

Abstract
This work establishes that Kupffer cell release of platelet activating factor (PAF), a lipidic molecule with pro-inflammatory and vasoactive signaling properties, dictates dose-limiting siRNA nanocarrier-associated toxicities. High-dose intravenous injection of siRNA-polymer nano-polyplexes (si-NPs) elicited acute, shock-like symptoms in mice, associated with increased plasma PAF and consequently reduced PAF acetylhydrolase (PAF-AH) activity. These symptoms were completely prevented by prophylactic PAF receptor inhibition or Kupffer cell depletion. Assessment of varied si-NP chemistries confirmed that toxicity level correlated to relative uptake of the carrier by liver Kupffer cells and that this toxicity mechanism is dependent on carrier endosome disruptive function. 4T1 tumor-bearing mice, which exhibit increased circulating leukocytes, displayed greater sensitivity to these toxicities. PAF-mediated toxicities were generalizable to commercial delivery reagent in vivo-jetPEI® and an MC3 lipid formulation matched to an FDA-approved nanomedicine. These collective results establish Kupffer cell release of PAF as a key mediator of siRNA nanocarrier toxicity and identify PAFR inhibition as an effective strategy to increase siRNA nanocarrier tolerated dose.
Keywords: Nanoparticle toxicity, nucleic acid nanocarriers, platelet activating factor, Kupffer cells
Introduction
With the recent first-in-class FDA approval of Onpattro in August 2018, nanoparticle-based delivery of siRNA has entered an era of clinical reality.(1) However, cationic carrier components continue to face toxicological challenges that hinder their clinical development. In fact, Alnylam Pharmaceuticals, the company that developed Onpattro, has abandoned its cationic DLin-MC3-DMA carrier lipids for their pipeline therapeutics; the Onpattro formulation requires immunosuppressive steroid pretreatment in patients to reduce potential injection related toxicities.(2)
For cationic siRNA-polymer nano-polyplexes (si-NPs), there is a correlation between transfection efficiency and toxicity which poses a challenge for maximizing the therapeutic window of siRNA nanocarriers.(3) After intravenous injection, nanomaterials inevitably accumulate in cells of the mononuclear phagocyte system (MPS), especially in the liver.(4) Complement adsorption to nanoparticle surfaces can increase MPS clearance while also inducing inflammatory reactions.(3, 5) Aggregation of cationic si-NPs in the lungs can furthermore induce severe clotting resulting in acute, fatal toxicity in mice.(3, 6–8) Steric shielding of polyplexes with materials such as PEG is often considered a panacea to these toxicities.(3, 5, 8, 9) However, PEGylated nanoparticles can still be toxic at high doses and cause poorly-understood allergy-like adverse reactions even in the absence of prior nanomaterial exposure (lacking IgE antibodies).(4, 10) CALAA-01, a PEGylated cyclodextrin-based siRNA carrier, was discontinued from clinical development in part due to infusion-related innate immune responses.(1, 11)
We previously developed novel siRNA-complexing polymers (PMPC-DB) containing a high molecular weight (20kDa) zwitterionic corona composed of a poly(2-methacryloyloxyethyl phosphorylcholine) (PMPC) homopolymer.(12) The core is composed of a poly(dimethylaminoethyl methacrylate-co-butyl methacrylate) (DMAEMA-co-BMA) 50:50 random copolymer which provides optimal endosomolytic behavior while reducing cytotoxicity.(13, 14) PMPC-DB si-NPs have improved circulation half-life compared to similar si-NPs with smaller molecular weight coronas and achieve greater tumor cell uptake compared to similar si-NPs with 20kDa PEG coronas.(12) When used at an ideal polymer:siRNA (N:P) formulation ratio and stabilized by “dual hydrophobization”, implemented by palmitate conjugation to the siRNA cargo, this si-NP is well-tolerated in long-term, multi-dose regimens.(15)
Despite this favorable profile, we have empirically observed that the zwitterionic si-NPs have notably narrower therapeutic window than the PEGylated si-NPs, manifested by acute toxicities in mice when intravenously delivered in a sub-optimal formulation (elevated dose, high N:P ratio, and absence of stabilizing hydrophobic functionalization of the siRNA). Mice intravenously treated under these conditions exhibit lethargy and prostrated body positions within 5–15 minutes and succumb within 1 hour of injection.
We hypothesized that these rapid, shock-like toxicities associated with si-NP intravenous injection may be associated with platelet activating factor (PAF). PAF is a potent phospholipid signaling molecule that is produced by, and is an effector of macrophages, neutrophils, and platelets that triggers vasodilation, vascular permeability, and leukocyte degranulation.(16–20) PAF is associated with multiple systemic inflammatory responses, including sepsis and anaphylactic shock, and it has been associated with acute toxicities caused by intravenous adenovirus injection.(21, 22) The only characterized instances of PAF involvement in nanoparticle-related toxicity to our knowledge are a report relating PAF to propagating antibody-mediated effects from repeat injection of immunogenic PEGylated liposomes(23) and another on liposome encapsulated hemoglobin where the mechanism for this toxicity was not understood.(24) The potential role of PAF in dose limiting toxicity of siRNA nanomedicines, an important and clinically-emerging class of therapeutics, has not been explored. The potential cell/tissue source and the nanoparticle characteristics that influence PAF release has also not been rigorously studied for synthetic nanomedicines to our knowledge. Here, we were interested to establish whether PAF broadly dictates dose limiting toxicities for siRNA nanomedicines. We also sought to determine the nanoparticle (surface chemistry and endosome disruption functionality) and animal pathophysiological (tumor-bearing or not) features that potentiate PAF effects. We also wished to explore the cell and tissue source of PAF, with the hypothesis that PAF activity is mediated by Kupffer cells, specialized liver macrophages,(4, 25, 26) which are known to internalize a large percentage of nanomedicines and to be a rich source of PAF.(27)
Results and Discussion
The PMPC-DB polymers used for this study were confirmed for structure by 1H-NMR (Supplemental Figure S1)(12) and were free of endotoxin contamination prior to initiation of all studies (Supplemental Figure S2). An si-NP formulation was prepared by complexing unmodified siRNA with PMPC-DB polymers at a 20:1 charge ratio (defined as protonated nitrogen to phosphate ratio), yielding monodisperse si-NPs with average hydrodynamic diameter of 100 (Supplemental Figure S3). This formulation was purposefully defined and used at a high dose (1.2 mg/kg siRNA, 73.6 mg/kg polymer), known to induce adverse events.
We pre-treated BALB/c mice with either saline or ABT-491intravenously 10 minutes prior to injecting mice with si-NPs. ABT-491 is a potent and highly specific small-molecule inhibitor of the PAF receptor (PAFR).(20, 28) Of five mice pre-treated with saline, four mice experienced fatality within 1 hour of injection (Figure 1a). Mice pre-treated with ABT-491 experienced no fatalities and no behavioral changes. Thirty minutes after si-NP injection, mice treated with si-NPs only (no PAFR inhibitor) exhibited gross pathology changes characteristic of shock and hypotension, including reddened intestines, congested (darkened) livers, and vasodilation (Figure 1b, Supplemental Figure S4). Mice pre-treated with ABT-491 did not experience these changes and appeared phenotypically similar to saline-treated mice.
Figure 1. PAFR inhibitor ABT-491 rescues acute toxicities associated with high-dose si-NP injection.
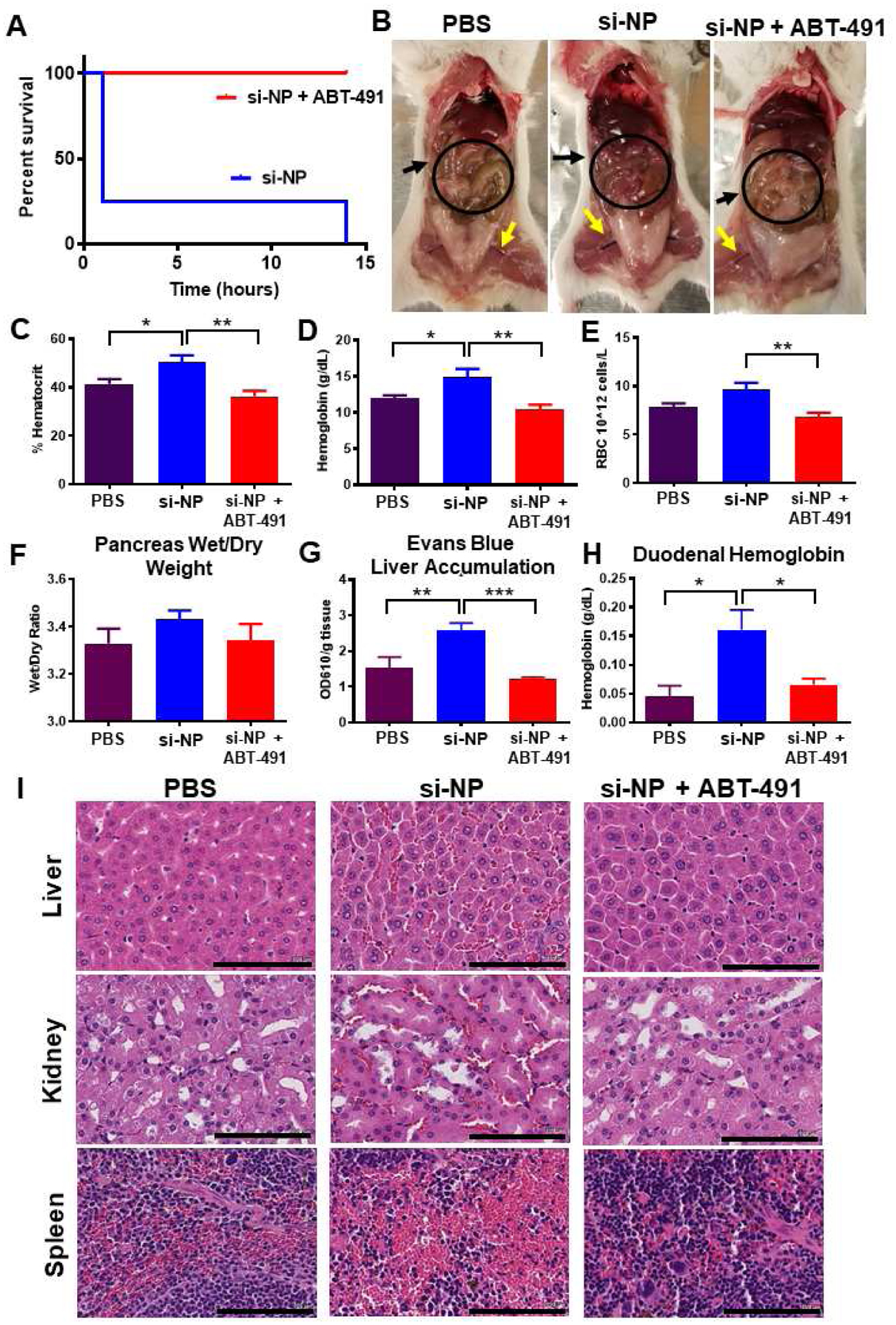
A) Upon intravenous injection of si-NPs at high N:P ratio (high polymer dose) and 1.2 mg/kg siRNA, 4 out of 5 mice experienced fatalities within 1 hour, but mortality was fully prevented by pre-injection of ABT-491 10 minutes prior to injection of the same si-NP dose (n=5 mice per group). B) Gross pathology demonstrates vasodilatory pathology in major blood vessels (yellow arrows) and reddening of intestines (black arrows, circles) in mice treated with si-NPs. C-E) Mice injected with high-dose si-NPs experienced significant increases in blood hematocrit (C), hemoglobin (D), and red blood cell concentration (E), all of which were abrogated by ABT-491 pretreatment. F) Pancreas wet weight vs. dry weight with 1.2 mg/kg si-NPs (n=5–7), saline, or si-NPs with ABT-491 pre-treatment. G) Evans Blue concentration (as a marker for vascular leakiness) in mouse livers. H) Duodenum hemoglobin concentration (n=5–7, * p<0.05, ** p<0.01, *** p<0.001). I) H&E staining of mouse liver, kidney, and spleen after i.v. injection demonstrating red blood cell congestion in each tissue. Scale bars = 100 μm. All measures were made 30 minutes after injection.
