

Abstract
In preparation for cell division, the genome must be copied with high fidelity. However, replisomes often encounter obstacles, including bulky DNA lesions caused by reactive metabolites and chemotherapeutics, as well as stable nucleoprotein complexes. Here, we discuss recent advances in our understanding of TRAIP, a replisome-associated E3 ubiquitin ligase that is mutated in microcephalic primordial dwarfism. In interphase, TRAIP helps replisomes overcome DNA interstrand crosslinks and DNA-protein crosslinks, whereas in mitosis it triggers disassembly of all replisomes that remain on chromatin. We describe a model to explain how TRAIP performs these disparate functions and how they help maintain genome integrity.
Keywords: E3 ubiquitin ligase, DNA interstrand crosslink repair, DNA-protein crosslink repair, Mitotic DNA replication, Replication termination
TRAIP: a new player in the preservation of genomic integrity
During DNA replication, replisomes often encounter impediments (“replication stress”) that threaten genomic integrity [1]. Cells possess various means to respond to these challenges, including mechanisms to protect and restart stalled replication forks [2], replication-coupled DNA repair pathways [3], and DNA damage checkpoints that stabilize stalled forks, regulate the firing of dormant origins, and increase the time frame to rescue stalled forks and/or repair DNA damage [4]. Failure of these pathways results in persistence of unreplicated DNA into mitosis, resulting in anaphase bridges, chromosome mis-segregation, and gross chromosomal rearrangements [5, 6]. Mitosis thus becomes the last chance to prevent large-scale chromosome alterations that could result from incompletely replicated DNA.
A recent series of studies has uncovered several key roles of the E3 ubiquitin ligase TRAIP (TRAF-interacting protein) in how cells respond to replication impediments and cope with unreplicated DNA during mitosis. This review highlights how TRAIP helps replication forks overcome specific obstacles during S phase but then triggers the wholesale disassembly of any replisome that remains on chromatin in mitosis. We propose a molecular model to explain how TRAIP’s selective action during interphase transforms into a general replisome disassembly role in mitosis. Finally, how failures of TRAIP function might lead to primordial dwarfism, genomic instability, and cancer is discussed. In general terms, the new work on TRAIP deepens our appreciation of the intimate link between the response to replication stress and signaling by ubiquitin and the related small ubiquitin-like modifier (SUMO) (reviewed in [7-10]).
TRAIP is a component of the DNA replication machinery
As its name indicates, TRAIP was initially discovered as a novel interactor of the TNF receptor/TRAF signaling complex [11]. While TRAIP was proposed to regulate nuclear factor-kappa B signaling [11, 12], proteins in this pathway that are ubiquitylated by TRAIP have not been identified [13, 14]. More recently, a potential link between TRAIP and the DNA damage response emerged when three children with microcephalic primordial dwarfism were found to have homozygous TRAIP mutations [15]. This phenotype is a form of Seckel syndrome, a genetically heterogenous autosomal recessive disorder caused by mutations in any one of ten genes functioning in the DNA damage response or centrosome function [15, 16]. Notably, one of these mutations, R18C, resides in TRAIP's RING domain and compromises TRAIP’s ubiquitin ligase activity [15, 17]. TRAIP-deficient cells proliferate slowly and exhibit hypersensitivity to the DNA interstrand crosslink (ICL)-inducing agent, mitomycin C. TRAIP-depleted cells display a diverse array of other phenotypes including accumulation in the G2 phase, diminished activation of the DNA damage response, micronucleation, and gross chromosomal rearrangements [15, 18, 19]. Together, the data indicate that TRAIP plays broad roles in the maintenance of genomic integrity.
TRAIP is usually concentrated in nucleoli [18-20], but colocalizes with PCNA at sites of DNA damage and upon replication stress [15, 18-20]. This recruitment to stressed replication forks is mediated by a PCNA-interacting protein (PIP)-box motif at the C-terminus of TRAIP [18, 19]. However, TRAIP lacking its PIP box is still functional in tissue culture cells and in Xenopus egg extracts (detailed below) [17, 19]. It is conceivable that the PCNA interaction is dispensable under certain experimental conditions (e.g. TRAIP overexpression). Adding to the intrigue, analysis of DNA replicating in Xenopus egg extracts and isolation of proteins on nascent DNA (iPOND) in mammalian cells suggests that TRAIP resides at replication forks in the absence of exogenous DNA damage [17, 19, 21, 22]. Although this association could reflect de novo TRAIP recruitment to forks that encounter endogenous replication stress, TRAIP increases the replisome’s physical footprint [17], consistent with TRAIP traveling constitutively with each replication fork. These observations raise the question of which proteins TRAIP ubiquitylates at replication forks and how their modification contributes to the maintenance of genome stability.
The function of TRAIP in S phase
Regulating replication-coupled ICL repair
Silencing TRAIP sensitizes cells to mitomycin C [19], suggesting that TRAIP plays a role in the repair of ICLs, highly cytotoxic DNA lesions that impede replisome progression. Convergence of two replisomes on either side of an ICL initiates at least three distinct pathways that resolve ("unhook") the ICL and allow completion of DNA replication (Figure 1A). Acetaldehyde-ICLs appear to be directly reversed by an unknown enzyme [23], whereas psoralen/UV-A and abasic site ICLs are unhooked when the NEIL3 glycosylase cuts one of the two glycosyl bonds that form the ICL [24]. The third repair pathway involves the 22 FANC proteins that are mutated in Fanconi anemia, a human disease characterized by cellular sensitivity to ICL-inducing agents, short stature and other congenital abnormalities, bone marrow failure, and cancer predisposition [25]. The FANC proteins unhook ICLs through incisions of the phosphodiester backbone on either side of the crosslink, followed by repair of the resulting double-strand break by homologous recombination [26]. Since the Fanconi anemia repair mechanism promotes incisions adjacent to the ICL, it can unhook any ICL regardless of its chemical structure. Despite this, the majority of psoralen and abasic ICLs are repaired by NEIL3 [24, 27], suggesting that the Fanconi anemia pathway might function primarily as a back-up. This prioritization of direct unhooking avoids double-strand breaks that could lead to chromosomal rearrangements.
Figure 1. Replication-coupled Interstrand crosslink (ICL) and DNA-protein crosslink (DPC) repair pathways.
