

Abstract
The essential human O-linked β-N-acetylglucosamine (O-GlcNAc) transferase (OGT) is the sole enzyme responsible for modifying thousands of intracellular proteins with the monosaccharide O-GlcNAc. This unique modification plays crucial roles in human health and disease, but the substrate recognition of OGT remains poorly understood. Intriguingly, the only human enzyme reported to remove this modification, O-GlcNAcase (OGA), is O-GlcNAc modified. Here, we exploited a GlcNAc electrophilic probe (GEP1A) to rapidly screen OGT mutants in a fluorescence assay that can discriminate between altered OGT-sugar and -protein substrate binding to help elucidate the binding mode of OGT toward OGA protein substrate. Since OGT tetratricopeptide repeat (TPR) domain plays a key role in OGT-OGA binding, we screened 30 OGT TPR mutants, which revealed 15 “ladder like” asparagine or aspartate residues spanning TPRs 3-7 and 10-13.5 that affect OGA O-GlcNAcylation. By applying a truncated OGA construct, we found that OGA’s N-terminal region or pseudo histone acetyltransferase domain is not required for its O-GlcNAcylation, suggesting OGT functionally interacts with OGA through its catalytic and/or stalk domains. This work represents the first effort to systemically investigate each OGT TPR and our findings will facilitate the development of new strategies to investigate the role of substrate-specific O-GlcNAcylation.
Keywords: O-GlcNAc transferase (OGT), O-GlcNAcase (OGA), substrate recognition
Graphical Abstract
1. Introduction
O-linked β-N-acetyl glucosamine modification (O-GlcNAcylation) is an essential, dynamic, and reversible posttranslational modification carried out by a single pair of opposing human enzymes, O-GlcNAc transferase (OGT) and O-GlcNAcase (OGA) [1–5]. OGT adds O-GlcNAc from sugar donor uridine diphosphate N-acetylglucosamine (UDP-GlcNAc) to serine or threonine residues of protein substrates, while OGA removes it [6]. Currently, over a thousand intracellular proteins are reported to be O-GlcNAcylated [7–13], many of which play intricate roles in various biological processes [5,14–16], including transcription [17,18], translation [19–22], and signal transduction [15,23,24]. Aberrant O-GlcNAcylation is closely linked to diseases, such as cancer [25–28], diabetes [29–31]. X-linked intellectual disability [32–34], and Alzheimer’s disease [35–37]. To better understand O-GlcNAcylation and the role of OGT, a number of chemical tools [38–44], including OGT inhibitors, such as OSMI [45,46], Ac45SGlcNAc [47], and ESI [48] that target the catalytic pocket of OGT, have been developed. While providing many useful insights, these inhibitors reduce global O-GlcNAc level and struggle to deconvolute the role of O-GlcNAcylation on individual proteins [49]. Thus, elucidating OGT protein substrate recognition is critical for expanding our understanding of O-GlcNAc (dys)regulation and promoting the development of new strategies to overcome these limitations.
Human OGT is a multidomain protein containing an N-terminal tetratricopeptide repeat (TPR) domain, and a C-terminal catalytic domain that is split by an intervening domain (Fig. S1a) [50]. The TPR domain, which is not found in any other glycosyltransferases, contains 13.5 TPRs that form an extended α-helical tunnel leading directly to OGT active site and facilitates protein substrate recognition [51]. As the full-length OGT is not amenable for structural characterization, the substrate binding information of OGT was mainly derived from a truncated construct OGT4.5, which lacks N-terminal TPRs 1-9 and can O-GlcNAcylate peptide substrates but shows minimal activity toward protein substrates [39,52]. The crystal structures of OGT4.5 complexed with peptide substrates demonstrated that a series of “ladder like” asparagine and aspartate residues line the concave surface of TPRs 10-13.5 and respectively form polar interactions with the backbone or sidechains of different peptide substrates [53–56]. Subsequent microarray and proteomic studies with full-length OGT revealed that members of the asparagine ladder on TPRs 10-13.5 are essential for global O-GlcNAcylation while residues of the aspartate ladder on TPRs 12-13 help OGT discriminate against certain protein substrates [57,58]. Aside from these few residues on the C-terminal region of the TPR domain, which were mostly studied as double [58] or quintuple [57] mutants, the functional role of OGT residues along the entire 13.5 TPRs toward protein substrates remains largely unexplored due to a number of challenges. One is the lack of structural information of full-length OGT with a protein substrate, which has stymied efforts to rapidly identify OGT residues that modulate protein binding within TPRs 1-9. Furthermore, previous reports revealed that OGT follows an ordered bi-bi mechanism, where OGT first binds UDP-GlcNAc and then the protein substrate prior to catalyzing sugar transfer [56]. OGT binding of UDP-GlcNAc can affect subsequent protein substrate binding, thus traditional binding assays, such as isothermal titration calorimetry (ITC) [59] and surface plasmon resonance (SPR) [60], usually cannot efficiently distinguish whether an OGT residue is functionally important for sugar or protein substrate binding, posing a significant challenge for studying OGT protein substrate recognition. Additionally, the TPR domain contains around 500 residues [51], some of whose mutation render OGT inactive [39,52], presenting another hurdle for applying traditional methods to characterize OGT residues along the 13.5 TPRs.
To help address these issues, our lab recently developed GlcNAc Electrophilic Probes (GEPs) [39]. GEP1A (Fig. S2a), an analogue of UDP-GlcNAc in this class, can be transferred to protein substrates by OGT for glycosylation, while reacting specifically with a unique cysteine (C917) in the OGT active site when OGT-protein binding and/or sugar transfer is impaired (Fig. S2b). Leveraging the azide handle on GEP1A for click-chemistry conjugation to an alkyne fluorophore, this assay offers an innovative platform to quickly screen OGT mutants, even those with no activity, and characterize the potential role of corresponding residues in sugar binding or protein substrate binding/sugar transfer, based on the unique labeling patterns on OGT and its protein substrate (Fig. S2c) [39].
OGA is the sole enzyme responsible for removing O-GlcNAcylation and interestingly, is also a protein substrate of OGT [61–63]. OGA contains an N-terminal catalytic domain, a stalk domain, and a C-terminal pseudo histone acetyltransferase (HAT) domain [64]. The only reported O-GlcNAcylation site of OGA is located on a flexible linker region in the stalk domain (Fig. S1b) [62]. OGA interacts with OGT to form the O-GlcNAczyme, which is expected to be tightly regulated to prevent a futile O-GlcNAc cycle [65]. The interaction of OGT and OGA has profound implications in dynamic O-GlcNAc regulation, not only for these two opposing enzymes themselves, but also for numerous protein substrates involved in various biological processes. However, how OGT interacts with OGA remains largely unknown.
Here we applied the GEP1A assay to investigate the substrate recognition of OGT toward OGA protein substrate, with a focus on characterizing the functional role of inward and outward facing residues spanning the length of the TPR domain. Our study demonstrates the application of GEP1A assay for rapidly screening and identifying functionally important OGT residues in protein substrate recognition, a challenging task for many traditional methods. Furthermore, we identified 15 “ladder like” residues that each modulate OGT-OGA interaction in one of the three unique categories, laying a foundation for designing inhibitors to disrupt OGT-OGA interaction, understanding O-GlcNAc (dys)regulation, and developing new strategies to investigate the precise role of O-GlcNAcylation on different substrates.
2. Methods
2.1. GEP1A synthesis
GEP1A synthesis and characterization were performed as previously described [39].
2.2. OGA and OGT mutant cloning and protein purification
OGA_D175N and WT OGT were used as DNA templates with primers listed in Table S1 to generate truncated OGA (OGA(60-704)D175N, based on the numbering of the full-length human protein) and OGT mutants, respectively. OGA(60-704)D175N was subcloned into a pET-SUMO vector, while OGT mutants remained in a pET24b vector. Plasmid DNA purified from transformed Escherichia coli strain XL10-Gold was confirmed using Sanger sequencing before being transformed into E. coli BL21(DE3) competent cells for protein expression. Cells were grown at 37 °C in Luria–Bertani (LB) medium until reaching an OD600 of 0.6, where the culture was induced with 0.3 mM isopropyl β-D-1-thiogalactopyranoside (IPTG) at 16 °C for 16 h. Cells were pelleted, resuspended in TBS buffer (20 mM Tris pH 8.0, 150 mM NaCl, for OGT) or HBS buffer (20 mM HEPES pH 7.0, 150 mM NaCl, for OGA) supplemented with 1 mM phenylmethylsulfonyl fluoride (PMSF), and then lysed using an ultra-high-pressure cell disrupter (Emulsiflex-C5, Canada) at 4 °C. The supernatant was incubated for 2 h at 4 °C on an affinity chromatography Ni-NTA column (Qiagen), and then eluted with either TBS buffer with 250 mM imidazole for OGT or HBS buffer with 250 mM imidazole for OGA. After adding Tris(3-hydroxypropyl)phosphine (THP) for a final concentration of 0.5 mM, OGA(60-704)D175N was digested by Sumo protease to cleave the N-terminal 6×His–SUMO tag. Proteins were further purified by size-exclusion chromatography (Superdex 200 increase 10/300, GE Healthcare) in TBS buffer with 0.5 mM THP for OGT or HBS buffer with 0.5 mM THP for OGA. The retention time of OGT mutants was closely monitored to verify that no significant change in conformation or protein folding compared to WT had occurred (Fig. S3). Purified protein was then concentrated, quantified by Bradford assay and SDS-PAGE, flash frozen, and stored at −80 °C. OGA(60-704)D175N was stored at 4 °C and used directly for the assays.
