

Abstract
Carbohydrate chemistry is an essential component of the glycosciences and is fundamental to their progress. This perspective takes the position that carbohydrate chemistry, or glycochemistry, has reached three crossroads on the path to the transformation of the glycosciences, and illustrates them with examples from the author’s and other laboratories. The first of these potential inflexion points concerns the mechanism of the glycosylation reaction and the role of protecting groups. It is argued that the experimental evidence supports bimolecular SN2-like mechanisms for typical glycosylation reactions over unimolecular ones involving stereoselective attack on naked glycosyl oxocarbenium ions. Similarly, it is argued that the experimental evidence does not support long range stereodirecting participation of remote esters through bridged bicyclic dioxacarbenium ions in organic solution in the presence of typical counterions. Rational design and improvement of glycosylation reactions must take into account the role of the counterion and of concentration. A second crossroads is that between mainstream organic chemistry and glycan synthesis. The case is made that the only real difference between glycan and organic synthesis is the formation of C-O rather than C-C bonds, with diastereocontrol, strategy, tactics and elegance being of critical importance in both areas: mainstream organic chemists should feel comfortable taking this fork in the road, just as carbohydrate chemists should travelling in the opposite direction. A third crossroads is that between carbohydrate chemistry and medicinal chemistry, where there are equally many opportunities for traffic in either direction. The glycosciences have advanced enormously in the last decade or so, but the creativity, input and ingenuity of scientists from all fields is needed to address the many sophisticated challenges that remain, not the least of which is the development of a broader and more general array of stereospecific glycosylation reactions.
Graphical Abstract
Introduction
The critical role of carbohydrates and their conjugates across all domains of life, revealed over the course of the last several decades, has led to a considerable upswing of interest in the glycosciences, and their essential component, glycochemistry. In the United States this culminated in a study by the National Research Council of the National Academies of Sciences and of Engineering, and the publication of a report entitled “Transforming Glycoscience: A Roadmap for the Future (2012).1 In this report glycochemistry, traditionally known as carbohydrate chemistry, is considered to have lagged behind the glycosciences in so far as the predictable synthesis of glycosidic bonds and consequently of the many kinds of glycoconjugates required to better enable glycobiologists remains a challenge. Indeed, with regard to glycan synthesis the NRC report concluded that “Glycochemistry needs to be brought into the mainstream of synthetic organic chemistry rather than kept as a specialized field practiced by only a handful of glycochemists” and that “New synthetic methods need to be brought to bear on glycan synthesis, and the creativity of the entire synthesis community needs to be leveraged to solve the long-standing and vexing problems of stereoselective, regioselective syntheses with simple, high yielding reactions”. These laudable and important goals were taken up by the National Institutes of Health, which responded by setting up a Glycoscience program under its Common Fund umbrella,2 one of whose initiatives targeted the development of methods and technologies for synthesizing biomedically relevant carbohydrates.
As a mainstream organic chemist drawn to glycochemistry,3 when a beginning academic with no previous experience in the area, I take this opportunity to present my personal perspective on a field that I consider to be at multiple crossroads. These crossroads are both internal to the field, where they relate to the way we think about glycosylation reactions, and external to it, where the issue is more one of removing perceived but artificial barriers between carbohydrate chemistry and organic and medicinal chemistry.
I begin at the internal crossroads with the glycosylation mechanism, the heart of glycochemistry, as I believe that the field is currently at a stage at which it must decide whether to take an evidence-based track or continue to follow a more traditional path that, according to the NRC report, clearly has not yielded the desired results. Turning to overlap with the broader field of organic chemistry in general, I continue with the challenge of stereocontrolled synthesis of complex glycans, which provides ample but rarely exploited opportunity for mainstream organic chemists to take a side road and exhibit their creativity in glycochemistry. Finally, I address carbohydrate chemistry as a source of inspiration for problems in organic and medicinal chemistry, drawing on examples from my own laboratory, to show how traffic at this crossroads can travel in all directions.
Crossroads 1. The glycosylation mechanism
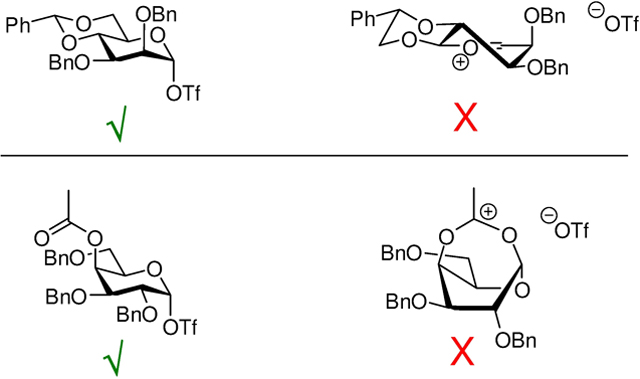
Consideration of reaction mechanisms is one of the best means of generating hypotheses in organic chemistry. In carbohydrate chemistry this leads inevitably to the general glycosylation reaction mechanism (Scheme 1),4 which is a specific instance of Winstein’s ion pair theory of nucleophilic substitution.5 Glycosidic bond synthesis is therefore very much a bridge between carbohydrate chemistry and mainstream organic chemistry, and there should be no barrier to cross-fertilization. The mechanism presented (Scheme 1), particularly with its emphasis on associative SN2-like mechanisms, is an extrapolation of ideas presented by Rhind-Tutt and Vernon in 1960,6 the group of Schuerch and others in the mid-1970’s,7, 8 and deployed by Lemieux and coworkers in their halide ion-catalyzed synthesis of α-glucopyranosides.9 The Web of Science10 reveals Lemieux’s paper on halide ion catalyzed glycosylations and associative mechanisms9 to be the third most highly cited one in the opus of this influential and revered glycoscientist,11 behind only those describing the Lemieux-Johnson oxidation,12 and the seminal correlation of NMR coupling constant data to configuration in six membered rings.13 In this light it is something of a paradox that pure SN1 mechanisms based on the concept of naked oxacarbenium ions as reaction intermediates continue to be so widely and mostly uncritically adopted.
Scheme 1.
General Glycosylation Mechanism.
The prevailing oxacarbenium ion-centric perspective of glycosylation reactions is easy to understand in light of the stability of resonance delocalized oxacarbenium ions vis à vis carbenium ions as featured in many organic textbooks, and because it allows interpretation of facial selectivity by familiar stereoelectronic principles.14, 15 Unfortunately, this oxacarbenium ion-based perspective does not reflect the true nature of glycosyl oxacarbenium ions, which is dominated by the multiplicity of electron-withdrawing, destabilizing C-O bonds. We have collected and discussed at length the evidence for and against glycosyl oxacarbenium ions as long-lived intermediates, will only briefly recap the broad lines here, and refer the reader to these earlier reviews for in-depth coverage.4, 16–20 Similarly, in support of covalent intermediates and SN2-like reactions, we have presented the extensive evidence for the nucleophilicity of the trifluoromethanesulfonate counterion common to most modern glycosylation reactions.21 This Perspective necessarily emphasizes the role of the triflate counterion, because it is so widespread in modern glycosylation reactions. However, it should be clear that other less nucleofugal counterions play directly analogous roles. The main difference in changing from a given covalent glycosyl triflate to an obviously more stable glycosyl bromide, for example, is that higher reaction temperatures are required to overcome the greater activation barriers. This change in counterion, and consequent change in reaction temperature, does not change the overall picture of the glycosylation mechanism as a set of equilibria (Scheme 1), as beautifully illustrated by Bennett and coworkers in their studies on glycosyl arenesulfonates as opposed to triflates.22 I make no attempt in this Perspective to distinguish between classical SN2 mechanisms, with direct displacement of a leaving group by a nucleophile from the anomeric position of a glycosyl donor, and kinetically indistinguishable SN2-like mechanisms, in which the nucleophile attacks a transient glycosyl oxacarbenioum ion – counterion pair in equilibrium with the covalent glycosyl donor. Rather the emphasis is on the distinction between bimolecular SN2-type mechanisms emphasizing the role of the counterion and reaction concentration, and classical unimolecular SN1 mechanisms with naked oxacarbenium ions as actual intermediates.
Like simple carbenium ions,23, 24 oxacarbenium ions are inductively destabilized by C-O bonds in proportion to the number and proximity of those bonds, as apparent in the Wong and Ley relative reactivity indexes of glycosyl donors.25, 26 Consequently, “typical” hexopyranosyl oxacarbenium ions have only a fleeting existence in organic solvents, ionic liquids, and superacidic media, preferring instead to form covalent adducts with the obligate counterion and/or solvent.4
Synthetic carbohydrate chemists have long espoused and exploited the concept of armed and disarmed glycosyl donors,26 introduced by Lemieux’s former student Fraser-Reid,27 and of which the Wong and Ley indexes25, 26 are extensions. According to this terminology, glycosyl donors protected by ethers are “armed” and can be activated selectively in the presence of donors protected by the more electron-withdrawing esters, which are termed disarmed. Unfortunately, however convenient this terminology may be, it obscures the fact that all C-O bonds are inductively electron-withdrawing and destabilizing toward oxacarbenium ions: all glycosyl oxacarbenium ions are destabilized and disarmed with respect to the parent tetrahydropyrylium oxacarbenium ion.
NMR spectra of glycosyl oxacarbenium ions were only reported in the peer-reviewed literature for the first time in 2016, with generation by dissolution of glycopyranosyl acetates in superacidic HF/SbF5. The 3,4,6-tri-O-acetyl-2-deoxy-glucopyranosyl ion 2 was the only pure oxacarbenium ion observed with 13C and 1H chemical shifts of δ 229 and δ 8.89, respectively, at the anomeric position.28 This ion was shown to be protonated on each of the three acetate esters, which took up pseudoequatorial positions around the oxacarbenium ion, and favoured an envelope conformation (4E) (Scheme 2). For the 3,4,6-tri-O-acetyl-2-bromo-2-deoxyglucopyranosyl ion 4, 13C and 1H anomeric chemical shifts of δ 198 and δ 8.36, respectively, were observed indicative of participation by the vicinal C-Br bond in a dissymmetric cyclic bromonium ion. In the 2,3,4,6-tetra-O-acetyl gluocpyranosyl and 2-acetamido-3,4,6-tri-O-acetyl-2-deoxy glucopyranosyl series the only ions observed were those resulting from participation by the proximal acetoxy or acetamido groups. 2-Azido-3,4,6-tri-O-acetyl-2-deoxy glucopyranosyl acetate did not undergo fragmentation under the reaction conditions, which was considered to be due to the protonation of the azido group and its consequent enhanced electron-withdrawing ability. Later, cyclic bromonium ion formation was also observed in the 3,4,6-tri-O-acetyl-2-bromo-2-deoxygalactopyranosyl series, where line broadening that prevented observation of the anomeric carbon resonance in the 13C NMR spectrum was attributed to a stabilizing orbital overlap between the bromine atom and C1.29 In the 3,4,6-tri-O-acetyl-2-fluoro-2-deoxyglucopyranosyl series, carrying a strongly electron-withdrawing but non-participating fluoride substituent at the 2-position, an adduct 6 with the SbF6− ion was observed, characterized by a chair conformation of the pyranose ring, anomeric 13C and 1H chemical shifts of δ 101 and δ 4.55, respectively, extensive line broadening at −40 °C, and 1JCF coupling to a single fluoride in the ligand at 25 °C of 24 Hz (Scheme 2). In contrast 2-fluorotetrahydropyrylium ion was readily observed under the same conditions with anomeric 13C and 1H chemical shifts of δ 227 and δ 8.79, respectively. These observations in the 2-deoxy-2-fluoro series emphasize the destabilizing effect of more remote esters on oxacarbenium ions and underline the possibility of participation by even the relatively non-nucleophilic hexafluoroantimonate anion.29 As most common promoters of glycosylation reactions incorporate the triflate counterion, activated glycosyl donors are mostly found in the form of covalent glycosyl triflates, many of which have been spectroscopically characterized4, 19, 30 since our early studies.31, 32 It follows that if the highly nucleofugal triflate, and the “non-nucleophilic” hexafluoroantimonate, perchlorate33, tetrafluoroborate,34 and possibly even triflimide35 anions form stable covalent adducts with typical glycosyl oxacarbenium ions other less nucleofugal counterions do so to an even greater extent.
Scheme 2.

Formation of oxacarbenium ions and a covalent glycosyl hexafluoroantimonate in superacid solution
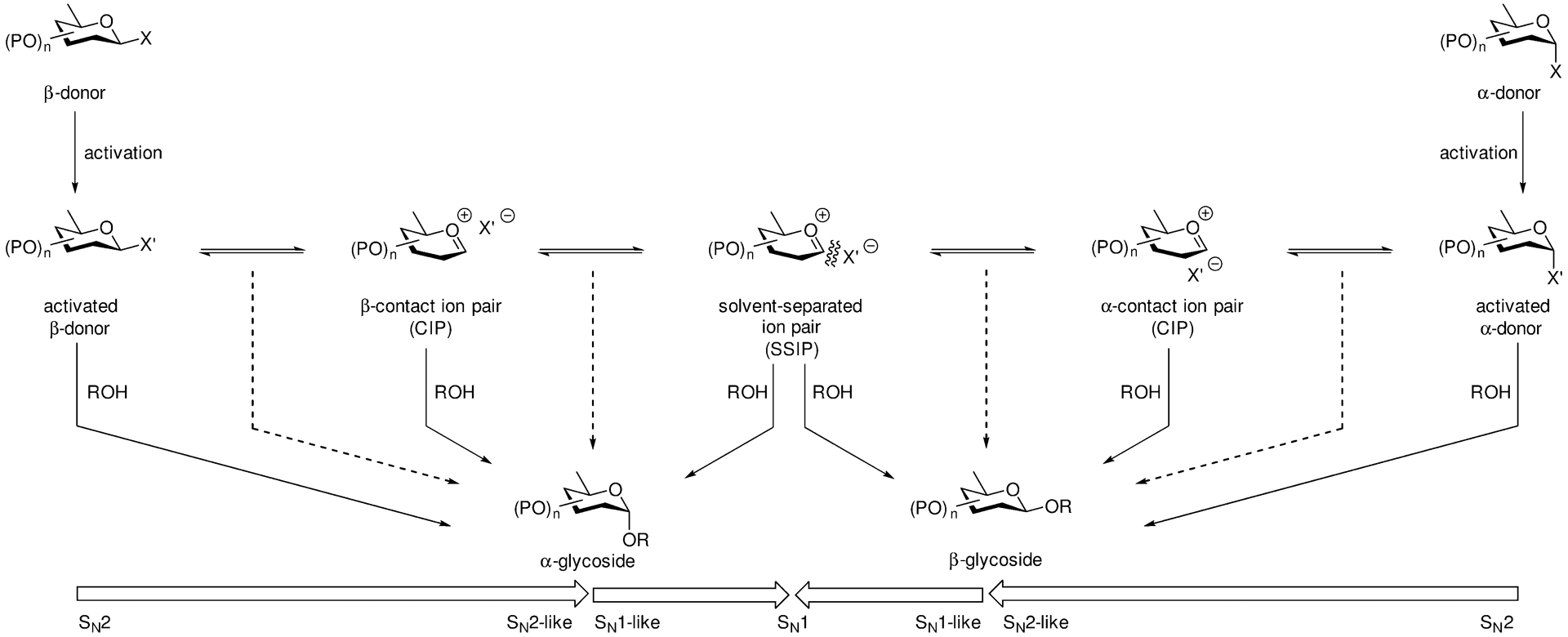
In addition to the failure to observe glycosyl oxacarbenium ions in organic solution, and to the very limited range of such ions observable even in superacid media,28, 29 an ever increasing number of studies, using kinetic isotope effect measurements, cation clock methods, NMR-based kinetic methods, and improved computational methods for ion pairs, have found bimolecular kinetics for a range of O-glycosylation reactions.4, 22, 32, 36–53 This accumulation of evidence supporting bimolecular reaction mechanisms stands in contrast to the very limited kinetic support for unimolecular glycosylation reactions proceeding by free oxacarbenium ions.4, 36, 38 The predominance of bimolecular mechanisms necessarily incorporates the possibility of Curtin-Hammett kinetics arising from rapid equilibration of anomeric pairs of activated donors (Scheme 1). However, both the position of these equilibria and the rate of exchange will be system and counterion dependent,32, 44 such that variation of counterions is a viable path to reaction optimization.
Evidence of the importance of counterions in the chemistry of even simple oxacarbenium ions is apparent from matching and mismatching effects in cationic polymerization,54 and especially alkylation of vinyl ethers where it is clears that, even if not covalently bound, the cation and anion are tightly associated in the confines of an ion pair 9 (Scheme 3).55
Scheme 3.

Counterion-Directed Addition of Enol Ethers to the Tetrahydrofuranylium Ion.
Overall, the accumulating evidence that most O-glycosylation reactions subjected to kinetic analysis proceed by bimolecular pathways with loosely associated transition states is substantial, suggesting that it is time for carbohydrate chemists to deemphasize the concept of naked oxacarbenium ions as intermediates and focus on the more evidence-based bimolecular mechanisms, for it is likely there that the path to more reproducible and stereoselective reactions will be found.
This is not to deny that oxacarbenium ions have any role to play at all, or are unsupported by any evidence. Indeed, work by Codée, van der Marel and their coworkers employing the silanes and allylstannanes as acceptors, building on the earlier studies of the Woerpel group,14, 56 is strongly supportive of oxacarbenium ion-centric mechanisms for glycosylation of such weak nucleophiles.57, 58 It must be stressed, however, that the computational work underpinning these studies does not include counterions and consequently can only find mechanisms based on naked oxacarbenium ions. Accordingly, caution must be exercised in extrapolating such studies of C-glycosylations and reductions to O-glycosylations of the far more nucleophilic alcohols, as stressed by Woerpel,56 and underlined by our work comparing C- and O-glycosylations in the sialic and ulosonic acid series.59 Kinetic isotope effect measurements point to the formation of the minor axial O-glycosides in the 4,6-O-benzylidene-directed mannopyranosylation reaction by an SN1 mechanism.36 Further support for the transient existence of an oxocarbenium ion in the formation of these minor isomers derives from the use of intramolecular Sakurai reactions as cation clocks. Thus, activation of either sulfoxide 11 or trichloroacetimidate 12 with triflic anhydride or TMS triflate, respectively, resulted in a ~1:2 mixture of the expected cis-fused product 16, and the unexpected trans-fused product 17 (Scheme 4). This result was interpreted as requiring population of a B2,5 conformation 15 of the oxacarbenium ion and consequently of a lifetime sufficient to attain it under the reaction conditions.37, 38 It is stressed, however, that α-mannosylation is an exception, with the evidence supporting associative mechanisms for all other systems investigated in these studies.36, 38
Scheme 4.
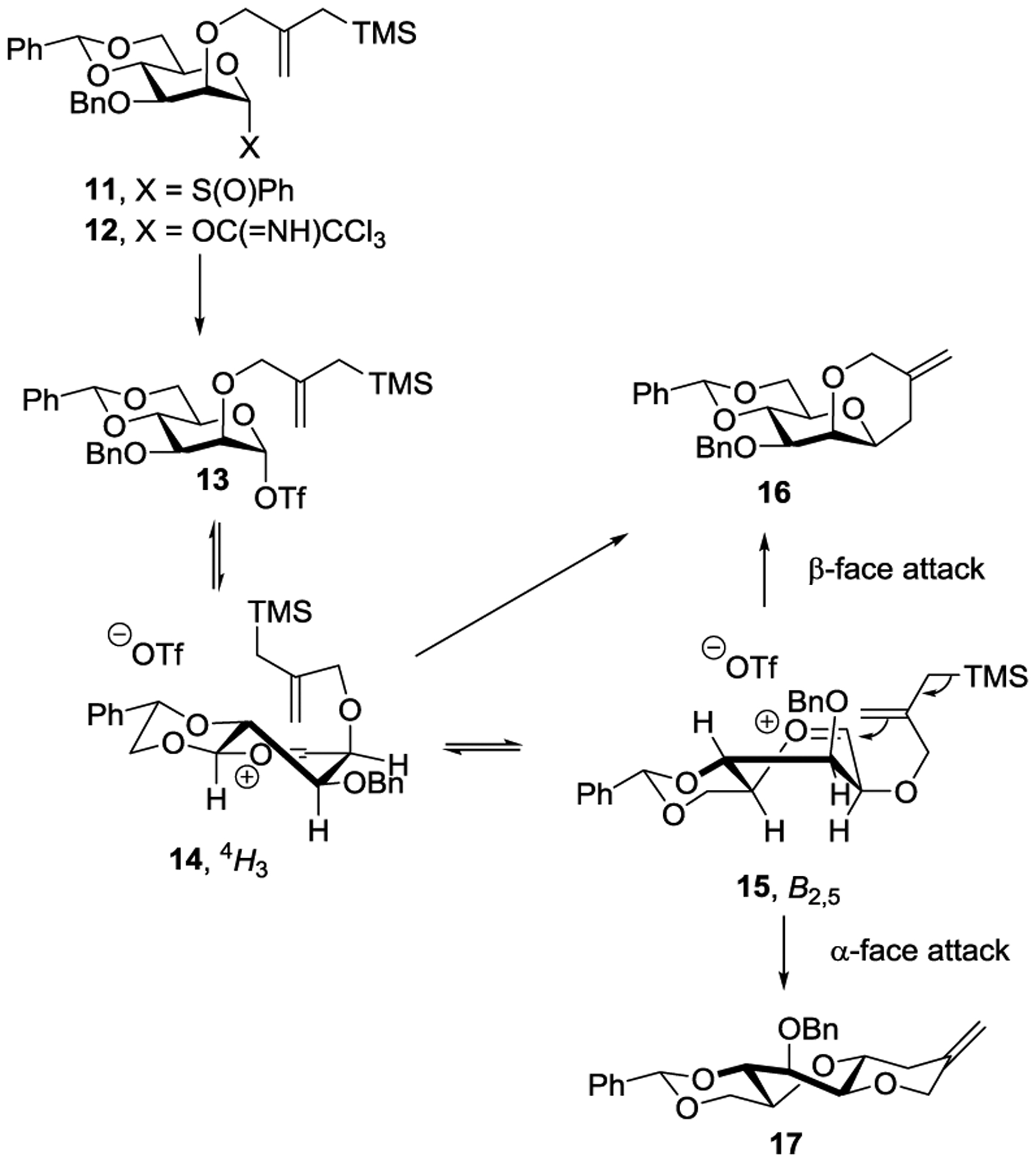
Intramolecular Trapping of a Transient Oxacarbenium Ion.
In the broader context, SN1-like mechanisms are more likely in glycosylations with furanosyl donors because of the reduced number of electron-withdrawing C-O bonds and the greater ease of accommodation of an sp2 center in a five as opposed to a six-membered saturated ring. Consequently, it is in furanosylation reactions that the future observation of oxacarbenium ions in organic solution is most likely.
A related concept to that of oxacarbenium ions, is the notion of dioxacarbenium ions as intermediates in reactions involving stereodirecting participation by ester groups. We have surveyed the evidence for and against participation by neighboring and more remote ester groups in glycosylation recently60 and cover only the main points here.