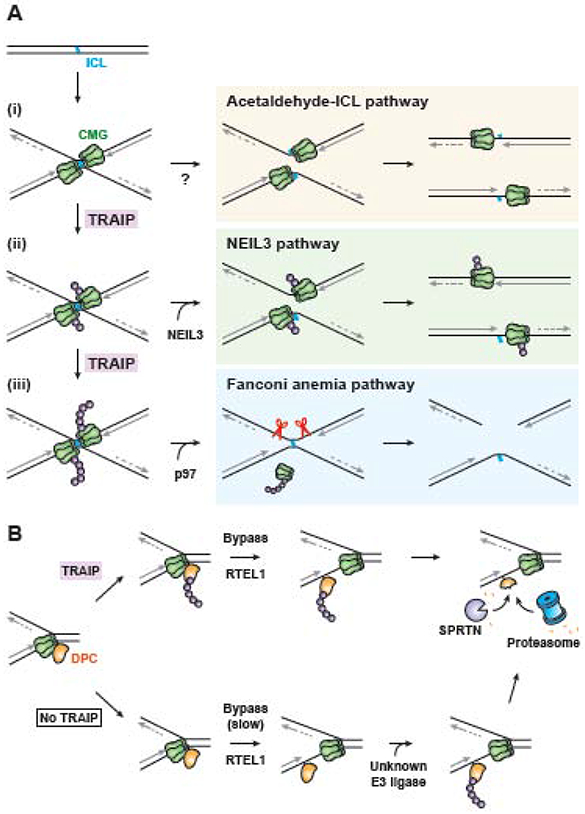
A. In Xenopus egg extracts, three pathways of ICL repair require the convergence of two replication forks at the crosslink. Fork convergence triggers ubiquitylation of the replisome’s CDC45/MCM2-7/GINS (CMG) helicase by TRAIP (ubiquitin shown as purple spheres). Acetaldehyde-ICL repair is depicted as being initiated before CMG ubiquitylation (i), but this remains to be confirmed. Ubiquitin chains as short as one or two ubiquitins may recruit the NEIL3 glycosylase to directly unhook psoralen and abasic ICLs, allowing completion of DNA replication (ii). If NEIL3 fails to unhook the crosslink, TRAIP continues to extend the ubiquitin chains (iii). Long ubiquitin chains trigger CMG unloading by the p97 segregase, which allows Fanconi anemia pathway endonucleases to unhook the ICL. The resulting double-strand break is repaired by homologous recombination.
B. Proteins crosslinked to the leading strand template hinder progression of the CMG helicase. Upon fork collision, TRAIP ubiquitylates the DPC (top arrow). After the accessory helicase RTEL1 promotes CMG bypass of the lesion, the DPC is ubiquitylated by a second, unknown E3 ubiquitin ligase. DPC ubiquitylation marks the lesion for proteolysis by the proteasome. The DPC-specific protease SPRTN also contributes to proteolysis of both ubiquitylated and unmodified DPCs. Note that at DPC-fork collisions, TRAIP activity does not require fork convergence and CMG is not ubiquitylated. In the absence of TRAIP, the initial ubiquitylation of the DPC is delayed, as is CMG bypass, but bypass does eventually occur, followed by CMG ubiquitylation by an unknown E3 ubiquitin ligase and proteolysis by SPRTN and the proteasome.
How is ICL repair pathway choice regulated? In Xenopus egg extracts, convergence of two forks at the crosslink is critical for all three forms of ICL repair [23, 24, 28]. Fork convergence triggers ubiquitylation of the stalled CMG helicases by TRAIP [17]. Short ubiquitin chains on CMG are sufficient to promote recruitment of the NEIL3 glycosylase via a ubiquitin-binding domain within NEIL3 (Figure 1A,ii). In contrast, longer ubiquitin chains are required to trigger CMG removal from chromatin by the p97 segregase, which in turn allows endonucleases to access DNA and initiate the incisions that unhook the ICL (Figure 1A,iii) [29-32]. Thus, NEIL3 is recruited to ICLs first, and if it unhooks the lesion, repair occurs without formation of a double-strand break. If NEIL3 cannot act (such as at cisplatin ICLs), ubiquitin chains grow, activating the Fanconi anemia pathway. Consistent with this model, psoralen ICL processing in cells generates Fanconi anemia pathway-dependent double-strand breaks only in the absence of NEIL3 [27]. Whether TRAIP-dependent CMG ubiquitylation also regulates the acetaldehyde-ICL pathway remains an important question. If not, it might suggest that acetaldehyde-ICL repair is engaged even before the NEIL3 pathway (Figure 1A,i).
The question arises why human TRAIP mutations cause dwarfism instead of classical Fanconi anemia. One possibility is that these TRAIP alleles retain enough function to support ICL repair, but are defective in other functions of TRAIP (see below) that suppress short stature. Importantly, mammalian cells possess a subpathway of ICL repair called "traverse." In this mechanism, single forks bypass ICLs [33, 34], yielding an X-shaped DNA structure that is probably processed by the FANC proteins, as seen after fork convergence [34]. If traverse does not require TRAIP, this mechanism might provide enough ICL repair function to prevent Fanconi anemia.
Ubiquitylating replisome-blocking protein obstacles
Given that TRAIP can trigger CMG unloading at converged forks, it is crucial to understand how CMG unloading is avoided at single forks to prevent disassembly of replisomes that have not completed replication. One hypothesis is that TRAIP’s ubiquitin ligase is activated only when forks converge, for example through TRAIP dimerization. While such a model is appealing, TRAIP has been found to act without fork convergence [35]. One such context is when single replisomes stall at DNA-protein crosslinks (DPCs) (Figure 1B). These lesions arise when chromatin-associated proteins become covalently crosslinked to DNA by chemotherapeutics or endogenous aldehydes [36]. DPCs attached to the leading strand template pose particularly challenging obstacles to DNA replication because they reside on CMG’s translocation strand [37]. Cells degrade DPCs through the action of the proteasome and the specialized protease SPRTN/DVC1 [35, 38-40] (Wss1 in yeast [41]). As with ICL repair, DPC repair is triggered by arrival of a replication fork, which leads to ubiquitylation of the DPC (Figure 1B) [42]. In Xenopus egg extracts, CMG first bypasses leading strand DPCs with the assistance of the RTEL1 DNA helicase [43]. After CMG has moved safely beyond the lesion, the DPC is degraded by SPRTN and the proteasome [35]. While DPCs are likely acted upon by multiple ubiquitin ligases, the modification that occurs specifically as a result of CMG collision with the lesion depends on TRAIP [35]. Although it is not required for SPRTN-mediated degradation, TRAIP-dependent DPC ubiquitylation appears to stimulate DPC proteolysis by the proteasome, probably by enhancing CMG bypass of the DPC [35, 42]. Whether TRAIP also promotes the ubiquitylation and clearance of non-covalent nucleoprotein barriers such as tightly bound proteins or transcribing RNA polymerases [44-46] has not been determined. However, such a function would be consistent with the observation that TRAIP-deficient cells exhibit fork asymmetry and deficient fork progression during replication stress [15, 18].
