

Abstract
Since its conception, the ribonuclease S complex (RNase S) has led to historic discoveries in protein chemistry, enzymology, and related fields. Derived by the proteolytic cleavage of a single peptide bond in bovine pancreatic ribonuclease (RNase A), RNase S serves as a convenient and reliable model system for incorporating unlimited functionality into an enzyme. Applications of the RNase S system in biomedicine and biotechnology have, however, been hindered by two shortcomings: (1) the bovine-derived enzyme could elicit an immune response in humans, and (2) the complex is susceptible to dissociation. Here, we have addressed both limitations in the first semisynthesis of an RNase S conjugate derived from human pancreatic ribonuclease and stabilized by a covalent interfragment cross-link. We anticipate that this strategy will enable unprecedented applications of the “RNase–S” system.
Graphical Abstract
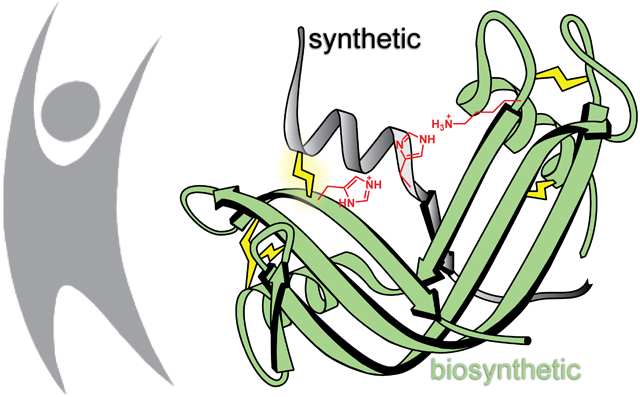
Semisynthesis allows access to precisely modified variants of otherwise native enzymes and other proteins. By combining recombinant DNA expression and organic synthesis, noncanonical residues can be incorporated for the precise interrogation and manipulation of protein structure and function.1 The first enzyme to be prepared by merging biosynthesis and chemical synthesis was ribonuclease S (RNase S). RNase S is a noncovalent complex that forms upon the association of two fragments of bovine pancreatic ribonuclease (RNase A; UniProtKB Q9BEC3) that are generated upon proteolysis with subtilisin. Alone, the S-peptide (1–20) and S-protein (21–124) fragments are catalytically inactive, but they associate spontaneously to reconstitute the enzymic active site and restore ribonucleolytic activity (Figure 1).2-5 A truncated variant of S-peptide (residues 1–15) also forms an active complex.6 By preparation of synthetic variants of S-peptide, a wide array of chemical modifications have been incorporated into RNase S. The ensuing data have enhanced our understanding of protein structure, protein folding, protein–protein interactions, and enzymology.3-5,7 S-peptide can even serve as an affinity tag for the purification and detection of recombinant proteins.8-11 In recent studies, semisynthetic RNase S variants have been used to stabilize α-helices via peptide stapling,12,13 control peptide–protein binding with photoswitchable peptides,14 and validate l-tellurienylalanine as a phenylalanine isostere.15
Figure 1.

Traditional production of semisynthetic RNase S. Step 1: Proteolytic digestion of RNase A to cleave S-peptide (residues 1–20) and yield catalytically inactive S-protein (residues 21–124). Step 2: Reversible association of S-protein and a synthetic S-peptide variant to produce catalytically active, semisynthetic RNase S. Images were produced with PyMOL software and PDB entries 1jvt21 and 1rnu.22
RNase 1 (UniProtKB P07998), which is the human homologue of RNase A, has demonstrable utility in biomedical contexts. In particular, an engineered variant of RNase 1 exhibits specific toxicity for cancer cells and is in clinical trials as a chemotherapeutic agent.16,17 RNase 1 also has considerable antiviral activity,18 and manifests antibacterial activity in the presence of a pore-forming peptide.19 To date, however, RNase S has not been employed in the clinic. Indeed, such applications are discouraged by the potential immunogenicity of the bovine-derived complex in humans.20 Hence, we reasoned that an RNase S of human origin could have an expanded scope.
