara H, Role of Apoptosis in the Disappearance of Infiltrated and Proliferated Interstitial Cells After Myocardial Infarction, Circ. Res 82 (1998) 1130–1138. 10.1161/01.RES.82.11.1130. [DOI] [PubMed] [Google Scholar]
- [34].Zhao W, Lu L, Chen SS, Sun Y, Temporal and spatial characteristics of apoptosis in the infarcted rat heart, Biochem. Biophys. Res. Commun 325 (2004) 605–611. 10.1016/j.bbrc.2004.10.064. [DOI] [PubMed] [Google Scholar]
- [35].Coppé JP, Desprez PY, Krtolica A, Campisi J, The senescence-associated secretory phenotype: The dark side of tumor suppression, Annu. Rev. Pathol. Mech. Dis 5 (2010) 99–118. 10.1146/annurev-pathol-121808-102144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Zhu F, Li Y, Zhang J, Piao C, Liu T, Li HH, Du J, Senescent cardiac fibroblast is critical for cardiac fibrosis after myocardial infarction., PLoS One. 8 (2013). 10.1371/journal.pone.0074535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Frangogiannis NG, Cardiac fibrosis: Cell biological mechanisms, molecular pathways and therapeutic opportunities, Mol. Aspects Med 65 (2019) 70–99. 10.1016/j.mam.2018.07.001. [DOI] [PubMed] [Google Scholar]
- [38].Bassat E, Mutlak YE, Genzelinakh A, Shadrin IY, Baruch Umansky K, Yifa O, Kain D, Rajchman D, Leach J, Riabov Bassat D, Udi Y, Sarig R, Sagi I, Martin JF, Bursac N, Cohen S, Tzahor E, The extracellular matrix protein agrin promotes heart regeneration in mice, Nature. 547 (2017) 179–184. 10.1038/nature22978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Pakshir P, Alizadehgiashi M, Wong B, Coelho NM, Chen X, Gong Z, Shenoy VB, McCulloch C, Hinz B, Dynamic fibroblast contractions attract remote macrophages in fibrillar collagen matrix, Nat. Commun 10 (2019) 1850 10.1038/s41467-019-09709-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Perbellini F, Watson SA, Bardi I, Terracciano CM, Heterocellularity and Cellular Cross-Talk in the Cardiovascular System, Front. Cardiovasc. Med 5 (2018). 10.3389/fcvm.2018.00143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Fountoulaki K, Dagres N, Iliodromitis EK, Cellular Communications in the Heart, Card. Fail. Rev 1 (2015) 64–68. 10.15420/cfr.2015.1.2.64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Takahashi K, Tanabe K, Ohnuki M, Narita M, Ichisaka T, Tomoda K, Yamanaka S, Induction of Pluripotent Stem Cells from Adult Human Fibroblasts by Defined Factors, Cell. 131 (2007) 861–872. 10.1016/j.cell.2007.11.019. [DOI] [PubMed] [Google Scholar]
- [43].Takahashi K, Yamanaka S, Induction of Pluripotent Stem Cells from Mouse Embryonic and Adult Fibroblast Cultures by Defined Factors, Cell. 126 (2006) 663–676. 10.1016/j.cell.2006.07.024. [DOI] [PubMed] [Google Scholar]
- [44].Davis RL, Weintraub H, Lassar AB, Expression of a single transfected cDNA converts fibroblasts to myoblasts, Cell. 51 (1987) 987–1000. 10.1016/0092-8674(87)90585-X. [DOI] [PubMed] [Google Scholar]
- [45].Ieda M, Fu J-DD, Delgado-Olguin P, Vedantham V, Hayashi Y, Bruneau BG, Srivastava D, Direct Reprogramming of Fibroblasts into Functional Cardiomyocytes by Defined Factors, Cell. 142 (2010) 375–386. 10.1016/j.cell.2010.07.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [46].Qian L, Huang Y, Spencer CI, Foley A, Vedantham V, Liu L, Conway SJ, Fu JD, Srivastava D, In vivo reprogramming of murine cardiac fibroblasts into induced cardiomyocytes, Nature. 485 (2012) 593–598. 