

Abstract
CDKL5 deficiency disorder (CDD) is an infantile epileptic encephalopathy presenting with early-onset seizures, intellectual disability, motor impairment, and autistic features. The disorder has been linked to mutations in the X-linked CDKL5, and mouse models of the disease recapitulate several aspects of CDD symptomology, including learning and memory impairments, motor deficits, and autistic-like features. Although early-onset epilepsy is one of the hallmark features of CDD, evidence of spontaneous seizure activity has only recently been described in Cdkl5-deficient heterozygous female mice, but the etiology, prevalence, and sex-specificity of this phenotype remain unknown. Here, we report the first observation of disturbance-associated seizure-like events in heterozygous female mice across two independent mouse models of CDD: Cdkl5 knockout mice and CDKL5 R59X knock-in mice. We find that both the prevalence and severity of this phenotype increase with aging, with a median onset around 28 weeks of age. Similar seizure-like events are not observed in hemizygous knockout male or homozygous knockout female littermates, suggesting that X-linked cellular mosaicism is a driving factor underlying these seizure-like events. Together, these findings not only contribute to our understanding of the effects of CDKL5 loss on seizure susceptibility, but also document a novel, pre-clinical phenotype for future therapeutic investigation.
Keywords: CDKL5, seizures, mouse models of epilepsy, neurodevelopmental disorders, X-linked mosaicism
Introduction
Pathogenic variants in the X-linked gene encoding cyclin-dependent kinase-like 5 (CDKL5) have been associated with the severe childhood epileptic encephalopathy known as CDKL5 deficiency disorder (CDD)(Bahi-Buisson and Bienvenu, 2012; Fehr et al., 2013; Kalscheuer et al., 2003; Tao et al., 2004; Weaving et al., 2004). CDD is characterized by a heterogeneous array of clinical symptoms including early-onset seizures, marked hypotonia, autistic features, and severe neurodevelopmental impairment(Demarest et al., 2019; Olson et al., 2019). The disorder predominantly affects young females heterozygous for mutations in the X-linked CDKL5, with an overall incidence of one per forty-two thousand live births, making it one of the most common genetic causes of epilepsy in children(Demarest et al., 2019; Symonds et al., 2019). Despite the constellation of phenotypes presented in CDD, the severe, early-onset seizures in particular drastically impact quality of life, with ninety percent of patients displaying seizures by three months of age. Eighty percent of children with CDD have daily seizures, and fewer than half report more than two months of seizure freedom(Olson et al., 2019). Furthermore, children with epilepsy that presents before 3 years of age have been shown to carry a high burden of behavioral and cognitive comorbidities, and this risk increases with increased seizure incidence(Berg et al., 2008; Berg et al., 2012). This high burden of refractory seizures in CDD make it a particularly debilitating aspect of the disorder for both patients and their families, highlighting the pressing need to develop effective therapeutics for seizure management.
Several mouse models of CDD have been established and exhibit many of the cardinal phenotypes of the disorder, including autistic-like behaviors, impaired motor control, and poor learning and memory(Zhou et al., 2017; Zhu and Xiong, 2019). The first Cdkl5 knockout mouse was generated and characterized in 2012, mimicking a reported CDD splice-site mutation resulting in skipping of exon 6 and loss of CDKL5 function(Archer et al., 2006; Wang et al., 2012). Although hippocampal electroencephalographic (EEG) recordings in postnatal day (P) 70-90 male mice of this line demonstrated disruptions in event-related neuronal oscillations in response to auditory stimuli, chronic video-EEG recordings revealed no spontaneous seizure activity. Furthermore, basal EEG patterns and power distribution across various oscillation frequencies were unchanged in hemizygous knockout male mice compared to littermate controls. In 2014, Amendola et al. developed and characterized another Cdkl5 knockout mouse (exon 4 deletion) in which EEG recordings also revealed no spontaneous epileptiform activity in hemizygous male knockouts(Amendola et al., 2014). More recently, an exon 2 deletion mouse of Cdkl5 as well as the CDKL5 R59X knock-in, mimicking a CDD-associated nonsense mutation resulting in loss of CDKL5 function, were created. Although these animals display an increased sensitivity to NMDA-induced seizures, no overt spontaneous seizure activity was reported(Okuda et al., 2017; Tang et al., 2019). Recently, spontaneous epileptic spasms were observed in aged heterozygous female mice carrying an exon 6 deletion (Cdkl5 knockout) and CDKL5 R59X knock-in(Mulcahey et al., 2020). However, the etiology, prevalence, and age-dependence of this phenotype remain unknown.
The lack of a robust seizure phenotype in CDD mouse models has so far been attributed to neural network differences between human and mouse brains(Zhou et al., 2017), but the majority of these studies have been limited to hemizygous male mice(Amendola et al., 2014; Wang et al., 2012). Despite the lack of overt, behavioral seizures in animal models of CDD reported to date, several groups have observed synaptic and circuit-level hyperexcitability in various brain regions as well as altered sensitivity to specific chemoconvulsants. CDKL5 is most highly expressed in neurons of the forebrain, and selective loss of CDKL5 from forebrain excitatory neurons results in increased frequency of miniature excitatory post-synaptic currents (mEPSC) in the hippocampal CA1 region(Tang et al., 2017). Interestingly, selective loss from GABAergic forebrain neurons also recapitulates this increased mEPSC frequency coupled with circuit hyperexcitability in the form of aberrant paired-pulse facilitation(Tang et al., 2019). In this same region, loss of CDKL5 was found to result in enhanced long-term potentiation by a misregulation of postsynaptic GluN2B-containing NMDA receptor localization, and in line with this, knockout mice exhibit significant hyperexcitability to NMDA(Okuda et al., 2017; Tang et al., 2019). Although high-dose kainic acid induces overt seizures in Cdkl5 hemizygous knockout males at similar latency to wild-type littermates, the mean duration of the resultant epileptic EEG bursts is reported to be longer(Amendola et al., 2014). Cdkl5 knockout mice have also been reported to display decreased latency to the first stages of seizure progression upon low-dose pentylenetetrazol administration(Yennawar et al., 2019). These findings suggest that seizure susceptibility may in fact be altered in the absence of CDKL5, but perhaps only under certain environmental conditions or specific periods of development and aging.
Here, we report the occurrence of overt, myoclonic and tonic-clonic behavioral seizure-like events in two distinct mouse models of CDD: a Cdkl5 knockout carrying a deletion of exon 6 and the CDKL5 R59X knock-in, both resulting in loss of CDKL5 function. These events are specific to female mice heterozygous for mutations in Cdkl5, and not seen in hemizygous knockout males, homozygous knockout females, or wild-type littermates. Both the frequency and severity of this seizure-like activity increased with aging, with a median age of onset around 28 weeks of age. The lack of overt, similar seizure-like events in hemizygous male knockout and homozygous female knockout mice suggests that X-linked mosaicism of Cdkl5 deficiency likely drives this specific phenotype in mouse models of CDD.
Materials and Methods
Mouse strains.
The CDKL5 R59X knock-in(Tang et al., 2019) and Cdkl5 knockout(Wang et al., 2012) lines were generated as previously described, and have since been deposited at Jackson Laboratories (R59X: Stock No. 028856; Cdkl5 KO: Stock No. 021967).
Animal husbandry.
Experiments were conducted in accordance with the ethical guidelines of the National Institutes of Health and with the approval of the Institutional Animal Care and Use Committee of the University of Pennsylvania. Mice were group-housed in cages of two to five in a twelve-hour light/dark cycle with food and water provided ad libitum. Each breeding cage contains one male (C57BL/6J or Cdkl5R59X/y, R59X/y) and one to two females (Cdkl5R59X/+, R59X/+, or Cdkl5KO/+, KO/+). All progeny were genotyped either with the previously reported PCR strategies(Tang et al., 2019; Wang et al., 2012) or with qPCR by Transnetyx, Inc (Cordova, TN). Male littermates (Cdkl5R59X/y with Cdkl5+/y; Cdkl5KO/y with Cdkl5+/y) and female littermates (Cdkl5+/+ and/or Cdkl5R59X/R59X with Cdkl5R59X/+; Cdkl5KO/+ with Cdkl5+/+) were weaned at 3 weeks of age and housed together.
Seizure-like behavior monitoring.
Mice were monitored for disturbance-associated seizures twice a week, during varying times between 8AM to 7PM. Times of day with no extra disturbances occurring (such as cage changing) were chosen. Cages were swiftly and gently pulled out of the rack and moved onto the cage changing station, one cage at a time. Cage lids were immediately removed, and mice were mildly disturbed by a gentle touch of the hand, frequently to separate each mouse from huddling. Mice in the cage were then closely observed for about 3 minutes for the occurrence of any seizure-like events and scored according to the modified Racine scale (see Table 1, adapted from Velíšková et al., 1990). Stage 0: no changes in behavior; Stage 1: myoclonic jerks with sudden and repetitive movement of the head and neck with or without tail stiffening; Stage 2: atypical (unilateral or incomplete) clonic seizure; Stage 3: clonic seizure with forelimb clonus and rearing; Stage 4: tonic-clonic seizure with an initial wild run and subsequent loss of righting reflex; Stage 5: tonic-clonic seizure with full extension of fore- and hind-limbs. To streamline data collection, analysis, and presentation, all seizures ranging from Racine stages 1 to 2 were termed ‘myoclonic seizure-like events’ and events resembling Racine stages 3 to 5 were grouped as ‘severe seizure-like events.’
Table 1.