2.3. GEP1A fluorescence assay
To evaluate the role of selected OGT residues toward OGA protein substrate, 1.2 μM OGT WT or mutant was incubated for 30 min at 37 °C with 15 μM OGA_D175N or OGA(60-704)D175N and 25 μM GEP1A in TBS buffer with 0.5 mM THP for a final volume of 5 μL. Click chemistry reagents were consecutively mixed and immediately added to each sample with 1 mM CuSO4, 0.1 mM Tris[(1-benzyl-1H-1,2,3-triazol-4-yl)methyl]amine (TBTA), 50 μM fluor 488-alkyne, and 1 mM sodium ascorbate as the final concentrations. The click chemistry reaction was performed at RT for 30 min in the dark and then quenched using SDS loading buffer and boiled for 5 min at 95 °C. Samples were separated on an SDS-PAGE gel and subjected to fluorescence detection and subsequent imaging for relative quantitation after Coomassie Blue staining using Azure Biosystems c600 imager. Band intensity was relatively quantified using ImageJ (v1.8.0_112). OGA relative background labeling was calculated from sample without OGT (negative control) and subtracted from each sample before the GEP1A-modified proteins were normalized to the WT OGT sample. As we observed minimal OGT background labeling, OGT relative labeling was directly calculated by normalization to the WT OGT sample. The experiment was performed in triplicate.
2.4. Radiolabeled kinetic assay for OGT mutants
The radiolabeled kinetic assay was performed similarly as described in ref. [39]. In brief, purified OGT was incubated with 100 μM UDP-[3H]GlcNAc (specific activity 92 Ci/mol, PerkinElmer NET434250UC) and indicated concentrations of purified OGA protein (full-length or truncated) in TBS buffer at 37 °C for 30 min. When full-length OGA was used as the protein substrate, OGT WT, N84A, D152A, N155A, N186A, N321A/N322A, N390A, and D420A were used at 40 nM, while N424A and N458A were used at 80 nM. When truncated OGA was used as the protein substrate, OGT WT, D152A, and N155A were used at 15 nM, while N424A and N458A were used at 30 nM. The reactions were stopped by applying each onto the nitrocellulose membrane (Bio-Rad 1620251), air-dried, and washed 5 min in PBS buffer for four times. The O-GlcNAcylation level of OGA protein in each reaction represented by the radioactivity of each nitrocellulose membrane was counted by a Beckman LS6000TA Scintillation Counter. A reaction without OGT was counted as background. Another reaction without wash was counted as the total 100 μM of UDP-GlcNAc input to calculate the O-GlcNAcylation level of OGA in each reaction. The experiment was performed in triplicate, and the data were analyzed by GraphPad PRISM v5 (GraphPad Software).
2.5. Statistical analysis
All data shown are mean values with the error bars representing ± s.d.. Statistical significance was determined using a Student’s t-test, with significance indicated at *P < 0.05, **P < 0.01.
3. Results
3.1. Selection of OGT mutants from entire TPR region for probing OGT protein substrate recognition
In this study, we selected 30 TPR residues (Fig. 1 and Table S2) to evaluate their roles in regulating OGT protein substrate recognition using mutagenesis and GEP1A fluorescence assay. We were inspired to investigate the full Asn- and Asp-ladder, which spans nearly the entire 13.5 TPR region [51], by recent reports demonstrating that a few Asn- and Asp-ladder residues positioned relatively near (2-5 nm) the OGT active site affect its substrate recognition [57,58]. To similarly investigate the entire TPR and enhance our coverage of TPRs 1-3 and 7 that either lack apparent Asn- and Asp-ladder residues or proper characterization, we selected OGT residues E16, H19, S53, E91, and N223 by applying the following parameters: the residues 1) reside within the inner lumen of the TPR and roughly align with the asparagine or aspartate ladder, 2) are unlikely to influence the overall structure of the TPR domain, which was determined by measuring potential bond proximities to nearby residues using PDB model 3PE3 [52] and 1W3B [51], and 3) can potentially form protein-protein interactions. Meanwhile, we applied similar criteria and selected K73, K206, R311, E345, and Q406 that are relatively evenly distributed throughout the TPR domain to determine whether outside facing TPR residues play a role in protein substrate recognition (Fig. 1 and Table S2).
Fig. 1.
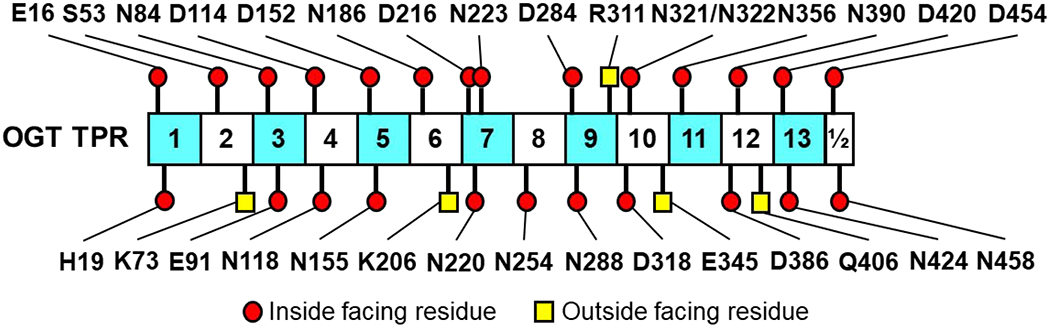
Distribution schematic of the 30 OGT TPR residues selected for this study. Individual TPRs are labeled with the corresponding TPR repeat numbers and alternately colored cyan or white. Residues selected for mutation are labeled and depicted as red circles or yellow squares based on their orientation within the TPR domain in the PDB 3PE3 [52] and 1W3B [51].
3.2. Asparagine and aspartate ladder residues across TPRs 3-13.5 modulate OGA glycosylation
OGA hydrolyzes O-GlcNAcylation and is also O-GlcNAc modified by OGT at serine residue 405 [62]. However, how these two critical O-GlcNAc cycling enzymes interact with each other is largely unknown, despite previous studies showing that the TPR domain is essential for OGA O-GlcNAcylation [39]. To identify functionally important OGT residues for protein substrate recognition, we applied the GEP1A assay to screen the 30 selected OGT mutants against OGA_D175N protein substrate, a catalytically deficient mutant to avoid potential removal of GEP1A (a GlcNAc analogue) but can be similarly O-GlcNAcylated as wild-type (WT) OGA. The functional role of OGT residues was evaluated using the predicted readout of the GEP1A assay (Fig. S2c) with over 20% change relative to WT OGT considered as significant, since we have demonstrated the assay fits within a 17% deviation range [39]. We incubated each OGT mutant with OGA_D175N and GEP1A. Following click chemistry conjugation with an alkyne-fluorophore and in-gel fluorescence detection, we found that residues in the asparagine ladder, N321A/N322A, N356A, N424A, and N458A mutants from TPR 10, 11, 13, and 13.5, respectively, all showed a significant increase in relative OGT labeling and decrease in relative OGA labeling (Fig. 2a–b, Fig. 3), demonstrating that these residues are important for binding OGA and/or sugar transfer. In contrast, N390A from TPR 12 showed minimal changes in labeling of both proteins (Fig. 2a, Fig. 3), suggesting that not all C-terminal TPR asparagines within the Asn-ladder are important for facilitating the glycosylation of a given substrate. To determine whether the more N-terminal located Asn-ladder residues play a role in recognizing OGA, we tested Asn-ladder mutants spanning TPRs 3-9. Surprisingly, N254 and N288 from TPR 8-9 had a negligible impact on modifying OGA, while N84, N118, N155, N186, and N220 from TPR 3-7 were critical for OGT-OGA binding and/or sugar transfer (Fig. 2a–b, Fig. 3). Thus, these asparagines spanning the inner surface of nearly the entire TPR domain are potentially essential for OGT protein substrate recognition. Furthermore, as these 10 functionally important asparagines all enhance OGA glycosylation, the Asn-ladder appears to form more generic interactions with protein substrates, rather than playing a fine-tuning role.
Fig. 2.