10.1038/nature11044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [47].Nam Y-J, Song K, Olson EN, Heart repair by cardiac reprogramming, Nat. Med 19 (2013) 413–415. 10.1038/nm.3147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [48].Song K, Nam Y-J, Luo X, Qi X, Tan W, Huang GN, Acharya A, Smith CL, Tallquist MD, Neilson EG, Hill JA, Bassel-Duby R, Olson EN, Heart repair by reprogramming non-myocytes with cardiac transcription factors, Nature. 485 (2012) 599–604. 10.1038/nature11139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [49].Zhao Y, Londono P, Cao Y, Sharpe EJ, Proenza C, O’Rourke R, Jones KL, Jeong MY, Walker LA, Buttrick PM, McKinsey TA, Song K, High-efficiency reprogramming of fibroblasts into cardiomyocytes requires suppression of pro-fibrotic signalling, Nat. Commun 6 (2015) 8243 10.1038/ncomms9243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [50].Wang L, Liu Z, Yin C, Asfour H, Chen O, Li Y, Bursac N, Liu J, Qian L, Stoichiometry of Gata4, Mef2c, and Tbx5 influences the efficiency and quality of induced cardiac myocyte reprogramming, Circ. Res 116 (2015) 237–244. 10.1161/CIRCRESAHA.116.305547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [51].Liu Z, Wang L, Welch JD, Ma H, Zhou Y, Vaseghi HR, Yu S, Wall JB, Alimohamadi S, Zheng M, Yin C, Shen W, Prins JF, Liu J, Qian L, Single-cell transcriptomics reconstructs fate conversion from fibroblast to cardiomyocyte, Nature. 551 (2017) 100–104. 10.1038/nature24454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [52].Zhang J, Tao R, Campbell KF, Carvalho JL, Ruiz EC, Kim GC, Schmuck EG, Raval AN, da Rocha AM, Herron TJ, Jalife J, Thomson JA, Kamp TJ, Functional cardiac fibroblasts derived from human pluripotent stem cells via second heart field progenitors, Nat. Commun 10 (2019) 1–15. 10.1038/s41467-019-09831-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [53].Hao Z, Lei T, Mengcheng S, Chengyi T, Haodi W, Mingxia G, P. DT, W. JC, Generation of Quiescent Cardiac Fibroblasts From Human Induced Pluripotent Stem Cells for In Vitro Modeling of Cardiac Fibrosis, Circ. Res 125 (2019) 552–566. 10.1161/CIRCRESAHA.119.315491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [54].Khalil H, Kanisicak O, Prasad V, Correll RN, Fu X, Schips T, Vagnozzi RJ, Liu R, Huynh T, Lee SJ, Karch J, Molkentin JD, Fibroblast-specific TGF-β-Smad2/3 signaling underlies cardiac fibrosis, in: J. Clin. Invest., American Society for Clinical Investigation, 2017: pp. 3770–3783. 10.1172/JCI94753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [55].Bujak M, Ren G, Kweon HJ, Dobaczewski M, Reddy A, Taffet G, Wang XF, Frangogiannis NG, Essential role of Smad3 in infarct healing and in the pathogenesis of cardiac remodeling, Circulation. 116 (2007) 2127–2138. 10.1161/CIRCULATIONAHA.107.704197. [DOI] [PubMed] [Google Scholar]
- [56].Kong P, Shinde AV, Su Y, Russo I, Chen B, Saxena A, Conway SJ, Graff JM, Frangogiannis NG, Opposing actions of fibroblast and cardiomyocyte smad3 signaling in the infarcted myocardium, Circulation. 137 (2018) 707–724. 10.1161/CIRCULATIONAHA.117.029622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [57].Divakaran V, Adrogue J, Ishiyama M, Entman ML, Haudek S, Sivasubramanian N, Mann DL, Adaptive and maladptive effects of SMAD3 signaling in the adult heart after hemodynamic pressure overloading, Circ. Hear. Fail 2 (2009) 633–642. 10.1161/CIRCHEARTFAILURE.108.823070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [58].Bagchi RA, Roche P, Aroutiounova N, Espira L, Abrenica B, Schweitzer R, Czubryt MP, The transcription factor scleraxis is a critical regulator of cardiac fibroblast phenotype, BMC Biol. 14 (2016) 3–6. 10.1186/s12915-016-0243-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [59].Bagchi RA, Czubryt MP, Synergistic roles of scleraxis and Smads in the regulation of collagen 1α2 gene expression, Biochim. Biophys. Acta - Mol. Cell Res 1823 (2012) 1936–1944. 10.1016/j.bbamcr.2012.07.002. [DOI] [PubMed] [Google Scholar]