Modified Racine scale used for behavioral seizure scoring (adapted from Velíšková et al., 1990)
| Seizure Stage | Behavioral Expression | Grouping for analysis | Video Example |
|---|---|---|---|
| 0 | No changes in behavior | ||
| 1 | Myoclonic jerks with sudden and repetitive movement of the head and neck with or without tail stiffening | Myoclonic | Suppl Video 1A–C |
| 2 | Atypical (unilateral or incomplete) clonic seizure | Myoclonic | Suppl Video 2 |
| 3 | Clonic seizure with forelimb clonus and rearing | Severe | Suppl Video 3A–C |
| 4 | Tonic-clonic seizure with an initial wild run and subsequent loss of righting reflex | Severe | Suppl Video 4A–C |
| 5 | Tonic-clonic seizure with full extension of fore- and hind-limbs | Severe | Suppl Video 5 |
PTZ-induced seizure scoring.
Seizures were induced in male and female mice at either around postnatal day 60 or 150 by subcutaneous administration of 80–100 mg·kg−1 pentylenetetrazol (PTZ; Sigma-Aldrich; P6500) in sterile, 0.9% PBS. A pilot cohort of wild-type and Cdkl5 knockout mice at about P60 were tested at various doses of PTZ to establish a sufficient dose for maximum seizure score induction in wild-type and establish an effect size for this phenotype in males and females that could then be applied across ages. Seizure progression was scored using the same modified Racine scale as described above for seizure monitoring (Stages 0-5). After drug injection, maximum seizure scores were recorded within a 45 min observation period.
Determination of estrous cycle stage.
Mice observed having disturbance-associated seizures underwent vaginal lavage immediately after they resumed normal activities to check for estrous cycle stage. Each mouse was picked up by the base of the tail and 20ul of saline was gently expelled into the vaginal cavity using a pipette, aspirated back five times, and then dispensed onto a microscopic slide. Dry smears were then evaluated under the microscope and scored for the estrous cycle stage according to the presence/absence and percentage of neutrophils, nucleated epithelial cells and cornified epithelial cells(Caligioni, 2009).
Proestrus stage: predominance of nucleated epithelial cells; Estrus stage: predominance of cornified epithelial cells; Metestrus stage: predominance of leucocytes with a small amount of nucleated and/or cornified epithelial cells; Diestrus stage: predominance of leucocytes.
Two-Channel EEG implantation surgery.
Experiments were conducted in accordance with the approval of the Institutional Animal Care and Use Committee of the University of Pennsylvania. Mice were stereotaxically implanted with an electrode assembly under continuous isoflurane anesthesia. The electrode assembly consisted of 4 leads: 2 surface electrodes attached to miniature skull screws placed over the left and right frontal cortices (from Bregma: A/P +1.0 mm, M/L ± 1.5 mm); and a reference and a ground electrode also attached to miniature skull screws directly behind Lambda on either side of midline (from Bregma: A/P – −5.2 mm, M/L ± 1.5 mm);. Silver wires (0.13 mm diameter) were attached to each electrode and connected to an 8-pin headmount (Pinnacle Technology Inc, Lawrence, KS). The entire assembly was secured on the skull with dental cement (Ortho-Jet, Lang Dental, Wheeling, IL). Mice were given at least 72 hours post-surgery to recover prior to being placed in a Plexiglas recording cage (Pinnacle Technology Inc, Lawrence, KS). EEG waveform was amplified by a preamplifier with a gain of 10 pV (Pinnacle Technology Inc, Lawrence, KS) at the head of the animal before being passed through a low-torque commutator to the Sirenia Acquisition system (Pinnacle Technology Inc, Lawrence, KS) for final-stage amplification and filtering. EEG was sampled at 2000 Hz in a 12-hour light/dark cycle with food and water provided ad libitum. Mice in the monitoring cages were mildly disturbed by a gentle touch of the hand at varying times and intervals throughout the monitoring period to mimic the seizure monitoring interactions as described above. Video was watched by a reviewer familiar with the appearance of the disturbance-associated seizures. EEG traces were processed with the application of a 1 Hz high-pass filter, 500 Hz low-pass filter, and 60 Hz notch filter and visualized within Serenia Basic (Pinnacle Technology Inc, Lawrence, KS).
Four-Channel EEG implantation surgery.
EEG recordings were performed as previously described in the Mulcahey et al. study(Mulcahey et al., 2020). All experiments were conducted in accordance with the approval of the Institutional Animal Care and Use Committee of the Children’s Hospital of Philadelphia. The electrode assembly consisted of 6 leads: 2 surface electrodes attached to miniature skull screws placed over the left and right frontal cortices (from Bregma: A/P −1.2 mm, M/L ±1.1 mm); a double-depth electrode in right hippocampus (A/P −2.2 mm, M/L +1.2 mm, D/V −1.3 mm); and a reference and a ground electrode directly behind Lambda on either side of midline. Teflon-coated silver wires (0.13 mm diameter) were attached to each electrode and connected to a 6-pin pedestal (Plastics One, Roanoke, VA). The entire assembly was secured on the skull with dental cement (Ortho-Jet, Lang Dental, Wheeling, IL). All surgeries were performed by an experimenter blinded to genotype. Video-EEG recording from freely moving mice was conducted as previously described in Mulcahey et al.(Mulcahey et al., 2020). Each mouse underwent video-EEG recording for at least 11 days across 8 wild type mice, 7 R59X/+ mice, and 9 KO/+ mice. During this period, each mouse underwent a routine cage cleaning each week at approximately 9:00-11:00 AM (lights out at 1:00 PM). Cage cleanings alternated between “deep” cleans (mouse is removed from recording cage while recording cage is cleaned) and “quick” cleans (mouse is lifted, a temporary floor is placed in the recording cage, a clean floor and clean bedding are placed in the cage). In addition to cage cleanings, veterinary staff checked each recording cage each morning before 11:00 AM. Veterinary staff only opened the cages (1) if the mouse needed food or water or (2) if the mouse appeared unhealthy. At the conclusion of video-EEG recording, the mice were removed from the recording apparatus.
Eight-channel EEG implantation surgery.
Electroencephalographic mouse studies were performed as previously described using 8 electrodes (hippocampal, bilateral parietal, bilateral motor, bilateral visual electrodes) using the following coordinates (from Bregma – Bilateral motor: 0.5 mm Anterior-Posterior (A-P) and 1.0 mm medial-lateral (M-L); bilateral parietal: −0.7 mm A-P, 3.0 mm M-L; bilateral visual −3.5 A-P, 2.0 mm M-L; hippocampus: −2.2 mm A-P, 2.0 mm M-L, and 1.7 mm ventral). (Marsh et al., 2009; Simonet et al., 2015). All experiments were conducted in accordance with the approval of the Institutional Animal Care and Use Committee of the Children’s Hospital of Philadelphia. After 24–48 h of recovery in their home cage the animals were transferred to the monitoring cage and recorded using a 32-channel Intan extracellular amplifier (RHD2000 USB Interface Board, Intan Technologies) using either DataWave software or importing the raw data into Matlab. The recordings were sampled at 2000 Hz, with no on-line filtering. Electrode impedances were tested prior to all recordings. All animals were recorded for at least 72 h. Other than when being placed in the chambers or removed, mice were not handled during the monitoring.
Statistical analyses.
Similar sample sizes for all seizure monitoring across genotypes was chosen. For PTZ seizure induction experiments, we tested a pilot cohort of wild-type and Cdkl5 knockout animals at various doses of PTZ to establish an effect size for this phenotype. This enabled proper power analysis to determine the appropriate sample size and dose for our experimental cohorts (n=6-9 for males, n=8-10 for females). Maximum seizure scores were statistically analyzed using the Mann-Whitney test for significance. Statistical analyses were performed using Prism (GraphPad). All data sets were analyzed using the Shapiro-Wilk test for normality. All survival curves were compared using a log-rank (Mantel-Cox) test to test for significance. All graphs are plotted using Prism (GraphPad). In our figures, * is used to denote all 0.01<p<0.05, ** for 0.001<p<0.01, *** for 0.0001<p<0.001, and **** for p<0.0001.
Results
Female mice heterozygous for mutations in Cdkl5 exhibit seizure-like events in an age-dependent manner
The severity and prevalence of seizures in patients with CDD has motivated numerous efforts to model and examine seizure-related phenotypes in Cdkl5-deficient mice(Amendola et al., 2014; Okuda et al., 2018; Tang et al., 2019; Tang et al., 2017; Wang et al., 2012; Yennawar et al., 2019). Initial reports in several mouse models of CDD failed to recapitulate the spontaneous seizure phenotype commonly presented in CDD patients. However, the majority of these studies, including our own, have focused on hemizygous knockout male mice. Furthermore, the majority of these behavioral or functional assays have been confined to relatively young adult mice (postnatal days (P) 50-120).
Given that Cdkl5 is X-linked, we maintain and generate experimental mice by crossing wild-type males to females heterozygous for Cdkl5 mutations (e.g., R59X). In the process of breeding R59X heterozygous females (Cdkl5RS9X/+ or R59X/+), we observed that numerous aged (>P200) females displayed abnormal movements following cage opening/disturbance. Upon observing these abnormal movements, which we felt resembled seizure-like behaviors, we sought to establish a methodology to consistently and quantitatively score these events. We adapted a modified Racine scale widely used to describe the progress and severity of chemoconvulsant-induced seizures (Table 1), and set up a process of regular monitoring (~3 minutes per cage, 2–3 times a week) and seizure scoring along this scale for our R59X/+ breeder and non-breeder females as well as wild-type littermate controls(Velíšková et al., 1990). Over the span of fourteen months, upon cage opening or handling, we have documented behavioral events that we believe resemble seizures beginning as early as 17 weeks of age.
