

Abstract
Prognosis for patients with recurrent and/or metastatic head and neck squamous cell carcinoma (HNSCC) remains poor. Development of more effective and less toxic targeted therapies is necessary for HNSCC patients. Checkpoint kinase 1 (CHK1) plays a vital role in cell cycle regulation and is a promising therapeutic target in HNSCC. Prexasertib, a CHK1 inhibitor, induces DNA damage and cell death, however, its effect on the tumor immune microenvironment (TIME) is largely unknown. Therefore, we evaluated a short-term and long-term effects of prexasertib in HNSCC and its TIME. Prexasertib caused increased DNA damage and cell death in vitro and significant tumor regression and improved survival in vivo. The gene expression and multiplex immunohistochemistry (mIHC) analyses of the in vivo tumors demonstrated increased expression of genes that are related to T-cell activation and increased immune cell trafficking, and decreased expression of genes that related to immunosuppression. However, increased expression of genes related to immunosuppression emerged over time suggesting evasion of immune surveillances. These findings in gene expression analyses were confirmed using mIHC which showed differential modulation of TIME in the tumor margins and as well as cores over time. These results suggest that evasion of immune surveillance, at least in part, may contribute to the acquired resistance to prexasertib in HNSCC.
Keywords: Head and neck squamous cell carcinoma (HNSCC), Prexasertib, Multiplex immunohistochemical staining, Gene expression, Tumor immune microenvironment
1. INTRODUCTION
Head and neck squamous cell carcinoma (HNSCC) is the sixth most common cancer worldwide with a global estimate of more than 650,000 new cases and 330,000 deaths annually.1 The common risk factors for HNSCC are the consumption of tobacco and alcohol, and human papillomavirus (HPV) infection.2,3 Despite improvements in multi-modality therapies including radiation, surgery, chemotherapy, and most recently immunotherapy, the five-year survival rate of HNSCC patients remains poor due to its recurrence and metastasis.4 Therefore, novel therapies are urgently needed for HNSCC.
The host immune system plays a central role in cancer development, and the nascent transformed cells can be initially removed by activation of innate and adaptive immune responses. The cellular components of innate immune response include macrophages, dendritic cells (DCs), nature killer (NK) cells, neutrophils, eosinophils, and mast cells whereas the adaptive immune response involves B-cells and T-cells. Increasing evidence suggests that the tumor immune microenvironment (TIME) of HNSCC is immunosuppressive, and the tumor-mediated immune suppression may play an important role in HNSCC progression and treatment resistance.5,6 The balance of anti-tumor effector cells such as CD8+ cytotoxic T-cells, CD4+ helper T-cells, and NK cells, and immunosuppressive cells such as FOXP3+ regulatory T-cells (Treg) and myeloid-derived suppressor cells (MDSC) determines whether an individual’s TIME is associated with productive anti-tumor immunity or evasion of immune surveillance by tolerance.7
Checkpoint kinase 1 (CHK1) plays a vital role in cell cycle regulation and is a promising therapeutic target in HNSCC.8 CHK1 is a key regulatory protein that gets activated by the ataxia-telangiectasia and RAD3-related protein (ATR) upon DNA damage.9 Prexasertib (LY2606368) is a selective ATP competitive small molecule inhibitor of CHK1 and has limited activity against CHK2. Prexasertib mediated CHK1 inhibition results in abrogation of cell cycle arrest, leading to double-strand DNA breaks, increased replication stress and eventually leading to catastrophic cell death.10 The effects of prexasertib in cancer cells as a single agent or in combination with other agents have been evaluated in clinical and preclinical models in different cancer types including HNSCC,11–14 however, we have a limited understanding about its effect on the TIME. A recent study has reported that prexasertib treatment results in modulation of TIME by increasing cytotoxic T-cell infiltration and activation.15
In this study, we hypothesized that prexasertib mediated killing of cancer cells would result in release of tumor specific neoantigens, which would be recognized by the host immune system and trigger anti-tumor immune response. To test our hypothesis, we used syngeneic mouse model of HNSCC and observed that treatment with prexasertib resulted in significant tumor regression and improved survival. Interestingly, within few days of prexasertib treatment, we found increased expression of markers for T-cell activation, cytokines and chemokines, and an increased infiltration of inflammatory DCs, all associated with pro-inflammatory TIME. As the treatment continued for three weeks, we observed complete response in some tumors, however, a subset of mice harbored persistent tumors that suggest escaping the host immune surveillance. In these tumors, we found an increased expression and infiltration of several immunosuppressive markers including Tregs and tumor-associated neutrophils. In summary, our results suggest that prexasertib treatment initially recruits pro-inflammatory cells following immune activation by prexasertib-induced cell death. However, treatment resistant cells may emerge over time due to a switch from pro-inflammatory to immunosuppressive TIME.
2. MATERIALS AND METHODS
2.1. Cell lines
For this study, we used two mouse cell lines; 1) mouse tonsil epithelial cells (MTECs) transformed with HRAS expression and short hairpin RNA (shRNA) against Ptpn13 (protein tyrosine phosphatase, nonreceptor type 13) to model non-HPV-related HNSCC (MTE-Ras) and 2) MTECs transformed with HRAS and E6/E7 expression to model HPV-related HNSCC (MEER). Both cell lines were obtained from Dr. John Lee.16,17 Short tandem repeat analysis was performed to authenticate cell lines before use. The cells were periodically monitored for mycoplasma.
2.2. Western blot analysis
Lysates were prepared from mouse cell lines or tumors using M-PER lysis buffer with addition of Roche Complete Protease Inhibitor Cocktail tablet (Sigma Millipore), Phosphatase Inhibitor Cocktails 2 and 3 (Sigma Millipore), DTT (Sigma Millipore) and serine protease inhibitor AEBSF (Sigma Millipore). Protein concentration was determined by the bicinchoninic acid (BCA) Assay Kit (Thermo Fisher Scientific). Proteins from each sample were resolved under reducing conditions using NuPAGE Bis-Tris Precast gels (Novex; Invitrogen), transferred to PVDF membrane (Sigma Millipore), and immunostained overnight using antibodies detailed in Supplemental File. Blots were developed by chemiluminescence using the ECL Western blotting Detection System (GE Healthcare). Immunoblot signals were detected using Amersham Hyperfilm. Densitometric quantification for the immunoblots was done by using ImageJ, and the numbers represent protein intensity after normalizing with GAPDH or Actin. The additional methods for proliferation, colony formation, apoptosis, and cell cycle assays are also described in Supplemental File.
2.3. In vivo tumor growth inhibition
All experiments involving mice were done according to a protocol reviewed and approved by the Institutional Animal Care and Use Committee at the University of South Florida. 0.5×106 MTE-Ras cells or MEER cells in PBS were injected subcutaneously into the right flank of 5–6 week old male C57BL/6 mice. When tumor volumes arrived at approximately 80–100mm3 in size (80mm3 for MEER and 100mm3 for MTE-Ras; Day 0), the mice were randomly divided into control (20% captisol) and prexasertib treatment (10 mg/kg prexasertib) groups. Prexasertib was administered (Day 1) subcutaneously twice daily for 3 days, followed by four days of rest (1 cycle = 7 days) and repeated for an additional two cycles. Tumor size and body weight were measured biweekly and compared between control and drug treated groups. Tumor volumes were measured using calipers (volume=0.52 × length × width2). The following criteria were considered as survival endpoints: death or requiring euthanasia per the protocol due to tumor volume of >2000mm3, signs of lethargy or pain, weight loss >20% baseline body weight, and/or tumor ulceration >0.5cm. Tumor tissues were collected upon euthanasia. Tumors were snap-frozen in liquid nitrogen for further analysis or fixed in 10% buffered formalin and processed for paraffin embedding and sectioning. GraphPad Prism was used to generate survival curves.
