

Abstract
Introduction:
Cardiac fibrosis is a major factor in development of cardiovascular disease (CVD) and arrhythmia. Cardiac fibroblasts (CFs) are collagen-producing cells that regulate extracellular matrix (ECM) homeostasis. A complex signaling network has been defined linking environmental stress to changes in CF function and cardiac fibrosis. Signal Transducer and Activator of Transcription 3 (STAT3) has emerged as a critical integrator of pro-fibrotic signals in CFs downstream of several established signaling networks.
Areas covered:
This article provides an overview of STAT3 function in CFs and its involvement in coordinating pro-fibrotic signaling involving a vast web of intracellular signaling molecules and transcription factors. We highlight recent work elucidating a critical role for the fibroblast cytoskeleton in maintaining spatial and temporal control of STAT3-related signaling in CFs. Finally, we discuss potential opportunities and obstacles for therapeutic targeting of STAT3 to modulate cardiac fibrosis and arrhythmias. Relevant publications on the topic were identified through Pubmed.
Expert opinion:
Therapeutic targeting of STAT3 for CVD and arrhythmias presents unique challenges and opportunities. Thus, it is critical consider the multimodal and dynamic nature of STAT3 signaling. Going forward, it will be beneficial to consider ways to maintain balanced STAT3 function, rather than large scale perturbations in STAT3 function.
Keywords: Heart Failure, Fibrosis, Arrhythmia, Cardiac Fibroblasts, STAT3, Cytoskeleton, Spectrin
1. Introduction
Cardiovascular disease (CVD) remains the leading cause of death in the United States with cardiac arrhythmia as an important driver of mortality [1]. While mechanisms for arrhythmia in the setting of CVD remain to be fully elucidated and are undoubtedly multifactorial, increased cardiac fibrosis is a common finding across cardiac disease states [e.g. myocardial infarction (MI), heart failure, atrial fibrillation] with proposed roles in the development of cardiac electrical and mechanical dysfunction. Cardiac fibroblasts (CFs) are populous cell types in the heart derived from progenitor cells of the embryonic epicardial and endothelial/endocardial regions. CFs maintain homeostasis of the extracellular matrix (ECM), which in turn provides structural support for cardiomyocytes, distributes mechanical forces, and modulates electrical conduction through the myocardium [2–5]. Heterogeneous and plastic in nature, CFs are highly adaptable to extracellular signals (e.g. growth factors, cytokines/chemokines, neurohormones, and mechanical forces) [6]. In the normal (non-diseased) heart, CFs regulate structural integrity of the myocardium through a dynamic balance of ECM deposition and degradation. However under conditions of chronic biomechanical and/or neurohumoral stress, CFs undergo a phenotypic transition resulting in a more proliferative and contractile cell type (myofibroblast) with enhanced secretion of ECM proteins disrupting normal organization and coupling of cardiomyocytes to promote arrhythmogenic slow and discontinuous conduction [7]. Underlying these phenotypic changes are dramatic alterations in expression of genes encoding for contractile and pro-fibrotic proteins, including alpha-smooth muscle actin (α-SMA), fibrillar collagen types I and III, connective tissue growth factor (cTGF), and periostin [5, 8–10]. While the genetic and phenotypic changes observed in myofibroblasts contribute to myocardial repair and preservation of structural integrity following injury, dysregulated myofibroblast activity contributes to excessive reactive and/or reparative, and/or perivascular fibrosis leading to adverse left ventricular remodeling and diastolic dysfunction [11].
A host of neurohumoral and biomechanical extracellular stress signals promote fibroblast activation with important outstanding questions about how these signals are transduced into changes in gene expression and cell funciton. In this vein, a complex web of intracellular signaling pathways has been defined for mediating the response of CFs to stress signals [12, 13]. Growing evidence supports the multifunctional transcription factor, STAT3, as a critical integrator of pro-fibrotic signals in CFs downstream of several established signaling networks. Here, we provide an overview of STAT3 function in CFs and its involvement in coordinating pro-fibrotic signaling involving a vast network of intracellular signaling molecules, transcription factors, etc. We also highlight recent work elucidating a critical role for the fibroblast cytoskeleton in maintaining spatial and temporal control of STAT3-related signaling in cardiac fibroblasts. Finally, we discuss the potential opportunities and obstacles for therapeutic targeting of STAT3 to modulate fibrosis in heart. Relevant publications were identified via Pubmed using related keywords without constraint on date of publication although recent publications (within past 5 years) received extra consideration.
2. STAT3 signaling in cardiac cells
Signal transducer and activator of transcription 3 (STAT3) is a multifunctional transcription factor with a high degree of pleiotropy and identified roles in regulating gene programs important for inflammation, hypertrophy, proliferation, growth, differentiation, migration, ECM synthesis, fibrosis, and survival. This 89 kDa protein is ubiquitously expressed and found in a variety of cardiac cells, including fibroblasts, myocytes, endothelial cells, and immune cells. STAT3 is composed of six distinct domains, including amino N-terminal, coiled-coiled, DNA binding, linker, src homology 2 (SH2), and transactivation domains (Table 1) [14]. With pleiotropic functions depending on the source and wide variety of stimulus duration, STAT3 is regulated by multiple different signaling cascades to control an array of biological processes (e.g. inflammation, fibrosis) [15, 16].
Table 1:
Function of STAT3 Domains.
| Domain | Function [14, 18, 101] |
|---|---|
| NH2 | Formation of higher order complexes |
| CCD | Protein-protein interaction |
| Receptor binding | |
| Nuclear export | |
| DBD | Recognition of GAS |
| Nuclear transport | |
| LD | Interaction with specific tyrosine-phosphorylated residues of activated STAT3 |
| Formation of STAT3 homo or heterodimers | |
| SH2 | Formation of STAT3 dimerization through intramolecular phosphorylated tyrosine- SH2 interaction |
| Recruitment of specific receptors | |
| Transactivation domain | Regulatory phosphorylation |
NH2: (Amino) N-terminal domain; CCD: Coiled-coiled domain; DBD: DNA binding domain; LD: Linker domain; SH2: Src homology 2 domain; GAS: γ-activated sequences.
STAT3 activation typically occurs through a cascade of events beginning with activation of a receptor complex by its associated ligand [17–19]. Perhaps the best characterized receptor system upstream of STAT3 involves activation of the receptor glycoprotein-130 (gp130) by cytokines in the interleukin-6 (IL-6) family. Other STAT3-associated receptor systems include growth factor receptor Tyr kinases, the leptin receptor and the angiotensin II (AngII) type 1 receptor. Once the receptor system is activated, it recruits Janus kinase (JAK) and tyrosine kinase 2 (Tyk2) to an intracytoplasmic region allowing for phosphorylation of the receptor and subsequent binding of STAT3. Once bound to the receptor complex, STAT3 is phosphorylated by JAK at tyrosine705 (Tyr705) in the transactivation domain. Following detachment from the complex, phosphorylated STAT3 undergoes homo- or hetero-dimerization, allowing for nuclear translocation and regulation of target genes. There is evidence that STAT3 is also subject to phosphorylation at alternative sites, notably serine 727 (Ser727). While the upstream regulators and physiological effects of STAT3 Try705 phosphorylation are relatively well characterized, much less is known about STAT3 phosphorylation at Ser727. Previous studies have revealed that serine/threonine kinases (e.g. protein kinase c [PKC], mitogen-activated protein kinase [MAPK], extracellular signal-regulated kinase 1/2 [ERK1/2], and c-Jun N-terminal kinase [JNK]) are involved in the phosphorylation of Ser727 that promote the recruitment of various other transcriptional cofactors (e.g. histone acetyltransferase p300) [18]. Interestingly, Ser727 phosphorylation has been shown to be involved in STAT3 regulation of mitochondrial function [20]. Specifically, STAT3 Ser727 phosphorylation has been linked to the regulation of respiration and electron transport chain, production of ATP and reactive oxygen species (ROS), and permeability of mitochondrial pore opening [17, 18, 21, 22]. Interestingly, through the use of murine models with ablation of Ser727 on STAT3 (Ser-to-Ala mutation, S727A mice), it has been shown that STAT3 transcriptional activity is essential for maintaining healthy cardiomyocyte myofibril morphology and systolic cardiac function in the setting of hypertension [23].
While canonical activation of STAT3 involves phosphorylation at Y705, alternative (non-canonical) pathways for STAT3 activation have been identified involving translocation of unphosphoylated STAT3 (U-STAT3) to the nucleus to regulate gene transcription. Specifically, it has been proposed that STAT3 lacking the phosphorylation site at Try705 (Y705F-STAT3) forms a complex with unphosphoylated nuclear factor kappa-light-chain-enhancer of activated B cells (NF-κB) to induce gene transcription [24]. In fact, U-STAT3 has been associated with modulation of chromatin stability and gene programming to promote inflammation and cardiac hypertrophy through osteopontin and regulator of G-protein coupled receptor (GPCR) signaling 2 [20, 25]. Aside from phosphorylation, STAT3 activity has also been shown to be regulated by site-specific acetylation downstream of CD44 [26]. Thus, several alternative pathways for STAT3 activation have been identified with pathophysiological implications to be determined.