The most frequent seizure-like events observed consisted of myoclonic jerks involving sudden and repetitive movement of the head and neck with or without tail stiffening (Racine Stage 1, Supplementary Video 1A, 1B, 1C). Tonic-clonic events with an initial appearance of wild running followed by loss of the righting reflex (Racine Stage 4, Supplementary Video 4A, 4B, 4C) were the second most frequent seizure-like event observed. On rare occasions, the tonic-clonic events would progress into extension of fore- and hind-limbs, reaching Racine Stage 5 (Supplementary Video 5). Clonic events with forelimb clonus and rearing were also relatively rare (Racine Stage 3, Supplementary Video 3A, 3B, 3C). To streamline data collection, analysis, and presentation, all events ranging from Racine stages 1 to 2 were termed ‘myoclonic seizures-like events’ and events resembling Racine stages 3 to 5 were grouped as ‘severe seizure-like events.’ Mice were only scored as having exhibited a seizure-like event after two separate seizure-like events had been observed (typically Racine Stage 1). By 20 weeks of age, we found that 12% of R59X/+ females had displayed a second seizure-like event, and this increased to 70% of R59X/+ females exhibiting at least 2 seizures-like events by 40 weeks of age. We have not observed similar events (or similar behaviors of any type) in wild-type (+/+) females, hemizygous knockout male mice (R59X/y), or wild-type male mice (+/y) across the age groups (Figure 1A). Notably, we also found that R59X/+ females presented an age-dependent shift in chemoconvulsant-induced seizures upon high-dose pentylenetetrazol (PTZ) administration. Both R59X/+ heterozygous females and R59X/y hemizygous males demonstrate a resilience to PTZ-induced seizures when compared to wild-type littermate controls at P60 (Supplemental Figure 1A & 1C). With aging, R59X/y males carry a similar resilience to PTZ-induced seizures when compared to +/y littermates (P150; Supplemental Figure 1B), however, R59X/+ females no longer show a resilience to PTZ at this older age (Supplemental Figure 1D). This sex-specific, age-dependent shift in PTZ-sensitivity provides further evidence that Cdkl5 heterozygous mutant female mice display a change in seizure susceptibility with aging that is not mirrored by hemizygous male knockout animals or wild-type littermate controls.
Figure 1. Disturbance-associated seizure-like events in heterozygous CDD female mice.
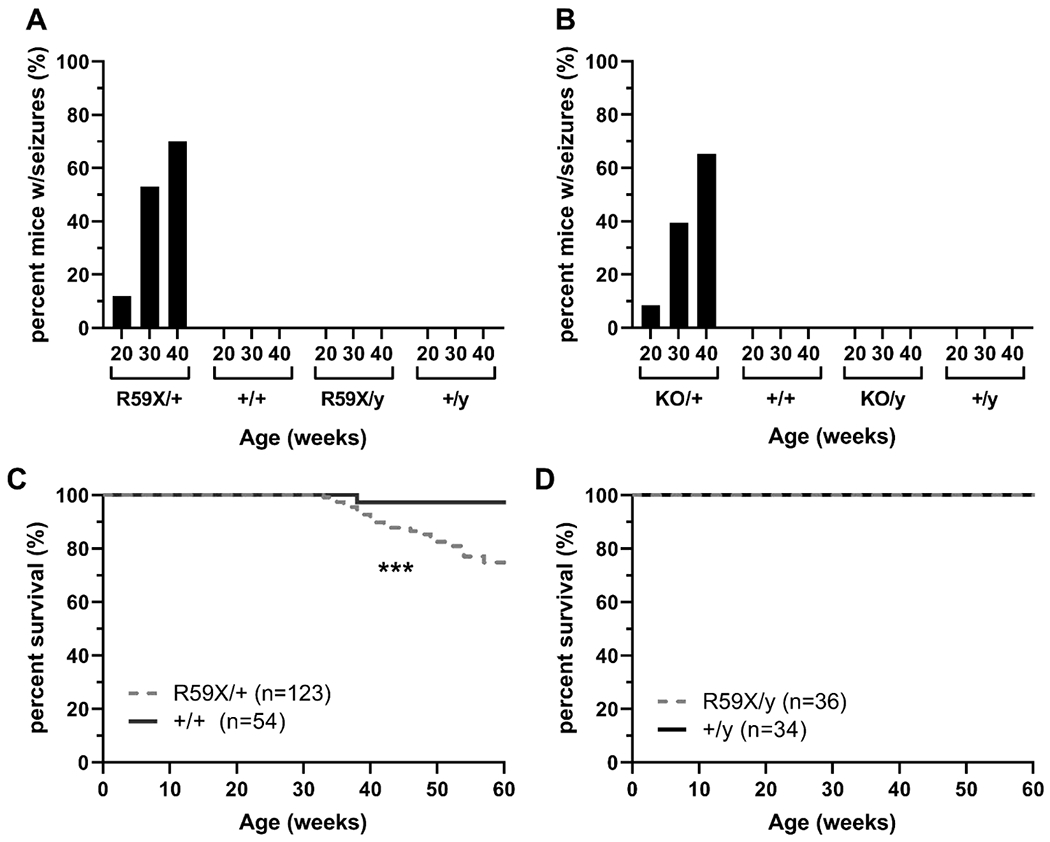
(A) Percentage of CdklRS9X (R59X) mice and littermates demonstrating seizure-like events at 20, 30, and 40 weeks of age. Cdkl5R59X/+ (R59X/+): 12% at 20 weeks (n=31); 53% at 30 weeks (n=28); 70% at 40 weeks (n=20). Cdkl5+/+ (+/+): 0% at 20 weeks (n=21); 0% at 30 weeks (n=19); 0% at 40 weeks (n=12). Cdkl5R59/y (R59X/y): 0% at 20 weeks (n=33); 0% at 30 weeks (n=24); 0% at 40 weeks (n=10). Cdkl5+/y (+/y): 0% at 20 weeks (n=34); 0% at 30 weeks (n=22); 0% at 40 weeks (n=10). (B) Percentage of Cdkl5KO (KO) mice and littermates displaying seizure-like events at 20, 30, and 40 weeks of age. Cdkl5KO/+ (KO/+): 8.57% at 20 weeks (n=35); 39.39% at 30 weeks (n=33); 65.22% at 40 weeks (n=23). Cdkl5+/+ (+/+): 0% at 20 weeks (n=34); 0% at 30 weeks (n=9); 0% at 40 weeks (n=5). Cdkl5KO/y (KO/y): 0% at 20 weeks (n=18); 0% at 30 weeks (n=8); 0% at 40 weeks (n=5). Cdkl5+/y (+/y): 0% at 20 weeks (n=20); 0% at 30 weeks (n=8); 0% at 40 weeks (n=7). (C) Longevity of R59X/+ (gray, dotted line; n=123) and +/+ (black, solid line; n=54) females. Mantel-Cox (log-rank) test, p=0.0007. (D) Longevity of R59X/y (gray, dotted line; n=36) and +/y (black, dotted line; n=34) males. Mantel-Cox (log-rank) test, ns. *p<0.05, **p<0.01, ***p<0.001, and ****p<0.0001.
As no laboratory had previously documented seizures in any CDD mouse model at the time, we attempted to confirm our findings in an independent Cdkl5 knockout line lacking Cdkl5 exon 6 (hereafter referred to as KO). Indeed, by 12 weeks of age we had observed at least two of these similar events in 8.7% of KO/+ females, and this increased to 65% of KO/+ females by 40 weeks of age. Again, no seizure-like activity was observed in wild-type (+/+) females, hemizygous knockout male mice (KO/y), or wild-type male mice (+/y) across any of the age groups (Figure 1B).
To rule out the possibility that this seizure-like phenotype is exclusive to the CDKL5 colonies we have maintained in our animal vivarium, we obtained additional R59X/+females (Stock No. 028856) and KO/KO females (Stock No. 003724) from the Jackson Laboratory, crossed them to newly obtained C57BL/6J males (Stock No. 000664), and subsequently monitored their progeny twice a week for seizure-like events as described above. 10 out of 13 R59X/+ females (77%), but no +/+ female or R59X/y male littermates, were observed to have at least one seizure-like event by 30 weeks of age. Similarly, KO/+ females, but no +/+ female or KO/y male littermates, displayed seizure-like events by 30 weeks of age. We therefore concluded that the phenotype we had detected was indeed due to loss of Cdkl5 function.
Cdkl5 heterozygous mutant females have a reduced life span
While monitoring R59X/+ female mice, we observed that several mice reaching Racine Stage 5 events would die after reaching the tonic phase of the seizure-like event. We found that on average, R59X/+ females have a significantly decreased lifespan compared to their wild-type littermate (+/+) females with significantly more R59X/+ females dying between 40-50 weeks of age (Figure 1C), possibly as a result of infrequent but lethal seizures. In contrast, no significant change in life span was found between R59X/y males and their wild-type (+/y) littermates (Figure 1D). These results suggest that the seizure-like events occurring in R59X/+ females may decrease life span.
Seizure-like event frequency and severity in Cdkl5 heterozygous females increases with age
Of the R59X/+ females observed starting at 12 weeks of age, the median age of onset of a second observed myoclonic seizure-like event (Racine Stages 1 to 2) was 28 weeks. More severe seizure-like events (Racine Stages 3 to 5) also occurred more frequently with increasing age, with a median onset of 47 weeks of age (Figure 2A). In addition to this increasing seizure-like event severity, the frequency of all observed seizure-like events also increased with age (Figure 2B).
Figure 2. Prevalence and severity of disturbance-associated seizure-like events in R59X/+ females increases with age.