The enhanced stability of a dioxacarbenium ion relative to a comparable mono-oxacarbenium ion is beyond doubt, and is emphasized by Woerpel’s work on the readily observable 1-alkoxyglycosyl oxacarbenium ions.61, 62 Likewise, few would doubt that the equilibrium between an acetoxytetrahydropyrylium ion 18 and the corresponding dioxacarbenium ion 19, resulting from participation by the ester, favours the latter (Scheme 5a). However, inclusion of the inevitable counterion in the picture complicates matters. This is illustrated for the common triflate counterion with covalent glycosyl triflates 20 being more stable than oxacarbenium – triflate ion pairs. The important equilibrium in organic solution is therefore not that of the acyloxy-substituted oxacarbenium ion 18 with the cyclic dioxacarbenium ion 19, but rather that of the equilibrium of between the cyclic dioxacarbenium ion 19 and the covalent triflate 20 (Scheme 5b) or its equivalent with the particular counterion employed. .
Scheme 5.
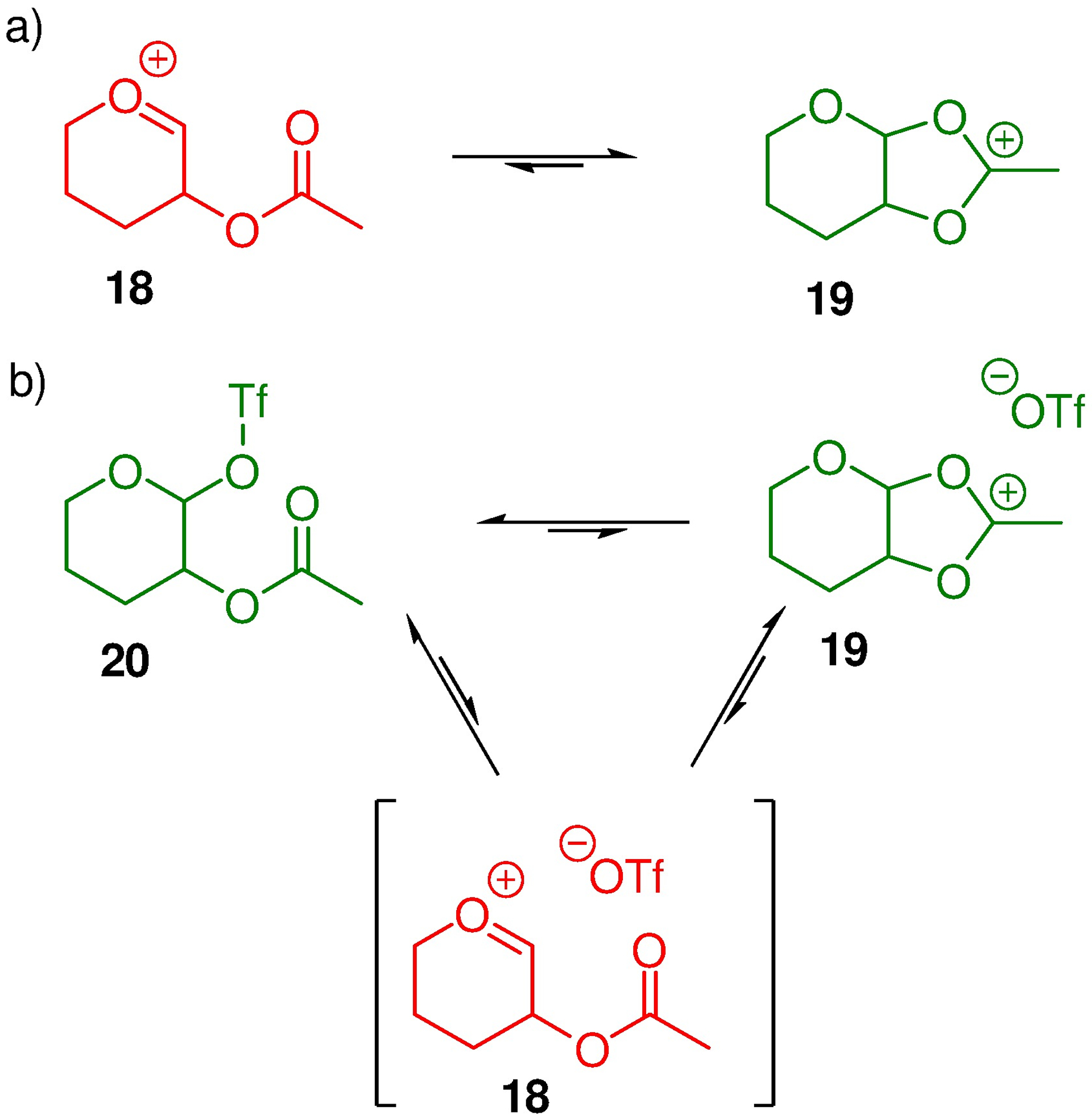
Equilibrating Oxacarbenium and Dioxacarbenium Ions and Glycosyl Triflates
The problem of the covalent adduct/cyclic dioxacarbenium ion equilibrium is a subtle one whose answer depends on the nature of the full substituent array. Thus, while my laboratory showed by 13C NMR spectroscopy that pentopyranosyl tri-O-benzoyl xylopyranosyl donors 21 cleanly afford a fused dioxalenium ion 22 on activation at low temperature (Scheme 6a),63 work from the Huang and Williams laboratories revealed a more complex scenario in hexopyranosyl donors. Thus, in the galactopyranose series, Huang and coworkers found that a 3,4,6-tri-O-benzyl-2-O-benzoyl donor 23 cleanly gave the dioxalenium ion 24 on activation at −60 °C in deuteriochloroform, whereas the corresponding tetra-O-benzoyl donor 25 gave only the galactosyl triflate 26 (Scheme 6b and c). The protecting group dependent outcome of these reactions was attributed to the greater electron-withdrawing effect of the benzoate esters destabilizing the positive charge on the corresponding dioxalenium ion. Likewise, the differing outcomes between the xylopyranosyl series (Scheme 6a) and the galactopyranosyl series (Scheme 6c) can be attributed to the presence of the additional electron-withdrawing C-O bond in the latter case. In the tetra-O-acetylglucopyranose system a temperature dependent equilibrium of the glycosyl triflate 27 and the fused dioxalenium ion 28 was observed, in the form of a 1:1 mixture at −60 °C, but completely favouring the fused ion at 0 °C (Scheme 6d).64 On activating a 3,4,6-tri-O-acetyl-2-O-mesitoyl galactosyl donor in deuteriochloroform at −60 °C followed by warming to 0 °C, Williams and coworkers recorded the NMR spectra of a 1:1 mixture of the glycosyl triflate 29 and the fused dioxalenium ion 30, whereas under the same conditions the tetra-O-mesitoyl donor 31 showed only the fused dioxalenium ion 32 (Scheme 6e and f).65 Whatever the reason(s) for these protecting group dependent shifts in equilibria, it is difficult to escape the conclusion that even stereodirecting participation by a vicinal ester through a five-membered fused dioxalenium ion is a relatively borderline event in the presence of the triflate or other less nucleofugal counterions. It follows that the frequently reported but unanticipated less than perfect selectivity observed in neighboring group-directed glycosylation reactions is likely the consequence of competition between two or more energetically similar mechanisms.
Scheme 6.
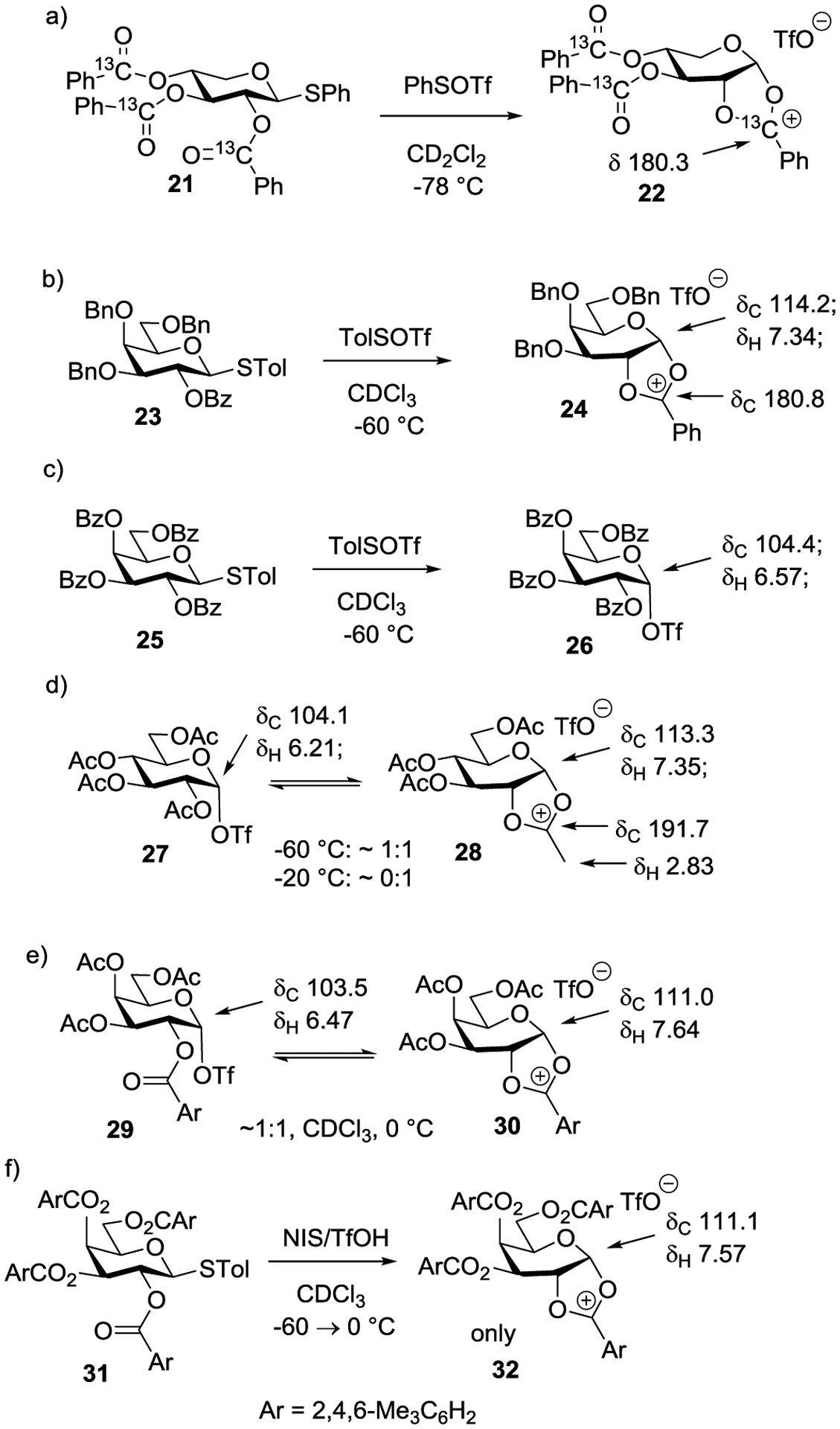
Protecting Group Dependence of Dioxalenium Ion Formation in Neighboring Group Participation.
I now turn to participation by more remote esters, through bridged bicyclic dioxacarbenium ions. This phenomenon, which has been postulated since the 1970’s,66, 67 including by my own laboratory,68 and which continues to find strong support, does not hold up to objective scrutiny with the possible exception of axial esters at the 3-position. From a thermodynamic perspective five-membered cyclic 1,3-dioxalenium ions are more stable than six-membered cyclic 1,3-dioxenium ions as has been amply demonstrated by Paulsen and coworkers69 and others.70 A helpful comparison is the relative stability of γ- versus δ-lactones,71, 72 or more closely of five- versus six-membered cyclic carbonates: the former are commonly used as protecting groups for 1,2-diols,73–75 while the latter are much harder to install and constitute a readily polymerizable function group.76 I focus here on participation by axial esters at the 4-position, which is the most commonly-evoked form of remote participation, although the arguments advanced are valid for participation by any distal ester.
It is a fact25, 26 that the more remote an ester is from the anomeric center, the less it influences anomeric reactivity by its electron-withdrawing effect (the less disarming it is), from which it is appreciated that oxacarbenium 33 is more stable than 34 (Figure 1). At the same time the bridged bicyclic dioxacarbenium ion 35 is clearly less stable than its fused isomer 36. Therefore, participation by the distal ester is less favoured than by the vicinal ester (Kdistal < Kvicinal), which, as we have seen is itself borderline in many cases and dependent on the protecting group array.
Figure 1.
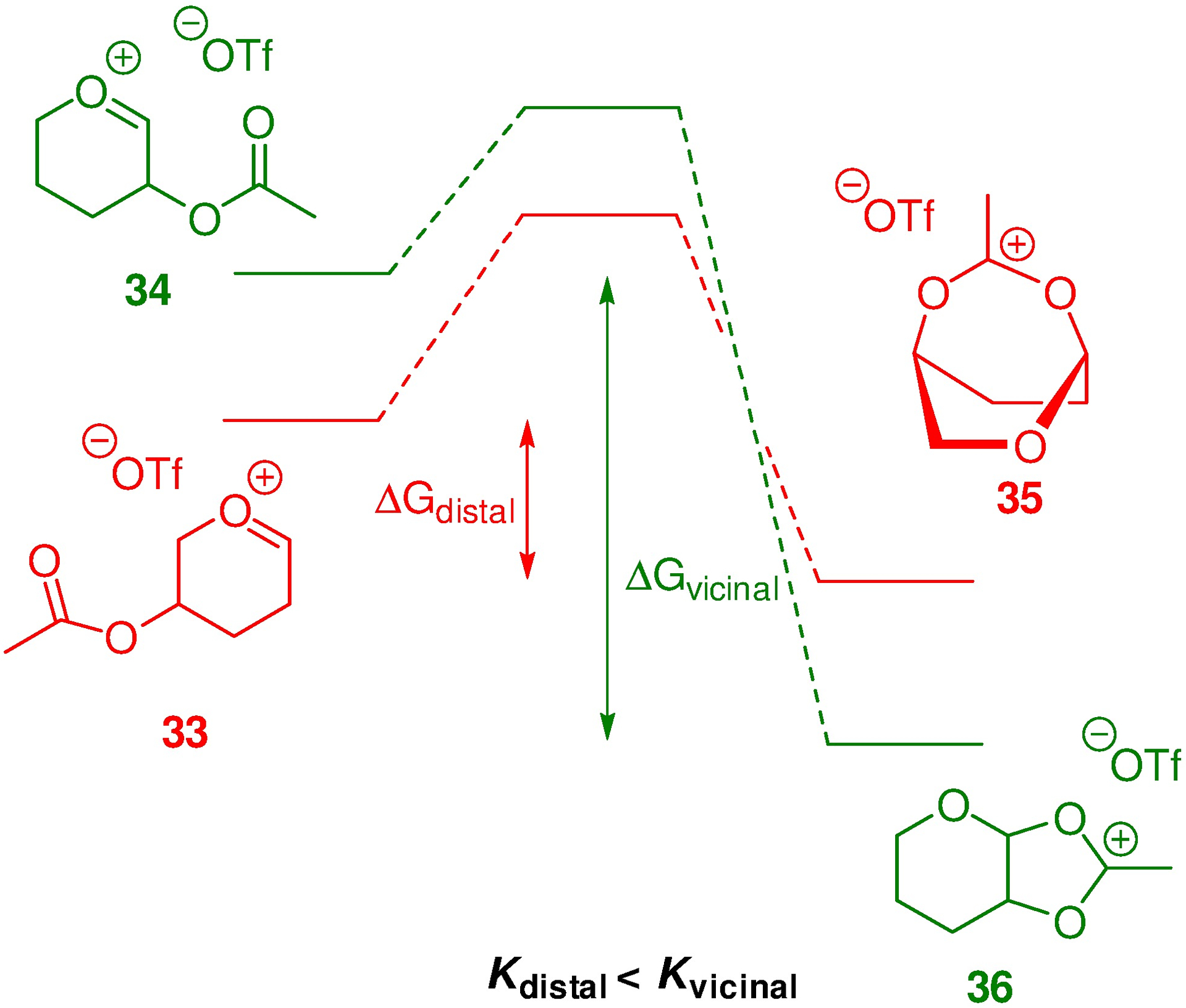
Relative Energetics of Participation by Vicinal and Distal Esters (Not to Scale).
It is noteworthy that while Blériot, Jiménez-Barbero, Thibeadeau and their collaborators observed the clean formation of the fully protonated fused dioxalenium ion 38 from 37 in superacid media, only the fully protonated bromine-stabilized oxacarbenium ion 40 was observed on subjection of 39 to the same conditions (Scheme 7). Parallel observations were made in the 3,4,6-tri-O-acetyl-2-bromo-2-deoxygalactopyranoside series where no bridging from the 4-position was observed. It was argued that protonation of the remote ester prevents cyclization, but it must be noted that similar protonation did not prevent cyclization from the 2-position in 37: an alternative explanation is that remote participation is simply not thermodynamically favoured.29
Scheme 7.
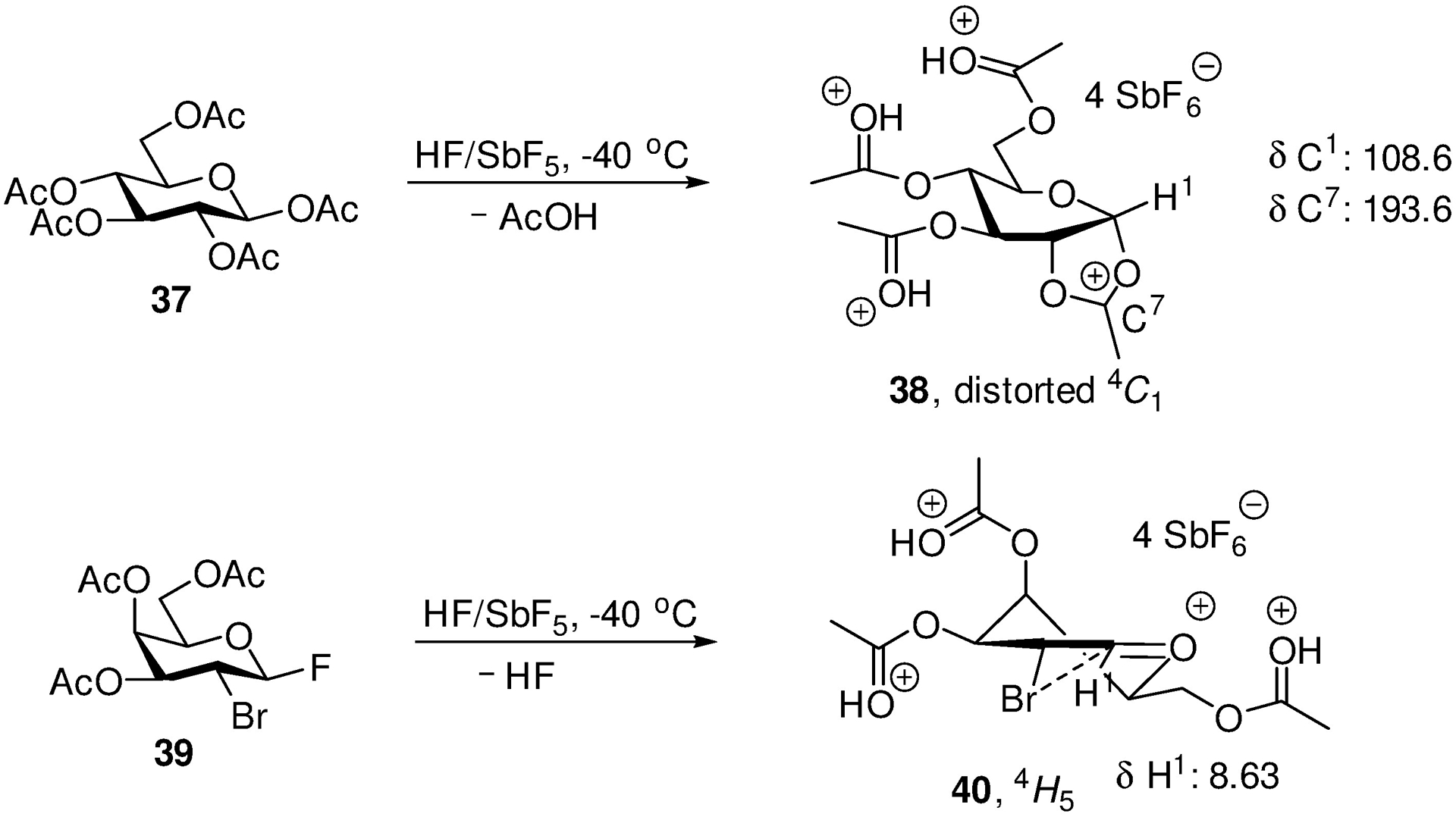
Formation of a Fused but not a Bridged Dioxacarbenium Ion in Superacidic Medium
Turning to the kinetics of remote participation, important considerations are ester conformation, which strongly favours eclipsing of the C-H bond by the carbonyl group, and the barrier to rotation about the C-O bond, which has been computed at ~ 13 kcal.mol−1 (Figure 2 a).77, 78 Again taking the example of axial esters at the 4-position, remote participation therefore requires population of a high energy conformation 42 of the ester (Figure 2b). Lest it be argued that the conformation of the ester is influenced by the presence of the sp2 center in the oxacarbenium ion, attention is drawn to the corresponding cyclohexyl series. Kleinpeter and coworkers have demonstrated that, as in carbohydrates, esters have a greater propensity to adopt axial positions than ethers, which they attribute to hyperconjugative stabilization of the electron-deficient axial C-O bond by vicinal axial C-H bonds.79 Pertinently, this phenomenon was shown to be more significant in 4-aryloxycyclohexanones than in the analogous cyclohexanes. 4-(p-Chlorobenzoyloxy)cyclohexanone crystallized with the ester in the axial position, but nevertheless in the standard ground state conformation of esters with the carbonyl group eclipsing the C-H bond 44 (Figure 2c).80 This phenomenon was attributed to electrostatic stabilization of the ketone sp2 C by the ester; any such stabilization from the ground state ester conformation is necessarily provided by the electron density on the oxygen atom directly attached to C4, rather than by the carbonyl oxygen.
Figure 2.