- [60].Koitabashi N, Danner T, Zaiman AL, Pinto YM, Rowell J, Mankowski J, Zhang D, Nakamura T, Takimoto E, Kass DA, Pivotal role of cardiomyocyte TGF-β signaling in the murine pathological response to sustained pressure overload, J. Clin. Invest 121 (2011) 2301–2312. 10.1172/JCI44824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [61].Davis J, Burr AR, Davis GF, Birnbaumer L, Molkentin JD, A TRPC6-Dependent Pathway for Myofibroblast Transdifferentiation and Wound Healing In Vivo, Dev. Cell 23 (2012) 705–715. 10.1016/j.devcel.2012.08.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [62].Richardson WJ, Holmes JW, Emergence of Collagen Orientation Heterogeneity in Healing Infarcts and an Agent-Based Model, Biophys. J 110 (2016) 2266–2277. 10.1016/j.bpj.2016.04.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [63].Xiao Y, Hill MC, Li L, Deshmukh V, Martin TJ, Wang J, Martin JF, Hippo pathway deletion in adult resting cardiac fibroblasts initiates a cell state transition with spontaneous and self-sustaining fibrosis, Genes Dev. 33 (2019) 1491–1505. 10.1101/gad.329763.119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [64].Liu ZP, Wang Z, Yanagisawa H, Olson EN, Phenotypic modulation of smooth muscle cells through interaction of Foxo4 and Myocardin, Dev. Cell 9 (2005) 261–270. 10.1016/j.devcel.2005.05.017. [DOI] [PubMed] [Google Scholar]
- [65].Small EM, Thatcher JE, Sutherland LB, Kinoshita H, Gerard RD, Richardson JA, Dimaio JM, Sadek H, Kuwahara K, Olson EN, Myocardin-related transcription factor-a controls myofibroblast activation and fibrosis in response to myocardial infarction, Circ. Res 107 (2010) 294–304. 10.1161/CIRCRESAHA.110.223172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [66].Small EM, The Actin-MRTF-SRF gene regulatory axis and myofibroblast differentiation, J. Cardiovasc. Transl. Res 5 (2012) 794–804. 10.1007/s12265-012-9397-0. [DOI] [PubMed] [Google Scholar]
- [67].Kuwahara K, Wang Y, McAnally J, Richardson JA, Bassel-Duby R, Hill JA, Olson EN, TRPC6 fulfills a calcineurin signaling circuit during pathologic cardiac remodeling, J. Clin. Invest 116 (2006) 3114–3126. 10.1172/JCI27702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [68].Francisco J, Zhang Y, Jeong JI, Mizushima W, Ikeda S, Ivessa A, Oka S, Zhai P, Tallquist MD, Del Re DP, Blockade of Fibroblast YAP Attenuates Cardiac Fibrosis and Dysfunction Through MRTF-A Inhibition, JACC Basic to Transl. Sci 5 (2020) 931–945. 10.1016/j.jacbts.2020.07.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [69].Perestrelo AR, Silva AC, La Cruz JO, Martino F, Horváth V, Caluori G, Polanský O, Vinarský V, Azzato G, Marco D, Žampachová V, Skládal P, Pagliari S, Rainer A, Pinto-do- P, Multiscale Analysis of Extracellular Matrix Remodeling in the Failing Heart, Circ. Res (2020). 10.1161/CIRCRESAHA.120.317685. [DOI] [PubMed] [Google Scholar]