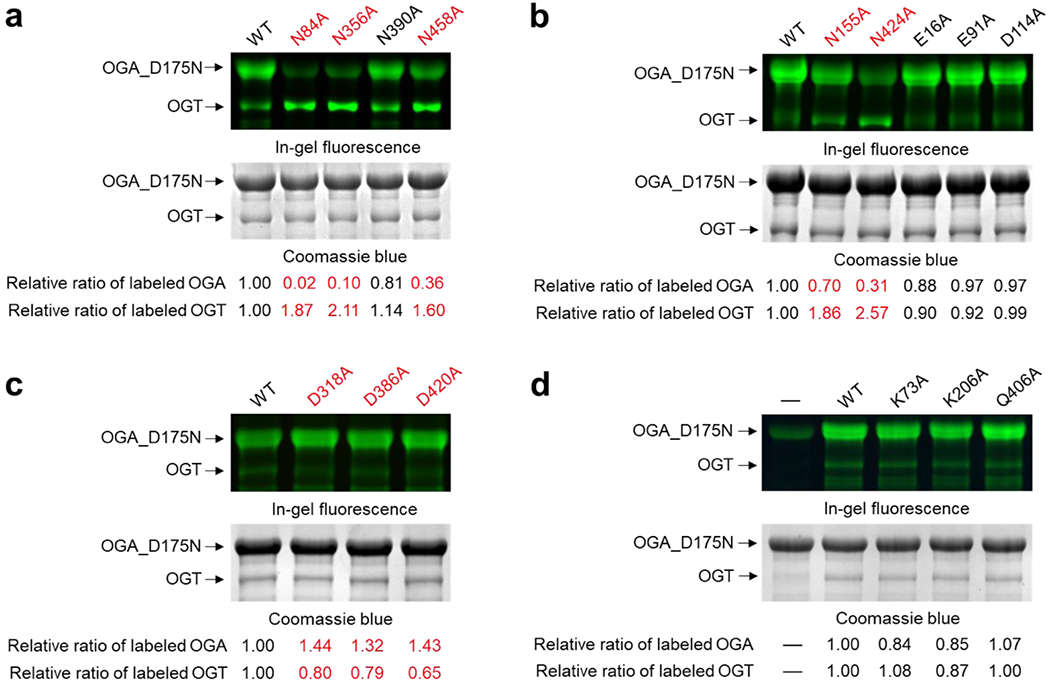
Representative gels of the GEP1A fluorescence assay for full-length OGA_D175N with OGT mutants: (a-c) residues facing inside of TPR domain, and (d) residues facing outside of TPR domain. Red color highlighted the OGT mutants showing significant changes in the relative labeling of OGT or OGA (Δ ≥ 20%). For a-d, top panel is the fluorescent gel detection of reacted samples underwent click chemistry with an alkyne fluorophore, while the bottom panel is the same gel after Coomassie blue staining to detect the protein loading amounts. The relative ratio of labeled OGA and OGT was calculated by dividing the fluorescence intensity by the corresponding Coomassie blue staining and normalizing their values to the WT control. WT, wild-type OGT. Experiments were repeated in triplicate and the results were summarized in Fig. 3.
Fig. 3.
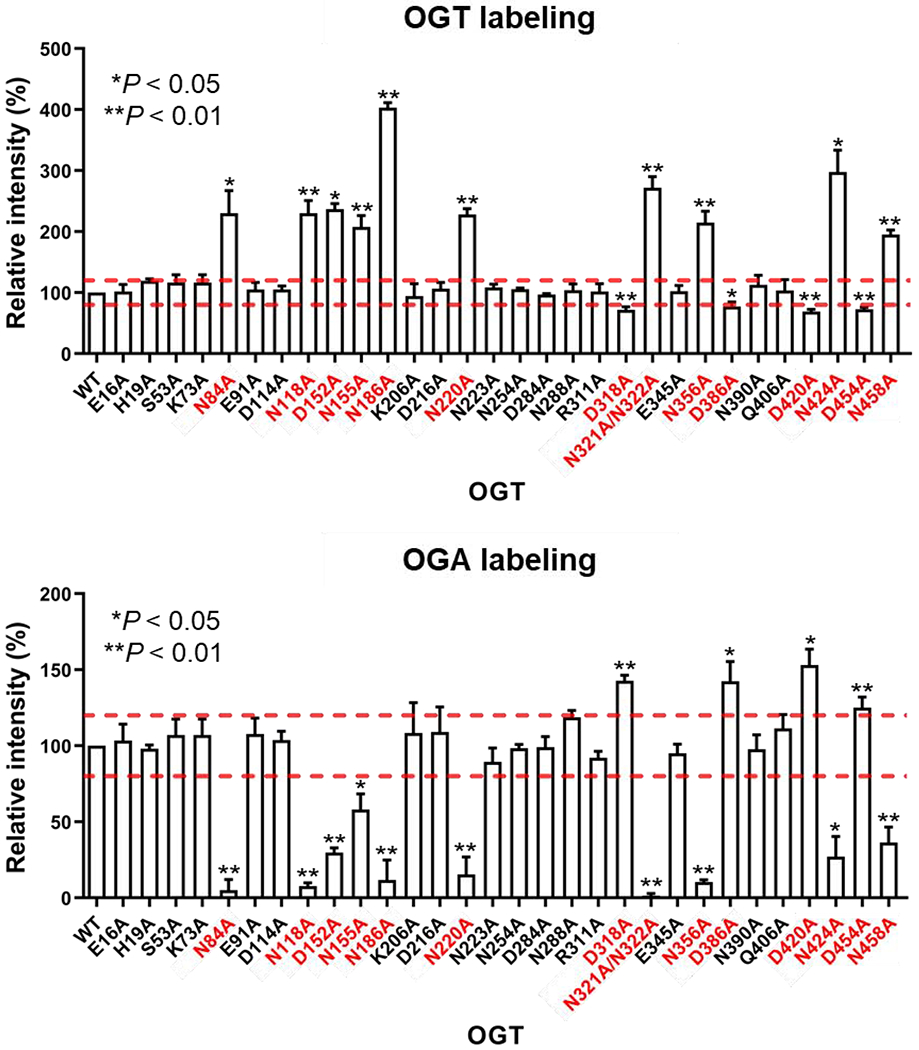
Quantification of the relative intensities of GEP1A modified OGT mutants (top) and full-length OGA_D175N (bottom) after normalization to the wild-type (WT) OGT control. Mutant patterns were deemed significant and highlighted in red if changes relative to WT OGT control reaction were above or below the dashed lines (Δ ≥ 20%) with *P < 0.05 and **P < 0.01. Statistical analysis was performed by a Student’s two-tailed t-test (n = 3 independent experiments). Error bars represent ± s.d.. Representative gels are shown in Fig. 2.
Inspired by the recent report on OGT D386 and D420 residues that help discriminate protein substrates [58], we further screened all eight aspartate ladder residues using GEP1A fluorescence assay to evaluate their functional role toward OGA-D175N. In stark contrast with the above identified asparagine mutants, we found that D318A, D386A, D420A, and D454A in TPR 10, 12, 13, and 13.5 all demonstrated significantly increased OGA labeling and decreased OGT labeling compared to WT (Fig. 2c, Fig. 3), suggesting that these aspartate residues impede OGA glycosylation. Interestingly, D318, D420, and D454 are each adjacent to a neighboring asparagine (N321/N322, N424, and N458) that facilitates OGA glycosylation. We did not find any other Asp-ladder mutants in the TPR deviated significantly from WT OGT, except D152A, which showed impaired protein binding and/or sugar transfer, akin to the above described asparagine mutants. D152 is also situated by a neighboring asparagine (N155) that similarly affected OGA modification. Taken together, these data provide strong evidence supporting that the aspartate ladder plays an important role in fine-tuning OGT substrate specificity, as we identified aspartates that either hinder or enhance O-GlcNAcylation of OGA.
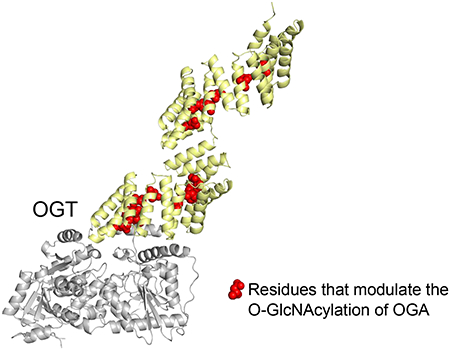
After screening Asn- and Asp-ladder residues and assessing their importance, we were curious whether residues facing outside the TPR domain or within the first two TPRs would also play a functional role in OGA glycosylation. We similarly tested several residues throughout the TPR convex surface (K73, K206, R311, E345, and K406), but did not find any outside facing residues that significantly altered OGA O-GlcNAcylation (Fig. 2d, Fig. 3). Furthermore, inward facing residues E16, H19, S53, E91, and N223 from TPRs 1-3 and 7 were found to also be dispensable for modifying OGA (Fig. 2b, Fig. 3). Through these screens a total of 15 functionally important residues, all belonging to the Asn- or Asp-ladder, were identified on the concave surface of each 13.5 TPRs except TPR 1, 2, 8, and 9, forming an OGA interaction groove and supporting the model that OGT interacts with its protein substrate through the entire TPR lumen (Fig. 4, Video S1). Furthermore, of the 30 mutants tested, only members of the asparagine and aspartate ladder impacted OGA glycosylation, underscoring their importance in OGT substrate recognition or catalysis.