Mice treated with si-NPs experienced significant increases in blood hematocrit percentage, hemoglobin concentration, and red blood cell concentration at 30 minutes post-injection compared to saline-treated or ABT-491-pre-treated mice (Figure 1c–e). These blood hemoconcentration symptoms were indicative of shock, wherein an increase in vascular permeability results in leakage of plasma fluid into surrounding tissues. Hemoconcentration is also one of the hallmark side effects of intravenous PAF administration.(29)
Shock-induced vascular permeability can lead to edema in organs and GI tract effects, including increased duodenal hemoglobin.(21) We noted increases (though not statistically significant) in the pancreas wet/dry weight ratio for si-NP-treated mice compared to ABT-491-pre-treated mice (Figure 1f). We further confirmed these symptoms of tissue edema by injecting mice with Evans Blue dye five minutes after si-NP treatment and measuring dye absorbance in liver homogenates after si-NP treatment. Evans Blue dye binds to the serum protein albumin and can assess aberrant vascular permeability and plasma leakage into tissues.(30) Evans Blue dye absorbance was elevated in si-NP-treated mice compared to saline-treated mice, while ABT-491-pre-treated mice did not have increases (Figure 1g). These results indicate that ABT-491 pre-treatment rescued mice from si-NP-induced vascular permeability. si-NP-treated mice also had severe red blood cell congestion in their duodenums as indicated by nearly four-fold higher hemoglobin concentrations than saline-treated or ABT-491-pre-treated mice (Figure 1h).
Histologically, these results were confirmed by evidence of significant congestion in multiple organs of si-NP-treated mice (Figure 1i). Notably increased amounts of red blood cells were visible in the liver, kidney, and spleen of these mice. By comparison, mice pre-treated with ABT-491 did not experience histologic changes compared to saline-treated mice.
The combined data in Figure 1 suggests that PAF plays a significant role in the acute toxicities caused by high-dose si-NP injection. Inhibition of PAFR completely rescued mice from si-NP-associated toxicities such that there were no phenotypical or pathological differences observed between ABT-491+si-NP-treated mice and saline-treated mice. Additionally, we have demonstrated that the acute toxicities resulting from si-NP treatment are symptomatic of shock-like conditions including vasodilation, hemoconcentration, and vascular edema. These symptoms became apparent upon first exposure to si-NP treatments and are thus not consistent with anaphylactic, IgE-mediated shock. Plasma levels of select proinflammatory cytokines (IL-1β, TNF-α) were measured 15 minutes following si-NP injection (Supplementary Figure S5). Though there was a significant difference in TNF-α levels between saline and si-NP injected mice, neither group displayed dangerously high levels of any of the cytokines.(31, 32) In comparison, mice began displaying adverse symptoms as early as 5 minutes post si-NP injection, suggesting that pro-inflammatory cytokine release is not the cause of these toxicities. We also confirmed that acute toxicities observed upon si-NP administration were not dependent on mouse strain. Study of strain is relevant because while BALB/c mice are generally more prone to Th-2 - type responses, C57BL/6 are Th-1 biased and can result in different pharmacokinetic profiles for the same nanoparticles.(33) Shock-like toxicities seen with BALB/c mice were also consistently observed in C57BL/6 mice injected with the same si-NP (Supplemental Figure S6a–e). Plasma aspartate aminotransferase (AST), alanine aminotransferase (ALT), and blood urea nitrogen (BUN) levels were also quantified in these mice and were similar to levels seen in saline-treated mice (Supplemental Figure S7). These data suggest that the toxic vasoactive effects of PAF are more acute than any organ long-term, treatment-associated damage that may occur.
To further confirm the role of PAF in si-NP-associated toxicities, we measured the activity of plasma PAF acetylhydrolase (PAF-AH) in si-NP-treated mice compared to saline treated mice. PAF itself is a lipid mediator with an extremely short half-life of 30 seconds to 5 minutes.(16, 34) It is rapidly degraded by PAF-AH, making it difficult to accurately measure without advanced analytical methods. PAF-AH activity, on the other hand, is an alternative to direct measurement of PAF. Measured activity of PAF-AH in plasma is inversely correlated with PAF levels; PAF-AH becomes saturated, and therefore, measurable PAF-AH activity decreases in the presence of increased concentrations of PAF. (16, 35–37)
Fifteen minutes post-si-NP treatment, mice experienced significantly decreased plasma PAF-AH activity, going from around 0.08 umol/min/mL (saline-treated mice) to 0.04 umol/min/mL (si-NP treated mice)(p<0.01). (Figure 2a). These data, combined with the significant prophylactic effects of the PAFR inhibitor, indicate that intravenous administration of si-NPs induces systemic release of PAF. Significant increases in PAF itself were also detected by ELISA in the blood of si-NP treated mice compared to saline-treated controls--- up to 8 ng/mL (Supplemental Figure S8). This value is below the quantity of purified PAF that must be delivered exogenously to cause toxicity in vivo.(38) Due to rapid PAF degradation in plasma, the ELISA measurement likely underestimates the PAF concentration and is a less reliable method to gauge PAF levels from in vivo samples than measuring PAF-AH activity.
Figure 2. si-NPs stimulate PAF release, as measured through PAF-AH exhaustion, and do not directly agonize the PAF receptor.
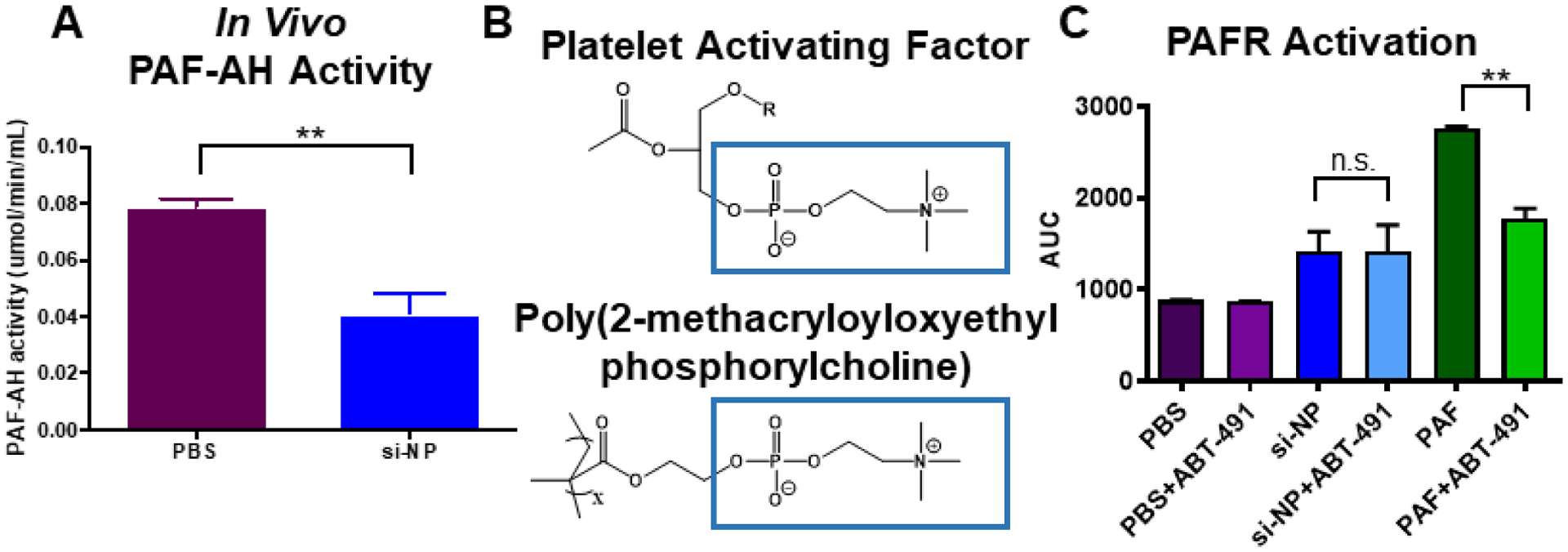
A) Mice injected with si-NPs display decreased PAF-AH activity in blood samples 15 minutes after injection. B) The si-NP PMPC corona comprises zwitterionic phosphorylcholine that contains similar structure to PAF. C) PAF activates the PAFR through a mechanism that is inhibited by ABT-491 pre-treatment in Chem-1 cells overexpressing PAFR, but PMPC-based si-NPs do not (** p<0.01).