2.4. Multiplex immunohistochemistry (mIHC)
To perform mIHC, formalin-fixed paraffin-embedded (FFPE) mouse tissues were sectioned at 4 microns onto charged AutoFrost IHC enhanced coated hydrophilic slides and baked for 1 hour at 60°C. The slides were then deparaffinized in xylene and rehydrated in gradient ethanol [3x (100%, 90%, 70%) and water; 5 min each]. Firstly, nuclei were stained using Hematoxylin # SH26 (Fisher Scientific) and stained slides were covered with micro cover glass # 48393081 (VWR) using distilled water to prevent tissue drying, and immediately scanned using digital pathology scanner Aperio CS2 (Leica Biosystems). Slides were destained from hematoxylin by washing with 1% HCL for 1 min and following this antigen retrieval was performed with citrate buffer (pH=6.0) # ab93678 (Abcam) or EDTA (pH=8.0) # ab93678 (Abcam), using steamer (95–100◦C) for 30 min. The slides were incubated in 3% hydrogen peroxide # H325–500 (Fisher Scientific) twice for 15 min each to block endogenous peroxidase activity followed by serum # 55984 (MP Biomedicals) incubation for 20 min at room temperature (RT). The first primary antibody was applied for 60 min at RT or 4◦C overnight followed by three washes using wash buffer # K8007 (Dako). The slides were incubated with appropriate secondary universal immuno-peroxidase polymer, anti-rabbit # 414141F (Nichirei Bioscience) or anti-rat # 414131F (Nichirei Bioscience) antibody for 30 min, followed by washing × 3, and the chromogenic signal was detected using AEC # 415184F (Nichirei Bioscience). Slides were scanned using Aperio CS2 as described above. For subsequent staining with the next antibody, slides were washed in gradient ethanol (70%, 90%, 100%; 2 min each and 100%, 90%, 70%, water; 5 min each), followed by antigen retrieval for 30 min and serum blocking 15 min and the above staining procedure was repeated seven times.
For mIHC, two slides were stained for each tumor. The first slide was stained sequentially with the following markers – Hematoxylin, FOXP3 # 12653 (Cell Signaling Technlogy), CD8 # 98941 (Cell Signaling Technology), GranzymeB (GrzB) # ab4059 (Abcam), B220 # NB100–77420 (Novus Biologicals), CD3 # ab5690 (Abcam) and CD45 # 70257 (Cell Signaling Technology). The second slide was stained sequentially with the following markers - Hematoxylin, PD-L1 # 64988 (Cell Signaling Technology), Ly-6G # ab25377 (Abcam), CD11c # 97585 (Cell Signaling Technology), F4/80 # ab111101 (Abcam) and CD11b # ab133357 (Abcam). To optimize the sequence of antibodies, a single slide was subsequently stained with the same antibody for at least eight times and evaluated potential antigen loss upon each sequential staining using the protocol as described above.
2.5. TCGA HNSCC Cohort
The Pan-Cancer Cohort of the Cancer Genome Atlas (TCGA) HNSCC normalized RNA-seq (processed version L4, data_RNA_Seq_V2_expression_median.txt) and clinical dataset were downloaded from the cBioPortal (n = 523 samples). 415 HPV-negative patients were selected and included in the analysis. CHK1 expression for these patients were Z-transformed (mean = 0, standard deviation = 1), and a patient is classified as CHK1 high (CHK1-hi) expression if Z-score > 1 (n=66) and CHK1 low (CHK1-lo) expression if Z-score < −1 (n=59). Gene Set Enrichment Analysis (GSEA) was performed by comparing the CHK1-hi and CHK1-lo gene expressed patients.
2.6. GSEA
GSEA was performed using the GSEA toolkit version 4.018 against Hallmark of Cancer and prexasertib-induced immune signature. False discovery rate (FDR) and p-value for the enriched pathways were estimated by performing 1000 gene-set permutations, and gene sets with FDR <10% were considered as significant in this study.
3. RESULTS
3.1. Prexasertib increases DNA damage and cell death in HNSCC
To validate the on-target effects of prexasertib in HNSCC, we first examined the inhibitory effects of prexasertib on CHK1. We used two cell lines, MTE-Ras cells to model non-HPV-related HNSCC and MEER cells to model HPV-related cancer and test any distinct effects on E6 and E7 driven HNSCC. We performed immunoblot analysis to compare total CHK1 and phosphorylated CHK1 (S296) levels following prexasertib treatment and found a decrease in CHK1 phosphorylation at S296, indicating that prexasertib inhibited CHK1 activation in MTE-Ras cells (Figure 1A) and MEER cells (Figure 1B). Interestingly, we also found a decrease in total CHK1 levels following prexasertib treatment (Figure 1A,B). Similar observation has been made before in HPV-negative UM-SCC1 HNSCC cell line.19
FIGURE 1.
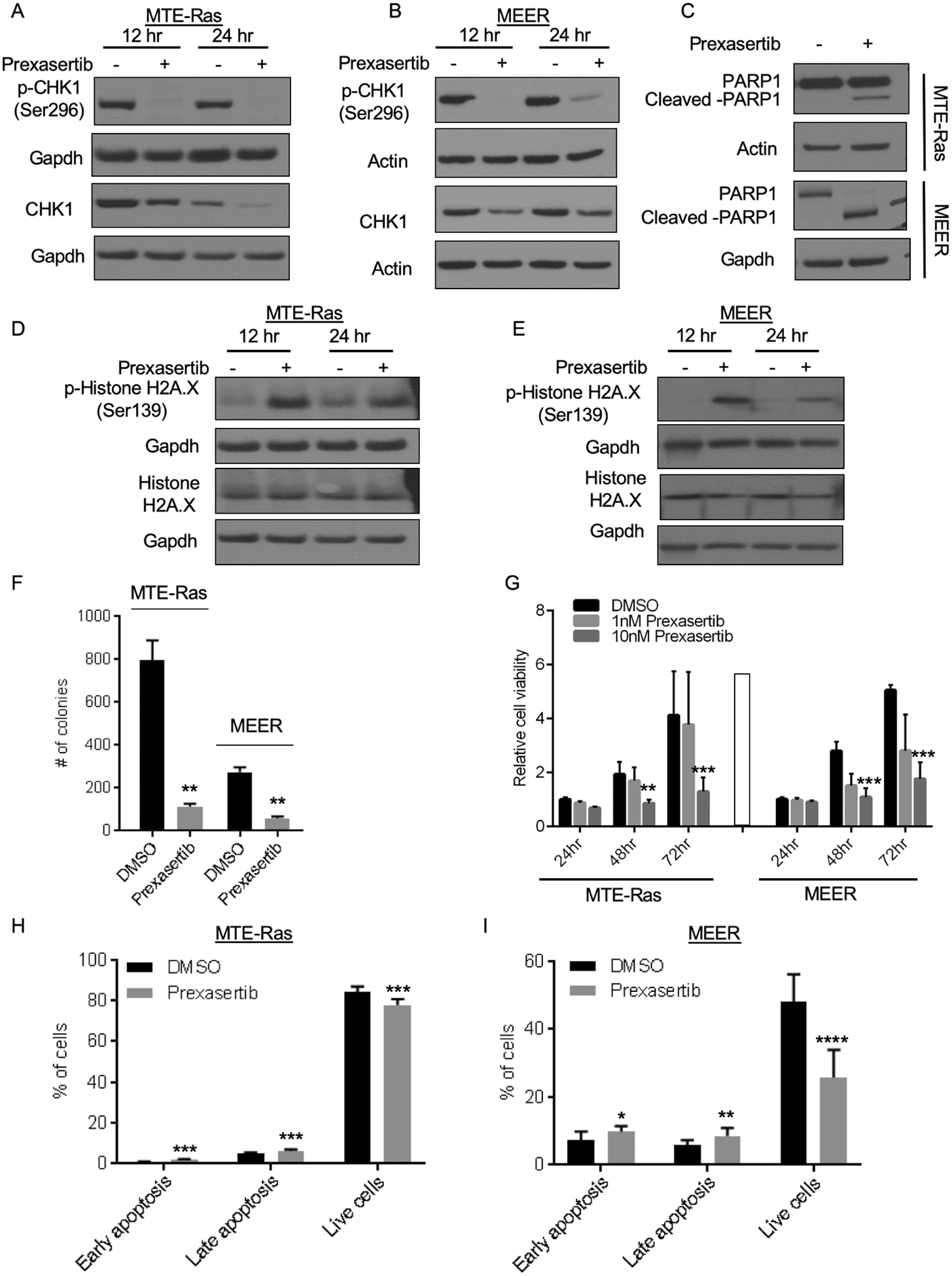
Prexasertib causes DNA damage in the cancer cells. (A,B) Immunoblot analysis showing prexasertib is a CHK1 inhibitor and treatment with prexasertib abolished CHK1 activity as measured by reduced p-CHK1 (Ser296) in MTE-Ras (A) and MEER (B) cells. GAPDH and Actin are used a loading controls. (C) Immunoblot analysis shows that prexasertib induces DNA damage in MTE-Ras and MEER cells as measured by increased levels of PARP1 (C) and p-Histone H2A.X (D,E) These are representative western blots from at least 3 biological replicates. (F) MTE-Ras or MEER cells were treated with DMSO or 10nM prexasertib for 24 hours and colony formation assays were performed. Error bars represent standard deviation (SD) from three independent wells of a representative biological replicate. (G) Cell proliferation assays were performed by treating MTE-Ras or MEER cells with DMSO or 1nM or 10nM prexasertib for the indicated times. Error bars represent SD from four biological replicates and each biological replicate had four technical replicates. (H,I) MTE-Ras cells were treated with DMSO or 10nM prexasertib and MEER cells were treated with DMSO or 5nM prexasertib for 48 hours and the percentage of apoptotic cells was determined by performing Annexin V staining in conjunction with PI staining followed by FACS analysis. Error bars represent SD from three biological replicates and each biological replicate had three-four technical replicates.