- [70].Del Re DP, Yang Y, Nakano N, Cho J, Zhai P, Yamamoto T, Zhang N, Yabuta N, Nojima H, Pan D, Sadoshima J, Yes-associated protein isoform 1 (Yapl) promotes cardiomyocyte survival and growth to protect against myocardial ischemic injury, J. Biol. Chem 288 (2013) 3977–3988. 10.1074/jbc.M112.436311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [71].Xin M, Kim Y, Sutherland LB, Murakami M, Qi X, McAnally J, Porrello ER, Mahmoud AI, Tan W, Shelton JM, Richardson JA, Sadek HA, Bassel-Duby R, Olson EN, Hippo pathway effector Yap promotes cardiac regeneration, Proc. Natl. Acad. Sci. U. S. A 110 (2013) 13839–13844. 10.1073/pnas.1313192110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [72].Schelbert EB, Hsu LY, Anderson SA, Mohanty BD, Karim SM, Kellman P, Aletras AH, Arai AE, Late gadolinium-enhancement cardiac magnetic resonance identifies postinfarction myocardial fibrosis and the border zone at the near cellular level in ex vivo rat heart, Circ. Cardiovasc. Imaging 3 (2010) 743–752. 10.1161/CIRCIMAGING.108.835793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [73].Orn S, Manhenke C, Anand IS, Squire I, Nagel E, Edvardsen T, Dickstein K, Effect of Left Ventricular Scar Size, Location, and Transmurality on Left Ventricular Remodeling With Healed Myocardial Infarction, Am. J. Cardiol 99 (2007) 1109–1114. 10.1016/j.amjcard.2006.11.059. [DOI] [PubMed] [Google Scholar]
- [74].Kyoi S, Otani H, Matsuhisa S, Akita Y, Tatsumi K, Enoki C, Fujiwara H, Imamura H, Kamihata H, Iwasaka T, Opposing effect of p38 MAP kinase and JNK inhibitors on the development of heart failure in the cardiomyopathic hamster, Cardiovasc. Res 69 (2006) 888–898. 10.1016/j.cardiores.2005.11.015. [DOI] [PubMed] [Google Scholar]
- [75].See F, Thomas W, Way K, Tzanidis A, Kompa A, Lewis D, Itescu S, Krum H, P38 mitogen-activated protein kinase inhibition improves cardiac function and attenuates left ventricular remodeling following myocardial infarction in the rat, J. Am. Coll. Cardiol 44 (2004) 1679–1689. 10.1016/j.jacc.2004.07.038. [DOI] [PubMed] [Google Scholar]
- [76].Yamagami K, Oka T, Wang Q, Ishizu T, Lee J-K, Miwa K, Akazawa H, Naito AT, Sakata Y, Komuro I, Pirfenidone exhibits cardioprotective effects by regulating myocardial fibrosis and vascular permeability in pressure-overloaded hearts, Am. J. Physiol. Circ. Physiol 309 (2015) H512–H522. 10.1152/ajpheart.00137.2015. [DOI] [PubMed] [Google Scholar]
- [77].Nguyen DT, Ding C, Wilson E, Marcus GM, Olgin JE, Pirfenidone mitigates left ventricular fibrosis and dysfunction after myocardial infarction and reduces arrhythmias, Hear. Rhythm 7 (2010) 1438–1445. 10.1016/j.hrthm.2010.04.030. [DOI] [PubMed] [Google Scholar]
- [78].Wang Y, Wu Y, Chen J, Zhao S, Li H, Pirfenidone attenuates cardiac fibrosis in a mouse model of TAC-induced left ventricular remodeling by suppressing NLRP3 Inflammasome formation, Cardiol. 126 (2013) 1–11. 10.1159/000351179. [DOI] [PubMed] [Google Scholar]
- [79].Yamazaki T, Yamashita N, Izumi Y, Nakamura Y, Shiota M, Hanatani A, Shimada K, Muro T, Iwao H, Yoshiyama M, The antifibrotic agent pirfenidone inhibits angiotensin II-induced cardiac hypertrophy in mice, Hypertens. Res 35 (2012) 34–40. 10.1038/hr.2011.139. [DOI] [PubMed] [Google Scholar]
- [80].Newby LK, Marber MS, Melloni C, Sarov-Blat L, Aberle LH, Aylward PE, Cai G, De Winter RJ, Hamm CW, Heitner JF, Kim R, Lerman A, Patel MR, Tanguay JF, Lepore JJ, Al-Khalidi HR, Sprecher DL, Granger CBI, Losmapimod, a novel p38 mitogen-activated protein kinase inhibitor, in non-ST-segment elevation myocardial infarction: A randomised phase 2 trial, Lancet. 384 (2014) 1187–1195. 10.1016/S0140-6736(14)60417-7. [DOI] [PubMed] [Google Scholar]