We next sought to test the hypothesis that si-NPs themselves might be capable of agonizing the PAF receptor. The PMPC corona of these si-NPs is made up of zwitterionic, phosphorylcholine moieties, and the chemical structure of PAF contains a prominent phosphorylcholine (Figure 2b). Additionally, there have been examples in nature of bacteria using PAF-mimicry to engage the PAF receptor. For example, Streptococcus pneumoniae uses phosphorylcholine moieties in its cell wall to bind PAFR, block PAF signaling, and prevent inflammatory neutrophil reactions.(18, 39) We therefore treated Chem-1 cells overexpressing recombinant human PAFR with si-NPs and used an intracellular calcium dye to measure fluorescence from calcium influx (indicative of receptor activation). Addition of purified PAF caused a significant increase in cytosolic calcium that was reduced by pre-treatment with ABT-491. Addition of si-NPs caused a slight increase in fluorescent signal, but the effect was not reduced by ABT-491 pre-treatment (Figure 2c). These data suggest that, despite some chemical similarity, zwitterionic phosphorylcholine moieties on the si-NP surface do not directly act on PAFR but instead stimulate systemic release of PAF, which then acts on PAFR to mediate vasoactive changes that cause si-NP toxicities. This effect is also consistent with studies showing that release of PAF causes dose-dependent increases in vascular permeability and hemoconcentration.(19, 29)
PAF can also be released when cells of the MPS are activated by phagocytosing material.(19) Nanoparticles in the 100 nm size range are well-known to be cleared from circulation by the MPS, particularly by liver Kupffer cells.(4) Kupffer cells reside intravascularly in liver sinusoids and serve as a phagocytic microbial filter for the blood, removing nanoparticle and microbe-sized materials. To test whether Kupffer cells are responsible for si-NP treatment associated PAF release and consequent toxicities, we utilized clodronate liposomes to selectively deplete Kupffer cells in mice.
Pre-treatment of mice with clodronate liposomes (Clod) prevented shock toxicities associated with intravenous si-NP injections (Figure 3). Clod-pre-treated mice receiving si-NPs did not experience significant elevations in hematocrit, duodenal hemoglobin, or ELISA-detectable blood PAF levels (Figure 3a–c), relative to Clod-treated mice receiving saline. These data indicate that macrophages of the MPS are primary mediators of PAF-related shock toxicities associated with si-NP injection. Liposomal clodronate selectively depletes both liver and splenic macrophages, so contributions of splenic macrophages cannot be ruled out. However, our previous work has shown that only 2–5% of injected dose of si-NPs distributes to the spleen while closer to 50% of injected dose distributes to the liver (12, 15), suggesting that Kupffer cells are the primary PAF source. Our combined data indicate that Kupffer cells, upon stimulation with intravenously delivered si-NPs, release PAF into the blood. PAF release activates downstream PAF-associated vasoactive signals and consequently causes hypotension, edema, and tissue congestion.
Figure 3. Pre-treatment with clodronate liposomes abrogates si-NP-related PAF elevation and toxicities.
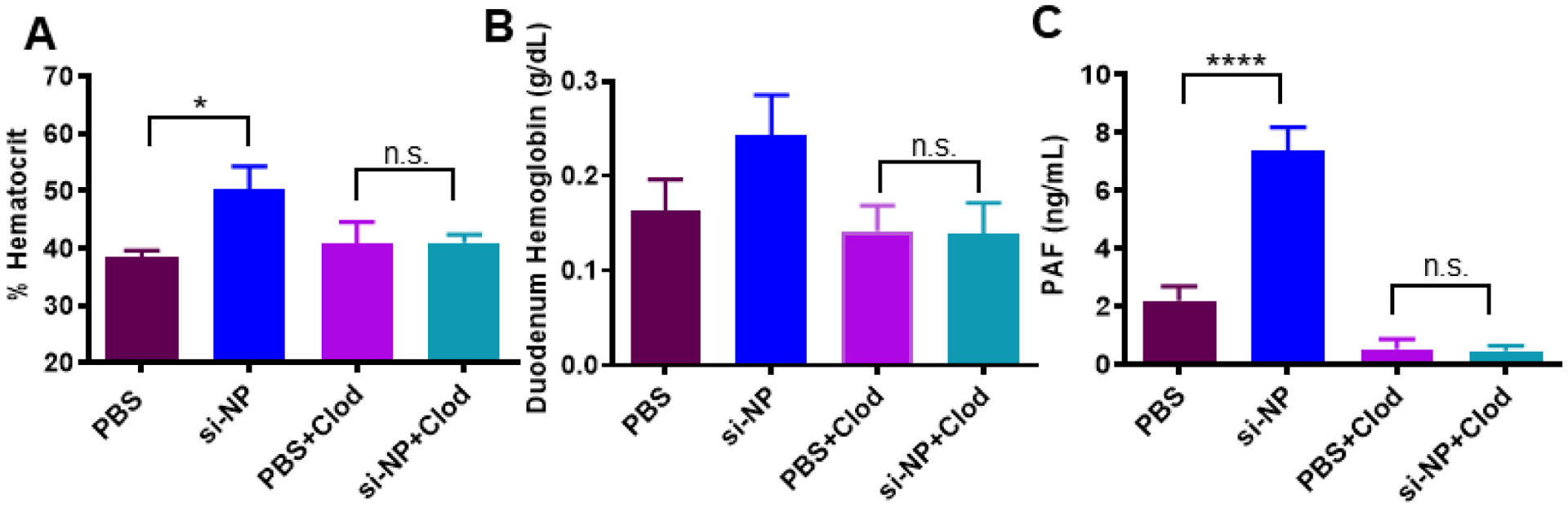
A) Mice pre-treated with Clod liposomes 24 hours prior to injection do not experience significant elevations in blood hematocrit 30 min after intravenous si-NP injection. B) Mice pre-treated with Clod liposomes do not experience significant increases in duodenal hemoglobin after intravenous si-NP injection. (n=4–5) C) Clod pre-treatment prevents increase in blood PAF levels after si-NP injection. Note that PBS and si-NP group data are reiterated from Supplemental Figure S8. (n=3–5, *p<0.05, ****p<0.0001).
We hypothesized that if PAF-related symptoms are nonspecifically initiated by Kupffer cell interactions with nanomaterials, then other polymeric nanoparticles would also stimulate PAF-related symptoms. In order to better understand the impact of si-NP structure on si-NP-associated toxicities, we compared our zwitterionic, phosphorylcholine surface si-NPs (zwitter si-NP) to si-NPs with a 20kDa PEG surface (matched molecular weight of PEG and PMPC polymer blocks) and the same endosomolytic DB core (PEG si-NP) (Figure 4a, polymer characteristics Supplemental Figure S9A, S9C). We also compared these si-NPs to a noncationic, non-endosomolytic zwitterionic micelle (zwitter micelle) composed of a phosphorylcholine (PMPC) surface and a purely hydrophobic (BMA) core (Figure 4a, polymer characteristics Supplemental Figure S9B, S9C). These micelles were equivalent in size to both PEG si-NPs and zwitter si-NPs, but, due to their non-cationic nature, did not complex with siRNA and do not have pH-dependent membrane disruptive activity for endosome escape (Supplemental Figure S9D).
Figure 4. PAF-associated toxicities correlate with level of Kupffer cell delivery and are dependent on endosome-disruptive activity.
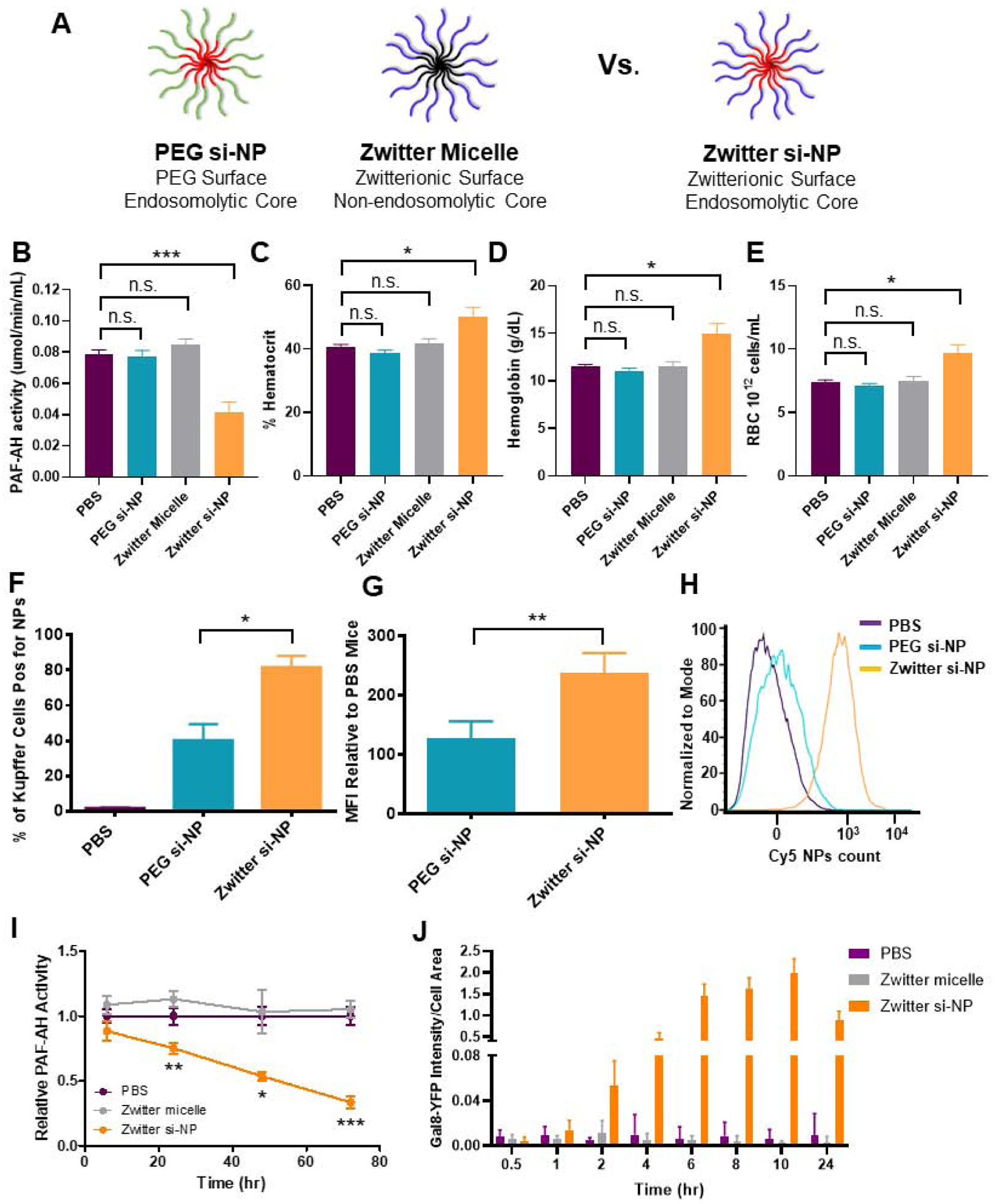
A) Schematic of zwitter si-NP, PEG si-NP, and zwitter micelle structures tested. B) Plasma PAF-AH activity from mice 15 min after receiving 1.2 mg/kg siRNA dose of each si-NP (n=4–5 per group, polymer dose: 73.6 mg/kg zwitter si-NP, 74.6 mg/kg PEG si-NP, 73.6 mg/kg zwitter micelle). Note that PBS and zwitter si-NP data are reiterated from Figure 2a. C-E) Hematocrit, hemoglobin, and red blood cell concentration values from mice 30 min after receiving a 1.2 mg/kg siRNA dose of each treatment. F) Percent of mouse liver Kupffer cells positive for Cy5 si-NPs 20 min after intravenous administration. G) Mean fluorescence intensity (MFI) of Cy5 PEG and zwitter si-NPs in mouse Kupffer cells relative to Kupffer cells of saline-treated mice (n=5). H) Representative histograms of Kupffer cell uptake of fluorescent si-NPs after intravenous delivery. I) PAF-AH activity in BMDM supernatants treated with 100 nM zwitter si-NPs or equivalent polymer concentration zwitter micelles. Significance indicated for differences between zwitter si-NP and zwitter micelle. J) Gal8 recruitment measured in GAL8-YFP expressing cells treated with 100 nM zwitter si-NPs or zwitter micelles. (* p<0.05, ** p<0.01, *** p<0.001).