Traditionally, S-protein is generated by treating intact, folded RNase A with subtilisin, a bacterial serine protease.2 Proteolytic cleavage occurs within a surface loop bordering the active site (residues 15–25), primarily between residues Ala20 and Ser21. Because the bovine and human pancreatic ribonucleases have 68% sequence identity, we inferred that the same strategy could be employed to generate an S-protein fragment from RNase 1. To the contrary, however, treating RNase 1 with subtilisin under similar conditions does not result in the formation of the corresponding S-protein and S-peptide fragments.23 This resistance arises because the key surface loop (residues 15–25) of RNase 1 is sterically incompatible with the subtilisin active site due to a conformational change induced by Pro19.23,24 Moreover, S-protein does not fold properly in the absence of S-peptide,25 and thus cannot be produced simply by heterologous expression.
There have been unsuccessful attempts to express the native RNase 1 21–127 fragment in Escherichia coli. Some success has been achieved through the digestion of an engineered chimeric bovine/human ribonuclease.20,26 As an alternative strategy, we imagined that enterokinase, which is a highly specific protease, could be employed to produce the S-protein fragment directly from recombinant RNase 1. Specifically, we envisaged that the enterokinase recognition sequence (DDDDK↓) could be installed via site-directed mutagenesis at the boundary of the S-peptide and S-protein fragments.27 Proteolytic cleavage, which occurs at the C-terminus of the recognition site, would release native S-protein along with S-peptide fused to the enterokinase recognition sequence, DDDDK.
In addition to circumventing issues associated with immunogenicity, enhancing the stability of the RNase S complex is crucial to enabling therapeutic applications. Despite being highly specific, the noncovalent interactions between the S-protein and S-peptide fragments render the complex prone to dissociation (Kd = 49 nM), particularly at low concentrations.28 Additionally, RNase S has greater conformational entropy than does intact RNase A, resulting in decreased thermodynamic stability and higher susceptibility toward denaturation and proteolysis.28,29 Using a strategy employed to enhance the conformational stability of intact ribonucleases,30,31 we previously described bovine S-protein and S-peptide variants that were able to form a disulfide cross-link (Cys4–Cys118) at the fragment interface.27 By restricting conformational flexibility, this additional disulfide bond was found to improve thermodynamic stability and increase resistance to proteolysis. Crucially, the disulfide cross-link was not highly detrimental to the ribonucleolytic activity of the enzyme. Given the similar three-dimensional structures of RNase A and RNase 1, we hypothesized that a cross-linking strategy could also stabilize an RNase S complex derived from RNase 1. Toward this end, we report herein a novel semisynthetic strategy for the preparation of human RNase–S, where the “–” highlights the covalent cross-link between the two fragments.
We designed the requisite variant of RNase 1 with an enterokinase cleavage site inserted between residues S20 and S21 (Figure 2A). As a precaution against contaminant activity arising from undigested enzyme, we replaced His12, which is a key active-site residue within S-peptide, with alanine. This substitution reduces catalytic activity by >104-fold without causing structural perturbations.32-35 We introduced an additional substitution, V118C, to serve as a conjugation handle for disulfide cross-linking with synthetic S-peptide variants. The ensuing RNase 1 variant was produced and purified as described previously.27 Following purification, we protected the unpaired thiol of Cys118 by reaction with 5,5′-dithio-(2-nitrobenzoic acid) (DTNB) to form a mixed disulfide with 2-nitro-5-thiobenzoic acid (NTB) as a safeguard against adventitious oxidation during storage. To produce the S-protein fragment, the RNase 1 variant (1 mg/mL in 20 mM Tris–HCl buffer, pH 7.4, containing 50 mM NaCl and 2 mM CaCl2) was treated with enterokinase (1 U/0.1 mg), resulting in efficient, sequence-specific cleavage of the protein in <6 h (Figure 2B). The H12A S-peptide fragment bearing the C-terminal enterokinase recognition sequence (DDDDK) was then discarded. We separated the fragments with semipreparative reversed-phase HPLC, and isolated purified V118C(NTB) S-protein (see: Supporting Information).
Figure 2.

(A) Map of the amino acid sequence of the RNase 1 variant. The locations of the enzymic active-site residues, native disulfide bonds, substitutions, and the inserted enterokinase recognition sequence are shown. (B) SDS–PAGE analysis of the digestion of the RNase 1 variant with enterokinase (1 U per 0.1 mg) to yield V118C S-protein and a variant peptide.