While STAT3 regulates survival and growth in both cardiac myocytes and fibroblasts, cell-specific roles for STAT3 have been identified in heart. In CFs, STAT3 regulates proliferation, differentiation, and ECM synthesis. Specifically, constitutive STAT3 activation via IL-6 stimulation promotes collagen biosynthesis and myofibroblast activation while STAT3 suppression decreases ECM deposition [27]. In contrast to its role in CFs, STAT3 suppresses pro-fibrotic pathways in cardiomyocytes. For example, STAT3 activation in cardiomyocytes via interleukin-11 (IL-11) prevents extensive post infarct fibrosis and adverse ventricular remodeling [18]. In fact, overexpression of STAT3 in cardiomyocytes induces hypertrophic growth, provides protective effects from ischemia/reperfusion injury, and increases vascular endothelial growth factor (VEGF) to promote capillary formation [28]. Furthermore, constitutive STAT3 activation in cardiomyocytes after MI results in dramatic hypertrophic changes, increased recruitment of inflammatory cells (leukocytes and macrophages), elevated left ventricular rupture rates, scar dilation, and pro-fibrotic gene expression [14, 28]. Beyond regulating the function of individual cells, STAT3 also supports communication between cell types in the heart. For example, it has been observed that conditioned media from cultured cardiomyocytes with STAT3-specific deletion enhances CF proliferation [18]. While it is clear that STAT3 signaling plays a major role in regulating cardiac function, the precise mechanisms by which STAT3 integrates stress signals to regulate cardiac cell function remain to be elucidated.
3. Synergistic role of TGF-β/SMAD and STAT3 signaling in regulating fibrosis
The transforming growth factor-β/SMAD (TGF-β/SMAD) signaling axis is among the most studied pathways for transcriptional regulation of CF phenotype and fibrosis. Briefly, mechanical tension promotes secretion of TGF-β into the ECM from the latent complex and subsequent binding to its cell surface receptor consisting of a heterodimeric complex involving TGF-β receptor type I (TGFβRI) and II (TGFβRII). The bound receptor promotes phosphorylation of SMAD2/3 transcription factors by TGFβRI allowing SMAD2/3 association with SMAD4 and their nuclear translocation [8, 29]. Previous work has shown that nuclear translocation of SMAD2/3-SMAD4 promotes myofibroblast differentiation through upregulation of several target proteins including α-SMA, TGF-β, collagen type I, and Periostin (Figure 1). Specifically, CF-specific deletion of TGFβRI and TGFβRII or SMAD3, but not SMAD2, in murine models reduces pressure-overload induced fibrotic expression of the fibrotic target genes Postn, Col1a1, and Col3a1. Additionally, CF-specific deletion of TGFβRI and TGFβRII, but not SMAD2/3 mitigates cardiac hypertrophic response to pressure overload. This suggests that TGF-β/SMAD signaling axis is specific for modulating the fibrotic response while different SMAD-independent pathways regulate hypertrophy. Finally, deletion of SMAD2/3 or TGFβRI and TGFβRII from CFs inhibits fibrotic gene expression and myofibroblast transition and ultimately abrogates ECM remodeling [30]. Aside from SMAD proteins, other transcriptional regulators including myocardin-related transcription factor (MRTF), serum response factor (SRF), and p38 have been shown to promote fibrosis through TGF-β-dependent non-canonical activation [31–34].
Figure 1: Synergistic roles of STAT3, TGF-β/SMAD, and YAP/TAZ signaling in regulating ECM remodeling and cardiac fibroblast proliferation and differentiation from biochemical and mechanical stress.
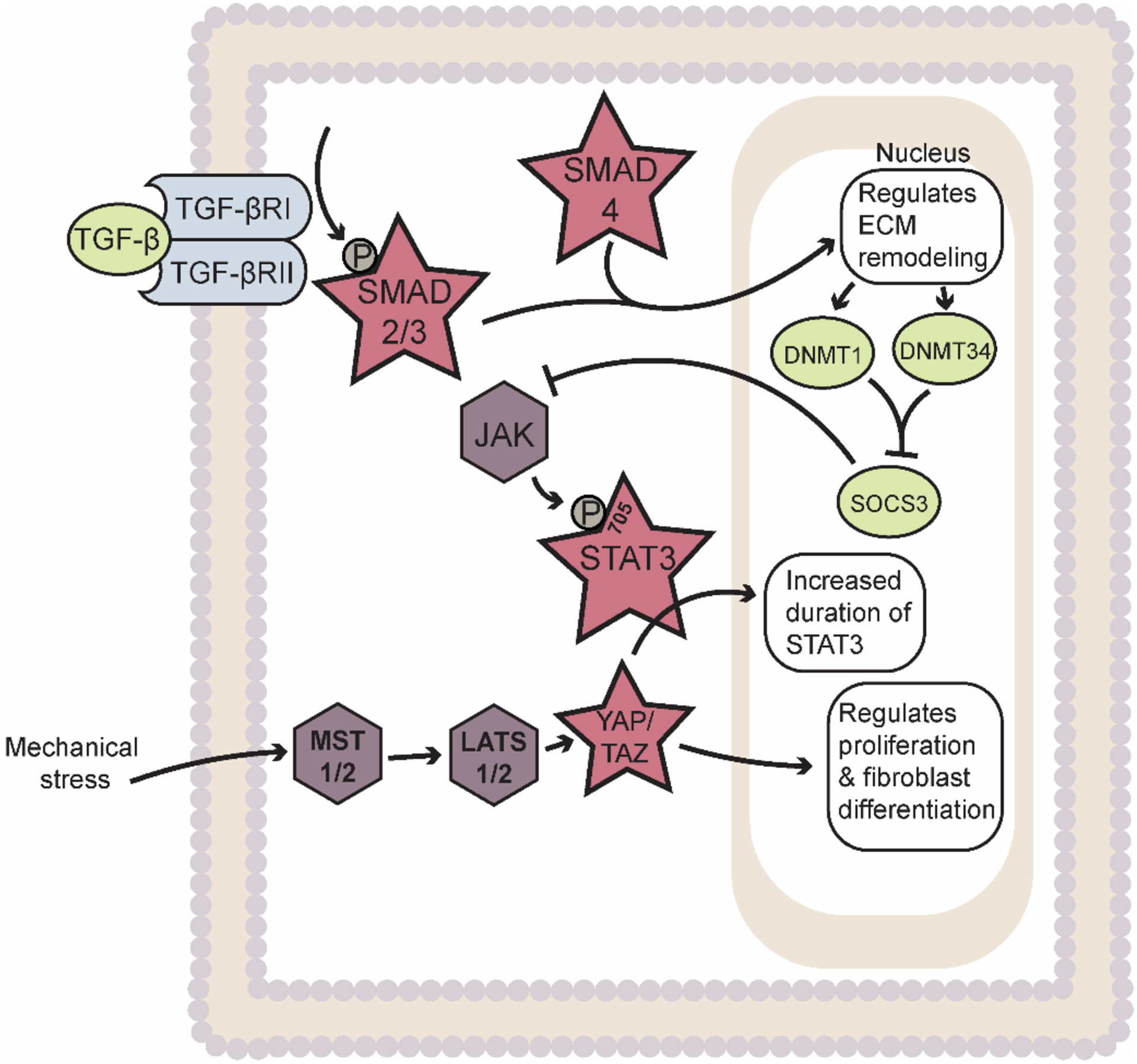
Abbreviations: ECM – Extracellular matrix; TGF-β – Transforming growth factor-β; TGFβRI – TGF-β receptor type I; TGFβRII – TGF-β receptor type II; JAK – Janus Kinase; STAT3 – Signal Transducer and Activator of Transcription 3; YAP – Yes-associated protein; TAZ – PDZ-binding motif; DNMT1 – DNA Methyltransferase 3A; DNA Methyltransferase 3A; SOCS3 – Suppressor of cytokine signaling 3; MST 1/2 – Mammalian sterile 20-like kinases 1 and 2; LATS 1/2 – Large tumor suppressors 1 and 2. Star indicates transcription factors. Rhombus indicates kinases. Ovals indicate other factors (e.g. cytokines, enzymes, proteins, etc.).
Several studies have identified STAT3 as a critical downstream node in TGF-β-dependent regulation of the response to acute/chronic stress. Interestingly, recent data indicate considerable feedback between STAT3 and TGF-β signaling. Namely, TGF-β has been shown to induce phosphorylation of STAT3 at Tyr705 and subsequent STAT3 activation in mouse models of systemic sclerosis. In fact, it was shown that pharmacological or genetic inhibition of STAT3 suppressed TGF-β-dependent activation and collagen secretion in human and mouse dermal fibroblast [35]. A novel epigenetic pathway was further elucidated in systemic sclerosis whereby TGF-β induces upregulation of DNA methyltransferase 1 and 3A (DNMT1 and DNMT3A) in a SMAD-dependent manner to inhibit expression of suppressor of cytokine signaling 3 (SOCS3). The silencing of SOCS3 in dermal fibroblasts was shown to promote STAT3 activation to promote myofibroblast activation, collagen deposition, and fibrosis [36]. At the same time, IL-6 trans signaling was found to drive a STAT3-dependent pathway in systemic sclerosis via an indirect mediation through the TGF-β signaling axis promoting SMAD3 activation and fibrosis via gremlin-1 protein, a profibrotic bone morphogenic protein (BMP) antagonist [37]. In the setting of cardiac disease, it was revealed that erythropoietin-producing hepatoma interactor B2 (ephrinB2) serves as a profibrotic transmembrane signaling molecule in cardiac fibrosis by modulating the interaction of STAT3 and TGF-β/SMAD signaling [38]. Finally, the pleiotropic hormone, relaxin, has been reported to play an anti-fibrotic role in CFs by attenuating TGF-β1-dependent cardiac fibrosis. Mechanistically, it was revealed that TGF-β1 inhibition via relaxin occurs through a STAT3-dependent regulation to reverse pathological cardiac fibrosis [39]. While conventionally viewed as distinct signaling pathways, emerging data reveals that TGF-β and STAT3 work synergistically to regulate the fibrotic response.
