

Summary
Machine learning can infer how protein sequence maps to function without requiring a detailed understanding of the underlying physical or biological mechanisms. It’s challenging to apply existing supervised learning frameworks to large-scale experimental data generated by deep mutational scanning (DMS) and related methods. DMS data often contain high dimensional and correlated sequence variables, experimental sampling error and bias, and the presence of missing data. Notably, most DMS data do not contain examples of negative sequences, making it challenging to directly estimate how sequence affects function. Here, we develop a positive-unlabeled (PU) learning framework to infer sequence-function relationships from large-scale DMS data. Our PU learning method displays excellent predictive performance across ten large-scale sequence-function data sets, representing proteins of different folds, functions, and library types. The estimated parameters pinpoint key residues that dictate protein structure and function. Finally, we apply our statistical sequence-function model to design highly stabilized enzymes.
Keywords: protein-sequence function relationships, deep mutational scanning, protein engineering, statistical learning, supervised learning, positive-unlabeled learning
Graphical Abstract
eTOC blurb
The quantity of protein sequence-function data is growing rapidly with advances in high-throughput experimentation. Song et al. present a machine learning approach to infer sequence-function relationships from large-scale data generated by deep mutational scanning. The learned models capture important aspects of protein structure and function, and can be applied to design new and enhanced proteins.
Introduction
A protein’s sequence of amino acids encodes its function. This “function” could refer to a protein’s natural biological function, or it could also be any other property including binding affinity toward a particular ligand, thermodynamic stability, or catalytic activity. A detailed understanding of how these functions are encoded would allow us to more accurately reconstruct the tree of life, diagnose genetic diseases before they manifest symptoms, and design new proteins with useful properties. The mapping from protein sequence to function is extraordinarily complex because it involves thousands of molecular interactions that are dynamically coupled across multiple length and time scales.
Machine learning can infer how protein sequence encodes function without needing to understand the underlying biophysical mechanisms (Yang et al. 2019, Mazurenko et al. 2020). These learning methods can be broadly categorized into unsupervised and supervised depending on whether the data points are labeled. In the protein context, unsupervised methods learn from examples of sequences that share some common function/property, while supervised methods learn from sequence-function examples. Unsupervised methods are often trained on natural sequence data derived from large genomic databases, and effectively learn the rules of folding/function for a given protein family (Morcos et al. 2011, Hopf et al. 2017, Riesselman et al. 2018). In contrast, supervised methods are trained directly on sequence-function examples, and therefore, can learn the mapping to a particular protein property or set of properties. This capability to predict a target protein property is important in protein engineering, which seeks to design and optimize non-natural protein functions. Supervised models have been used to rationally engineer proteinases with improved activity at elevated temperatures, cytochrome P450s with enhanced stability, carbonic anhydrases for industrial carbon capture, and bacteriorhodopsins for optogenetics (Liao et al. 2007, Romero et al. 2013, Alvizo et al. 2014, Bedbrook et al. 2019).
The accuracy and resolution of statistical models improve with increasing data; however, existing supervised methods cannot learn from large-scale sequence-function data generated by deep mutational scanning (DMS) and related methods. DMS combines high-throughput screening and next-generating DNA sequencing to experimentally map sequence-function relationships for thousands to millions of protein variants (Fowler & Fields 2014, Boucher et al. 2014, Weile & Roth 2018). In principle, DMS data should provide rich sequence-function information for training supervised models. However, learning from DMS data is challenging due to its scale and dimensionality, correlations between sequence variables, sampling error caused by low numbers of observations, and missing/low quality sequence information. In addition, most DMS data sets do not contain negative sequence examples because these sequences are difficult or impossible to obtain using high-throughput screening/selection methods. These negative sequences are important to directly infer how sequence maps to function. Hence DMS data are neither amenable to supervised learning due to the lack of negative sequences, nor unsupervised learning since many sequences have positive labels.
In this paper, we present a supervised learning framework for inferring sequence-function relationships from large-scale data generated by deep mutational scanning (DMS). We categorize DMS data as positive-unlabeled (PU) data because it contains examples of both positive sequences and sequences without labels. Learning from PU data has applications in domains such as text mining, gene identification, marketing, and ecological modeling (Liu et al. 2003, Mordelet & Vert 2011, Yi et al. 2017, Ward et al. 2009). We develop a PU learning method that models a protein’s function as an unobserved latent variable, and then infers how sequence maps to this latent function by maximizing an observed likelihood function. Our learned PU models displayed excellent predictive ability and stability across ten diverse DMS data sets. The PU model’s parameters describe how amino acid substitutions affect protein function, and the significance of these parameter estimates can be evaluated using statistical hypothesis testing. We demonstrate the extrapolative power of the learned sequence-function mapping by designing enzymes with increased thermostability.
Results
A statistical framework for learning sequence-function relationships
Supervised learning methods can infer how sequence maps to function from a set of experimental sequence-function examples. However, it’s challenging to apply existing learning methods to large-scale data generated by deep mutational scanning (DMS) due to the lack of negative sequence examples. We use the term DMS to broadly refer to any experiment that maps sequence-function relationships using a combination of gene mutagenesis, high-throughput screening/selections, and next-generation DNA sequencing. An overview of a standard DMS experiment is illustrated in Figure 1a. This section describes the DMS data generation process, introduces key statistical variables, and proposes a positive-unlabeled approach to learn from DMS data.
Figure 1.
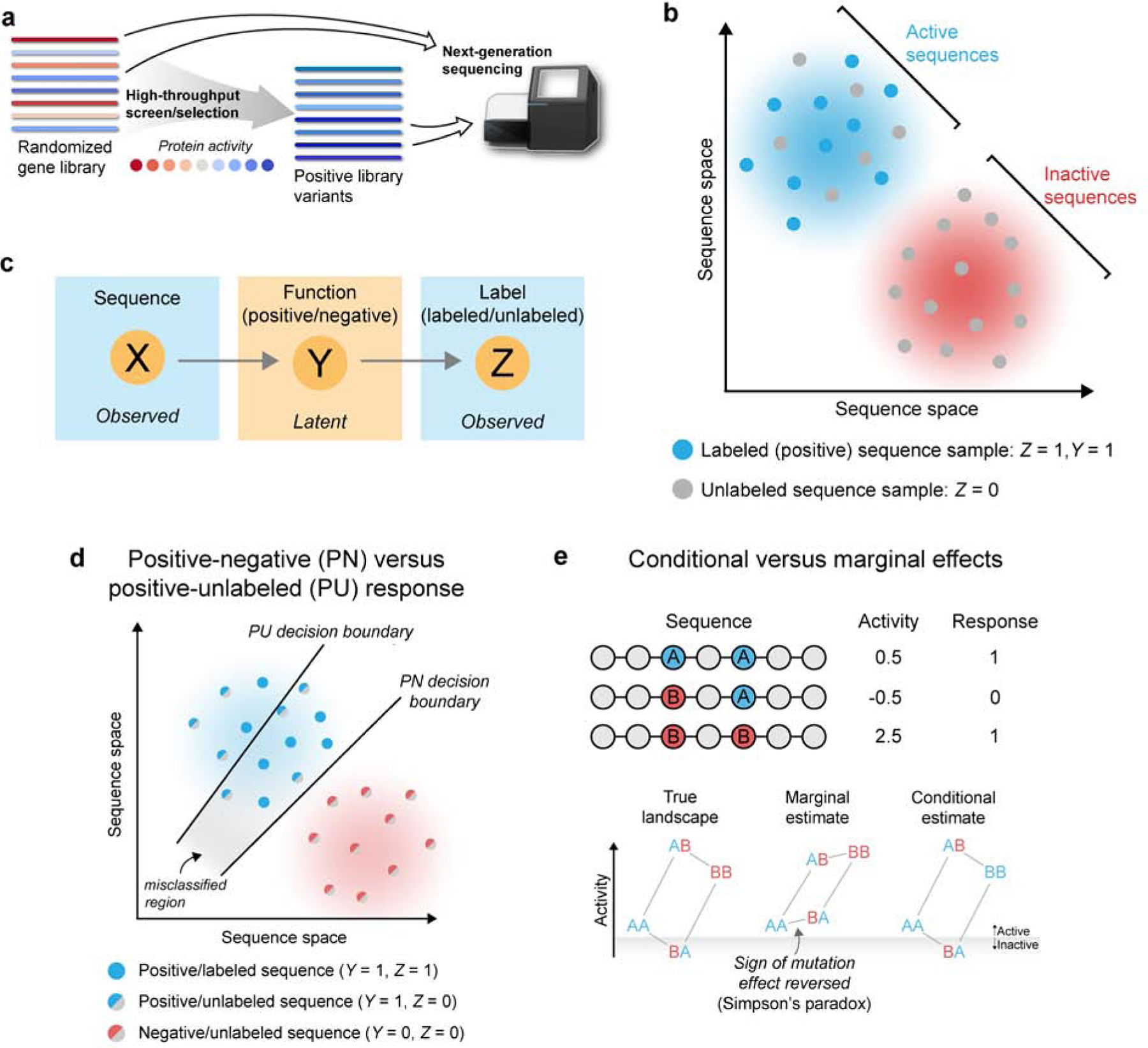
Positive-unlabeled learning from deep mutational scanning (DMS) data. (a) Overview of a typical DMS experiment. DMS experiments start with a large library of gene variants that display a range of activities. The gene library is then expressed and passed through a high-throughput screen or selection that isolates the positive variants. The activity threshold to be categorized as positive will depend on the details of the particular high-throughput screen/selection. It is often difficult or impossible to experimentally isolate negative sequences. Genes from the initial library and the isolated positive variants are then extracted and analyzed using next-generation DNA sequencing. DMS experiments generate thousands to millions of sequence examples from both the initial and positive sets. (b) DMS experiments sample sequences from protein sequence space. The resulting data contain positive labeled sequence examples (Y = 1, Z = 1), and unlabeled sequence examples (Z = 0) that contain a mixture of active and inactive sequences. (c) The relationships between variables representing protein sequences (X), latent function (Y), and the observed labels (Z). Y is not directly observed in DMS experiments and must be inferred from X and Z. (d) PU learning models the true positive-negative (PN) response, while enrichment-based estimates capture the positive-unlabeled (PU) response. Modeling the PU response gives rise to a decision boundary that is shifted toward the positive class, resulting in positive sequences that are misclassified as negative. (e) PU learning estimates the conditional effect of a mutation, while site-wise enrichment estimates the marginal effect. Marginal estimates are biased and in extreme cases can result in a sign reversal phenomenon known as Simpson’s paradox. In the example, we consider amino acid substitutions A→B at two independent sites in a protein. If we observe sequences AA, BA, and BB, the marginal estimate will reverse the sign of substitution A→B at the first position. The marginal model will also misclassify sequence BA as positive, even though it was observed to be negative. In contrast, the conditional estimate correctly models the true protein function landscape.
