

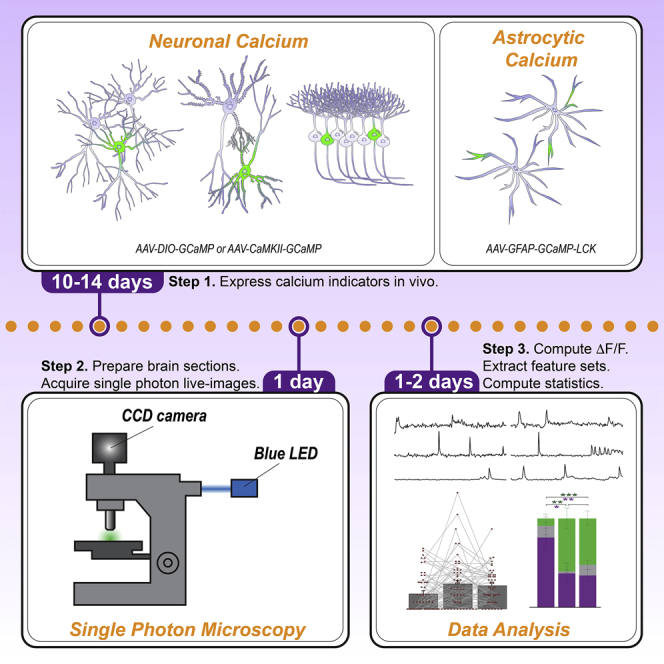
Summary
Confocal, multiphoton, or other advanced microscopy techniques produce high-quality datasets of calcium activity in live tissue. However, researchers without access to such expensive equipment can still produce meaningful observations from single-photon datasets. Here, we describe a protocol to extract meaningful features of both somatic neuronal and membranous astrocytic calcium dynamics obtained from charge-coupled device (CCD)-based camera setups, typical of electrophysiology rigs and highly relevant for investigating neuronal and astrocytic involvement in brain circuitry.
For complete details on the use and execution of this protocol, please refer to Asrican et al. (2020).
Subject areas: Cell biology, Microscopy, Neuroscience
Graphical Abstract
Highlights
-
•
In vivo expression of Ca2+ indicator GCaMP in astrocytes and neurons in mouse brain
-
•
Camera-based single-photon methodologies can provide meaningful calcium data
-
•
Region-based analyses extract relevant neuronal and astrocytic feature sets
-
•
Protocol is applicable without access to confocal or multiphoton microscopy
Confocal, multiphoton, or other advanced microscopy techniques produce high-quality datasets of calcium activity in live tissue. However, researchers without access to such expensive equipment can still produce meaningful observations from single-photon datasets. Here, we describe a protocol to extract meaningful features of both somatic neuronal and membranous astrocytic calcium dynamics obtained from charge-coupled device (CCD)-based camera setups, typical of electrophysiology rigs and highly relevant for investigating neuronal and astrocytic involvement in brain circuitry.
Before you begin
Neuronal calcium has long been associated with the levels of activity, and recording somatic calcium is a reasonable proxy for action potential frequency. However, in astrocytes, recent indications are that calcium signaling in the processes may be functionally more relevant to synaptic functions than the dynamics of the astrocyte soma (Bindocci et al., 2017; Khakh and McCarthy, 2015). Here, we use AAV delivery of GCaMP6 or GCaMP7 constructs in genetically defined neuronal populations, or use a LCK-tagged GCaMP6 construct (Shigetomi et al., 2010) to target the plasma membrane of astrocytes. Along with delivery of floxed chemogenetic DREADDs (Armbruster et al., 2007), we stimulate neuronal circuitry in CCK-Cre mice, and use a scientific-grade complementary metal-oxide-semiconductor (sCMOS) camera to acquire fluorescence datasets in hippocampal CCK interneurons, granule cells, mossy cells, or astrocytes.
Viral injection
Timing: 2 weeks prior
-
1.
Acquire male and female recombinant mice of about 2–6 months of age. CCK-Cre mice (Jax) will be used in this example to demonstrate stimulation of local CCK interneurons and imaging of neuronal and astrocyte calcium.