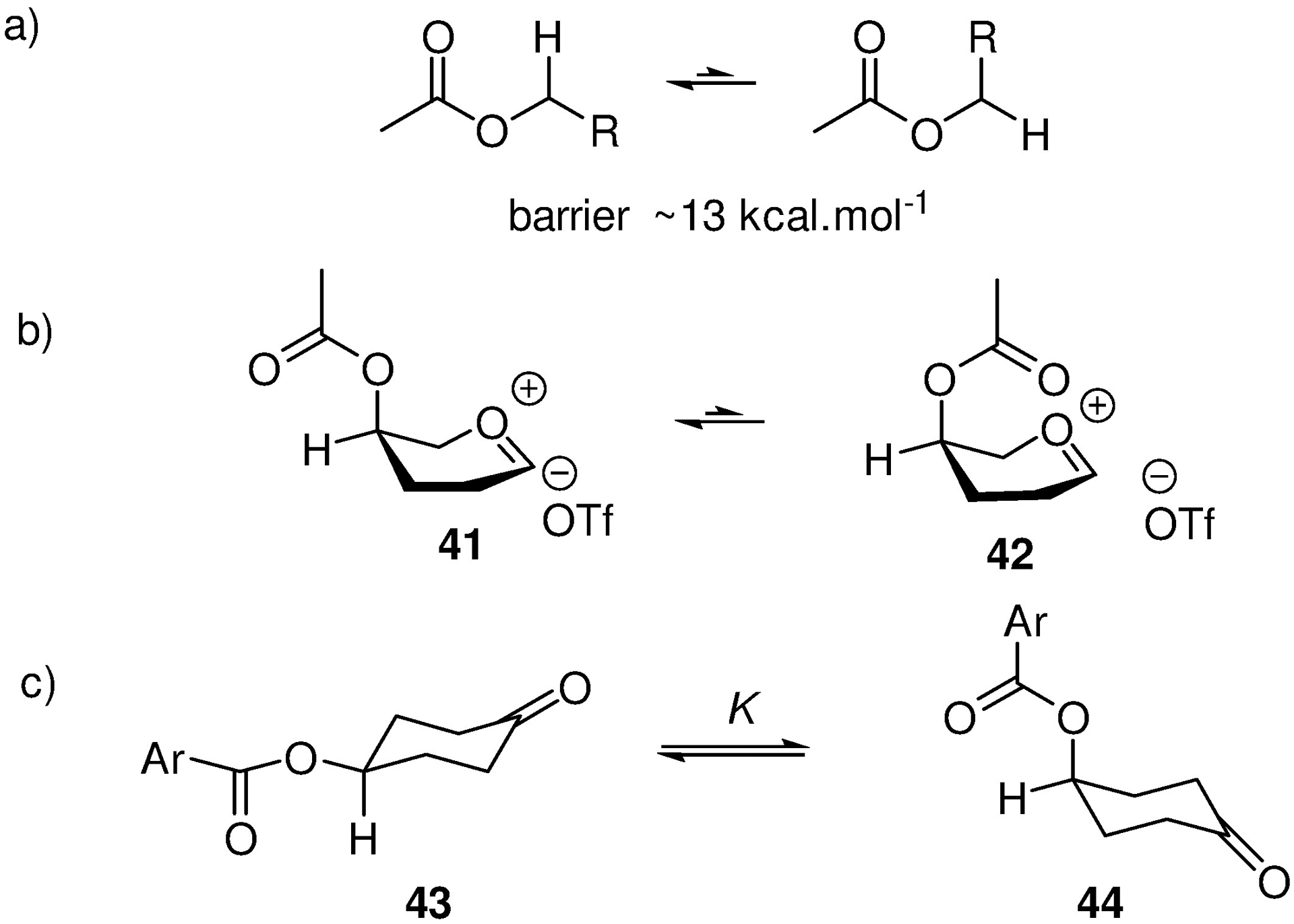
Conformational Equilibria of Esters
Overall participation by remote esters is necessarily slow as it must convolute multiple unfavourable processes before closure to a bridged bicyclic ion. These processes include dissociation of the activated covalent donor, conformational reorganization of the oxacarbenium ion, and conformational reorganization of the ester (Scheme 8). A parallel argument can be made against remote participation with direct displacement of the anomeric leaving group by the ester, without the need for the intermediacy of an oxacarbenium ion, and is supported by the current lack of evidence of anchimeric assistance in glycosylation by remote esters.60
Scheme 8.
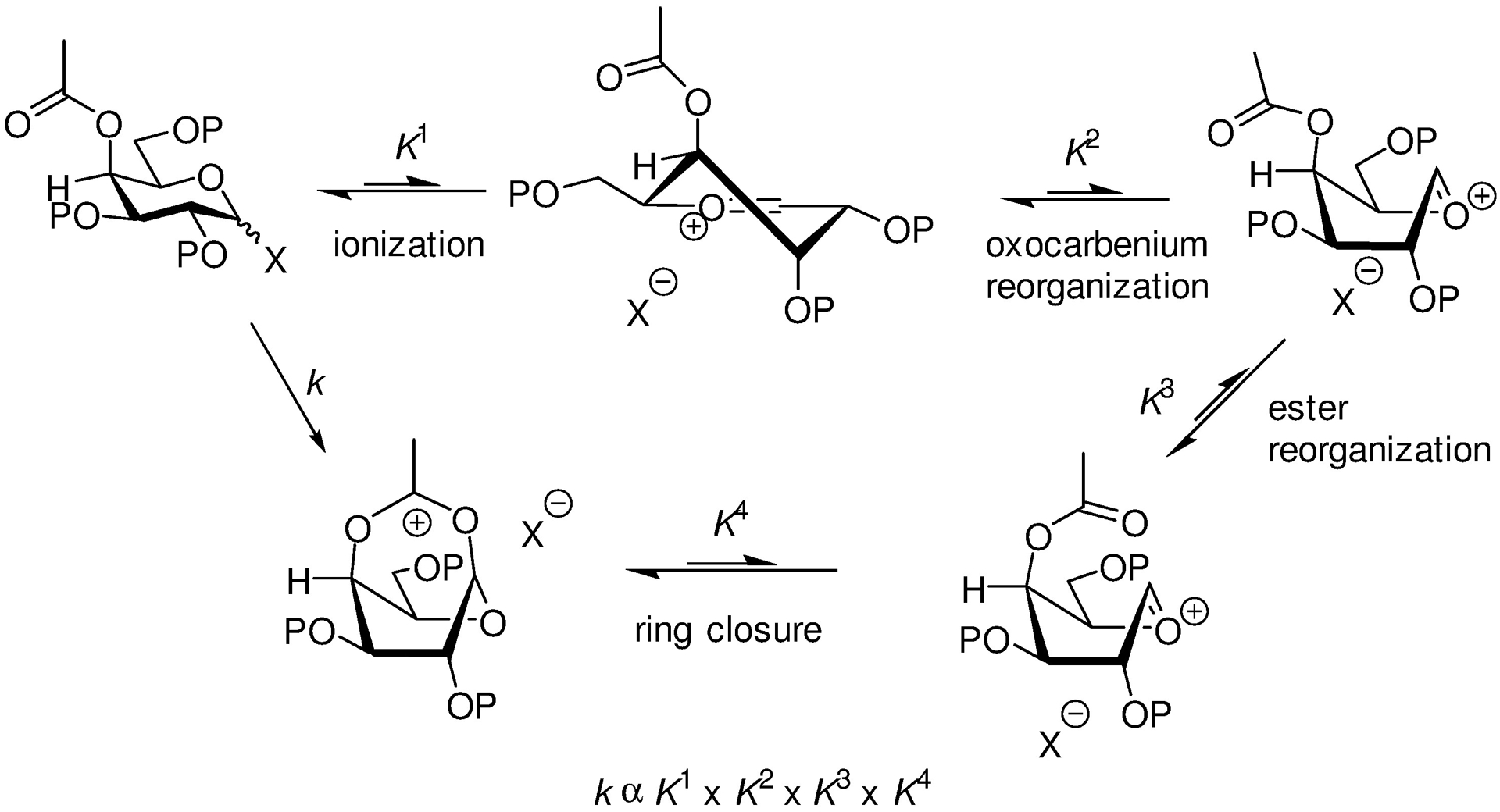
Participation by Remote Esters is Kinetically Disfavoured by a Multiplicity of Unfavourable Equilibria
We introduced multiple probes for long range participation, but only found evidence in support of this phenomenon by axial esters at the 3-position.81 For the galactose 4-position, three separate probes failed to trap a bridged intermediate. One of these, use of 18O-water as nucleophile to interrupt the process at the level of the bridging dioxacarbenium, is presented in Scheme 9. The product 47 was satisfactorily explained by direct attack of the labelled water at the anomeric center: no evidence was found for the anticipated kinetic trapping of the bridged ion 49 in the form of the orthoacid 50, or of its fragmentation products 51 and 52.
Scheme 9.
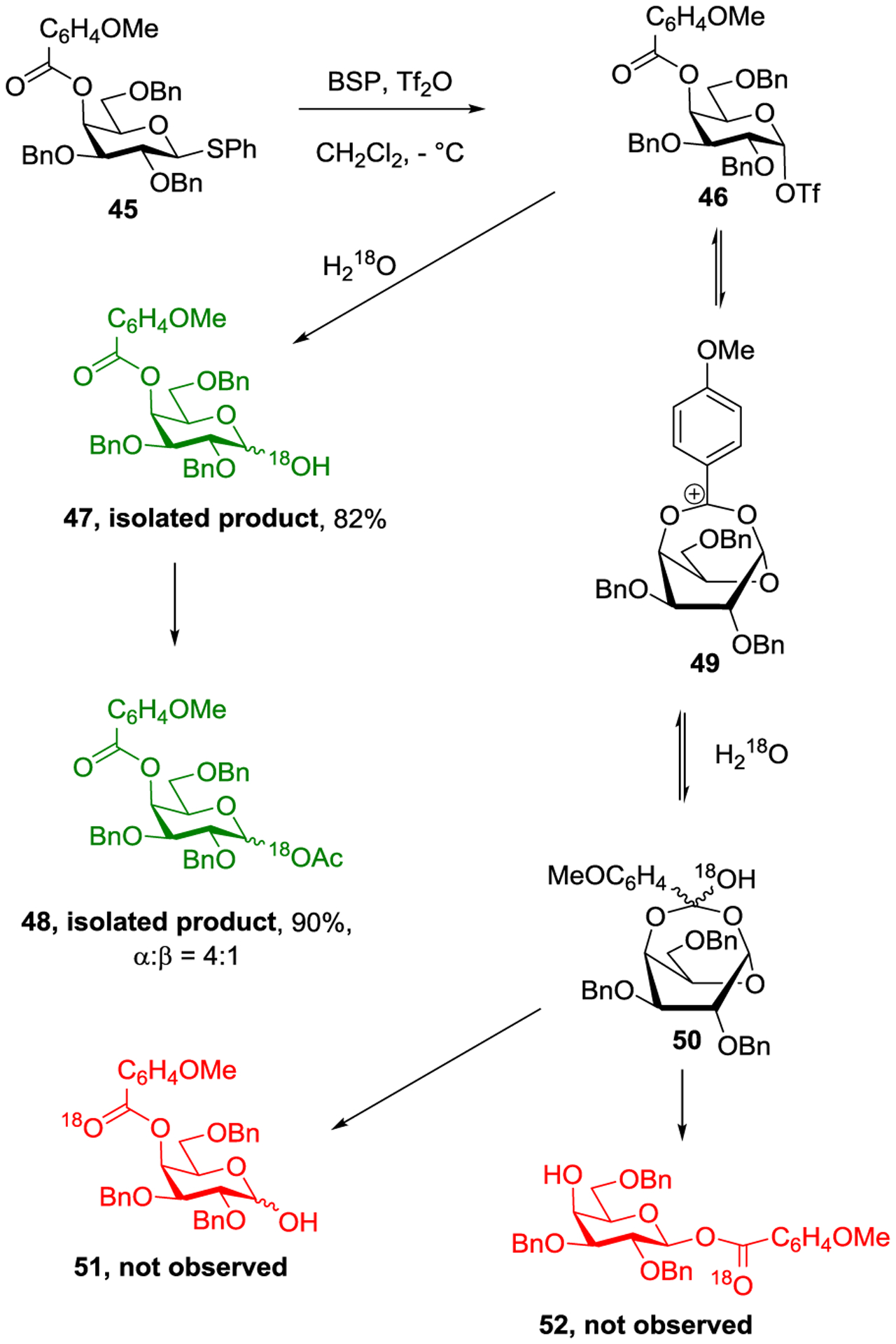
18O Labelling Experiment Argues Against Remote Participation
The strongest evidence in favour of remote participation is drawn from papers by the Nifantiev and Boons laboratories according to which the α-directing effect of substituted benzoate esters at the fucopyranose and galactopyranose 4-position is maximized by the presence of electron-donating substituents in the p-position of the ester, suggesting increased stabilization of a bridged dioxacarbenium ion 53 (Figure 3a).82, 83 However, drawing on the parallel with 4-(p-chlorobenzoyloxy)cyclohexanone (Figure 2b), it is evident60 that the same substituent effect also supports through space stabilization of positive charge at the anomeric position by the ester from its ground state conformation 54 (Figure 3b).
Figure 3.
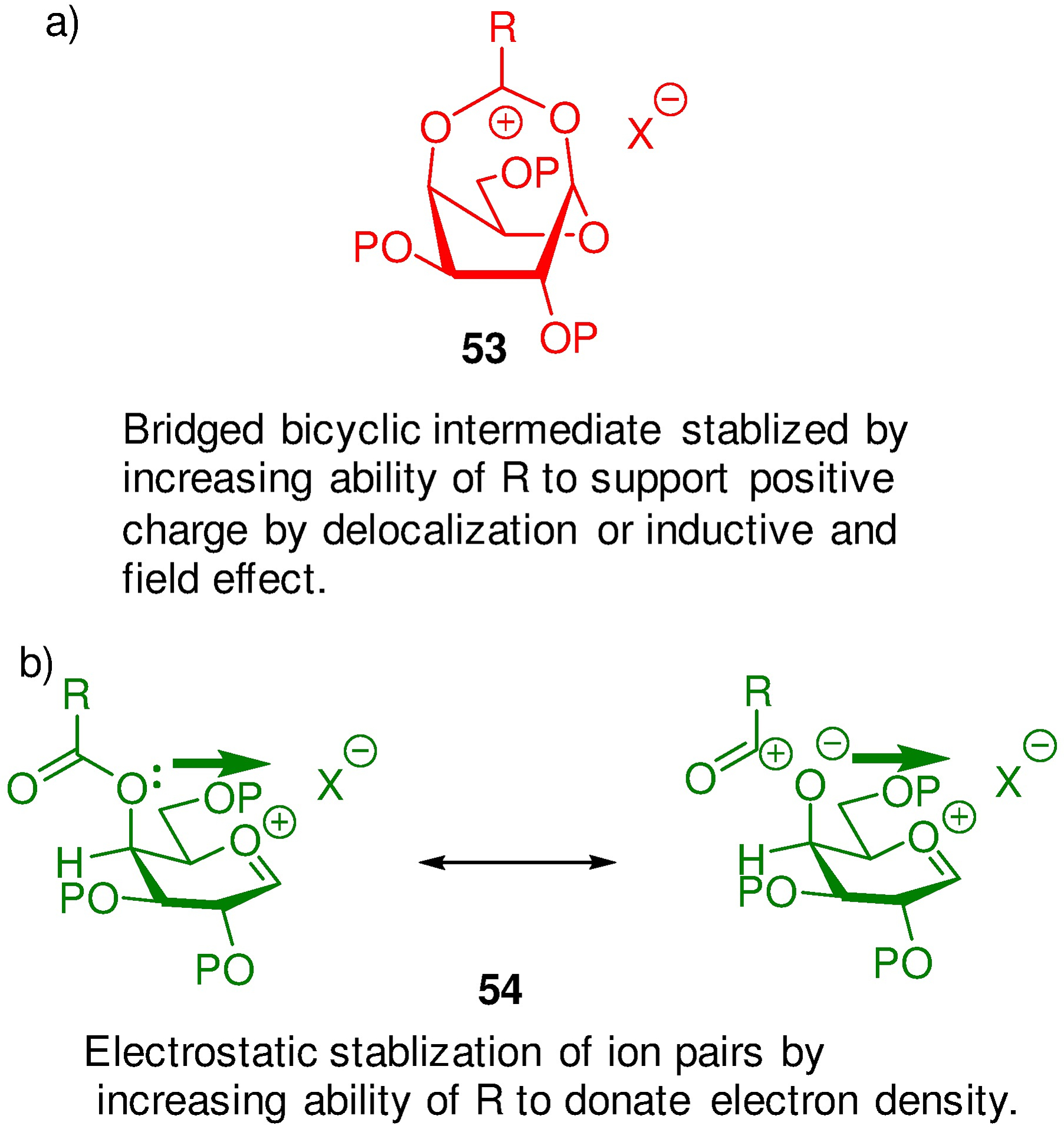
Two Rationalizations of the Influence of p-Substituent Effects in Axial Benzoate Esters in Glycosylation.
The work of Yu and coworkers on the isolation and characterization of a bridged intermediate 56 from the 4-position of a peracetylated glucopyranosyl donor 55 (Scheme 10)84 provides a rare demonstration of remote participation by an ester under actual glycosylation conditions. It must be noted, nevertheless, that the system employs the very weakly nucleophilic triflimide counterion and not the more common and more nucleophilic triflate,21 and that the skeleton provides a ready intramolecular trap of the bridged ion which removes it from the equilibrium. Multiple examples of bridged bicyclic products arising from trapping of transient glycosyl oxacarbenium ions by remote functionality are collected in our review,60 but these typically employ more nucleophilic groups than carboxylate esters and so cannot be considered as evidence of participation by esters in the course of actual glycosylation reactions.
Scheme 10.
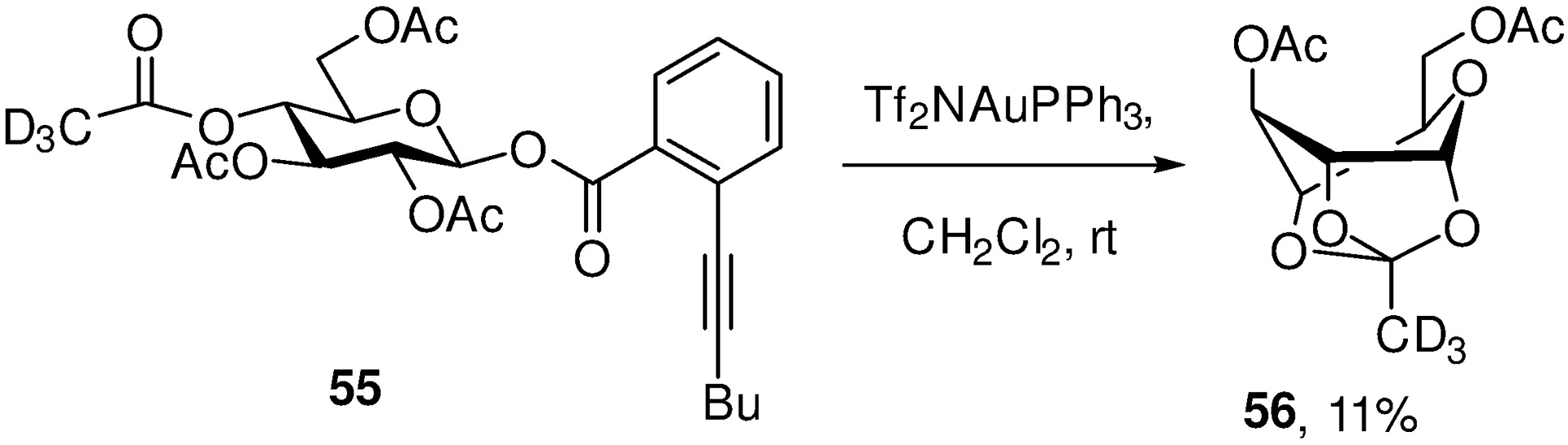
Intramolecular Trapping of a Bridged Ion in the Presence of the Weakly Nucleophilic Triflimide Counterion.
Bridging ions have recently been characterized by two laboratories using coupled mass spectrometric-infrared spectroscopic methods, and advanced as evidence of long range participation in glycosylation.85–87 However, while these studies demonstrate the formation of bicyclic ions in the gas phase at low temperature, counterions are excluded by necessity such that the results have little bearing on solution phase glycosylation reactions.
Overall, I conclude that carbohydrate chemistry has arrived at a crossroads. One fork consists of continued adherence to the longstanding but demonstrably deficient models of naked oxacarbenium and bridged dioxacarbenium ions, and unimolecular reaction kinetics, while the second follows the mounting experimental evidence and incorporates the role of counterions and covalent donors, and consequently of associative mechanisms with bimolecular characteristics. Empirical models for the prediction and optimization of glycosylation reactions, currently under development, will only be of true value when concentration and counterions are fully incorporated in to the parameters.88 Bimolecular mechanisms provide a rationale for the frequently stated extreme sensitivity of glycosylation reactions to the presence of water: simply, water is involved in the rate determining step of bimolecular hydrolysis. Bimolecular kinetics also account for the poor yields and selectivity observed in the assembly of larger glycans, where recourse is typically made to more dilute solutions. Finally, the less than perfect selectivity seen in many neighboring group directed glycosylations can be understood as the consequence of the borderline nature of participation and competition with direct glycosylation under bimolecular kinetics.
Crossroads 2. Stereocontrolled Glycan Synthesis as a Challenge for Synthetic Organic Chemists and Carbohydrate Chemists Alike.
Synthetic organic chemists have traditionally been inspired by the invention of highly diastereoselective and enantioselective methods of C-C bond formation for application in practical, elegant syntheses of natural and unnatural products, pharmaceuticals and the like. Glycan synthesis differs from mainstream organic chemistry only in so far as it revolves around the formation of C-O bonds at the anomeric center rather than the formation of C-C bonds: excellent diastereoselectivity and efficiency, and even elegance remain essential requirements and indeed are still the main challenges. In what are typically reactions between two chiral partners, the concept of double asymmetric induction and stereochemical matching and mismatching is always present.89, 90 As such carbohydrate chemistry poses many significant challenges to organic chemists, which go beyond the oft-repeated need for extreme dryness (hardly a challenge for practitioners of carbanion chemistry). Moreover, these challenges are not limited to glycosidic bond formation by the classical route but offer many opportunities for creativity in terms of alternate routes,91–94 streamlining of processes by removal of the need for protecting groups,46, 95 catalyst development,96 etc. Indeed, my own entry into the field involved adapting diastereoselective radical reactions to stereocontrolled 2-deoxy-β-glycoside synthesis.3
With regard to more specific classes of glycan, the chemical synthesis of the N-glycans has advanced tremendously,97, 98 but is likely to be eclipsed by chemoenzymatic methods, particularly as a wider array of glycosyltransferases becomes available.99 Automated oligosaccharide synthesis has reached a level at which preparation of linear glycans of previously unimaginable length can now be accomplished.100 The bacterial glycans on the other hand offer a very broad selection of unusual targets to challenge the imagination and creativity of the organic chemists. Some of these targets are of great complexity such that strategy and tactics are as important as the actual reactions used to assemble them.101, 102 A prime example is provided by the hyper-branched chorella virus N-glycan 57, tackled by the Lowary and Ye laboratories,103, 104 with Lowary’s successful sequence of bond formation around the core, beginning with red and following the colors of the rainbow, highlighted in Figure 4a. Yu’s synthesis of the pregnane glycoside periploside A 58, with its multiplicity of 2,6-dideoxy-β-glycosides and a unique type of spirocyclic orthoester, is a further illustration of the triumph of strategy over structural complexity (Figure 4b).105 Pleasingly, the ring-closing formation of the yellow bond employed chemistry developed in our laboratories in a different context.106 Clearly, C-O bond formation in the context of glycan synthesis offers many challenges to the intrepid organic chemist. More obvious opportunities for mainstream organic chemists to gain an entry in glycochemistry arise from the enormous numbers of glycosylated natural products, whose syntheses should not be halted at the level of the aglycones.