Fig. 4.
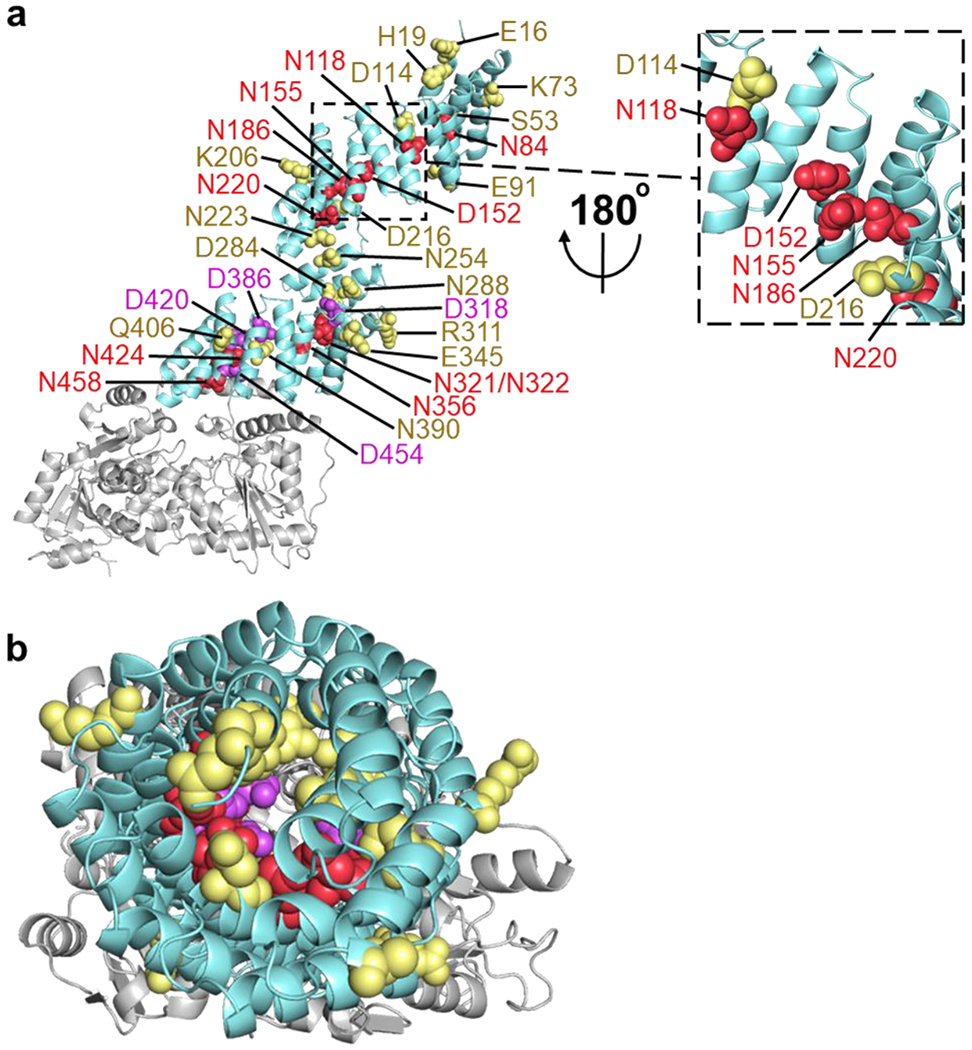
Inner asparagine and aspartate residues extending throughout OGT TPR lumen regulate OGA glycosylation. (a) Full-length human OGT structure was generated by overlaying the crystal structures of OGT4.5 (PDB code 3PE3) [52] and the first 11.5 TPRs (PDB code 1W3B) [51]. Residues that facilitated protein binding and/or sugar transfer for OGA are shown as red spheres, while residues that hindered protein binding and/or sugar transfer for OGA are colored purple. Functionally disposable residues for the glycosylation of full-length OGA are depicted as yellow spheres. (b) Top-down (N-terminal to C-terminal) view of the same OGT model and coloring as a.
To validate the GEP1A assay results and further characterize members of the Asn- and Asp-ladder, we selected a number of OGT mutants for radiolabeled kinetic analysis whose relative activity toward OGA protein substrate was enhanced (D420A), unchanged (N390A), impaired (N84A, D152A, N155A, N186A, N424A, and N458A), or undetected (N321A/N322A). In strong agreement with the GEP1A assay results, each mutant that showed impaired protein binding and/or sugar transfer had a two- to 10-fold reduction in catalytic efficiency (kcat/Km) relative to the wild-type OGT (Fig. 5), while mutants with enhanced or unchanged protein binding and/or sugar transfer had either a moderate increase or an insignificant change, correspondingly, to their kcat/Km (Fig. S4). Though previously unstudied, we hypothesized that N84, D152, and N155 were important for OGA protein binding rather than sugar transfer since these residues are quite far (~8 nm) from the OGT active site according to the structural model of the full-length OGT. Supporting this, mutation of N84, D152, and N155 to alanine showed a two- to five-fold increase in Michaelis constant (Km), while the change in the turnover number (kcat) was negligible. In further agreement with the GEP1A assay results, counts of OGT N321A/N322A obtained by kinetic experiments were too low to determine kinetic parameters, supporting its classification as a dead mutant for OGA O-GlcNAcylation (Fig. S4). Surprisingly, OGT N186A, N424A, and N458A each had a notable increase in Km, but also a significant decrease in kcat, distinguishing them from the other OGT mutants. Hence, while OGT N84A, D152A, N155A, N186A, D420A, N424A, and N458A all affect protein binding to a varying degree, the last four mutants also affect the rate that OGA is O-GlcNAcylated.
Fig. 5.
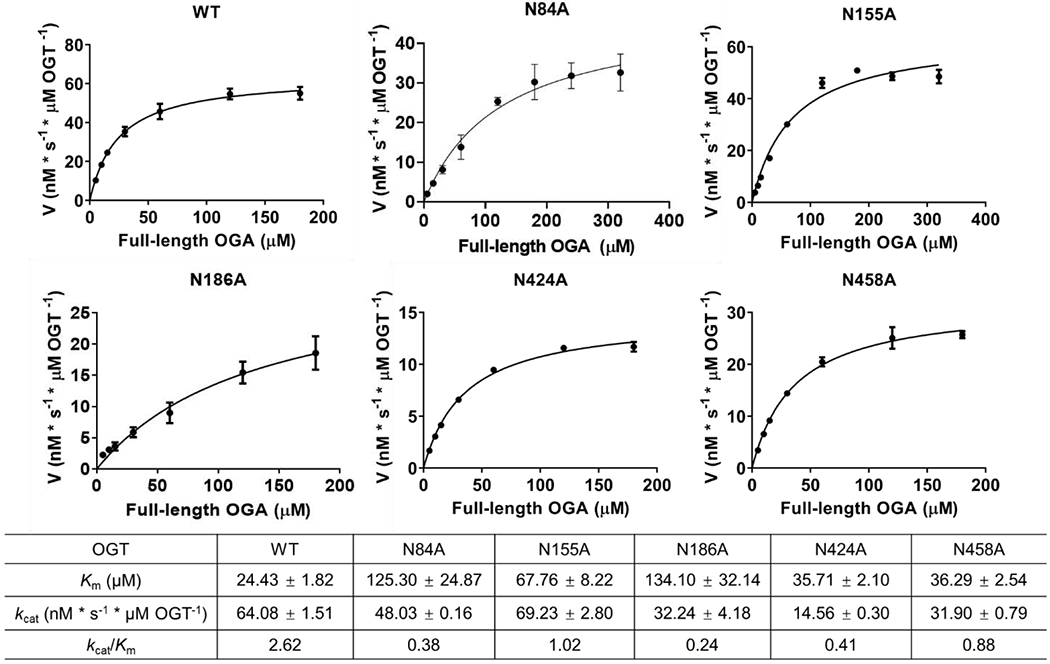
Kinetic studies validating the results of the GEP1A assay for OGT mutants toward full-length OGA_D175N protein substrate using radiolabeled UDP-[3H]GlcNAc as the sugar donor. Error bar represents ± s.d. (n = 3 independent experiments).