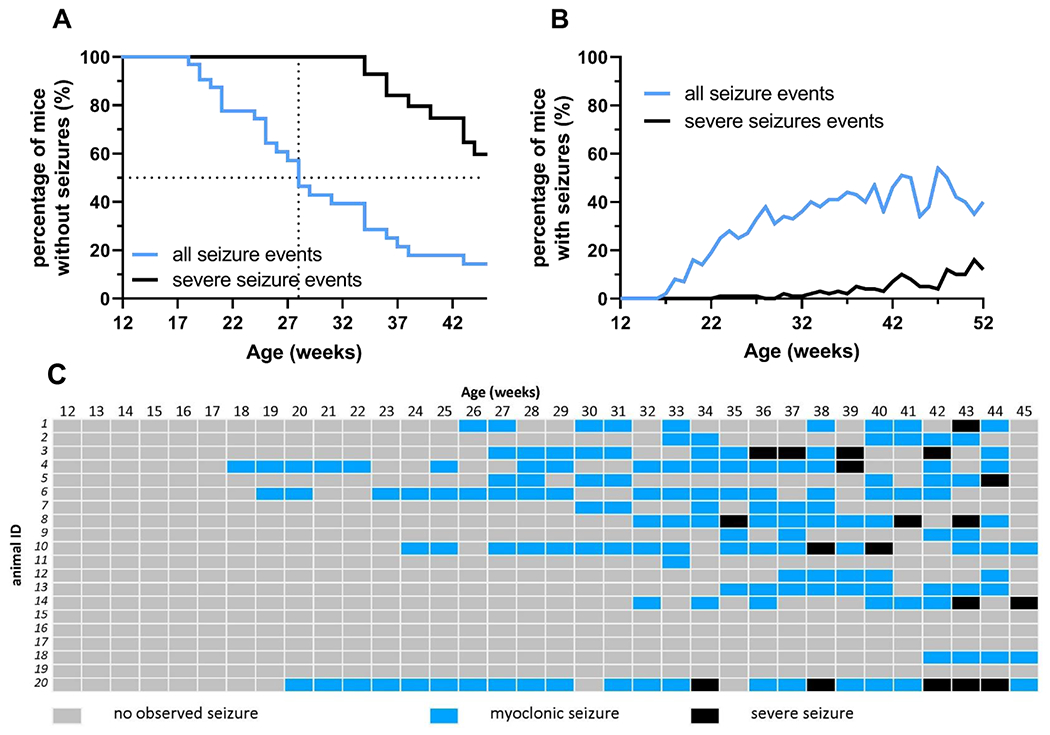
(A) Percentage of Cdkl5RS9X/+ (R59X/+) females presenting with any seizure-like event (Racine Stages 1 to 5; black line) and percentage of R59X/+ females presenting with at least one severe seizure-like event (Racine Stages 3 to 5; blue line) between 12 weeks to 45 weeks of age (n=20). Median age of onset for second observed seizure-like event was 28 weeks; median age of onset for severe seizure-like events was 47 weeks of age. (B) Percentage of R59X/+ females observed having at least one seizure-like event (Racine Stages 1 to 5; black line) and percentage of R59X/+ females observed having at least one severe seizure-like event (Racine Stages 3 to 5; blue line) any given week between 12 to 52 weeks of age (n=25).
(C) Event record of 20 Cdkl5RS9X/+ females observed from 12 weeks to 45 weeks of age. Gray: no seizure-like events observed during that week. Blue: at least one myoclonic seizure-like event (Racine Stages 1 to 2), but no severe seizure-like event, observed during that week. Black: at least one severe seizure-like event (Racine Stages 3 to 5) observed during that week.
It should be noted that there is significant individual variability within R59X/+ females for both the onset and the frequency of these seizure-like events. During our observations, some females displayed their first disturbance-associated seizure-like event as early as 17 weeks of age while others did not display an overt seizure-like event until 49 weeks of age or ever. For some R59X/+ mice, disturbance-associated seizure-like events occurred every week for a time span of a few months, while for other mice no events were observed after two initial seizure-like events. A summary of 20 R59X/+ females monitored from 12 to 45 weeks of age for all seizure-like events demonstrates this trend of age-dependency and variability (Figure 2C).
We similarly tracked the age-dependent development of seizure-like events in an independent Cdkl5 mutant line to validate our findings. In KO/+ females, the median age of onset of a second observed myoclonic seizure-like event (Racine Stages 1 to 2) was 32 weeks, with a first seizure-like event observed as early as 16 weeks of age in some mice. More severe seizure-like events (Racine Stages 3 to 5) occurred in KO/+ females with a median age of onset of 57 weeks (Figure 3A). Similar to the R59X/+ females, the likelihood of both myoclonic seizure-like events and a severe seizure-like events in KO/+ females increased with age (Figure 3B). We monitored 17 KO/+ females from 12 weeks until 42-45 weeks of age and recorded all seizure-like events, and found similar individual variability in both age-of-onset and event frequency as reported above for R59X/+ (Figure 3C).
Figure 3. Prevalence and severity of disturbance-associated seizure-like events in KO/+ females increases with age.
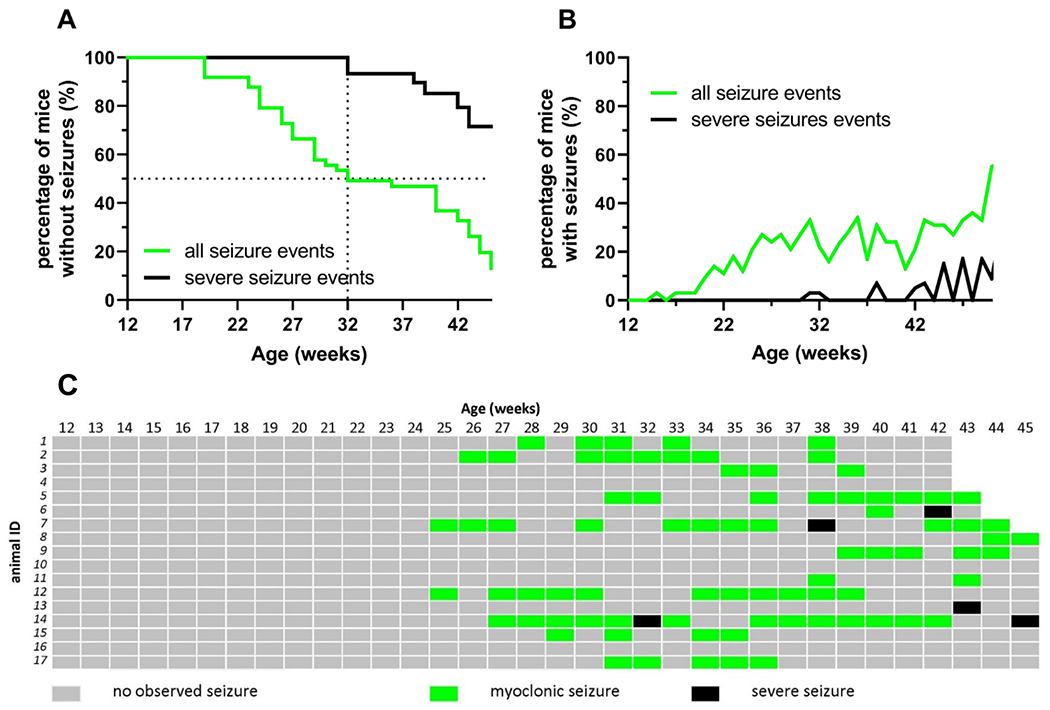
(A) Percentage of Cdkl5KO/+ (KO/+) females presenting with any seizure-like event (Racine Stages 1 to 5; black line) and percentage of KO/+ females presenting with at least one severe seizure-like event (Racine Stages 3 to 5; green line) between 12 weeks to 45 weeks of age (n=10). (B) Percentage of KO/+ females observed having at least one seizure-like event (Racine Stages 1 to 5; black line) and percentage of KO/+ females observed having at least one severe seizure-like event (Racine Stages 3 to 5; green line) any given week between 12 to 50 weeks of age (n>=10 at any given time point). Median age of onset for second observed seizure-like event was 32 weeks; median age of onset for severe seizure-like events was 57 weeks of age. (C) Event record of 17 KO/+ females observed from 12 weeks to 45 weeks of age. Gray: no seizure-like event observed during that week. Green: at least one myoclonic seizure-like event (Racine Stages 1 to 2), but no severe seizure-like event, observed during that week. Black: at least one severe seizure-like event (Racine Stages 3 to 5) observed during that week.
We recognize that this variability may also be a result of our sampling method of only observing seizure-like events 2-3 times a week upon cage disturbance, and that our reported incomplete penetrance of ~70% may not accurately reflect the actual incidence of seizure-like events amongst heterozygous female carriers of Cdkl5 mutations. In an attempt to capture EEG correlates of these seizure-like events, we performed EEG implantation surgery utilizing 2 cortical surface electrodes and video monitoring in R59X/+ female mice as described in the methods section (n=13). Mice under EEG surveillance with time-locked video were occasionally disturbed by a gentle touch of the hand at varying times and intervals throughout the monitoring period to mimic the seizure monitoring interactions. No Racine stage 3-5 handling-associated generalized tonic-clonic seizure-like events could be provoked across 40 handling events (average of 3 attempts/mouse). We watched over 700 hours of recorded video from 7 mice that had previously exhibited Racine stage 3-5 events (average of 102 hours/mouse) without observing any noticeable Racine stage 3-5 seizures. We also recorded over 145 hours of video-EEG data across 4 wild-type female and wild-type male mice without detection of similar seizure-like events as reported prior to surgery and implantation. Using this technique, we detected spontaneous epileptic spasms in R59X/+ female mice as recently described in the same mouse lines(Mulcahey et al., 2020), correlating with brief behavioral spasms we had previously observed, as well as interictal spikes during behavioral arrest without an otherwise behavioral correlate (Figure 4; Supplementary Videos 6 & 7).