The P values were determined by performing paired t test (F) or 2way ANOVA (G) or Welch’s t test (H,I). *p<0.05; ** p<0.01; ***p<0.0001.
We next examined DNA damage by measuring PARP1 levels following prexasertib treatment. PARP1 immunoblot analysis revealed PARP1 cleavage at 24 hours in both cell lines (Figure 1C), indicating presence of DNA damage and replication stress in response to prexasertib treatment. To further evaluate the amount of DNA damage caused by prexasertib treatment, we measured the γH2AX levels, a marker of DNA double-strand break and found significant increase in γH2AX levels in both cell lines (Figure 1D,E). In addition, we observed subsequent apoptosis indicated by increased cleaved caspase-3 after prexasertib treatment (Fig.S1A,1B). Together, these experiments confirm that prexasertib causes DNA damage and apoptosis in HNSCC.
Furthermore, we assessed the effects of prexasertib on cancer cell survival by performing a colony formation assay after 24 hours of prexasertib treatment. In both MTE-Ras and MEER cells, we observed that prexasertib treatment significantly impacted the ability of cells to grow and form colonies (Figure 1F; Fig.S1C) indicating that prexasertib effects are similar in both cell lines under these conditions.
To further assess the cytotoxicity of prexasertib, we performed cell proliferation assays. Prexasertib treatment of MTE-Ras or MEER cells resulted in significant decrease in cell proliferation at 48 hours that continued to 72 hours, suggesting that prexasertib exerts anti-proliferative effects against these cells (Figure 1G). Given the key role of CHK1 in regulating G2/M phase of cell cycle, we evaluated the effect of prexasertib mediated CHK1 inhibition on the different phases of cell cycle. We treated the MTE-Ras and MEER cells with prexasertib and examined the effect on cell cycle by flow cytometry. We observed that prexasertib treatment resulted in a significant increase in percentage of cells arrested in G2/M phase of cell cycle and a corresponding decrease in the G1 phase in both MTE-Ras and MEER cells (Fig.S1D,1F). These results indicate that prexasertib leads to an increase in G2/M accumulation and thus prevents the re-entry of cells into G1 phase possibly due to increased DNA damage. To further evaluate the cells undergoing apoptosis, we performed AnnexinV and PI staining. In both MTE-Ras cells (Figure 1H) and MEER (Figure 1I), we observed a significant increase in percentage of apoptotic cells and a significant decrease in the percentage of live cells. Altogether, these results indicate that prexasertib-mediated CHK1 inhibition induces DNA damage, cell cycle arrest, and eventually leads to apoptotic cell death.
3.2. Prexasertib treatment reduces HNSCC tumor growth and improves survival in vivo
To test the efficacy of prexasertib in vivo, MTE-Ras cells were implanted subcutaneously in immunocompetent C57BL/6 mice (control n=13, prexasertib n=12) and were allowed to form tumors. Tumor-bearing mice with an average tumor size of 100mm3 were treated with prexasertib twice a day for 3 days per week for 3 weeks (Day 1; Fig.S2A). During the course of treatment, three mice were randomly removed at Day 4, Day 23, and survival endpoints (as described in Materials and Methods) from both the groups for molecular analyses (Fig.S2A). We first confirmed the on-target effect of prexasertib in vivo by performing an immunoblot analysis for p-CHK1 and total CHK1 and observed that prexasertib treatment successfully inhibited CHK1 in vivo (Figure 2A; Fig.S2B). Moreover, cleaved caspase-3 levels were increased in the prexasertib-treated tumors (Figure 2A; Fig.S2B).
FIGURE 2.
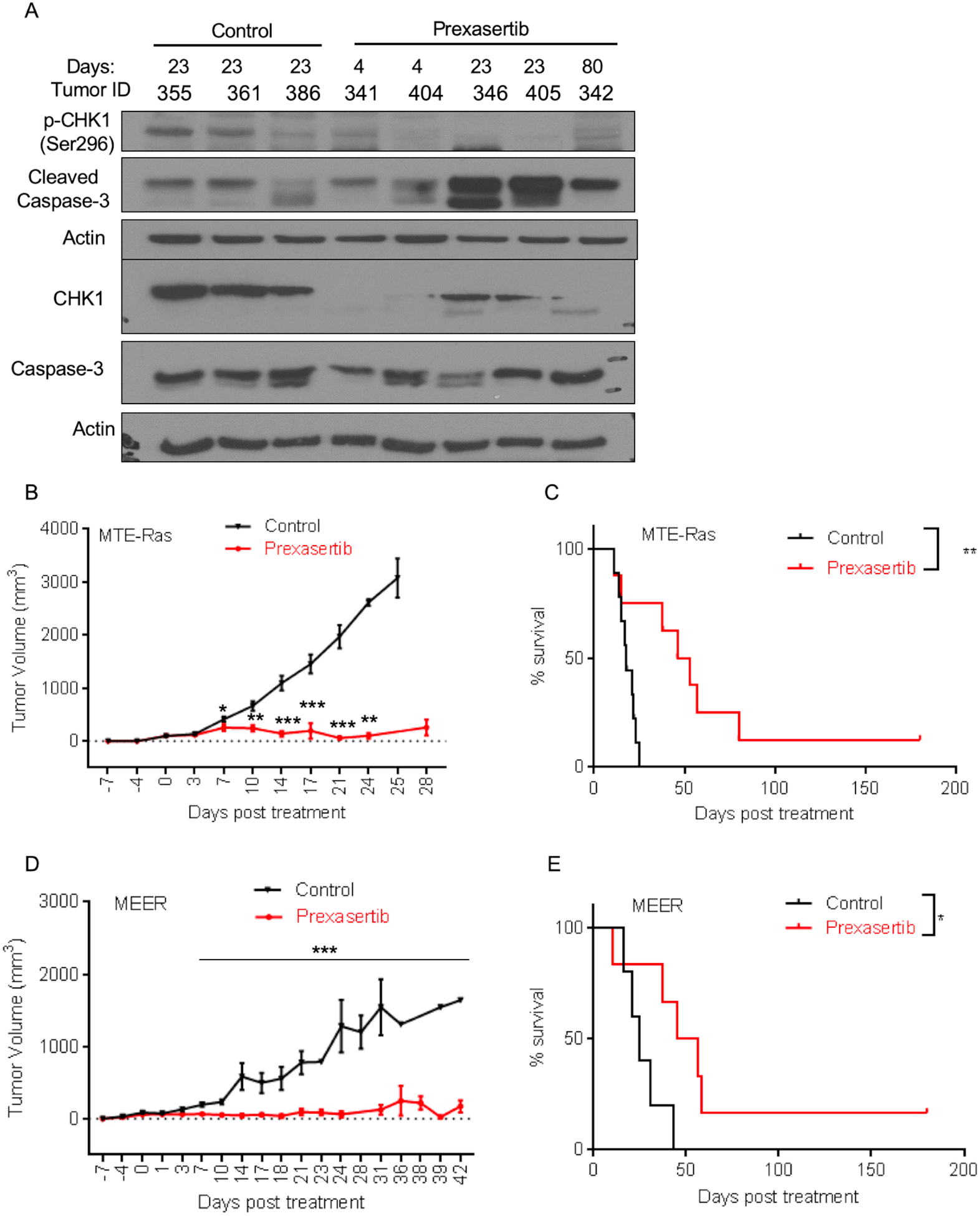
Prexasertib causes significant tumor regression and increases survival in a syngeneic mouse model of HNSCC. (A) Immunoblot analysis of p-CHK1(Ser296) and cleaved caspase-3 in the MTE-Ras implanted mice tumors. (B,D) Tumor volume changes (mean ± SEM) in mice injected with MTE-Ras (B) or MEER (D) cells and treated with control or prexasertib. (C,E) Survival of mice injected with MTE-Ras (C) or MEER (E) cells and treated with control or prexasertib. Error bars represent SD from individual mice. The P values were determined by performing Mann-Whitney test. *p<0.05; ** p<0.01; ***p<0.0001.