A protein’s biochemical activity Ai is a function of its amino acid sequence, i.e. Ai = f(xi), where xi is a vector that specifies a protein’s amino acid sequence (see Methods Details). Now suppose a protein sequence can be categorized as active or inactive depending on whether its activity Ai falls above/below a defined activity threshold t. There is also some error in experimentally determining whether a sequence is active or inactive. We define a protein’s experimentally measured functional response as:
| (1) |
where ϵi is the random error associated with experimentally characterizing the ith sequence. We say a sequence is “positive” if its experimentally measured activity exceeds the threshold t (i.e. Yi = 1) and “negative” otherwise. Note that we make subtle distinction between the terms active/inactive and positive/negative: we use active/inactive to describe the true functional state of a protein and positive/negative to indicate the result of an experimental measurement.
A DMS experiment starts with an initial library of sequences that each maps to a particular activity value and can be categorized as active or inactive. We refer to this initial library as “unlabeled” because it contains an unknown mixture of active and inactive sequences. A high-throughput screen/selection then samples this initial unlabeled library to obtain examples of positive sequences. Notably, it is often difficult or impossible to isolate negative sequences because most experimental methods are designed to identify positive sequences (e.g. growth selections). We refer to the sampled positive sequences as “labeled” because they are known to have positive labels. The sequences within the initial unlabeled set and positive labeled set are then determined using next-generation sequencing. The final data contains nu sequences sampled from the unlabeled set and np sequences sampled from the positive labeled set (Figure 1b).
We aim to learn from DMS data to understand how amino acid sequence maps to function (i.e. infer f). If the data consisted of (Xi, Yi) pairs, we could simply train a binary classifier such as a logistic regression model or a multilayer perceptron. However, DMS experiments do not reveal the true functional response (Y), but instead only provide examples of positive sequences. The lack of negative sequence examples results in a mis-specified binary classification problem and makes it challenging to directly infer how sequence maps to function.
We propose a positive-unlabeled approach to learn from DMS data. Positive-unlabeled (PU) learning estimates how input variables affect the positive-negative response from positive and unlabeled data (Liu et al. 2003, Elkan & Noto 2008, Song & Raskutti 2018). We introduce a binary variable Z that specifies whether a sequence is labeled (Z = 1) or unlabeled (Z = 0). DMS experiments effectively generate (Xi, Zi) pairs with an unobserved functional response Yi (Figure 1c). From the set-up of the problem, Z and Y are closely related. In particular, all labeled sequences are positive, and the proportion of positive sequences in the unlabeled set is the same as that of the initial library. In other words,
where π is the proportion of positive sequences in the initial library. We aim to infer f using the observed examples , where n = np + nu denotes the total number of sequence examples.
Algorithms for large-scale positive-unlabeled learning
Sequence-function data obtained by deep mutational scanning (DMS) typically contains sequence examples from the initial (unlabeled) library and positive sequences (Figure 1). We develop algorithms for learning the sequence-function mapping from this positive-unlabeled sequence data. Our approach utilizes the distributional relationship between X, Y, and Z to infer the latent functional response yi from the observed labels and amino acid sequences , and then infer the f that best describes the latent responses .
We model f as a linear function of amino acid sequence f(x) = Σj, aa xj, aaaj, aa, and use a logistic function to describe the probability that a sequence is positive:
| (2) |
where x⊤θ = Σj, aa xj, aaaj, aa − t represents a relative activity level of amino acid sequence x with respect to the activity threshold t in (1), and θ parameterizes the effect of making an amino acid substitution from a defined reference sequence (see Methods Details). The model’s parameters (θ) are closely related to the site-wise enrichment scores that are commonly used to analyze DMS data (Bloom 2015, Klesmith & Hackel 2019, Wrenbeck et al. 2017, Abriata et al. 2016). However, site-wise enrichment is a biased estimator for θ because it is derived from positive-unlabeled data and makes strong assumptions about the independence between sequence positions. We derive the mathematical relationship between site-wise enrichment and a mutation’s true effect (θ) in the Methods Details.
We take a likelihood-based approach for estimating θ from the observed examples . To account for the fact that the true responses are latent, we use an observed likelihood that is a product of the marginalized probabilities (Ward et al. 2009)
| (3) |
for , where Si ∈ (0, 1) is an indicator variable representing whether the ith example is present in the data. We use the maximum likelihood approach to estimate θ. In particular, we minimize the negative observed log-likelihood and define the estimated coefficients as
| (4) |
The negative observed log-likelihood, −log ℓ(θ), is a non-convex function of θ. Obtaining a global minimizer of a non-convex function is in general a challenging problem, so the feasibility of obtaining is not immediate. We previously found similar classes of problems can be solved when the likelihood function is calculated with sufficiently large sample size n (Song et al. 2019). In these cases, any stationary point is the global minimizer with high probability. Since our sequence-function data sets typically contain millions of observations, we can find the maximum likelihood estimate by identifying a stationary point of the negative observed log-likelihood.
We solved this optimization problem using the Majorization-Minimization (MM) algorithm to obtain a stationary point of the negative observed log-likelihood function (Eqn 3). Note that the likelihood (Eqn 3) involves the hyperparameter π, the proportion of positive sequences in the unlabeled set. π was experimentally determined for some data sets, and we used this value for the hyperparameter if it was available. Otherwise, we carried out a grid search over π values and chose the π value that maximized the area under the receiver operating characteristic curve. We used the learned model parameters to calculate p-values to test whether amino acid substitutions have a significant impact on protein function. These p-values were adjusted using the Benjamini-Hochberg (BH) procedure to account for multiple hypothesis testing (Methods Details). An overview of our data processing, parameter estimation, and model analysis workflow is provided in Supplemental Figure 2.
Relationship between learned PU model parameters and site-wise enrichment scores
There is a close connection between our PU model’s parameters and the site-wise enrichment scores that are commonly used to analyze deep mutational scanning (DMS) data (Bloom 2015, Klesmith & Hackel 2019, Wrenbeck et al. 2017, Abriata et al. 2016). Both quantities evaluate how amino acid substitutions affect a protein’s functional response (i.e. estimate ). However, our PU model provides a consistent estimate of because it directly models the true positive-negative (PN) response and considers the conditional effects of amino acid substitutions. In this section, we define site-wise enrichment scores, contrast them with the PU model parameters, and identify two different sources of bias in their estimate of .
Site-wise enrichment scores are calculated using marginal amino acid frequencies:
| (5) |
where P and U are the positive and unlabeled sets of sequences, respectively. Ej, aa compares the prevalence of amino acid aa with the reference sequence (typically wild-type) in the positive and unlabeled sets. A residue with a negative enrichment score is underrepresented in the positive set, and therefore associated with decreased protein activity. Conversely, a residue with a positive enrichment score is associated with the increased protein activity. These enrichment scores provide a simple and convenient method to compare frequencies before/after selection and estimate the effects of amino acid substitutions.
Enrichment and our PU learning method capture different response variables related to protein function (Figure 1d). Enrichment models the positive-unlabeled (PU) response, and since all labeled sequences are positive, this is equivalent to modeling a sequence’s label Z. In contrast, our PU learning method directly models the positive-negative (PN) response by inferring the latent function Y from observed labels Z. The presence of positive sequences in the unlabeled set causes enrichment-based methods to provide attenuated estimates of an amino acid substitution’s effect (see Methods Details). This leads to a decision boundary that is shifted toward the positive class and results in misclassified sequences. Our PU learning method models the true PN response, and thus provides an unbiased estimate of a substitution’s effect .
The second key difference between PU model parameters and enrichment is related to marginal versus conditional effects. Our PU learning method estimates the effect of an amino acid substitution from a defined sequence background, typically wild-type. This is a conditional estimate because the effect of the mutation is conditioned on all other sites in the protein. This conditional effect provides an unbiased estimate of an amino acid substitution’s effect . In contrast, site-wise enrichment estimates the effect of an amino acid substitution in combination with averaged effects from all other sites in the data set. These marginal estimates include the true effect , in addition to indirect effects from other sequence positions. These indirect effects lead to bias in the estimate of (see Methods Details). In more extreme instances, marginal estimates can reverse the sign of an effect and lead to incorrect conclusions about whether a substitution is beneficial or deleterious (Figure 1e). This effect reversal is referred to as Simpson’s paradox.