Figure 4. R59X/+ female mice present with epileptic spasms.
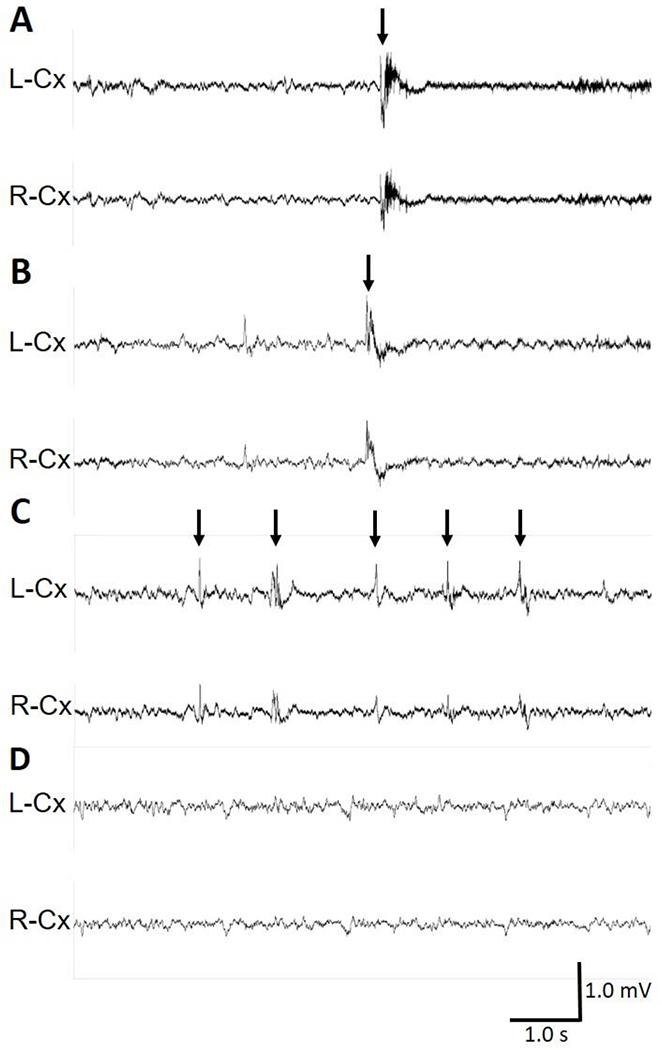
(A-D) Representative 10-s traces of intracranial EEG recorded in wild-type (+/+) and R59X/+ female mice. (A) Epileptic spasm in a R59X/+ heterozygous female mouse occurring during sleep, characterized by bilateral sharp-wave activity. Also see Supplementary Video 6 of this event. (B) Epileptic spasm in a different R59X/+ heterozygous female mouse occurring during sleep, characterized by bilateral spike/polyspike-wave activity. Also see Supplementary Video 7 of this event. (A-B) Arrows indicate time of spasm onset. (C) Interictal spiking during a period of behavioral arrest in a R59X/+ mouse. Arrows indicate interictal spikes. (D) Normal EEG during sleep in a wild type mouse. Vertical scale bar represents 1 mV. Horizontal scale bar represents 1.0 s in all traces. L-Cx: left cortical lead, R-Cx: right cortical lead.
Given the concern that inability to observe the seizure-like behaviors could be due to altered seizure semiology following EEG lead implantation, we utilized a different EEG implantation technique with 2 cortical surface electrodes and a double-depth electrode in right hippocampus in 7 R59X/+ and 9 KO/+ heterozygous females mice, as well as 8 wild-type female mice. Each mouse underwent video-EEG recording for at least 11 days with several handling events as described in the methods. Over the entire duration of over 2,300 hours of video-EEG monitoring for all 24 mice, the technicians who handled the mice observed no Racine stage 1-5 handling-provoked events (myoclonic jerks or generalized tonic-clonic seizure-like events). We also utilized a third EEG implantation technique with 8 electrodes (hippocampal, bilateral parietal, bilateral motor, bilateral visual electrodes) and failed to detect any myoclonic events or handling-provoked generalized tonic-clonic seizures while working with these mice.
To confirm the quality of our 2 cortical surface electrode EEG method and verify that the method does not suppress all generalized tonic-clonic seizures, we administered 100 mg·kg−1 PTZ subcutaneously at the conclusion of video monitoring. All mice (n=13/13) exhibited typical PTZ-induced seizures including sharp spikes, spike-wave, and polyspike discharges, typical characteristic of PTZ-induced seizures (Supplemental Figure 2).
Heterozygous mosaic, but not homozygous or hemizygous, loss of CDKL5 underlies spontaneous seizure-like event development
We were intrigued by the sex-specificity of our observed seizure-like phenotype and wanted to dissect the contributing factors that may underlie this segregation of seizure-like presentation to R59X/+ and KO/+ females but not R59X/y or KO/y hemizygous male knockouts. Since sex hormones are well known to affect the seizure threshold of both epileptic humans and mice, we examined whether estrous cycle or androgen hormones may be contributing to the sex-specificity of this phenotype(Tauboll et al., 2015). We collected vaginal lavage samples from R59X/+ and KO/+ females immediately upon observing disturbance-associated seizure-like events in order to test for estrous cycle stage at the time of event occurrence. Following a seizure-like event, we identified seizing females in all four stages of the mouse estrous cycle (Figure 5A), suggesting that the levels of sex hormones or estrous cycle stage are unlikely to be triggering factors for the seizure-like events we observed. We also compared the median age of event onset between breeder and nulligravid R59X/+ females and did not observe any significant differences (Figure 5B).
Figure 5. Seizure-like events are confined to Cdkl5 heterozygous female knockout mice and are independent of estrous cycle and parity.
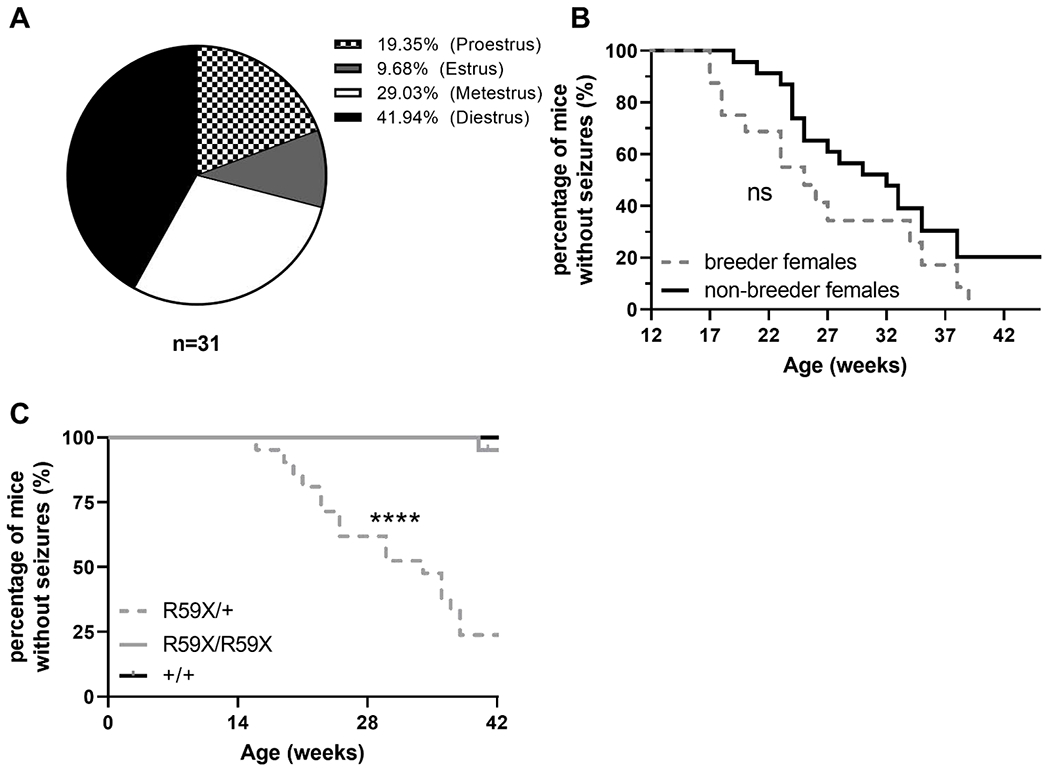
(A) Estrous cycle stages of females displaying disturbance-associated seizures. Proestrus: 19.35% (n=6); estrous: 9.68% (n=3); metestrus: 29.03% (n=9); diestrus: 41.94% (n=13). (B) Percentage of breeder (gray, dotted line; n=16) and nulligravid (black, solid line; n=23) R59X/+ females not exhibiting seizure-like events between 12 to 45 weeks of age. Mantel-Cox (log-rank test), ns. (C) Percentage of R59X/+ females (gray, dotted line; n=21), R59X/R59X females (gray, solid line; n=21), and +/+ females (black line; n=9) not exhibiting seizure-like events between 12 to 42 weeks of age. Mantel-Cox (log-rank) test, p<0.0001. *p<0.05, **p<0.01, ***p<0.001, and ****p<0.0001.