-
2.AAVs are available from commercial sources, and can be injected into brain regions of interest, such as into the hilus of the dentate gyrus.
-
a.Obtain and mix viral aliquots. Typical stocks from Addgene are on the order of 1 × 1012 to 1 × 1013 viral genomes/mL.
-
i.AAV2-hSyn-DIO-hM3Dq-mCherry (Addgene) is a Cre-dependent version of an excitatory DREADD virus, which in our case will be expressed in CCK type interneurons for circuit stimulation. Aliquot and store at −80°C.
-
ii.AAV2/5-GFAP-GCaMP6-LCK (Addgene), uses the GFAP promoter to restrict expression of the GCaMP6 indicator to astrocyte cell types. Aliquot and store at −80°C.
-
iii.AAV9-Syn-DIO-jGCaMP7f (Addgene) is a Cre-dependent version of the newer version of GCaMP, which in our case will be expressed in CCK type interneurons. We have found that GCaMP7 works better than GCaMP6 in these cells. Aliquot and store at −80°C.
-
iv.AAVDJ-CaMKII-GCaMP6s (Stanford) expresses GCaMP6 in most glutamatergic neurons, and in dentate gyrus, and will express in granular cells as well as hilar mossy cells.
-
i.
-
b.Mix a small volume (∼5–10 μL) of one of the following combinations of AAVs: All vials must be kept on ice.
-
i.DIO-hM3Dq with GFAP-GCaMP6-LCK. Use a final concentration of 1.4 × 1011 of the DREADD, and 3 × 1012 of the GFAP-GCaMP-LCK virus in sterile PBS.
-
ii.DIO-hM3Dq with DIO-GCaMP7. Use a final dilution of concentration of 1.4 × 1011 of the DREADD, and 1.3 × 1012 of the DIO-GCaMP7 virus in sterile PBS.
-
iii.DIO-hM3Dq with CaMKII-GCaMP6. Use a final concentration of 1.4 × 1011 of the DREADD, and 5 × 1011 of the CaMKII-GCaMP6 virus in sterile PBS.
-
i.
-
a.
-
3.Inject viral constructs into CCK-Cre mice using a stereotaxis apparatus. A surgical scope with adjustable zoom, and digital display that reports coordinates of the stereotaxis arm is needed to properly target the injection.
-
a.Prepare the animal.
-
i.Anesthetize animals according to institutional guidelines. Inhalable isoflurane at regulated concentration and rate is recommended. Determine deep state of sleep by non-reactivity to a forceful toe pinch.
-
ii.Affix the animal to stereotaxis apparatus that employs a bite plate and ear bars. Adjust the head position to be approximately straight and level. Continuous isoflurane should be administered by nose cone while in the stereotaxis apparatus. A heat pad under the animal can assist in maintaining body temperature. Breathing rate should be periodically observed, and adjust the anesthesia concentration as needed.
-
iii.Shave, or use depilatory cream to remove hair on the scalp.
-
iv.Follow institutional guidelines on sterile surgical techniques (autoclaving tools, betadine scrubbing, and ethanol cleaning of skin at surgical site).
-
v.Make a single ∼1.5 cm incision along the midline of the head to open the scalp and underlying layers, exposing the top of the skull. This should expose a region slightly larger than the area running from bregma to lambda.
-
vi.Mount a motorized drill in a vertical position with a surgical bit on the stereotaxic arm. Approach the skull, and use the tip of the drill bit at the skull surface to confirm correct alignment and levelness of the animal. Adjust the animal position until:
- Movement of the drill bit in “Y” direction follows the midline (yaw is correct).
- The “Z” position at bregma and lambda are equivalent (pitch is level).
- The “Z” position at the surface of the scull at deflections of about 2 mm left and right of the midline at a point midway between bregma and lambda are equivalent (roll is level).
-
i.
-
b.Deliver virus.
-
i.Drill a small 1 mm burr hole in each hemisphere at AP −2.0 mm, ML ± 1.5 mm from Bregma.
-
ii.Mount a micropipette pulled to a fine tip into a Hamilton syringe, and fill with corn oil. Micropipettes can be created by heating capillary glass on a micropipette forge. Tips can be snapped with fine forceps to a diameter that allows extrusion of liquid.