A model for TRAIP action in S phase
As described above, when a replisome collides with a protein barrier such as a DPC, TRAIP ubiquitylates the barrier but does not trigger ubiquitylation or unloading of the “host” CMG with which it travels [35]. In contrast, when forks converge at an ICL, CMG undergoes ubiquitylation. One model to explain these observations is that TRAIP’s RING domain is rigidly positioned on the replisome’s leading edge like the hood ornament on an automobile. In this way, when TRAIP recruits an E2 conjugating enzyme, it can transfer ubiquitin to any protein obstacle located ahead of the replisome, whether this is a DPC or a converging CMG (Figure 2, “S phase”) [17]. In the model proposed, CMG ubiquitylation at ICLs occurs strictly in trans. Whether these ubiquitylation events involve a constitutively active TRAIP that travels with each replisome or TRAIP molecules recruited de novo upon fork stalling remains to be determined. Either way, a critical feature of the model is that during S phase, TRAIP is unable to ubiquitylate its host CMG. This constraint prevents ubiquitylation of single replisomes engaged in replication, which would cause premature replisome disassembly and fork collapse.
Figure 2. Model for TRAIP function in S phase.
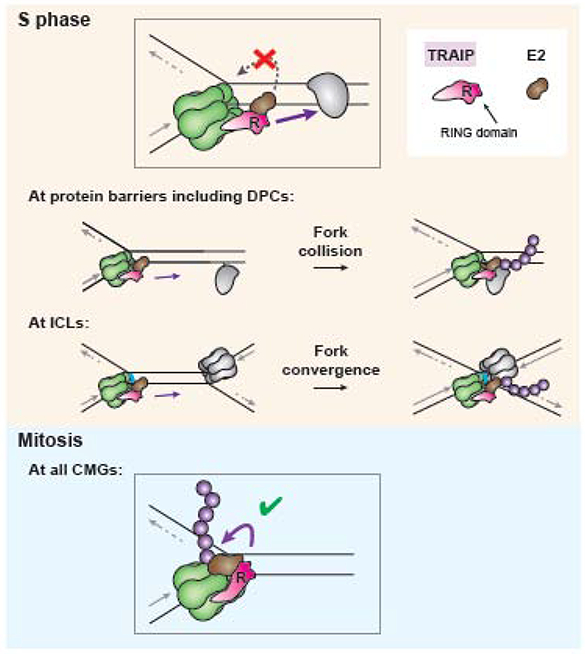
During unperturbed DNA replication in S phase, TRAIP is assembled with the replisome with its RING domain positioned to ubiquitylate any protein encountered by the replisome (inset; purple arrow signifies direction of ubiquitylation). Such proteins include DNA-protein crosslinks (DPCs) and CDC45/MCM2-7/GINS (CMG) helicases when forks converge at interstrand crosslinks (ICLs) (“S phase," center, gray). TRAIP ubiquitylation of the host replisome on which it is assembled (“S phase,” inset, gray broken arrow) does not occur, possibly because rigid positioning of the RING domain prevents it from bringing its E2 ubiquitin conjugating enzyme (brown) into contact with CMG. During mitosis, TRAIP undergoes a conformational change, allowing it to ubiquitylate its host CMG (“Mitosis,” purple arrow).
An alternative model is that TRAIP indiscriminately ubiquitylates proteins in the vicinity of the replication fork. In this scenario, deubiquitylating enzymes (DUBs), some of which act at the replisome [47-50], would be required to erase modifications of host replisome proteins while allowing ubiquitylation of fork barriers such as DPCs. Moreover, to account for CMG ubiquitylation at ICLs, it would be necessary to invoke DUB inhibition or displacement specifically upon fork convergence at these lesions. A drawback of the DUB model is that inadvertent DUB dissociation prior to fork convergence would lead to premature replisome ubiquitylation and disassembly. Thus, the hood ornament model provides the most attractive explanation of how TRAIP function is constrained at the fork.
TRAIP functions during mitosis
A transformation of TRAIP function during mitosis
While TRAIP avoids ubiquitylating its host CMG in interphase extracts, its activity appears to be strikingly altered in mitosis. When egg extracts are driven into a mitotic state by addition of cyclin B1-CDK1, TRAIP ubiquitylates its host CMG in single replisomes stalled at various types of fork barriers, triggering their ubiquitylation and disassembly by p97 (Figure 3A,i-ii) [51]. The fact that this TRAIP-dependent CMG ubiquitylation occurs in the absence of fork convergence suggests that in mitosis, TRAIP gains the capacity to ubiquitylate any CMG on chromatin. How mitotic CMG unloading preserves genome stability is discussed below.
Figure 3. Cell cycle regulation of TRAIP specificity.
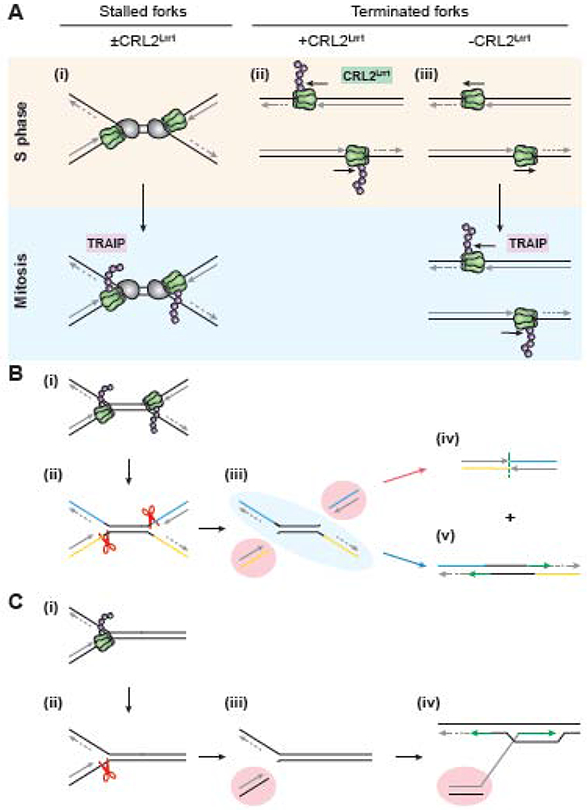
A. (i) During S phase, the CDC45/MCM2-7/GINS (CMG) helicases of stalled but unconverged replisomes are not ubiquitylated. However, if these stalled CMGs persist into mitosis, they undergo TRAIP-dependent ubiquitylation and subsequent unloading by p97. (ii) Terminated CMGs are normally ubiquitylated in S phase by the Cul2-based RING E3 ubiquitin ligase CRL2Lrr1. (iii) in the absence of CRL2Lrr1, terminated CMGs remain unmodified and persist on chromatin until mitosis, when they are ubiquitylated by TRAIP and unloaded.