3.3. Key asparagine and aspartate ladder residues of OGT do not functionally interact with the N-terminal region or HAT domain of OGA
OGA S405 residue is in a disordered loop of the stalk domain and is the only reported O-GlcNAcylation site of OGA [62,66]. However, it remained unclear if other distal regions of OGA are important for interacting with OGT and its glycosylation. Since the structure and function of the OGA N-terminal region (residues 1-59) and pseudo histone acetyltransferase (HAT) domain (residues 705-916) are largely unknown [50,67], we were curious whether these two regions functionally interact with the above identified asparagine or aspartate residues important for full-length OGA_D175N glycosylation. Thus, we generated OGA(60-704)D175N, which lacks the N-terminal region and the HAT domain to test against the 10 identified OGT mutants with impaired protein binding and/or sugar transfer. As OGA(60-704)D175N is a new substrate for the GEP1A assay, we first validated the principle of this assay towards OGA(60-704)D175N using OGT K842A and OGT4.5, which have known deficiencies in sugar or protein substrate binding, respectively. OGT C917S, which lacks the cysteine covalently modified by GEP1A, was used to demonstrate that OGT background labeling was minimal (Fig. S2b, Fig. S5). Next, we applied the GEP1A assay to screen OGA(60-704)D175N against the 10 OGT mutants that impaired protein binding and/or sugar transfer for full-length OGA-D175N: N84A, N118A, D152A, N155A, N186A, N220A, N321A/N322A, N356A, N424A, and N458A. Interestingly, the general labeling patterns for all of these OGT mutants were similar to their full-length OGA counterparts (Fig. S6, Fig. 6), suggesting that the identified OGT TPR residues interact with OGA protein mainly through OGA residues 60-704 rather than the N-terminal region or HAT domain.
Fig. 6.
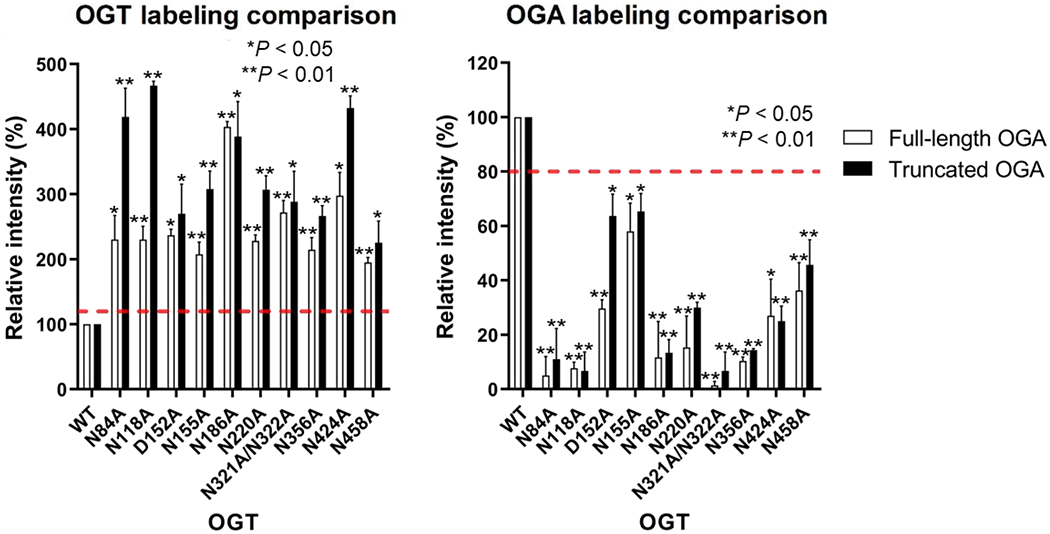
Labeling comparison between full-length OGA_D175N and truncated OGA(60-704)D175N with OGT mutants that show impaired protein binding and/or sugar transfer. Relative quantitation of fluorescence intensities was derived from gel scans for full-length and truncated OGA after normalizing the protein amount to the OGT wild-type (WT) sample using the Coomassie blue staining (Fig. 2 and Fig. S6). OGA background labeling was calculated using a no OGT negative control and was subtracted from the other samples. Statistical analysis was performed by a Student’s two-tailed t-test (n = 3 independent experiments). Error bars represent ± s.d.; red dashed lines represent the minimum change in GEP1A labeling for a result to be significantly different from WT OGT.
We further validated the GEP1A assay results by radiolabeled kinetic experiments with OGT D152A, N155A, N424A, and N458A using OGA(60-704)D175N as the protein substrate (Fig. S7). Intriguingly, the catalytic efficiency (kcat/Km) of each OGT mutant tested was at least two-fold higher with truncated OGA than full-length OGA, suggesting that truncated OGA is a better substrate for OGT (Fig. 5 and Fig. S7). Similarly to the radiolabeled kinetic results with full-length OGA, the relative Km significantly increased for OGT D152A and N155A, though modestly for N424A and N458A. Furthermore, the kcat drastically decreased for OGT N424A and N458A, but the kcat of OGT D152A negligibly differed from the WT control. While we observed a ~50% increase in kcat for OGT N155A with truncated but not full-length OGA (Fig. 5 and Fig. S7), this may be related to a faster release of glycosylated OGA(60-704)D175N, which is in line with the more drastic increase in the Km of OGT N155A with truncated than full-length OGA. Taken together, these findings support that the identified OGT TPR residues regulate interactions with OGA protein substrate through the catalytic and/or stalk domain rather than the N-terminal or HAT domain.
4. Discussion
To date, how OGT recognizes various protein substrates remains largely unknown, which has hindered the understanding of O-GlcNAc dynamic regulation and its study in disease. Using OGA as a model protein substrate, we report here the first investigation that spans the full-length OGT TPR domain, probing each of the 13.5 TPRs to screen a total of 30 residues including the full Asn- and Asp-ladder along with additional residues on the concave surface of TPRs 1-3 and 7 and the convex surface of TPRs 2, 6, 9, 10, and 12. Applying the GEP1A fluorescence assay toward full-length OGA protein substrate, we identified 15 functionally important residues, seven of which have only been investigated here, all belonging to the Asn- or Asp-ladder that span TPRs 3-7 and 10-13.5 and modulate OGT-protein binding and/or sugar transfer (Fig. 2 and Fig. 3).
Notably, 10 of the 15 functionally important residues are asparagines (N84, N118, N155, N186, N220, N321, N322, N356, N424, and N458) located on TPRs 3-7, 10, 11, 13, and 13.5 that encompass 77% of the full OGT Asn-ladder and facilitate OGT-OGA binding and/or sugar transfer. In contrast, the remaining three residues of the Asn-ladder (N254, N288, and N390) are dispensable for OGA glycosylation (Fig. 2a and Fig. 3). By attaining the first kinetic parameters for OGT Asn-ladder mutants N84A, N155A, N186A, N424A, and N458A with protein substrate (Fig. 5), we discovered that N186, N424, and N458 residues significantly affect (≥ 50% change) the rate that OGA is O-GlcNAcylated (kcat). As sugar binding was not altered for any Asn-ladder mutant, the drastic decrease in kcat for OGT N186A, N424A, and N458A is likely caused by impaired protein binding or sugar transfer. Since N424 and N458 residues are relatively close (2 nm) to the active site in the structure model [52], we expect that they affect sugar transfer, while N84, N155, and N186 residues, which are further (> 8 nm) from the active site and showed a more dramatic Km change, are expected to affect protein binding.
Intriguingly, most members of the Asn-ladder are juxtaposed with a “ladder like” aspartate that typically plays a different role in modulating OGA O-GlcNAcylation. By screening all eight members of the Asp-ladder, of which only D386, D420, and D454 were previously investigated [58], we identified three unique categories of aspartates that either: 1) facilitate OGT-OGA binding (D152), 2) are dispensable in OGA glycosylation (D114, D216, and D284), or 3) hinder OGA modification (D318, D386, D420, and D454) (Fig. 2c and Fig. 3). Potentially explaining its distinct role, D152 is the only Asp-ladder residue that is not located on the edge of the TPR concave surface (Fig. 4a). Instead, we expect that D152 interacts with OGA similarly to important Asn-ladder residues since D152 is positioned directly in between N118 and N186, and the sequence comparison showed that it can be either an aspartate (45.9% conservation) or asparagine (48.1% conservation) in OGT homolog proteins [58]. Besides the previously reported D386 and D420 that hinder O-GlcNAcylation on a population of proteins in the proteome, we further discovered that D318 and D454 also hamper OGA glycosylation. The broad implications of these residues and even the dispensable Asp-ladder residues (D114, D216, and D284) are worth further investigation.
From this and previous studies, the important roles of “ladder like” asparagines and aspartates in modulating OGT interactions with protein substrates have become evident. We were more intrigued to learn that this Asn- and Asp-ladder substrate recognition motif is poorly conserved across helix-turn-helix (α-solenoid) containing proteins [68,69]. The initial analysis of OGT TPR structure drew comparison to importin α- and β-catenin [51,70], whose Armadillo (ARM)-repeats have an Asn-ladder centrally positioned on their concave surfaces but lack an Asp-ladder [71,72]. Proteins BBS4 and BBS8, which have similar TPR domains as OGT (RMSD: 3.696 and 3.774, respectively), contain substantially fewer “ladder like” asparagines and negatively charged residues on their concave surface [73,74]. In another case, the TPR domain of G-protein-signaling modulator 2 protein (LGN) contains an Asn- and Asp-ladder positioned similarly to OGT ladders, where co-crystal structures of LGN with four different peptide substrates showed contacts between the Asn-ladder and peptide [75–78] reminiscent of OGT-peptide complexes [53–56]. However, unlike in OGT, the Asp-ladder of LGN is too far (≥ 5 Å) from bound peptides to play a role in substrate discrimination. Thus, OGT could be a unique member in α-solenoid protein family to apply both Asn- and Asp-ladders in substrate recognition.