To conjugate V118C S-protein with synthetic S-peptide (residues 1–20 of the RNase 1 sequence), we envisioned the formation of a stabilizing disulfide bond between Cys118 and a cysteine residue incorporated into a synthetic S-peptide. A de novo designed disulfide bridge between residues 4 and 118 of intact RNase 1 has been reported to enhance the thermodynamic stability of the intact protein.31 In this strategy, however, replacing Arg4 with a cysteine residue eliminates a charged residue on the periphery of phosphoryl group-binding subsites36,37 and could diminish ribonucleolytic activity (Figure S6).38 To avoid this pitfall, we examined the three-dimensional structure of RNase 1 (PDB entry 1z7x39) to identify an alternative site for a cross-link within the S-peptide sequence. We found that the Cα–Cα distances from residue 118 to Arg4 and Ala5 are 5.3 and 4.8 Å, respectively (Figure S7). Since the mean Cα–Cα distance in a cystine residue is 5.6 Å,40 we hypothesized that an A5C substitution would enable facile disulfide cross-linking without a significant alteration of conformation or charge.
We assembled human RNase–S by a convergent scheme (Figure 3A). Specifically, we prepared A5C S-peptide by standard Fmoc-strategy solid-phase peptide synthesis and purified the peptide by preparative reversed-phase HPLC, obtaining a high overall yield (see: Supporting Information). Immediately prior to conjugation, we prepared a solution of the lyophilized peptide (420 μM in phosphate-buffered saline, pH 8.0, containing 40 mM EDTA) and treated the solution with immobilized tris(carboxyethyl)phosphine to ensure complete reduction of the peptide. After 30 min at 37 °C, we added the A5C S-peptide solution to a solution of NTB-protected V118C S-protein (8.1 μM in 0.2 M sodium acetate buffer, pH 5.5), and agitated the resulting solution gently at 37 °C. The solution gradually turned slightly yellow, indicative of the release of the NTB2− dianion, which provided a convenient visual indication that the conjugation reaction was proceeding. After only 1 h, we found the formation of disulfide-linked RNase–S to be complete by MALDI–TOF mass spectrometry. The conjugate was separated from residual excess A5C S-peptide by cation-exchange chromatography and judged to be pure by SDS–PAGE and Q-TOF mass spectrometry (see: Supporting Information).
Figure 3.

(A) Scheme for the semisynthesis of human RNase–S. Image was produced with PyMOL software and PDB entry 1z7x.39 (B) Graph showing the ribonucleolytic activity of RNase–S (5 nM) with a fluorogenic substrate, 6-FAM–dArUdAdA–6-TAMRA (200 nM), in 0.10 M DEPC-treated41 OVS-free42 MES–NaOH buffer, pH 6.0, containing NaCl (0.10 M); λex 493 nm, λem 515 nm. The assay was performed in triplicate.
Next, we subjected the purified RNase–S conjugate to a fluorogenic assay of ribonucleolytic activity to confirm that the active site had been reconstituted correctly. In this assay, the fluorescence of the substrate, 6-FAM–dArUdAdA–6-TAMRA, increases by 200-fold upon cleavage of its single RNA residue by a ribonuclease.43 Gratifyingly, the semisynthetic RNase–S conjugate displayed robust activity, with kcat/KM = 3.6 × 105 M−1s−1 (Figure 3B). This value is substantial, though somewhat lower than that of wild-type RNase 1 under the same conditions (kcat/KM = 9.5 × 106 M−1s−1). Notably, these assays were conducted with an enzymic concentration (5 nM) at which the noncovalent bovine RNase S complex is largely dissociated and thus inactive.28
Then, we used differential scanning fluorimetry44 to assess the thermostability of the RNase–S conjugate. We found that RNase–S has a Tm value of (44.5 ± 0.3) °C (Figure 4). The data were indicative of a two-state transition, that is, the absence of an intermediate between the native and denatured states. As a comparator, we also assessed the thermostability of wild-type RNase 1. We found a Tm value of (55.9 ± 0.1) °C, which is indistinguishable from one that we reported previously.45 Thus, RNase–S has lower conformational stability than does RNase 1, but its Tm value is well above physiological temperature.
Figure 4.