B. (i) During mitosis, CMGs stalled at the boundaries of a common fragile site are ubiquitylated by TRAIP. (ii) Ubiquitylated CMGs are unloaded, which exposes the leading strand templates of the two forks to structure-specific endonucleases such as MUS81-EME1. (iii) Cleavage of the leading strand templates results in two broken ends (highlighted in red) and an intact strand (highlighted in blue). The two broken ends are repaired by end joining (iv), while the intact strand is restored by gap filling (green arrows) (v). These two repair processes result in the deletion of the unreplicated DNA and sister chromatid exchange, which are hallmarks of common fragile site (CFS) expression.
C. If a chromosome containing a stalled fork enters mitosis in the absence of a nearby converging fork (e.g. at a telomere) (i), TRAIP-mediated mitotic CMG unloading (ii) leads to leading strand template cleavage and a single-ended double-strand break (highlighted in red) (iii). This break could then be repaired by break-induced replication (iv).
The concept that TRAIP acts on all CMGs during mitosis is supported by experiments on replication termination. When CMGs converge at the end of DNA replication, they are ubiquitylated and unloaded by p97 (Figure 3A,iii) [52, 53]. In interphase, this ubiquitylation depends on the E3 ubiquitin ligase CRL2Lrr. TRAIP is not involved, probably because CMGs pass each other during termination [54, 55], which precludes the in trans ubiquitylation by TRAIP observed at ICLs. However, in worms and Xenopus egg extracts lacking CRL2Lrr1, CMGs are retained on chromatin until mitosis and are then ubiquitylated by TRAIP and unloaded by p97 (Figure 3A, iv-v) [51, 56-58]. Thus, mitotic ubiquitylation of terminated replisomes represents another situation where TRAIP ubiquitylates CMGs that are not in close contact. Together, these studies further support the notion that in mitosis, TRAIP triggers the unloading of any CMG from chromatin.
What underlies the dramatic increase in TRAIP’s appetite for unloading CMG during mitosis? Mitotic entry does not appear to trigger de novo recruitment of additional TRAIP molecules to the replisome [51]. Therefore, mitotic kinases could induce a conformational change in TRAIP that permits its RING domain to bring an E2 enzyme into contact with its host CMG (Figure 2, “Mitosis”). However, other mechanisms can also be envisioned, such as the displacement of inhibitors that suppress host CMG ubiquitylation in interphase, or mitosis-specific CMG modifications that make it a better TRAIP substrate. Structural analysis will likely be required to understand how the S phase constraint on TRAIP function is relieved in mitosis.
How does mitotic CMG unloading protect genomic stability?
The evidence discussed above indicates that TRAIP promotes unloading of terminated CMGs when the interphase pathway fails, but also CMGs associated with replication forks at incompletely replicated DNA [51, 56-58]. Incomplete DNA replication poses a serious threat to genomic stability. This is because, upon sister chromatid segregation, unreplicated DNA forms DNA bridges, leading to binucleated cells, aneuploidy, chromothripsis, and other gross chromosomal rearrangements [59-62]. The effect of persistently stalled forks and under-replicated DNA is exemplified by common fragile sites (CFSs), specific genomic loci frequently rearranged in cancer genomes. CFSs are replicated late in S phase and prone to under-replication due to the presence of difficult-to-replicate sequences, large transcriptionally active genes, and sparse origins of replication [63]. Under-replication at CFS is enhanced by mild replication stress, which induces high levels of anaphase ultrafine bridges, copy number variations, chromosomal rearrangements, and distinctive gaps and breaks on metaphase chromosomes (“CFS expression”). Cells experiencing replication stress undergo a specialized form of mitotic DNA synthesis (“MiDAS”) at known CFSs. Because MiDAS requires fork cleavage by the structure-specific endonuclease MUS81-EME1 [64-66], it has been interpreted as a form of break-induced replication that completes replication between the forks stalled at the CFS boundaries. But while break-induced replication would be consistent with the small deletions and microhomologies often found at expressed CFSs, it does not explain the high frequency of sister chromatid exchanges at these sites [63].
Experiments in Xenopus egg extracts suggest a new model of CFS expression. In mitotic extracts, CMG unloading at stalled forks is followed by quantitative fork breakage, consistent with previous observations of extensive chromosome breakage after S phase and mitotic cells are fused [67]. This breakage requires CMG unloading by TRAIP and p97 [51], suggesting that CMG normally protects the DNA it encircles. Upon mitotic entry, CMGs stalled at either end of a CFS undergo TRAIP-dependent unloading, which would expose the leading strand templates to cleavage by a structure-specific endonuclease such as MUS81-EME1 (Figure 3Bi-iii) [51]. Cleavage would result in one intact strand, which can be restored directly through gap filling (Figure 3B, blue arrow pathway), and two broken ends, which can be repaired by alternative end joining (Figure 3B, red arrow pathway). The result of these transactions is a sister chromatid exchange event and a small deletion corresponding to the unreplicated DNA, the hallmarks of CFS expression. If a converging fork is not available for end joining (as would be the case at a telomere), the single-ended double-strand break could be repaired via break-induced replication (Figure 3C). MiDAS could thus represent DNA synthesis involved in restoring the intact strand, joining the broken ends, and/or break-induced replication. Consistent with this interpretation, Sonneville et al. found that in mammalian cells, depletion of TRAIP suppresses MiDAS [57].