Figure 4.

Recently conquered, structurally-complex oligosaccharides a) chorella virus N-glycan 57 and b) periploside A 58: the bond formation sequence follows the colors of the rainbow.
Work on complex glycans from bacteria, plants and other sources in my laboratories spanning two decades107–119 is not covered here except for more recent studies on the bacterial sialosides, which led us to consider aspects of remote stereoinduction and to study the conformations of polyhydroxylated alkanes. Following development of the N- acetyl oxazolidinone protected sialic acid thioglycosides 59 and of the related 4,5-O-carbonate protected KDN thioglycosides 60 as highly equatorially selective donors (Figure 5),120–123 we were intrigued by the bacterial sialosides,124, 125 and by the manner the differing substitution patterns and configurations influence reactivity and selectivity. The axial C-N bond at the 5-position of pseudaminic acid 65 was of particular concern as it has the same spatial relationship to the anomeric center as the axial C-O bond at the 4-position of the galactopyranosides, which is well known to reduce selectivity.
Figure 5.
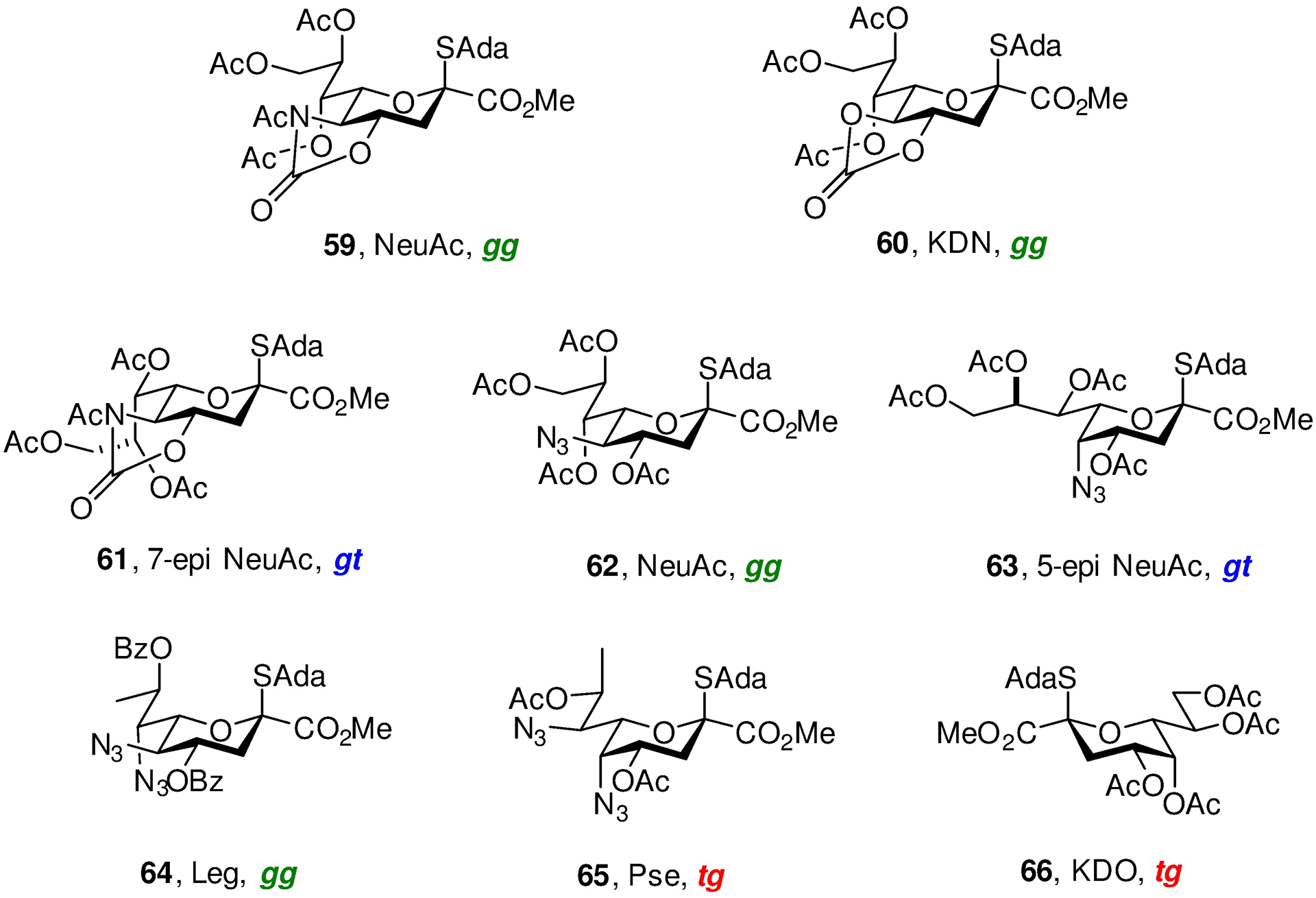
Glycosyl Donors in the Neuraminic Acid and Ulosonic Series
To address this question we studied the influence of configuration at the 5- and 7-positions on donor reactivity and/or selectivity, demonstrating the 7-epi 61 to be less reactive under our standard glycosylation conditions than the natural isomer 59.126 Turning to the 5-position we made the practically useful discovery that the 5-azido NeuAc donor 62 exhibited good to excellent equatorial selectivity, provided reactions were conducted at −78 °C,127 in contrast to the existing literature on comparable 5-azido-protected donors.128 Most revealing, however, was the finding that the 5-epi-azido donor 63 was exquisitely equatorially selective under the same conditions.127 These observations led to the realization that the relative configurations of the 5-, 6- and 7-positions controls the predominant conformation of the exocyclic bond in the sialic acids and to the prediction that the pseudaminic acid donor 65, with the 5,7-bis-epi configuration, would have the most disarming side chain conformation and be the most equatorially selective among the azido-protected series. These predictions were borne out by the gram-scale synthesis of both 64 and 65 from N-acetyl neuraminic acid and a thorough investigation of their selectivity, and provided the first equatorially selective synthesis of Leg and Pse bacterial glycoside components.129, 130 In passing, it is interesting to note that N-acetylneuraminic acid, a target for synthesis in the not too distant past,131 is now so cheap (<$5/g) that we employed it as starting material for our syntheses of 64 and 65, and indeed of the 4-aminoheptose core of the septicidins and spicamycins.132 Recognizing the pseudosymmetric relationship of Pse with KDO, we prepared the KDO donor 66 and demonstrated that it adopts the same (albeit pseudoenantiomeric) conformation about the exocyclic bond and is highly selective for the formation of equatorial glycosides under our standard conditions.133 When discussing the conformation of the exocyclic bond in carbohydrates, loosely called the side chain conformation, the three staggered conformations are described as trans,gauche (tg), gauche,gauche (gg), or gauche,trans (gt) where the first descriptor refers to the relationship of the C6–O6 bond to the C5–O5 bond, and the second to that of the C6–O6 bond to the C5–C4 bond (Figure 6). In the sialic and ulosonic acids the relationship of the C7–O7 bond to the endocyclic bonds from C6 are described analogously.
Figure 6.
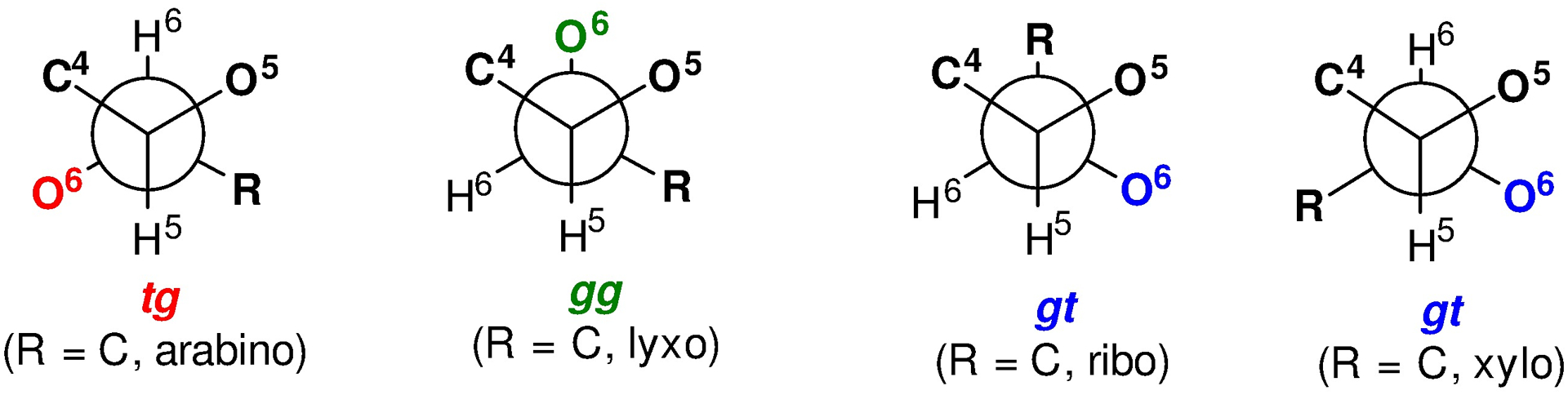
Definition of the Staggered Conformations about the Exocyclic Bond in the hexopyranosides (R = H) and higher pyranosides (R = C). The C6-O6 bond is either trans, or gauche to the C5-O5 bond and trans or gauche to the C5-C4 bond in that order.
Building on our work with the sialic acids, and literature work on the conformations of open chain forms of the pentose sugars, we assembled a predictive model of the conformation of the exocyclic bond in higher carbon sugars in general. This model applies the basic organic chemical principles of minimization of 1,3-interactions and the maximization of the gauche effect, and the relative configuration of the ring carbon bearing the side chain and the two flanking centers (arabino, lyxo, ribo or xylo) to predict the tg, gg, gt, and gt conformations of the side chain, respectively, as illustrated in Figure 7 for the 6-deoxyheptoses.134 This model should also be valid for any 2-(1-hydroxyalkyl)-3-hydroxytetrahydropyran or tetrahydrofuran and so represents a further crossroads with mainstream organic chemistry.
Figure 7.
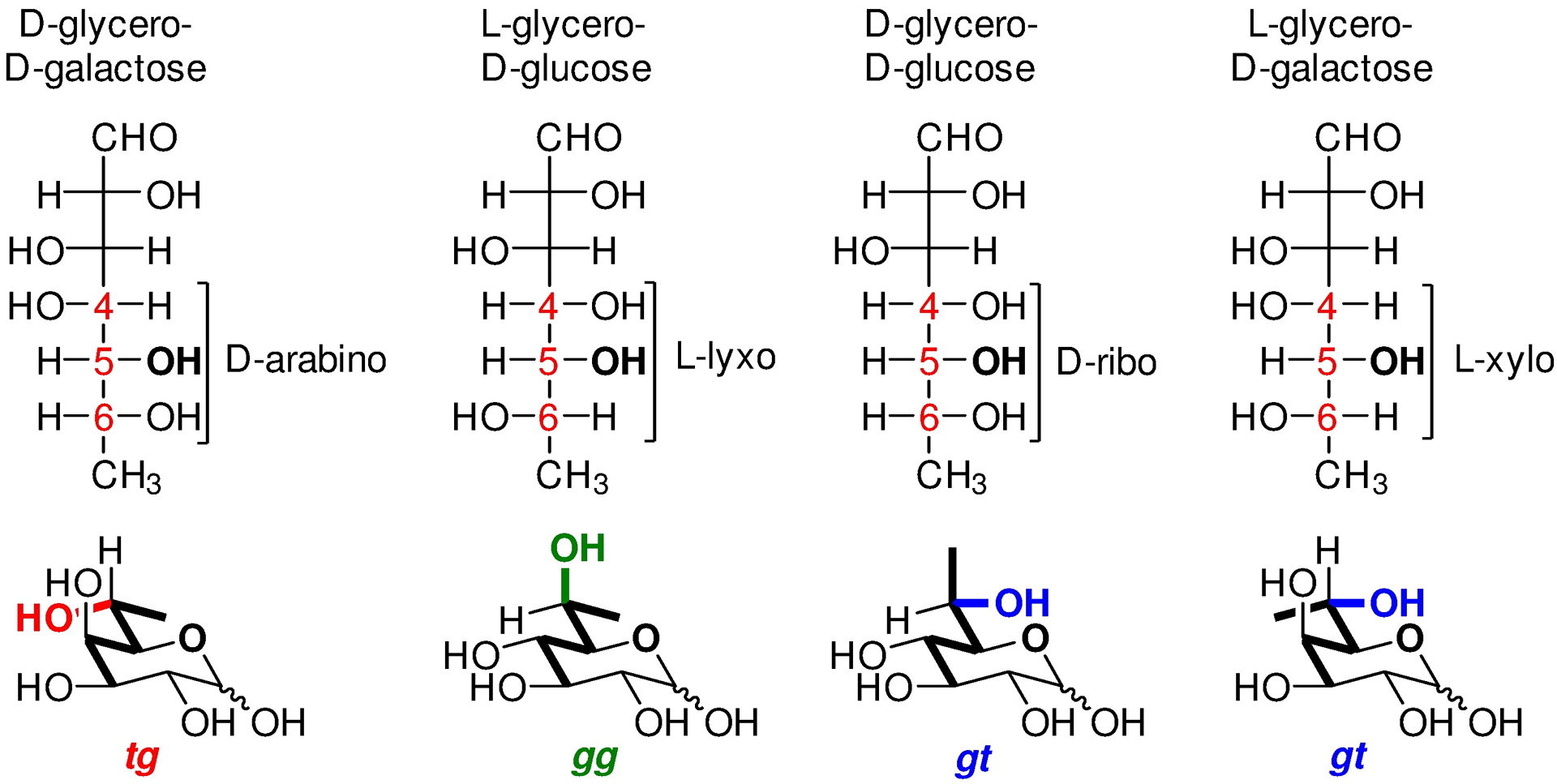
Predictive model for the conformation of the exocyclic bond in the higher carbon sugars (The pentose chain from C4–C7 is in bold).
Extrapolating from bicyclic model systems,135–137 the reduced reactivity of donors with the tg conformation derives from the maximization of the electron-withdrawing effect of the C6–O6 bond, which destabilizes partial postive charge on the ring oxygen in the course of the glycosylation reaction, thereby increasing the activiation barrier. This has the effect of shifting the key equilibria (Scheme 1) toward the covalent donor and away from ion pairs, and so of increasing the SN2 character of the glycosylation and consequently its stereoselectivity. At the other extreme the gg conformer is considered to provide through space electrostatic stabilization to positive charge at the anomeric locus and so to reduce SN2 character, much as the axial C4–O4 bond in galactopyranosides does. The gt conformation is intermediate between the gg and tg ones in its ability to stabilize positive charge electrostatically during the course of the reaction. According to this rationale the 7-epi-NeuAc donor 61, with the gt conformation of its side chain would be expected to be less reactive and more selective than the natural isomer 59 with its gg conformation. Indeed, 61 was less reactive than 59 to such an extent that the temperature had to be raised to promote an acceptable reaction rate, which acts negatively on the key equilibria and faciliates the dissociative mechanism leading to a reduction in selectivity.
Our understanding of the role played by side chain conformation in glycosylation reactions prompted us to ask if carbohydrate processing enzymes apply the same principles. The combination of conformational analysis and carbohydrate reactivity thus led us to take a new direction into mechanistic enzymology. To address this question we examined all structures of glycoside hydrolases (GHs) in the protein data base (PDB) with bound inhibitors and/or substrates and substrate analogs in the active site (−1) with a resolution of ≤2Å, with the aid of the Carbohydrate Active Enzymes Database (CAZy, http://www.cazy.org).138
After examination of more than 500 crystal structures we discovered that the α- and β-glucosidases and the β-mannosidases, and even the α- and β-glucosaminidases, show a very strong preference for binding their active site ligands with the gg conformation of the side chain, and do so by means of hydrogen bonding to the side chain hydroxyl group.139 Taking the glucoimidazole type glycosidase inhibitors as an example, of the nine structures in the PDB that met our criteria for resolution seven had the inhibitor bound with the gg conformation of the side chain (Scheme 11). A similar pattern was seen with the isofagamine class of inhibitors, where fourteen of the fifteen examples located bound the ligand with the gg conformation of the side chain (Scheme 11). These preferences for binding the gg conformation far exceed the population of the same conformation in free solution. The evidence is compelling that typical glucosidases have evolved to bind their substrates with the gg conformation of the side chain to gain additional transition state stabilization.
Scheme 11.
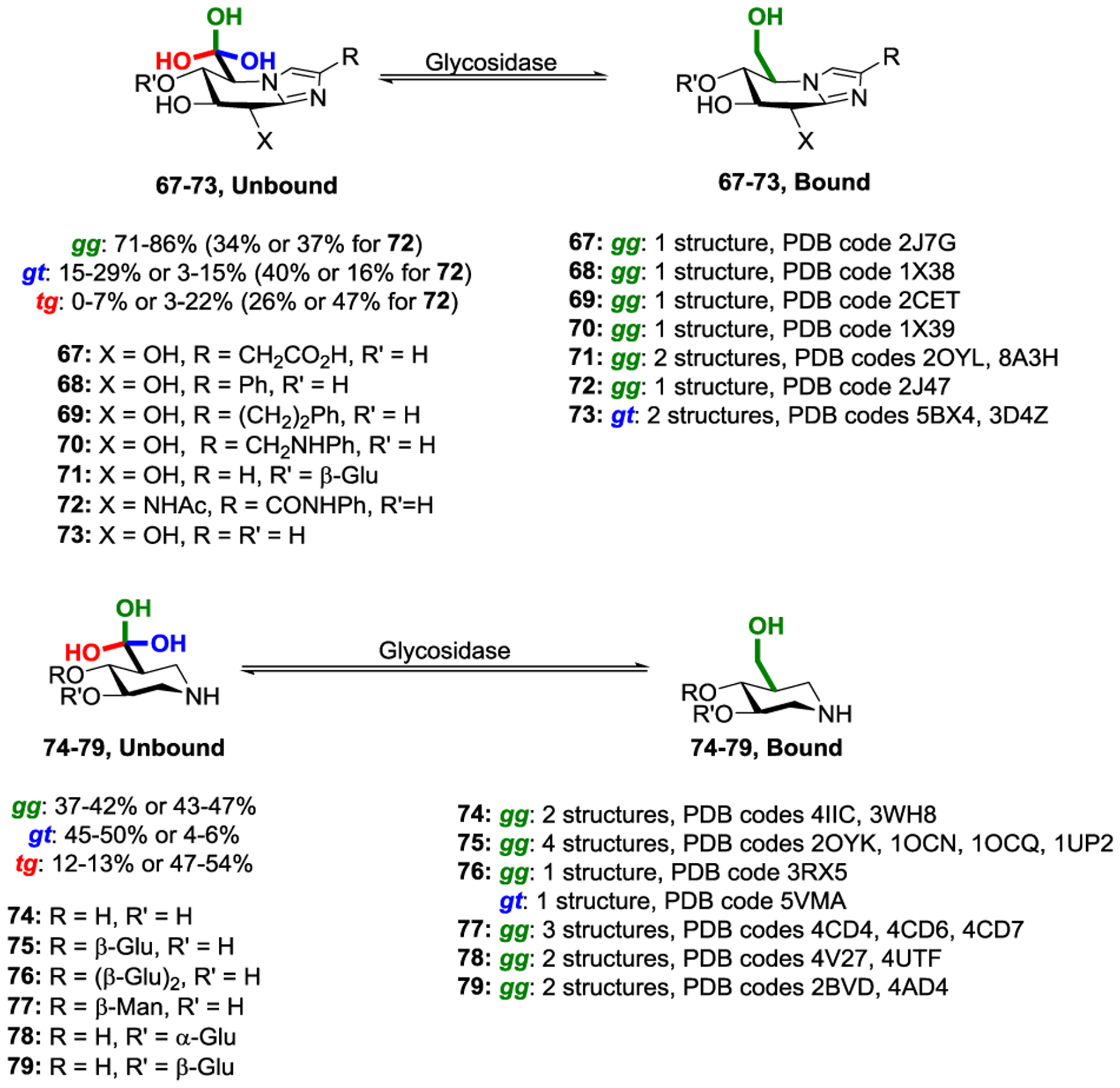
Restriction of glucoimidazole and isofagamine side chain conformation by GHs
The data revealed that the same pattern is found both at the level of bound transition state inhibitors and their close analogs, and even that of simple bound substrates and substrate analogs, suggesting that organization begins at the level of the enzyme-substrate complex and is maintained through the transition state all the way to the enzyme-product complex. This is illustrated by a series of crystallographic snapshots on the termite GH1 β-glucosidase for a) an enzyme • substrate complex, b) a complex with a pseudo-transition state inhibitor, and c) an enzyme • product complex (Scheme 12).140 This preference of glucopyranosidases and β-mannopyranosidases to bind their substrates with the gg conformation of the side chain and maintain it through the course of the hydrolysis suggests that these enzymes have evolved to preorganize their substrates in so that the substrate itself provides additional through space electrostatic stabilization of the positive charge at the transition state for its own hydrolysis.
Scheme 12.

Partial X-ray crystal structures of termite GH1 β-glucosidase with (a) cellobiose (PDB ID 3VIK), (b) 1-deoxynojirimycin (PDB ID 3VIG), and (c) a glucose-Hepes conjugate (PDB ID 3VIO)
The α-mannosidases at first sight do not make use of side chain control in the same manner, overall showing a ~50/50 ratio for ligand binding in the gg and gt conformations that mirrors their approximate population in free solution. However, breakdown of the data into CAZy hydrolase families, reveals that all 25 structures with the ligand bound in the gg conformation belong to GH families 63, 92, 99, and 125, while the 28 structures exhibiting the gt conformation all belong to families 38 and 47. Consequently, most α-mannosides have evolved to follow the general preference for the gg conformation of side chain, with only a limited number of families adopting a different trajectory. The neuraminidases all bind their ligands with the gg conformation of their side chains, but no conclusions can be drawn owing to the very high preference of neuraminic acids for this conformation in free solution.
The galactoside hydrolases display a different preference to their gluco and manno-congenors. This is due in large part to the instability of the gg conformation in the galactose series, which is populated to only ~15% in free solution. Most galactose-based CAZy families prefer to bind their ligands with the gt conformation of the side chain, which affords a degree of stabilization to positive charge at the transition state albeit less than that due to the gg conformation. It remains a matter of speculation as to why certain galactoside hydrolases bind their ligands in the deactivating tg conformation.