By further testing OGT residues essential for recognizing full-length OGA against a truncated OGA protein, we revealed that these “ladder like” asparagine and aspartate residues functionally interact with the stalk and/or catalytic domain of OGA, but not with the N-terminal region or HAT domain (Fig. 6 and Fig. S7). As the identified TPR residues only spanned the inside of TPRs 3-13.5, OGT may predominately rely on TPR residues on the concave rather than convex surface to recognize OGA protein (Fig. 5a). Supporting this substrate-binding mode, the identified functionally important OGT residues form a rotating path spanning nearly the entire TPR lumen, revealing an apparent binding tunnel leading directly to the active site (Fig. 5b and Video S1).
In summary, we applied the GEP1A fluorescence assay to quickly identify 15 TPR residues that impact OGT-OGA binding and/or sugar transfer, and further characterized eight of these residues by radiolabeled kinetic assay. Our results showed that OGT functionally binds OGA 60-704 region, rather than the HAT domain or N-terminal region, through the Asn- and Asp-ladder residues located even far away from OGT active site. This explains the essential role of the elongated TPR domain for OGT modifying protein substrates. To our knowledge, this is the first systematic investigation on OGT residues spanning the entire TPR domain, where we discovered four categories of residues, three of which uniquely modulate OGT-OGA protein interactions. These findings will significantly advance the understanding of OGT-protein substrate recognition, aid in the design of inhibitors to disrupt OGT-OGA interaction, and lay a foundation for developing novel strategies to investigate the role of O-GlcNAcylation on particular protein substrates of interest.
Supplementary Material
Highlights.
GlcNAc Electrophilic Probe in-gel fluorescence assay was applied to screen 30 OGT mutants against OGA protein substrate
D152 and ten Asn-ladder residues spanning OGT TPRs 3-13.5 facilitate O-GlcNAcylation of OGA
Four Asp-ladder residues closer to the OGT active site hamper OGT-OGA interaction
OGA HAT domain and N-terminal region are dispensable for its O-GlcNAcylation
Acknowledgement
We would like to acknowledge funding support from NIH R01 GM121718 and R01 GM126300.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Declaration of competing interest
The authors have no conflict of interest to declare.
References
- [1].Torres CR, Hart GW, Topography and polypeptide distribution of terminal N-acetylglucosamine residues on the surfaces of intact lymphocytes. Evidence for O-linked GlcNAc., J. Biol. Chem 259 (1984) 3308–3317. [PubMed] [Google Scholar]
- [2].Dong DL, Hart GW, Purification and characterization of an O-GlcNAc selective N-acetyl-beta-D-glucosaminidase from rat spleen cytosol, J. Biol. Chem 269 (1994) 19321–19330. [PubMed] [Google Scholar]
- [3].Haltiwanger RS, Blomberg MA, Hart GW, Glycosylation of nuclear and cytoplasmic proteins. Purification and characterization of a uridine diphospho-N-acetylglucosamine:polypeptide beta-N-acetylglucosaminyltransferase, J. Biol. Chem 267 (1992)9005–9013. [PubMed] [Google Scholar]
- [4].Gao Y, Wells L, Comer FI, Parker GJ, Hart GW, Dynamic O-glycosylation of nuclear and cytosolic proteins: cloning and characterization of a neutral, cytosolic beta-N-acetylglucosaminidase from human brain, J. Biol. Chem 276 (2001) 9838–9845. 10.1074/jbc.M010420200. [DOI] [PubMed] [Google Scholar]
- [5].Bond MR, Hanover JA, O-GlcNAc cycling: a link between metabolism and chronic disease, Annu. Rev. Nutr 33 (2013) 205–229. 10.1146/annurev-nutr-071812-161240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Vocadlo DJ, O-GlcNAc processing enzymes: catalytic mechanisms, substrate specificity, and enzyme regulation, Curr. Opin. Chem. Biol 16 (2012) 488–497. 10.1016/j.cbpa.2012.10.021. [DOI] [PubMed] [Google Scholar]
- [7].Holt GD, Hart GW, The subcellular distribution of terminal N-acetylglucosamine moieties. Localization of a novel protein-saccharide linkage, O-linked GlcNAc, J. Biol. Chem 261 (1986) 8049–8057. [PubMed] [Google Scholar]
- [8].Whelan SA, Hart GW, Identification of O-GlcNAc sites on proteins, Methods Enzymol. 415 (2006) 113–133. 10.1016/S0076-6879(06)15008-9. [DOI] [PubMed] [Google Scholar]
- [9].Lee A, Miller D, Henry R, Paruchuri VDP, O’Meally RN, Boronina T, Cole RN, Zachara NE, Combined antibody/lectin enrichment identifies extensive changes in the O-GlcNAc sub-proteome upon oxidative stress, J. Proteome Res 15 (2016) 4318–4336. 10.1021/acs.jproteome.6b00369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Woo CM, Lund PJ, Huang AC, Davis MM, Bertozzi CR, Pitteri SJ, Mapping and quantification of over 2000 O-linked glycopeptides in activated human T cells with isotope-targeted glycoproteomics (Isotag), Mol. Cell. Proteomics. 17 (2018) 764–775. 10.1074/mcp.RA117.000261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Qin K, Zhu Y, Qin W, Gao J, Shao X, Wang YL, Zhou W, Wang C, Chen X, Quantitative profiling of protein O-GlcNAcylation sites by an isotope-tagged cleavable linker, ACS Chem. Biol 13 (2018) 1983–1989. 10.1021/acschembio.8b00414. [DOI] [PubMed] [Google Scholar]
- [12].Hao Y, Fan X, Shi Y, Zhang C, Sun DE, Qin K, Qin W, Zhou W, Chen X, Next-generation unnatural monosaccharides reveal that ESRRB O-GlcNAcylation regulates pluripotency of mouse embryonic stem cells, Nat. Commun 10 (2019) 4065 10.1038/s41467-019-11942-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Darabedian N, Yang B, Ding R, Cutolo G, Zaro BW, Woo CM, Pratt MR, O-acetylated chemical reporters of glycosylation can display metabolism-dependent background labeling of proteins but are generally reliable tools for the identification of glycoproteins, Front. Chem 8 (2020) 318 10.3389/fchem.2020.00318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Varki A, Cummings RD, Esko JD, Stanley P, Hart GW, Aebi M, Darvill AG, Kinoshita T, Packer NH, Prestegard JH, Schnaar RL, Seeberger PH, eds., Essentials of glycobiology, 3rd ed., Cold Spring Harbor Laboratory Press, Cold Spring Harbor (NY), 2015. http://www.ncbi.nlm.nih.gov/books/NBK310274/ (accessed October 10, 2020). [PubMed] [Google Scholar]
- [15].Yang X, Qian K, Protein O-GlcNAcylation: emerging mechanisms and functions, Nat. Rev. Mol. Cell Biol 18 (2017) 452–465. 10.1038/nrm.2017.22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Liu C, Li J, O-GlcNAc: A sweetheart of the cell cycle and DNA damage response, Front. Endocrinol 9 (2018) 415 10.3389/fendo.2018.00415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Ranuncolo SM, Ghosh S, Hanover JA, Hart GW, Lewis BA, Evidence of the involvement of O-GlcNAc-modified human RNA polymerase II CTD in transcription in vitro and in vivo, J. Biol. Chem 287 (2012) 23549–23561. 10.1074/jbc.M111.330910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Lewis BA, O-GlcNAcylation at promoters, nutrient sensors, and transcriptional regulation, Biochim. Biophys. Acta 1829 (2013) 1202–1206. 10.1016/j.bbagrm.2013.09.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Zhu Y, Liu TW, Cecioni S, Eskandari R, Zandberg WF, Vocadlo DJ, O-GlcNAc occurs cotranslationally to stabilize nascent polypeptide chains, Nat. Chem. Biol 11 (2015) 319–325. 10.1038/nchembio.1774. [DOI] [PubMed] [Google Scholar]