-
iii.Remove drill attachment and replace with microinjection apparatus containing the Hamilton syringe.
-
iv.Pull up > 600 nL of virus into the tip of the micropipette using an injection pump.
-
v.Re-zero the coordinates at bregma, and approach burr holes. Inject virus using the following settings:
- Position AP −2.0 mm, ML ± 1.5 mm, DV −2.1 mm.
- Injection speed of 75 nL/min.
- Injection volume of 300 nL per injection site.
-
vi.Activate the pump to initiate the injection.
- Wait 5 min after cessation of pump to reduce leakage.
- Slowly retract the needle ∼0.1 mm.
- Wait another 1 min before carefully retracting the rest of the way.
-
i.
-
c.Close the wound, allow animal to recover, and monitor postoperatively according to institutional requirements.
-
a.
CRITICAL: Confirm accuracy of viral injections. Occasionally combining two viruses has unexpected effects, and the ratios of the two must be adjusted to get desired expression levels. It is suggested to try a variety of dilutions to find the concentrations that work best. GCaMP expression can be detected in fixed tissue using standard antibody staining against GFP if necessary. Expression of hM3Dq can be confirmed by presence red fluorescence due to the mCherry reporter.
Alternatives: An alternative to micropipette injection is to use an injection needle made for a Hamilton syringe (ex: 33GA). We have found, however, that appropriately small micropipettes induce less injury along the needle track due to the reduced diameter. Care must be taken to prevent clogging at the tip. Validate that the virus level is decreasing during the injection by observing movement of a small bubble at the interface of oil and virus.
Prepare stock solutions
-
4.Healthy brain slices require careful preparation of cutting solutions, recovery solutions, and recording solutions. These stocks can be prepared ahead of time, and then adjusted for osmolarity and pH on the day of recording.
-
a.Prepare 200 mL of 1 molar stocks of:
-
i.MgSO4
-
ii.CaCl2
-
iii.MgCl2
Note: Salt stocks can remain at 20°C–24°C for several months, but check for cloudiness or contamination. Remake as necessary. -
i.
-
b.Prepare the following solutions listed in the Materials and equipment section.
-
i.10× HEPES stock
-
ii.1× NMDG solution
-
iii.1× HEPES solution
-
iv.10× ACSF stock
-
i.
-
a.
Key resources table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Bacterial and virus strains | ||
| AAV2-hSyn-DIO-hM3Dq-mCherry | Addgene | RRID: AddGene_44361 |
| AAV2/5-GFAP-GCaMP6-LCK | Addgene | RRID: AddGene_52924 |
| AAVDJ-CaMKII-GCaMP6s | Stanford Vector Core | Cat# GVVC-AAV-88 |
| AAV9-Syn-DIO-jGCaMP7f-WPRE | Addgene | RRID: AddGene_104488 |
| Chemicals, peptides, and recombinant proteins | ||
| HEPES | Sigma | Cat# H3375 |
| N-Methyl-D-glucamine (NMDG) | Sigma | Cat# M2004 |
| Sodium ascorbate | Sigma | Cat# A7631 |
| Thiourea | Sigma | Cat# T8656 |
| Sodium pyruvate | Sigma | Cat# P5280 |
| N-Acetyl-L-cysteine | Sigma | Cat# A7250 |
| CNO | NIH | Cat# C-929 |
| TTX | Tocris | Cat# 1069 |
| Experimental models: organisms/strains | ||
| Mouse: CCK-IRES-Cre | Jackson Laboratory | RRID: IMSR_JAX:012706 |