In summary, our studies on the influence of side chain conformation on the reactivity of glycosyl donors led us to a crossroads with mechanistic enzymology and the understanding that gluco- and most mannoside hydrolases bind their substrates with the side chain preorganized in the gg conformation so as to provide additional transition state stabilization. These revelations should assist in the design of improved inhibitors for numerous carbohydrate processing enzymes and so potentially impact biomedical science. Careful examination of the exceptions to the broad rules observed in this study may afford insight into alternative modes of transition state stabilization, with potential for exploitation in selective inhibitor design.
Crossroads 3. Carbohydrate Chemistry as a Source of Problems in Organic Chemistry and an Entry into Medicinal Chemistry
Returning to the theme of glycosidic bonds, a decade ago we turned to the development of glycosidic bond surrogates for potential use in medicinal chemistry. We first focused on the phostones and phostone-phostone dimers 76 (Scheme 13) in which the anomeric carbon is replaced by a P=O moiety, and developed interesting chemistry based on the use of phosphine-boranes 74.141, 142
Scheme 13.
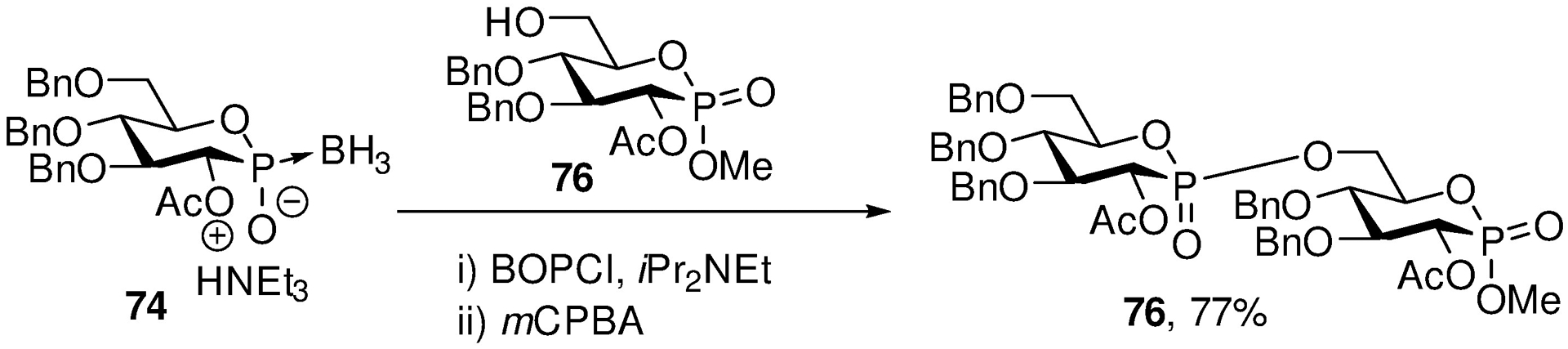
A Phostone Phostone Dimer Mimic of a Disaccharide
In parallel we conceived the idea of N-alkoxy polyhydroxypiperidines as convertible replacements for glycosidic bonds. This idea builds on the known barriers to pyramidal inversion of the nitrogen atom and of rotation about the N-O bond in hydroxylamines of ~15 kcal.mol−1, which coupled with the pKa of ~5.9 for the conjugate acids, indicates that at physiological pH trialkylhydroxylamines are essentially unprotonated and undergo rapid conformational inversion. We first developed a new route to the glycosidase inhibitor isofagamine based on a double ring-closing reductive amination protocol.143 This method was refined to provide a range of N-alkoxy polyhydroxypiperidines 80, which were used in VT NMR studies to determine the barrier of ~15 kcal.mol−1 to stereomutation at nitrogen (Scheme 14),144 that was consistent with literature barriers for simple trisubstituted hydroxylamines. We demonstrated the hydroxylamine functionality to be compatible with cleavage of benzyl ethers by BCl3, oxidative cleavage of p-methoxybenzyl and naphthylmethyl ethers with DDQ, and also to glycosylation conditions using thioglycoside donors and our benzenesulfinyl piperidine (BSP)/triflic anhydride145 coupling protocol (Scheme 15).
Scheme 14.
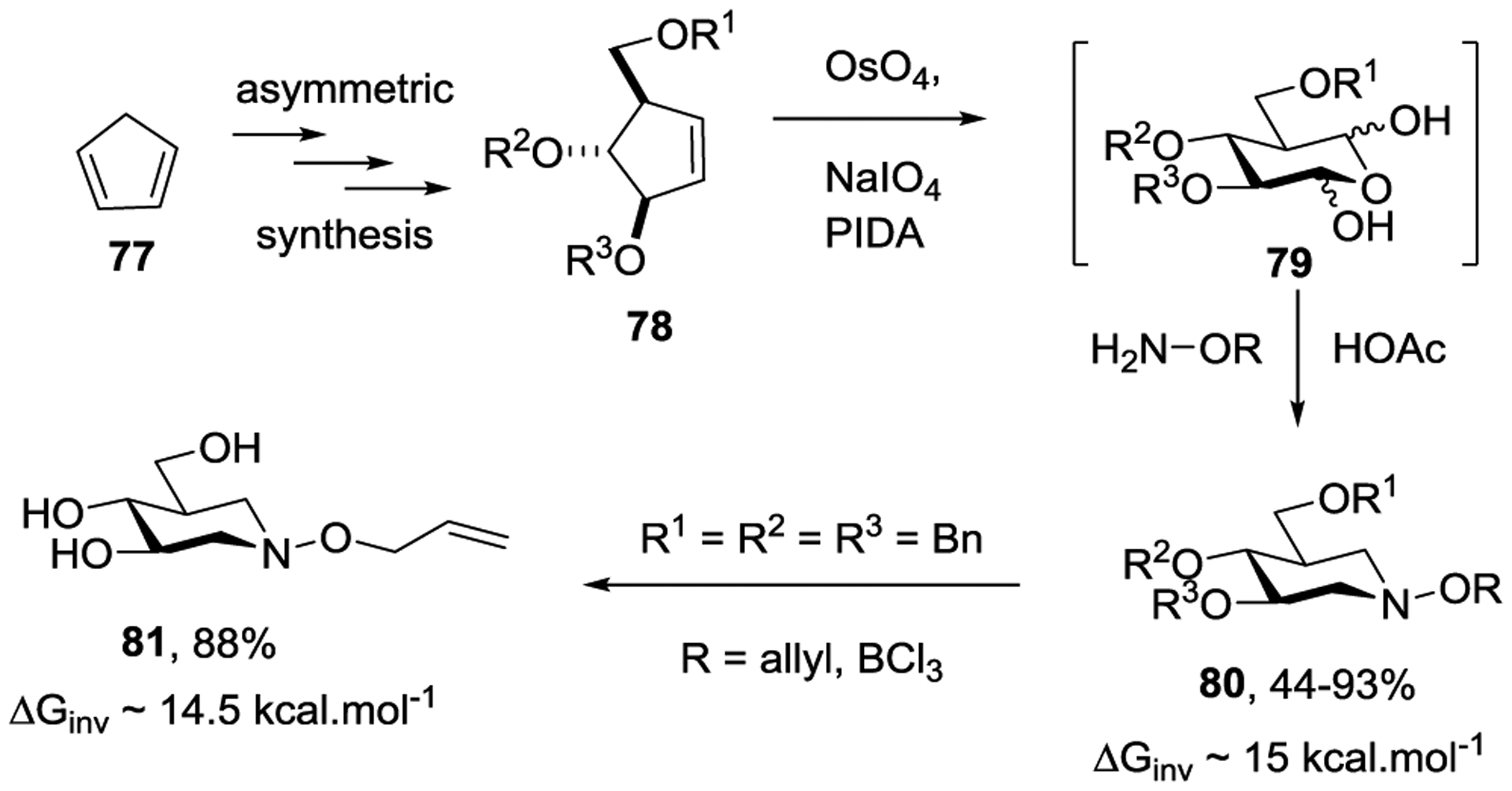
Asymmetric synthesis of polyhydroxy-N-alkoxypiperidines, with barriers to stereomutation of the hydroxylamine moiety.
Scheme 15.

Compatibility of the Hydroxylamine Moiety with Glycosylation.
With the fundamentals of the hydroxalog concept established, we turned to proofs of concept and focused initially on analogs of the immunostimulatory β-(1→3)-glucans. By iterative application of the chemistry outlined in Scheme 14, we synthesized a series of di- and trimeric (trihydroxy)piperidines linked by the hydroxylamine unit. Unfortunately, the yields in the ring-closing double reductive amination process diminished as a function of chain length and we were unable to prepare longer oligomers than 86 (Scheme 16).146 Nevertheless, we were able to isolate sufficient material to deprotect and screen for stimulation of phagocytosis and for inhibition of anti-CR3 or anti-Dectin-1 fluorescein isothiocyanate (FITC) conjugated antibody staining of human neutrophils and mouse macrophages, respectively, with Václav Vĕtvička at the University of Louisville.
Scheme 16.
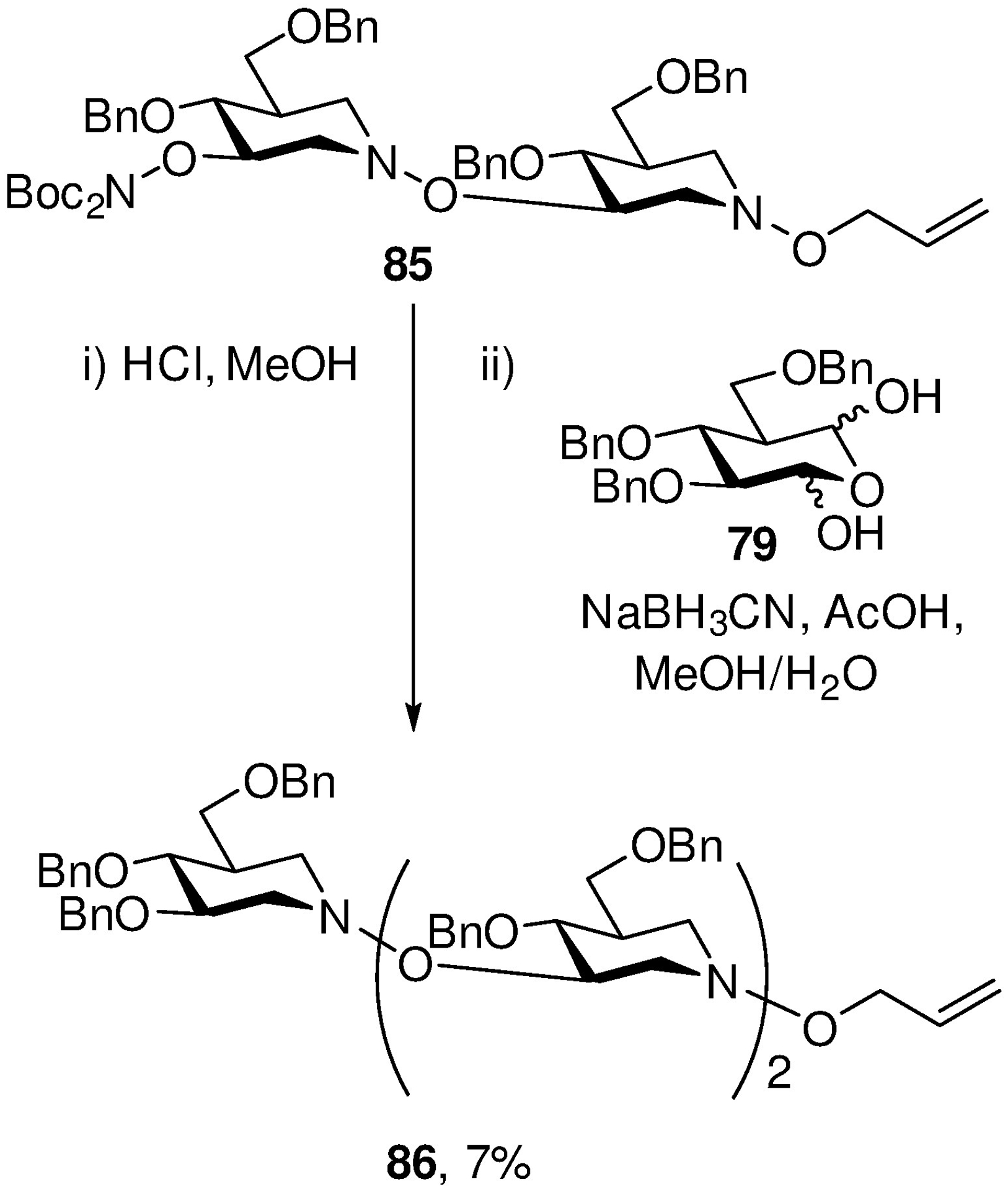
Final stages of the synthesis of a trimeric β-(1→3)-glucan hydroxalog
The hydroxalog 86 stimulated phagocytosis approximately twice as effectively as the natural substrate of the same length, laminaritriose 87 (Figure 8). Likewise, 86 was twice as effective at inhibition of anti-CR3-FITC staining of human neutrophils and of anti-Dectin-1-FITC staining of mouse macrophages than laminaritriose. Building on literature saturation transfer difference NMR-based models for the binding of laminarin, a natural β-(1→3)-glucan, to recombinant Dectin-1 (Figure 9a),147–149 we hypothesized that 86 adheres to the lectin domain of the receptor protein in a shallow hydrophobic pocket lined by the side chains of Trp 221 and His 223 (Figure 9b). Focusing on Dectin-1, we postulated that the enhanced activity of 86 arises from the replacement of the C2-OH bond and the ring oxygen of the natural substrate by a C-H bond and a methylene group, respectively, leading to enhanced hydrophobic interactions with the binding pocket. We further hypothesized that the swopping of the polar glycosidic bond for the relatively soft and polarizable hydroxylamine unit contributes to the enhanced interaction of the ligand with the receptor hydrophobic pocket.
Figure 8.
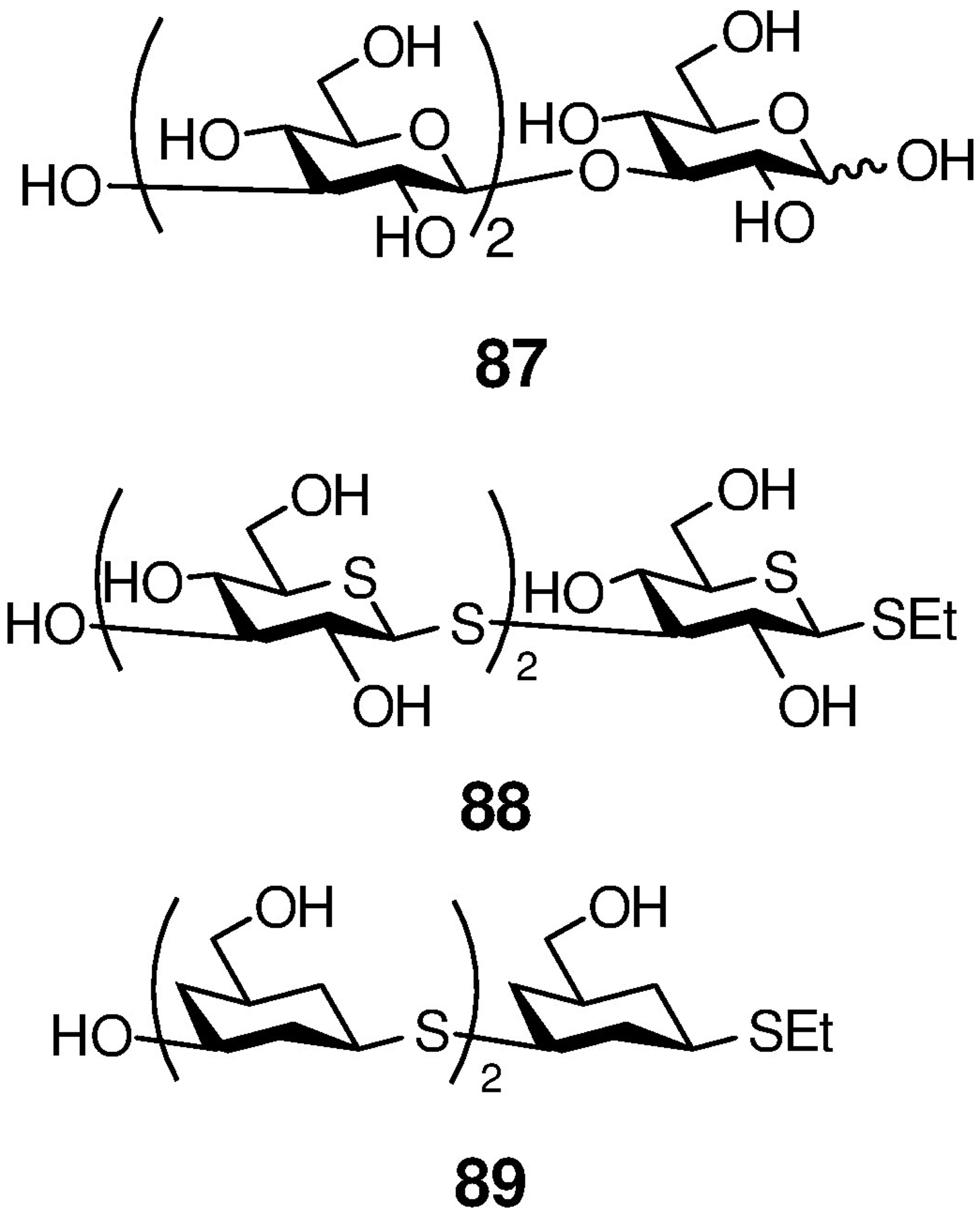
Laminaritriose 87 and the Thioether Mimetics 88 and 89
Figure 9.
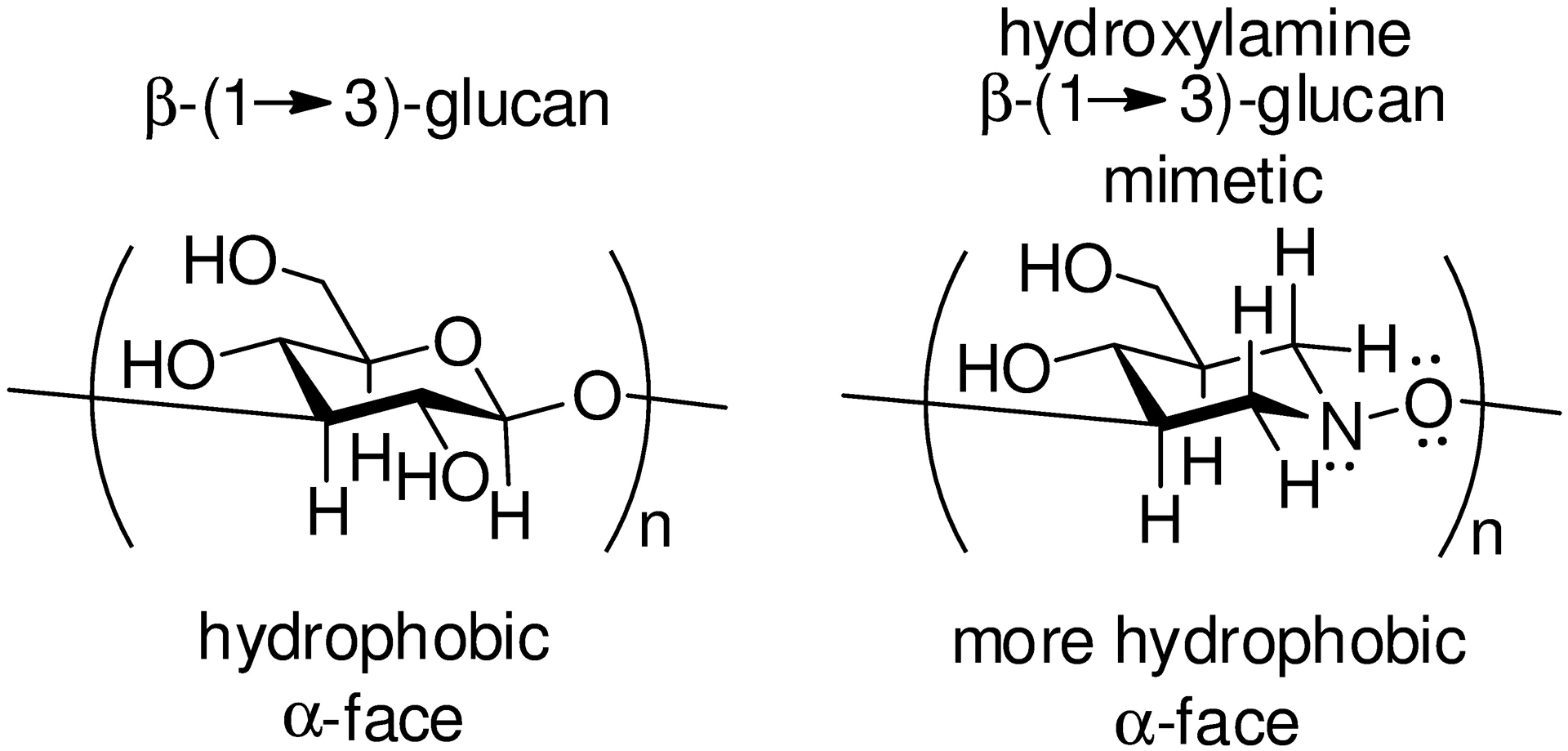
Increased Potential for Hydrophobic Interactions on the α-Face of Hydroxylamine 86 as Compared to β-(1→3)-Glucans.
Extrapolating from these hypotheses, and incorporating results from the Ferrières laboratory with simple thioglycoside analogs of the β-(1→3)-glucan motif,150 we synthesized the 1,5-dithialaminaritriose mimetic 88 (Figure 8) and found that it exhibited comparable activity to the hydroxalog in our screens.151 Finally, and again building on work by Ferrières and coworkers with 4-deoxyglycans,152, 153 we prepared the 2,4-dideoxy-thioether-linked trimer 89 (Figure 8) and showed that it too exhibits comparable activities to the trimeric hydroxalog.154 These preliminary studies suggest that multivalent constructs of the trimeric hydroxalog 86, and particularly of the synthetically more accessible 1,5-dithialaminaritriose 88, may have value.
The promising results obtained with 86 encouraged us to investigate the broader application of the hydroxalog concept and to consider the replacement of simple stereogenic centers with an adjacent methylene group (-CRH-CH2-) by a hydroxylamine moiety (-NR-O-) in natural products and drugs.155 Working with Fred Valeriote at the Henry Ford Cancer Institute in Detroit, we targeted the marine natural product kalkitoxin 90 for modification. We chose this target because of its selective nanomolar cytotoxicity against the human hepato-carcinoma cell line HepG2, and because of the established negative impact on cytoxicity of inversion of any one of its multiple stereogenic centers. Total synthesis, involving construction of the hydroxylamine unit by two back-to-back reductive amination sequences, afforded 10-aza-9-oxakalkitoxin 91, which showed a small increase in cytotoxicity toward HepG2 over the parent, and is the first kalitoxin analog with enhanced activity (Figure 10).156.
Figure 10.