In conclusion, the model described in Figure 3 suggests that if cells fail to complete replication before mitosis, TRAIP promotes biased strand breakage of stalled forks, thereby avoiding the catastrophic genomic instability that occurs from segregation of incompletely replicated chromosomes [51]. In agreement with this idea, TRAIP deletion results in dramatic accumulation of anaphase bridges in C. elegans under replication stress [57] and TRAIP-depleted cells exhibit increased micronucleation and gross chromosomal aberrations [18, 19].
TRAIP in human health and cancer
TRAIP-deficient human cells proliferate slowly, even in the absence of exogenous DNA damage [15]. This observation explains why TRAIP deficiency causes microcephalic primordial dwarfism, which involves a reduction of cellular proliferation during development [16]. A critical question is why loss of TRAIP compromises cell proliferation. The simplest explanation is that replication stress leads to cell cycle delay. For example, through delayed activation of the Fanconi anemia pathway, hypomorphic TRAIP mutations might elicit a G2 arrest, as seen in Fanconi patient cells (REF), while still supporting enough repair to prevent the other symptoms of Fanconi anemia. However, defects in other putative functions (see Box 1: “Other Proposed TRAIP functions”) may also disrupt normal cell cycle progression. A deeper understanding of TRAIP’s mechanism of action will be required to determine how it contributes to normal cellular proliferation.
Box 1: Other proposed TRAIP functions.
In addition to the functions discussed in the main text, TRAIP’s contribution to genomic stability may also derive from a role in double-strand break repair. TRAIP interacts with RNF20-RNF40, the E3 ubiquitin ligase complex responsible for monoubiquitylating histone H2B in vertebrates [71]. This interaction recruits TRAIP to double-strand breaks [71] and reciprocally, TRAIP depletion leads to a decrease in ionizing radiation-induced H2B monoubiquitylation [78]. H2B monoubiquitylation has been implicated in homologous recombination by promoting BRCA1 recruitment [79]. Intriguingly, TRAIP also interacts with the BRCA1 partner RAP80, and TRAIP depletion decreases RAP80 levels at double-strand breaks [71]. These results suggest that TRAIP functions in double-strand break repair, both upstream and downstream of H2B monoubiquitylation. Accordingly, TRAIP correlates highly with recombination genes in genome-wide CRISPR screens against genotoxic agents [80]. Curiously, there are conflicting data on whether TRAIP depletion sensitizes cells to ionizing radiation (compare for example [71] and [19]). Regardless, it is notable that TRAIP potentially promotes H2B ubiquitylation, a modification that has been implicated in many aspects of DNA replication and repair, including fork progression during unperturbed and stressed replication [81], checkpoint activation [82], nucleotide excision repair [83], and transcription [84]. Thus, TRAIP function could impinge on an array of chromatin-related processes, any of which could contribute to the defects in the DNA damage response and proliferation caused by TRAIP deficiency.
TRAIP has also been reported to function at kinetochores during mitosis and meiosis. TRAIP depletion leads to decreased stability of kinetochore-microtubule attachments [85] and lowered MAD2 levels at centromeres, resulting in diminished spindle assembly checkpoint function [85, 86]. Together, these defects lead to higher rates of chromosomal misalignment and segregation errors. The mechanisms by which TRAIP impacts kinetochore function and MAD2 localization remain unclear, and a caveat of these studies is that rescue of chromosomal segregation defects by exogenous TRAIP re-expression was not reported. Nevertheless, the findings could point to an additional mechanism by which chromosomal instability arises from loss of TRAIP function.
Genomic instability accelerates tumor progression and contributes to the development of cytotoxic chemotherapy resistance [68]. Chromosomal instability, apparent in TRAIP-depleted cells as micronuclei and chromosomal abnormalities [18, 19], is a hallmark of cancer cells [69, 70]. Given its roles in genome integrity maintenance, TRAIP loss-of-function mutations or decreased expression may be predicted to promote tumorigenesis. Indeed, TRAIP is expressed at significantly lower levels in human lung adenocarcinoma patient tissues relative to matched normal tissues [71]. However, cancer predisposition is not a feature of Seckel syndrome, including the disorder caused by TRAIP deficiency [16]. One possible reason is that the TRAIP patients were not followed long enough to detect cancer development. However, an alternative explanation is that the cellular proliferation deficiency in Seckel syndrome suppresses tumor progression [72]. In this view, TRAIP may be under positive selective pressure in tumor cells. Consistent with this notion, TRAIP expression is upregulated in non-small-cell lung cancer tissues, and overexpression is significantly associated with tumor metastasis and poor patient prognosis [73]. Furthermore, cancer cells with elevated replication stress and/or defective DNA repair may be particularly sensitive to TRAIP inhibition. Such synthetic lethal relationships have been identified with ATR [72, 74, 75], another Seckel syndrome gene, and ATR inhibitors are now being developed for cancer treatment [76, 77].
Concluding remarks
In the years since the link between TRAIP and microcephalic primordial dwarfism was found, considerable progress has been made toward understanding the mechanisms by which TRAIP promotes DNA replication and genome stability. Studies in cells and extracts described here have shown that by ubiquitylating proteins blocking the replication fork during S phase, and by triggering replisome unloading during mitosis, TRAIP promotes the completion of DNA replication and, failing this, minimizes catastrophic outcomes upon cell division. Understanding how TRAIP carries out these functions will ultimately require answering fundamental outstanding questions (see Outstanding Questions Box), such as how TRAIP interacts with the replisome and how its specificity changes during the cell cycle. Given TRAIP’s central roles in the response to replication stress, we expect that elucidating its molecular mechanisms will have important ramifications for the treatment of human disease.
Outstanding Questions.
How do TRAIP’s varied functions contribute to the preservation of genomic stability? Which function is most crucial for cellular fitness, viability, and suppression of dwarfism?
What is the structure of TRAIP within the replisome? Can this reveal how TRAIP ubiquitylates fork barriers and how TRAIP is prevented from prematurely disassembling its host replisome?
While TRAIP deficiency sensitizes cells to ICL-inducing agents, less is known about its contribution to cellular tolerance of DPCs. Is TRAIP’s function crucial for the resolution of all DPCs or a particular subset?
Which chromatin-associated proteins are ubiquitylated by TRAIP? Does TRAIP activity have particular consequences for specific replication contexts, such as at replication-transcription conflicts or at specific genomic loci?
What is the mechanism of TRAIP’s mitotic specificity switch?
Highlights.
TRAIP is an essential replisome-associated E3 ubiquitin ligase that preserves genomic integrity by promoting replication fork progression, especially during genotoxic stress.