Unlike zwitter si-NPs, neither PEG si-NPs nor zwitter micelles caused changes in in vivo plasma PAF-AH activity, hematocrit, hemoglobin, or red blood cell concentration when given intravenously at an equivalent dose (Figure 4b–d). Similarly, PEG si-NPs and zwitter micelles did not cause increases in organ congestion (Supplemental Figure S10). These data suggest that the combination of a phosphorylcholine-based si-NP surface and an endosomolytic core may induce PAF-mediated shock, since the endosomolytic core (PEG si-NP) and phosphorylcholine corona (zwitter micelle) individually did not induce the same symptoms in mice. These collective data indicate that a combination of Kupffer cell uptake and endosome disruption is necessary for PAF-related toxicities.
To better understand the increased sensitivity to zwitter si-NPs compared to PEG si-NPs, we measured fluorescent si-NP uptake in Kupffer cells. In mice receiving zwitter si-NPs, an average 82% of Kupffer cells took up si-NPs, while in mice receiving PEG si-NPs, only 40% of Kupffer cells took up the particles (Figure 4f). Additionally, mean fluorescent intensity was approximately two-fold higher in Kupffer cells from mice receiving zwitter si-NPs, indicating that this chemistry also achieved a higher amount of uptake per cell (Figure 4g–h) (Gating Supplemental Figure S11). These data suggest that si-NP PAF toxicities correlate to the level of acute delivery into Kupffer cells. We have previously shown that zwitter and PEG si-NPs do not adsorb complement proteins, have similar resistance to albumin adsorption, and also possess equivalent serum stability,(12) indicating that level of Kupffer cell internalization is the main driver of these toxicities. Increased cellular internalization of zwitter si-NPs by Kupffer cells is consistent with our previous observations that zwitter si-NPs were more highly internalized than PEG si-NPs by mouse tumor cells after intravenous injection.(12) Zwitter si-NPs suffer less from the “PEG dilemma” whereby increased PEGylation provides stability and improves systemic pharmacokinetics but comes at a significant cost to cell internalization and activity.(40) The increased uptake in zwitter si-NPs may result from differences in the physicochemical properties and molecular structures between PEG and zwitterionic PMPC. Water organizes in cage-like structures around PEG, but maintains its normal molecular organization around PMPC.(41) Additionally, the presence of phosphorylcholine moieties in PMPC may increase interactions with cell membranes, which contain phosphocholine-containing lipids.(42) Notably, we do anticipate that PEG si-NPs will be susceptible to PAF-associated toxicities when delivered above their maximum tolerated dose.
To further investigate the involvement of endosome escape in the PAF-related toxicity, we measured PAF-AH activity in mouse BMDMs treated with 100 nM of zwitter si-NPs and zwitter micelles. BMDMs are easy to obtain in high yields relative to Kupffer cells and have been shown to be excellent models for other aspects of the murine innate immune response, such as oxidative burst.(43) Cell supernatants were assayed for PAF-AH activity at 6, 24, 48, and 72 hours. Over time, PAF-AH activity in zwitter micelle-treated cells was similar to levels seen in PBS treated cells. In zwitter si-NP-treated cells, PAF-AH activity was decreased relative to control PBS treatment (Figure 4i). We furthermore confirmed that zwitter si-NPs do indeed induce endosome escape, while zwitter micelles do not, using a previously-described assay to quantify endosomal disruption via intracellular Galectin 8 (Gal8) tracking (14, 44). This assay utilizes cells engineered to express Gal8 fused with yellow fluorescent protein (YFP) to enable real-time Gal8 microscopic tracking in live cells. Under normal conditions, Gal8 is evenly dispersed in the cell cytosol. However, Gal8 has high affinity binding to glycosylation moieties located on the inner membrane of the endosome, and therefore, when endosome disruption occurs, Gal8 redistributes and concentrates into the endosomes. This redistribution can be visualized as bright fluorescent puncta via microscopy. Gal8-YFP expressing cells were treated with 100 nM zwitter si-NPs and zwitter micelles, and cells were imaged for resulting Gal8 redistribution. Gal8 intensity was increased in cells treated with zwitter si-NPs compared to cells treated with either PBS or zwitter micelles. In cells treated with zwitter si-NPs, puncta formation was maintained through imaging to 24 hours (Figure 4j, Supplemental Figure S12). In comparison, cells treated with zwitter micelles did not display recruitment of intense Gal8-YFP puncta (Figure 4j, Supplemental Figure S12). Together, these data suggest that endosomolytic function is a critical factor in triggering PAF-related toxicities.
The mechanistic association between endosomal escape and PAF release in macrophages merits further investigation. In accordance with our observations for zwitter si-NPs versus zwitter micelles, previous work has also demonstrated that adenovirus vectors, which are also capable of triggering PAF release from Kupffer cells, do not elicit innate immune responses when they are mutated to be endosomal escape-deficient.(21, 45) These authors found that viral endosome escape was linked to MAPK pathway activation and postulated that PAF release may also be triggered by activation of innate immune sensors such as TLRs and NLRP3 inflammasome.
We next investigated whether PAF-associated toxicities are a generalizable dose-limiting mechanism that is relevant to other types of polymeric, pH-responsive nucleic acid delivery vehicles. We injected normal mice with a subtoxic dose of 2 mg/kg of a commercially available polymeric in vivo transfection reagent, in vivo-jetPEI® (IVJP), which contains poly(ethylene imine) (PEI). PEI is considered a gold standard siRNA transfection reagent and is capable of triggering endosomolysis via the proton-sponge mechanism.(46) The 2 mg/kg dose did not reach the maximum tolerated dose in normal BALB/c mice (Supplemental Figure S13a-d).
We completed parallel experiments with IVJP on mice bearing 4T1 mammary tumors. These studies were designed to examine whether systemic inflammatory status of the mice affects sensitivity to PAF-related toxicities, as a follow up to previous empirical observations that this IVJP dose causes toxicity in tumor-bearing mice.(47) The metastatic 4T1 mammary tumor model induces a leukemoid reaction in mice, causing significant elevations in number of circulating neutrophils, monocytes, and other leukocytes (Figure 5a–b) through upregulation of 4T1 myeloid- and granulocytes-colony stimulating factors.(48) In human cancer patients, myeloid dysfunction and myelopoiesis is observed across many diverse tumor types, including breast, colon, pancreatic, prostate, and skin cancers.(49) Based on literature indicating that immune cells play a role in propagating and amplifying the vasoactive effects of PAF,(16–20) we hypothesized that PAF-associated toxicities would be enhanced in 4T1 tumor-bearing mice due to their increased number of circulating leukocytes.
Figure 5. PAF-related toxicities are generalizable to IVJP and MC3 LNP nanocarriers and are enhanced in systemically-inflamed 4T1 tumor-bearing mice.
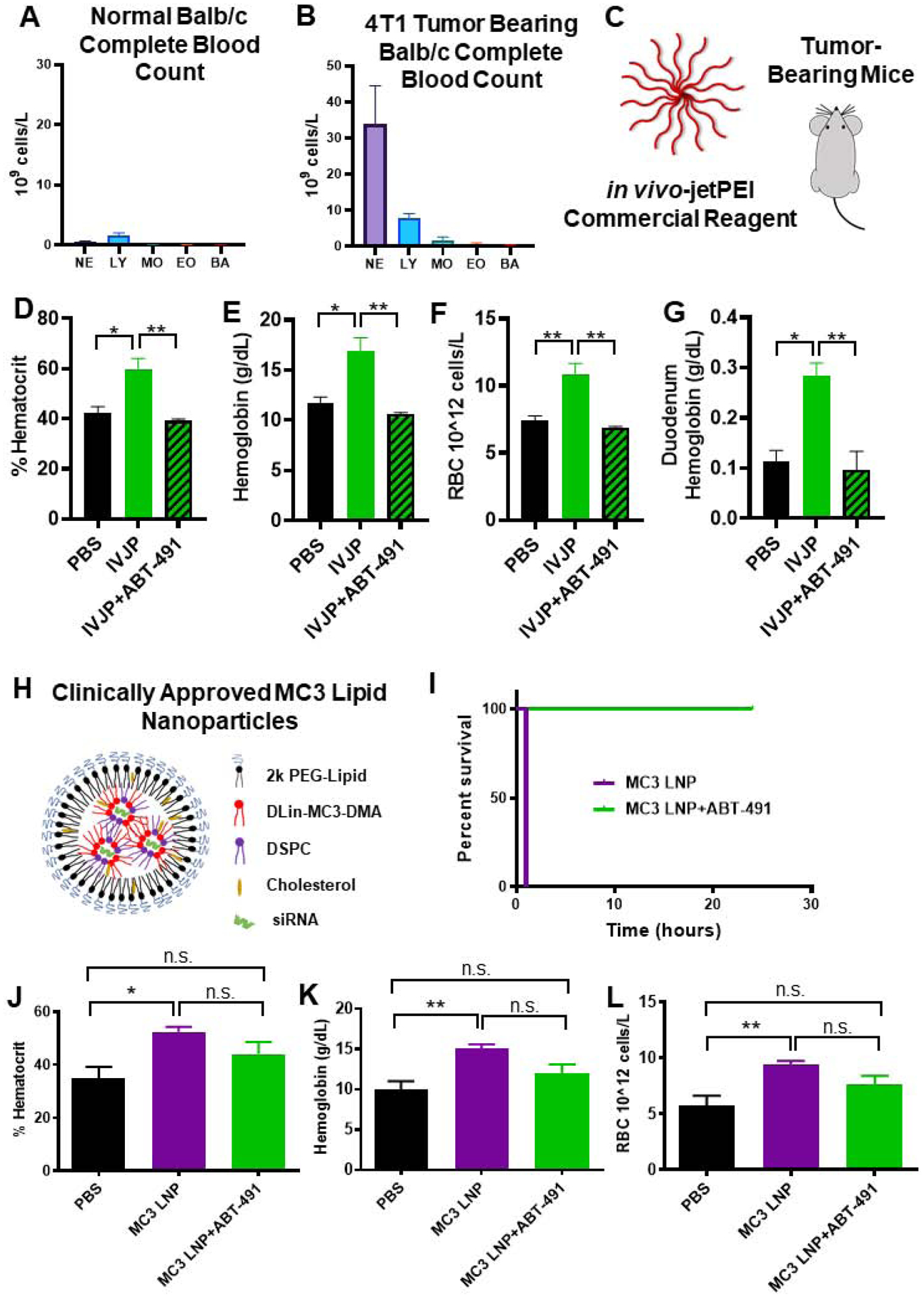
A-B) Comparison of leukocytes in complete blood counts of untreated normal BALB/c mice or 4T1 tumor-bearing mice. C) Tumor-bearing mice were injected with 2 mg/kg IVJP D-G) Blood hematocrit, hemoglobin, red blood cell concentration, and duodenum hemoglobin concentration for tumor-bearing mice treated with saline, IVJP, or ABT-491 pre-treatment in combination with IVJP (n=3 mice per group). H) Schematic illustrating components of Patisiran LNP formulation. I) Survival curve for mice injected intravenously with 5 mg/kg MC3 LNP formulation with either saline or ABT-491 pre-treatment. (n=2 mice per group) J-L) Hematocrit, hemoglobin, and red blood cell concentration values of mice treated with saline, MC3 LNPs, or MC3 LNPs with ABT-491 pre-treatment (n=4–5). Blood measurements were made 20 min after administration of MC3 LNPs. (* p<0.05, **p<0.01) NE, neutrophils; LY, lymphocytes; MO, monocytes; EO, eosinophils; BA, basophils.