Learning from large-scale sequence-function data
We applied our PU learning method to infer the sequence-activity mapping from ten large sequence-function data sets (see Supplemental Table 1). These data sets represent proteins of diverse folds/functions, were generated using different library mutagenesis methods, span several orders of magnitude in size, and have varying levels of missing sequence information. The PU models displayed excellent predictive ability on all ten data sets, with cross-validated area under the receiver operating characteristic curve (ROC-AUC) ranging from 0.68 to 0.98 (Figure 2ab). For comparison, we also evaluated predictions from structure-based (Alford et al. 2017) and unsupervised learning methods (Hopf et al. 2017, Riesselman et al. 2018). Rosetta, EVmutation, and DeepSequence all displayed substantially lower AUC values than the PU model (Figure 2b, Supplemental Figure 3a).
Figure 2.

Performance of the PU learning method across protein data sets. (a) Receiver operating characteristic (ROC) curves for the ten tested protein data sets. ROC curves were generated using 10-fold cross-validation and corrected to account for PU data (See Methods Details and Supplemental Figure 1). (b) The PU model’s corrected ROC-AUC values range from 0.68 to 0.98, and outperform structure-based (Rosetta) and unsupervised learning methods (EVmutation and DeepSequence). (c) A statistical comparison between PU model predictions and site-wise enrichment. The PU model outperformed enrichment on all ten tested data sets, with p < 10−9.
Our PU learning method estimates how mutations affect a protein’s functional response. This PU estimate is closely related to the site-wise enrichment scores that are commonly used to analyze DMS data. We compared the predictive ability of the PU model versus enrichment using a corrected cross-validation test. We found the PU model predictions were better than enrichment for all ten data sets, with p < 10−9 (Figure 2c). However, the PU models’ AUCs were only marginally higher than enrichment, with AUC differences ranging from 0.002 to 0.017 (Supplemental Figure 3b).
We evaluated the robustness of the learned PU models to data sampling and the hyperparameter π. We analyzed the stability of each model’s parameter estimates by calculating the coefficient of variation (CV) across different cross-validation folds for all significant parameters (i.e. BH-adjusted p < 0.05). The parameters displayed average absolute CVs ranging from 0.01 to 0.08 (Supplemental Figure 3c), indicating the estimates were highly insensitive to different training sets. We also evaluated the feature selection stability by computing the average fraction of commonly selected features across different cross-validation folds (Methods Details). We found the selected features were nearly identical across cross-validation folds for each data set (Supplemental Figure 3d). Finally, we tested how the choice of the hyperparameter π (experimentally determined or estimated) affects the learned PU models. We found the parameter values estimated using our chosen π value were highly correlated with parameters estimated across the entire range of π values tested (Supplemental Figure 3e).
Learned parameters relate to protein structure and function
The B1 domain of protein G (GB1) is a small 8 kDa alpha-beta roll that binds to IgG. We performed further analysis relating the learned GB1 model with protein G structure and function. The PU model’s coefficients describe how an amino acid substitution (mutation) affects the protein’s functional response (Eqn 7). A negative coefficient indicates that a substitution decreases protein activity, while a substitution with a positive coefficient increases activity. We found that most amino acid substitutions in GB1 are slightly deleterious, while a smaller subset is highly deleterious (Figure 3a). Each position in the amino acid sequence displayed a range of mutational effects (Figure 3b). Substitutions to proline are the most deleterious on average (presumably because they disrupt protein structure), followed by substitutions to the acidic amino acids. We found a site’s average mutational effect is highly dependent on its location in the three-dimensional structure. Sites with large average mutational effects tend to be located in either the protein core or the IgG binding interface (Figure 3bc).
Figure 3.
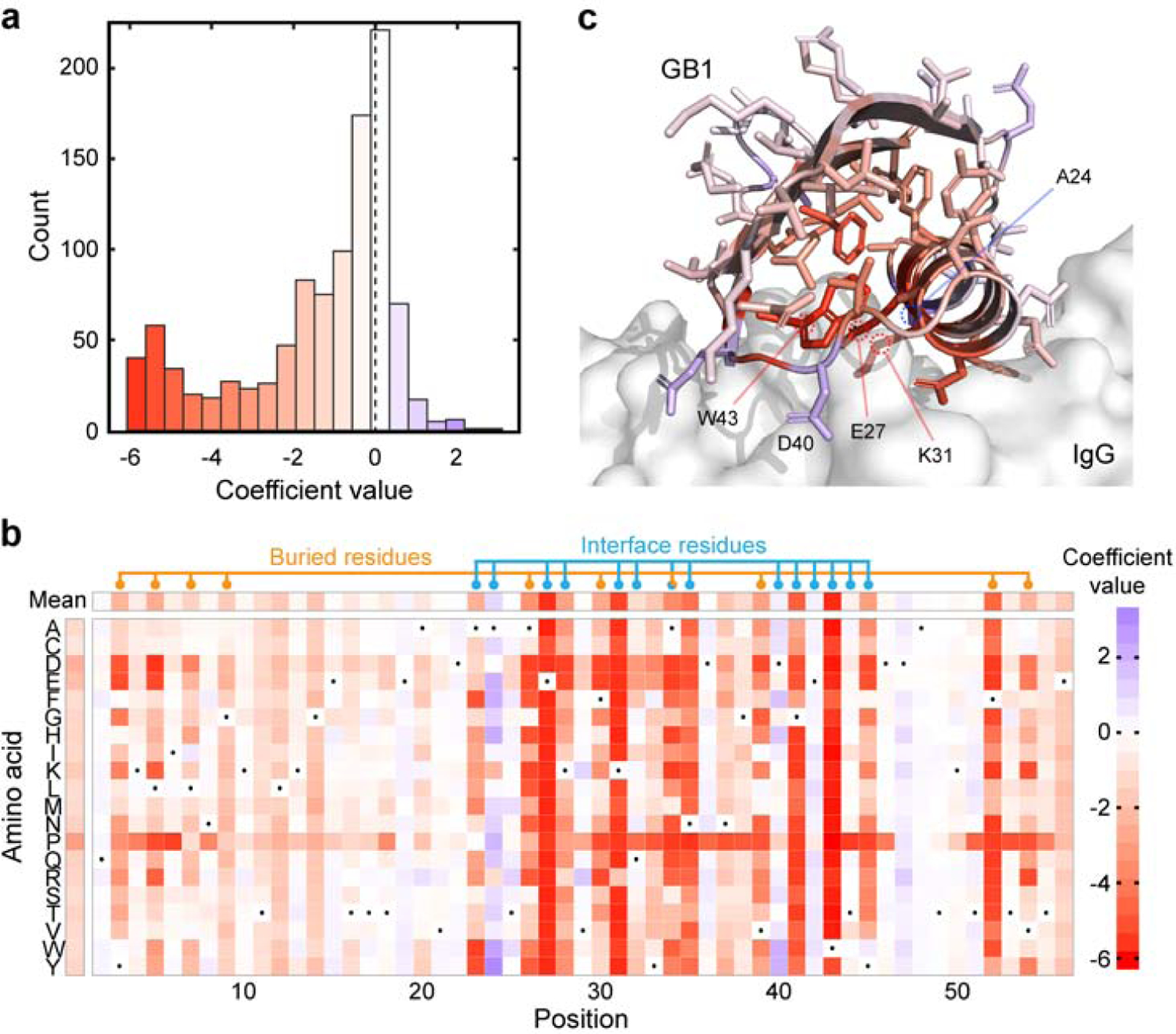
Model parameters relate to GB1 structure and function. (a) The distribution of model coefficients. Most coefficients have a relatively small magnitude, while a substantial fraction of coefficients have a large negative effect. (b) A heatmap of the GB1 model coefficients. The wild-type amino acid is depicted with a black dot. Buried and interface residues tend to have larger magnitude coefficients, indicating their important role in GB1 function. Buried and interface residues were determined from the protein G crystal structure (PDB ID: 1FCC). Buried residues were defined as having a relative solvent accessibility less than 0.1. Interface residues were defined as having a heavy atom within 4Å of IgG. (c) The site-wise average model coefficients mapped onto the protein G crystal structure (PDB ID: 1FCC). The IgG binding partner is depicted as a grey surface. Residues in the protein core and binding interface tend to have the largest average coefficients.
GB1 residues E27, K31, and W43 have the most negative average mutational effect, suggesting that many substitutions at these positions are highly deleterious. Consistent with the model results, these three residues are known to form key hydrogen bonds and salt bridges with the IgG ligand and make the largest contributions to the free energy of binding (Sauer-Eriksson et al. 1995, Sloan & Hellinga 1999). Residues A24 and D40 have the largest positive average mutational effect, with many substitutions that are predicted to increase GB1 activity. Both of these sites are located in the IgG-binding interface. Previous studies have identified residue position 24 to play a key role in IgG binding, and substitutions from A24 can increase binding affinity through improved ionic interactions (Sauer-Eriksson et al. 1995). Furthermore, high-affinity computationally designed protein G variants have substitutions at position 24 (Jha et al. 2014). The model parameters suggest that residue D40 prefers substitution to aromatic amino acids (Figure 3b). Inspection of the crystal structure suggests these mutations could form potential interactions (pi-pi, cation-pi) with a nearby histidine in IgG (Supplemental Figure 4).