| Software and algorithms | ||
| Micromanager | Micro-manager.org | RRID: SCR_000415 |
| FIJI | Image J | RRID: SCR_002285 |
| Igor Pro | Wavemetrics | RRID: SCR_000325 |
| Other | ||
| Small animal stereotaxis apparatus | Kopf | Cat# Model 942 |
| Stereo zoom microscope | Leica | Cat# M80 |
| Rotary drill | Foredom | Cat# MH-170 |
| Capillary glass | WPI | Cat# 1B100-4 |
| Microforge (microelectrode puller) | Narishige | Cat# PC-10 |
| Infusion pump | Kd Scientific | Cat# 78-8130 |
| Gastight syringe 5 μL | Hamilton | Cat# 80016 |
| 33 gauge needle (optional) | Hamilton | Cat# 7762-06 |
| Osmometer | Wescor | Cat# Vapro 5520 |
| pH meter with temperature probe | Fisher | Cat# Accumet AE150 |
| Semiautomatic vibratome slicer | Leica | Cat# VT1200S |
| SliceScope recording microscope | Scientifica | Cat# SliceScope Pro 1000 |
| 40× water fluorescence objective | Olympus | Cat# LUMPLFLN40XW |
| RC-27LD ultra quiet imaging chamber | Warner | Cat# 64-1532 |
| Slice anchor | Warner | Cat# 64-1551 |
| Accuflow flow regulator infusion set | EMED Technologies | Cat# FLOW-100 |
| Optimos sCMOS camera | Q-Imaging | Cat# 01-OPTIMOS-R-M-16-C |
| Blue LED (470 nm) | CoolLED | Cat# pE-100 |
Materials and equipment
10× HEPES stock (store at 4°C; lasts about 1 month)
| Reagent | Final (1×) concentration (mM) | 10× concentration (mM) | Amount for 1 L |
|---|---|---|---|
| KCl | 2.5 | 25 | 1.86 g |
| NaH2PO4 | 1.25 | 12.5 | 1.50 g |
| NaHCO3 | 30 | 300 | 25.2 g |
| HEPES | 20 | 200 | 47.6 g |
| ddH2O | – | – | Add to 1 L |
1× NMDG solution (for cutting; store at 4°C; lasts about 1 week). NMDG solution reduces excitotoxicity during the slicing procedure by replacement of sodium ions (Ting et al., 2014)
| Reagent | Stock (mM) | Final concentration (mM) | Amount for 1 L |
|---|---|---|---|
| 10× HEPES stock | 10× | 1× | 100 mL |
| NMDG | – | 92 | 17,960 mg |
| Glucose | – | 25 | 4,505 mg |
| Sodium ascorbate | – | 5 | 990 mg |
| Thiourea | – | 2 | 152.2 mg |
| Sodium pyruvate | – | 3 | 330 mg |
| MgSO4 | 1,000 | 10 | 10 mL |
| CaCl2 | 1,000 | 0.5 | 0.5 mL |
| ddH2O | – | – | ∗Add to 900 mL |
Note: NMDG solution comes out very alkaline. Adjust the stock to about pH 7.5–8 using HCl. This typically requires about 10–12 mL of 6 N HCl. Final adjustments to pH will be made on day of recording.
Note: Prepare NMDG slightly overconcentrated (bring ddH2O to about 900 mL when making this stock). Final adjustments to volume will be made on the day of recording based on osmolarity.
1× HEPES Holding (for recovery. Store at 4°C; lasts about 1 week) (Ting et al., 2014)
| Reagent | Stock (mM) | Final concentration (mM) | Amount for 1 L |
|---|---|---|---|
| 10× HEPES stock | 10× | – | 100 mL |
| NaCl | – | 92 | 5,372 mg |
| Glucose | – | 25 | 4,505 mg |
| Sodium ascorbate | – | 5 | 990 mg |
| Thiourea | – | 2 | 152.2 mg |
| Sodium pyruvate | – | 3 | 330 mg |
| MgSO4 | 1,000 | 2 | 2 mL |
| CaCl2 | 1,000 | 2 | 2 mL |
| ddH2O | – | – | ∗Add to 900 mL |
Note: Prepare HEPES Holding slightly overconcentrated (bring ddH2O volume to only 900 mL when making this stock). Final adjustments to volume will be made on the day of recording based on osmolarity.