In 4T1 tumor-bearing mice, IVJP treatment significantly increased blood hematocrit, hemoglobin, red blood cell concentration, and duodenum hemoglobin compared to saline-treated mice (Figure 5c–g), indicating PAF-induced toxicities in these mice. Pre-treatment with ABT-491 completely abrogated these blood changes, confirming involvement of PAFR in IVJP-related toxicity. Additionally, IVJP treatment in tumor-bearing mice significantly decreased plasma PAF-AH activity compared to saline-treated, tumor-bearing mice (Supplemental Figure S13e). In non-tumor bearing mice, no changes in PAF-AH activity occurred for either treatment group (Supplemental Figure S13e). Our data indicate that PAF-associated toxicities are generalizable to other types of pH-responsive, polymeric nanoparticle systems and that nanoparticle doses that are in a tolerable range for healthy animals can trigger PAF toxicities in scenarios where systemic tumor-associated inflammation is present.
We last sought to assess the generalizability of this mechanism to lipid-based nucleic acid delivery systems. We tested a similar lipid nanoparticle (LNP) formulation to clinically-approved Patisiran, including the ionizable lipid DLin-MC3-DMA, at a high dose that induces toxicity (Figure 5h). While this LNP formulation is reported to be tolerated in non-tumor-bearing mice at doses as high as 15 mg/kg,(50) it induced rapid fatality in 4T1 tumor-bearing mice at 5 mg/kg (54.38 mg/kg DLin-MC3-DMA dose) (Figure 5i). Pre-treatment of these mice with the PAFR inhibitor ABT-491 completely prevented fatalities (Figure 5i). Complete blood count measurements taken 20 minutes after LNP injection revealed increased hematocrit, hemoglobin, and red blood cell concentrations (Figure 5j–l). Mice pre-treated with ABT-491 prior to LNP injection did not experience significant increases in hemoconcentration compared to saline-treated mice. These data indicate that inhibition of the PAF receptor prevents LNP-induced toxicities in tumor-bearing mice and suggest that PAF has broad implications in siRNA nanomedicine-related toxicities. The LNP finding also has significant clinical implications, as Patisiran and other LNP-based formulations are either currently FDA-approved or in clinical trials.
The increased sensitivity to PAF signaling in 4T1 tumor-bearing mice was associated with the presence of increased numbers of circulating leukocytes, highlighted by a 22-fold increase in monocytes and a 61-fold expansion of circulating neutrophils (Figure 5a–b). These results are consistent with previous data showing that 4T1 tumors sensitize mice to anaphylactoid-type reactions in response to adenovirus treatment.(51) PAF is known to activate monocyte secretion of inflammatory cytokines and to promote chemoattraction, adhesion, and degranulation of neutrophils, all of which can affect vascular permeability(52–56). Our collective data indicate that Kupffer cells are stimulated by high doses of nucleic acid-bearing nanomedicines to release PAF, which in turn acts through circulating leukocytes as an intermediate that produces vasoactive agents (such as nitric oxide)(57–60) and consequent shock-like toxicities (Figure 6).
Figure 6.
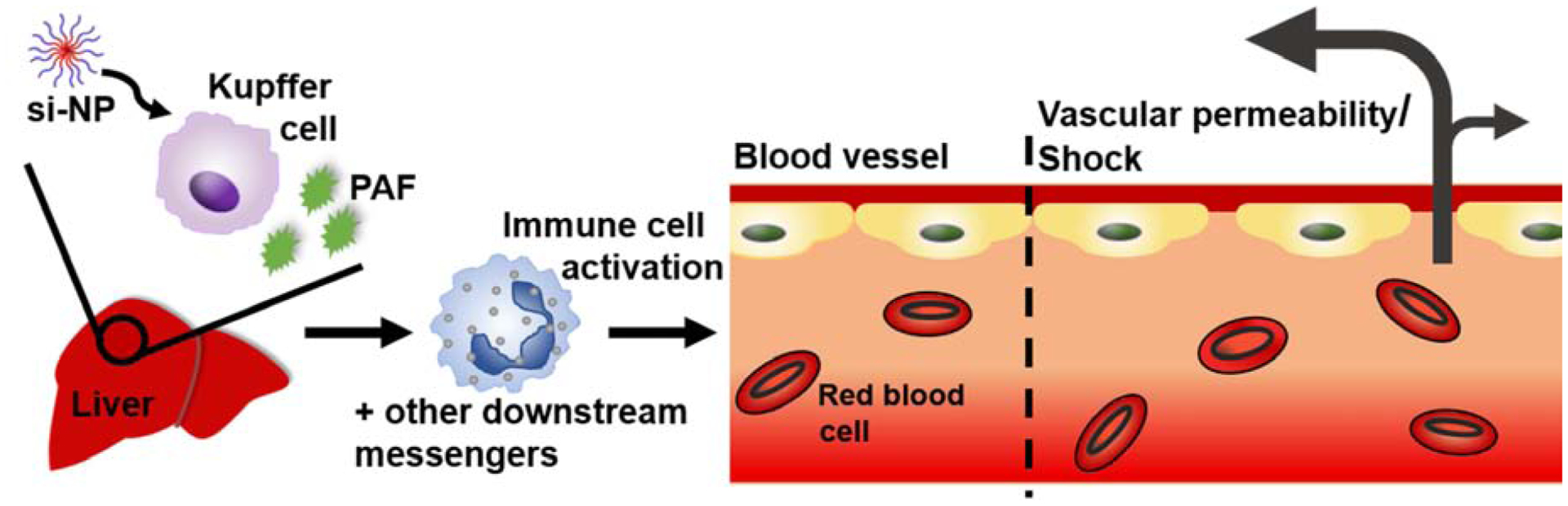
Schematic of proposed mechanism for PAF-mediated effects upon intravenous administration of nucleic acid nanocarriers.
Our findings have important potential clinical implications, particularly for the use of siRNA nanomedicines in cancer treatment. The increased sensitivity of tumor-bearing animals to PAF-mediated toxicity may carry over to human cancer patients, who often exhibit myeloid dysfunction, elevated platelet counts, and an increased risk of clotting due to the secretion of pro-clotting factors by tumor macrophages.(48, 49, 61) While PAFR is present on all of the same cell types in humans as in rodents, humans additionally possess PAFR on platelets.(16) Thus, nanoparticle and PAF-mediated toxicities are expected to be similar or potentially even more pronounced in humans, particularly in cancer patients who are in a hyper-coagulative state. Indeed, adverse events reported in the Phase 1a/1b clinical trial for cancer patients receiving cyclodextrin-based siRNA carrier CALAA-01 included hypersensitivity and GI effects such as ischemic colitis and edema as well as events such as thrombocytopenia and tachycardia, all of which can be potentially related to effects caused by PAF.(11)
The broadly-applicable role of PAF in dose-limiting toxicities of siRNA nanomedicines shown here may also provide new clues on toxicity observations in previous studies. For example, multiple studies in rodents have reported rapid fatalities within the first hour of injection of PEI polyplexes, although this lethality is often blamed on pulmonary clotting due to cationic polymer components.(7, 8) However, other in-depth studies of in vivo PEI-mediated toxicities have shown evidence of liver necrosis, shock, and smaller aggregates of CD11b+ cells and platelets, without major lung obstruction.(6) The connection between PAF release and nucleic acid nanocarrier uptake by Kupffer cells may extend beyond endosomolytic, siRNA-bearing nanoparticles. In humans, adenovirus and nanoparticle Doxil injections have been associated with adverse events including hypotension and decreased cardiac output, consistent with effects of PAF.(62, 63) The potential implications with Doxil are supported by the noted PAF involvement in the toxicity profile of liposome-encapsulated hemoglobin,(24) and in anaphylactic (antibody-mediated) reactions following repeated administration of PEGylated liposomes.(23) Though not previously explored, it will be interesting to study whether Kupffer cell production of PAF dictates the therapeutic window for these and other preclinical and clinically-approved nanomedicines.
Prophylactic treatment protocols are already the norm in clinically used nanomedicines. Onpattro, the only FDA-approved siRNA nanomedicine, requires pre-treatment of patients with corticosteroids to dampen immune activation.(1) While no PAFR antagonists have yet been clinically approved in the U.S., rupatadine is a combination anti-histamine and PAFR inhibitor approved for use in other countries.(17, 21) PAFR inhibitors such as ABT-491 or rupatadine may find new clinical benefit by making nanomedicines safer and to potentially enable use of higher, more efficacious doses by expanding their therapeutic index.