Statistics-based protein design
Our PU learning method provides a quantitative description of the sequence-function mapping. The model also captures statistical uncertainties arising from undersampling and correlated sequence variables. Here, we develop a protein design framework that leverages this statistical sequence-function information.
We trained a PU model on a Bgl3 deep mutational scan that had been performed at an elevated temperature (Bgl3-HT, Supplemental Table 1) (Romero et al. 2015). Under these experimental conditions, the positive class corresponds to Bgl3 sequences with a high thermal tolerance, and therefore the model should learn how amino acid substitutions affect thermostability. The learned PU model displayed excellent predictive ability (Corrected AUC of 0.72, Supplemental Figure 5a).
We applied the PU model to design Bgl3 variants based on either coefficient magnitudes or p-values (Figure 4a). The coefficient-based design (Bgl.cf) contained ten amino acid substitutions corresponding to the ten largest positive coefficients. The p-value-based design (Bgl.pv) contained ten substitutions corresponding to the ten positive coefficients with the smallest p-values. We also designed a sequence that contained the ten substitutions with the largest enrichment scores (Bgl.en). The Bgl.cf and Bgl.en designs contained six common substitutions, while the substitutions in Bgl.pv were distinct from the other two (Supplemental Figure 5b). The substitutions within these three designs are generally distributed throughout the protein structure (Figure 4b); however, there appears to be some bias for the coefficient/enrichment designs to choose substitutions in the termini.
Figure 4.

Applying the PU model to design enhanced proteins. (a) A plot of model coefficients versus p-values. Sequences were designed to combine ten mutations with the largest coefficient values, smallest p-values, or largest enrichment scores. (b) The positions chosen by the three design methods are mapped onto the Bgl3 protein structure. The structure is based on the Bgl3 crystal structure (PDB ID: 1GNX) and missing termini/loops were built in using MODELLER (Sali & Blundell 1993). (c) Thermostability curves for wild-type Bgl3 and the three designed proteins. T50 values were estimated by fitting a sigmoid function to the fraction of active enzyme. Note the curve for Bgl.en is shown in yellow and falls directly behind the orange Bgl.cf curve.
We experimentally characterized the thermostability of wild-type Bgl3 and the three designed enzymes. All three designed sequences were stabilized relative to wild-type Bgl3 (Figure 4c). The coefficient and enrichment-based designs displayed modest stability increases (~ 2 °C), while the p-value-based design was almost 12 °C more stable than wild-type Bgl3.
Discussion
We have presented a supervised learning framework to infer the mapping from protein sequence to function from large-scale sequence-function data. We applied a positive-unlabeled (PU) learning approach to address the lack of negative sequence examples typically encountered in deep mutational scanning data. Our PU learning method models a protein’s true functional response as an unobserved latent variable, and then estimates how sequence maps to this latent response by maximizing the observed likelihood. Our approach leverages established statistical methods and hypothesis testing to evaluate the significance of sequence features and predictions. The PU models displayed excellent predictive ability and robustness across ten diverse protein data sets. The learned model parameters capture important aspects of protein structure and function, and can be used to design new and enhanced proteins.
We compared the PU model’s predictive ability to established structure-based and unsupervised learning methods including Rosetta, EVmutation, and DeepSequence. This is a rather unequal comparison because the PU model is trained directly on the DMS data, while the other methods are trained on peripherally related sequence/structure data. As expected, the PU model displayed substantially higher predictive performance than structure-based or unsupervised methods. These other methods have the distinct advantage that they can make reasonable predictions in the absence of DMS data, while our PU model requires DMS data for training. The relative performance of these various predictive methods is likely dependent on the particular protein activity that’s being modeling. We expect Rosetta to capture protein activities related to folding and stability; while EVmutation and DeepSequence may capture preservation of native function and the associated biophysical properties. Along these lines, supervised methods that learn the mapping to a particular property are required to model and predict non-natural protein properties. The ability to predict non-natural properties is essential for designing new proteins with behaviours beyond naturally evolved biological function.
There is a close connection between our PU model’s parameters and the enrichment scores commonly used to evaluate DMS experiments. Both methods estimate how amino acid substitutions affect a protein’s functional response. However, there are two key differences in these estimates: (1) enrichment-based methods estimate the positive-unlabeled response, while our method directly estimates the positive-negative response, and (2) enrichment-based methods estimate marginal amino acid effects and therefore make strong assumptions about the independence between sequence positions. In theory, the parameters estimated using our PU learning method should provide a more accurate and less biased estimate of how amino acid substitutions affect function. We found the PU model had greater predictive ability than enrichment on all ten protein data sets tested. While the differences in predictive performance were small (AUC differences < 0.02), these differences were statistically significant (p < 10−9) in all cases. These results suggest the learned PU model is better overall, but the predictions may not be much different from enrichment. The greatest advantage of the PU model over enrichment is the ability to perform statistical hypothesis testing to evaluate the significance of the model parameters. Hypothesis testing allows us to have confidence in the parameter estimates and predictions, and is thus essential for protein design.
We applied the learned PU models to design beta-glucosidases with improved thermal tolerance. We compared design strategies based on enrichment, PU model coefficients, and p-values. We found that enrichment and coefficient-based methods chose similar substitutions, and the resulting designs had modest increases in thermostability. In contrast, the p-value-based design contained a distinct set of substitutions and was significantly stabilized relative to the wild-type parent sequence. These results suggest that it’s better to design sequences containing high confidence substitutions rather than including uncertain substitutions with the largest magnitudes. In principle, this protein engineering strategy could be implemented iteratively, where a DMS data set is generated from an initial parent sequence, these data are used to design an improved sequence, then a new DMS data set is generated around this improved sequence, and the process is repeated. This iterative sequence optimization approach is similar to directed evolution, however, it fully leverages sequence-function information at each generation, allowing it to take larger jumps in protein sequence space.
There are several interesting extensions of the PU learning framework presented here. In this work we only considered a linear sequence-function mapping. In theory, our modeling framework could be extended to include pairwise or even higher order interactions between residues. These models would account for epistatic interactions between sites, and could possibly be used to determine contacting residues in the protein’s three-dimensional structure. We performed preliminary tests to evaluate whether we could model interactions in DMS data, and found the massive increase in system variables made the computations intractable in most cases. Future work could explore more efficient algorithms for learning from high-dimensional interaction models. Another interesting area to explore is multi-response models that consider several protein properties simultaneously. For example, we could model the Bgl3 room temperature and high temperature data sets simultaneously to directly resolve the residues responsible for protein stability. Finally, our PU modeling approach used a point estimate for a hyperparameter π. A more integrated modeling framework could account for uncertainty in π estimates and how that propagates to model coefficients and p-values.
We applied our PU learning framework to model protein sequence-function relationships. In principle, similar approaches could be used to model genotype-phenotype mappings across any level of biological organization. Positive-unlabeled data arise whenever a population of genetic variants (generated via mutagenesis, crossbreeding, etc.) is passed through a phenotypic screen/selection, and the genotypes from the before/after populations are determined using high-throughput DNA sequencing. This general format has been used to experimentally map genotype-phenotype relationships for promoters/regulatory sequences (Kosuri et al. 2013, Holmqvist et al. 2013), metabolic pathways (Ghosh & Landick 2016), microbial and mammalian genomes (Ehrenreich et al. 2010, Robins et al. 2013, Price et al. 2018, Findlay et al. 2018), and microbial communities (Kehe et al. 2019, Hsu et al. 2019).
A quantitative understanding of the mapping between protein sequence and function is important for describing natural evolution, diagnosing and treating human disease, and designing customized proteins. Advances in experimental technology have enabled researchers to map sequence-function relationships on an unprecedented scale and resolution. The resulting data are challenging to analyze because they’re typically massive, high-dimensional, contain missing sequence information, and lack negative sequence examples. Our PU learning framework provides a principled way of analyzing large-scale sequence-function data to yield biochemical insights and make quantitative predictions.
STAR Methods
Resource Availability
Lead Contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Philip Romero (promero2@wisc.edu).
Materials Availability
Plasmids generated in this are available from the Lead Contact.
Data and Code Availability
This paper analyzes existing, publicly available data. These datasets’ accession numbers are provided in the Key Resource Table and Supplemental Table 1. The PU learning code is publicly available on GitHub: https://github.com/RomeroLab/pudms The scripts used to analyze the data sets and generate the figures reported in this paper are available on GitHub: https://github.com/RomeroLab/PU-learning-paper-analysis Any additional information required to reproduce this work is available from the Lead Contact.”