4. Role of STAT3 in integrin-mediated mechanotransduction
The actin cytoskeleton plays critical roles in activating signaling cascades in response to mechanical signals through a variety of established/canonical associated partners. Among the best characterized cytoskeleton-dependent signaling pathways involved in CF activation involve integrins, transmembrane receptors that function to connect the ECM on the exterior of the cell membrane to the interior cytoskeleton and facilitate transmission of mechanical forces. Specifically, integrins sense traction forces exerted on the ECM as part of a focal adhesion protein complex comprised of talin, vinculin, paxillan, integrin linked kinase, focal adhesion kinase (FAK), kindlin, parvin, and actinin. This complex supports actin polymerization and connection to actin stress fibers allowing for signal propagation across the plasma membrane to the actin cytoskeleton [40–43]. Structurally, integrins assemble as heterodimers of α- and β-subunits composed of extracellular, transmembrane spanning, and cytoplasmic domains. Metazoans express 18 α- and 8 β-subunits with 24 different α and β combinations [44]. Multiple studies have revealed that the α1β1, α2β1, α11β1, αvβ3, and αvβ5 integrin subunits in CFs play critical roles in regulating fibrillar collagen adhesion, morphology, spreading, collagen contractility, proliferation, and migration [45–48]. Integrins αvβ3, and αvβ5 have been found to regulate myofibroblast transition through a TGF-β1-dependent activation in human cardiac fibroblast [48], while the β3 chain is required for cytoskeletal stress fiber rearrangement in CFs upon stimulation with platelet derived growth factor (PDGF) [47]. Finally, computational modeling demonstrated that integrins associate with more than 150 biomolecules and directly activate non-receptor tyrosine-protein kinases (e.g. FAK and Src) and signaling pathways (e.g. TGF-β, mitogen activated protein kinases [MAPK] and p38) to promote ECM production and fibrosis [12, 13].
While integrins serve as regulatory nodes in response to mechanical stimuli, STAT3 resides downstream from integrin activation. Interestingly, β1-integrin upregulation through cyclic stretch of human renal proximal epithelial (HK-2) cells was found to enhance c-Src activity that subsequently promotes STAT3 phosphorylation at Tyr705 to increase production of TGF-β1 and fibronectin [49]. Similarly, a role for STAT3 was identified in activation of primary human lung fibroblasts in the setting of usual interstitial pneumonia (UIP)/idiopathic pulmonary fibrosis (IPF). Specifically, it was revealed that lung fibroblasts from patients with UIP/IPF exert high basal levels of phospho-STAT3, particularly in areas with dense levels of fibrosis. Enhanced STAT3 activity was found to suppress β3-integrin gene expression and αVβ3 integrin surface expression on the cell surface compared to that with low basal p-STAT3 levels that enhanced resistance of IPF lung fibroblasts to staurosporine-induced apoptosis and TGF-β1 responsiveness [50] (Figure 2). While an interaction between integrins and STAT3 has been clearly defined in renal and pulmonary fibrosis, a similar relationship remains to be defined in cardiac fibrosis. Moreover, while breakthroughs have been made in characterizing the upstream integrin-related complexes and their force sensing mechanism, continued effort is needed to understand the downstream effect of STAT3 in CFs.
Figure 2: Interplay of integrin, YAP/TAZ, and STAT3 mediated mechanosensing to modulate ECM production, integrin production, and cell morphology and growth.
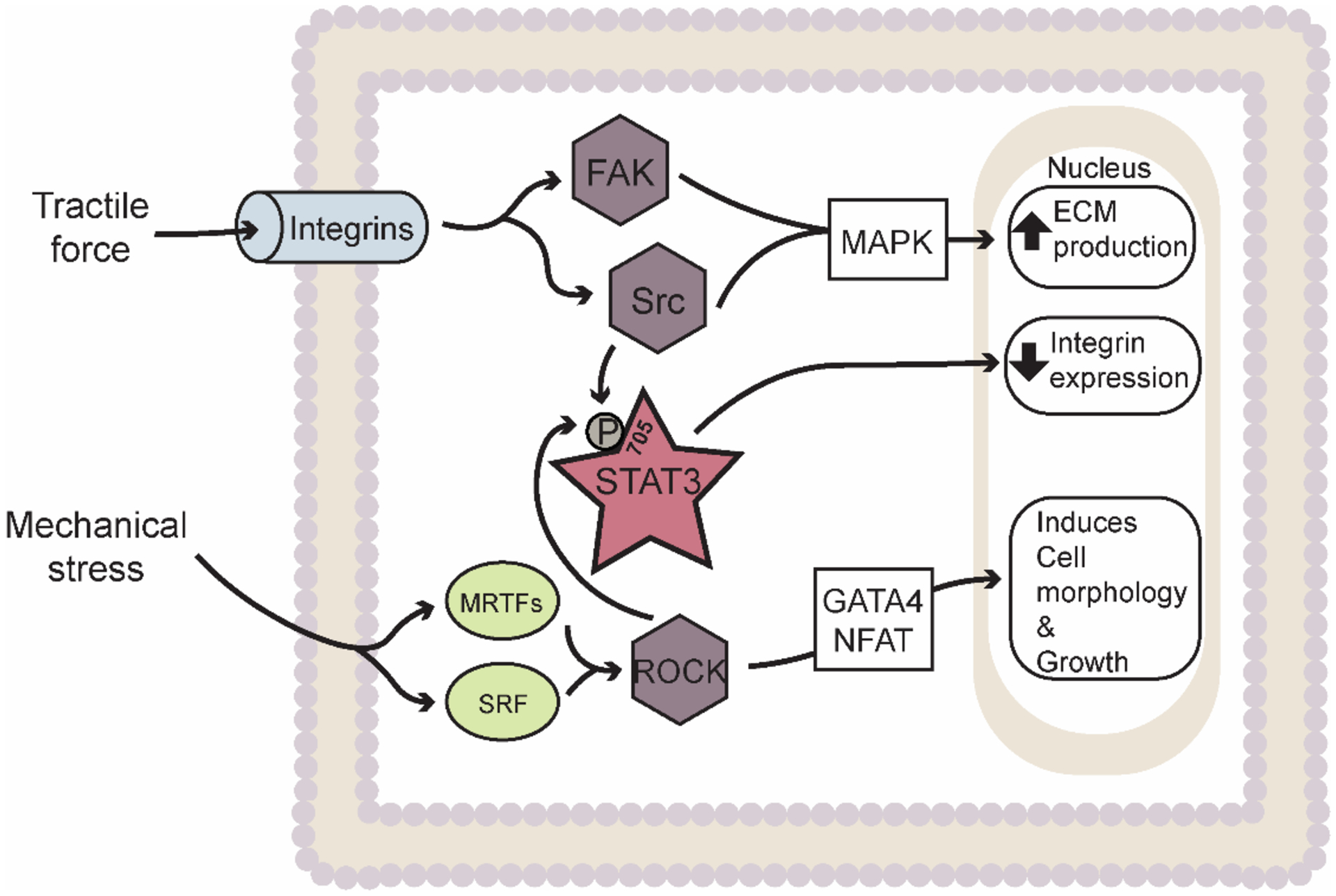
Abbreviations: ECM – Extracellular matrix; FAK – Focal adhesion kinase; STAT3 – Signal Transducer and Activator of Transcription 3; ROCK – Rho associated protein kinase; MRTF – Myocardin-related transcription factor; SRF – Serum response factor. Star indicates transcription factors. Rhombus indicates kinases. Ovals indicate other factors (e.g. cytokines, enzymes, proteins, etc.).
5. STAT3 involvement with YAP/TAZ-mediated mechanosensing and fibrosis
Yes-associated protein (YAP) and transcriptional coactivator with PDZ-binding motif (TAZ) represents another signaling axis important for regulation CF function in response to mechanical stress stimuli (e.g. substrate stiffness and hemodynamic load). Similar to TGF-β, YAP/TAZ signaling regulates CF function and the two pathways appear to be cooperative in nature. Specifically, YAP/TAZ are crucial transcriptional regulators that modulate CF proliferation and differentiation under the negative regulation of the HIPPO signaling pathway (traditionally viewed as a key regulator of organ growth and development through a serine/threonine kinase cascade) [51, 52]. Briefly, upon activation of HIPPO via the mammalian sterile 20-like kinases 1 and 2 (MST1/2) and large tumor suppressors 1 and 2 (LATS1/2) axis in response to the upstream cytoskeletal tension, YAP/TAZ activity is inhibited where it is unable to translocate into the nucleus to interact with its binding partner, YAP/TAZ-transcriptional enhanced associate domain (TEAD) [53, 54]. Previous studies demonstrated that cytoskeletal tension and F-actin assembly modulate the nuclear translocation and activity of YAP/TAZ in epithelial cells and lung fibroblasts that drive proliferation and profibrotic responses [55, 56]. In heart, YAP has been shown to play essential role in the phenotypic transition of fibroblast morphology in response to mechanical stress. For example, it was reported that AngII-mediated YAP activation enhanced α-SMA expression and collagen volume fraction, thus promoting contractile myofibroblast differentiation in a murine model of dilated cardiomyopathy [57]. Moreover, actin cytoskeletal disruption/remodeling (direct or via increased cyclic adenosine monophosphate [cAMP] activity) downregulated exchange protein directly activated by cAMP 2 (EPAC2) gene expression leading to suppression of YAP/TAZ/TEAD activity to modulate CF morphology and cell spreading [58]. Despite the effort to understand the role of YAP/TAZ in the differentiation process, much less is known about how upstream stress signals affect this pathway. Previous work suggests that perhaps these transcriptional co-activators are linked with SMAD3 from the TGF-β signaling pathways, however further studies are needed to clarify the relationship between TGF-β/SMAD3 and YAP/TAZ [12]. Finally, it has been reported that YAP activity is crucial for the upregulation and stabilization of cytoskeletal components in fibroblasts associated with tumorigenicity [59]. It will be exciting for future studies to identify a similar link between YAP and the actin-associated cytoskeleton CF during chronic stress conditions in heart.