10× ACSF Stock (for recording. Store at 4°C; lasts about 2 months).
| Reagent | Final (1×) concentration (mM) | 10× concentration (mM) | Amount for 1 L |
|---|---|---|---|
| NaCl | 125 | 1,250 | 73.05 g |
| KCl | 2.5 | 25 | 1.863 g |
| NaH2PO4 | 1.25 | 12.5 | 1.499 g |
| NaHCO3 | 26 | 260 | 21.84 g |
| D-Glucose | 20 | 200 | 36.03 g |
| ddH2O | – | – | Add to 1 L |
Step-by-step method details
Acute brain slice preparation
Preparing tissue for calcium imaging follows the exact same protocol as for electrophysiological recordings with the exception of the addition of 12 mM N-acetyl-L-cysteine (NAC) to the NMDG and HEPES holding solutions. This has the benefit of preserving glutathione production and improves calcium imaging in acute brain slices (Ting et al., 2014).
-
1.Final preparation of slicing solutions. (∼ 25 min)
-
a.Measure 180 mL of NMDG stock solution and 90 mL of HEPES holding solution.
-
b.Mix in 392 and 196 mg of NAC to NMDG and HEPES solutions respectively.
-
c.Bubble solutions with 95% O2 and 5% CO2 for 10 min.
-
d.pH the NMDG using HCl and/or KOH to 7.24
-
e.pH the HEPES solution using HCl and/or NaOH to 7.24.
-
f.Adjust osmolarity to 300 mOsm.
-
i.Measure the slightly overconcentrated solutions with an osmometer
-
ii.Reduce osmolarity by adding volumes of ddH2O.
For NMDG; volume needed (mL) ∼= (Osm × 180/300 – 180 ) × 0.9 For HEPES; volume needed (mL) ∼= (Osm × 90/300 – 90) × 0.9 -
iii.Typically this will bring the final volume to ∼ 200 mL of NMDG, and 100 mL of HEPES.
-
i.
-
a.
-
2.Dissection and Sectioning. (∼ 45 min)
-
a.Setup dissection tools and vibratome and water bath.
-
i.Bring water bath to 35°C.
-
ii.Assemble useful tools: including, large scissors (for removing head), small scissors (for cutting skin and skull), blunt forceps, spatula, single edge razor blade; tray and Styrofoam lid for pinning and dissection; syringe for cardiac perfusion.
-
iii.Pack vibratome outer-chamber with ice. Fill inner-chamber with 100 mL of the ice-cold NMDG solution. Bubble with 95% O2 and 5% CO2.
-
iv.Place remaining 100 mL of ice-cold NMDG in slice-holding chamber, and keep on ice. Bubble with 95% O2 and 5% CO2. Reserve a small amount for cardiac perfusion.
-
i.
-
b.Anesthetize the research animal according to institutional requirements. Vaporized isoflurane will anesthetize the mice and slow the breathing rate. Animals should be alive, but non-responsive to serious toe pinch. An inverted 15 mL centrifuge tube containing a tissue moistened with additional isoflurane placed over the animal’s snout will ensure a continued anesthetized state during cardiac perfusion procedure.
-
c.Pin animal, expose heart, puncture right atrium and perfuse ∼10 mL of ice-cold NMDG solution into left ventricle.
-
d.Quickly remove head, open skull, and remove brain directly into the ice-cold NMDG. Let sit for 1 min.
-
e.Remove to a towel and bisect hemispheres. Block brain for semi-transverse sections (about 22.5 degrees from coronal, and angled slightly off vertical, inward/downward) with single edge razor blade.
-
f.Dab on towel, and glue the cut cerebellar side to the vibratome plate. Insert plate into vibratome.
-
g.Slice at 280 μm thickness, using speed ∼ 0.16–0.20 mm/s and amplitude of 1.6 mm. Slow sectioning while cutting through the hippocampus will preserve integrity of the hippocampal layers.
-
h.Gently collect sections containing hippocampus into a slice holder containing ice-cold NMDG using inverted/cut transfer pipette, or gentle paintbrush.
-
a.
-
3.Warming. (∼ 10 min)
-
a.Transfer the NMDG containing slice holder with sections from the ice, into the 35°C water bath.
-
b.Continue bubbling in the water bath for 8–10 min. Do not over-warm. It is recommended to shield from light to preserve fluorescence.
-
a.
-
4.Recovery. (∼ 1.5 h)
-
a.Remove holder from water bath.