An additional factor that differs between heterozygous knockout females and hemizygous males is the X-linked mosaicism exclusively present in heterozygous female knockouts. To investigate whether mosaicism may be underlying the seizure-like phenotype segregation between our male and female CDD mice, we generated a cohort of mice by breeding R59X/+ females to +/y and R59X/y males. To date, we have monitored 21 homozygous knockout females (R59X/R59X) together with 21 heterozygous knockout females (R59X/+) and 9 wild-type (+/+) littermate controls beginning at 12 weeks of age. 18 out of 21 (86%) R59X/+ females were observed to display at least two disturbance-associated seizure-like events with a median age-of-onset of 34 weeks, while no seizure-like events were recorded in any +/+ littermate controls (Figure 5C). Surprisingly, only 1 out of 21 R59X/R59X female littermates was observed to display a seizure-like event, arising significantly later in life (>40 weeks of age; Figure 5C). This supports the idea that X-linked mosaicism of Cdkl5 deficiency may be a driving factor of the disturbance-associated and spontaneous seizure-like events observed in female models of CDD.
Discussion
Despite the presence of multiple behavioral and neurophysiological impairments, the extent to which mouse models of CDD exhibit spontaneous seizures remains to be explored further. This is the first report of disturbance-associated seizure-like phenotypes in mouse models of CDD. The disturbance-associated seizure-like activity appears to be specific to female mice heterozygous for mutations in Cdkl5, with no hemizygous male knockout, homozygous female knockout, or wild-type littermate control mice displaying epileptic activity to our knowledge. In contrast to humans with CDD, we find that our female CDD mice display a delayed seizure phenotype onset, at a median age around 30 weeks (averaged across two independent Cdkl5 mutant lines). We speculate that this delayed-onset as well as the variable presentation between individual animals (Figures 2C and 3C) may account for the absence of previous reports of seizures across various heterozygous female models of CDD.
Our initial observations in R59X/+ females led us to investigate and validate our seizure-like findings in an independent Cdkl5 knockout line (KO/+) with similar age-of-onset and incidence, suggesting that this phenotype is indeed linked to mutations in Cdkl5 and will likely be shared across additional mouse models of CDD. Myoclonic seizures, as well as generalized tonic-clonic seizures, have been widely described in CDD patients, reflecting a potential similarity between human CDD epilepsy and the events we observe in our mice(Olson et al., 2019). Furthermore, we find that the segregation of this seizure-like phenotype exclusively to heterozygous female knockouts (and not hemizygous males or homozygous females) highlights X-linked mosaicism as a driving factor for seizure presentation in mouse models of CDD.
Late-onset of disturbance-associated seizure-like events in female CDD models
The delayed onset of seizure-like events in our CDD mice reflects a key distinction between the human CDD patient seizure phenotype and that of our models. The median age of onset for a second observed seizure-like event in our R59X/+ and KO/+ females was 28 weeks and 32 weeks of age, respectively; considered well into the middle-aged stage of the lifespan of C57BL/6 mice(Flurkey et al., 2007). In contrast, CDD patients display early-onset seizures with a median age of onset at six weeks of age and with 90% developing seizures by three months of age(Fehr et al., 2013; Olson et al., 2019). As previously discussed, one factor that could be underlying this variability between human and mouse CDD seizure onset in this study is the behavioral method of observation for seizure-like events. By sampling events in our mouse models 2-3 times a week, it is possible we may have missed any spontaneous seizure-like events occurring at earlier ages, especially given the frequency of seizure-like events appears to increase with age. It is also plausible that earlier-occurring seizures in our mice may not have had recognizable behavioral correlates. The prevalence, frequency, and semiology of spontaneous epileptic spasms are characterized in a recent publication using chronic video-EEG recording in aged, Cdkl5 mutant mice(Mulcahey et al., 2020). In future studies, a more detailed characterization of seizure progression and onset in younger animals via chronic, video-EEG will allow for a detailed dissection of the temporal progression of this seizure phenotype.
In CDD patients, typical seizure progression appears to follow a pattern progressing from early onset and at times pharmacoresponsive seizures (Stage I; onset 1-10 weeks), to epileptic encephalopathy with infantile spasms (Stage II; onset 6 months to 3 years), and subsequently refractory, multifocal and myoclonic epilepsy (Stage III; onset 5-7 years)(Bahi-Buisson et al., 2008). This final stage of refractory epilepsy appears to continue long-term in most patients, highlighting an increasing seizure severity with age that human CDD patients also appear to display. Developmental differences between human and mouse brains may underlie these phenotypic differences in age of onset. As such, a more in-depth focal analysis of the cell type or circuit of origin for these epileptic events in the future will provide clarity on the nature of this delayed-onset phenotype in mice. Our findings of a second seizure-like behavior in addition to the recently described epileptic spasms complements the previous findings and suggest that similar to CDD patients, mouse models of CDD exhibit multiple seizure semiologies.
Disturbance-associated nature of CDD mouse seizure phenotypes
Seizures are known to be precipitated by a myriad of factors including stress, menses, photic stimulation, etc.(Engel Jr., 2012). Our initial observances of seizure-like events in our aged CDD female mice occurred upon cage opening or disturbance, suggesting that specific environmental triggers may precipitate seizure presentation in our model. Although mice were often gently separated by hand for easier observation, disturbance by touching was not required for seizure-like events to occur. These events may have been invoked by the movement of the cage during transfer to the changing station, sound from the ventilation of the changing station hood, brighter light, exposure to human odor, or a combination of multiple of the above factors. It remains unclear what aspects of cage disturbance specifically trigger the associated seizure-like events we observed in our R59X/+ and KO/+ females. Interestingly, some CDD patients have been reported to have reflex seizures, or seizures that are predictably triggered by specific stimuli, suggesting a precedence for this phenotype in humans(Peikes et al., 2019; Solazzi et al., 2018). While it seems likely that these observed events are seizures, we were unfortunately unable to electrographically capture these events despite using three different EEG implantation techniques. Failing to capture these events could be due to the rarity and unpredictability of the events in individual mice, inability to replicate an appropriate environmental trigger while in our video-EEG system, altered semiology of seizure-like events, or a possible blunting effect from the surgery itself. The unpredictability of seizure events in chronic epilepsy is cited as a major factor affecting quality of life, thus, further work into the potential triggering factors for CDD-associated seizures could provide valuable insight into environmental influences on seizure development and potential avenues for mitigation(Devinsky et al., 1995).
Mosaic loss of CDKL5 drives the overt presentation of seizure-like events
CDD affects both male and female patients, albeit at varying frequencies, and hemizygous male Cdkl5 knockout mice have been able to recapitulate several of the cardinal phenotypes presented in CDD patients(Akamine et al., 2018; Amendola et al., 2014; Liang et al., 2019; Mirzaa et al., 2013; Okuda et al., 2018; Sartori et al., 2009; Szafranski et al., 2015; Van Esch et al., 2007; Wang et al., 2012; Yennawar et al., 2019). However, in our study, no overt seizure-like activity was observed in either R59X/y or KO/y male mice, at least at the ages we focused on, and previously reports in various hemizygous CDD knockout males have also not observed spontaneous seizures(Amendola et al., 2014; Okuda et al., 2018; Wang et al., 2012; Yennawar et al., 2019). Both of our CDD mouse models (Cdkl5RS9Xand Cdkl5 knockout) have been generated and maintained in the C57BL/6J strain for at least 10 generations – an inbred strain of mouse known to confer increased seizure resistance, thus potentially occluding the presentation of spontaneous seizures in other Cdkl5 knockout mice(Ferraro et al., 2010; McKhann et al., 2003). Despite this, previous attempts by our group and others at backcrossing our Cdkl5 lines to less seizure-resistant strains (e.g., DBA/2J, FVB, 129) have not precipitated the manifestation of noticeable spontaneous seizure events in hemizygous Cdkl5 knockout males (data not shown)(Amendola et al., 2014). This suggests that genetic background is likely not causal to the lack of overt behavioral seizures in hemizygous male CDD models.
We propose the differences we observe between hemizygous male and heterozygous female mouse models of CDD may be due to the mosaicism of CDKL5 loss caused by X-chromosome inactivation in R59X/+ and KO/+. This hypothesis is supported by the lack of an overt seizure-like phenotype in either hemizygous male knockout (R59X/y; KO/y) or homozygous female knockout (R59X/R59X; KO/KO) mice. Furthermore, estrous cycle and androgen hormone levels appear to be unrelated to event incidence across female mice. Interestingly, another epileptic encephalopathy, caused by mutations in the X-linked PCDH19, is well known for causing seizures predominantly in heterozygous females and mosaic males(Kolc et al., 2019). The mechanism underlying PCDH19-related epilepsy has been traced to abnormalities in cellular adhesion and consequent cell sorting in animal models(Bartnik et al., 2011; Pederick et al., 2018). While it remains unclear whether epilepsy in CDD is definitively related to mosaicism, mosaic exon deletions and mosaic de novo point mutations have been reported in both male and female CDD patients and may be an underappreciated phenomenon(Stosser et al., 2018).
Together, our findings demonstrate the first report of overt, disturbance-associated seizure-like events in Cdkl5 deficient mice and provide the community with newfound face validity for existing CDD models. Although we were not able to capture the events describe in this study on video-EEG, our video records suggest that they closely resemble the spectrum of behavioral seizure phenotypes described by the Racine scale. In addition, our video-EEG recoding confirmed the recently reported presence of epileptic spasms, coupled with bilateral sharp-wave activity, in Cdkl5 heterozygous female mice. Our current and future work aim to further dissect both the focal circuits (or multi-focality) and continuum of severity underlying seizures presented in these CDD mouse models. Investigating the circuit origin/nature of this epilepsy phenotype in mice will continue to highlight mechanisms by which loss of CDKL5 drives network imbalance and circuit hyperexcitability.