TRAIP initiates the NEIL3 and Fanconi anemia DNA interstrand crosslink repair pathways and regulates the choice between them by ubiquitylating the CMG helicases of replisomes stalled at the lesion.
TRAIP ubiquitylates DNA-protein crosslink fork barriers, marking the lesion for degradation.
TRAIP triggers disassembly of replisomes that remain on chromatin in mitosis, enabling processing events that prevent chromosomal instability.
According to the model proposed here, replisome-associated TRAIP ubiquitylates protein barriers ahead of the fork in S phase, whereas in mitosis, TRAIP also triggers ubiquitylation and disassembly of all replisomes remaining on DNA.
Acknowledgements:
Studies on TRAIP in the Walter and Pellman Laboratories are supported by American Cancer Society postdoctoral fellowship 131415-PF-17-168-01-DMC to R.A.W and NIH grants 2R01 HL098316-09 to J.C.W. and 5R01 CA213404-22 to D.S.P. D.S.P. and J.C.W. are Howard Hughes Medical Institute Investigators. J.C.W. is an American Cancer Society Research Professor.
Glossary
- CMG helicase
A DNA helicase consisting of the subunits CDC45, MCM2-7, and the GINS complex, that unwinds DNA at replication forks by passing the leading strand template of the parental DNA through its central pore, thus driving progression of the replisome.
- CRL2Lrr1
A member of the Cullin-Rbx family of E3 ubiquitin ligases, consisting of the subunits Cul2, Rbx1, Lrr1, Elongin B, and Elongin C, which has been implicated in ubiquitylating CMGs upon replication termination to trigger replisome disassembly.
- DNA interstrand crosslink (ICL)
A DNA lesion in which the two strands of DNA become covalently linked, preventing the unwinding of DNA that is required for DNA replication and transcription.
- DNA-protein crosslink (DPC)
A form of DNA damage in which a protein becomes covalently crosslinked to DNA, potentially blocking chromatin-associated processes, such as the translocation of DNA helicases.
- E3 ubiquitin ligase
Eukaryotic enzymes that catalyze the covalent attachment of ubiquitin to substrate proteins.
- p97 segregase
Also known as Cdc48 in yeast or VCP, a hexameric AAA+ ATPase that functions with various adaptor proteins to recognize and unfold polyubiquitylated substrates by translocating them through its central pore.
- Replication forks
The splayed DNA structure where DNA synthesis occurs.
- Replication termination
The process during which converging replication forks meet, replisomes are disassembled, and daughter duplexes are decatenated.
- Replisome
The collection of proteins, including DNA helicase, DNA polymerases, and primase, that carry out DNA replication.
- RING domain
A zinc-coordinating domain commonly found in E3 ubiquitin ligases that facilitates transfer of ubiquitin from an E2 ubiquitin conjugating enzyme to the target substrate.
- Ubiquitylation
A post-translational modification in which the amino group of a lysine side chain or the terminal amino group of a protein is conjugated to the carboxyl terminus of the 8 kDa protein ubiquitin. Monoubiquitylation refers to the attachment of one ubiquitin, whereas conjugation of the modifying ubiquitin with additional ubiquitin molecules is termed polyubiquitylation.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Zeman MK and Cimprich KA, Causes and consequences of replication stress. Nat Cell Biol, 2014. 16(1): p. 2–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Rickman K and Smogorzewska A, Advances in understanding DNA processing and protection at stalled replication forks. J Cell Biol, 2019. 218(4): p. 1096–1107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Cortez D, Replication-Coupled DNA Repair. Mol Cell, 2019. 74(5): p. 866–876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Saldivar JC, Cortez D, and Cimprich KA, The essential kinase ATR: ensuring faithful duplication of a challenging genome. Nat Rev Mol Cell Biol, 2017. 18(10): p. 622–636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Fernández-Casañas M and Chan KL, The Unresolved Problem of DNA Bridging. Genes (Basel), 2018. 9(12). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Bizard AH and Hickson ID, Anaphase: a fortune-teller of genomic instability. Curr Opin Cell Biol, 2018. 52: p. 112–119. [DOI] [PubMed] [Google Scholar]
- 7.Bergink S and Jentsch S, Principles of ubiquitin and SUMO modifications in DNA repair. Nature, 2009. 458(7237): p. 461–7. [DOI] [PubMed] [Google Scholar]
- 8.Yates M and Maréchal A, Ubiquitylation at the Fork: Making and Breaking Chains to Complete DNA Replication. Int J Mol Sci, 2018. 19(10). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.García-Rodríguez N, Wong RP, and Ulrich HD, Functions of Ubiquitin and SUMO in DNA Replication and Replication Stress. Front Genet, 2016. 7: p. 87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Villa-Hernández S, Bueno A, and Bermejo R, The Multiple Roles of Ubiquitylation in Regulating Challenged DNA Replication. Adv Exp Med Biol, 2017. 1042: p. 395–419. [DOI] [PubMed] [Google Scholar]
- 11.Lee SY, Lee SY, and Choi Y, TRAF-interacting protein (TRIP): a novel component of the tumor necrosis factor receptor (TNFR)- and CD30-TRAF signaling complexes that inhibits TRAF2-mediated NF-kappaB activation. J Exp Med, 1997. 185(7): p. 1275–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Regamey A, et al. , The tumor suppressor CYLD interacts with TRIP and regulates negatively nuclear factor kappaB activation by tumor necrosis factor. J Exp Med, 2003. 198(12): p. 1959–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Besse A, et al. , TRAF-interacting protein (TRIP) is a RING-dependent ubiquitin ligase. Biochem Biophys Res Commun, 2007. 359(3): p. 660–4. [DOI] [PubMed] [Google Scholar]
- 14.Chapard C, Hohl D, and Huber M, The role of the TRAF-interacting protein in proliferation and differentiation. Exp Dermatol, 2012. 21(5): p. 321–6. [DOI] [PubMed] [Google Scholar]