-
b.Carefully discard the NMDG solution, and replace with 20°C–24°C, bubbled HEPES holding solution. Ensure that sections are submerged and loosely separated. Continue bubbling at 20°C–24°C for 1–1.5 h. Take care that bubbling is not so vigorous as to cause turbulence/tumbling of the sections. Continue shielding from light with sheets of aluminum foil.
-
a.
Calcium imaging
The techniques to keep brain slices alive and heathy for calcium imaging are identical to those used for electrophysiological recordings. Illumination of tissue with a blue LED and collecting green fluorescence is done using a standard GFP filter-set.
The sCMOS camera used here is controlled via Micromanager software, which can also interface with the SliceScope microscope stage (Scientifica). Many popular microscope cameras, such as Hamamatsu or Photometrics cameras, use CCD or sCMOS sensors that offer high sensitivity and speed, and are excellent for detecting fluorescent calcium signals. Most devices are already compatible with the Micromanager software (https://micro-manager.org/wiki/Device_Support).
-
5.Prepare the 1× ACSF recording solution. Calcium events are very numerous in astrocytes, while very sparse in granule cells. It is often beneficial to adjust the levels of Ca2+ and Mg2+ depending on which cells you will observe to slightly increase or decrease network excitability.
-
a.Combine in ddH2O
-
i.For Astrocytes:
Reagent Stock (mM) Final concentration (mM) Amount for 500 mL 10× ACSF stock 10× 1× 50 mL CaCl2 1,000 1.8 900 μL MgCl2 1,000 1.5 750 μL ddH2O – – Add to 500 mL -
ii.For granule cells and mossy cells:
Reagent Stock (mM) Final concentration (mM) Amount for 500 mL 10× ACSF stock 10× 1× 50 mL CaCl2 1,000 2.2 1,100 μL MgCl2 1,000 1.1 550 μL ddH2O – – Add to 500 mL -
iii.For CCK Cells:
Reagent Stock (mM) Final concentration (mM) Amount for 500 mL 10× ACSF stock 10× 1× 50 mL CaCl2 1,000 2 1,000 μL MgCl2 1,000 1.3 650 μL ddH2O – – Add to 500 mL
-
i.
-
b.Bubble for at least 10 min at 20°C–24°C with 95% O2 and 5% CO2.
-
c.pH to 7.24.
-
d.Adjust osmolarity to ∼ 305 mOsm.
-
e.Begin perfusion of ACSF on the microscope, continuing to bubble, and use an IV dripper and flow regulator to control the perfusion rate to about 1–2 mL/min.
-
a.
-
6.Identify regions to image.
-
a.Place a single slice in upright microscope recording chamber. Secure with a slice anchor.
-
b.Locate the dentate gyrus with a 10× air objective.
-
c.Switch to a long-working distance water-dipping objective with 40× magnification. Confirm the presence of mCherry+ virally targeted CCK neurons containing DREADD construct, and green GCaMP expression in neurons or astrocytes. GCaMP has a low baseline fluorescence, which can be observed as a diffuse green signal. Typically, spread of injected AAV2/5-GCaMP is not spatially restricted as tightly as AAV2-hM3Dq virus, and expression can be found in cells beyond the hilar region, presumably due to differences in capsid or infectability of different cell types. See Figure 1 for typical live-fluorescence appearance of hM3Dq-mCherry and GCaMP expression in astrocytes or neurons in the dentate gyrus.
-
a.
-
7.Set up an imaging session that acquires data at two different XY stage positions and extends for several different depths into the slice. Adjust the number of cycles to last approximately 10 min.Note: Calcium events in acute slice preparation can last several seconds at 20°C–24°C, depending on the specific GCaMP variant that is utilized (fast or slow) and the cell type. Since the frame-rates of most cameras greatly exceed the duration of these calcium events, some time resolution can be sacrificed in order to gain additional image planes. Adjustment of specific imaging parameters may be required for your application.The main limitation on time resolution of image acquisition is the refocusing and movement of the XY translational stage. The following parameters worked well for a Scientifica microscope and Micromanager software and gave an effective time resolution of 2.85 s. While some fast events may be missed, this is acceptable for detecting the majority of slow buffered calcium events.