- [81].O’Donoghue ML, Glaser R, Cavender MA, Aylward PE, Bonaca MP, Budaj A, Davies RY, Dellborg M, Fox KAA, Gutierrez JAT, Hamm C, Kiss RG, Kovar F, Kuder JF, Im KA, Lepore JJ, Lopez-Sendon JL, Ophuis TO, Parkhomenko A, Shannon JB, Spinar J, Tanguay JF, Ruda M, Steg PG, Theroux P, Wiviott SD, Laws I, Sabatine MS, Morrow DA, Effect of losmapimod on cardiovascular outcomes in patients hospitalized with acute myocardial infarction: A randomized clinical trial, JAMA - J. Am. Med. Assoc 315 (2016) 1591–1599. 10.1001/jama.2016.3609. [DOI] [PubMed] [Google Scholar]
- [82].Muchir A, Wu W, Choi JC, Iwata S, Morrow J, Homma S, Worman HJ, Abnormal p38α mitogen-activated protein kinase signaling in dilated cardiomyopathy caused by lamin A/C gene mutation, Hum. Mol. Genet 21 (2012) 4325–4333. 10.1093/hmg/dds265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [83].Nguyen CDK, Yi C, YAP/TAZ Signaling and Resistance to Cancer Therapy, Trends in Cancer. 5 (2019) 283–296. 10.1016/j.trecan.2019.02.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [84].Liang M, Yu M, Xia R, Song K, Wang J, Luo J, Chen G, Cheng J, Yap/Taz deletion in gli+ cell-derived myofibroblasts attenuates fibrosis, J. Am. Soc. Nephrol 28 (2017) 3278–3290. 10.1681/ASN.2015121354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [85].Szeto SG, Narimatsu M, Lu M, He X, Sidiqi AM, Tolosa MF, Chan L, De Freitas K, Bialik JF, Majumder S, Boo S, Hinz B, Dan Q, Advani A, John R, Wrana JL, Kapus A, Yuen DA, YAP/TAZ are mechanoregulators of TGF-b-smad signaling and renal fibrogenesis, J. Am. Soc. Nephrol 27 (2016) 3117–3128. 10.1681/ASN.2015050499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [86].Santos DM, Pantano L, Pronzati G, Grasberger P, Probst CK, Black KE, Spinney JJ, Hariri LP, Nichols R, Lin Y, Bieler M, Seither P, Nicklin P, Wyatt D, Tager AM, Medoff BD, Screening for YAP inhibitors identifies statins as modulators of fibrosis, Am. J. Respir. Cell Mol. Biol 62 (2020) 479–492. 10.1165/rcmb.2019-0296OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [87].Haak AJ, Kostallari E, Sicard D, Ligresti G, Choi KM, Caporarello N, Jones DL, Tan Q, Meridew J, Espinosa AMD, Aravamudhan A, Maiers JL, Britt RD, Roden AC, Pabelick CM, Prakash YS, Nouraie SM, Li X, Zhang Y, Kass DJ, Lagares D, Tager AM, Varelas X, Shah VH, Tschumperlin DJ, Selective YAP/TAZ inhibition in fibroblasts via dopamine receptor D1 agonism reverses fibrosis, Sci. Transl. Med 11 (2019) 6296 10.1126/scitranslmed.aau6296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [88].Fu JD, Stone NR, Liu L, Spencer CI, Qian L, Hayashi Y, Delgado-Olguin P, Ding S, Bruneau BG, Srivastava D, Direct reprogramming of human fibroblasts toward a cardiomyocyte-like state, Stem Cell Reports. 1 (2013) 235–247. 10.1016/j.stemcr.2013.07.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [89].Wada R, Muraoka N, Inagawa K, Yamakawa H, Miyamoto K, Sadahiro T, Umei T, Kaneda R, Suzuki T, Kamiya K, Tohyama S, Yuasa S, Kokaji K, Aeba R, Yozu R, Yamagishi H, Kitamura T, Fukuda K, Ieda M, Induction of human cardiomyocyte-like cells from fibroblasts by defined factors, Proc. Natl. Acad. Sci. U. S. A 110 (2013) 12667–12672. 10.1073/pnas.1304053110. [DOI] [PMC free article] [PubMed] [Google Scholar]