As compared to the control group, in the prexasertib treatment group we observed a statistically significant decrease in the tumor volume after the first cycle of prexasertib (Day 7) (Figure 2B). After two cycles (Day 14), we found a statistically significant 8-fold decrease including complete responses in 2 of 12 mice (Figure 2B,C; Fig.S2C). Interestingly, after three cycles of prexasertib (Days 23–25), 9 of 12 mice survived, and 4 had a complete response with a significant 30-fold decrease in the tumor volume as compared to the control group. Whereas in the control group, 7 of 13 mice survived at Day 21, and they all reached the survival endpoints by Day 25. We continue to monitor the tumor growth and mice survival in the prexasertib-treated group up to 180 days and at that time point there were 3 of 12 mice survived with a complete response (Figure 2C). Consistent with the tumor data, we found that the overall survival rate in the prexasertib-treated group was significantly higher than in the control group.
We next performed an in vivo study by subcutaneously implanting MEER in the immunocompetent C57BL/6 mice (control n=12, prexasertib n=12). Tumor-bearing mice with an average tumor size of 80mm3 were treated with prexasertib, and the same treatment regime was followed as described above (Fig.S2A). After one cycle of prexasertib treatment, we found statistically significant decrease in the tumor volume, similar to what we observed in the tumors with MTE-Ras cells. Similarly, after two cycles of prexasertib treatment, there was a significant 13-fold decrease in tumor volume as compared to control mice. After three cycles of prexasertib (Day 21), we found a significant 21-fold decrease in the tumor volume in prexasertib-treated group (n=10) compared to the control group (n=9; Figure 2D). All the mice in the control group reached the survival endpoint at Day 42, whereas 6 mice had a complete response at Day 180 (end of experiment) in the prexasertib-treated group (Figure 2E; Fig.S2D). These results are similar to what we observed with MTE-Ras. Altogether, our in vivo data show that prexasertib treatment significantly reduces tumor volume and increases mice survival. Importantly, prexasertib treatment was efficacious in both HPV-related and HPV-unrelated syngeneic mouse models for HNSCC.
3.3. Prexasertib treatment induces changes in the expression of immune-related genes
To understand the effects of prexasertib on the TIME, MTE-Ras tumors from the control and prexasertib-treated groups sacrificed at Day 4 and Days 22–80 were evaluated for differential expression of immune-related genes. Using a mouse immune profiling Quantigene Plex (QGP) panel (Table S1), we examined changes related to inflammation, specifically genes representing T-cell activation, cytokines and chemokines, immunosuppressive enzymes, and different immune inhibitory as well as co-stimulatory markers. We found that prexasertib treatment resulted in significant changes in the expression of these immune-related genes. At Day 4, we observed statistically significant upregulation of transcripts associated with pro-inflammatory TIME including T-cell activation [Perforin-1 (Prf1), Granzyme A (Gzma)], cytokines and chemokines [Cxcl1, Ccl22, Ccl2, Cxcl10, Interleukin 18 (Il18)], IFN type 1 responsive genes [Interferon Regulatory Factor 7 (Irf7), 2’−5’-Oligoadenylate Synthetase 3 (Oas3)], and CD68 macrophages (Figure 3A,B). Interestingly, we observed statistically significant downregulation of inhibitory factor T-cell immunoreceptor with Ig and an immune costimulatory ITIM Domains (Tigit), and vascular endothelial growth factor A (Vegfa). We also observed downregulation of Foxp3 which is a marker of immunosuppressive Tregs (Figure 3A,B).
FIGURE 3.
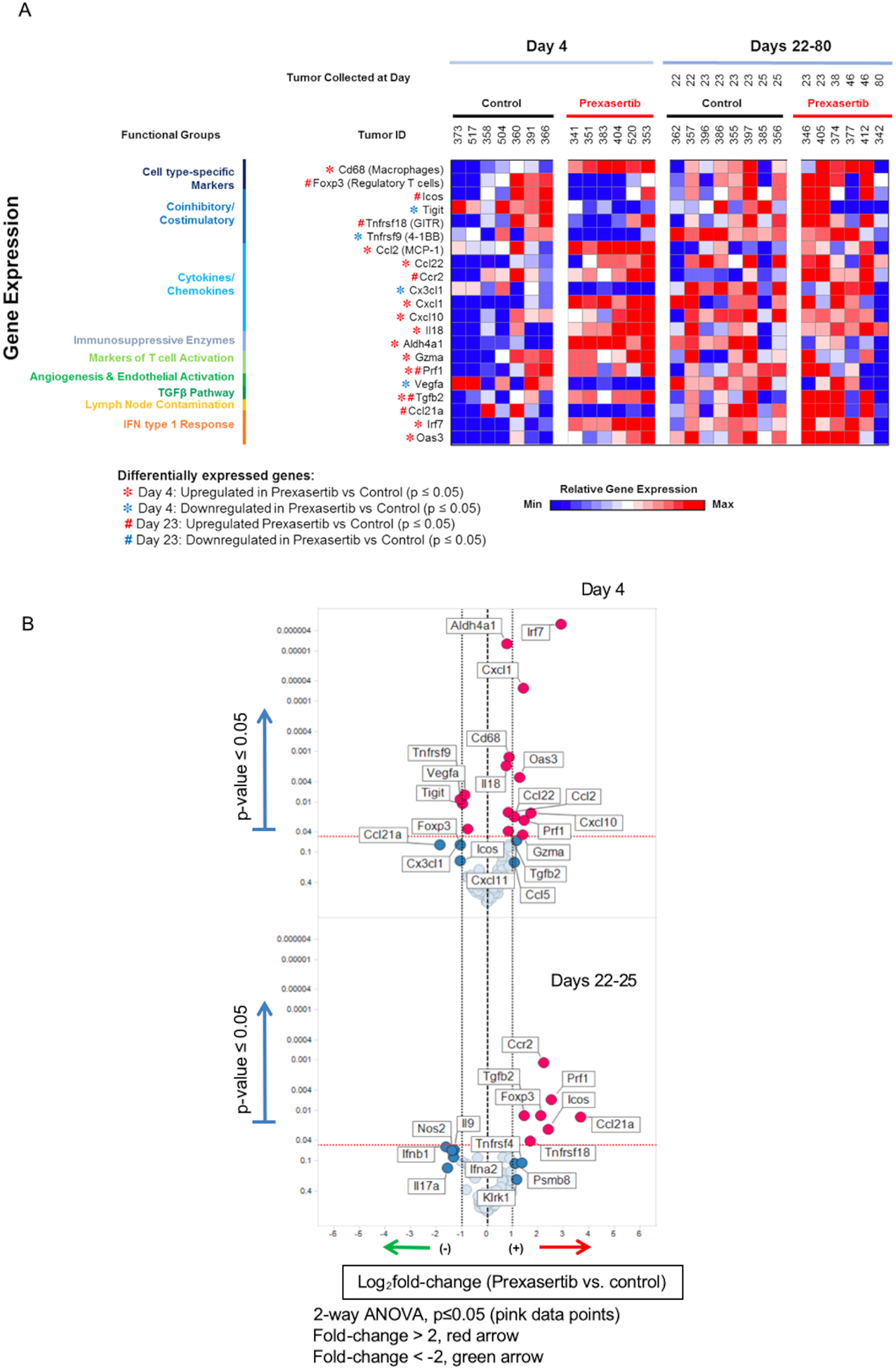
Overview of immune-related prexasertib resistance signature. (A) Heat map showing gene expression results from mice tumors treated with control or prexasertib. The set of 21 gene presented here is defined as immune-related prexasertib resistance signature. Tumor samples were collected at Day 4 and Days 22–80 and expression analysis was performed based on immune profiling Quantigene Plex panel. (B) Volcano plots show log2 fold-change of gene expression in the prexasertib vs control groups with differentially expressed genes that display greater than 2-fold difference with p < 0.05 (two-way ANOVA) compared with control group.