- 15.Harley ME, et al. , TRAIP promotes DNA damage response during genome replication and is mutated in primordial dwarfism. Nat Genet, 2016. 48(1): p. 36–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Klingseisen A and Jackson AP, Mechanisms and pathways of growth failure in primordial dwarfism. Genes Dev, 2011. 25(19): p. 2011–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Wu RA, et al. , TRAIP is a master regulator of DNA interstrand crosslink repair. Nature, 2019. 567(7747): p. 267–272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Feng W, et al. , TRAIP regulates replication fork recovery and progression via PCNA. Cell Discov, 2016. 2: p. 16016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Hoffmann S, et al. , TRAIP is a PCNA-binding ubiquitin ligase that protects genome stability after replication stress. J Cell Biol, 2016. 212(1): p. 63–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Chen Y, et al. , Nucleolar residence of the seckel syndrome protein TRAIP is coupled to ribosomal DNA transcription. Nucleic Acids Res, 2018. 46(19): p. 10119–10131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Dewar JM, et al. , CRL2(Lrr1) promotes unloading of the vertebrate replisome from chromatin during replication termination. Genes Dev, 2017. 31(3): p. 275–290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Wessel SR, et al. , Functional Analysis of the Replication Fork Proteome Identifies BET Proteins as PCNA Regulators. Cell Rep, 2019. 28(13): p. 3497–3509 e4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Hodskinson MR, et al. , Alcohol-derived DNA crosslinks are repaired by two distinct mechanisms. Nature, 2020. 579(7800): p. 603–608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Semlow DR, et al. , Replication-Dependent Unhooking of DNA Interstrand Cross-Links by the NEIL3 Glycosylase. Cell, 2016. 167(2): p. 498–511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Taylor AMR, et al. , Chromosome instability syndromes. Nat Rev Dis Primers, 2019. 5(1): p. 64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Ceccaldi R, Sarangi P, and D'Andrea AD, The Fanconi anaemia pathway: new players and new functions. Nat Rev Mol Cell Biol, 2016. 17(6): p. 337–49. [DOI] [PubMed] [Google Scholar]
- 27.Li N, et al. , Cooperation of the NEIL3 and Fanconi anemia/BRCA pathways in interstrand crosslink repair. Nucleic Acids Res, 2020. 48(6): p. 3014–3028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Zhang J, et al. , DNA interstrand cross-link repair requires replication-fork convergence. Nat Struct Mol Biol, 2015. 22(3): p. 242–247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Klein Douwel D, et al. , XPF-ERCC1 acts in Unhooking DNA interstrand crosslinks in cooperation with FANCD2 and FANCP/SLX4. Mol Cell, 2014. 54(3): p. 460–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Long DT, et al. , BRCA1 promotes unloading of the CMG helicase from a stalled DNA replication fork. Mol Cell, 2014. 56(1): p. 174–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Fullbright G, et al. , p97 Promotes a Conserved Mechanism of Helicase Unloading during DNA Cross-Link Repair. Mol Cell Biol, 2016. 36(23): p. 2983–2994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Twomey EC, et al. , Substrate processing by the Cdc48 ATPase complex is initiated by ubiquitin unfolding. Science, 2019. 365(6452). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Huang J, et al. , The DNA Translocase FANCM/MHF Promotes Replication Traverse of DNA Interstrand Crosslinks. Mol Cell, 2013. 52(3): p. 434–446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Mutreja K, et al. , ATR-Mediated Global Fork Slowing and Reversal Assist Fork Traverse and Prevent Chromosomal Breakage at DNA Interstrand Cross-Links. Cell Rep, 2018. 24(10): p. 2629–2642 e5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Larsen NB, et al. , Mechanism of replication-coupled DNA-protein crosslink proteolysis by SPRTN and the proteasome Mol Cell, 2019. 73(3): p.574–588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Stingele J, Bellelli R, and Boulton SJ, Mechanisms of DNA-protein crosslink repair. Nat Rev Mol Cell Biol, 2017. 18(9): p. 563–573. [DOI] [PubMed] [Google Scholar]
- 37.Fu YV, et al. , Selective Bypass of a Lagging Strand Roadblock by the Eukaryotic Replicative DNA Helicase. Cell, 2011. 146(6): p. 931–941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Lopez-Mosqueda J, et al. , SPRTN is a mammalian DNA-binding metalloprotease that resolves DNA-protein crosslinks. Elife, 2016. 5: e21491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Vaz B, et al. , Metalloprotease SPRTN/DVC1 Orchestrates Replication-Coupled DNA-Protein Crosslink Repair. Mol Cell, 2016. 64(4): p. 704–719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Stingele J, et al. , Mechanism and Regulation of DNA-Protein Crosslink Repair by the DNA-Dependent Metalloprotease SPRTN. Mol Cell, 2016. 64(4): p. 688–703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Stingele J, et al. , A DNA-dependent protease involved in DNA-protein crosslink repair. Cell, 2014. 158(2): p. 327–38. [DOI] [PubMed] [Google Scholar]
- 42.Duxin JP, et al. , Repair of a DNA-protein crosslink by replication-coupled proteolysis. Cell, 2014. 159(2): p. 346–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Sparks J, et al. , The CMG helicase bypasses DNA protein cross-links to facilitate their repair. Cell, 2019. 176(1-2): p. 167–181.e21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Wang Y, Vujcic M, and Kowalski D, DNA replication forks pause at silent origins near the HML locus in budding yeast. Mol Cell Biol, 2001. 21 (15): p. 4938–48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Krings G and Bastia D, Sap1p binds to Ter1 at the ribosomal DNA of Schizosaccharomyces pombe and causes polar replication fork arrest. J Biol Chem, 2005. 280(47): p. 39135–42. [DOI] [PubMed] [Google Scholar]
- 46.Hamperl S and Cimprich KA, Conflict Resolution in the Genome: How Transcription and Replication Make It Work. Cell, 2016. 167(6): p. 1455–1467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Lecona E, et al. , USP7 is a SUMO deubiquitinase essential for DNA replication. Nat Struct Mol Biol, 2016. 23(4): p. 270–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Álvarez V, et al. , PCNA Deubiquitylases Control DNA Damage Bypass at Replication Forks. Cell Rep, 2019. 29(5): p. 1323–1335.e5. [DOI] [PubMed] [Google Scholar]
- 49.Lim KS, et al. , USP1 Is Required for Replication Fork Protection in BRCA1-Deficient Tumors. Mol Cell, 2018. 72(6): p. 925–941.e4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Hernández-Pérez S, et al. , USP37 deubiquitinates Cdt1 and contributes to regulate DNA replication. Mol Oncol, 2016. 10(8): p. 1196–206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Deng L, et al. , Mitotic CDK promotes replisome disassembly, fork breakage, and complex DNA rearrangements. Mol Cell, 2019. 73(5): p. 915–929.e6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Moreno SP, et al. , Polyubiquitylation drives replisome disassembly at the termination of DNA replication. Science, 2014. 346(6208): p. 477–81. [DOI] [PubMed] [Google Scholar]