Conclusion
Overall, this work provides compelling evidence that PAF is the key factor in determining dose-limiting toxicities of polymer- and lipid-based siRNA nanomedicines. We provide new insights that PAF release is dependent on level of delivery of endosome-escaping nanocarriers to Kupffer cells and that the presence of systemic inflammation, associated with higher circulating immune cells, predisposes mice to PAF-mediated toxicities. We have also shown that PAF-related toxicities are generalizable to other types of polymeric and lipid nanocarriers, including commercially available IVJP and cationic lipid nanoparticles that match FDA-approved Patisiran, indicating that this may be an unrealized and under-studied toxicity mechanism for other types of particles. Inhibition of PAFR prior to nanoparticle administration can completely prevent acute shock-associated toxicities in mice, suggesting that PAFR is a viable prophylactic target that can be used to broaden the therapeutic window and improve clinical management of adverse events related to siRNA nanomedicines.
Materials and Methods
All materials were purchased from Sigma-Aldrich (St. Louis, MO, USA) or Fisher Scientific (Waltham, MA, USA) unless indicated otherwise. siRNAs used in these studies were purchased from Integrated DNA Technologies (Coralville, IA, USA) and consisted of a sense strand (5’-CAAUUGCACUGAUAAUGAACUCC[dT][dC]-3’) and antisense strand (5’-GAGGAGUUCA[mU]U[mA]A[mU]U[mG][mU][mU]-3’) [Note: m=2’O-methyl modification; d=chimeric DNA base]. In flow cytometry experiments, Cy-5 labeled DNA oligonucleotides (IDT) were used instead of siRNA, and consisted of sense strand (5’-dGdTdCdAdGdAdAdAdTdAdGdAdAdAdCdTdGdGdTdCdAdTdC-3’) and antisense strand (5’-[Cy5]dGdAdTdGdAdCdCdAdGdTdTdTdCdTdAdTdTdTdCdTdGdAdC-3’).
Polymer Synthesis and Characterization (NMR, GPC, DLS)
Polymers were made using Reversible Addition Fragmentation Chain Transfer (RAFT) polymerization using a chain transfer agent (CTA) of 4-(ethylsulfanylthiocarbonyl)sulfanylpentanoic acid (ECT). All monomers except 2-methacryloyloxyethyl phosphorylcholine were passed twice through activated alumina columns to remove inhibitors. PMPC-DB polymers (poly(2-methacryloyloxyethyl phosphorylcholine (PMPC))-b-(dimethylaminoethyl methacrylate (DMAEMA)-co-butyl methacrylate (BMA))) were synthesized by first creating a random copolymer of 50% DMAEMA and 50% BMA monomers polymerized from ECT in dioxane under nitrogen (30-minute purge) for 24 h at 65°C using azobisisobutyronitrile (AIBN) as an initiator (1:10 CTA:AIBN ratio). Equivalent molar amounts of DMAEMA and BMA were added to target an overall degree of polymerization (DP) of 210 repeating units overall (anticipating ~ 70% conversion and final DP of 150–160). The crude reaction was precipitated in ice cold pentane three times to remove monomers and dried under vacuum overnight. DP was confirmed using 1H-NMR and GPC. From this purified first block (DB-ECT), the PMPC hydrophilic, si-NP surface-forming block was polymerized in methanol for 24 hours at 65°C under nitrogen (30-minute purge) with a 1:5 CTA:AIBN ratio. A DP of 75 was targeted to achieve an approximately 20,000 g/mol corona block (anticipating 90–95% conversion). The crude reaction mixture was then dialyzed overnight in methanol and then in deionized water for one day. The polymer was lyophilized, and PMPC block degree of polymerization was confirmed with 1H-NMR. The control PMPC-BMA polymer (zwitter micelle) was similarly synthesized, starting from a PBMA homopolymer. The PMPC block was polymerized in pure ethanol with all other conditions the same to improve solubility of the PBMA macroCTA. The 20kPEG DB polymer (PEG si-NP) was synthesized and characterized as described previously.(12) All NMR spectra are featured in Supplemental Figures S1, S9a-b, S14. Polymers were evaluated for endotoxin using a Chromogenic LAL Endotoxin Assay Kit (GenScript, Nanjing, China).
si-NP Formulations
Zwitter si-NPs and PEG si-NPs were formulated as described previously.(12) Briefly, polymers were complexed with siRNA at a 20:1 N:P ratio (protonated nitrogens on polymer: anionic phosphates on siRNA) in 10 mM citrate buffer at pH 4. Polymers were initially dissolved at 30 mg/mL in pure ethanol and then diluted to 3 mg/mL in the pH 4 citrate buffer. After 30 minutes, samples were supplemented by addition of a 5x volume excess of 10 mM phosphate buffer at pH 8 to achieve a final pH of 7.4. PMPC-BMA micelles were dissolved in ethanol (5% of the final si-NP solution volume) and added dropwise to PBS for a final concentration equivalent to the polymer concentration of PMPC-DB for delivering 1.2 mg/kg siRNA (roughly 18 mg/mL). For in vivo preparations, polyplexes and micelles were sterile filtered (0.45 μm filters) and then concentrated to the appropriate volume (1.2 mg/kg dose in 100 μL injections) in Amicon spin filters (MWCO 50,000 kDa).
PAFR Inhibition Studies
Mice (BALB/c, C57BL/6) were purchased from Jackson Laboratories (Bar Harbor, ME) or Charles River Laboratories (Wilmington, MA). For PAFR inhibition studies, mice were pre-injected with either 50 μL saline or 50 μL of ABT-491 (0.05 mg/mL in PBS −/−). Ten minutes after the first injection, mice were then injected with 1.2 mg/kg si-NPs (PMPC-DB polyplexes). A separate cohort was injected with saline only. For hematology and pathology studies, mice were sacrificed 30 minutes following si-NP injection, and blood was harvested by cardiac puncture using EDTA as an anti-coagulant. Blood samples were analyzed for complete blood counts (including hematocrit, hemoglobin, and red blood cell measures) by the Vanderbilt Translational Pathology Shared Resource using a Forcyte Analyzer (Oxford Science, CT, USA). Plasma chemistry (AST, ALT, BUN) was also analyzed by Translation Pathology Shared Resource using the Alfa Wasserman ACE Alera system. Plasma cytokines (IL-1b, IL-6, TNFa) were assayed by the Vanderbilt Hormone and Analytical Services Core using Multiplex Luminex technology. Mouse organs were removed, except for the pancreas and duodenum, and were fixed in 10% formalin for histologic analysis.
The pancreas was weighed after removal, frozen, lyophilized, and weighed again for pancreas wet/dry weight analysis. The duodenum was washed with a saline solution containing 0.16 mg/mL heparin to remove excess blood. The tissue was then weighed and homogenized in 1 mL of saline/heparin solution. The tissue was centrifuged at 10,000 xg for 15 minutes at 4°C. Supernatants were removed and assayed for hemoglobin content using a Hemoglobin Colorimetric Assay Kit (Cayman Chemical No 700540) according to manufacturer instructions. Fixed tissues were embedded in paraffin and stained with hematoxylin and eosin by the Vanderbilt Translational Pathology Shared Resource. For Evans Blue measurements of vascular leakiness, mice were injected with 50 μL of a 0.5% (wt/v%) Evans Blue solution in saline that had been sterile-filtered through a 0.45 μm filter 2 minutes after si-NP (or saline) injection. After sacrificing mice at 30 minutes post-injection, livers were removed, weighed, and then incubated in 1 mL formamide for 24 hours at room temperature. A 50 uL aliquot of formamide for each sample was then transferred to a 96-well plate, and Evans Blue absorbance was measured at 610 nm using a plate reader (Tecan, Mannedorf, Switzerland).
PAF-AH Activity Assay
For in vivo studies, mice were intravenously injected with nanoparticles and blood was collect at 15 minutes post-injection. Mouse EDTA-treated blood samples were centrifuged at 1,000xg for 10 min at 4°C and plasma was isolated for analysis. Mouse PAF-AH activity kits were purchased from Cayman Chemical (No 760901). Plasma samples were assayed in duplicate according to the supplier’s protocol using 10 μL samples in each well.
For in vitro PAF-AH activity assays, mouse bone marrow-derived macrophages (BMDMs) were extracted from the femurs and tibias of healthy female FVB mice sacrificed at 4–8 weeks of age. Bone marrow was flushed out with DMEM using a 5 mL syringe and collected in DMEM on ice. The cell suspension was centrifuged at 1000xg for 5 min, and the pellet was resuspended in 2 mL ACK Lysis buffer and incubated for 2 minutes on ice. The lysis solution was then diluted in 20 mL warm DMEM (37°C) and centrifuged again at 1000xg for 5 minutes. BMDMs were then re-suspended in 10 mL of BMDM media (DMEM, 10% FBS, 1% PenStrep, 1% L-glutamine, 14% (1:1 v/v) L929 media week 1 and week 2 media [created by culturing L929 murine fibroblast cells in complete DMEM and collecting media after 7 days of incubation [week 1] and again 7 days later [week 2]]). The cells were then seeded in a 24-well plate at 2.5×105 cells per well. At 4 days after seeding, media was removed, and cells were treated with 100 nM si-NPs. Cell supernatants were sampled at 6, 24, 48, and 72 hours post-treatment and assayed using the PAF-AH activity kit.
Gal8-YFP Endosomal Disruption Imaging
Gal8-YFP measurement were carried out as previously described.(14, 44) Gal8-YFP expressing MDA-MB-231 cells were seeded at 2,000 cells/well in opaque half-area 96-well plates. After allowing cells to adhere for 24 hours, cells were treated with 100 nM si-NPs or equivalent polymer concentration zwitter micelles. Cells were imaged for Gal8 recruitment by automated fluorescent microscopy (Nikon C1si+ confocal microscope system on a Nikon Eclipse Ti-0E inverted microscope base, Plan Apo VC 20× objective, Galvano scanner, and 408/488/543 dichroic mirror). Wells were imaged by a software-controlled motorized stage that moved the plate between images. Cells were imaged at 30 minutes, and at 1, 2, 4, 6, 8,10, and 24 hours. Recruited Gal8 was identified by a MATLAB script recognizing Gal8-YFP fluorescent puncta and intensity was normalized to total cell area.
Clodronate Liposome Studies
Clodronate liposome pre-treatment was used to deplete Kupffer cells.(64) For animal studies involving clodronate pre-treatment, half of the mice were injected both i.v. and i.p. with 100 μL of Clodronate Liposomes (Cedarlane, Burlington, Canada) 24 hours prior to si-NP treatments. Both normal mice and clodronate-treated mice were then either injected with si-NPs or saline 24 hours later and sacrificed 30 minutes after si-NP injection. Organs and blood were collected and processed as described above.
PAF ELISA
Mouse EDTA-treated blood samples were centrifuged at 3,000xg for 10 min at 4°C and plasma was isolated for ELISA analysis. Mouse PAF ELISA kits were purchased from LifeSpan Biosciences (Seattle, WA, USA). Plasma samples were assayed in duplicate according to kit manual using 50 μL samples in each well.