Supplementary Material
Supplemental Figure 1. Heterozygous Cdkl5 knockout females demonstrate an age-dependent shift in PTZ-induced seizure susceptibility. (A-D) Maximum seizure score recorded in response to high-dose pentylenetetrazol (PTZ) administration scored by the modified Racine scale reported in Table 1. (A) R59X/y males at postnatal day (P) 60 and (B) P150 display a resilience to PTZ-induced seizures upon high-dose subcutaneous administration of PTZ (for P60 males: n=9 +/y, n=12 R59X/y all receiving 90 mg·kg−1 PTZ subcutaneously; for P150 males: n=6 +/y, n=7 R59X/y all receiving 100 mg·kg−1 PTZ subcutaneously). (C) R59X/+ females demonstrate a significant resilience to PTZ-induced seizures measured by maximum seizure score when compared to +/+ littermate controls at P60. (D) Aged, P150, R59X/+ females no longer display a resilience to PTZ-induced seizures when compared to +/+ littermate controls (for P60 females: n=8 +/+, n=10 R59X/+; for P150 females: n=10 +/+, n=11 R59X/+ where all females received 80 mg·kg−1 PTZ subcutaneously). Mann-Whitney test *p<0.05, **p<0.01, ***p<0.001.
Supplemental Figure 2. PTZ-induced seizure in R59X/+ mouse. Representative 10-s trace of intracranial EEG recorded in R59X/+ mouse following subcutaneous 100 mg·kg−1 pentylenetetrazol (PTZ) administration demonstrating characteristic sharp spikes and spike-wave complexes as indicated by arrows.
Supplemental Video 1. Disturbance-associated seizure-like events of Racine stage 1 in Cdkl5R59X/+
(a) Cdkl5RS9X/+ female at 70 weeks of age displaying a seizure-like event of Racine stage 1 in a home cage environment. Mouse of interest is highlighted using a white arrow with a red asterisk appearing on the animal upon event occurrence.
(b) Cdkl5R59X/+ female at 59 weeks of age displaying a seizure-like event of Racine stage 1 in a home cage environment. Mouse of interest is highlighted using a white arrow with a red asterisk appearing on the animal upon event occurrence.
(c) Cdkl5R59X/+ female at 26 weeks of age displaying a seizure-like event of Racine stage 1. A red asterisk appears on the animal upon event occurrence.
Supplemental Video 2. Disturbance-associated seizure-like event of Racine stage 2 in Cdkl5R59X/+
Cdkl5R59X/+ female at 56 weeks of age displaying a seizure-like event of Racine stage 2 in a home cage environment. Mouse of interest is highlighted using a red arrow with a red asterisk appearing on the animal upon event occurrence.
Supplemental Video 3. Disturbance-associated seizure-like events of Racine stage 3 in Cdkl5R59X/+
(a) Cdkl5RS9X/+ female at 71 weeks of age displaying a seizure-like event of Racine stage 3 in a home cage environment. A red asterisk appears on the animal-of-interest upon event occurrence, with the Racine stage progression noted in the bottom of the video.
(b) Cdkl5RS9X/+ female at 63 weeks of age displaying a seizure-like event of Racine stage 3 in a home cage environment. A red asterisk appears on the animal-of-interest upon event occurrence, with the Racine stage progression noted in the bottom of the video.
(c) Cdkl5RS9X/+ female at 50 weeks of age displaying a seizure-like event of Racine stage 3. A red asterisk appears on the animal-of-interest upon event occurrence, with the Racine stage progression noted in the bottom of the video.
Supplemental Video 4. Disturbance-associated seizure-like events of Racine stage 4 in Cdkl5R59X/+
(a) Cdkl5RS9X/+ female at 60 weeks of age displaying a seizure-like event of Racine stage 4 in a home cage environment. A red asterisk appears on the animal-of-interest upon event occurrence, with the Racine stage progression noted in the bottom of the video.
(b) Cdkl5R59X/+ female at 71 weeks of age displaying a seizure-like event of Racine stage 4 in a home cage environment. A red asterisk appears on the animal-of-interest upon event occurrence, with the Racine stage progression noted in the bottom of the video.
(c) Cdkl5R59X/+ female at 58 weeks of age displaying a seizure-like event of Racine stage 4 in a home cage environment. A red asterisk appears on the animal-of-interest upon event occurrence, with the Racine stage progression noted in the bottom of the video.
Supplemental Video 5. Disturbance-associated seizure-like event of Racine stage 5 in Cdkl5R59X/+
Cdkl5R59X/+ female at 26 weeks of age displaying a seizure-like event of Racine stage 5 in a home cage environment. A red asterisk appears on the animal-of-interest upon event occurrence.
Supplemental Video 6. Video-EEG recording of epileptic spasm in R59X/+ heterozygous female. Video-EEG recording clip corresponding to the event in Fig. 4A. The first portion of the video represents real-time playback of the epileptic spasm at 1× speed. The second portion represents manual frame-by-frame viewing of the event. Note that the onset of the behavioral spasm occurs immediately after the onset of the ictal event on EEG.
Supplemental Video 7. Video-EEG recording of epileptic spasm in R59X/+ heterozygous female. Video-EEG recording clip corresponding to the event in Fig. 4B. The first portion of the video represents real-time playback of the epileptic spasm at 1× speed. The second portion represents manual frame-by-frame viewing of the event. Note that the onset of the behavioral spasm occurs immediately after the onset of the ictal event on EEG.
Acknowledgments
This work was supported by funding from The Loulou Foundation (Z.Z.), International Foundation for CDKL5 Research (Z.Z.), the Intellectual and Developmental Disabilities Research Center (IDDRC) at CHOP/PENN U54HD086984 (Z.Z., D.A.C., and E.D.M.), R01NS102731 (Z.Z.), F31NS101762 (B.T.), T32GM008638 (A.C.E.), and F30NS100433 (S.T.). The authors thank Joshua A. Edmondson for video compilation and editing. The authors declare no competing financial interests.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Conflict of Interest Statement: The authors declare no conflicts of interest with this study.
References
- Akamine S, et al. , 2018. A male case with CDKL5-associated encephalopathy manifesting transient methylmalonic acidemia. Eur J Med Genet. 61, 451–454. [DOI] [PubMed] [Google Scholar]
- Amendola E, et al. , 2014. Mapping pathological phenotypes in a mouse model of CDKL5 disorder. PLoS One. 9, e91613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Archer HL, et al. , 2006. CDKL5 mutations cause infantile spasms, early onset seizures, and severe mental retardation in female patients. J Med Genet. 43, 729–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bahi-Buisson N, Bienvenu T, 2012. CDKL5-Related Disorders: From Clinical Description to Molecular Genetics. Mol Syndromol. 2, 137–152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bahi-Buisson N, et al. , 2008. The three stages of epilepsy in patients with CDKL5 mutations. Epilepsia. 49, 1027–37. [DOI] [PubMed] [Google Scholar]
- Bartnik M, et al. , 2011. Early-onset seizures due to mosaic exonic deletions of CDKL5 in a male and two females. Genet Med. 13, 447–52. [DOI] [PubMed] [Google Scholar]
- Berg AT, et al. , 2008. Global cognitive function in children with epilepsy: a community-based study. Epilepsia. 49, 608–14. [DOI] [PubMed] [Google Scholar]
- Berg AT, et al. , 2012. Age at onset of epilepsy, pharmacoresistance, and cognitive outcomes: a prospective cohort study. Neurology. 79, 1384–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caligioni CS, 2009. Assessing reproductive status/stages in mice. Curr Protoc Neurosci. Appendix 4, Appendix 4I. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Demarest S, et al. , 2019. Severity Assessment in CDKL5 Deficiency Disorder. Pediatr Neurol. 97, 38–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Devinsky O, et al. , 1995. Development of the quality of life in epilepsy inventory. Epilepsia. 36, 1089–104. [DOI] [PubMed] [Google Scholar]
- Engel J Jr., 2012. Seizures and Epilepsy. Oxford University Press. [Google Scholar]
- Fehr S, et al. , 2013. The CDKL5 disorder is an independent clinical entity associated with early-onset encephalopathy. Eur J Hum Genet. 21, 266–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferraro TN, et al. , 2010. Confirmation of multiple seizure susceptibility QTLs on chromosome 15 in C57BL/6J and DBA/2J inbred mice. Physiol Genomics. 42A, 1–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Flurkey K, et al. , 2007. PohnB6F1: a cross of wild and domestic mice that is a new model of extended female reproductive life span. J Gerontol A Biol Sci Med Sci. 62, 1187–98. [DOI] [PubMed] [Google Scholar]
- Kalscheuer VM, et al. , 2003. Disruption of the serine/threonine kinase 9 gene causes severe X-linked infantile spasms and mental retardation. Am J Hum Genet. 72, 1401–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kolc KL, et al. , 2019. A systematic review and meta-analysis of 271 PCDH19-variant individuals identifies psychiatric comorbidities, and association of seizure onset and disease severity. Mol Psychiatry. 24, 241–251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liang JS, et al. , 2019. Phenotypic manifestations between male and female children with CDKL5 mutations. Brain Dev. 41, 783–789. [DOI] [PubMed] [Google Scholar]
- Elia M, Ferri MF,R, Spalletta A, Bottitta M, Calabrese G, Carotenuto M, Musumeci SA, Lo Giudice M, Fichera M, 2008. CDKL5 mutations in boys with severe encephalopathy and early-onset intractable epilepsy. Neurology. 71, 997–999. [DOI] [PubMed] [Google Scholar]
- McKhann GM 2nd, et al. , 2003. Mouse strain differences in kainic acid sensitivity, seizure behavior, mortality, and hippocampal pathology. Neuroscience. 122, 551–61. [DOI] [PubMed] [Google Scholar]