Similar to YAP/TAZ, the JAK/STAT3 signaling pathway has been found to respond to mechanical stress [14]. For example, mechanical stretch was found to activate JAK/STAT3 in conjunction with AngII secretion through paracrine/autocrine fashion in rat cardiomyocytes [60]. Furthermore, studies have explored the relationship between YAP/TAZ and STAT3 signaling related to oncology and angiogenesis. In endothelial cells, it was revealed that binding of YAP to STAT3 interrupts the chromosome region maintenance 1 (CRM1) – mediated STAT3 nuclear export and therefore prolongs STAT3 nuclear accumulation (Figure 1) to promote angiogenesis and vessel remodeling through transcriptional regulation of angiopoietin-2 [61]. Du et al. investigated the miR-205/YAP1 signaling axis in breast tumorigenesis and linked its role in activating breast normal fibroblasts into cancer-associated fibroblasts. Specifically, it was found that YAP1 promotes elevated expression of IL-11 and IL-15 that in turns stimulates STAT3 signaling in vascular endothelial cells to enhance tumor angiogenesis and metastasis [62]. Finally, studies in genetic mouse models of pancreatitis ductal adenocarcinoma (PDAC) have identified a novel mechanism by which oncogenetic KRas induces acinar-to-ductal metaplasia (ADM) through activation of YAP/TAZ and changes in JAK/STAT3-dependent transcriptional gene regulation [63]. While great strides have been made in linking YAP/TAZ and STAT3 signaling, their connection in relevance to cardiovascular disease needs further exploration.
6. Interplay of non-canonical STAT3 and MRTF/ROCK-dependent signaling in cardiac fibrosis
Another example of the importance of the cytoskeleton in mediating extracellular signal transduction in fibroblasts comes from the non-canonical effects of myocardin-related transcription factors (MRTFs). MRTFs are mechanosensitive coactivators of the serum-response factor (SRF) that interact with the actin cytoskeleton and associate with the Rho associated protein kinase (ROCK) signaling cascade to alter cell morphology [64] (Figure 2). Interestingly, studies in mouse models subjected to AngII stimulation or MI have shown that MRTF-A promotes pathological scar formation, collagen accumulation, and increases the myofibroblast population by responding to TGF-β1 through ROCK-dependent activation [31]. Moreover, MRTF-A has also been reported to directly bind with monomeric G-actin and prevent actin polymerization into F-actin polymers until mechanical stimuli exerted on the cell activates ROCK signaling to release MRTF-A to allow polymerization for CF contractility [65, 66]. While important advances have been made in understanding how changes in mechanical load alter this process, future studies still need to be performed on understanding how neurohumoral stress mediates the association of MRTF-A and the actin cytoskeleton.
Along with its association with MRTF-A, ROCKs alone have received attention in modulating CF activity. In fact, ROCKs are known to regulate apoptosis, growth, proliferation, migration and actin cytoskeleton remodeling [67]. In particular, ROCK2 has been revealed to be an important regulator of cardiac hypertrophy, fibrosis, and diastolic dysfunction. Intriguingly, a recent study demonstrated that murine models with fibroblast-specific deletion of ROCK2 with AngII stimulation exhibited diminished left ventricle (LV) wall thickness and fibrosis with improved isovolumetric relaxation corresponding to decreased expression of CTGF and fibroblast growth factor 2 (FGF2) [68]. Furthermore, an RNAi screen to identify a mechanosensitive ROCK-JAK2-STAT3 network for modulation of myofibroblast activation revealed that constitutive STAT3 phosphorylation at Tyr705 drives pro-fibrotic gene expression, contractility, and ECM secretion via a ROCK and JAK2-dependent fashion in a stiff matrix environment [69]. While great strides have been made in linking canonical to non-canonical signaling pathways in understanding cardiac fibrosis, the same is essential for merging different non-canonical mechanism to precisely understand fibroblast biology.
7. Emerging role for spectrin-based cytoskeleton in control of STAT3 signaling
While previous efforts have focused on the actin cytoskeleton as a force transducer, there is growing appreciation for a role of cytoskeletal proteins in dynamic signaling with important implications for cardiac pathophysiology. Mounting data support spectrins as critical cytoskeleton proteins with active roles in signal transduction pathways important for fibrosis [70]. Structurally, spectrins assemble as large, fibrous tetramers from heterodimers comprised of α- and β-subunits containing triple-helical spectrin repeats that give rise to their flexibility. β-spectrins serve to link the actin-associated cytoskeleton with the membrane bound protein via ankyrin adaptor proteins to maintain structural integrity and membrane stability [71–74]. Early studies found that the βII-spectrin subunit isoform (embryonic liver fodrin [ELF]) associated with the TGF-β receptor complex and SMAD3/4 to regulate their nuclear translocation, thereby contributing to regulation of cardiac development and function [75]. In fact, ELF deficiency promotes mislocalization of SMAD3/4 and loss of TGF-β-dependent transcriptional response. It was further discovered that downregulation of βII-spectrin reduces expression levels of SMAD1, SMAD2, and SMAD4 along with decreased phosphorylation of SMAD1/5 and SMAD2 in cardiomyocytes [76]. Subsequent studies revealed that TGF-β induces degradation of αII- and βII-spectrin isoforms subunits in human dermal fibroblasts although a functional role for spectrins in TGF-β-dependent signaling has not been established [77]. Finally, βII-spectrin has been proposed to coordinate a feedback mechanism to regulate STAT3 and SMAD3 protein expression in hepatocellular carcinoma through the cAMP-response element-binding proteins, ATF3 and CREB2 [78].
More recently, a direct role for another a-spectrin isoform betaIV-spectrin has been identified in regulating STAT3 signaling in heart (Figure 3) [15, 79]. In cardiomyocytes, βIV-spectrin was initially found to organize a macromolecular complex with calcium/calmodulin-dependent protein kinase II (CaMKII) and the cardiac voltage-gated Na+ channel Nav1.5 at the intercalated disc to regulate membrane excitability [80]. Subsequent studies found that βIV-spectrin is essential for the spatial and temporal control of STAT3 signaling by directly regulating its subcellular localization and activity in cardiomyocytes. STAT3 dysregulation induced by CaMKII-dependent loss of βIV-spectrin resulted in maladaptive remodeling, fibrosis, and decreased cardiac function in a mouse model of chronic pressure overload [15]. Interestingly, this same βIV-spectrin/STAT3 complex was found to regulate the phenotype and activity of CFs [79]. Specifically, genetic and acquired loss of βIV-spectrin function in mouse CFs promoted STAT3 nuclear accumulation and transcriptional activity that subsequently alters gene expression to induce CF activation to enhance fibrosis and cardiac dysfunction. Interestingly, CFs subjected to TGF-β stimulation in vitro showed a decrease in βIV-spectrin levels [79]. Consistent with these findings, previous work has demonstrated that isolated CFs subjected to either transverse aortic banding, AngII, or electrical field stimulation significantly upregulate CaMKII expression that enhance proliferation and ECM secretion [e.g. matrix metalloproteinase (MMP)-1,2,9 and collagen I/III] [81, 82]. Together these studies demonstrate that the spectrin-based cytoskeleton not only forms a structural platform essential for the coordination of macromolecular complexes, but also as an important functional regulator for fine-tuning cellular response. While emphasis has been devoted to canonical players in understanding their response to neurohumoral and mechanical stress, the non-canonical spectrin cytoskeleton pathway has become a critical stress sensor for the modulation of genetic programming for cardiac fibrosis.
Figure 3: Spectrin-based cytoskeleton serves as a regulatory platform to control STAT3 signaling and cardiac fibroblast function.