- [20].Qin W, Lv P, Fan X, Quan B, Zhu Y, Qin K, Chen Y, Wang C, Chen X, Quantitative time-resolved chemoproteomics reveals that stable O-GlcNAc regulates box C/D snoRNP biogenesis, Proc. Natl. Acad. Sci. U. S. A 114 (2017) E6749–E6758. 10.1073/pnas.1702688114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Li X, Zhu Q, Shi X, Cheng Y, Li X, Xu H, Duan X, Hsieh-Wilson LC, Chu J, Pelletier J, Ni M, Zheng Z, Li S, Yi W, O-GlcNAcylation of core components of the translation initiation machinery regulates protein synthesis, Proc. Natl. Acad. Sci. U. S. A 116 (2019) 7857–7866. 10.1073/pnas.1813026116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Zhu Y, Willems LI, Salas D, Cecioni S, Wu WB, Foster LJ, Vocadlo DJ, Tandem bioorthogonal labeling uncovers endogenous cotranslationally O-GlcNAc modified nascent proteins, J. Am. Chem. Soc 142 (2020) 15729–15739. 10.1021/jacs.0c04121. [DOI] [PubMed] [Google Scholar]
- [23].Hart GW, Nutrient regulation of signaling and transcription, J. Biol. Chem 294 (2019) 2211–2231. 10.1074/jbc.AW119.003226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Pedowitz NJ, Batt AR, Darabedian N, Pratt MR, MYPT1 O-GlcNAc modification regulates sphingosine-1-phosphate mediated contraction, Nat. Chem. Biol (2020). 10.1038/s41589-020-0640-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Ferrer CM, Sodi VL, Reginato MJ, O-GlcNAcylation in cancer biology: linking metabolism and signaling, J. Mol. Biol 428 (2016) 3282–3294. 10.1016/j.jmb.2016.05.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Gu Y, Mi W, Ge Y, Liu H, Fan Q, Han C, Yang J, Han F, Lu X, Yu W, GlcNAcylation plays an essential role in breast cancer metastasis, Cancer Res. 70 (2010) 6344–6351. 10.1158/0008-5472.CAN-09-1887. [DOI] [PubMed] [Google Scholar]
- [27].Slawson C, Hart GW, O-GlcNAc signalling: implications for cancer cell biology, Nat. Rev. Cancer. 11 (2011)678–684. 10.1038/nrc3114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Lynch TP, Ferrer CM, Jackson SR, Shahriari KS, Vosseller K, Reginato MJ, Critical role of O-Linked β-Ν-acetylglucosamine transferase in prostate cancer invasion, angiogenesis, and metastasis, J. Biol. Chem 287 (2012) 11070–11081. 10.1074/jbc.M111.302547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Lehman DM, Fu DJ, Freeman AB, Hunt KJ, Leach RJ, Johnson-Pais T, Hamlington J, Dyer TD, Arya R, Abboud H, Göring HHH, Duggirala R, Blangero J, Konrad RJ, Stem MP, A single nucleotide polymorphism in MGEA5 encoding O-GlcNAc-selective N-acetyl-beta-D glucosaminidase is associated with type 2 diabetes in Mexican Americans, Diabetes. 54 (2005) 1214–1221. 10.2337/diabetes.54.4.1214. [DOI] [PubMed] [Google Scholar]
- [30].Shin SJ, Chen CH, Kuo WC, Chan HC, Chan HC, Lin KD, Ke LY, Disruption of retinoid homeostasis induces RBP4 overproduction in diabetes: O-GlcNAcylation involved, Metabolism. 113 (2020) 154403 10.1016/j.metabol.2020.154403. [DOI] [PubMed] [Google Scholar]
- [31].Chatham JC, Young ME, Zhang J, Role of O-linked N-acetylglucosamine (O-GlcNAc) modification of proteins in diabetic cardiovascular complications, Curr. Opin. Pharmacol 57 (2020) 1–12. 10.1016/j.coph.2020.08.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Hewagama A, Gorelik G, Patel D, Liyanarachchi P, McCune WJ, Somers E, Gonzalez-Rivera T, Cohort Michigan Lupus, Strickland F, Richardson B, Overexpression of X-linked genes in T cells from women with lupus, J. Autoimmun 41 (2013) 60–71. 10.1016/j.jaut.2012.12.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Vaidyanathan K, Niranjan T, Selvan N, Teo CF, May M, Patel S, Weatherly B, Skinner C, Opitz J, Carey J, Viskochil D, Gecz J, Shaw M, Peng Y, Alexov E, Wang T, Schwartz C, Wells L, Identification and characterization of a missense mutation in the O-linked β-N-acetylglucosamine (O-GlcNAc) transferase gene that segregates with X-linked intellectual disability, J. Biol. Chem 292 (2017) 8948–8963. 10.1074/jbc.M116.771030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Pravata VM, Muha V, Gundogdu M, Ferenbach AT, Kakade PS, Vandadi V, Wilmes AC, Borodkin VS, Joss S, Stavridis MP, van Aalten DMF, Catalytic deficiency of O-GlcNAc transferase leads to X-linked intellectual disability, Proc. Natl. Acad. Sci. U. S. A 116 (2019) 14961–14970. 10.1073/pnas.1900065116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Liu F, Iqbal K, Grundke-Iqbal I, Hart GW, Gong CX, O-GlcNAcylation regulates phosphorylation of tau: a mechanism involved in Alzheimer’s disease, Proc. Natl. Acad. Sci. U. S. A 101 (2004) 10804–10809. 10.1073/pnas.0400348101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Yuzwa SA, Vocadlo DJ, O-GlcNAc and neurodegeneration: biochemical mechanisms and potential roles in Alzheimer’s disease and beyond, Chem. Soc. Rev 43 (2014) 6839–6858. 10.1039/c4cs00038b. [DOI] [PubMed] [Google Scholar]
- [37].Ryan P, Xu M, Davey AK, Danon JJ, Mellick GD, Kassiou M, Rudrawar S, O-GlcNAc modification protects against protein misfolding and aggregation in neurodegenerative disease, ACS Chem. Neurosci 10 (2019) 2209–2221. 10.1021/acschemneuro.9b00143. [DOI] [PubMed] [Google Scholar]
- [38].Chuh KN, Zaro BW, Piller F, Piller V, Pratt MR, Changes in metabolic chemical reporter structure yield a selective probe of O-GlcNAc modification, J. Am. Chem. Soc 136 (2014) 12283–12295. 10.1021/ja504063c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Hu CW, Worth M, Fan D, Li B, Li H, Lu L, Zhong X, Lin Z, Wei L, Ge Y, Li L, Jiang J, Electrophilic probes for deciphering substrate recognition by O-GlcNAc transferase, Nat. Chem. Biol 13 (2017) 1267–1273. 10.1038/nchembio.2494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Lewis YE, Galesic A, Levine PM, De Leon CA, Lamiri N, Brennan CK, Pratt MR, O-GlcNAcylation of α-synuclein at serine 87 reduces aggregation without affecting membrane binding, ACS Chem. Biol 12 (2017) 1020–1027. 10.1021/acschembio.7b00113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Darabedian N, Gao J, Chuh KN, Woo CM, Pratt MR, The metabolic chemical reporter 6-Azido-6-deoxy-glucose further reveals the substrate promiscuity of O-GlcNAc transferase and catalyzes the discovery of intracellular protein modification by O-Glucose, J. Am. Chem. Soc 140 (2018) 7092–7100. 10.1021/jacs.7bl3488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Li J, Li Z, Duan X, Qin K, Dang L, Sun S, Cai L, Hsieh-Wilson LC, Wu L, Yi W, An isotope-coded photocleavable probe for quantitative profiling of protein O-GlcNAcylation, ACS Chem. Biol 14 (2019) 4–10. 10.1021/acschembio.8b01052. [DOI] [PubMed] [Google Scholar]
- [43].Ramirez DH, Aonbangkhen C, Wu HY, Naftaly JA, Tang S, O’Meara TR, Woo CM, Engineering a proximity-directed O-GlcNAc transferase for selective protein O-GlcNAcylation in cells, ACS Chem. Biol 15 (2020) 1059–1066. 10.1021/acschembio.0c00074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [44].Gorelik A, van Aalten DMF, Tools for functional dissection of site-specific O-GlcNAcylation, RSC Chem. Biol 1 (2020) 98–109. 10.1039/D0CB00052C. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [45].Ortiz-Meoz RF, Jiang J, Lazarus MB, Orman M, Janetzko J, Fan C, Duveau DY, Tan ZW, Thomas CJ, Walker S, A small molecule that inhibits OGT activity in cells, ACS Chem. Biol 10 (2015) 1392–1397. 10.1021/acschembio.5b00004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [46].Martin SES, Tan ZW, Itkonen HM, Duveau DY, Paulo JA, Janetzko J, Boutz PL, Törk L, Moss FA, Thomas CJ, Gygi SP, Lazarus MB, Walker S, Structure-based evolution of low nanomolar O-GlcNAc transferase inhibitors, J. Am. Chem. Soc 140 (2018) 13542–13545. 10.1021/jacs.8b07328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [47].Gloster TM, Zandberg WF, Heinonen JE, Shen DL, Deng L, Vocadlo DJ, Hijacking a biosynthetic pathway yields a glycosyltransferase inhibitor within cells, Nat. Chem. Biol 7 (2011) 174–181. 10.1038/nchembio.520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [48].Worth M, Hu CW, Li H, Fan D, Estevez A, Zhu D, Wang A, Jiang J, Targeted covalent inhibition of O-GlcNAc transferase in cells, Chem. Commun 55 (2019) 13291–13294. 10.1039/c9cc04560k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [49].Estevez A, Zhu D, Blankenship C, Jiang J, Molecular interrogation to crack the case of O-GlcNAc, Chem.-Eur. J 26 (2020) 12086–12100. 10.1002/chem.202000155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [50].Joiner CM, Li H, Jiang J, Walker S, Structural characterization of the O-GlcNAc cycling enzymes: insights into substrate recognition and catalytic mechanisms, Curr. Opin. Struct. Biol 56 (2019) 97–106. 10.1016/j.sbi.2018.12.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [51].Jinek M, Rehwinkel J, Lazarus BD, Izaurralde E, Hanover JA, Conti E, The superhelical TPR-repeat domain of O-linked GlcNAc transferase exhibits structural similarities to importin [alpha], Nat. Struct. Mol. Biol 11 (2004) 1001–1007. 10.1038/nsmb833. [DOI] [PubMed] [Google Scholar]