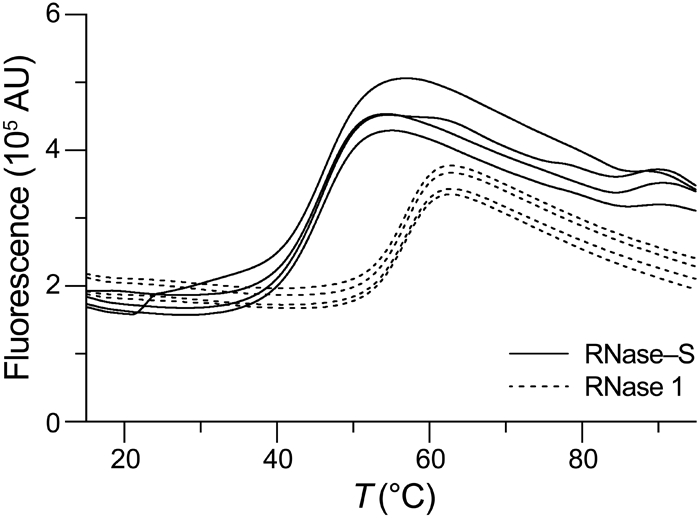
Thermal denaturation profiles of human RNase–S and human RNase 1 obtained by differential scanning fluorimetry using SYPRO Orange, λex (470 ± 15) nm, and λem (586 ± 10) nm. Assays were performed in quadruplicate.
In contrast to the four native disulfide bonds, Cys5–Cys118 is likely to be more solvent accessible and might, therefore, be more susceptible to reduction in the folded conformation.31 To gauge the stability of the 5–118 cross-link in a reducing environment like the cytosol, we treated RNase–S with glutathione (1 mM, 10:1 GSH/GSSG) at 37 °C and monitored reduction by SDS–PAGE (see: Supporting Information). After 1 h, we could detect some dissociated S-peptide from reduction of the Cys5–Cys118 bond (Figure 5). Still, intact RNase–S survived for up to 4 h.46
Figure 5.
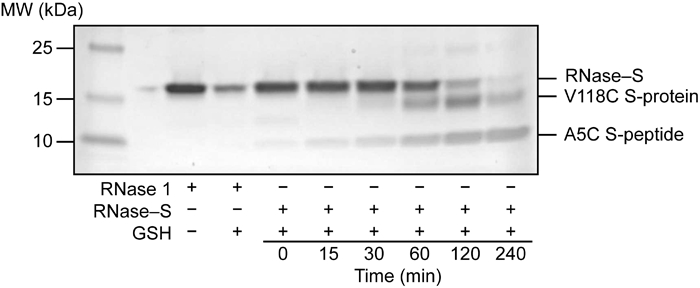
SDS–PAGE analysis of the reduction stability of human RNase–S.
Finally, we realized that this semisynthetic strategy offers convenient access to active human ribonucleases bearing homogeneous, noncanonical modifications. To demonstrate this capability, we prepared exemplar S-peptide variants bearing N-terminal alkyne, biotin, and fluorescein moieties (Figure 6A) and conjugated the modified peptides to V118C S-protein as described above (see: Supporting Information). We isolated the resulting homogeneously tagged RNase–S variants by cation- exchange chromatography and characterized them by Q–TOF mass spectrometry. We detected alkynyl RNase–S after a Cu(I)-catalyzed alkyne–azide cycloaddition (CuAAC) with 5-TAMRA–azide and SDS–PAGE (Figure 6B), biotinylated RNase–S with an antibiotin immunoblot (Figure 6C), and fluorescein-tagged RNase–S with fluorescence imaging following SDS–PAGE (Figure 6D). All three conjugates had high, nearly indistinguishable ribonucleolytic activity (Table S1). Notably, with the requisite fragments in hand, each RNase–S conjugate could be assembled, purified, and used for an application in less than 1 day. These examples highlight the efficiency and flexibility of our semisynthetic strategy, which offers unprecedented opportunities to produce an unlimited variety of precisely modified variants of an active human ribonuclease.
Figure 6.

(A) A5C S-peptide bearing N-terminal pendants. (B) Coomassie staining and fluorescence imaging of alkynyl RNase–S following CuAAC with 5-TAMRA–azide and SDS–PAGE. (C) Antibiotin immunoblot analysis of biotinylated RNase–S. (D) Coomassie staining and fluorescence imaging of fluorescein-tagged RNase–S after SDS–PAGE. WT, wild-type RNase 1.