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Bacterial and Virus Strains | ||
| E coli BL21(DE3) | Lucigen | Cat# 60401–1 |
| Biological Samples | ||
| Chemicals, Peptides, and Recombinant Proteins | ||
| Golden Gate Assembly Kit | New England Biolabs | Cat#: E1601 |
| LB Broth | Fisher Scientific | Cat#: BP9723 |
| LB Agar | Fisher Scientific | Cat#: BP1425 |
| QIAprep Spin Miniprep Kit | Qiagen | Cat#: 27104 |
| MagicMedia E. coli Expression Medium | ThermoFisher Scientific | Cat#: K6803 |
| BugBuster® 10X Protein Extraction Reagent | Millipore | Cat#: 70921 |
| rLysozyme Solution | Millipore | Cat#: 71110 |
| DNase I | New England Biolabs | Cat#: M0303 |
| Potassium phosphate | Sigma Aldrich | Cat#: P5655 |
| 4-Methylumbelliferyl β-D-glucopyranoside | Sigma Aldrich | Cat#: M3633 |
| Critical Commercial Assays | ||
| Deposited Data | ||
| Protein G domain B1 data set | Olson et al. 2014 | SRR2054943,SRR2054944 |
| Pyrrolidine Ketide Synthase data set | Wrenbeck et al. 2019 | SRR8238223,SRR8238221,SRR8238219,SRR8238217,SRR8238222,S RR8238220,SRR8238218,SRR8238216 |
| Levoglucosan kinase data set | Wrenbeck et al. 2019 | SRR8238225,SRR8238229,SRR8238231,SRR8238227,SRR8238224,S RR8238228,SRR8238230,SRR8238226 |
| Small ubiquitin-related modifier 1 data set | Weile et al. 2017 | SRR5680627,SRR5680629,SRR5680640,SRR5680625,SRR5680634,S RR5680638 |
| SUMO-conjugating enzyme UBC9 data set | Weile et al. 2017 | SRR5680697,SRR5680703,SRR5680713,SRR5680715,SRR5680731,S RR5680699,SRR5680705,SRR5680707,SRR5680727,SRR5680733 |
| Thiamin pyrophosphokinase 1 data set | Weile et al. 2017 | SRR5680677,SRR5680635,SRR5680674,SRR5680718,SRR5680717,S RR5680693,SRR5680694,SRR5680669,SRR5680632,SRR5680722,SR R5680719,SRR5680689,SRR5680692,SRR5680670 |
| Hemagglutinin data set | Doud & Bloom 2016 | SRR3113656,SRR3113660 |
| Beta-glucosidase data set | Romero et al. 2015 | SRR11472224, SRR11472223, SRR11472221, SRR11472220, SRR11472219, SRR11472215, SRR11472218, SRR11472216, SRR11472214, SRR11472217, SRR11472222 |
| 1-deoxy-D-xylulose-5-phosphate synthase data set | Romero Lab, UW-Madison | SRR11467347,SRR11467346 |
| Rocker data set | Romero Lab, UW-Madison | SRR12767727,SRR12767726 |
| Protein G domain B1 protein structure | Protein Data Bank | PDB ID: 1FCC |
| Beta-glucosidase protein structure | Protein Data Bank | PDB ID: 1GNX |
| Experimental Models: Cell Lines | ||
| Experimental Models: Organisms/Strains | ||
| Oligonucleotides | ||
| Recombinant DNA | ||
| Beta-glucosidase gene (bgl3) from Streptomyces sp. strain QM-B814 | GenBank | GenBank: CAA82733.1 |
| Software and Algorithms | ||
| Bowtie2 | Langmead & Salzberg 2012 | http://bowtie-bio.sourceforge.net/bowtie2 |
| Rosetta | Alford et al. 2017 | https://www.rosettacommons.org/software/ |
| EVmutation | Hopf et al. 2017 | https://marks.hms.harvard.edu/evmutation/ |
| DeepSequence | Riesselman et al. 2018 | https://github.com/debbiemarkslab/DeepSequence |
| HMMER | Wheeler & Eddy 2013 | http://hmmer.org/ |
| PU learning code | This Paper | https://github.com/RomeroLab/pudms |
| scripts used to analyze the data sets and generate the figures | This Paper | https://github.com/RomeroLab/PU-learning-paper-analysis |
| Other | ||
Method Details
Linear sequence-function model
We model the sequence-function mapping f as a linear function of amino acid sequence. Suppose we have a protein of length L and a protein’s activity can be described as the sum of individual amino acid contributions:
| (6) |
where xi, j, aa ∈ {0, 1} is a binary variable that specifies whether sequence i has amino acid aa at position j, aj, aa specifies the contribution of amino acid aa at position j to protein activity, [L] is the set of all positions in the sequence (1, …, L), and is the set of all 20 amino acids and the stop codon (A, V, …, *). We note that each sequence position takes on one and only one amino acid option ( for all i, j) and therefore the model is over-parameterized. We introduce a reduced model:
| (7) |
where refj is a reference amino acid state for the jth position. This reference sequence is typically the wild-type parent sequence that was used to make the DMS library. It can be shown with simple algebra that , and therefore parameterizes the effect of making an amino acid substitution from the reference state refj to aa at position j.
Our linear sequence-function model can be specified in vector notation as:
| (8) |
where xi is a one-hot encoded vector that specifies a protein’s amino acid sequence:
| (9) |
and the vector θ contains the model parameters:
Here we see that the intercept term parameterizes the activity of the reference sequence relative to the activity threshold t.
Site-wise enrichment captures marginal effects, which are biased
Here we demonstrate that site-wise enrichment scores capture marginal mutational effects that are in general biased for estimating an amino acid substitution’s true effect . Suppose the true positive-negative responses (yi) were available. Enrichment scores can be calculated from this positive-negative (PN) response as:
| (10) |
where P and N are the positive and negative sets of sequences. We demonstrate that are in general biased for .
Recall the logistic model from Eqn 2 in the main text:
| (11) |
where and is a one-hot encoded vector of the sequence i. Maximizing the likelihood function will produce consistent estimates for . Consistent estimates will approach the true value of as the number of data points increases. It is a well-known result in Statistics that the maximum likelihood estimator provides a consistent estimate.
Site-wise enrichment scores calculated from the true PN response (yi) are equivalent to the maximum likelihood estimates (MLE) from a logistic model that considers only one site at a time:
| (12) |
where . Note how this equation only includes terms related to the jth position, in contrast to Eqn 11, which sums over all j ∈ [L].
We demonstrate that the MLE of γj, aa is equal to the enrichment score calculated from the true positive-negative responses . The maximum likelihood estimate solves likelihood equations
and for all aa ≠ refj,
That is,
| (13) |
and for all aa ≠ refj,
| (14) |
where |A| denotes the size of A. Solving Eqn 13 and 14 for γj, aa,
Thus .
In addition, in general because the estimate for γj, aa does not consider other sites and is therefore subject to omitted variable bias (e.g. see Lee (1982)). From this we can conclude that site-wise enrichment scores calculated from the true positive-negative response () provide biased estimates for .
There exists one exception when sequence positions are independent given each response class. In this case, and enrichment scores from the positive-unlabeled response provide consistent estimates . Class conditional independence between positions is unlikely to hold for any DMS data set due to biophysical interactions between sites, correlated sequence variables in multi-mutant sequences, and the nonlinear threshold in the response.
Enrichment-based methods provide attenuated estimates of a mutation’s effect due to latent positive sequences
Here we demonstrate that enrichment scores calculated from positive-unlabeled (PU) responses provide biased estimates of a mutation’s effect. Consider the population enrichment score calculated from the positive-negative (PN) responses:
| (15) |
which correspond to an amino acid substitution’s true marginal effect, i.e. the effect of changing site j from refj to amino acid aa while allowing all other positions to vary. Also consider the population enrichment score calculated from PU responses
| (16) |
We consider to be the true marginal effect, and show that enrichment calculated from the PU response is not equivalent (i.e. ). We also demonstrate that provides an attenuated estimate of the true mutational effect due to latent positive sequences in the unlabeled set.
Since labeled sequences are positive, for any k ∈ (A, V, …, *) we have
On the other hand,
Therefore,
| (17) |
where . Applying (17) to (16) with k ∈ {refj, aa},
To ease notation, let
Then,
Note if and only if . In other words, if and only if . Since by definition, is equivalent to . That is, if and only if . Therefore, underestimates the effect amino acid substitutions with positive effects () and overestimates substitutions with negative effects.
Data preprocessing
We obtained the ten large-scale sequence-function data sets from previously published work (Olson et al. 2014, Wrenbeck et al. 2019, Weile et al. 2017, Doud & Bloom 2016, Romero et al. 2015) and the Sequence Read Archive (SRA) (Leinonen et al. 2011). The details of each data set and their SRA accession numbers are available in Supplemental Table 1, and an overview of the data processing is summarized in Supplemental Figure 2. For each data set, we obtained raw FASTQ files for both unlabeled and positive sequences, and mapped these reads to a reference sequence using Bowtie2 (Langmead & Salzberg 2012). We translated the aligned gene sequences to amino acid sequences, and filtered the data sets to remove any amino acid substitutions that were observed less than ten times.
We used mode imputation to fill in any missing sequence information. Many of the analyzed data sets consisted of partial sequencing fragments (either tiled or random) because the entire gene was too long to cover with a paired-end Illumina read. The remainder of the sequence positions were unobserved. We used mode imputation to replace this missing sequence information. For nearly all DMS data sets, mode imputation simply replaces unobserved positions with the wild-type amino acid.
We converted protein sequence observations to a design matrix X using one-hot encoding. Each row of the design matrix X has the form in (9), where the reference amino acid sequence is taken to be the most frequent amino acid at each position (usually corresponding to the wild-type sequence). The DXS data set consisted of eight recombined gene fragments from one of the four DXS parent sequences (E. coli, B. subtilis, Z. mobilis, P. trichocarpa). These chimeric DXS sequences can be represented as an ordered sequence of “blocks” that indicates which parent the gene fragment was inherited from. We chose the E. coli DXS as the reference and generated dummy variables for each block change from the reference. Each data set resulted in two design matrices corresponding to unlabeled and positive sequences.