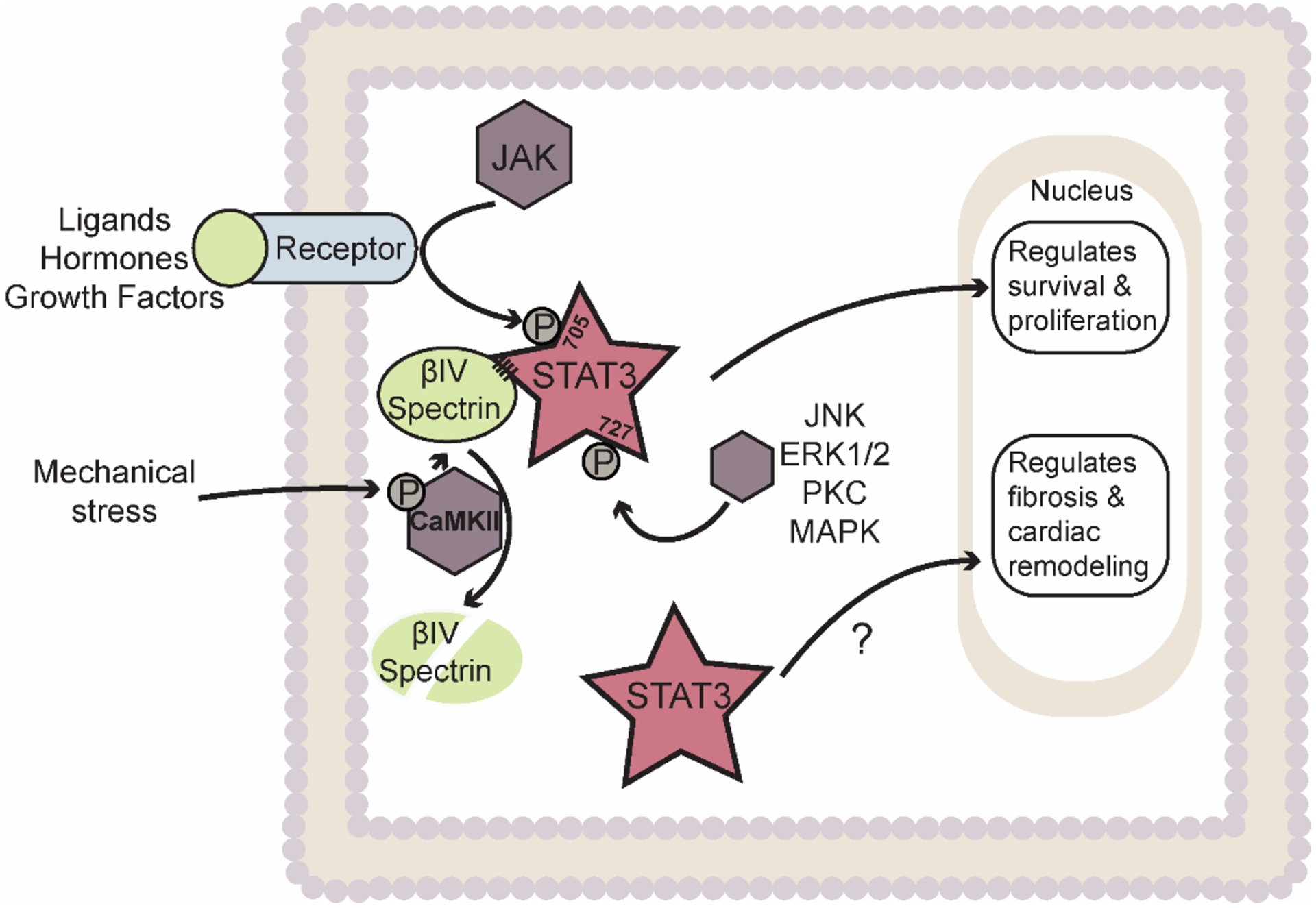
STAT3 normally resides in a complex with βIV-spectrin allowing for regulation at Tyr 705 by Janus kinase (JAK) or Ser 727 by several other kinases. Chronic stress induces degradation of betaIV-spectrin allowing for non-canonical activation of STAT3 (phosphorylation status to be determined) and regulation of maladaptive remodeling. Abbreviations: ECM – Extracellular matrix; STAT3 – Signal Transducer and Activator of Transcription 3; CaMKII – Calcium/calmodulin-dependent protein kinase II; JNK – c-Jun N-terminal kinase; ERK1/2 – Extracellular signal-regulated kinase 1/2; PKC – Protein kinase C; MAPK – Mitogen-activated protein kinase. Star indicates transcription factors. Rhombus indicates kinases. Ovals indicate other factors (e.g. cytokines, enzymes, proteins, etc.).
8. Role of STAT3 in modulating susceptibility to cardiac arrhythmia
Life-threatening cardiac arrhythmias are the main cause of sudden cardiac arrest attributing to over 366,000 deaths per year in the United States among patients with CVD [1]. As already discussed, STAT3 is as an important mediator of ECM synthesis, collagen accumulation, and cardiac fibrosis [14]. Excess ECM and collagen accumulation is a substrate directly linked to electrical conduction slowing, and arrhythmia vulnerability through reentry. A number of studies have identified a specific link between STAT3 dysregulation and the most common sustained arrhythmia, atrial fibrillation (AF). In fact, a positive feedback loop was identified involving STAT3 activation and upregulation of miR-21 to promote atrial fibrosis and atrial arrhythmia in a rat model of post-operative AF [83]. Similarly, atrial enlargement and electrical and fibrotic remodeling were observed in mice following MI and these changes were prevented by treatment with the STAT3 inhibitor S3I-201 [84]. Additional studies in large animal models have associated increased STAT3 activity with development of AF [84, 85], while patients with sustained AF show increased STAT3 protein expression compared to those in sinus rhythm [86]. STAT3 has also been implicated as a key mediator in development of atrial fibrosis and AF downstream of TGF-beta/CD44 activation [87]. Consistent with the link between STAT3, fibrosis and arrhythmia, increased arrhythmic events (e.g. premature ventricular contractions, sinus pauses, and ventricular tachycardia) have been observed in genetic and acquired mouse models of βIV-spectrin/STAT3 disruption, which associated with changes in the extent of fibrotic remodeling [15, 79]. Indirect activation of STAT3 in a SOCS3 knockout mouse model also revealed electrical abnormalities including disrupted Ca2+ handling, triggered activity, and premature ventricular contractions [88]. Although growing data support a direct role for STAT3 signaling in modulating susceptibility to arrhythmia, in general there is a lack of data using electrical function or arrhythmia as a primary endpoint. Instead, most studies have focused on STAT3 effects on contractile function and/or fibrosis, which serves as a substrate for arrhythmia but is not a direct assessment. Thus, there is a need for further studies explicitly evaluating electrical function directly following STAT3 intervention, as well as more translational models with STAT3 targeting intervention to evaluate the full scope of STAT3 dependent pathways on cardiac electrical remodeling.
9. Opportunities and challenges for therapeutic targeting of STAT3 signaling to modulate fibrosis.
A major challenge for targeting STAT3 to treat cardiac disease stems from the fact that cardiac dysfunction has been associated with both gain and loss of STAT3 function. In fact, numerous investigations involving genetic or pharmacologic manipulation of STAT3 or associated proteins have demonstrated that both downregulation and upregulation of STAT3 activity gives rise to pathology. For example, cardiac specific deletion of STAT3 in the mouse results in fibrosis, impaired cardiac function, ventricular remodeling, and increased mortality [89–93]. Consistent with these findings, reduced STAT3 activity has been linked to a wide range of risk factors for cardiovascular disease (e.g. diabetes, aging, obesity) [94]. At the same time, transgenic expression of constitutively active STAT3 leads to increased mortality in mice following MI through enhanced inflammation, increased wall rupture, scar dilation, and heart failure [28], indicating that upregulation of STAT3 activity also drives pathology.
The complex dependence of cardiac function on STAT3 function is likely due to a host of factors, including the highly pleiotropic and multifunctional nature of STAT3 signaling. Furthermore, STAT3 is ubiquitously expressly with disparate roles in distinct cardiac cell types (e.g. myocytes, fibroblasts, immune cells). Myocytes, for example, may benefit from STAT3 dependent cell-survival mechanisms, while fibroblasts may experience pathologic proliferation and fibrosis. Additional complexity stems from the potential for recruitment of alternative cell signaling pathways depending on the mode of STAT3 activation. For example, one model of genetically induced STAT3 activity employed a phosphomimetic mutation of the gfp130 receptor (Y757F), which disrupts recruitment of SOCS3, a negative feedback regulator of STAT3 activation. While this mutation effectively activates STAT3, recruitment of SOCS3 is a critical part of additional pathway regulation, including PI3K/AKT [28] and MAPK pathways [88, 95], with evidence of for roles in subsequent inflammatory signaling. This indicates that with STAT3 activation comes parallel recruitment of several signaling cascades that may be critical for distinguishing beneficial from pathologic STAT3 activity. Importantly, ligands like LIF and IL-11 may recruit such pathways in parallel with STAT3 that may contribute to these improved phenotypes. Likewise, STAT3 inhibition by S3I-201 may directly target STAT3 dependent signaling, while preserving signaling of such parallel signaling cascades. Finally, observations of disparate roles for STAT3 signaling dependent on its phosphorylation status between its 705 and 727 site have been observed, with completely unphosphorylated STAT3 (U-STAT3) representing a transcriptionally active STAT3 with heightened pathologic and inflammatory roles [28, 96, 97]. However, it remains to be seen what specific mechanisms regulate STAT3 phosphorylation patterns and how such mechanisms contribute to disease development. Overall, it appears that while it is not yet clear whether STAT3 activation or inhibition should be the therapeutic goal, the answer will likely require careful monitoring and characterization of which cell signaling pathways are recruited in parallel with STAT3, the potential dependence of specific ligands which recruit these additional pathways, and finally the steady-state phosphorylation status of STAT3 and resulting differences in pathologic phenotypes.
Despite the complexity and potential pitfalls of targeting STAT3 for therapeutic benefit in heart, mounting data provide reasons to be optimistic. The most frequently used pharmacologic inhibitor of STAT3 has been the molecule S3I-201, a cell permeable compound that interacts with the STAT3-SH2 domain, competing for the interaction of STAT3 with the phosphorylated gp130 receptor, prohibiting STAT3 Tyr705 phosphorylation by JAK. Ultimately, the compound leads to reduced activation, dimerization, and transcriptional activity of STAT3. Applications of this drug have been shown to reduce fibrosis and improve cardiac electrical and mechanical function in a wide range of animal models of CVD, including MI, pressure overload, sleep apnea, and AF [15, 83, 84, 98, 99]. Thus, there is hope that under the right conditions STAT3 may be targeted effectively for therapeutic benefit although the translational potential of these findings remain to be defined.
10. Conclusion
There is a growing appreciation in the field in understanding the synergistic role of STAT3 signaling with established networks that drive cardiac fibroblast activation and fibrosis in the heart. Recent studies have also highlighted the spectrin-based cytoskeleton in controlling STAT3 signaling to modulate gene reprogramming as a result of various stress signals. These studies collectively bring forth exciting opportunities and challenges for therapeutic targeting of STAT3 in a tightly balanced fashion to regulate cardiac fibrosis. Based on the growing body of literature, strategies for targeting STAT3 either through activation or inhibition may serve to be beneficial in the setting of CVD.