CONCLUSIONS
Sixty years after the description of bovine RNase S, we employed recombinant DNA technology and a sequence-specific protease to achieve the first preparation of its human homologue. Strategic incorporation of an unpaired cysteine residue into the S-protein sequence enabled disulfide cross-linking with a chemically synthesized S-peptide fragment to produce an RNase–S conjugate. Conveniently, the disulfide conjugation reaction proceeds rapidly in mild conditions and without any exogenous reagents. High ribonucleolytic activity confirmed the successful reconstitution of the enzymic active site. The semisynthetic nature of this strategy enables synthetic S-peptide variants bearing a plethora of modifications to be incorporated into an active catalyst of RNA cleavage. We anticipate that this highly efficient semisynthetic strategy will find broad utility in preparing modified human RNase–S variants that enable novel applications in biotechnology and biomedicine.
Supplementary Material
Acknowledgments
Funding
E.C.W. was supported by a Graduate Research Fellowship from the NSF. This work was supported by Grant R01 CA073808 (NIH).
ABBREVIATIONS
- CuAAC
Cu(I)-catalyzed alkyne–azide cycloaddition
- DEPC
diethyl pyrocarbonate
- DTNB
5,5′-dithio-(2-nitrobenzoic acid)
- FAM
carboxyfluorescein
- MES
2-(N-morpholino)ethanesulfonic acid
- NTB
2-nitro-5-thiobenzoic acid
- OVS
oligo(vinylsulfonic acid)
- PAGE
polyacrylamide gel electrophoresis
- PDB
protein data bank
- RNase
ribonuclease
- SDS
sodium dodecyl sulfate
- TAMRA
carboxytetramethylrhodamine
- Tris
tris(hydroxymethyl)aminomethane
Footnotes
Supporting Information
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acs.bioconjchem.0c00557.
Experimental procedures, Table S1, and Figures S1–S24 (PDF)
The authors declare no competing financial interest.
REFERENCES
- (1).Thompson RE; Muir TW Chemoenzymatic semisynthesis of proteins. Chem. Rev 2020, 120, 3051–3126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (2).Richards FM; Vithayathil PJ The preparation of subtilisin-modified ribonuclease and the separation of the peptide and protein components. J. Biol. Chem 1959, 234, 1459–1465. [PubMed] [Google Scholar]
- (3).Richards FM; Wyckoff HW Bovine pancreatic ribonuclease. The Enzymes 1971, 4, 647–806. [Google Scholar]
- (4).Richards FM Linderstrøm-Lang and the Carlsberg Laboratory: The view of a postdoctoral fellow in 1954. Protein Sci. 1992, 1, 1721–1730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (5).Richards FM Whatever happened to the fun? An autobiographical investigation. Annu. Rev. Biophys. Biomol. Struct 1997, 26, 1–25. [DOI] [PubMed] [Google Scholar]
- (6).Taylor HC; Richardson DC; Richardson JS; Wlodawer A; Komoriya A; Chaiken IM “Active” conformation of an inactive semi-synthetic ribonuclease-S. J. Mol. Biol 1981, 149, 313–317. [DOI] [PubMed] [Google Scholar]
- (7).Genz M; Sträter N Posttranslational incorporation of noncanonical amino acids in the RNase S system by semisynthetic protein assembly. Methods Mol. Biol 2014, 1216, 71–87. [DOI] [PubMed] [Google Scholar]
- (8).Kim J-S; Raines RT Ribonuclease S-peptide as a carrier in fusion proteins. Protein Sci. 1993, 2, 348–356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (9).Kim J-S; Raines RT Peptide tags for a dual affinity fusion system. Anal. Biochem 1994, 219, 165–166. [DOI] [PubMed] [Google Scholar]
- (10).Karpeisky MY; Senchenko VN; Dianova MV; Kanevsky VY Formation and properties of S-protein complex with S-peptide–containing fusion protein. FEBS Lett. 1994, 339, 209–212. [DOI] [PubMed] [Google Scholar]
- (11).Raines RT; McCormick M; Van Oosbree TR; Mierendorf RC The S·Tag fusion system for protein purification. Methods Enzymol. 2000, 326, 362–376. [DOI] [PubMed] [Google Scholar]