PU model training
We trained PU models on the unlabeled and positive sequence sets for each protein data set. We computed the observed likelihood (Eqn 3) for a given data set and a hyperparameter π. We found a stationary point of the negative observed log-likelihood using a Majorization-Minimization (MM) algorithm (Ortega & Rheinboldt 1970, Lange et al. 2000). Specifically, starting from an initial parameter value which corresponds to the null model (no features in the model), we obtained a quadratic majorizer of the negative log-likelihood function at the current parameter value and updated the current parameter with a minimizer of the quadratic majorizer function. Since the majorizer function is greater than the negative likelihood at all points, the minimizer decreases the function value of the negative likelihood compared to the function value evaluated at the current parameter value, i.e. the minimizer increases the likelihood value. This process was repeated until convergence. For the implementation of this process, we have used the PUlasso R package from the Comprehensive R Archive Network (CRAN) (Song & Raskutti 2018), setting the regularization parameter λ = 0 to fit the un-penalized model.
The hyperparameter π was either determined experimentally or tuned to maximize the model’s classification performance. For hyperparameter tuning, we used twenty log-spaced π values ranging from 10−3 to 0.5. For each π value, we trained a model on 90% of the data set, used the model to make predictions on the remaining 10%, and generated a receiver operating characteristic (ROC) curve for the predictions using the labeled/unlabeled response Z. π sets an upper limit on this labeled/unlabeled ROC curve (Supplemental Figure 1), and in some instances the observed ROC curve exceeded this upper limit. These values of π were determined to be infeasible because they resulted in true positive rates greater than the oracle classifier. We selected the π value which resulted in the highest ROC-AUC value among feasible π values.
Aggregating models from multiple replicates
In some instances we had data from multiple replicates that needed to be combined. For example, the Bgl3 high-temp data set had two experimental replicates (R = 2). We trained models on each individual replicate and then aggregated these results into a single model with estimated coefficients and variance-covariance matrix . Let be the estimated coefficients and the variance-covariance matrix of the coefficients from the ith replicate. Here was computed as an inverse of the estimated Fisher information at . The aggregated coefficient and variance-covariance matrix were calculated as follows:
| (18) |
where
We note that the aggregated variance matrix has two components: for the variation in within each replicate and for the variation across different replicates, i.e. . Thus the form of in Eqn 18 is a classical variance decomposition with the extra 1/R factor to account for the finite R (see e.g. Carpenter & Kenward (2013)).
Evaluating and comparing model predictive ability
We used the area under the receiver operating characteristic curve (ROC-AUC) to evaluate the predictive ability of each model. With PU data, we don’t have negative examples and therefore we can’t directly calculate a model’s false positive rate (FPR). Instead, we used the labeled-unlabeled response (Z) to calculate the false positive rate (FPRPU) and true positive rate (TPRPU), and then performed a correction (Jain et al. 2017) to obtain ROC curves and ROC-AUC values:
This PU ROC curve correction is illustrated in Supplemental Figure 1. We obtained corrected ROC curves and ROC-AUC values for each of the ten cross-validation folds, and then averaged over folds to obtain the model’s corrected ROC curve and ROC-AUC value.
We used a corrected repeated cross-validation test to compare the predictive ability between the PU model and site-wise enrichment (Bouckaert & Frank 2004). This test controls inflated type 1 error caused by data overlaps in cross-validation folds (Dietterich 1998, Nadeau & Bengio 2003) and also has high replicability (Bouckaert & Frank 2004). The test involves running K-fold cross validation for R independent runs and comparing models using a corrected test statistic. For each run i = 1, …, R, we split the data randomly into K sub-samples and fit one model for each fold j = 1, …, K. We used R = K = 10 as recommended by the authors (Bouckaert & Frank 2004). Let ROC-AUC(M)ij be the corrected ROC-AUC value for model M from the ith run and jth cross-validation fold. For the enrichment-based predictions, we used an additive model that summed all individual enrichment scores. We define the difference between the models dij ≔ ROC-AUC(PU model)ij − ROC-AUC(enrich)ij and the standard deviation of this difference . The test statistic t was calculated as follows
and was compared with the t distribution with K · R − 1 degrees of freedom.
Predictions using Rosetta, EVmutation, and DeepSequence
We made protein function predictions using established structure-based and unsupervised learning methods including Rosetta, EVmutation, and DeepSequence (Alford et al. 2017, Hopf et al. 2017, Riesselman et al. 2018). For Rosetta modeling, we searched the Protein Data Bank to identify the structure most similar to the DMS data’s reference sequence and used this as a template for Rosetta comparative modeling using the default options (Song et al. 2013). We sampled 500 random sequences from both the unlabeled and positive sequence sets, built Rosetta models for these 1000 sequences, and calculated the Rosetta energy for each. We used these calculated Rosetta energies to create ROC curves classifying unlabeled and positive sets, and corrected these ROC curves to account for PU data.
We made predictions using EVmutation and DeepSequence models for each protein data set. We created family multiple sequence alignments (MSAs) using jackhmmer (Wheeler & Eddy 2013) to query the DMS data set’s reference sequence against the UniRef90 sequence database (Suzek et al. 2015). For DXS, we chose the E. Coli parent as the jackhmmer reference sequence. For Rocker, we had to relax the inclusion threshold (jackhmmer domE option set to 10000) to include additional sequences because Rocker is a de novo designed protein. We filtered the jackhmmer results to remove amino acid insertions relative to the reference sequence and also removed any resulting sequences that had less than 50% coverage over the reference sequence. We trained EVmutation and DeepSequence models on these curated MSAs using the default options. For EVmutation, we scored all sequences in each data set, except for GB1, where we sampled 106 random sequences and DXS, where we sampled 104 random sequences. For DeepSequence, we scored 104 random sequences from each data set. We used the EVmutation and DeepSequence scores to create corrected ROC curves for each data set.
Statistical hypothesis testing
We performed hypothesis tests to determine which features “significantly” affect protein function. We calculated the Z statistic zj to test whether a feature j affects protein function or not (i.e. H0 : θj = 0):
where is the estimated variance-covariance matrix of , computed as the inverse of the estimated Fisher information. We obtained p-values under the null hypothesis that θj = 0 and computing tail probabilities:
where Z is a standard normal variable. These p-values were then adjusted using the Benjamini-Hochberg (BH) procedure to account for multiple hypothesis testing and control the false discovery rate (Benjamini & Hochberg 1995). We considered a feature to be significant if its BH-adjusted p-value was less than 0.05.
Evaluating PU model stability
We used 10-fold cross-validation to evaluate the stability of the fitted PU model’s parameter estimates and selected features. We calculated the coefficient of variation for each feature j across different cross-validation folds:
| (19) |
where is an estimated coefficient for the jth feature from the ith cross-validation fold. The absolute value of this coefficient of variation is a measure of a coefficient’s relative variability (Supplemental Figure 3d).
We evaluated feature selection stability by comparing the set of selected features across different cross-validation folds. We defined a selection stability measure (SS) as the average fraction of common selected features:
where k is the selection size, i and j are different cross-validation folds, and is the set containing features with the k smallest p-values from the ith fold. We computed SSk for k = 1, …, K, where K was chosen to be the number of the significant features (BH-adjusted p < 0.05). We then averaged SSk over k to obtain the average feature selection stability . A value of that approaches one indicates that many common features are selected across different cross-validation folds (Supplemental Figure 3d).
Beta-glucosidase cloning, expression, and characterization
We designed the genes encoding the Bgl3 variants by making codon substitutions into a base Bgl3 gene sequence. If there were multiple codon options for an amino acid, we chose the particular codon randomly from a set of 31 codons that are optimized for expression in E coli (Boël et al. 2016). We ordered the designed genes as gBlocks from IDT and cloned them into a protein expression vector (pET22b) using GoldenGate cloning. We verified the sequences of all genes using Sanger sequencing with the T7 promoter and T7 terminator primers.
We expressed the Bgl3 variants shaking at 30 °C in a 5-mL MagicMedia (Invitrogen) culture overnight. We then pelleted the expression culture by centrifugation and froze at −20 °C. We resuspended the cell pellets in lysis buffer [0.3× BugBuster (Novagen), 30 kU/mL rLysozyme (Novagen), and 50 U/mL DNase I (New England Biolabs) in 100 mM potassium phosphate, pH 7.2] and performed serial dilutions to determine the linear range of the enzyme assay. We then diluted all samples in 100 mM potassium phosphate, pH 7.2 to be within the linear range and have similar end-point activities.
We arrayed the diluted cell extracts into 96-well PCR plates and heated the samples over multiple temperatures (50–75 °C) for 15 min using a gradient thermocycler. After the heat step, we quantified the remaining functional enzyme by adding the fluorogenic substrate 4-methylumbelliferyl-β-D-glucopyranoside (Sigma) to a final concentration of 1 mM. We monitored the reaction progress by fluorescence spectroscopy (372 nm excitation/445 nm emission), and determined the rate by fitting a linear function to the progress curves. We normalized all rates to enzyme samples that had been incubated at room temperature (25 °C). The T50 (temperature where 50% of the protein is inactivated in 15 min) was determined by fitting a shifted sigmoid function to the thermal inactivation curves. All measurements were performed in at least duplicate with the mean T50 values reported.