In contrast, at Days 22–25, we observed a shift in these immune-related gene expressions from pro-inflammatory to immunosuppressive TIME. We found an increased expression of Foxp3, Icos, and Tnfrsf18(Gitr). Foxp3 and Gitr are key markers of Tregs that has immunosuppressive function, and Icos stabilizes FOXP3.20,21 We also found increased expression of chemokine receptor Ccr2 and cancer-derived soluble immunosuppressive factor transforming growth factor beta-2 (Tgfb2) (Figure 3A,B). Ccr2 recruits tumor-associated macrophages and other inflammatory monocytes promoting metastasis.22,23 Tgfb2, an immunosuppressive cytokine, promotes N2 tumor-associated neutrophil polarization creating a pro-tumor phenotype24 and also impairs the NK cell mediated anti-tumor effects.25 Considering these tumors are prexasertib resistant indicated by their persistence over time as opposed to the tumors with complete response, these immunosuppressive TIME may provide the mechanism to evade the immune surveillance by Days 22–25. By Day 80, there was no effects of prexasertib in TIME resembling the expression profile of the control tumors at Day 4.
Overall, we identified a set of 21 genes to be significantly differentially regulated in response to prexasertib treatment (p < 0.05), and we called this immune signature to be an immune-related prexasertib resistance signature (Figure 3A). This signature indicates that treatment of mouse tumors with prexasertib results in an acute activation of pro-inflammatory genes; however, additional increase in expression of immunosuppressive genes by potentially recruiting immunosuppressive immune cells over time may allow survival of prexasertib resistant cells by evasion of immune surveillance in TIME.
3.4. Prexasertib converts immune desert to inflamed tumor phenotype by modulating both innate and adaptive immunities
To further understand the effects of prexasertib treatment on the TIME, we performed mIHC on FFPE tissues collected at different time points. We measured the expression of CD45, CD3, CD8, FOXP3, B220, GrzB, CD11b, CD11c, LY-6G, F4/80, and PD-L1. We characterized the T-cell population for cytotoxic T-cells (CD3+CD8+), Tregs (CD3+CD8−FOXP3+), NKT-cells (CD3+CD8−GrzB+), NK cells (CD3−CD8−GrzB+) and B-cells (CD45+B220+). Similarly, the myeloid population was characterized as macrophages (CD11b+F4/80+), neutrophils (CD11b+Ly-6G+), and inflammatory DC (CD11b+CD11c+). The mIHC data were obtained from three separate regions of interest (ROIs) selected within the tumor core and tumor margin, and differences in immune cell counts between treatment groups were analyzed using a statistical modeling. There were a few mouse tumor samples with less than ten detectable B-cells in any compartment, and there was a high background with F4/80; therefore, these markers were not incorporated into subsequent analyses.
In response to prexasertib treatment at Day 4, we found a statistically significant increase in the number of inflammatory DC (2.3-fold) infiltrating the tumor margins (Figure 4A; Table S2). We also observed an increased number of cytotoxic T-cells, Tregs, PD-L1+ and neutrophils, suggesting increased cancer antigen presentation by recruitment of inflammatory DCs and subsequent increased T-cell trafficking. We did not observe much significant differences inside the tumor core at this early time point (Figure 4A; Table S3); however, the differences observed at the tumor margins highlighted a trend towards increased immune infiltrations in response to prexasertib treatment.
FIGURE 4.
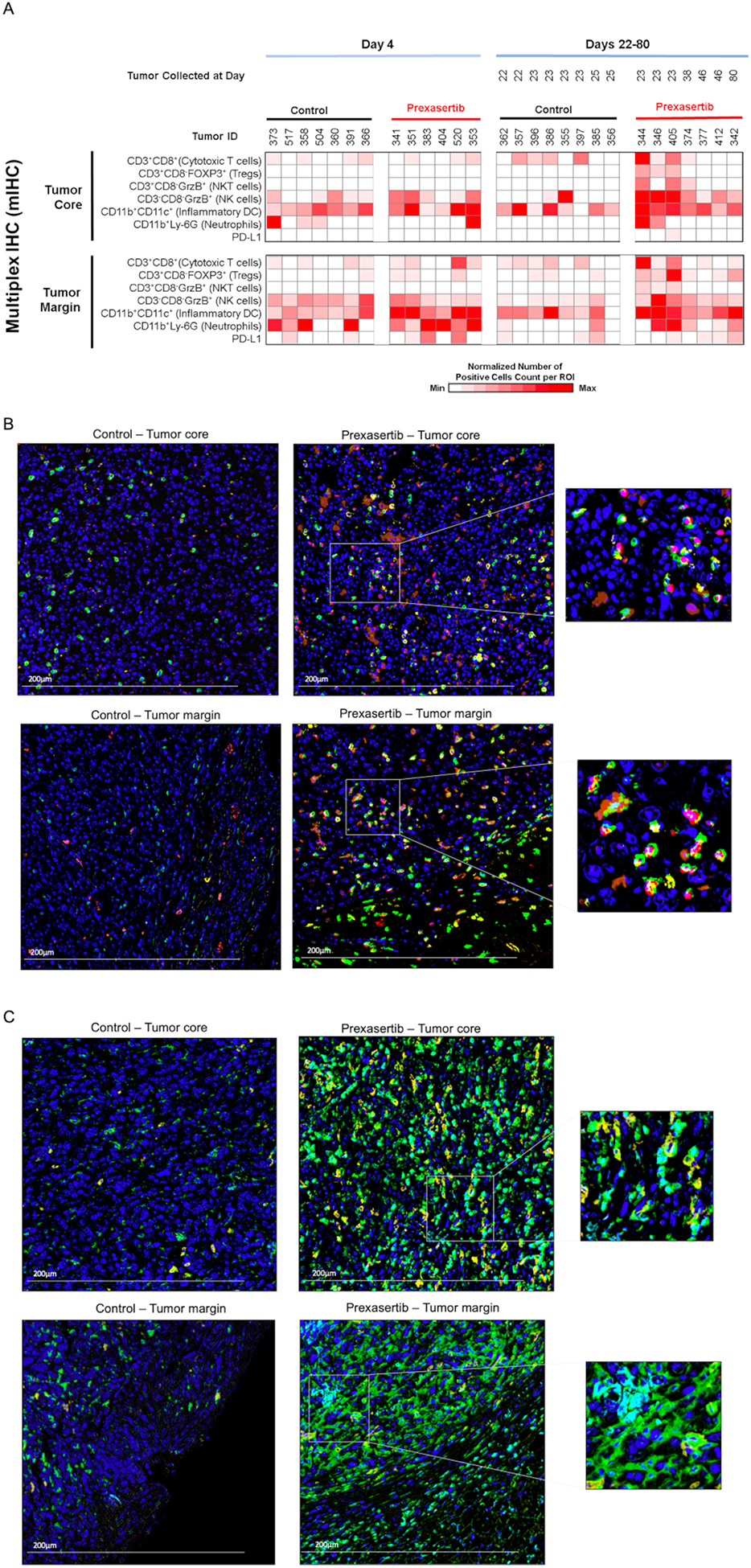
Prexasertib treatment increases tumor infiltration of lymphoid and myeloid immune markers as measured by mIHC analysis. (A) Heat map showing quantification of mIHC signal (as described in the methods section) from mice tumors treated with control or prexasertib and the samples were collected at indicated times. (B,C) Representative images of mIHC staining demonstrating lymphoid (B) and myeloid (C) markers. Tumor core (top panels) and margin (bottom panels) with lymphocyte density from control mice and prexasertib treated mice. A representative enlarged area is presented to show overlapping markers. All images are at 20x magnification, and the scale bar is 200 μm. The colors represent the following staining for lymphoid markers (B) blue = nuclei (hematoxylin), green = CD3, yellow = CD8, red=FOXP3, brownish orange = GrzB; for myeloid markers (C) blue = nuclei (hematoxylin), green = CD11b, yellow = CD11c, cyan=LY-6G.