11. Expert Opinion
The central role of STAT3 in modulating cardiac cell function (including both cardiomyocytes and fibroblasts) and heart function downstream of several parallel signaling networks raises the prospect that therapeutic targeting of STAT3 may be beneficial in the setting of CVD. Importantly, STAT3 activity is regulated in a complex way with activation modes that are not fully understood. Notably, a multitude of additional post-translational modifications including Ser727 phosphorylation, methylation, and acetylation, reveal an impact for recruiting additional transcriptional co-factors potentiating gene expression as well as the possibility of entirely unique transcriptional targets. Aside from the conventional phosphorylated nature of STAT3 activity, there has been a growing appreciation in the field for identifying the precise mechanistic role of U-STAT3 in cardiac cells. Furthermore, observations of disparate roles for STAT3 signaling dependent on its phosphorylation status have been observed, with U-STAT3 representing heightened pathologic and inflammatory roles [28, 96, 97]. Overall, it appears that defining physiological versus pathologic states for STAT3 signaling will require careful monitoring of specific ligands recruiting STAT3 activation, specific cell signaling pathways recruited in parallel with STAT3, and finally the steady-state phosphorylation status of STAT3. Moreover, significant observations for mitochondrial expression of STAT3 has identified actions important for respiration and redox sensing. Thus, STAT3 activation and inhibition have both proven beneficial in different settings. Going forward, it will be important to delineate the role for STAT3 in specific disease states to better inform therapeutic strategies. Specifically, it will be important to consider ways to restore normal STAT3 function, rather than large scale perturbations in STAT3 function (e.g. complete knockdown/inhibition or activation). For example, it may be that therapy should seek to maintain a balanced state of STAT3 activity across the full injury time course in order to optimize the full therapeutic potential. The injury response following MI illustrates this potential scenario well, where distinct phases of inflammatory and injury response characterize the early and late remodeled scar suggesting the potential necessity of STAT3 activity in some circumstances but a deliberately inhibited role in others. In such cases, it is easy to envision how transgenic models which remove this temporal regulation would lead to heightened disease signaling. Consistently, the artificial activation and deregulation of STAT3 [18, 28, 88] as well as its genetic ablation [100] associate with cardiac disease. Conversely, studies which instead recruit STAT3 activity through ligand activation are able to more reliably show improved cardiac function and cardioprotection following injury [90, 91], again, supporting the notion of therapeutic STAT3 activation, but achieved through a more balanced means. Thus, it is interesting to consider the possibility that more nuanced approaches to perturbing STAT3 signaling may yield breakthroughs for treating a diverse CVD patient population.
As delineated throughout this article, although a great deal has been elucidated about mechanisms governing STAT3 biology and physiological function, critical gaps in knowledge remain that provide opportunities for uncovering novel therapeutic avenues. The link between STAT3 and the spectrin-based cytoskeleton is one such area that may prove particularly beneficial for defining ways to maintain balanced STAT3 activity in the face of chronic cardiac stress. Specifically, it will be important to define the structural requirements for direct interaction between STAT3 and the spectrin cytoskeleton along with the precise steps leading to STAT3 activation and gene changes following stress-induced disruption of the spectrin cytoskeleton. As the spectrin-based pathway for controlling STAT3 signaling continues to evolve for the next 5 years, it will be critical to integrate this network with the canonical cascades for regulation of fibrosis, cardiac function and arrhythmia, including TGF-β/SMAD, integrin mediated mechanotransduction, YAP/TAZ-mediated mechanosensing, and MRTF/ROCK-dependent signaling. Finally, it will be critical to better understand the pleiotropic actions of STAT3 across disparate cardiac cell types, which may point to more effective ways of tuning the cardiac response to stress.
Article Highlights.
STAT3 signaling regulates cardiac fibrosis and susceptibility to cardiac arrhythmia.
STAT3 resides downstream of several canonical pathways linked to cardiac fibroblast gene reprogramming.
Spectrin-based cytoskeleton maintains spatial and temporal control of STAT3-related signaling in cardiac fibroblasts.
STAT3 plays a multimodal role with its activation and inhibition both having beneficial effects in different settings.
STAT3 may serve as an effective therapeutic target to modulate cardiac fibrosis and function in cardiac disease.
Funding
This work was supported by National Institutes of Health grants R01-HL135096 and R01-HL134824 to TJH.
List of Abbreviations
- CVD
Cardiovascular disease
- CFs
Cardiac fibroblasts
- ECM
Extracellular matrix
- STAT3
Signal transducer and activator of transcription 3
- cTGF
Connective tissue growth factor
- SH2
Src homology 2
- NH2
Amino N-terminal
- CCD
Coiled-coiled domain
- DBD
DNA binding domain
- LD
Linker domain
- GAS
γ-activated sequences
- IL-6
Interleukin-6
- AngII
Angiotensin II
- JAK
Janus Kinase
- Tyr705
Tyrosine 705
- Ser727
Serine727
- PKC
Protein kinase C
- MAPK
Mitogen-activated protein kinase
- ERK1/2
Extracellular signal-regulated kinase 1/2
- JNK
c-Jun N-terminal kinase
- ROS
Reactive oxygen species
- U-STAT3
Unphosphoylated STAT3
- NF-κB
Nuclear factor kappa-light-chain-enhancer of activated B cells
- GPCR
G protein-coupled receptor
- IL-11
Interleukin-11
- TGF-β
Transforming growth factor-β
- TGFβRI
TGF-β receptor type I
- TGFβRII
TGF-β receptor type II
- MRTF
Myocardin-related transcription factor
- SRF
Serum response factor
- SS
Systemic sclerosis
- DNMT1
DNA methyltransferase 1
- DNMT3A
DNA methyltransferase 3A
- SOCS3
Suppressor of cytokine signaling 3
- BMP
Bone morphogenic protein
- EphrinB2
Erythropoietin-producing hepatoma interactor B2
- FAK
Focal adhesion kinase
- PDGF
Platelet derived growth factor
- MAPK
Mitogen activated protein kinases
- UIP
Usual interstitial pneumonia
- IPF
Idiopathic pulmonary fibrosis
- YAP
Yes-associated protein
- TAZ
PDZ-binding motif
- MST1/2
Mammalian sterile 20-like kinases 1 and 2
- LATS1/2
Large tumor suppressors 1 and 2
- TEAD
Transcriptional enhanced associate domain
- cAMP
Cyclic adenosine monophosphate
- EPAC2
Exchange protein directly activated by cAMP 2
- CRM1
Chromosome region maintenance 1
- PDAC
Pancreatitis ductal adenocarcinoma
- ADM
Acinar-to-ductal metaplasia
- SRF
Serum-response factor
- ROCK
Rho associated protein kinase
- MI
Myocardial Infarction
- FGF2
Fibroblast growth factor 2
- CaMKII
Calcium/calmodulin-dependent protein kinase II
- MMP
Matrix metalloproteinase
- ELF
Embryonic liver fodrin
Footnotes
Declaration of interest
The authors have no relevant affiliations or financial involvement with any organization or entity with a financial interest in or financial conflict with the subject matter or materials discussed in the manuscript. This includes employment, consultancies, honoraria, stock ownership or options, expert testimony, grants or patents received or pending, or royalties.