- [52].Lazarus MB, Nam Y, Jiang J, Sliz P, Walker S, Structure of human O-GlcNAc transferase and its complex with a peptide substrate, Nature. 469 (2011) 564–567. 10.1038/nature09638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [53].Lazarus MB, Jiang J, Kapuria V, Bhuiyan T, Janetzko J, Zandberg WF, Vocadlo DJ, Herr W, Walker S, HCF-1 is cleaved in the active site of O-GlcNAc transferase, Science. 342 (2013) 1235–1239. 10.1126/science.1243990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [54].Rafie K, Raimi O, Ferenbach AT, Borodkin VS, Kapuria V, van Aalten DMF, Recognition of a glycosylation substrate by the O-GlcNAc transferase TPR repeats, Open Biol. 7 (2017) 170078 10.1098/rsob.170078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [55].Pathak S, Alonso J, Schimpl M, Rafie K, Blair DE, Borodkin VS, Albarbarawi O, van Aalten DMF, The active site of O-GlcNAc transferase imposes constraints on substrate sequence, Nat. Struct. Mol. Biol 22 (2015) 744–750. 10.1038/nsmb.3063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [56].Lazarus MB, Jiang J, Gloster TM, Zandberg WF, Whitworth GE, Vocadlo DJ, Walker S, Structural snapshots of the reaction coordinate for O-GlcNAc transferase, Nat. Chem. Biol 8 (2012) 966–968. 10.1038/nchembio.1109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [57].Levine ZG, Fan C, Melicher MS, Orman M, Benjamin T, Walker S, O-GlcNAc transferase recognizes protein substrates using an asparagine ladder in the tetratricopeptide repeat (TPR) superhelix, J. Am. Chem. Soc 140 (2018) 3510–3513. 10.1021/jacs.7b13546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [58].Joiner CM, Levine ZG, Aonbangkhen C, Woo CM, Walker S, Aspartate residues far from the active site drive O-GlcNAc transferase substrate selection, J. Am. Chem. Soc 141 (2019) 12974–12978. 10.1021/jacs.9b06061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [59].Pierce MM, Raman CS, Nall BT, Isothermal titration calorimetry of protein-protein interactions, Methods. 19 (1999) 213–221. 10.1006/meth.1999.0852. [DOI] [PubMed] [Google Scholar]
- [60].Drescher DG, Selvakumar D, Drescher MJ, Analysis of protein interactions by surface plasmon resonance, Adv. Protein Chem. Struct. Biol 110 (2018) 1–30. 10.1016/bs.apcsb.2017.07.003. [DOI] [PubMed] [Google Scholar]
- [61].Lazarus BD, Love DC, Hanover JA, Recombinant O-GlcNAc transferase isoforms: identification of O-GlcNAcase, yes tyrosine kinase, and tau as isoform-specific substrates, Glycobiology. 16 (2006)415–421. 10.1093/glycob/cwj078. [DOI] [PubMed] [Google Scholar]
- [62].Khidekel N, Ficarro SB, Clark PM, Bryan MC, Swaney DL, Rexach JE, Sun YE, Coon JJ, Peters EC, Hsieh-Wilson LC, Probing the dynamics of O-GlcNAc glycosylation in the brain using quantitative proteomics, Nat. Chem. Biol 3 (2007) 339–348. 10.1038/nchembio881. [DOI] [PubMed] [Google Scholar]
- [63].Rexach JE, Rogers CJ, Yu SH, Tao J, Sun YE, Hsieh-Wilson LC, Quantification of O-glycosylation stoichiometry and dynamics using resolvable mass tags, Nat. Chem. Biol 6 (2010) 645–651. 10.1038/nchembio.412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [64].Alonso J, Schimpl M, van Aalten DMF, O-GlcNAcase: promiscuous hexosaminidase or key regulator of O-GlcNAc signaling?, J. Biol. Chem 289 (2014) 34433–34439. 10.1074/jbc.R114.609198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [65].Whisenhunt TR, Yang X, Bowe DB, Paterson AJ, Van Tine BA, Kudlow JE, Disrupting the enzyme complex regulating O-GlcNAcylation blocks signaling and development, Glycobiology. 16 (2006) 551–563. 10.1093/glycob/cwj096. [DOI] [PubMed] [Google Scholar]
- [66].Li B, Li H, Lu L, Jiang J, Structures of human O-GlcNAcase and its complexes reveal a new substrate recognition mode, Nat. Struct. Mol. Biol 24 (2017) 362–369. 10.1038/nsmb.3390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [67].King DT, Males A, Davies GJ, Vocadlo DJ, Molecular mechanisms regulating O-linked N-acetylglucosamine (O-GlcNAc)-processing enzymes, Curr. Opin. Chem. Biol 53 (2019) 131–144. 10.1016/j.cbpa.2019.09.001. [DOI] [PubMed] [Google Scholar]
- [68].Goebl M, Yanagida M, The TPR snap helix: a novel protein repeat motif from mitosis to transcription, Trends Biochem. Sci 16 (1991) 173–177. 10.1016/0968-0004(91)90070-c. [DOI] [PubMed] [Google Scholar]
- [69].Sikorski RS, Boguski MS, Goebl M, Hieter P, A repeating amino acid motif in CDC23 defines a family of proteins and a new relationship among genes required for mitosis and RNA synthesis, Cell. 60 (1990) 307–317. 10.1016/0092-8674(90)90745-z. [DOI] [PubMed] [Google Scholar]
- [70].Zeytuni N, Zarivach R, Structural and functional discussion of the tetra-trico-peptide repeat, a protein interaction module, Structure. 20 (2012) 397–405. 10.1016/j.str.2012.01.006. [DOI] [PubMed] [Google Scholar]
- [71].Fontes MR, Teh T, Kobe B, Structural basis of recognition of monopartite and bipartite nuclear localization sequences by mammalian importin-alpha, J. Mol. Biol 297 (2000) 1183–1194. 10.1006/jmbi.2000.3642. [DOI] [PubMed] [Google Scholar]
- [72].Sun J, Weis WI, Biochemical and structural characterization of β-catenin interactions with nonphosphorylated and CK2-phosphorylated Lef-1, J. Mol. Biol 405 (2011) 519–530. 10.1016/j.jmb.2010.11.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [73].Klink BU, Gatsogiannis C, Hofnagel O, Wittinghofer A, Raunser S, Structure of the human BBSome core complex, eLife. 9 (2020) e53910 10.7554/eLife.53910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [74].Singh SK, Gui M, Koh F, Yip MC, Brown A, Structure and activation mechanism of the BBSome membrane protein trafficking complex, eLife. 9 (2020) e53322 10.7554/eLife.53322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [75].Pan Z, Shang Y, Jia M, Zhang L, Xia C, Zhang M, Wang W, Wen W, Structural and biochemical characterization of the interaction between LGN and Frmpd1, J. Mol. Biol 425 (2013) 1039–1049. 10.1016/j.jmb.2013.01.003. [DOI] [PubMed] [Google Scholar]
- [76].Zhu J, Wen W, Zheng Z, Shang Y, Wei Z, Xiao Z, Pan Z, Du Q, Wang W, Zhang M, LGN/mInsc and LGN/NuMA complex structures suggest distinct functions in asymmetric cell division for the Par3/mInsc/LGN and Gαi/LGN/NuMA pathways, Mol. Cell 43 (2011) 418–431. 10.1016/j.molcel.2011.07.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [77].Yuzawa S, Kamakura S, Iwakiri Y, Hayase J, Sumimoto H, Structural basis for interaction between the conserved cell polarity proteins Inscuteable and Leu-Gly-Asn repeat-enriched protein (LGN), Proc. Natl. Acad. Sci. U. S. A 108 (2011) 19210–19215. 10.1073/pnas.1110951108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [78].Culurgioni S, Alfieri A, Pendolino V, Laddomada F, Mapelli M, Inscuteable and NuMA proteins bind competitively to Leu-Gly-Asn repeat-enriched protein (LGN) during asymmetric cell divisions, Proc. Natl. Acad. Sci. U. S. A 108 (2011) 20998–21003. 10.1073/pnas.1113077108. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.