Multiplex IHC analysis of the tumor margins from Days 22–25 revealed increased infiltration of cytotoxic T-cells (3.3-fold), Tregs (3.7-fold), NK cells (2.5-fold), NKT-cells (5.6-fold), inflammatory DCs (2.3-fold) and neutrophils (5.8-fold) in response to prexasertib treatment (Figure 4A–C; Table S2). Within the tumor core, we found a statistically significant increase in the immune cells creating immunosuppressive TIME including increased numbers of Tregs (11.6-fold), NKT-cells (12.6-fold), NK cells (6.5-fold) and neutrophils (24-fold). While there was a borderline statistically significant increase in cytotoxic T-cells (2.8-fold) but also increased PD-L1+ cells (1.8-fold) indicating exhausted T-cells in prexasertib treated samples (Figure 4A–C; Table S3).
Next, we evaluated changes in the number of immune cells from Day 4 to Days 22–25 in the prexasertib-treated samples. We found a statistically significant increase in Tregs (2.4-fold) and NKT (5-fold) at the tumor margins (Table S2). Whereas, inside the tumor core, we found statistically significant increased numbers of NK cells (2.4-fold), NKT-cells (7.2-fold), cytotoxic T-cells (7.6-fold), Tregs (7.5-fold), neutrophils (10-fold) and PD-L1+ cells (2.2-fold) (Table S3), indicating that prexasertib treatment resulted in establishment of immunosuppressive TIME over time. Overall, mIHC results are consistent with the gene expression analyses showing initial increase in pro-inflammatory immune cell trafficking at the tumor margin at Day 4 but subsequent emergence of immunosuppressive response by further recruiting immunosuppressive immune cells and T cell exhaustion in the tumor margins and cores of the prexasertib resistant tumors at Days 22–25.
3.5. CHK1 as a predictive marker of prexasertib response
We next hypothesized that tumors with low CHK1 expression in HNSCC patients might resemble the immunosuppressive expression profile of prexasertib resistant tumors, potentially suggesting an association with the immunosuppressive TIME. To test this hypothesis, we interrogated the immune-related prexasertib resistance signature against the HNSCC TCGA data. In this analysis, we focused on the HPV-negative cohort, and we divided the cohort into CHK1-hi (z-score > 1, n=66) and CHK1-lo (z < −1, n= 59) expression groups. We performed GSEA using the MSigDB Hallmarks of Cancer and the immune-related prexasertib resistance signature. Using FDR <0.1 as the cut-off, we found that CHK1-hi patients are enriched with biological pathways including G2/M checkpoint, DNA repair, and E2F targets (Figure 5A). As CHK1 is a key regulator of cell cycle, these results are consistent with the current understanding of cell cycle regulation. Whereas, CHK1-lo patients were enriched with myogenesis, KRAS signaling, hypoxia, p53 pathway, and immune-related prexasertib resistance signature (Figure 5A). The enrichments plots of E2F targets and immune-related prexasertib resistance signature in TCGA CHK1-hi and -lo groups are shown in Figure 5B. The interactions of the genes within the immune-related prexasertib resistance signature clustered by the STRING database (k-means cluster, k=4) are shown in Figure 5C. An enrichment of the immune-related prexasertib resistance signature in the CHK1-lo expression cohort suggests that these patients may have the immunosuppressive TIME and may benefit from immune checkpoint inhibitors or MDSC targeted agents.
FIGURE 5.
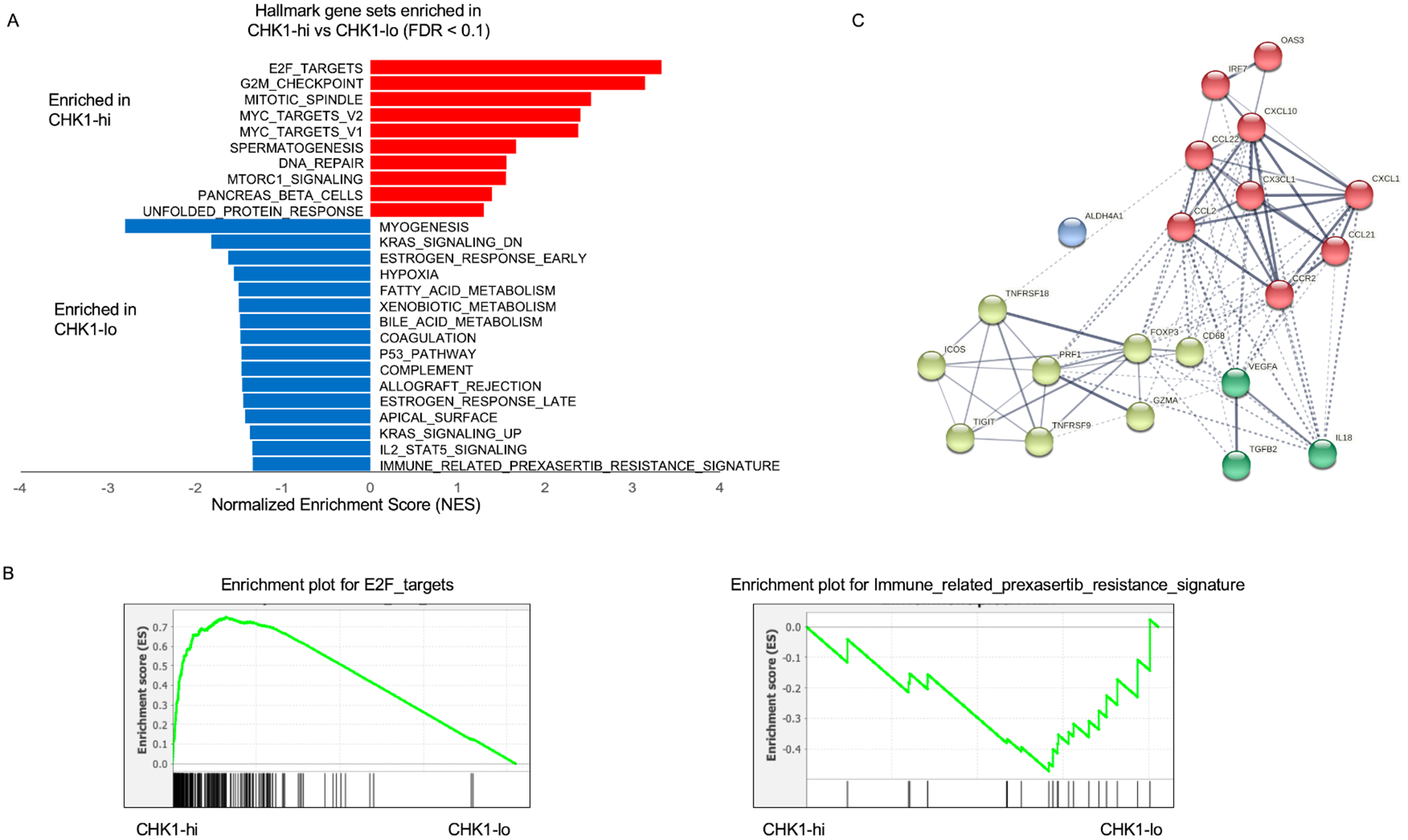
Association of CHK1 gene abundances with cancer-related biological pathways. (A) GSEA of hallmark pathways including immune-related prexasertib resistance signature, associated with CHK1-lo and CHK1-hi gene expression in the TCGA cohort. (B) Enrichment plot of E2F targets and immune-related prexasertib resistance signature in TCGA CHK1-hi and -lo groups. (C) The interactions of the genes in the immune-related prexasertib resistance signature as clustered by the STRING database.