- (12).Assem N; Ferreira DJ; Wolan DW; Dawson PE Acetone-linked peptides: A convergent approach for peptide macrocyclization and labeling. Angew. Chem., Int. Ed 2015, 54, 8665–8668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (13).Grison CM; Burslem GM; Miles JA; Pilsl LKA; Yeo DJ; Imani Z; Warriner SL; Webb ME; Wilson AJ Double quick, double click reversible peptide “stapling”. Chem. Sci 2017, 8, 5166–5171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (14).Jankovic B; Gulzar A; Zanobini C; Bozovic O; Wolf S; Stock G; Hamm P Photocontrolling protein–peptide interactions: From minimal perturbation to complete unbinding. J. Am. Chem. Soc 2019, 141, 10702–10710. [DOI] [PubMed] [Google Scholar]
- (15).Vurgun N; Nitz M Validation of l-tellurienylalanine as a phenylalanine isostere. ChemBioChem 2020, 1136–1139. [DOI] [PubMed] [Google Scholar]
- (16).Strong LE; Kink JA; Pensinger D; Mei B; Shahan M; Raines RT Efficacy of ribonuclease QBI-139 in combination with standard of care therapies. Cancer Res. 2012, 72 (Suppl. 1), 1838. [Google Scholar]
- (17).Strong LE; Kink JA; Mei B; Shahan MN; Raines RT First in human phase I clinical trial of QBI-139, a human ribonuclease variant, in solid tumors. J. Clin. Oncol 2012, 30 (15, Suppl.), TPS3113. [Google Scholar]
- (18).Lu L; Li J; Moussaoui M; Boix E Immune modulation by human secreted RNases at the extracellular space. Front. Immunol 2018, 9, 1012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (19).Eller CH; Raines RT Antimicrobial synergy of a ribonuclease and a peptide secreted by human cells. ACS Infect. Dis 2020, 6, 3083–3088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (20).Backer MV; Gaynutdinov TI; Gorshkova II; Crouch RJ; Hu T; Aloise R; Arab M; Przekop K; Backer JM Humanized docking system for assembly of targeting drug delivery complexes. J. Controlled Release 2003, 89, 499–511. [DOI] [PubMed] [Google Scholar]
- (21).Vitagliano L; Merlino A; Zagari A; Mazzarella L Reversible substrate-induced domain motions in ribonuclease A. Proteins 2002, 46, 97–104. [DOI] [PubMed] [Google Scholar]
- (22).Kim EE; Varadarajan R; Wyckoff HW; Richards FM Refinement of the crystal structure of ribonuclease S. Comparison with and between the various ribonuclease A structures. Biochemistry 1992, 31, 12304–12314. [DOI] [PubMed] [Google Scholar]
- (23).Pous J; Mallorquí-Fernández G; Peracaula R; Terzyan SS; Futami J; Tada H; Yamada H; Seno M; de Llorens R; Gomis-Rüth FX; Coll M Three-dimensional structure of human RNase 1∆N7 at 1.9 Å resolution. Acta Crystallogr. D Biol. Crystallogr 2001, 57, 498–505. [DOI] [PubMed] [Google Scholar]
- (24).Gupta V; Muyldermans S; Wyns L; Salunke DM The crystal structure of recombinant rat pancreatic RNase A. Proteins 1999, 35, 1–12. [DOI] [PubMed] [Google Scholar]
- (25).Kato I; Anfinsen CB On the stabilization of ribonuclease S-protein by ribonuclease S-peptide. J. Biol. Chem 1969, 244, 1004–1007. [PubMed] [Google Scholar]
- (26).Gaynutdinov TI; Myshkin E; Backer JM; Backer MV Chimeric ribonuclease as a source of human adapter protein for targeted drug delivery. Protein Eng. 2003, 16, 771–775. [DOI] [PubMed] [Google Scholar]
- (27).Watkins RW; Arnold U; Raines RT Ribonuclease S redux. Chem. Commun 2011, 47, 973–975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (28).Connelly PR; Varadarajan R; Sturtevant JM; Richards FM Thermodynamics of protein–peptide interactions in the ribonuclease S system studied by titration calorimetry. Biochemistry 1990, 29, 6108–6114. [DOI] [PubMed] [Google Scholar]
- (29).Catanzano F; Giancola C; Graziano G; Barone G Temperature-induced denaturation of ribonuclease S: A thermodynamic study. Biochemistry 1996, 35, 13378–13385. [DOI] [PubMed] [Google Scholar]