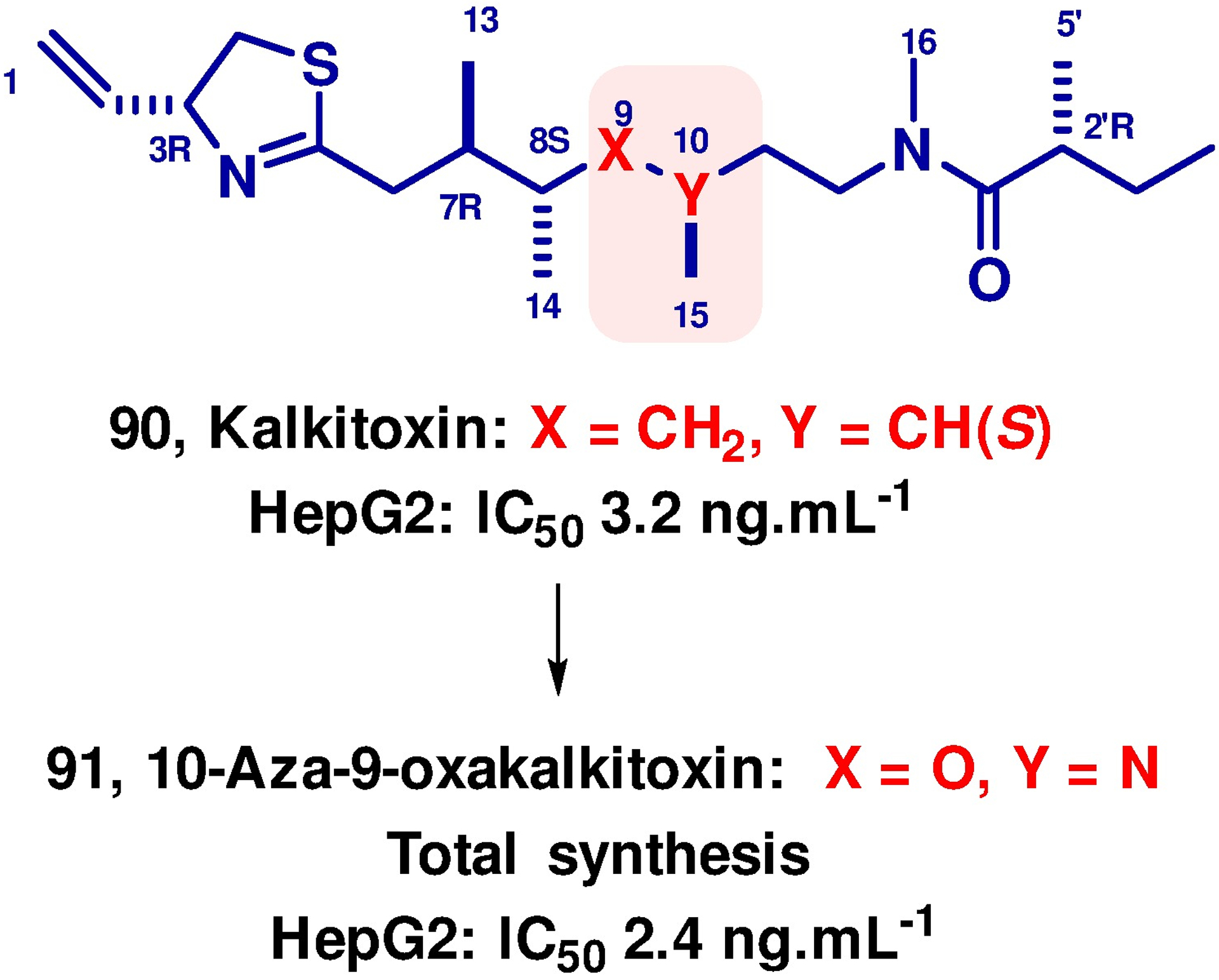
Hydroxaloging of Kalkitoxin
With the potential of hydroxalogs established, we turned to improved synthetic methods for their construction, first drawing an analogy with Rychnovsky’s ether synthesis.157 Accordingly, readily accessible N-hydroxy secondary amines were condensed with carboxylic acids to give O-acyl-N,N-dialkyl hydroxylamines 92, which were reduced with DIBAL in dichloromethane before quenching with acetic anhydride. This gave a series of O-(1-hydroxyalkyl) N,N-dialkyl hydroxylamines 93, which were then further reduced with triethylsilane in the presence of boron trifluoride etherate. Alternatively, the intermediate reduction products were reacted with suitable C-nucleophiles, again in the presence of BF3.OEt2, to give the more highly branched 94 (Scheme 17).155, 158 The availability of further trisubstituted hydroxylamines by this method provided the opportunity to measure additional barriers to stereomutation of the N-O functionality. We focused on the N-alkoxypiperidines and pyrrolidines and determined barriers of 15–16 kcal.mol−1 for the piperidines and 12.3 kcal.mol−1 for N-(3-phenylpropyloxy)pyrrolidine.155
Scheme 17.
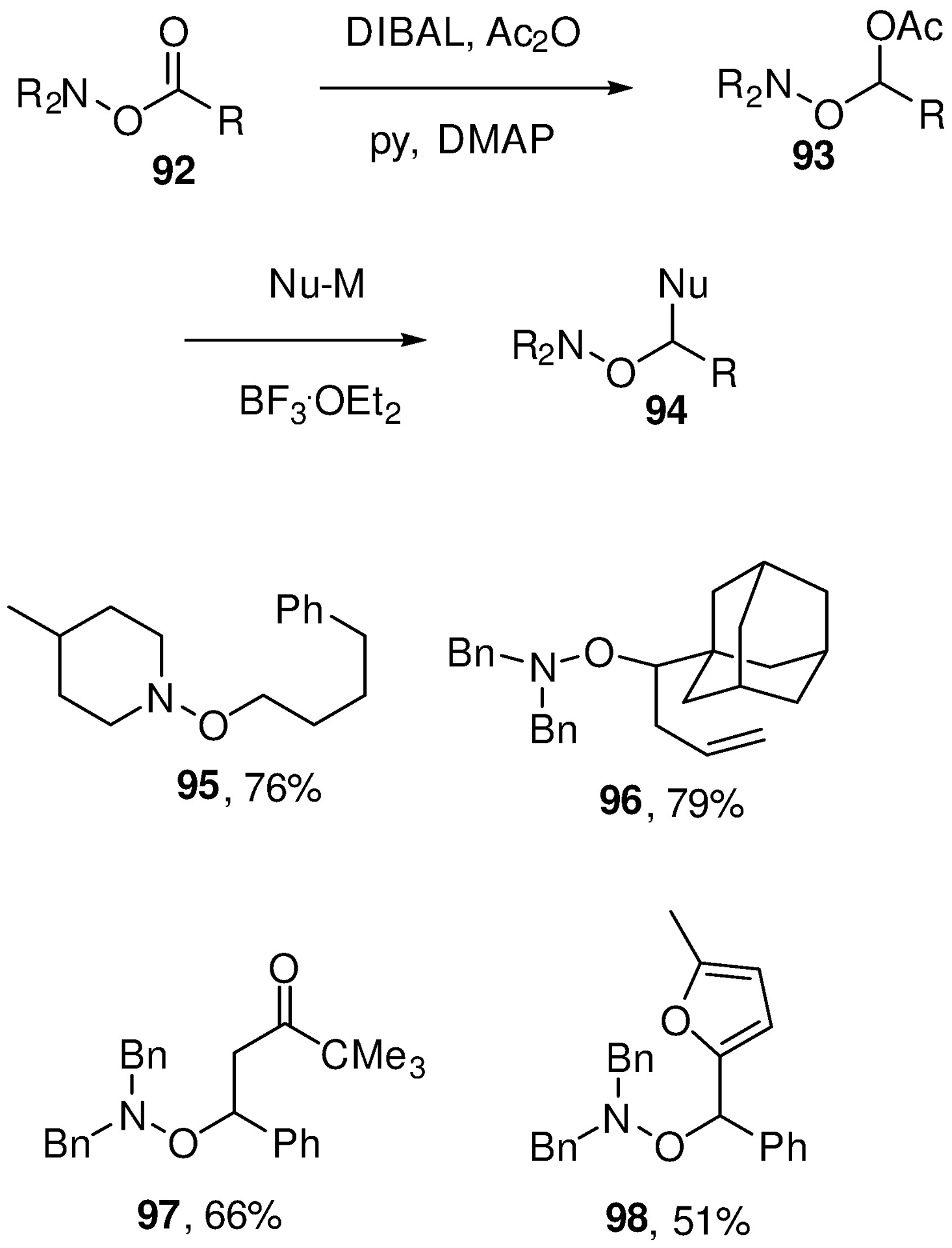
Preparation of Hydroxalogs from O-acyl-N,N-dialkyl hydroxylamines
Next, given the breadth of commercially available secondary amines and alcohols, we targeted methods based on the formation of N-O bonds. Again ether forming reactions, this time by the Dussault and Herzon laboratories,93, 159 provided the inspiration and led directly to the development of a new reaction involving reaction of a magnesium amide 99 with an alkyl 2-methyl-2-tetrahydropyranyl peroxide 100; a small selection of the numerous examples conducted on scales of up to 1 g is presented (Scheme 18). Clearly, given the relative ease of preparation of the peroxides from alcohols, this chemistry constitutes a powerful convergent method for the synthesis of diverse hydroxalogs.160 The hydroxalog concept is an prima facie example of an idea with its origins firmly in carbohydrate chemistry that has changed direction at the crossroads with mainstream organic chemistry and now enriches the fields of synthetic methodology and medicinal chemistry.
Scheme 18.
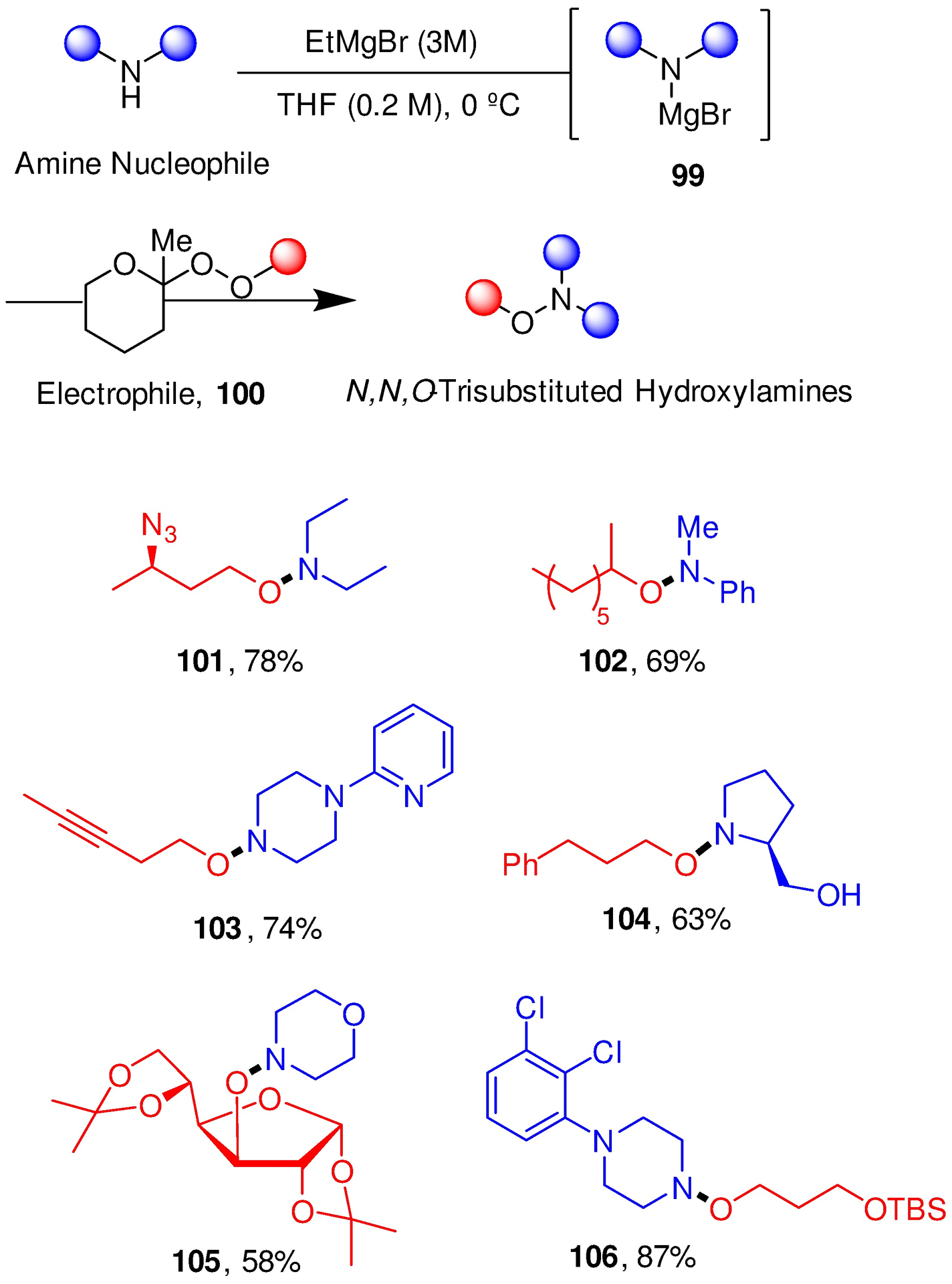
Hydroxalog synthesis by N-O Bond Formation
A more obvious example of the application of our expertise in glycochemistry to the medicinal chemistry arena, arose from discussions with Andrea Vasella at the ETH and Erik Böttger at the University of Zurich resulting in a collaboration targeting the development of much-needed, improved and less toxic aminoglycoside antibiotics for the treatment of ESKAPE pathogens.161 Building on observations on the beneficial influence of ethers at the 4’-position of paromomycin on selectivity for the inhibition of bacterial ribosomes over their human counterparts, albeit with something of a reduction in activity,162 we progressed via paromomycin 4’-O-glycosides and conformationally restricted analogs,163, 164 ultimately arriving at 4’-deoxy-4’-C-propylparomomycin, which we dubbed propylamycin 116.165 The synthesis of this next generation aminoglycoside, of which we prepared 30 g over eleven steps from the parent paromomycin 107, exploits basic knowledge of carbohydrate protecting groups to achieve the necessary regioselectivity, and a large scale diastereoselective radical reaction for the installation of the C-C bond (Scheme 19). Propylamycin is a rare aminoglycoside analog with increased in vitro and in vivo antibacterial activity vis à vis the parent coupled with reduced ototoxicity in an animal model. Crucially in the current era of multidrug resistant bacterial infections, the 4’-deoxy-4’-propyl modification also restricts the action of some of the aminoglycoside-modifying enzymes that aminoglycoside-producing bacteria secret to protect themselves from their own aminoglycosides, and which have subsequently evolved into the main resistance determinants.
Scheme 19.
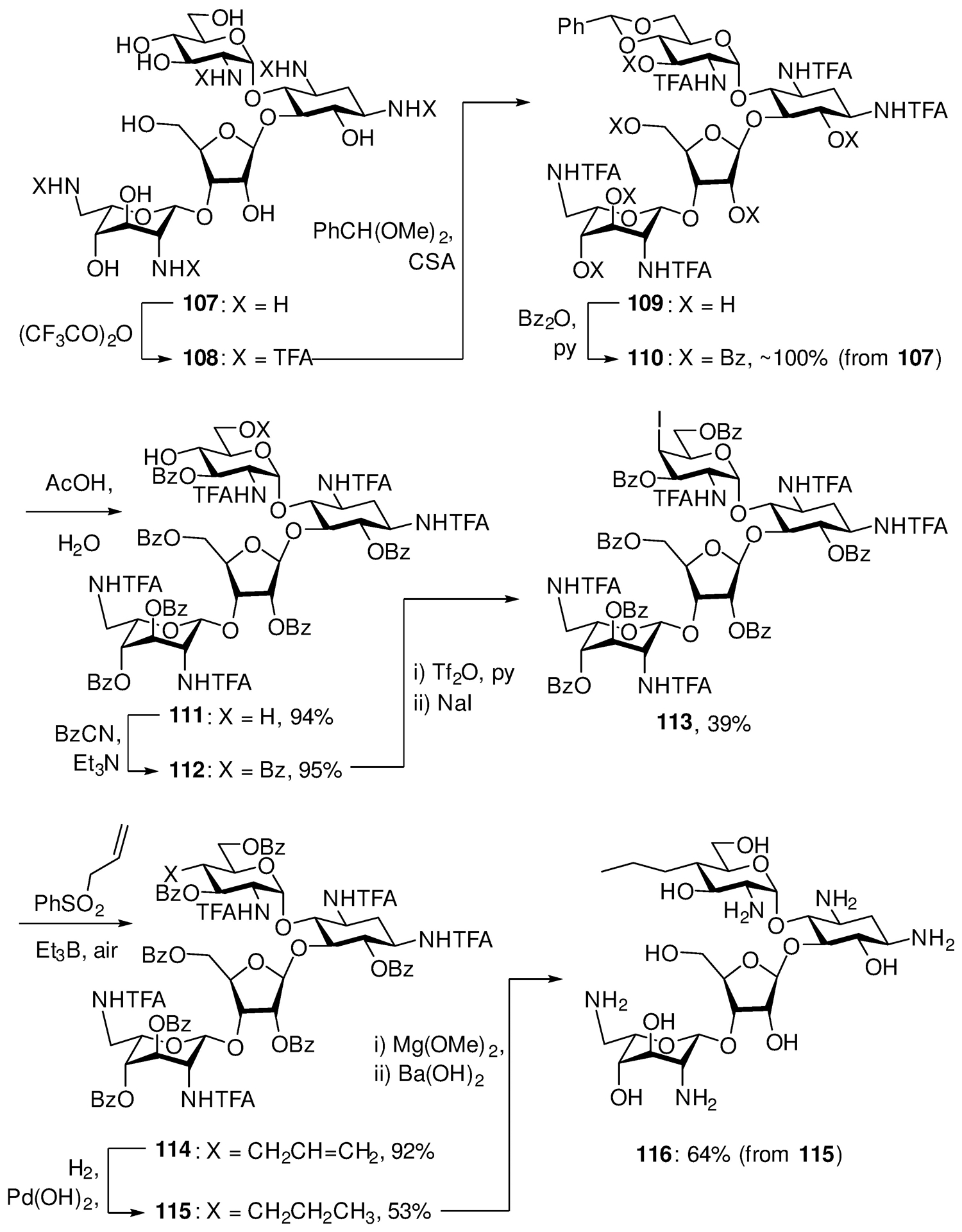
Synthesis of Propylamycin 116
A more obvious example of the application of carbohydrate chemistry to medicinal chemistry and antibacterial design is found in our development of the 5-O-glycosylapramycin derivatives, or apralogs.166 The goal was to develop an analog of the atypical monosubstituted 2-deoxystreptamine type aminoglycoside apramycin 117 that retained its excellent toxicity and minimal resistance profile while improving activity. Ultimately, we arrived at 5-O-(5-amino-3-O-aminoethyl-5-deoxy)-β-D-ribofuranosyl apramycin 123 which was prepared from the parent in only six robust and scalable linear steps including a classical Helferich type glycosylation (Scheme 20).167 This advanced apralog displays across the board improved activity against ESKAPE pathogens when compared to the parent, is approximately twice as active in a mouse model, retains full activity in the presence of the aminoglycoside acetyltransferase (3)-IV that is the only known AME activing on the parent, and has reduced cochleotoxicity in an ex-vivo assay.
Scheme 20.

Synthesis of the Advanced Apralog 123
Conclusions
The 2012 NRC report on the glycosciences identified the need for improved methods for glycan synthesis, and for cross fertilization of glycochemistry by mainstream organic chemists. I have endeavoured to show that knowledge of the basic mechanisms of glycosylation has advanced significantly in recent years such that organic chemists willing to look past the concepts of naked oxacarbenium ion intermediates and willing to embrace bimolecular mechanisms should experience no greater difficulty moving into glycan synthesis than into any other field. Subsequently, mostly using examples from my laboratories, I have attempted to show that carbohydrate chemistry provides fertile grounds for creative organic chemists to exercise their imagination, not the least by fully completing syntheses of natural products rather than stopping at the level of the aglycones, and by participating in the discovery of much-needed stereospecific glycosylation reactions. Finally, again using examples from my own laboratories, I have attempted to show that traffic between organic and medicinal chemistry, and carbohydrate chemistry moves in both directions on a two-way street: carbohydrate chemists should be more open to applying their ideas in mainstream organic and medicinal chemistry. In closing, I apologize to the many giants who have preceded me in the journey from organic to carbohydrate chemistry and back for not having cited their work in this personal perspective. May there be many more in the years to come.
Acknowledgments
Thanks are due to the many coworkers and collaborators who have contributed enormously to the development of all projects in my laboratory, past and present, at the interface between organic and carbohydrate chemistry. Abundant thanks are due to the NIH for continued support of our programs, currently through grants GM62160, GM125271, and AI123352.
References
- 1.Walt DR; Aoki-Kinoshita KF; Bendiak B; Bertozzi CR; Boons G-J; Darvill A; Hart GW; Kiessling LL; Lowe J; Moon R; Paulson J; Sasisekharan R; Varki AP; Wong C-H Transforming Glycoscience: A Roadmap for the Future (2012); National Research Council: Washinggton DC, 2012. [Google Scholar]
- 2.Glycoscience. https://commonfund.nih.gov/Glycoscience.
- 3.Crich D; Ritchie TJ, Stereoselective Free Radical Reactions in the Preparation of 2-Deoxy-β-D-glucosides. J. Chem. Soc., Chem. Commun 1988, 1461–1463. [Google Scholar]
- 4.Adero PO; Amarasekara H; Wen P; Bohé L; Crich D, The Experimental Evidence in Support of Glycosylation Mechanisms at the SN1-SN2 Interface. Chem. Rev 2018, 118, 8242–8284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Winstein S; Clippinger E; Fainberg AH; Heck R; Robinson GC, Salt Effects and Ion Pairs in Solvolysis. Common Ion Rate Depression and Exchange of Anions During Acetolysis. J. Am. Chem. Soc 1956, 78, 328–335. [Google Scholar]
- 6.Rhind-Tutt AJ; Vernon CA, Nucleophilic Substitution in 2,3,4,6-Tetra-O-methyl Glycopyranosyl Chlorides. J. Chem. Soc 1960, 4637–4644. [Google Scholar]
- 7.Eby R; Schuerch C, The Use of 1-O-Tosyl-D-glucopyranose Derivatives in α-D-Glucoside Synthesis. Carbohydr. Res 1974, 34, 79–90. [Google Scholar]
- 8.Wallace JE; Schroeder LR, Koenigs-Knorr Reactions. Part II. A Mechanisitc Study of Mercury (II) Cyanide-promoted Reactions of 2,3,4,6-Tetra-O-methyl-α-D-glucopyranosyl Bromide with Cyclohexanol in Benzene-Nitromethane. J. Chem. Soc., Perkin Trans 2 1976, 1632–1636. [Google Scholar]
- 9.Lemieux RU; Hendriks KB; Stick RV; James K, Halide Ion Catalyzed Glycosidation Reactions. Synthesis of α-Linked Disaccharides. J. Am. Chem. Soc 1975, 97, 4056–4062. [Google Scholar]
- 10. [Sept 30, 2020]; https://apps.webofknowledge.com/ Accessed.