4. DISCUSSION
Targeting cell cycle and DNA damage response in HNSCC has been extensively studied in the past.11,26,27 Multiple preclinical studies have confirmed the radiosensitizing effects of different CHK1 inhibitors, either administered alone or in combination with PARP inhibitors, or cetuximab and/or radiotherapy.19,28–30 However, most of these studies focus on the effects on cancer cells without evaluating the effects on the TIME. Here, by using a syngeneic mouse model of HNSCC, we report a previously unexplored role of a CHK1 inhibitor, prexasertib, in modulating the TIME. Our data show that treatment of in vivo tumors with prexasertib results in decreased tumor volume and improved survival. We found that prexasertib treatment resulted in activating pro-inflammatory genes converting immune desert phenotype to inflamed tumor phenotype in the in vivo syngeneic mouse model. However, the tumor cells acquired resistance to prexasertib over time and evaded the anti-tumor immune response, partly due to increased immunosuppressive immune cells and T cell exhaustion. This acquired resistance over a period resulted in regrowth of the tumors and resulted in mortality in the treatment group.
An anti-cancer immune response required for effective killing of cancer cells is well summarized in the sequential steps of cancer-immunity cycle.31 In our study, we anticipated and evaluated if similar series of cancer-immunity cycle occur in response to prexasertib. The first step of cancer-immunity cycle is cell death induced by chemotherapy, immunotherapy or targeted therapy. Dying cancer cells swell, burst, and release damage-associated molecular patterns and/or neoantigens.32 We found that prexasertib treatment resulted in increased DNA damage and apoptosis leading to cancer cell death in vitro and in vivo. In the following step, the released neoantigens presented on the surface of antigen presenting cells are recognized by the receptors on DCs and leads to immune cell activation.33 In our data, we observed increased expression of pro-inflammatory transcripts and a decrease in immunosuppressive genes at Day 4. We found increased expression of Prf1 and Gzma genes which are known to be mostly expressed by cytotoxic T-cells and NK cells and plays important roles in cancer cell death.34 We also observed increased expression of chemokines, Cxcl1, Ccl2 and Cxcl10, which represent the pathogen response-like chemokine (PARC) signature, typically triggered by immunogenic dying cancer cells and leads to neutrophil recruitment.35 Our Day 4 mIHC data further support the activation of immune response by significantly increased infiltration of DCs at the tumor margin. In addition, we observed decreased expression of Tigit and Vegfa transcripts, both of them play important roles in the development of an immunosuppressive TIME. TIGIT can inhibit the immunity cycle at multiple steps including antigen-presentation by DC and inhibition of CD8 T-cell mediated and/or NK cell mediated tumor killing.36 VEGF-A enhances expression of PD-1 and other inhibitory checkpoints involved in CD8+ T cell exhaustion.37
Unfortunately, we also observed evidence of immune surveillance evasion over time resulting in acquired resistance to prexasertib. To date, several mechanisms of prexasertib resistance have been published including but not limited to mutations in the target gene, activation of compensatory signaling pathways, and immune evasion.38 Recently, prexasertib-mediated acquired resistance is reported to be associated with components of innate immunity in a panel of pan-cancer cell line models and in vivo carcinoma and sarcoma models.39 In our study, we also provide evidence that prexasertib-mediated acquired resistance is, at least in part, due to immune evasion. In the expression data, we observed increased expression of several immunosuppressive regulatory genes including Foxp3, Icos, Gitr, Ccr2 and Tgfb2. Similarly, in our mIHC data, we found an increased infiltration of FOXP3+ T-regs, tumor-associated neutrophils, and tumor-associated NK cells, reflecting immunosuppressive and pro-tumorigenic TIME. The current knowledge of acquired resistance through evasion of immune surveillances is limited and is an area of active research for many. In our future study, we aim to further investigate the mechanisms of acquired resistance and additional factors to switch from pro-inflammatory to immunosuppressive TIME after establishing inflamed tumor phenotype.
Furthermore, the interaction between the tumor and immune cells as well as other stromal cells needs to be further delineated. For example, in our study, it is unclear which cells in the tumor are responsible for the differential expression of the immune response genes because the expression analyses were performed from bulk tumors rather than single cell analyses, and a significant number of these genes are expressed by numerous cell types. It is also unclear whether these prexasertib-induced changes are solely immunogenic response to cell death agnostic to cytotoxic agents or specific to the CHK1 inhibition by prexasertib. Further evaluation of these unresolved questions will require single cell analyses and comparison of prexasertib effects with other commonly used cytotoxic agents in HNSCC. Our analyses of the CHK1-hi and CHK1-lo HNSCC suggest that de novo CHK1 expression of tumors may significantly contribute to the changes in the TIME beyond a generic response to cell death.
In addition, our study provides an insight for future development of targeted agents such as prexasertib with immunotherapeutic agents. In previous clinical trials, continuous treatment of prexasertib resulted in treatment-emergent adverse events including neutropenia, leukopenia, anemia, thrombocytopenia, and fatigue.40,41 These toxicities hindered further clinical development in absence of predictive biomarkers. For future studies, prexasertib might be investigated by intermittent administration to introduce cell death and prime TIME followed by immune modulating agents as a sequential therapy. Here, we uncovered several immune markers that are significantly upregulated in response to prexasertib and can potentially be combined with prexasertib such as anti-PD-L1, anti-Tregs, NK-cell-targeted, or anti-neutrophil-targeted agents for future clinical trials although one has to be careful as many of these immune cells play a context dependent dual pro-tumor or anti-tumor roles.42,43
In conclusion, we found that prexasertib mediated CHK1 inhibition resulted in increased DNA damage and cell death in cancer cells and increased survival in vivo. These anti-tumor effects are mediated by an early increase in the expression and infiltration of the pro-inflammatory immune cells resulting in inflamed tumor phenotype. However, we also observed a transition from pro-inflammatory to immune tolerant TIME over time as some of the tumor cells acquired resistance. Overall, our study signified the importance of TIME evaluation in the development of targeted agents such as prexasertib and identified potential immunotherapeutic agents that can be evaluated with prexasertib for the future clinical investigation.
Supplementary Material
ACKNOWLEDGEMENT
We appreciate the staffs at the Moffitt Animal Facility, Tissue Core, and Flow Cytometry Core for their contribution.
Funding information
This study is partly funded by Eli Lilly and Company. It is also supported in part by James and Esther King Biomedical Research Grant (7JK02) and the H. Lee Moffitt Cancer Center and Research Institute, an NCI-designated Comprehensive Cancer Center (P30-CA076292). The content is solely the responsibility of the authors and does not necessarily represent the official views of the Grant Sponsor or the H. Lee Moffitt Cancer Center & Research Institute.
Footnotes
COI statement
Christine H. Chung and Robbert J. C. Slebos have received honoraria from Bristol Myers Squibb, AstraZeneca, and Lilly Oncology for serving in ad hoc scientific advisory boards and research funding for a preclinical study from Lilly Oncology. Dr. Jose Conejo-Garcia has stock options and research funding from Compass Therapeutics and Anixa Bioscience, and receives honorarium from Leidos, Compass Therapeutics and Anixa Bioscience. All other authors declare no conflict of interest.
DATA AVAILABILITY STATEMENT
Data available on request from the authors.