Imaging settings Value # of time points 210 frame interval (delay) (minimum) # of Z-planes 5 Z-Step Size 15 μm # XY-positions 2 -
a.Power the camera and stage and then start the Micromanager software. If not already done, setup and save a configuration file that can interact with:
-
i.The CCD camera.
-
ii.The XY stage position.
-
iii.The Z focus position.
-
i.
-
b.Set camera parameters for 100 ms exposure with no binning.
-
c.Open the Multi-Dimensional Acquisition Panel in MicroManager.
-
d.Enable “Time Points,” and set the image count to 210 images, 1 ms interval.
-
e.Enable “Z-Stacks (Slices)” and Select “Relative Z” mode. Adjust settings to use 5 imaging planes by entering the following parameters:
-
i.Enter 0 for “Start Z.”
-
ii.Enter -60 for “End Z.”
-
iii.Enter 15 for “Step size.”
-
i.
-
f.Disable “Channels” checkbox.
-
g.Enable “Multiple Positions (XY)”, and open the “Stage Position List.”
-
h.Start “Live View” and turn on the blue LED.
-
i.Move the XY Stage to the surface of the slice in a field of view of interest. Push “Mark” to store it as Position0.
-
j.Move the XY Stage to a second, non-overlapping region of the tissue. Mark it as Position1.
-
k.Stop Live View and turn off the LED.
-
l.Minimize room lights or cover the microscope cage with black-cloth. Allow the slice to equilibrate for 5–10 min to reduce mechanical drift. Recheck positioning to ensure stability of the slice.
-
a.
-
8.Acquire calcium data.
-
a.Once baseline drugs are added for 5–10 min (for example, 1 μM TTX) and slice is confirmed to be mechanically stable, turn on the blue LED at the lowest setting that provides reasonable signal. As the fluorescent signal does indeed bleach over time, and cellular activities may be susceptible to phototoxicity, it is critically important not to over-illuminate the tissue.
-
b.Push “Acquire!” to start collecting Data_1.
-
c.When complete, turn off the LED and briefly review the dataset. Save the data to disk with an appropriate filename.
-
d.Add CNO to the ACSF reservoir to begin circuit stimulation of CCK cells. Wait 10 min to allow drugs to reach the slice and begin to activate DREADD receptors.
-
e.When time has nearly expired, recheck the stage position and focus, and readjust the stored positions to match the beginning of Data_1 as needed.
-
f.Push “Acquire!” to start collecting Data_2, and save as above.
-
g.Wash out drugs and change the slice to continue with additional samples.
-
a.
Optional: Include various pharmacological agents such as glutamate receptor antagonists in the bath starting at baseline to help determine mechanisms of action. For examples, see Asrican et al., (2020).
Optional: Some researchers may wish to acquire calcium data during the 10 min delay in step 8d, or perhaps an additional 10 min period of washout. In this case there will be a total of 3–4 datasets from each slice.
Note: A highly sensitive CCD camera is useful when imaging calcium, so that the intensity of the illumination light can be minimized. An Optimos sCMOS camera from Q-Imaging is used in this example. While not used here, TTL control of the LED, or addition of a mechanical shutter in the light path can prevent excitation of the sample during non-imaging episodes of the acquisition, such as during focusing or stage-translation.
Note: Halogen lamps as illumination sources have some intrinsic fluctuations in intensity and result in signal disturbances when sensitive cameras are used. LEDs are typically much more reliable in terms of constant output and have the advantage of also permitting control via TTL signals.
Figure 1.

Typical fluorescence appearance in live tissue of hM3Dq-mCherry targeted CCK interneurons along with GCaMP constructs targeted to astrocytes, mossy cells/granule cells, or CCK interneurons from CCK-Cre animals
(Top) Diffuse and low basal level fluorescence in astrocytes due to the targeting of GCaMP6 construct to the fine processes with the LCK construct. (Middle) CaMKII-GCaMP6 targets glutamatergic cells of the DG including mossy cells and granule cells. (Bottom) CCK neurons expressing both hM3Dq and GCaMP7, with partial colocalization. GCL, granule cell layer; scale bar, 50 μm. See Asrican et al. (2020) for additional examples and resolutions.