Quantification and Statistical Analysis
In Figure 2b, the PU model’s Corrected AUC error bars were calculated by taking the standard deviation of AUCs across the ten cross-validation folds. In Figure 2c, we used a corrected repeated cross-validation test to compare the predictive ability between the PU model and site-wise enrichment (Bouckaert & Frank 2004). The full details of this test are given in the Methods Details section. Statistical significance for PU model coefficients was determined using Benjamini-Hochberg (BH) adjusted p-values (Benjamini & Hochberg 1995). We considered a coeffcient to be significant if its BH-adjusted p-value was less than 0.05.
Supplementary Material
Supplemental Table 1: Deep mutational scanning data sets used for PU learning, related to STAR Methods
Highlights.
Statistical learning framework to infer the mapping from protein sequence to function
The models learn key aspects of protein structure and function from large-scale data
The learned models are predictive and can be applied to design new proteins
Acknowledgments
We would like to acknowledge funding support from the NIH grants R35 GM119854 and R01 GM131381, and the NSF grant DMS-1811767.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Declaration of Interests
The authors declare no competing interests.
References
- Abriata LA, Bovigny C & Dal Peraro M (2016), ‘Detection and sequence/structure mapping of biophysical constraints to protein variation in saturated mutational libraries and protein sequence alignments with a dedicated server’, BMC Bioinformatics 17(1). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alford RF, Leaver-Fay A, Jeliazkov JR, O’Meara MJ, DiMaio FP, Park H, Shapovalov MV, Renfrew PD, Mulligan VK, Kappel K, Labonte JW, Pacella MS, Bonneau R, Bradley P, Dunbrack RL, Das R, Baker D, Kuhlman B, Kortemme T & Gray JJ (2017), ‘The Rosetta All-Atom Energy Function for Macromolecular Modeling and Design’, Journal of Chemical Theory and Computation 13(6), 3031–3048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alvizo O, Nguyen LJ, Savile CK, Bresson JA, Lakhapatri SL, Solis EOP, Fox RJ, Broering JM, Benoit MR, Zimmerman SA, Novick SJ, Liang J & Lalonde JJ (2014), ‘Directed evolution of an ultrastable carbonic anhydrase for highly efficient carbon capture from flue gas’, Proceedings of the National Academy of Sciences 111(46), 16436–16441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bedbrook CN, Yang KK, Robinson JE, Mackey ED, Gradinaru V & Arnold FH (2019), ‘Machine learning-guided channelrhodopsin engineering enables minimally invasive optogenetics’, Nature Methods 16(11), 1176–1184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benjamini Y & Hochberg Y (1995), ‘Controlling the false discovery rate: A practical and powerful approach to multiple testing’, J. R. Stat. Soc. Series B Stat. Methodol 57(1), 289–300. [Google Scholar]
- Bloom JD (2015), ‘Software for the analysis and visualization of deep mutational scanning data’, BMC Bioinformatics 16(1). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boël G, Letso R, Neely H, Price WN, Wong KH, Su M, Luff JD, Valecha M, Everett JK, Acton TB, Xiao R, Montelione GT, Aalberts DP & Hunt JF (2016), ‘Codon influence on protein expression in E. coli correlates with mRNA levels’, Nature 529(7586), 358–363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boucher JI, Cote P, Flynn J, Jiang L, Laban A, Mishra P, Roscoe BP & Bolon DN (2014), ‘Viewing protein fitness landscapes through a next-gen lens’, Genetics. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bouckaert RR & Frank E (2004), Evaluating the replicability of significance tests for comparing learning algorithms, in ‘Advances in Knowledge Discovery and Data Mining’, Springer; Berlin Heidelberg, pp. 3–12. [Google Scholar]
- Carpenter J & Kenward M (2013), Multiple Imputation and its Application, John Wiley & Sons. [Google Scholar]
- Dietterich TG (1998), ‘Approximate statistical tests for comparing supervised classification learning algorithms’, Neural Comput. 10(7), 1895–1923. [DOI] [PubMed] [Google Scholar]
- Doud MB & Bloom JD (2016), ‘Accurate measurement of the effects of all amino-acid mutations on influenza hemagglutinin’, Viruses 8(6). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ehrenreich IM, Torabi N, Jia Y, Kent J, Martis S, Shapiro JA, Gresham D, Caudy AA & Kruglyak L (2010), ‘Dissection of genetically complex traits with extremely large pools of yeast segregants’, Nature 464(7291), 1039–1042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elkan C & Noto K (2008), Learning classifiers from only positive and unlabeled data, in ‘Proceedings of the 14th ACM SIGKDD International Conference on Knowledge Discovery and Data Mining’, KDD ‘08, ACM, New York, NY, USA, pp. 213–220. [Google Scholar]
- Findlay GM, Daza RM, Martin B, Zhang MD, Leith AP, Gasperini M, Janizek JD, Huang X, Starita LM & Shendure J (2018), ‘Accurate classification of BRCA1 variants with saturation genome editing’, Nature 562(7726), 217–222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fowler DM & Fields S (2014), ‘Deep mutational scanning: a new style of protein science’, Nature Methods 11, 801–807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghosh IN & Landick R (2016), ‘OptSSeq: High-Throughput Sequencing Readout of Growth Enrichment Defines Optimal Gene Expression Elements for Homoethanologenesis’, ACS Synthetic Biology 5(12), 1519–1534. [DOI] [PubMed] [Google Scholar]
- Holmqvist E, Reimegård J & Wagner EGH (2013), ‘Massive functional mapping of a 5-UTR by saturation mutagenesis, phenotypic sorting and deep sequencing’, Nucleic Acids Research 41(12). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hopf TA, Ingraham JB, Poelwijk FJ, Schärfe CPI, Springer M, Sander C & Marks DS (2017), ‘Mutation effects predicted from sequence co-variation’, Nature Biotechnology 35(2). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hsu RH, Clark RL, Tan JW, Ahn JC, Gupta S, Romero PA & Venturelli OS (2019), ‘Microbial Interaction Network Inference in Microfluidic Droplets’, Cell Systems 9(3), 229–242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jain S, White M & Radivojac P (2017), Recovering true classifier performance in positive-unlabeled learning, in ‘Thirty-First AAAI Conference on Artificial Intelligence’. [Google Scholar]
- Jha RK, Gaiotto T, Bradbury AR & Strauss CE (2014), ‘An improved Protein G with higher affinity for human/rabbit IgG Fc domains exploiting a computationally designed polar network’, Protein Engineering, Design and Selection 27(4), 127–134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kehe J, Kulesa A, Ortiz A, Ackerman CM, Thakku SG, Sellers D, Kuehn S, Gore J, Friedman J & Blainey PC (2019), ‘Massively parallel screening of synthetic microbial communities’, Proceedings of the National Academy of Sciences of the United States of America 116(26), 12804–12809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klesmith JR & Hackel BJ (2019), ‘Improved mutant function prediction via PACT: Protein Analysis and Classifier Toolkit’, Bioinformatics 35(16), 2707–2712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kosuri S, Goodman DB, Cambray G, Mutalik VK, Gao Y, Arkin AP, Endy D & Church GM (2013), ‘Composability of regulatory sequences controlling transcription and translation in Escherichia coli’, Proceedings of the National Academy of Sciences of the United States of America 110(34), 14024–14029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lange K, Hunter DR & Yang I (2000), ‘Optimization transfer using surrogate objective functions’, J. Comput. Graph. Stat 9(1), 1–20. [Google Scholar]
- Langmead B & Salzberg SL (2012), ‘Fast gapped-read alignment with Bowtie 2’, Nature Methods 9(4), 357–359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee L-F (1982), ‘Specification error in multinomial logit models: Analysis of the omitted variable bias’, J. Econom 20(2), 197–209. [Google Scholar]
- Leinonen R, Sugawara H & Shumway M (2011), ‘The Sequence Read Archive’, Nucleic Acids Research 39(Database), D19–D21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liao J, Warmuth MK, Govindarajan S, Ness JE, Wang RP, Gustafsson C & Minshull J (2007), ‘Engineering proteinase K using machine learning and synthetic genes’, BMC Biotechnology 7(1), 16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu B, Dai Y, Li X, Lee WS & Yu PS (2003), Building text classifiers using positive and unlabeled examples, in ‘Third IEEE International Conference on Data Mining’, pp. 179–186. [Google Scholar]
- Mazurenko S, Prokop Z & Damborsky J (2020), ‘Machine-learning-guided directed evolution for protein engineering’, ACS Catalysis 10(2), 1210–1223. [Google Scholar]
- Morcos F, Pagnani A, Lunt B, Bertolino A, Marks DS, Sander C, Zecchina R, Onuchic JN, Hwa T & Weigt M (2011), ‘Direct-coupling analysis of residue coevolution captures native contacts across many protein families’, Proceedings of the National Academy of Sciences 108(49), E1293–E1301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mordelet F & Vert J-P (2011), ‘ProDiGe: Prioritization of disease genes with multitask machine learning from positive and unlabeled examples’, BMC Bioinformatics 12, 389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nadeau C & Bengio Y (2003), ‘Inference for the generalization error’, Mach. Learn 52(3), 239–281. [Google Scholar]