REFERENCES
- 1.Bray F, Ferlay J, Soerjomataram I, et al. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J Clin. 2018;68(6): 394–424. [DOI] [PubMed] [Google Scholar]
- 2.Blot WJ, McLaughlin JK, Winn DM, et al. Smoking and drinking in relation to oral and pharyngeal cancer. Cancer Res. 1988;48(11): 3282–3287. [PubMed] [Google Scholar]
- 3.Ragin CC, Modugno F, Gollin SM. The epidemiology and risk factors of head and neck cancer: a focus on human papillomavirus. J Dent Res. 2007;86(2): 104–114. [DOI] [PubMed] [Google Scholar]
- 4.Sacco AG, Cohen EE. Current Treatment Options for Recurrent or Metastatic Head and Neck Squamous Cell Carcinoma. J Clin Oncol. 2015;33(29): 3305–3313. [DOI] [PubMed] [Google Scholar]
- 5.Bhardwaj N Harnessing the immune system to treat cancer. J Clin Invest. 2007;117(5): 1130–1136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.de la Iglesia JV, Slebos RJ, Martin-Gomez L, et al. Effects of tobacco smoking on the tumor immune microenvironment in head and neck squamous cell carcinoma. Clin Cancer Res. 2020;26(6): 1474–1485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Chen DS, Mellman I. Elements of cancer immunity and the cancer-immune set point. Nature. 2017;541(7637): 321–330. [DOI] [PubMed] [Google Scholar]
- 8.Dai Y, Grant S. New insights into checkpoint kinase 1 in the DNA damage response signaling network. Clin Cancer Res. 2010;16(2): 376–383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Zhao H, Piwnica-Worms H. ATR-mediated checkpoint pathways regulate phosphorylation and activation of human Chk1. Mol Cell Biol. 2001;21(13): 4129–4139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.King C, Diaz HB, McNeely S, et al. LY2606368 Causes Replication Catastrophe and Antitumor Effects through CHK1-Dependent Mechanisms. Mol Cancer Ther. 2015;14(9): 2004–2013. [DOI] [PubMed] [Google Scholar]
- 11.Hong DS, Moore K, Patel M, et al. Evaluation of Prexasertib, a Checkpoint Kinase 1 Inhibitor, in a Phase Ib Study of Patients with Squamous Cell Carcinoma. Clin Cancer Res. 2018;24(14): 3263–3272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Lowery CD, Dowless M, Renschler M, et al. Broad Spectrum Activity of the Checkpoint Kinase 1 Inhibitor Prexasertib as a Single Agent or Chemopotentiator Across a Range of Preclinical Pediatric Tumor Models. Clin Cancer Res. 2019;25(7): 2278–2289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Lowery CD, VanWye AB, Dowless M, et al. The Checkpoint Kinase 1 Inhibitor Prexasertib Induces Regression of Preclinical Models of Human Neuroblastoma. Clin Cancer Res. 2017;23(15): 4354–4363. [DOI] [PubMed] [Google Scholar]
- 14.Zeng L, Nikolaev A, Xing C, Della Manna DL, Yang ES. CHK1/2 Inhibitor Prexasertib Suppresses NOTCH Signaling and Enhances Cytotoxicity of Cisplatin and Radiation in Head and Neck Squamous Cell Carcinoma. Mol Cancer Ther. 2020;19(6): 1279–1288. [DOI] [PubMed] [Google Scholar]
- 15.Sen T, Rodriguez BL, Chen L, et al. Targeting DNA Damage Response Promotes Antitumor Immunity through STING-Mediated T-cell Activation in Small Cell Lung Cancer. Cancer Discov. 2019;9(5): 646–661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Cicchini L, Westrich JA, Xu T, et al. Suppression of Antitumor Immune Responses by Human Papillomavirus through Epigenetic Downregulation of CXCL14. MBio. 2016;7(3): e00270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Spanos WC, Hoover A, Harris GF, et al. The PDZ binding motif of human papillomavirus type 16 E6 induces PTPN13 loss, which allows anchorage-independent growth and synergizes with ras for invasive growth. J Virol. 2008;82(5): 2493–2500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Subramanian A, Tamayo P, Mootha VK, et al. Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc Natl Acad Sci U S A. 2005;102(43): 15545–15550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Zeng L, Beggs RR, Cooper TS, Weaver AN, Yang ES. Combining Chk1/2 Inhibition with Cetuximab and Radiation Enhances In Vitro and In Vivo Cytotoxicity in Head and Neck Squamous Cell Carcinoma. Mol Cancer Ther. 2017;16(4): 591–600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Landuyt AE, Klocke BJ, Colvin TB, Schoeb TR, Maynard CL. Cutting Edge: ICOS-Deficient Regulatory T Cells Display Normal Induction of Il10 but Readily Downregulate Expression of Foxp3. J Immunol. 2019;202(4): 1039–1044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Ronchetti S, Ricci E, Petrillo MG, et al. Glucocorticoid-induced tumour necrosis factor receptor-related protein: a key marker of functional regulatory T cells. J Immunol Res. 2015;2015(171520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Franklin RA, Liao W, Sarkar A, et al. The cellular and molecular origin of tumor-associated macrophages. Science. 2014;344(6186): 921–925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Qian BZ, Li J, Zhang H, et al. CCL2 recruits inflammatory monocytes to facilitate breast-tumour metastasis. Nature. 2011;475(7355): 222–225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Fridlender ZG, Sun J, Kim S, et al. Polarization of tumor-associated neutrophil phenotype by TGF-beta: “N1” versus “N2” TAN. Cancer Cell. 2009;16(3): 183–194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Batlle E, Massague J. Transforming Growth Factor-beta Signaling in Immunity and Cancer. Immunity. 2019;50(4): 924–940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Morgan MA, Parsels LA, Maybaum J, Lawrence TS. Improving the efficacy of chemoradiation with targeted agents. Cancer Discov. 2014;4(3): 280–291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Glorieux M, Dok R, Nuyts S. Novel DNA targeted therapies for head and neck cancers: clinical potential and biomarkers. Oncotarget. 2017;8(46): 81662–81678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Guster JD, Weissleder SV, Busch CJ, et al. The inhibition of PARP but not EGFR results in the radiosensitization of HPV/p16-positive HNSCC cell lines. Radiother Oncol. 2014;113(3): 345–351. [DOI] [PubMed] [Google Scholar]
- 29.Busch CJ, Kriegs M, Laban S, et al. HPV-positive HNSCC cell lines but not primary human fibroblasts are radiosensitized by the inhibition of Chk1. Radiother Oncol. 2013;108(3): 495–499. [DOI] [PubMed] [Google Scholar]
- 30.Gadhikar MA, Sciuto MR, Alves MV, et al. Chk1/2 inhibition overcomes the cisplatin resistance of head and neck cancer cells secondary to the loss of functional p53. Mol Cancer Ther. 2013;12(9): 1860–1873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Chen DS, Mellman I. Oncology meets immunology: the cancer-immunity cycle. Immunity. 2013;39(1): 1–10. [DOI] [PubMed] [Google Scholar]
- 32.Hernandez C, Huebener P, Schwabe RF. Damage-associated molecular patterns in cancer: a double-edged sword. Oncogene. 2016;35(46): 5931–5941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Yarchoan M, Johnson BA 3rd, Lutz ER, Laheru DA, Jaffee EM. Targeting neoantigens to augment antitumour immunity. Nat Rev Cancer. 2017;17(4): 209–222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Martinez-Lostao L, Anel A, Pardo J. How Do Cytotoxic Lymphocytes Kill Cancer Cells? Clin Cancer Res. 2015;21(22): 5047–5056. [DOI] [PubMed] [Google Scholar]
- 35.Garg AD, Vandenberk L, Fang S, et al. Pathogen response-like recruitment and activation of neutrophils by sterile immunogenic dying cells drives neutrophil-mediated residual cell killing. Cell Death Differ. 2017;24(5): 832–843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Manieri NA, Chiang EY, Grogan JL. TIGIT: A Key Inhibitor of the Cancer Immunity Cycle. Trends Immunol. 2017;38(1): 20–28. [DOI] [PubMed] [Google Scholar]
- 37.Voron T, Colussi O, Marcheteau E, et al. VEGF-A modulates expression of inhibitory checkpoints on CD8+ T cells in tumors. Journal of Experimental Medicine. 2015;212(2): 139–148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Lackner MR, Wilson TR, Settleman J. Mechanisms of acquired resistance to targeted cancer therapies. Future Oncol. 2012;8(8): 999–1014. [DOI] [PubMed] [Google Scholar]
- 39.Blosser WD, Dempsey JA, McNulty AM, et al. A pan-cancer transcriptome analysis identifies replication fork and innate immunity genes as modifiers of response to the CHK1 inhibitor prexasertib. Oncotarget. 2020;11(3): 216–236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Hong D, Infante J, Janku F, et al. Phase I Study of LY2606368, a Checkpoint Kinase 1 Inhibitor, in Patients With Advanced Cancer. J Clin Oncol. 2016;34(15): 1764–1771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Lee JM, Nair J, Zimmer A, et al. Prexasertib, a cell cycle checkpoint kinase 1 and 2 inhibitor, in BRCA wild-type recurrent high-grade serous ovarian cancer: a first-in-class proof-of-concept phase 2 study. Lancet Oncol. 2018;19(2): 207–215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Bassani B, Baci D, Gallazzi M, et al. Natural Killer Cells as Key Players of Tumor Progression and Angiogenesis: Old and Novel Tools to Divert Their Pro-Tumor Activities into Potent Anti-Tumor Effects. Cancers (Basel). 2019;11(4): 461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Granot Z Neutrophils as a Therapeutic Target in Cancer. Front Immunol. 2019;10(1710. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.