- 11.Lemieux RU, Explorations with Sugars: How Sweet It Was American Chemical Society: Washington DC, 1990; p 185. [Google Scholar]
- 12.Pappo R; Allen DS; Lemieux RU; Johnson WS, Osmium Tetroxide-Catalyzed Periodate Oxidation of Olefinic Bonds. J. Org. Chem 1956, 21, 478–479. [Google Scholar]
- 13.Lemieux RU; Kullnig RK; Bernstein HJ; Schneider WG, Configurational Effects on the Proton Magnetic Resonance Spectra of 6-Membered Ring Compounds. J. Am. Chem. Soc 1958, 80, 6098–6105. [Google Scholar]
- 14.Smith DM; Woerpel KA, Electrostatic Interactions in Cations and their Importance in Biology and Chemistry. Org. Biomol. Chem 2006, 4, 1195–1201. [DOI] [PubMed] [Google Scholar]
- 15.Hagen B; van der Vorm S; Hansen T; van der Marel GA; Codée JDC, Stereoselective Glycosylations - Additions to Oxocarbenium Ions In Selective Glycosylations: Synthetic Methods and Catalysts, Bennett CS, Ed. Wiley-VCH: Weinheim, 2017; pp 3–28. [Google Scholar]
- 16.Bohé L; Crich D, A Propos of Glycosyl Cations and the Mechanism of Chemical Glycosylation. CR Chimie 2011, 14, 3–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Bohé L; Crich D, A Propos of Glycosyl Cations and the Mechanism of Chemical Glycosylation; the Current State of the Art. Carbohydr. Res 2015, 403, 48–59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Crich D, Mechanism of a Chemical Glycosylation. Acc. Chem. Res 2010, 43, 1144–1153. [DOI] [PubMed] [Google Scholar]
- 19.Aubry S; Sasaki K; Sharma I; Crich D, Influence of Protecting Groups on the Reactivity and Selectivity of Glycosylation: Chemistry of the 4,6-O-Benzylidene Protected Mannopyranosyl Donors and Related Species. Top. Curr. Chem 2011, 301, 141–188. [DOI] [PubMed] [Google Scholar]
- 20.Crich D, Methodology Development and Physical Organic Chemistry: A Powerful Combination for the Advancement of Glycochemistry. J. Org. Chem 2011, 76, 9193–9209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Dhakal B; Bohé L; Crich D, Trifluoromethanesulfonate Anion as Nucleophile in Organic Chemistry. J. Org. Chem 2017, 82, 9263–9269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Zhou M-H; Wilbur DJ; Kwan EE; Bennett CS, Matching Glycosyl Donor Reactivity to Sulfonate Leaving Group Ability Permits SN2 Glycosylations J. Am. Chem. Soc 2019, 141, 16743–16754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Lambert JB; Mark HW, Increased Electron Demand in the Solvolysis of Secondary 2-Norbornyl Tosylates. J. Am. Chem. Soc 1978, 100, 2501–2505. [Google Scholar]
- 24.Kirmse W; Mrotzeck U, Zerfall von 2-Oxa-5- und 6-Norbornandiazonium-Ionen. Chem. Ber 1988, 121, 485–492. [Google Scholar]
- 25.Zhang Z; Ollmann IR; Ye X-S; Wischnat R; Baasov T; Wong C-H, Programmable One-Pot Oligosaccharide Synthesis. J. Am. Chem. Soc 1999, 121, 734–753. [Google Scholar]
- 26.Douglas NL; Ley SV; Lucking U; Warriner SL, Tuning Glycoside Reactivity: New Tool for Efficient Oligosaccharide Synthesis. J. Chem. Soc., Perkin Trans 1 1998, 51–65. [Google Scholar]
- 27.Fraser-Reid B, From Sugar to Splenda: A Personal and Scientic Journey of a Carbohydrate Chemist and Expert Witness Springer-Verlag: Berlin, 2012; p 216. [Google Scholar]
- 28.Martin A; Arda A; Désiré J; Martin-Mingot A; Probst N; Sinaÿ P; Jiménez-Barbero J; Thibaudeau S; Blériot Y, Catching Elusive Glycosyl Cations in a Condensed Phase with HF/SbF5 Superacid. Nat. Chem, 2016, 8, 186–191. [DOI] [PubMed] [Google Scholar]
- 29.Lebedel L; Ardá A; Martin A; Désiré J; Mingot A; Aufiero M; Font NA; Gilmour R; Jiménez-Barbero J; Blériot Y; Thibaudeau S, Structural and Computational Analysis of 2-Halogeno-Glycosyl Cations in the Presence of a Superacid: an Expanisve Platform. Angew. Chem. Int. Ed 2019, 58, 13758–13762. [DOI] [PubMed] [Google Scholar]
- 30.Frihed TG; Bols M; Pedersen CM, Mechanisms of Glycosylation Reactions Studied by Low-Temperature Nuclear Magnetic Resonance. Chem. Rev 2015, 115, 4963–5013. [DOI] [PubMed] [Google Scholar]
- 31.Crich D; Sun S, Are Glycosyl Triflates Intermediates in the Sulfoxide Glycosylation Method? A Chemical and 1H, 13C, and 19F-NMR Spectroscopic Investigation. J. Am. Chem. Soc 1997, 119, 11217–11223. [Google Scholar]
- 32.Santana AG; Montalvillo-Jiménez L; Diaz-Casado L; Corzana F; Merino P; Cañada FJ; Jiménez-Osés G; Jiménez-Barbero J; Gomez AM; Asensio JL, Dissecting the Essential Role of Anomeric β-Triflates in Glycosylation Reactions. J. Am. Chem. Soc 2020, 142, 12501–12514. [DOI] [PubMed] [Google Scholar]
- 33.Igarashi K; Honma T; Irisawa J, Reaction of Glycosyl Chlorides with Silver Perchlorate. Carbohydr. Res 1970, 15, 329–337. [Google Scholar]
- 34.Igarashi K; Honma T; Irisawa J, Reaction of Glycosyl Chlorides with Silver Tetrafluoroborate. Carbohydr. Res 1970, 13, 49–55. [Google Scholar]
- 35.Yamago S; Yamada H; Maruyama T; Yoshida J.-i., Iterative Glycosylation of 2-Deoxy-2-aminothioglyocsides and its Application to the Combinatorial Synthesis of Linear Oligoglucosamines. Angew. Chem. Int. Ed 2004, 43, 2145–2148. [DOI] [PubMed] [Google Scholar]
- 36.Huang M; Garrett GE; Birlirakis N; Bohé L; Pratt DA; Crich D, Dissecting the Mechanisms of a Class of Chemical Glycosylation Using Primary 13C Kinetic Isotope Effects. Nat. Chem 2012, 4, 663–667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Huang M; Retailleau P; Bohé L; Crich D, Cation Clock Permits Distinction Between the Mechanisms of α- and β-O- and β-C-Glycosylation in the Mannopyranose Series; Evidence for the Existence of a Mannopyranosyl Oxocarbenium Ion. J. Am. Chem. Soc 2012, 134, 14746–14749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Adero PO; Furukawa T; Huang M; Mukherjee D; Retailleau P; Bohé L; Crich D, Cation Clock Reactions for the Determination of Relative Reaction Kinetics in Glycosylation Reactions: Applications to Gluco- and Mannopyranosyl Sulfoxide and Trichloroacetimidate Type Donors. J. Am. Chem. Soc 2015, 137, 10336–10345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Kwan EE; Park Y; Besser HA; Anderson TL; Jacobsen EN, Sensitive and Accurate 13C Kinetic Isotope Effect Measurements Enabled by Polarization Transfer. J. Am. Chem. Soc 2017, 139, 43–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Crich D; Chandrasekera NS, Mechanism of 4,6-O-Benzylidene Directed ß-Mannosylation as Determined by a-Deuterium Kinetic Isotope Effects. Angew. Chem. Int. Ed 2004, 43, 5386–5389. [DOI] [PubMed] [Google Scholar]
- 41.Banait NS; Jencks WP, Reactions of Anionic Nucleophiles with α-D-Glucopyranosyl Fluoride in Aqueous Solution through a Concerted, ANDN (SN2) Mechanism. J. Am. Chem. Soc 1991, 113, 7951–7958. [Google Scholar]
- 42.E.-Badri MH; Willenbring D; Tantillo DJ; Gervay-Hague J, Mechanistic Studies on the Stereoselective Formation of β-Mannosides from Mannosyl Iodides using α-Deuterium Kinetic Isotope Effects. J. Org. Chem 2007, 72, 4663–4672. [DOI] [PubMed] [Google Scholar]
- 43.Fang T; Gu Y; Huang W; Boons G-J, Mechanism of Glycosylation of Anomeric Sulfonium Ions. J. Am. Chem. Soc 2016, 138, 3002–3011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.D’Angelo KA; Taylor MS, Borinic Acid Catalyzed Stereo- and Regioselective Couplings of Glycosyl Methanesulfonates. J. Am. Chem. Soc 2016, 138, 11058–11067. [DOI] [PubMed] [Google Scholar]
- 45.Khomutnyk YY; Argüelles AJ; Winschel GA; Sun Z; Zimmerman PM; Nagorny P, Studies of the Mechanism and Origins of Enantioselectivity for the Chiral Phosphoric Acid-Catalyzed Stereoselective Spiroketalization Reactions. J. Am. Chem. Soc 2016, 138, 444–456. [DOI] [PubMed] [Google Scholar]
- 46.Tanaka M; Nakagawa A; Nishi N; Iijima K; Sawa R; Takahashi D; Toshima K, Boronic-Acid-Catalyzed Regioselective and 1,2-cis-Stereoselective Glycosylation of Unprotected Sugar Acceptors via SNi‑Type Mechanism. J. Am. Chem. Soc 2018, 140, 3644–3651. [DOI] [PubMed] [Google Scholar]
- 47.Pelletier G; Zwicker A; Allen CL; Schepartz A; Miller SJ, Aqueous Glycosylation of Unprotected Sucrose Employing Glycosyl Fluorides in the Presence of Calcium Ion and Trimethylamine. J. Am. Chem. Soc 2016, 138, 3175–3182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Li Q; Levi SM; Jacobsen EN, Highly Selective β-Mannosylations and β-Rhamnosylations Catalyzed by Bis-thiourea. J. Am. Chem. Soc 2020, 142, 11865–11872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Issa JP; Bennett CS, A Reagent-Controlled SN2-Glycosylation for the Direct Synthesis of b-Linked 2-Deoxy Sugars. J. Am. Chem. Soc 2014, 136, 5740–5744. [DOI] [PubMed] [Google Scholar]
- 50.Hosoya T; Kosma P; Rosenau T, Contact Ion Pairs and Solvent-Separated Ion Pairs from D-Mannopyranosyl and D-Glucopyranosyl Triflates. Carbohydr. Res 2015, 401, 127–131. [DOI] [PubMed] [Google Scholar]
- 51.Hosoya T; Kosma P; Rosenau T, Theoretical Study on the Effects of a 4,6-O-Diacetal Protecting Group on the Stability of Ion Pairs from D-Mannopyranosyl and D-Glucopyranosyl Triflates. Carbohydr. Res 2015, 411, 64–69. [DOI] [PubMed] [Google Scholar]
- 52.Hosoya T; Takano T; Kosma P; Rosenau T, Theoretical Foundation for the Presence of Oxacarbenium Ions in Chemical Glycoside Synthesis. J. Org. Chem 2014, 79, 7889–7894. [DOI] [PubMed] [Google Scholar]
- 53.Sletten ET; Tu Y-J; Schlegel HB; Nguyen HM, Are Brønsted Acids the True Promoter of Metal Triflate Catalyzed Glycosylations? A Mechanistic Probe into 1,2-cis-Aminoglycoside Formation by Nickel Triflate. ACS Catal 2019, 9, 2110–2123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Varner TP; Teator AJ; Reddi Y; Jacky PE; Cramer CJ; Leibfarth FA, Mechanistic Insight into the Stereoselective Cationic Polymerization of Vinyl Ethers. J. Am. Chem. Soc 2020, 142, 17175–17186. [DOI] [PubMed] [Google Scholar]
- 55.Lee S; Kaib PSJ; List B, Asymmetric Catalysis via Cyclic, Aliphatic Oxocarbenium Ions. J. Am. Chem. Soc 2017, 139, 2156–2159. [DOI] [PubMed] [Google Scholar]
- 56.Krumper JR; Salamant WA; Woerpel KA, Correlations Between Nucleophilicities and Selectivities in the Substitutions of Tetrahydropyran Acetals. J. Org. Chem 2009, 74, 8039–8050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.van der Vorm S; Hansen T; Overkleeft HS; van der Marel GA; Codée JDC, The Influence of Acceptor Nucleophilicity on the Glycosylation Reaction Mechanism. Chem. Sci 2017, 8, 1867–1875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Hansen T; Lebedel L; Remmerswaal WA; van der Vorm S; Wander DPA; Somers M; Overkleeft HS; Filippov DV; Désiré J; Mingot A; Bleriot Y; van der Marel GA; Thibaudeau S; Codée JDC, Defining the SN1 Side of Glycosylation Reactions: Stereoselectivity of Glycopyranosyl Cations. ACS Cent. Sci 2019, 5, 781–788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Onobun E; Crich D, Synthesis of 3‐Deoxy‐D‐manno‐oct‐2‐ulosonic Acid (KDO) and Pseudaminic Acid C‐Glycosides. J. Org. Chem 2020, 85, 16035–16042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Hettikankanamalage A; Lassfolk R; Ekholm F; Leino R; Crich D, Mechanisms of Stereodirecting Participation and Ester Migration from Near and Far in Glycosylation and Related Reactions. Chem. Rev 2020, 120, 7104–7151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Chamberland S; Ziller JW; Woerpel KA, Structural Evidence that Alkoxy Substituents Adopt Electronically Preferred Pseudoaxial Orientations in Six-Membered Ring Dioxocarbenium Ions. J. Am. Chem. Soc 2005, 127, 5322–5323. [DOI] [PubMed] [Google Scholar]
- 62.Yang MT; Woerpel KA, The Effect of Electrostatic Interactions on Conformational Equilibria of Multiply Substituted Tetrahydropyran Oxocarbenium Ions. J. Org. Chem 2009, 74, 545–553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Crich D; Dai Z; Gastaldi S, On the Role of Neighboring Group Participation and Ortho Esters in b-Xylosylation: 13C-NMR Observation of a Bridging 2-Phenyl-1,3-dioxalenium Ion. J. Org. Chem 1999, 64, 5224–5229. [DOI] [PubMed] [Google Scholar]
- 64.Zeng Y; Wang Z; Whitfield D; Huang X, Installation of Electron-Donating Protective Groups, A Strategy for Glycosylating Unreactive Thioglycosyl Acceptors using the Preactivation-Based Glycosylation Method. J. Org. Chem 2008, 73, 7952–7962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Williams RJ; McGill NW; White JM; Williams SJ, Neighboring Group Participation in Glycosylation Reactions by 2,6-Disubstituted 2-O-Benzoyl groups: A Mechanistic Investigation. J. Carbohydr. Chem 2010, 29, 236–263. [Google Scholar]
- 66.Dejter-Juszynski M; Flowers HM, A Stereoselective Synthesis of 2-Acetamido-2-deoxy-6-O-α-L-fucopyranosyl-D-glucose. Carbohydr. Res 1972, 23, 41–45. [Google Scholar]
- 67.Dejter-Juszynski M; Flowers HM, The Effect of Participating Groups on the Stereochemistry of Disaccharide Formation. Carbohydr. Res 1973, 61–74. [Google Scholar]
- 68.Crich D; Cai W; Dai Z, Highly Diastereoselctive α-Mannosylation in the Absence of Participating Protecting Groups. J. Org. Chem 2000, 65, 1291–1297. [DOI] [PubMed] [Google Scholar]
- 69.Paulsen H, Transformations of Carboxonium Compounds in Carbohydrate and Polyol Chemistry Pure & Appl. Chem 1975, 41, 69–92. [Google Scholar]
- 70.Larsen JW; Ewing S, Relative Heats of Formation of Cyclic Oxonoim Ions in Sulfuric Acid. J. Am. Chem. Soc 1971, 93, 5107–5111. [Google Scholar]
- 71.Wiberg KB; Waldron RF, Enthalpies of Hydrolysis, Reduction, and Formation of the C4–C13 Monocyclic Lactones. Strain Energies and Conformations. J. Am. Chem. Soc 1991, 113, 7697–7705. [Google Scholar]
- 72.Houk KN; Jabbari A; Hall HK; Alemán C, Why δ-Valerolactone Polymerizes and γ-Butyrolactone Does Not. J. Org. Chem 2008, 73, 2674–2678. [DOI] [PubMed] [Google Scholar]
- 73.Greene TW; Wuts PGM, Protective Groups in Organic Synthesis 3rd ed.; Wiley: New York, 1999; p 779. [Google Scholar]
- 74.Crich D; Vinod AU; Picione J, The 3,4-O-Carbonate Protecting Group as a ß-Directing Group in Rhamnopyranosylation in Both Homogeneous and Heterogeneous Glycosylations as Compared to the Chameleon-Like 2,3-O-Carbonates. J. Org. Chem 2003, 68, 8453–8458. [DOI] [PubMed] [Google Scholar]
- 75.Schuler M; Tatibouët A, Strategies Toward Protection of 1,2- and 1,3-Diols in Carbohydrate Chemistry In Protecting Groups: Strategies and Application in Carbohydrate Chemistry, Vidal S, Ed. Wiley: Weinheim, 2019; pp 307–335. [Google Scholar]
- 76.Mikami K; Lonnecker AT; Gustafson TP; Zinnel NF; Pai P-J; Russell DH; Wooley KL, Polycarbonates Derived from Glucose via an Organocatalytic Approach. J. Am. Chem. Soc 2013, 135, 6826–6829. [DOI] [PubMed] [Google Scholar]
- 77.Bjarnov E; Hocking WH, The Structure of the Other Rotamer of Formic Acid. cis-HCOOH. Z. Naturforsch 1978, 33A, 610–618. [Google Scholar]
- 78.Wang X; Houk KN, Theoretical Elucidation of the Origin of the Anomalously High Acidity of Meldrum’s Acid. J. Am. Chem. Soc 1988, 110, 1870–1872. [Google Scholar]
- 79.Kleinpeter E; Taddei F; Wacker P, Electronic and Steric Substituent Influences on the Conformational Equilibria of Cyclohexyl Esters: The Anomeric Effect Is Not Anomalous! Chem. Eur. J 2003, 9, 1360–1368. [DOI] [PubMed] [Google Scholar]
- 80.Kleinpeter E; Werner P; Linker T, Synthesis and NMR Spectroscopic Conformational Analysis of Benzoic Acid Esters of Mono- and 1,4-Dihydroxycyclohexane, 4-Hydroxycyclohexanone and the -Ene Analogue - The More Polar the Molecule the More Stable the Axial Conformer. Tetrahedron 2017, 73, 3801–3808. [Google Scholar]
- 81.Crich D; Hu T; Cai F, Does Neighboring Group Participation by Non-Vicinal Esters Play a Role in Glycosylation Reactions? Effective Probes for the Detection of Bridging Intermediates. J. Org. Chem 2008, 73, 8942–8953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Gerbst A. g.; Ustuzhanina NE; Grachev AA; Tsvetkov DE; Khatuntseva EA; Nifant’ev NE, Effect of the Nature of Protecting Group at O-4 on Stereoselectivity of Glycosylation by 4-O-Substituted 2,3-Di-O-benzylfucosyl Bromides. Mendeleev Commun 1999, 9, 114–116. [Google Scholar]
- 83.Demchenko AV; Rousson E; Boons G-J, Stereoselective 1,2-cis-Galactosylation Assisted by Remote Neighboring Group Participation and Solvent Effects. Tetrahedron Lett 1999, 40, 6523–6536. [Google Scholar]
- 84.Ma Y; Lian G; Li Y; Yu B, Identification of 3,6-Di-O-acetyl-1,2,4-O-orthoacetyl-α-D-glucopyranose as a Direct Evidence for the 4-O-Acyl Group Participation in Glycosylation. Chem. Commun 2011, 47, 7515–7517. [DOI] [PubMed] [Google Scholar]
- 85.Elferink H; Severijnen ME; Martens J; Mensink RA; Berden G; Oomens J; Rutjes FPJT; Rijs AM; Boltje TJ, Direct Experimental Characterization of Glycosyl Cations by Infrared Ion Spectroscopy. J. Am. Chem. Soc 2018, 140, 6034–6038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Hansen T; Elferink H; van Hengst JMA; Houtuijs KJ; Remmerswaal WA; Kromm A; Berden G; van der vorm S; Rijs AM; Overkleeft HS; Filippov DV; Ruthjes FPJT; van der Marel GA; Martens J; Oomens J; Codée JDC; Boltje TJ, Characterization of Glycosyl Dioxolenium Ions and Their Role in Glycosylation Reactions. Nat Commun 2020, 11, 2664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Matrianski M; Mucha E; Greis K; Moon S; Pardo A; Kirschbaum C; Thomas DA; Meijer G; von Helden G; Gilmore k.; Seeberger PH; Pagel K, Remote Participation During Glycosylation Reactions of Galactose Building Blocks: Direct Evidence from Cryogenic Vibrational Spectroscopy. Angew. Chem. Int. Ed 2020, 59, 6166–6171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Chatterjee S; Moon S; Hentschel F; Gilmore K; Seeberger PH, An Empirical Understanding of the Glycosylation Reaction. J. Am. Chem. Soc 2018, 140, 11942–11953. [DOI] [PubMed] [Google Scholar]
- 89.Bohé L; Crich D, Double Diastereoselection, Regioselectivity, and the Importance of Donor-Acceptor Complementarity in the Stereoselectivity of Glycosylation Reactions. Trends. Glycosci. Glycotech 2010, 22, 1–15. [Google Scholar]
- 90.Spijker NM; van Boeckel CAA, Double Stereodifferentiation in Carbohydrate Coupling Reactions: The Mismatched interaction of Donor and Acceptor as an Unprecedented Factor Governing the α/β-Ratio of Glycoside Formation. Angew. Chem. Int. Ed 1991, 30, 180–183. [Google Scholar]
- 91.Yang T; Zhu F; Walczak MA, Stereoselective Oxidative Glycosylation of Anomeric Nucleophiles with Alcohols and Carboxylic Acids. Nat. Commun 2018, 9, 3650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Yu F; Dickson JL; Loka RS; Xu HH; Schaugaard RN; Schlegel HB; Luo L; Nguyen HM, Diastereoselective sp3 C–O Bond Formation via Visible Light-Induced, Copper-Catalyzed Cross-Couplings of Glycosyl Bromides with Aliphatic Alcohols ACS Catal 2020, 10, 5990–6001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Hoang KM; Lees NR; Herzon SB, Programmable Synthesis of 2‑Deoxyglycosides. J. Am. Chem. Soc 2019, 141, 8098–8103. [DOI] [PubMed] [Google Scholar]