PAFR Activation Studies
Ready-to-Assay Platelet Activating Factor Receptor Cells (Millipore Sigma, Burlington, MA) (Chem-1 host cells overexpressing PAFR GPCR) were thawed and seeded at 14,000 cells per well in a 384-well plate according to manufacturer instructions. Cells were then washed with HBSS (20 mM HEPES) and loaded with FLIPR Calcium 6 dye (dissolved in HBSS [20mM HEPES, 2.5 mM probenecid] and incubated for 2 hours according to FLIPR Calcium 6 kit instructions. si-NPs were prepared at 50 nM siRNA (final in-well concentrations) in a 384-well plate, and ABT-491 was prepared at 135 μM in a separate plate. Fluorescence from calcium influx was assayed using the Vanderbilt High Throughput Screening Facility, on a Panoptic instrument (WaveFront Biosciences, Franklin, TN). Five μL of ABT-491 or saline were first added to the cells, incubated for 5 minutes, and then 12.5 μL of si-NP preparations, saline, or PAF positive control (28.3 μg/mL stock) were added to the cells, and fluorescence was recorded once per second for 20 minutes following si-NP addition.
In Vivo Kupffer Cell Uptake
Wild type BALB/c mice (Jackson Laboratories) were injected with either saline or with zwitter si-NPs or PEG si-NPs bearing 1.2 mg/kg Cy5-labeled DNA, prepared as described above. Twenty minutes after injection, mice were euthanized, perfused with PBS, and livers were removed and placed in 3 mL RPMI containing 5% FBS. An additional 3 mL of RPMI containing 4 mg/mL Collagenase II was added and incubated at 37°C for 30 minutes on an orbital shaker at 200 rpm. After digest, livers were filtered through 100 μm filters and hepatocytes were pelleted at 50xg for 2 min. Supernatants were collected and spun at 1200xg for 8 minutes. Pelleted cells were resuspended in 10 mL of clear 1X HBSS. A Percoll gradient was prepared consisting of a bottom layer of 20 mL 50% Percoll and a middle layer of 20 mL of 25% Percoll. Cells in HBSS suspension were then gently added to the top of the gradient. Samples were centrifuged 15 min at 800xg with no brakes. The buffy coat interface layer was removed carefully and placed in 40 mL of PBS containing 1% FBS. Cells were centrifuged 5 min at 500xg and then subjected to red blood cell lysis in 3 mL ACK buffer, after which time 20 mL of PBS (1% FBS) were added, and cells were again spun at 500xg for 5 min. Cells were resuspended in 400 uL FACS buffer containing Fc block (1:100) for 5 minutes, and passed through a 35 μm filter. Cells were centrifuged again at 500 xg for 5 minutes, and then resuspended in 200 uL FACS buffer. Samples were kept on ice.
Cells were immunolabeled using antibodies (Biolegend) reactive against F4/80 (PE; Clone BM8; 1:200), CD45 (PerCp-Cy5.5; Clone 30-F11; 1:4000), and CD11b (FITC; Clone M1/70; 1:200). Immediately prior to flow cytometry, cells were stained for viability with 4’,6-diamidino-2-phenylindole (DAPI, 0.25 μg/mL). Flow cytometry was performed on a MACSQuant Analyzer 10 (Miltenyi Biotec). Data analysis was performed using FlowJo software (FlowJo LLC, Ashland, OR). Kupffer cell gating was performed as described in Lynch et al and absence of non-specific antibody binding was confirmed using fluorescence-minus-one controls.(65)
Tumor Mice, IVJP, and LNPs
Seven-week-old female BALB/c mice were implanted with 1×105 4T1 cells per tumor (in 50:50 mixture of serum-free DMEM and Matrigel (Corning, #354234) in the inguinal mammary fatpad (2 tumors per mouse). Nanoparticle and micelle treatments were initiated once tumors had reached 150–300 mm3. All tissue processing/endpoint timing occurred as stated above for the PAFR inhibition studies. In vivo-jetPEI (IVJP) (Polyplus, NY, USA) was prepared according to manufacturer instructions, at a dose of 2 mg/kg siRNA. Lipid nanoparticles (LNPs) were prepared at a dose of 5 mg/kg siRNA, and consisted of DLin-MC3-DMA [(6Z,9Z, 28Z, 31Z)-Heptatriaconta-6,9,28,31-tetraen-19-yl 4-(dimethylamino) butanoate), (MedKoo Biosciences, Morrisville, NC)], DSPC (1,2-distearoyl-sn-glycero-3-phosphocholine, Avanti Polar Lipids Inc, AL, USA), Cholesterol (Avanti Polar Lipids Inc, AL, USA), and DMG-PEG 2000 (1,2-dimyristoyl-rac-glycero-3-methoxypolyethylene glycol-2000, Avanti), prepared at molar ratios of 50:10:37.5:2.5 and a total siRNA:lipid weight ratio of 0.06, as described in Yamamoto et al.(66) Lipids and siRNA were mixed in a NanoAssemblr (Precision NanoSystems, Vancouver, BC, CA), at a flow rate of 2 mL/min (1:2 flow ratio, lipids dissolved in ethanol at 33% of final volume, siRNA in sodium citrate buffer, pH 4). LNP formulation was dialyzed in sterile PBS −/− overnight prior to i.v. administration. Mice injected with LNPs were sacrificed 20 minutes after injection.
Supplementary Material
Acknowledgements
This work was supported by the DOD (DOD CDMRP OR130302), NIH (NIH R01 CA224241, NIH R01 EB019409, NIH R01 DK084246), NSF CAREER BMAT 1349604, the Vanderbilt Engineering and Immunity Pilot and Feasibility Grant, and the National Science Foundation (NSF GRF 1445197, NSF GRF 1937963).
The authors also acknowledge the assistance of the Vanderbilt Translational Pathology Shared Resource (TPSR), supported by NCI/NIH Cancer Center Support Grant 2P30 CA068485-14 and the Vanderbilt Mouse Metabolic Phenotyping Center Grant 5U24DK059637-13, for plasma chemistry and complete blood counts. The authors also acknowledge the Hormone Assay and Analytical Services Core (NIH grants DK059637 and DK02059) for plasma cytokine analysis.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Competing Interests
The authors declare no competing interests.
Data Availability
The raw/processed data required to reproduce these findings are available upon request.
References
- 1.Setten RL, Rossi JJ, Han S. P. A.-O. h. o. o., The current state and future directions of RNAi-based therapeutics. LID - 10.1038/s41573-019-0017-4 [doi]. [DOI] [PubMed] [Google Scholar]
- 2.Rizk M, Tüzmen Ş, Update on the clinical utility of an RNA interference-based treatment: focus on Patisiran. Pharmacogenomics and personalized medicine 10, 267–278 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Hall A, Lachelt U, Bartek J, Wagner E, Moghimi SM, Polyplex Evolution: Understanding Biology, Optimizing Performance. Mol Ther 25, 1476–1490 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Parhiz H et al. , Unintended effects of drug carriers: Big issues of small particles. Advanced Drug Delivery Reviews 130, 90–112 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Merkel OM et al. , In vitro and in vivo complement activation and related anaphylactic effects associated with polyethylenimine and polyethylenimine-graft-poly(ethylene glycol) block copolymers. Biomaterials 32, 4936–4942 (2011). [DOI] [PubMed] [Google Scholar]
- 6.Chollet P, Favrot A Mc Fau - Hurbin, Hurbin J-L A Fau - Coll, Coll JL, Side-effects of a systemic injection of linear polyethylenimine-DNA complexes. [DOI] [PubMed]
- 7.Ogris M, Brunner S, Schuller S, Kircheis R, Wagner E, PEGylated DNA/transferrin-PEI complexes: reduced interaction with blood components, extended circulation in blood and potential for systemic gene delivery. Gene Ther 6, 595–605 (1999). [DOI] [PubMed] [Google Scholar]
- 8.Merdan T et al. , PEGylation of Poly(ethylene imine) Affects Stability of Complexes with Plasmid DNA under in Vivo Conditions in a Dose-Dependent Manner after Intravenous Injection into Mice. Bioconjugate Chemistry 16, 785–792 (2005). [DOI] [PubMed] [Google Scholar]
- 9.Liu Y, Xu C-F, Iqbal S, Yang X-Z, Wang J, Responsive Nanocarriers as an Emerging Platform for Cascaded Delivery of Nucleic Acids to Cancer. Advanced Drug Delivery Reviews 115, 98–114 (2017). [DOI] [PubMed] [Google Scholar]
- 10.Moghimi SM, Complement Propriety and Conspiracy in Nanomedicine: Perspective and a Hypothesis. Nucleic Acid Ther 26, 67–72 (2016). [DOI] [PubMed] [Google Scholar]
- 11.Zuckerman JE et al. , Correlating animal and human phase Ia/Ib clinical data with CALAA-01, a targeted, polymer-based nanoparticle containing siRNA. Proceedings of the National Academy of Sciences 111, 11449–11454 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Jackson MA et al. , Zwitterionic Nanocarrier Surface Chemistry Improves siRNA Tumor Delivery and Silencing Activity Relative to Polyethylene Glycol. ACS Nano (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Nelson CE et al. , Balancing Cationic and Hydrophobic Content of PEGylated siRNA Polyplexes Enhances Endosome Escape, Stability, Blood Circulation Time, and Bioactivity in Vivo. ACS Nano 7, 8870–8880 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kilchrist KV et al. , Gal8 Visualization of Endosome Disruption Predicts Carrier-Mediated Biologic Drug Intracellular Bioavailability. ACS Nano 13, 1136–1152 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Jackson MA et al. , Dual carrier-cargo hydrophobization and charge ratio optimization improve the systemic circulation and safety of zwitterionic nano-polyplexes. Biomaterials 192, 245–259 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Gill P, Jindal NL, Jagdis A, Vadas P, Platelets in the immune response: Revisiting platelet-activating factor in anaphylaxis. Journal of Allergy and Clinical Immunology 135, 1424–1432 (2015). [DOI] [PubMed] [Google Scholar]
- 17.Upton J, Vadas P, Potential Therapeutic Strategies for Severe Anaphylaxis Targeting Platelet-Activating Factor and PAF Acetylhydrolase. Current Treatment Options in Allergy 1, 232–246 (2014). [Google Scholar]
- 18.Weijer S et al. , Improved Host Defense against Pneumococcal Pneumonia in Platelet-Activating Factor Receptor-Deficient Mice. The Journal of Infectious Diseases 189, 711–716 (2004). [DOI] [PubMed] [Google Scholar]
- 19.Iñarrea P, Alonso F, Sanchez-Crespo M, Platelet-activating factor: An effector substance of the vasopermeability changes induced by the infusion of immune aggregates in the mouse. Immunopharmacology 6, 7–14 (1983). [DOI] [PubMed] [Google Scholar]
- 20.Albert DH et al. , Pharmacology of ABT-491, a highly potent platelet-activating factor receptor antagonist. European Journal of Pharmacology 325, 69–80 (1997). [DOI] [PubMed] [Google Scholar]