Reviewer disclosures
Peer reviewers on this manuscript have no relevant financial or other relationships to disclose
References
Papers of special note have been highlighted as either of interest (•) or of considerable interest (••) to readers
- 1.Benjamin EJ, Muntner P, Alonso A, et al. Heart Disease and Stroke Statistics-2019 Update: A Report From the American Heart Association. Circulation 2019;139:e56–e528. [DOI] [PubMed] [Google Scholar]
- 2.Travers JG, Kamal FA, Robbins J, et al. Cardiac Fibrosis: The Fibroblast Awakens. Circ Res 2016;118:1021–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Kohl P, Gourdie RG. Fibroblast-myocyte electrotonic coupling: does it occur in native cardiac tissue? J Mol Cell Cardiol 2014;70:37–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Vasquez C, Benamer N, Morley GE. The cardiac fibroblast: functional and electrophysiological considerations in healthy and diseased hearts. J Cardiovasc Pharmacol 2011;57:380–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Tallquist MD, Molkentin JD. Redefining the identity of cardiac fibroblasts. Nat Rev Cardiol 2017;14:484–491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Humeres C, Frangogiannis NG. Fibroblasts in the Infarcted, Remodeling, and Failing Heart. JACC Basic Transl Sci 2019;4:449–467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Rohr S Myofibroblasts in diseased hearts: new players in cardiac arrhythmias? Heart Rhythm 2009;6:848–56. [DOI] [PubMed] [Google Scholar]
- 8.Davis J, Molkentin JD. Myofibroblasts: trust your heart and let fate decide. J Mol Cell Cardiol 2014;70:9–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Kong P, Christia P, Frangogiannis NG. The pathogenesis of cardiac fibrosis. Cell Mol Life Sci 2014;71:549–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Fu X, Khalil H, Kanisicak O, et al. Specialized fibroblast differentiated states underlie scar formation in the infarcted mouse heart. J Clin Invest 2018;128:2127–2143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Frangogiannis NG. Cardiac fibrosis: Cell biological mechanisms, molecular pathways and therapeutic opportunities. Mol Aspects Med 2019;65:70–99. [DOI] [PubMed] [Google Scholar]
- 12.Saucerman JJ, Tan PM, Buchholz KS, et al. Mechanical regulation of gene expression in cardiac myocytes and fibroblasts. Nat Rev Cardiol 2019;16:361–378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Zeigler AC, Richardson WJ, Holmes JW, et al. A computational model of cardiac fibroblast signaling predicts context-dependent drivers of myofibroblast differentiation. J Mol Cell Cardiol 2016;94:72–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Harhous Z, Booz GW, Ovize M, et al. An Update on the Multifaceted Roles of STAT3 in the Heart. Front Cardiovasc Med 2019;6:150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Unudurthi SD, Nassal D, Greer-Short A, et al. betaIV-Spectrin regulates STAT3 targeting to tune cardiac response to pressure overload. J Clin Invest 2018;128:5561–5572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Hutchins AP, Diez D, Miranda-Saavedra D. Genomic and computational approaches to dissect the mechanisms of STAT3’s universal and cell type-specific functions. JAKSTAT 2013;2:e25097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Haghikia A, Stapel B, Hoch M, et al. STAT3 and cardiac remodeling. Heart Fail Rev 2011;16:35–47. [DOI] [PubMed] [Google Scholar]
- 18.Haghikia A, Ricke-Hoch M, Stapel B, et al. STAT3, a key regulator of cell-to-cell communication in the heart. Cardiovasc Res 2014;102:281–9. [DOI] [PubMed] [Google Scholar]
- 19.Bousoik E, Montazeri Aliabadi H. “Do We Know Jack” About JAK? A Closer Look at JAK/STAT Signaling Pathway. Front Oncol 2018;8:287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Zouein FA, Altara R, Chen Q, et al. Pivotal Importance of STAT3 in Protecting the Heart from Acute and Chronic Stress: New Advancement and Unresolved Issues. Front Cardiovasc Med 2015;2:36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Wakahara R, Kunimoto H, Tanino K, et al. Phospho-Ser727 of STAT3 regulates STAT3 activity by enhancing dephosphorylation of phospho-Tyr705 largely through TC45. Genes Cells 2012;17:132–45. [DOI] [PubMed] [Google Scholar]
- 22.Wegrzyn J, Potla R, Chwae YJ, et al. Function of mitochondrial Stat3 in cellular respiration. Science 2009;323:793–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Zouein FA, Zgheib C, Hamza S, et al. Role of STAT3 in angiotensin II-induced hypertension and cardiac remodeling revealed by mice lacking STAT3 serine 727 phosphorylation. Hypertens Res 2013;36:496–503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Yang J, Liao X, Agarwal MK, et al. Unphosphorylated STAT3 accumulates in response to IL-6 and activates transcription by binding to NFkappaB. Genes Dev 2007;21:1396–408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Yue H, Li W, Desnoyer R, et al. Role of nuclear unphosphorylated STAT3 in angiotensin II type 1 receptor-induced cardiac hypertrophy. Cardiovasc Res 2010;85:90–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Lee JL, Wang MJ, Chen JY. Acetylation and activation of STAT3 mediated by nuclear translocation of CD44. J Cell Biol 2009;185:949–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Melendez GC, McLarty JL, Levick SP, et al. Interleukin 6 mediates myocardial fibrosis, concentric hypertrophy, and diastolic dysfunction in rats. Hypertension 2010;56:225–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Hilfiker-Kleiner D, Shukla P, Klein G, et al. Continuous glycoprotein-130-mediated signal transducer and activator of transcription-3 activation promotes inflammation, left ventricular rupture, and adverse outcome in subacute myocardial infarction. Circulation 2010;122:145–55. [DOI] [PubMed] [Google Scholar]
- 29.Biernacka A, Dobaczewski M, Frangogiannis NG. TGF-beta signaling in fibrosis. Growth Factors 2011;29:196–202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Khalil H, Kanisicak O, Prasad V, et al. Fibroblast-specific TGF-beta-Smad2/3 signaling underlies cardiac fibrosis. J Clin Invest 2017;127:3770–3783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Small EM, Thatcher JE, Sutherland LB, et al. Myocardin-related transcription factor-a controls myofibroblast activation and fibrosis in response to myocardial infarction. Circ Res 2010;107:294–304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Stempien-Otero A, Kim DH, Davis J. Molecular networks underlying myofibroblast fate and fibrosis. J Mol Cell Cardiol 2016;97:153–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Stambe C, Atkins RC, Tesch GH, et al. The role of p38alpha mitogen-activated protein kinase activation in renal fibrosis. J Am Soc Nephrol 2004;15:370–9. [DOI] [PubMed] [Google Scholar]
- 34.Wang L, Ma R, Flavell RA, et al. Requirement of mitogen-activated protein kinase kinase 3 (MKK3) for activation of p38alpha and p38delta MAPK isoforms by TGF-beta 1 in murine mesangial cells. J Biol Chem 2002;277:47257–62. [DOI] [PubMed] [Google Scholar]
- 35.Chakraborty D, Sumova B, Mallano T, et al. Activation of STAT3 integrates common profibrotic pathways to promote fibroblast activation and tissue fibrosis. Nat Commun 2017;8:1130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Dees C, Potter S, Zhang Y, et al. TGF-beta-induced epigenetic deregulation of SOCS3 facilitates STAT3 signaling to promote fibrosis. J Clin Invest 2020;130:2347–2363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.O’Reilly S, Ciechomska M, Cant R, et al. Interleukin-6 (IL-6) trans signaling drives a STAT3-dependent pathway that leads to hyperactive transforming growth factor-beta (TGF-beta) signaling promoting SMAD3 activation and fibrosis via Gremlin protein. J Biol Chem 2014;289:9952–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Su SA, Yang D, Wu Y, et al. EphrinB2 Regulates Cardiac Fibrosis Through Modulating the Interaction of Stat3 and TGF-beta/Smad3 Signaling. Circ Res 2017;121:617–627. [DOI] [PubMed] [Google Scholar]
- 39.Yuan Y, Zhang Y, Han X, et al. Relaxin alleviates TGFbeta1-induced cardiac fibrosis via inhibition of Stat3-dependent autophagy. Biochem Biophys Res Commun 2017;493:1601–1607. [DOI] [PubMed] [Google Scholar]
- 40.Civitarese RA, Kapus A, McCulloch CA, et al. Role of integrins in mediating cardiac fibroblast-cardiomyocyte cross talk: a dynamic relationship in cardiac biology and pathophysiology. Basic Res Cardiol 2017;112:6. [DOI] [PubMed] [Google Scholar]
- 41.Schroer AK, Merryman WD. Mechanobiology of myofibroblast adhesion in fibrotic cardiac disease. J Cell Sci 2015;128:1865–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Baker EL, Zaman MH. The biomechanical integrin. J Biomech 2010;43:38–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Chen C, Li R, Ross RS, et al. Integrins and integrin-related proteins in cardiac fibrosis. J Mol Cell Cardiol 2016;93:162–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Humphries JD, Byron A, Humphries MJ. Integrin ligands at a glance. J Cell Sci 2006;119:3901–3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Gullberg D, Gehlsen KR, Turner DC, et al. Analysis of alpha 1 beta 1, alpha 2 beta 1 and alpha 3 beta 1 integrins in cell--collagen interactions: identification of conformation dependent alpha 1 beta 1 binding sites in collagen type I. EMBO J 1992;11:3865–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Popova SN, Barczyk M, Tiger CF, et al. Alpha11 beta1 integrin-dependent regulation of periodontal ligament function in the erupting mouse incisor. Mol Cell Biol 2007;27:4306–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Balasubramanian S, Quinones L, Kasiganesan H, et al. beta3 integrin in cardiac fibroblast is critical for extracellular matrix accumulation during pressure overload hypertrophy in mouse. PLoS One 2012;7:e45076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Sarrazy V, Koehler A, Chow ML, et al. Integrins alphavbeta5 and alphavbeta3 promote latent TGF-beta1 activation by human cardiac fibroblast contraction. Cardiovasc Res 2014;102:407–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Hamzeh MT, Sridhara R, Alexander LD. Cyclic stretch-induced TGF-beta1 and fibronectin expression is mediated by beta1-integrin through c-Src- and STAT3-dependent pathways in renal epithelial cells. Am J Physiol Renal Physiol 2015;308:F425–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Pechkovsky DV, Prele CM, Wong J, et al. STAT3-mediated signaling dysregulates lung fibroblast-myofibroblast activation and differentiation in UIP/IPF. Am J Pathol 2012;180:1398–412. [DOI] [PubMed] [Google Scholar]