- Mirzaa GM, et al. , 2013. CDKL5 and ARX mutations in males with early-onset epilepsy. Pediatr Neurol. 48, 367–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mulcahey PJ, et al. , 2020. Aged heterozygous Cdkl5 mutant mice exhibit spontaneous epileptic spasms. Exp Neurol. 332, 113388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Okuda K, et al. , 2017. CDKL5 controls postsynaptic localization of GluN2B-containing NMDA receptors in the hippocampus and regulates seizure susceptibility. Neurobiol Dis. 106, 158–170. [DOI] [PubMed] [Google Scholar]
- Okuda K, et al. , 2018. Comprehensive behavioral analysis of the Cdkl5 knockout mice revealed significant enhancement in anxiety- and fear-related behaviors and impairment in both acquisition and long-term retention of spatial reference memory. PLoS One. 13, e0196587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olson HE, et al. , 2019. Cyclin-Dependent Kinase-Like 5 Deficiency Disorder: Clinical Review. Pediatr Neurol. 97, 18–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pederick DT, et al. , 2018. Abnormal Cell Sorting Underlies the Unique X-Linked Inheritance of PCDH19 Epilepsy. Neuron. 97, 59–66 e5. [DOI] [PubMed] [Google Scholar]
- Peikes T, et al. , 2019. Reflex Seizures in a Patient with CDKL5 Deficiency Disorder. Can J Neurol Sci. 46, 482–485. [DOI] [PubMed] [Google Scholar]
- Sartori S, et al. , 2009. A novel CDKL5 mutation in a 47,XXY boy with the early-onset seizure variant of Rett syndrome. Am J Med Genet A. 149A, 232–6. [DOI] [PubMed] [Google Scholar]
- Solazzi R, et al. , 2018. Diaper changing-induced reflex seizures in CDKL5-related epilepsy. Epileptic Disord. 20, 428–433. [DOI] [PubMed] [Google Scholar]
- Stosser MB, et al. , 2018. High frequency of mosaic pathogenic variants in genes causing epilepsy-related neurodevelopmental disorders. Genet Med. 20, 403–410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Symonds JD, et al. , 2019. Incidence and phenotypes of childhood-onset genetic epilepsies: a prospective population-based national cohort. Brain. 142, 2303–2318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Szafranski P, et al. , 2015. Neurodevelopmental and neurobehavioral characteristics in males and females with CDKL5 duplications. Eur J Hum Genet. 23, 915–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang S, et al. , 2019. Altered NMDAR signaling underlies autistic-like features in mouse models of CDKL5 deficiency disorder. Nat Commun. 10, 2655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang S, et al. , 2017. Loss of CDKL5 in Glutamatergic Neurons Disrupts Hippocampal Microcircuitry and Leads to Memory Impairment in Mice. J Neurosci. 37, 7420–7437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tao J, et al. , 2004. Mutations in the X-linked cyclin-dependent kinase-like 5 (CDKL5/STK9) gene are associated with severe neurodevelopmental retardation. Am J Hum Genet. 75, 1149–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tauboll E, et al. , 2015. Interactions between hormones and epilepsy. Seizure. 28, 3–11. [DOI] [PubMed] [Google Scholar]
- Van Esch H, et al. , 2007. Encephalopathy and bilateral cataract in a boy with an interstitial deletion of Xp22 comprising the CDKL5 and NHS genes. Am J Med Genet A. 143, 364–9. [DOI] [PubMed] [Google Scholar]
- Velíšková J, et al. , 1990. Ketamine suppresses both bicuculline- and picrotoxin-induced generalized tonic-clonic seizures during ontogenesis. Pharmacology Biochemistry and Behavior. 37, 667–674. [DOI] [PubMed] [Google Scholar]
- Wang IT, et al. , 2012. Loss of CDKL5 disrupts kinome profile and event-related potentials leading to autistic-like phenotypes in mice. Proc Natl Acad Sci U S A. 109, 21516–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weaving LS, et al. , 2004. Mutations of CDKL5 cause a severe neurodevelopmental disorder with infantile spasms and mental retardation. Am J Hum Genet. 75, 1079–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yennawar M, et al. , 2019. AMPA Receptor Dysregulation and Therapeutic Interventions in a Mouse Model of CDKL5 Deficiency Disorder. J Neurosci. 39, 4814–4828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou A, et al. , 2017. Molecular and genetic insights into an infantile epileptic encephalopathy - CDKL5 disorder. Front Biol (Beijing). 12, 1–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu Y-C, Xiong Z-Q, 2019. Molecular and Synaptic Bases of CDKL5 Disorder. Developmental Neurobiology. 79, 8–19. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplemental Figure 1. Heterozygous Cdkl5 knockout females demonstrate an age-dependent shift in PTZ-induced seizure susceptibility. (A-D) Maximum seizure score recorded in response to high-dose pentylenetetrazol (PTZ) administration scored by the modified Racine scale reported in Table 1. (A) R59X/y males at postnatal day (P) 60 and (B) P150 display a resilience to PTZ-induced seizures upon high-dose subcutaneous administration of PTZ (for P60 males: n=9 +/y, n=12 R59X/y all receiving 90 mg·kg−1 PTZ subcutaneously; for P150 males: n=6 +/y, n=7 R59X/y all receiving 100 mg·kg−1 PTZ subcutaneously). (C) R59X/+ females demonstrate a significant resilience to PTZ-induced seizures measured by maximum seizure score when compared to +/+ littermate controls at P60. (D) Aged, P150, R59X/+ females no longer display a resilience to PTZ-induced seizures when compared to +/+ littermate controls (for P60 females: n=8 +/+, n=10 R59X/+; for P150 females: n=10 +/+, n=11 R59X/+ where all females received 80 mg·kg−1 PTZ subcutaneously). Mann-Whitney test *p<0.05, **p<0.01, ***p<0.001.
Supplemental Figure 2. PTZ-induced seizure in R59X/+ mouse. Representative 10-s trace of intracranial EEG recorded in R59X/+ mouse following subcutaneous 100 mg·kg−1 pentylenetetrazol (PTZ) administration demonstrating characteristic sharp spikes and spike-wave complexes as indicated by arrows.
Supplemental Video 1. Disturbance-associated seizure-like events of Racine stage 1 in Cdkl5R59X/+
(a) Cdkl5RS9X/+ female at 70 weeks of age displaying a seizure-like event of Racine stage 1 in a home cage environment. Mouse of interest is highlighted using a white arrow with a red asterisk appearing on the animal upon event occurrence.
(b) Cdkl5R59X/+ female at 59 weeks of age displaying a seizure-like event of Racine stage 1 in a home cage environment. Mouse of interest is highlighted using a white arrow with a red asterisk appearing on the animal upon event occurrence.
(c) Cdkl5R59X/+ female at 26 weeks of age displaying a seizure-like event of Racine stage 1. A red asterisk appears on the animal upon event occurrence.
Supplemental Video 2. Disturbance-associated seizure-like event of Racine stage 2 in Cdkl5R59X/+
Cdkl5R59X/+ female at 56 weeks of age displaying a seizure-like event of Racine stage 2 in a home cage environment. Mouse of interest is highlighted using a red arrow with a red asterisk appearing on the animal upon event occurrence.
Supplemental Video 3. Disturbance-associated seizure-like events of Racine stage 3 in Cdkl5R59X/+
(a) Cdkl5RS9X/+ female at 71 weeks of age displaying a seizure-like event of Racine stage 3 in a home cage environment. A red asterisk appears on the animal-of-interest upon event occurrence, with the Racine stage progression noted in the bottom of the video.
(b) Cdkl5RS9X/+ female at 63 weeks of age displaying a seizure-like event of Racine stage 3 in a home cage environment. A red asterisk appears on the animal-of-interest upon event occurrence, with the Racine stage progression noted in the bottom of the video.
(c) Cdkl5RS9X/+ female at 50 weeks of age displaying a seizure-like event of Racine stage 3. A red asterisk appears on the animal-of-interest upon event occurrence, with the Racine stage progression noted in the bottom of the video.
Supplemental Video 4. Disturbance-associated seizure-like events of Racine stage 4 in Cdkl5R59X/+
(a) Cdkl5RS9X/+ female at 60 weeks of age displaying a seizure-like event of Racine stage 4 in a home cage environment. A red asterisk appears on the animal-of-interest upon event occurrence, with the Racine stage progression noted in the bottom of the video.
(b) Cdkl5R59X/+ female at 71 weeks of age displaying a seizure-like event of Racine stage 4 in a home cage environment. A red asterisk appears on the animal-of-interest upon event occurrence, with the Racine stage progression noted in the bottom of the video.
(c) Cdkl5R59X/+ female at 58 weeks of age displaying a seizure-like event of Racine stage 4 in a home cage environment. A red asterisk appears on the animal-of-interest upon event occurrence, with the Racine stage progression noted in the bottom of the video.
Supplemental Video 5. Disturbance-associated seizure-like event of Racine stage 5 in Cdkl5R59X/+
Cdkl5R59X/+ female at 26 weeks of age displaying a seizure-like event of Racine stage 5 in a home cage environment. A red asterisk appears on the animal-of-interest upon event occurrence.
Supplemental Video 6. Video-EEG recording of epileptic spasm in R59X/+ heterozygous female. Video-EEG recording clip corresponding to the event in Fig. 4A. The first portion of the video represents real-time playback of the epileptic spasm at 1× speed. The second portion represents manual frame-by-frame viewing of the event. Note that the onset of the behavioral spasm occurs immediately after the onset of the ictal event on EEG.
Supplemental Video 7. Video-EEG recording of epileptic spasm in R59X/+ heterozygous female. Video-EEG recording clip corresponding to the event in Fig. 4B. The first portion of the video represents real-time playback of the epileptic spasm at 1× speed. The second portion represents manual frame-by-frame viewing of the event. Note that the onset of the behavioral spasm occurs immediately after the onset of the ictal event on EEG.