- 21.Xu Z, Smith JS, Tian J, Byrnes AP, Induction of shock after intravenous injection of adenovirus vectors: a critical role for platelet-activating factor. Mol Ther 18, 609–616 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Schiedner G et al. , A hemodynamic response to intravenous adenovirus vector particles is caused by systemic Kupffer cell-mediated activation of endothelial cells. Hum Gene Ther 14, 1631–1641 (2003). [DOI] [PubMed] [Google Scholar]
- 23.Judge A, McClintock K, Phelps JR, MacLachlan I, Hypersensitivity and Loss of Disease Site Targeting Caused by Antibody Responses to PEGylated Liposomes. Molecular Therapy 13, 328–337 (2006). [DOI] [PubMed] [Google Scholar]
- 24.Rabinovici R, Rudolph AS, Yue TL, Feuerstein G, Biological responses to liposome-encapsulated hemoglobin (LEH) are improved by a PAF antagonist. Circulatory shock 31, 431–445 (1990). [PubMed] [Google Scholar]
- 25.MacParland SA et al. , Phenotype Determines Nanoparticle Uptake by Human Macrophages from Liver and Blood. ACS Nano 11, 2428–2443 (2017). [DOI] [PubMed] [Google Scholar]
- 26.Tavares AJ et al. , Effect of removing Kupffer cells on nanoparticle tumor delivery. Proceedings of the National Academy of Sciences 114, E10871 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Decker K, Biologically active products of stimulated liver macrophages (Kupffer cells). Eur J Biochem 192, 245–261 (1990). [DOI] [PubMed] [Google Scholar]
- 28.Albert DH et al. , The Role of Platelet-Activating Factor (PAF) and the Efficacy of ABT-491, a Highly Potent and Selective PAF Antagonist, in Experimental Allergic Rhinitis. Journal of Pharmacology and Experimental Therapeutics 284, 83 (1998). [PubMed] [Google Scholar]
- 29.Kelefiotis D, Vakirtzi-Lemonias C, In vivo responses of mouse blood cells to platelet-activating factor (PAF): role of the mediators of anaphylaxis. Agents Actions 40, 150–156 (1993). [DOI] [PubMed] [Google Scholar]
- 30.Au - Wick MJ, Au - Harral JW, Au - Loomis ZL, Au - Dempsey EC, An Optimized Evans Blue Protocol to Assess Vascular Leak in the Mouse. JoVE, e57037 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Seemann S, Zohles F, Lupp A, Comprehensive comparison of three different animal models for systemic inflammation. Journal of Biomedical Science 24, 60 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Cassel SL et al. , Inflammasome-independent IL-1β mediates autoinflammatory disease in Pstpip2-deficient mice. Proceedings of the National Academy of Sciences 111, 1072 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Jones SW et al. , Nanoparticle clearance is governed by Th1/Th2 immunity and strain background. The Journal of Clinical Investigation 123, 3061–3073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Cao H-C, Chen X-M, Xu W, Determination of platelet-activating factor by reverse phase high-performance liquid chromatography and its application in viral hepatitis. World journal of gastroenterology 11, 7364–7367 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Tsukioka K, Matsuzaki M, Nakamata M, Kayahara H, [Increased plasma levels of platelet-activating factor (PAF) and low serum PAF acetylhydrolase (PAFAH) activity in adult patients with bronchial asthma]. Arerugi = [Allergy] 42, 167–171 (1993). [PubMed] [Google Scholar]
- 36.Graham RM et al. , Plasma degradation of platelet-activating factor in severely ill patients with clinical sepsis. Crit Care Med 22, 204–212 (1994). [DOI] [PubMed] [Google Scholar]
- 37.Vadas P et al. , Platelet-Activating Factor, PAF Acetylhydrolase, and Severe Anaphylaxis. New England Journal of Medicine 358, 28–35 (2008). [DOI] [PubMed] [Google Scholar]
- 38.Ma X et al. , Epinephrine inhibits vascular hyperpermeability during platelet-activating factor- or ovalbumin-induced anaphylaxis. RSC Advances 7, 52762–52771 (2017). [Google Scholar]
- 39.Hergott CB et al. , Bacterial exploitation of phosphorylcholine mimicry suppresses inflammation to promote airway infection. The Journal of Clinical Investigation 125, 3878–3890 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Pelaz B et al. , Surface Functionalization of Nanoparticles with Polyethylene Glycol: Effects on Protein Adsorption and Cellular Uptake. ACS Nano 9, 6996–7008 (2015). [DOI] [PubMed] [Google Scholar]
- 41.Schlenoff JB, Zwitteration: Coating Surfaces with Zwitterionic Functionality to Reduce Nonspecific Adsorption. Langmuir 30, 9625–9636 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Li Z, Vance DE, Thematic Review Series: Glycerolipids. Phosphatidylcholine and choline homeostasis. Journal of Lipid Research 49, 1187–1194 (2008). [DOI] [PubMed] [Google Scholar]
- 43.Cunnick J, Kaur P, Cho Y, Groffen J, Heisterkamp N, Use of bone marrow-derived macrophages to model murine innate immune responses. J Immunol Methods 311, 96–105 (2006). [DOI] [PubMed] [Google Scholar]
- 44.Kilchrist KV, Tierney JW, Duvall CL, Genetically Encoded Split-Luciferase Biosensors to Measure Endosome Disruption Rapidly in Live Cells. ACS Sensors 5, 1929–1936 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Smith JS et al. , The role of endosomal escape and mitogen-activated protein kinases in adenoviral activation of the innate immune response. PloS one 6, e26755–e26755 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Behr J-P, The Proton Sponge: a Trick to Enter Cells the Viruses Did Not Exploit (1997), vol. 51, pp. 34–36. [Google Scholar]
- 47.Sarett SM et al. , Hydrophobic interactions between polymeric carrier and palmitic acid-conjugated siRNA improve PEGylated polyplex stability and enhance in vivo pharmacokinetics and tumor gene silencing. Biomaterials 97, 122–132 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.duPre SA, Hunter KW, Murine mammary carcinoma 4T1 induces a leukemoid reaction with splenomegaly: Association with tumor-derived growth factors. Experimental and Molecular Pathology 82, 12–24 (2007). [DOI] [PubMed] [Google Scholar]
- 49.Messmer MN, Netherby CS, Banik D, Abrams SI, Tumor-induced myeloid dysfunction and its implications for cancer immunotherapy. Cancer immunology, immunotherapy : CII 64, 1–13 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Kozauer N (2018) NDA 210922 Patisiran Cross-Discipline Team Leader Review. (Center for Drug Evaluation and Research, Food and Drug Administration, https://www.accessdata.fda.gov/drugsatfda_docs/nda/2018/210922Orig1s000MultiR.pdf). [Google Scholar]
- 51.Pande K et al. , Cancer-induced expansion and activation of CD11b+ Gr-1+ cells predispose mice to adenoviral-triggered anaphylactoid-type reactions. Molecular therapy : the journal of the American Society of Gene Therapy 17, 508–515 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Bonavida B, Mencia-Huerta JM, Braquet P, Effects of platelet-activating factor on peripheral blood monocytes: induction and priming for TNF secretion. Journal of lipid mediators 2 Suppl, S65–76 (1990). [PubMed] [Google Scholar]
- 53.Hauser CJ et al. , PAF-mediated Ca2+ influx in human neutrophils occurs via store-operated mechanisms. Journal of Leukocyte Biology 69, 63–68 (2001). [PubMed] [Google Scholar]
- 54.Bautista AP, Spitzer JJ, Platelet Activating Factor Stimulates and Primes the Liver, Kupffer Cells and Neutrophils to Release Superoxide Anion. Free Radical Research Communications 17, 195–209 (1992). [DOI] [PubMed] [Google Scholar]
- 55.Wang Q, Doerschuk CM, The Signaling Pathways Induced by Neutrophil-Endothelial Cell Adhesion. Antioxidants & Redox Signaling 4, 39–47 (2002). [DOI] [PubMed] [Google Scholar]
- 56.Peplow PV, Regulation of platelet-activating factor (PAF) activity in human diseases by phospholipase A2inhibitors, PAF acetylhydrolases, PAF receptor antagonists and free radical scavengers. Prostaglandins, Leukotrienes and Essential Fatty Acids (PLEFA) 61, 65–82 (1999). [DOI] [PubMed] [Google Scholar]
- 57.Ishii S, Shimizu T, Platelet-activating factor (PAF) receptor and genetically engineered PAF receptor mutant mice. Progress in Lipid Research 39, 41–82 (2000). [DOI] [PubMed] [Google Scholar]
- 58.Aliberti JC, Machado FS, Gazzinelli RT, Teixeira MM, Silva JS, Platelet-activating factor induces nitric oxide synthesis in Trypanosoma cruzi-infected macrophages and mediates resistance to parasite infection in mice. Infection and immunity 67, 2810–2814 (1999). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Szabó C et al. , Platelet-activating factor contributes to the induction of nitric oxide synthase by bacterial lipopolysaccharide. Circulation Research 73, 991–999 (1993). [DOI] [PubMed] [Google Scholar]
- 60.Howard AD, Erickson KL, The Induction and Augmentation of Macrophage Tumoricidal Responses by Platelet-Activating Factor. Cellular Immunology 164, 105–112 (1995). [DOI] [PubMed] [Google Scholar]
- 61.Frohlich E, Action of Nanoparticles on Platelet Activation and Plasmatic Coagulation. Curr Med Chem 23, 408–430 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Wolfram J et al. , Safety of Nanoparticles in Medicine. Current drug targets 16, 1671–1681 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Reid T, Warren R, Kirn D, Intravascular adenoviral agents in cancer patients: Lessons from clinical trials. Cancer Gene Therapy 9, 979 (2002). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Sturm E et al. , Kupffer cell depletion with liposomal clodronate prevents suppression of Ntcp expression in endotoxin-treated rats. Journal of Hepatology 42, 102–109 (2005). [DOI] [PubMed] [Google Scholar]
- 65.Lynch RW et al. , An efficient method to isolate Kupffer cells eliminating endothelial cell contamination and selective bias. J Leukoc Biol 104, 579–586 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Yamamoto Y et al. , siRNA Lipid Nanoparticle Potently Silences Clusterin and Delays Progression When Combined with Androgen Receptor Cotargeting in Enzalutamide-Resistant Prostate Cancer. Clin Cancer Res 21, 4845–4855 (2015). [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.