- 51.Boopathy GTK, Hong W. Role of Hippo Pathway-YAP/TAZ Signaling in Angiogenesis. Front Cell Dev Biol 2019;7:49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Halder G, Johnson RL. Hippo signaling: growth control and beyond. Development 2011;138:9–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Del Re DP. Beyond the Cardiomyocyte: Consideration of HIPPO Pathway Cell-Type Specificity. Circ Res 2018;123:30–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Sun M, Spill F, Zaman MH. A Computational Model of YAP/TAZ Mechanosensing. Biophys J 2016;110:2540–2550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Aragona M, Panciera T, Manfrin A, et al. A mechanical checkpoint controls multicellular growth through YAP/TAZ regulation by actin-processing factors. Cell 2013;154:1047–1059. [DOI] [PubMed] [Google Scholar]
- 56.Liu F, Lagares D, Choi KM, et al. Mechanosignaling through YAP and TAZ drives fibroblast activation and fibrosis. Am J Physiol Lung Cell Mol Physiol 2015;308:L344–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Jin B, Zhu J, Shi HM, et al. YAP activation promotes the transdifferentiation of cardiac fibroblasts to myofibroblasts in matrix remodeling of dilated cardiomyopathy. Braz J Med Biol Res 2018;52:e7914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Ebrahimighaei R, McNeill MC, Smith SA, et al. Elevated cyclic-AMP represses expression of exchange protein activated by cAMP (EPAC1) by inhibiting YAP-TEAD activity and HDAC-mediated histone deacetylation. Biochim Biophys Acta Mol Cell Res 2019;1866:1634–1649. [DOI] [PubMed] [Google Scholar]
- 59.Mo JS, Meng Z, Kim YC, et al. Cellular energy stress induces AMPK-mediated regulation of YAP and the Hippo pathway. Nat Cell Biol 2015;17:500–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Pan J, Fukuda K, Saito M, et al. Mechanical stretch activates the JAK/STAT pathway in rat cardiomyocytes. Circ Res 1999;84:1127–36. [DOI] [PubMed] [Google Scholar]
- 61.He J, Bao Q, Zhang Y, et al. Yes-Associated Protein Promotes Angiogenesis via Signal Transducer and Activator of Transcription 3 in Endothelial Cells. Circ Res 2018;122:591–605. [DOI] [PubMed] [Google Scholar]
- 62.Du YE, Tu G, Yang G, et al. MiR-205/YAP1 in Activated Fibroblasts of Breast Tumor Promotes VEGF-independent Angiogenesis through STAT3 Signaling. Theranostics 2017;7:3972–3988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Gruber R, Panayiotou R, Nye E, et al. YAP1 and TAZ Control Pancreatic Cancer Initiation in Mice by Direct Up-regulation of JAK-STAT3 Signaling. Gastroenterology 2016;151:526–39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Olson EN, Nordheim A. Linking actin dynamics and gene transcription to drive cellular motile functions. Nat Rev Mol Cell Biol 2010;11:353–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Janmey PA, Wells RG, Assoian RK, et al. From tissue mechanics to transcription factors. Differentiation 2013;86:112–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Huang X, Yang N, Fiore VF, et al. Matrix stiffness-induced myofibroblast differentiation is mediated by intrinsic mechanotransduction. Am J Respir Cell Mol Biol 2012;47:340–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Shimizu T, Liao JK. Rho Kinases and Cardiac Remodeling. Circ J 2016;80:1491–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Shimizu T, Narang N, Chen P, et al. Fibroblast deletion of ROCK2 attenuates cardiac hypertrophy, fibrosis, and diastolic dysfunction. JCI Insight 2017;2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Oh RS, Haak AJ, Smith KMJ, et al. RNAi screening identifies a mechanosensitive ROCK-JAK2-STAT3 network central to myofibroblast activation. J Cell Sci 2018;131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Unudurthi SD, Greer-Short A, Patel N, et al. Spectrin-based pathways underlying electrical and mechanical dysfunction in cardiac disease. Expert Rev Cardiovasc Ther 2018;16:59–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Baines AJ, Pinder JC. The spectrin-associated cytoskeleton in mammalian heart. Front Biosci 2005;10:3020–33. [DOI] [PubMed] [Google Scholar]
- 72.Bennett V, Baines AJ. Spectrin and ankyrin-based pathways: metazoan inventions for integrating cells into tissues. Physiol Rev 2001;81:1353–92. [DOI] [PubMed] [Google Scholar]
- 73.Bennett V, Lorenzo DN. Spectrin- and ankyrin-based membrane domains and the evolution of vertebrates. Curr Top Membr 2013;72:1–37. [DOI] [PubMed] [Google Scholar]
- 74.Hashemi SM, Hund TJ, Mohler PJ. Cardiac ankyrins in health and disease. J Mol Cell Cardiol 2009; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Tang Y, Katuri V, Dillner A, et al. Disruption of transforming growth factor-beta signaling in ELF beta-spectrin-deficient mice. Science 2003;299:574–7. [DOI] [PubMed] [Google Scholar]
- 76.Lim JA, Baek HJ, Jang MS, et al. Loss of beta2-spectrin prevents cardiomyocyte differentiation and heart development. Cardiovasc Res 2014;101:39–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Piersma B, Wouters OY, Bank RA. alphaII-spectrin and betaII-spectrin do not affect TGFbeta1-induced myofibroblast differentiation. Cell Tissue Res 2018;374:165–175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Lin L, Yao Z, Bhuvaneshwar K, et al. Transcriptional regulation of STAT3 by SPTBN1 and SMAD3 in HCC through cAMP-response element-binding proteins ATF3 and CREB2. Carcinogenesis 2014;35:2393–403. [DOI] [PubMed] [Google Scholar]
- 79.Patel NJ, Nassal DM, Greer-Short AD, et al. betaIV-Spectrin/STAT3 complex regulates fibroblast phenotype, fibrosis, and cardiac function. JCI Insight 2019;4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Hund TJ, Koval OM, Li J, et al. A betaIV spectrin/CaMKII signaling complex is essential for membrane excitability in mice. J Clin Invest 2010;120:3508–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Martin TP, Lawan A, Robinson E, et al. Adult cardiac fibroblast proliferation is modulated by calcium/calmodulin-dependent protein kinase II in normal and hypertrophied hearts. Pflugers Arch 2014;466:319–30. [DOI] [PubMed] [Google Scholar]
- 82.Zhang W, Chen DQ, Qi F, et al. Inhibition of calcium-calmodulin-dependent kinase II suppresses cardiac fibroblast proliferation and extracellular matrix secretion. J Cardiovasc Pharmacol 2010;55:96–105. [DOI] [PubMed] [Google Scholar]
- 83.Huang Z, Chen XJ, Qian C, et al. Signal Transducer and Activator of Transcription 3/MicroRNA-21 Feedback Loop Contributes to Atrial Fibrillation by Promoting Atrial Fibrosis in a Rat Sterile Pericarditis Model. Circ Arrhythm Electrophysiol 2016;9:e003396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Chen Y, Surinkaew S, Naud P, et al. JAK-STAT signaling and the atrial fibrillation promoting fibrotic substrate. Cardiovasc Res 2017;113:310–320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Tsai CT, Lin JL, Lai LP, et al. Membrane translocation of small GTPase Rac1 and activation of STAT1 and STAT3 in pacing-induced sustained atrial fibrillation. Heart Rhythm 2008;5:1285–93. [DOI] [PubMed] [Google Scholar]
- 86.Xue XD, Huang JH, Wang HS. Angiotensin II activates signal transducers and activators of transcription 3 via Rac1 in the atrial tissue in permanent atrial fibrillation patients with rheumatic heart disease. Cell Biochem Biophys 2015;71:205–13. [DOI] [PubMed] [Google Scholar]
- 87.Chang SH, Yeh YH, Lee JL, et al. Transforming growth factor-beta-mediated CD44/STAT3 signaling contributes to the development of atrial fibrosis and fibrillation. Basic Res Cardiol 2017;112:58. [DOI] [PubMed] [Google Scholar]
- 88.Yajima T, Murofushi Y, Zhou H, et al. Absence of SOCS3 in the cardiomyocyte increases mortality in a gp130-dependent manner accompanied by contractile dysfunction and ventricular arrhythmias. Circulation 2011;124:2690–701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Brar BK, Stephanou A, Liao Z, et al. Cardiotrophin-1 can protect cardiac myocytes from injury when added both prior to simulated ischaemia and at reoxygenation. Cardiovasc Res 2001;51:265–74. [DOI] [PubMed] [Google Scholar]
- 90.Zou Y, Takano H, Mizukami M, et al. Leukemia inhibitory factor enhances survival of cardiomyocytes and induces regeneration of myocardium after myocardial infarction. Circulation 2003;108:748–53. [DOI] [PubMed] [Google Scholar]
- 91.Obana M, Maeda M, Takeda K, et al. Therapeutic activation of signal transducer and activator of transcription 3 by interleukin-11 ameliorates cardiac fibrosis after myocardial infarction. Circulation 2010;121:684–91. [DOI] [PubMed] [Google Scholar]
- 92.Oba T, Yasukawa H, Hoshijima M, et al. Cardiac-specific deletion of SOCS-3 prevents development of left ventricular remodeling after acute myocardial infarction. J Am Coll Cardiol 2012;59:838–52. [DOI] [PubMed] [Google Scholar]
- 93.Obana M, Miyamoto K, Murasawa S, et al. Therapeutic administration of IL-11 exhibits the postconditioning effects against ischemia-reperfusion injury via STAT3 in the heart. Am J Physiol Heart Circ Physiol 2012;303:H569–77. [DOI] [PubMed] [Google Scholar]
- 94.Pipicz M, Demjan V, Sarkozy M, et al. Effects of Cardiovascular Risk Factors on Cardiac STAT3. Int J Mol Sci 2018;19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Zgheib C, Zouein FA, Kurdi M, et al. Differential STAT3 signaling in the heart: Impact of concurrent signals and oxidative stress. 2012;1:101–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Cheon H, Yang J, Stark GR. The functions of signal transducers and activators of transcriptions 1 and 3 as cytokine-inducible proteins. J Interferon Cytokine Res 2011;31:33–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Arany I, Reed DK, Grifoni SC, et al. A novel U-STAT3-dependent mechanism mediates the deleterious effects of chronic nicotine exposure on renal injury. Am J Physiol Renal Physiol 2012;302:F722–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Mir SA, Chatterjee A, Mitra A, et al. Inhibition of signal transducer and activator of transcription 3 (STAT3) attenuates interleukin-6 (IL-6)-induced collagen synthesis and resultant hypertrophy in rat heart. 2012;287:2666–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Bao Q, Zhang B, Suo Y, et al. Intermittent hypoxia mediated by TSP1 dependent on STAT3 induces cardiac fibroblast activation and cardiac fibrosis. Elife 2020;9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Enomoto D, Obana M, Miyawaki A, et al. Cardiac-specific ablation of the STAT3 gene in the subacute phase of myocardial infarction exacerbated cardiac remodeling. Am J Physiol Heart Circ Physiol 2015;309:H471–80. [DOI] [PubMed] [Google Scholar]
- 101.Kishore R, Verma SK. Roles of STATs signaling in cardiovascular diseases. JAKSTAT 2012;1:118–24. [DOI] [PMC free article] [PubMed] [Google Scholar]