- 94.Cuccarese MF; Li JJ; O’Doherty GA, De Novo Approaches to Monosaccharides and Complex Glycans In Modern Synthetic Methods in Carbohydrate Chemistry; From Monosaccharides to Complex Glycoconjugates, Werz DB; Vidal S, Eds. Wiley: Weinheim, 2014; pp 1–28. [Google Scholar]
- 95.Taylor MS, Catalysis Based on Reversible Covalent Interactions of Organoboron Compounds. Acc. Chem. Res 2015, 48, 295–305. [DOI] [PubMed] [Google Scholar]
- 96.Park Y; Harper KC; Kuhl N; Kwan EE; Liu RY; Jacobsen EN, Macrocyclic Bis-Thioureas Catalyze Stereospecific Glycosylation Reactions. Science 2017, 355, 162–166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Maki Y; Okamoto R; Izumi M; Murase T; Kajihara Y, Semisynthesis of Intact Complex-Type Triantennary Oligosaccharides from a Biantennary Oligosaccharide Isolated from a Natural Source by Selective Chemical and Enzymatic Glycosylation. J. Am. Chem. Soc 2016, 138, 3461–3468. [DOI] [PubMed] [Google Scholar]
- 98.Pawar S; Hsu LY; Narendar Reddy T; Ravinder M; Ren C-T; Lin Y-W; Cheng Y-Y; Lin T-W; Wang S-K; Wong C-H; Wu C-Y, Synthesis of Asymmetric N-Glycans as Common Core Substrates for Structural Diversification through Selective Enzymatic Glycosylation. ACS Chem. Biol 2020, 15, 2382–2394. [DOI] [PubMed] [Google Scholar]
- 99.Li T; Liu L; Wei N; Yang J-Y; Chapla DG; Moremen KW; Boons G-J, An Automated Platform for the Enzyme-Mediated Assembly of Complex Oligosaccharides. Nat. Chem 2019, 229–236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Guberman M; Seeberger PH, Automated Glycan Assembly: A Perspective. J. Am. Chem. Soc 2019, 141, 5581–5592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Wu Y; Xiong D-C; Chen S-C; Wang Y-S; Ye X-S, Total Synthesis of Mycobacterial Arabinogalactan Containing 92 Monosaccharide Units. Nat. Commun 2017, 8, 14851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Zhu Q; Shen Z; Chiodo F; Nicolardi S; Molinaro A; Silipo A; Yu B, Chemical Synthesis of Glycans Up To a 128-mer Relevant to the O-Antigen of Bacteroides vulgatus. Nat. Commun 2020, 11, 4142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Lin S; Lowary TL, Synthesis of a Highly Branched Nona-saccharide Chlorella Virus N-Glycan Using a “Counter-clockwise” Assembly Approach. Org. Lett 2020, 22, 7645–7649. [DOI] [PubMed] [Google Scholar]
- 104.Wang Y-S; Wu Y; Xiong D.-c.; Ye X-S, Total Synthesis of a Hyperbranched N-Linked Hexasaccharide Attached to ATCV-1 Major Capsid Protein without Precedent. Chin. J. Chem 2019, 37, 42–48. [Google Scholar]
- 105.Zhang X; Yu B, Tackling the Challenge of the Total Synthesis of Periploside A. Synlett 2018, 29, 1683–1692. [Google Scholar]
- 106.Crich D; Yang F, Phenylthiomethyl Glycosides: Convenient Synthons for the Formation of Azidomethyl and Glycosylmethyl Glycosides and Their Derivatives. Angew. Chem. Int. Ed 2009, 48, 8896–8899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Crich D; Dai Z, Direct Synthesis of b-Mannosides. β-D-Xyl-(1→2)-β-D-Man-(1→4)-α-D-Glc-OMe: a Trisaccharide Component of the Hyriopsis schlegelii Glycosphingolipid. Formation of an Orthoester from a Xylopyranosyl Sulfoxide. Tetrahedron 1999, 55, 1569–1580. [Google Scholar]
- 108.Crich D; Barba GR, Convergent, Stereoselective Synthesis of the Caloporoside Disaccharide. Tetrahedron Lett 1998, 39, 9339–9342. [Google Scholar]
- 109.Crich D; de la Mora MA; Cruz R, Synthesis of the Mannosyl Erythritol Lipid MEL A; Confirmation of the Configuration of the “meso-Erythritol” Moiety. Tetrahedron 2002, 58, 35–44. [Google Scholar]
- 110.Crich D; Banerjee A; Yao Q, Direct Chemical Synthesis of the ß-D-Mannans: the ß-(1→2)- and ß-(1→4)-Series. J. Am. Chem. Soc 2004, 126, 14930–14934. [DOI] [PubMed] [Google Scholar]
- 111.Crich D; Li W; Li H, Direct Chemical Synthesis of the ß-Mannans: Linear and Block Syntheses of the Alternating ß-(1→3)-ß-(1→4)-Mannan Common to Rhodotorula glutinis, Rhodotorula mucilaginosa, and Leptospira biflexa. J. Am. Chem. Soc 2004, 126, 15081–15086. [DOI] [PubMed] [Google Scholar]
- 112.Crich D; Banerjee A, Stereocontrolled Synthesis of the D- and L-Glycero-β-D-mannoheptopyranosides and their 6-Deoxy Analogs. Synthesis of Methyl α-L-Rhamnopyranosyl-(1→3)-D-glycero-β-D-manno-heptopyranosyl-(1→3)-6-deoxy-glycero-β-D-manno-heptopyranosyl-(1→4)-α-L-rhamnopyranoside, A Tetrasaccharide Subunit of the Lipopolysaccharide from Plesimonas shigelloides. J. Am. Chem. Soc 2006, 128, 8078–8086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Crich D; Bowers AA, Synthesis of a β-(1→3)-D-Rhamnotetraose by a One-Pot, Multiple Radical Fragmentation. Org. Lett 2006, 8, 4327–4330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Crich D; Vinogradova E, Synthesis of the Antigenic Tetrasaccharide Side Chain from the Major Glycoprotein of Bacillus anthracis Exosporium. J. Org. Chem 2007, 72, 6513–6520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Crich D; Wu B; Jayalath P, Convergent Synthesis of a β-(1→3)-Mannohexaose. J. Org. Chem 2007, 72, 6806–6815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Crich D; Li M, Block Synthesis of Tetra- and Hexasaccharides (β-D-glycero-D-manno-Hepp-(1→4)-[α-L-Rhap-(1→3)-β-D-glycero-D-manno-Hepp-(1→4)]n-α-L-Rhap-OMe (n = 1 and 2) Corresponding to Multiple Repeat Units of the Glycan from the Surface-Layer Glycoprotein from Bacillus thermoaerophilus. J. Org. Chem 2008, 73, 7003–7010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Crich D; Dudkin V, Confirmation of the Connectivity of 4,8,12,16,20-Pentamethylpentacosylphosphoryl β-D-Mannopyranoside, an Unusual β-Mannosyl Phosphoisoprenoid from Mycobacterium avium, through Synthesis. J. Am. Chem. Soc 2002, 124, 2263–2266. [DOI] [PubMed] [Google Scholar]
- 118.McCormick SP; Kato T; Maragos CM; Busman M; Lattanzio VMT; Galaverna G; Dall-Asta C; Crich D; Price NPJ; Kurtzman CP, Anomericity of T-2 Toxin Glucoside: Masked Mycotoxin in Cereal Crops. J. Agr. Food. Chem 2015, 63, 731–738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Crich D; Rahaman MY, Synthesis and Structural Verification of the Xylomannan Antifreeze Substance from the Freeze-Tolerant Alaskan Beetle Upis ceramboides. J. Org. Chem 2011, 76, 8611–8620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Crich D; Li W, α-Selective Sialylations at −78 °C in Nitrile Solvents with a 1-Adamantanyl Thiosialoside. J. Org. Chem 2007, 72, 7794–7797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Crich D; Li W, O-Sialylation with N-Acetyl-5-N,4-O-Carbonyl Protected Thiosialoside Donors in Dichloromethane; Facile and Selective Cleavage of the Oxazolidinone Ring. J. Org. Chem 2007, 72, 2387–2391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Crich D; Navuluri C, Efficient, Highly Stereoselective Synthesis of α-Keto-deoxy-D-glycero-D-galacto-nonulosonic Acid (KDN) Glycosides by Means of the 4,5-O-Carbonate Protecting Group. Angew. Chem. Int. Ed 2010, 49, 3049–3052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Crich D; Wu B, Stereoselective Iterative One Pot Synthesis of N-Glycolylneuraminic Acid-Containing Oligosaccharides. Org. Lett 2008, 10, 4033–4035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Knirel YA; Sheelev SD; Perepelov AV, Higher Aldulosonic Acids: Components of Bacterial Glycans. Mendeleev, Commun 2011, 21, 173–182. [Google Scholar]
- 125.Zunk M; Kiefel MJ, The Occurrence and Biological Significance of the α-Keto-Sugars Pseudaminic Acid and Legionaminic Acid within Pathogenic Bacteria. RSC Adv 2014, 4, 3413–3421. [Google Scholar]
- 126.Kancharla PK; Crich D, Influence of Side Chain Conformation and Configuration on Glycosyl Donor Reactivity and Selectivity as Illustrated by Sialic Acid Donors Epimeric at the 7-Position. J. Am. Chem. Soc 2013, 135, 18999–19007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Dhakal B; Buda S; Crich D, Stereoselective Synthesis of 5-Epi-α-Sialosides Related to the Pseudaminic Acid Glycosides. Reassessment of the Stereoselectivity of the 5-Azido-5-deacetamidosialyl Thioglycosides and Use of Triflate as Nucleophile in the Zbiral Deamination of Sialic Acids. J. Org. Chem 2016, 81, 10617–10630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.De Meo C; Priyadarshani U, C-5 Modifications in N-Acetyl-Neuraminic Acid: Scope and Limitations. Carbohydr. Res 2008, 343, 1540–1552. [DOI] [PubMed] [Google Scholar]
- 129.Popik O; Dhakal B; Crich D, Stereoselective Synthesis of the Equatorial Glycosides of Legionaminic Acid. J. Org. Chem 2017, 82, 6142–6152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Banik SM; Levina A; Hyde AM; Jacobsen EN, Lewis Acid Enhancement by Hydrogen-Bond Donors for Asymmetric Catalysis. Science 2017, 358, 761–764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Kiefel MJ; von Itzstein M, Recent Advances in the Synthesis of Sialic Acid Derivatives and Sialylmimetics as Biological Probes Chem. Rev 2002, 102, 471–490. [DOI] [PubMed] [Google Scholar]
- 132.Price NPJ; Furukawa T; Cheng F; Qi J; Chen W; Crich D, Biosynthesis of 4-Aminoheptose 2-Epimers, Core Structural Components of the Septacidins and Spicamycins. J. Antibiotics 2014, 67, 405–414. [DOI] [PubMed] [Google Scholar]
- 133.Ngoje P; Crich D, Stereocontrolled Synthesis of the Equatorial Glycosides of 3-Deoxy-D-manno-oct-2-ulosonic Acid (KDO): Role of Side Chain Conformation. J. Am. Chem. Soc 2020, 142, 7760–7764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Pirrone MG; Gysin M; Haldimann K; Hobbie SN; Vasella A; Crich D, Predictive Analysis of the Side Chain Conformation of the Higher Carbon Sugars: Application to the Preorganization of the Aminoglycoside Ring 1 Side Chain for Binding to the Bacterial Ribosomal Decoding A Site. J. Org. Chem 2020, 85, 16043–16059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Jensen HH; Nordstrøm LU; Bols M, The Disarming Effect of the 4,6-Acetal Group on Glycoside Reactivity: Torsional or Electronic. J. Am. Chem. Soc 2004, 126, 9205–9213. [DOI] [PubMed] [Google Scholar]
- 136.Moumé-Pymbock M; Furukawa T; Mondal S; Crich D, Probing the Influence of a 4,6-O-Acetal on the Reactivity of Galactopyranosyl Donors: Verification of the Disarming Influence of the trans-gauche Conformation of C5–C6 Bonds. J. Am. Chem. Soc 2013, 135, 14249–14255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Dharuman S; Crich D, Determination of the Influence of Side Chain Conformation on Glycosylation Selectivity Using Conformationally Restricted Donors. Chem. Eur. J 2016, 22, 4535–4542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Lombard V; Golaconda RH; Drula E; Coutinho PM; Henrissat B, The Carbohydrate-Active Enzymes Database (CAZy) in 2013. Nucleic Acid Res 2013, 42, D490–D495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Quirke JCK; Crich D, Glycoside Hydolases Restrict the Side Chain Conformation of their Substrates to Gain Additional Transition State Stabilization. J. Am. Chem. Soc 2020, 142, 16965–16973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Jeng W-Y; Wang N-C; Lin C-T; Chang W-J; Liu C-I; Wang AH, High-Resolution Structures of Neotermes koshunensis β-Glucosidase Mutants Provide Insights into the Catalytic Mechanism and the Synthesis of Glucoconjugates. Acta Crystallogr., Sect. B 2012, 68, 829–838. [DOI] [PubMed] [Google Scholar]
- 141.Ferry A; Guinchard X; Retailleau P; Crich D, Synthesis, Characterization and Coupling Reactions of Six-membered Cyclic P-Chiral Ammonium Phosphonite-boranes; Reactive H-Phosphinate Equivalents for the Stereoselective Synthesis of Glycomimetics. J. Am. Chem. Soc 2012, 134, 12289–12301. [DOI] [PubMed] [Google Scholar]
- 142.Ferry A; Malik G; Guinchard X; Retailleau P; Crich D, Alternative Synthesis of P-Chiral Phosphonite-borane Complexes; Application to the Synthesis of Phostone-Phostone Dimers. J. Org. Chem 2013, 78, 6858–6867. [DOI] [PubMed] [Google Scholar]
- 143.Malik G; Guinchard X; Crich D, Asymmetric Synthesis of Polyhydroxylated N-Alkoxypiperidines by Ring Closing Double Reductive Amination; Facile Preparation of Isofagomine and Analogues. Org. Lett 2012, 14, 596–599. [DOI] [PubMed] [Google Scholar]
- 144.Malik G; Ferry A; Guinchard X; Cresteil T; Crich D, N-O Bond as a Glycosidic Bond Surrogate; Synthetic Studies Toward Polyhydroxylated N-Alkoxypiperidines. Chem. Eur. J 2013, 19, 2168–2179. [DOI] [PubMed] [Google Scholar]
- 145.Crich D; Smith M, 1-Benzenesulfinyl Piperidine (BSP)/Trifluoromethanesulfonic Anhydride: A Potent Combination of Shelf-Stable Reagents for the Low Temperature Conversion of Thioglycosides to Glycosyl Triflates and for the Formation of Diverse Glycosidic Linkages. J. Am. Chem. Soc 2001, 123, 9015–9020. [DOI] [PubMed] [Google Scholar]
- 146.Ferry A; Malik G; Guinchard X; Vetvicka V; Crich D, Synthesis and Evaluation of Di- and Trimeric Hydroxylamine-Based β-(1→3)-Glucan Mimetics. J. Am. Chem. Soc 2014, 136, 14852–14857. [DOI] [PubMed] [Google Scholar]
- 147.Brown J; O’Callaghan CA; Marshall ASJ; Gilbert RJC; Siebold C; Gordon S; Brown GD; Jones EY, Structure of the Fungal b-Glucan-Binding Immune Receptor Dectin-1: Implications for Function. Protein Sci 2007, 16, 1042–1052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Sylla B; Guégan J-P; Wieruszeski J-M; Nugier-Chauvin C; Legentil L; Daniellou R; Ferrières V, Probing β-(1→3)-D-Glucans Interactions with Recombinant Human Receptors using High-Resolution NMR Studies. Carbohydr. Res 2011, 346, 1490–1494. [DOI] [PubMed] [Google Scholar]
- 149.Hanashima S; Ikeda A; Tanaka H; Adachi Y; Ohno N; Takahashi T; Yamaguchi Y, NMR Study of Short β-(1→3)-Glucans Provides Insights into the Structure and Interaction with Dectin-1. Glycoconj. J 2014, 31, 199–207. [DOI] [PubMed] [Google Scholar]
- 150.Sylla B; Legentil L; Saraswat-Ohri S; Vashishta A; Daniellou R; Wang H-W; Vetvicka V; Ferrières V, Oligo-β-(1→3)-glucans: Impact of Thio-Bridges on Immunostimulating Activities and the Development of Cancer Stem Cells. J. Med. Chem 2014, 57, 8280–8292. [DOI] [PubMed] [Google Scholar]
- 151.Liao X; Větvička V; Crich D, Synthesis and Evaluation of 1,5-Dithia-D-laminaribiose, Triose and Tetraose as Truncated β-(1→3)-Glucan Mimetics. J. Org. Chem 2018, 83, 14894–14904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Vetvicka V; Saraswat-Ohri S; Vashishta A; Descroix K; Jamois F; Yvin J-C; Ferrières V, New 4-Deoxy-(1→3)-β-D-Glucan-Based Oligosaccharides and Their Immunostimulating Potential. Carbohydr. Res 2011, 346, 2213–2221. [DOI] [PubMed] [Google Scholar]
- 153.Descroix K; Vetvicka V; Laurent I; Jamois F; Yvin J-C; Ferrières V, New Oligo-β-(1→3)-Glucan Derivatives as Immunostimulating Agents. Bioorg. Med. Chem 2010, 18, 348–357. [DOI] [PubMed] [Google Scholar]
- 154.Wen P; Vetvicka V; Crich D, Synthesis and Evaluation of Oligomeric Thioether-Linked Carbacyclic β-(1→3)-Glucan Mimetics. J. Org. Chem 2019, 84, 5554–5563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Dhanju S; Blazejewski BW; Crich D, Synthesis of Trialkylhydroxylamines by Stepwise Reduction of O-Acyl N,N-Disubstituted Hydroxylamines. Substituent Effects on the Reduction of O-(1-Acyloxyalkyl)hydroxylamines and on the Conformational Dynamics of N-Alkoxypiperidines. J. Org. Chem 2017, 82, 5345–5353. [DOI] [PubMed] [Google Scholar]
- 156.Dhanju S; Upadhyaya K; Rice CA; Pegan SD; Media J; Valeriote FA; Crich D, Synthesis, Cytotoxicity and Genotoxicity of 10-Aza-9-oxakalkitoxin, An N,N,O-Trisubstituted Hydroxylamine Analog, or Hydroxalog, of a Marine Natural Product. J. Am. Chem. Soc 2020, 142, 9147–9151. [DOI] [PubMed] [Google Scholar]
- 157.Kopecky DJ; Rychnovsky SD, Improved Procedure for the Reductive Acetylation of Acyclic Esters and a New Synthesis of Ethers. J. Org. Chem 2000, 65, 191–198. [DOI] [PubMed] [Google Scholar]
- 158.Dhanju S; Crich D, Synthesis of N N,O-Trisubstituted Hydroxylamines By Stepwise Reduction and Substitution of O-Acyl N,N-Disubstituted Hydroxylamines. Org. Lett 2016, 18, 1820–1823. [DOI] [PubMed] [Google Scholar]
- 159.Kyasa SK; Meier RN; Pardini RA; Truttmann TK; Kuwata KT; Dussault PH, Synthesis of Ethers via Reaction of Carbanions and Monoperoxyacetals J. Org. Chem 2015, 80, 12100–12114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Hill J; Hettikankanamalage AA; Crich D, Diversity-Oriented Synthesis of N,N,O-Trisubstituted Hydroxylamines from Alcohols and Amines by N-O Bond Formation. J. Am. Chem. Soc 2020, 142, 14820–14825. [DOI] [PubMed] [Google Scholar]
- 161.Böttger EC; Crich D, Aminoglycosides: Time for Resurrection of a Neglected Class of Antibacterials? ACS Infect. Dis 2020, 6, 168–172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162.Duscha S; Boukari H; Shcherbakov D; Salian S; Silva S; Kendall A; Kato T; Akbergenov R; Perez-Fernandez D; Bernet B; Vaddi S; Thommes P; Schacht J; Crich D; Vasella A; Böttger EC, Identification and Evaluation of Improved 4’-O-(Alkyl) 4,5-Disubstituted 2-Deoxystreptamines as Next Generation Aminoglycoside Antibiotics. mBio 2014, 5, 10.1128/mBio.01827-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163.Matsushita T; Chen W; Juskeviciene R; Teo Y; Shcherbakov D; Vasella A; Böttger EC; Crich D, Influence of 4′-O-Glycoside Constitution and Configuration on Ribosomal Selectivity of Paromomycin. J. Am. Chem. Soc 2015, 137, 7706–7717. [DOI] [PubMed] [Google Scholar]
- 164.Mandhapati AR; Yang G; Kato T; Shcherbakov D; Hobbie SN; Vasella A; Böttger EC; Crich D, Structure-Based Design and Synthesis of Apramycin-Paromomycin Analogues. Importance of the Configuration at the 6′-Position and Differences Between the 6′-Amino and Hydroxy Series. J. Am. Chem. Soc 2017, 139, 14611–14619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165.Matsushita T; Sati GC; Kondasinghe N; Pirrone MG; Kato T; Waduge P; Kumar HS; Cortes Sanchon A; Dobosz-Bartoszek M; Shcherbakov D; Juhas M; Hobbie SN; Schrepfer T; Chow CS; Polikanov YS; Schacht J; Vasella A; Böttger EC; Crich D, Design, Multigram Synthesis, and in Vitro and in Vivo Evaluation of Propylamycin: A Semisynthetic 4,5-Deoxystreptamine Class Aminoglycoside for the Treatment of Drug-Resistant Enterobacteriaceae and Other Gram-Negative Pathogens. J. Am. Chem. Soc 2019, 141, 5051–5061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166.Quirke JCK; Rajasekaran P; Sarpe VA; Sonousi A; Osinnii I; Gysin M; Haldimann K; Fang Q-J; Shcherbakov D; Hobbie SN; Sha S-H; Schacht J; Vasella A; Böttger EC; Crich D, Apralogs: Apramycin 5-O-Glycosides and Ethers with Improved Antibacterial Activity and Ribosomal Selectivity and Reduced Susceptibility to the Aminoacyltranserferase (3)-IV Resistance Determinant. J. Am. Chem. Soc 2020, 142, 530–544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167.Sonousi A; Quirke JCK; Waduge P; Janusic T; Gysin M; Haldimann K; Xu S; Hobbie SN; Sha S-H; Schacht J; Chow CS; Vasella A; Böttger EC; Crich D, An Advanced Apralog with Increased in-vitro and in-vivo Activity toward Gram-negative Pathogens and Reduced ex-vivo Cochleotoxicity. Chem. Med. Chem 2020, 10.1002/cmdc.202000726. [DOI] [PMC free article] [PubMed] [Google Scholar]