- 53.Maric M, et al. , Cdc48 and a ubiquitin ligase drive disassembly of the CMG helicase at the end of DNA replication. Science, 2014. 346(6208): p. 1253596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Dewar JM, Budzowska M, and Walter JC, The mechanism of DNA replication termination in vertebrates. Nature, 2015. 525(7569): p. 345–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Low E, et al. , The DNA replication fork suppresses CMG unloading from chromatin before termination. Genes Dev, 2020. 34(21-22): p. 1546–1558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Sonneville R, et al. , CUL-2LRR-1 and UBXN-3/FAF1 drive replisome disassembly during DNA replication termination and mitosis. Nat. Cell Biol, 2017. 19(5): p. 468–479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Sonneville R, et al. , TRAIP drives replisome disassembly and mitotic DNA repair synthesis at sites of incomplete DNA replication. Elife, 2019. 8: e48686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Priego Moreno S, et al. , Mitotic replisome disassembly depends on TRAIP ubiquitin ligase activity. Life Sci Alliance, 2019. 2(2): e201900390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Nielsen CF, et al. , PICH promotes sister chromatid disjunction and co-operates with topoisomerase II in mitosis. Nat Commun, 2015. 6: p. 8962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Maciejowski J, et al. , Chromothripsis and Kataegis Induced by Telomere Crisis. Cell, 2015. 163(7): p. 1641–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Liu S, et al. , Nuclear envelope assembly defects link mitotic errors to chromothripsis. Nature, 2018. 561(7724): p. 551–555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Umbreit NT, et al. , Mechanisms generating cancer genome complexity from a single cell division error. Science, 2020. 368(6488). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Glover TW, Wilson TE, and Arlt MF, Fragile sites in cancer: more than meets the eye. Nat Rev Cancer, 2017. 17(8): p. 489–501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Ying S, et al. , MUS81 promotes common fragile site expression. Nat Cell Biol, 2013. 15(8): p. 1001–7. [DOI] [PubMed] [Google Scholar]
- 65.Naim V, et al. , ERCC1 and MUS81-EME1 promote sister chromatid separation by processing late replication intermediates at common fragile sites during mitosis. Nat Cell Biol, 2013. 15(8): p. 1008–15. [DOI] [PubMed] [Google Scholar]
- 66.Minocherhomji S, et al. , Replication stress activates DNA repair synthesis in mitosis. Nature, 2015. 528(7581): p. 286–90. [DOI] [PubMed] [Google Scholar]
- 67.Rao PN, et al. , The Phenomenon of Premature Chromosome Condensation. 1982: p. 1–41. [Google Scholar]
- 68.Gaillard H, García-Muse T, and Aguilera A, Replication stress and cancer. Nat Rev Cancer, 2015. 15(5): p. 276–89. [DOI] [PubMed] [Google Scholar]
- 69.Thompson SL, Bakhoum SF, and Compton DA, Mechanisms of chromosomal instability. Curr Biol, 2010. 20(6): p. R285–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Hintzsche H, et al. , Fate of micronuclei and micronucleated cells. Mutat Res, 2017. 771 : p. 85–98. [DOI] [PubMed] [Google Scholar]
- 71.Soo Lee N, et al. , TRAIP/RNF206 is required for recruitment of RAP80 to sites of DNA damage. Nat Commun, 2016. 7: p. 10463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Murga M, et al. , Exploiting oncogene-induced replicative stress for the selective killing of Myc-driven tumors. Nat Struct Mol Biol, 2011. 18(12): p. 1331–1335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Li J, et al. , DCUN1D1 facilitates tumor metastasis by activating FAK signaling and up-regulates PD-L1 in non-small-cell lung cancer. Exp Cell Res, 2019. 374(2): p. 304–314. [DOI] [PubMed] [Google Scholar]
- 74.Toledo LI, et al. , A cell-based screen identifies ATR inhibitors with synthetic lethal properties for cancer-associated mutations. Nat Struct Mol Biol, 2011. 18(6): p. 721–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Hocke S, et al. , A synthetic lethal screen identifies ATR-inhibition as a novel therapeutic approach for POLD1-deficient cancers. Oncotarget, 2016. 7(6): p. 7080–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Boudny M and Trbusek M, ATR-CHK1 pathway as a therapeutic target for acute and chronic leukemias. Cancer Treat Rev, 2020. 88: p. 102026. [DOI] [PubMed] [Google Scholar]
- 77.Zhu H, et al. , Harnessing DNA Replication Stress for Novel Cancer Therapy. Genes (Basel), 2020. 11(9). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Han YG, et al. , TRAIP regulates Histone H2B monoubiquitination in DNA damage response pathways. Oncol Rep, 2019. 41(6): p. 3305–3312. [DOI] [PubMed] [Google Scholar]
- 79.Wu J, et al. , Histone ubiquitination associates with BRCA1-dependent DNA damage response. Mol Cell Biol, 2009. 29(3): p. 849–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Olivieri M, et al. , A Genetic Map of the Response to DNA Damage in Human Cells. Cell, 2020. 182(2): p. 481–496.e21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Trujillo KM and Osley MA, A role for H2B ubiquitylation in DNA replication. Mol Cell, 2012. 48(5): p. 734–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Giannattasio M, et al. , The DNA damage checkpoint response requires histone H2B ubiquitination by Rad6-Bre1 and H3 methylation by Dot1. J Biol Chem, 2005. 280(11): p. 9879–86. [DOI] [PubMed] [Google Scholar]
- 83.Tatum D and Li S, Evidence that the histone methyltransferase Dot1 mediates global genomic repair by methylating histone H3 on lysine 79. J Biol Chem, 2011. 286(20): p. 17530–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Pavri R, et al. , Histone H2B monoubiquitination functions cooperatively with FACT to regulate elongation by RNA polymerase II. Cell, 2006. 125(4): p. 703–17. [DOI] [PubMed] [Google Scholar]
- 85.Yuan YF, et al. , TRAIP is involved in chromosome alignment and SAC regulation in mouse oocyte meiosis. Sci Rep, 2016. 6: p. 29735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Chapard C, et al. , TRAIP is a regulator of the spindle assembly checkpoint. J Cell Sci, 2014. 127(Pt 24): p. 5149–56. [DOI] [PubMed] [Google Scholar]