- Olson CA, Wu NC & Sun R (2014), ‘A comprehensive biophysical description of pairwise epistasis throughout an entire protein domain’, Current Biology 24(22), 2643–2651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ortega JM & Rheinboldt WC (1970), Iterative Solution of Nonlinear Equations in Several Variables, SIAM. [Google Scholar]
- Price MN, Wetmore KM, Waters RJ, Callaghan M, Ray J, Liu H, Kuehl JV, Melnyk RA, Lamson JS, Suh Y, Carlson HK, Esquivel Z, Sadeeshkumar H, Chakraborty R, Zane GM, Rubin BE, Wall JD, Visel A, Bristow J, Blow MJ, Arkin AP & Deutschbauer AM (2018), ‘Mutant phenotypes for thousands of bacterial genes of unknown function’, Nature 557(7706), 503–509. [DOI] [PubMed] [Google Scholar]
- Riesselman AJ, Ingraham JB & Marks DS (2018), ‘Deep generative models of genetic variation capture the effects of mutations’, Nature Methods 15(10), 816–822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robins WP, Faruque SM & Mekalanos JJ (2013), ‘Coupling mutagenesis and parallel deep sequencing to probe essential residues in a genome or gene’, Proceedings of the National Academy of Sciences of the United States of America 110(9), E848–E857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Romero PA, Krause A & Arnold FH (2013), ‘Navigating the protein fitness landscape with Gaussian processes’, Proceedings of the National Academy of Sciences of the United States of America 110(3), E193–E201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Romero PA, Tran TM & Abate AR (2015), ‘Dissecting enzyme function with microfluidic-based deep mutational scanning’, Proceedings of the National Academy of Sciences of the United States of America 112(23), 7159–7164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sali A & Blundell TL (1993), ‘Comparative Protein Modelling by Satisfaction of Spatial Restraints’, Journal of Molecular Biology 234(3), 779–815. [DOI] [PubMed] [Google Scholar]
- Sauer-Eriksson AE, Kleywegt GJ, Uhlén M & Jones TA (1995), ‘Crystal structure of the C2 fragment of streptococcal protein G in complex with the Fc domain of human IgG’, Structure 3(3), 265–278. [DOI] [PubMed] [Google Scholar]
- Sloan DJ & Hellinga HW (1999), ‘Dissection of the protein G B1 domain binding site for human IgG Fc fragment’, Protein Science 8(8), 1643–1648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Song H, Dai R, Raskutti G & Barber RF (2019), ‘Convex and non-convex approaches for statistical inference with noisy labels’, ArXiv e-prints. [Google Scholar]
- Song H & Raskutti G (2018), ‘PUlasso: High-dimensional variable selection with presence-only data’, J. Am. Stat. Assoc pp. 1–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Song Y, Dimaio F, Wang RYR, Kim D, Miles C, Brunette T, Thompson J & Baker D (2013), ‘High-resolution comparative modeling with RosettaCM’, Structure 21(10), 1735–1742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suzek BE, Wang Y, Huang H, McGarvey PB & Wu CH (2015), ‘UniRef clusters: A comprehensive and scalable alternative for improving sequence similarity searches’, Bioinformatics 31(6). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ward G, Hastie T, Barry S, Elith J & Leathwick JR (2009), ‘Presence-only data and the em algorithm’, Biometrics 65(2), 554–563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weile J & Roth FP (2018), ‘Multiplexed assays of variant effects contribute to a growing genotype–phenotype atlas’, Human Genetics 137(9), 665–678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weile J, Sun S, Cote AG, Knapp J, Verby M, Mellor JC, Wu Y, Pons C, Wong C, Lieshout N, Yang F, Tasan M, Tan G, Yang S, Fowler DM, Nussbaum R, Bloom JD, Vidal M, Hill DE, Aloy P & Roth FP (2017), ‘A framework for exhaustively mapping functional missense variants’, Molecular Systems Biology 13(12), 957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wheeler TJ & Eddy SR (2013), ‘Nhmmer: DNA homology search with profile HMMs’, Bioinformatics 29(19). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wrenbeck EE, Bedewitz MA, Klesmith JR, Noshin S, Barry CS & Whitehead TA (2019), ‘An Automated Data-Driven Pipeline for Improving Heterologous Enzyme Expression’, ACS Synthetic Biology 8(3), 474–481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wrenbeck EE, Faber MS & Whitehead TA (2017), ‘Deep sequencing methods for protein engineering and design’, Current Opinion in Structural Biology 45, 36–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang KK, Wu Z & Arnold FH (2019), ‘Machine-learning-guided directed evolution for protein engineering’, Nature Methods 16(8), 687–694. [DOI] [PubMed] [Google Scholar]
- Yi J, Hsieh C-J, Varshney KR, Zhang L & Li Y (2017), Scalable Demand-Aware recommendation, in Guyon I, Luxburg UV, Bengio S, Wallach H, Fergus R, Vishwanathan S & Garnett R, eds, ‘Advances in Neural Information Processing Systems 30’, Curran Associates, Inc., pp. 2412–2421. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplemental Table 1: Deep mutational scanning data sets used for PU learning, related to STAR Methods
Data Availability Statement
This paper analyzes existing, publicly available data. These datasets’ accession numbers are provided in the Key Resource Table and Supplemental Table 1. The PU learning code is publicly available on GitHub: https://github.com/RomeroLab/pudms The scripts used to analyze the data sets and generate the figures reported in this paper are available on GitHub: https://github.com/RomeroLab/PU-learning-paper-analysis Any additional information required to reproduce this work is available from the Lead Contact.”
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Bacterial and Virus Strains | ||
| E coli BL21(DE3) | Lucigen | Cat# 60401–1 |
| Biological Samples | ||
| Chemicals, Peptides, and Recombinant Proteins | ||
| Golden Gate Assembly Kit | New England Biolabs | Cat#: E1601 |
| LB Broth | Fisher Scientific | Cat#: BP9723 |
| LB Agar | Fisher Scientific | Cat#: BP1425 |
| QIAprep Spin Miniprep Kit | Qiagen | Cat#: 27104 |
| MagicMedia E. coli Expression Medium | ThermoFisher Scientific | Cat#: K6803 |
| BugBuster® 10X Protein Extraction Reagent | Millipore | Cat#: 70921 |
| rLysozyme Solution | Millipore | Cat#: 71110 |
| DNase I | New England Biolabs | Cat#: M0303 |
| Potassium phosphate | Sigma Aldrich | Cat#: P5655 |
| 4-Methylumbelliferyl β-D-glucopyranoside | Sigma Aldrich | Cat#: M3633 |
| Critical Commercial Assays | ||
| Deposited Data | ||
| Protein G domain B1 data set | Olson et al. 2014 | SRR2054943,SRR2054944 |
| Pyrrolidine Ketide Synthase data set | Wrenbeck et al. 2019 | SRR8238223,SRR8238221,SRR8238219,SRR8238217,SRR8238222,S RR8238220,SRR8238218,SRR8238216 |
| Levoglucosan kinase data set | Wrenbeck et al. 2019 | SRR8238225,SRR8238229,SRR8238231,SRR8238227,SRR8238224,S RR8238228,SRR8238230,SRR8238226 |
| Small ubiquitin-related modifier 1 data set | Weile et al. 2017 | SRR5680627,SRR5680629,SRR5680640,SRR5680625,SRR5680634,S RR5680638 |
| SUMO-conjugating enzyme UBC9 data set | Weile et al. 2017 | SRR5680697,SRR5680703,SRR5680713,SRR5680715,SRR5680731,S RR5680699,SRR5680705,SRR5680707,SRR5680727,SRR5680733 |
| Thiamin pyrophosphokinase 1 data set | Weile et al. 2017 | SRR5680677,SRR5680635,SRR5680674,SRR5680718,SRR5680717,S RR5680693,SRR5680694,SRR5680669,SRR5680632,SRR5680722,SR R5680719,SRR5680689,SRR5680692,SRR5680670 |
| Hemagglutinin data set | Doud & Bloom 2016 | SRR3113656,SRR3113660 |
| Beta-glucosidase data set | Romero et al. 2015 | SRR11472224, SRR11472223, SRR11472221, SRR11472220, SRR11472219, SRR11472215, SRR11472218, SRR11472216, SRR11472214, SRR11472217, SRR11472222 |
| 1-deoxy-D-xylulose-5-phosphate synthase data set | Romero Lab, UW-Madison | SRR11467347,SRR11467346 |
| Rocker data set | Romero Lab, UW-Madison | SRR12767727,SRR12767726 |
| Protein G domain B1 protein structure | Protein Data Bank | PDB ID: 1FCC |
| Beta-glucosidase protein structure | Protein Data Bank | PDB ID: 1GNX |
| Experimental Models: Cell Lines | ||
| Experimental Models: Organisms/Strains | ||
| Oligonucleotides | ||
| Recombinant DNA | ||
| Beta-glucosidase gene (bgl3) from Streptomyces sp. strain QM-B814 | GenBank | GenBank: CAA82733.1 |
| Software and Algorithms | ||
| Bowtie2 | Langmead & Salzberg 2012 | http://bowtie-bio.sourceforge.net/bowtie2 |
| Rosetta | Alford et al. 2017 | https://www.rosettacommons.org/software/ |
| EVmutation | Hopf et al. 2017 | https://marks.hms.harvard.edu/evmutation/ |
| DeepSequence | Riesselman et al. 2018 | https://github.com/debbiemarkslab/DeepSequence |
| HMMER | Wheeler & Eddy 2013 | http://hmmer.org/ |
| PU learning code | This Paper | https://github.com/RomeroLab/pudms |
| scripts used to analyze the data sets and generate the figures | This Paper | https://github.com/RomeroLab/PU-learning-paper-analysis |
| Other | ||