- (30).Klink TA; Raines RT Conformational stability is a determinant of ribonuclease A cytotoxicity. J. Biol. Chem 2000, 275, 17463–17467. [DOI] [PubMed] [Google Scholar]
- (31).Futami J; Tada H; Seno M; Ishikami S; Yamada H Stabilization of human RNase 1 by introduction of a disulfide bond between residues 4 and 118. J. Biochem 2000, 128, 245–250. [DOI] [PubMed] [Google Scholar]
- (32).Thompson JE; Raines RT Value of general acid–base catalysis to ribonuclease A. J. Am. Chem. Soc 1994, 116, 5467–5468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (33).Eller CH; Lomax JE; Raines RT Bovine brain ribonuclease is the functional homolog of human ribonuclease 1. J. Biol. Chem 2014, 289, 25996–26006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (34).Thomas SP; Kim E; Kim J-S; Raines RT Knockout of the ribonuclease inhibitor gene leaves human cells vulnerable to secretory ribonucleases. Biochemistry 2016, 55, 6359–6362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (35).Lomax JE; Eller CH; Raines RT Comparative functional analysis of ribonuclease 1 homologs: Molecular insights into evolving vertebrate physiology. Biochem. J 2017, 474, 2219–2233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (36).Parés X; Nogués MV; de Llorens R; Cuchillo CM Structure and function of ribonuclease A binding subsites. Essays Biochem. 1991, 26, 89–103. [PubMed] [Google Scholar]
- (37).Cuchillo CM; Nogués MV; Raines RT Bovine pancreatic ribonuclease: Fifty years of the first enzymatic reaction mechanism. Biochemistry 2011, 50, 7835–7841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (38).Sorrentino S; Naddeo M; Russo A; D’Alessio G Degradation of double-stranded RNA by human pancreatic ribonuclease: Crucial role of noncatalytic basic amino acid residues. Biochemistry 2003, 42, 10182–10190. [DOI] [PubMed] [Google Scholar]
- (39).Johnson RJ; McCoy JG; Bingman CA; Phillips GN Jr.; Raines RT Inhibition of human pancreatic ribonuclease by the human ribonuclease inhibitor protein. J. Mol. Biol 2007, 368, 434–449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (40).Schmidt B; Ho L; Hogg PJ Allosteric disulfide bonds. Biochemistry 2006, 45, 7429–7433. [DOI] [PubMed] [Google Scholar]
- (41).Green MR; Sambrook J How to win the battle with RNase. Cold Spring Harb. Protoc 2019, 2019, pdb.top10185710.1101/pdb.top101857. [DOI] [PubMed] [Google Scholar]
- (42).Smith BD; Soellner MB; Raines RT Potent inhibition of ribonuclease A by oligo(vinylsulfonic acid). J. Biol. Chem 2003, 278, 20934–20938. [DOI] [PubMed] [Google Scholar]
- (43).Kelemen BR; Klink TA; Behlke MA; Eubanks SR; Leland PA; Raines RT Hypersensitive substrate for ribonucleases. Nucleic Acids Res. 1999, 27, 3696–3701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (44).Niesen FH; Berglund H; Vedadi M The use of differential scanning fluorimetry to detect ligand interactions that promote protein stability. Nat. Protoc 2007, 2, 2212–2221. [DOI] [PubMed] [Google Scholar]
- (45).Ressler VT; Raines RT Consequences of the endogenous N-glycosylation of human ribonuclease 1. Biochemistry 2019, 58, 987–996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (46).To prolong the half-life of the RNase–S conjugate even further, we attempted to generate a thioether–cross-linked variant. Specifically, we dehydrated A5C S-peptide to form A5Dha S-peptide, which we linked to V118C S-protein by a conjugate addition reaction. After 24 h, we did observe the formation of some of the desired thioether-linked RNase–S (see: Supporting Information). Unfortunately, the requisite pH of this reaction (≥7.5) led to dimerization of V118C S-protein and compromised the yield of the desired product. We therefore proceeded with the highly efficient disulfide-cross-linking strategy, which proceeds in nearly quantitative yield within 1 h.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.