Expected outcomes
Neurons and astrocytes respond to neuronal activity, and the resulting changes in frequencies and amplitudes of calcium signals can be quantified by region-based analysis algorithms. Datasets from Micromanager software are stored as OME-TIFFs using the open-source Bio Formats standard (Linkert et al., 2010) and can be opened using the FIJI extensions (Schindelin et al., 2012) in ImageJ. As datasets are very large (tens of GBs), a reasonably large and fast solid state drive (SSD) is needed to quickly store the data, and additional RAM may facilitate the analysis. Analysis here is accomplished with a combination of manual identification of responding regions in FIJI/ImageJ (see Figure 2), and quantification using custom procedures in Igor Pro. See Figures 3 and 4 for representative traces.
Figure 2.

Drawing regions of interest on an astrocytic domain using FIJI/ImageJ
(Left) Three frames of a calcium imaging time series show a calcium event arriving in the middle frame, and disappearing by the third frame. Top row shows a hand-drawn analysis region in yellow around the responding domain. Bottom, the same frames as above, but with an annular shaped surround region, drawn in blue, that will be used for background subtraction. Scale bar, 10 μm. (Right) Representative fluorescence traces from the corresponding region and surround ROIs.
Figure 3.

Analysis of time series calcium traces
(A) Depiction of raw fluorescence traces for 5 focal planes for the ROI, the annular background region, and the background-subtracted fluorescence (arbitrary units). X axis reports the datapoint number in the trace. Red indicates the focal plane defined as the “best” focal plane for this ROI.
(B) Calculation of ΔF/F values for the best focal plane defined as (F − F0)/F0. F0 is calculated by smoothing the background-subtracted fluorescence and then taking a running minimum.
(C) Preparation of the ΔF/F trace for peak detection. The ΔF/F trace is heavily smoothed to find the red shape of the trace. The difference between the ΔF/F and the smoothed trace reveals transients without any baseline shift. A two-cycle thresholding that considers the mean and standard deviation of the 5th through 95th percentile of datapoints reveals the signal component (red) above the noise. Peak detection algorithms can then proceed using this threshold.
Figure 4.

Quantification of calcium event frequency
(A) Representative ΔF/F traces from an astrocyte dataset, and (B) from a neuronal (granule cell) dataset. Traces are labeled with blue “plus” symbols whenever an event is detected by the peak detection algorithm. The number of detected peaks divided by the time gives the event frequency.
Quantification and statistical analysis
-
1.Open in FIJI/ImageJ.
-
a.Import a dataset in FIJI by using the File -> Import -> Bio Formats menu.
-
b.Select only one position to analyze a time.
-
c.Adjust brightness/contrast to enhance visibility of signal changes.
-
a.
-
2.Define regions of interest (ROIs). See Figure 2.
-
a.Open the ROI Manager from the Tool Menu.
-
b.Scroll through the time series to visually identify regions where there is an increase in fluorescence. Adjust the z-position to find the best plane at which the response is seen.
-
c.Draw an ROI around the region at the best focal plane using the freehand tool. For neurons, typically this will be a responding soma. For astrocytes, this area is less well defined, and may encompass a subcellular region of the processes of an astrocyte. Add the region to the ROI Manager.
-
d.Without changing the image display, draw an annular ROI that surrounds the original ROI. This will serve as a background subtraction region. Add this annulus to the ROI Manager.
-
e.Repeat steps 2.c and 2.d sequentially to form an alternating list of regions and surrounds, taking care to identify all active signals in the dataset.
-
f.Close the current dataset, and open the next one in that sequence (ex: Data_2), maintaining the previously drawn ROIs.
-
g.Continue adding additional ROIs and the annular ROIs to the ROI Manager until each of the datasets in the series has been scrutinized.
-
h.Once the entire sequence of datasets for a given position have had the regions defined, save the ROISet to disk by selecting the More -> Save buttons in the ROI Manager.
-
i.Select the More -> List buttons, to create a list of the Overlay Elements, which includes the z-stack position that is defined for each ROI. Save the Overlay Elements as a text file.
-
a.
Note: Manual definition of background regions can be somewhat subjective. It is possible that other calcium signals may be detected inside the annular background regions if care is not taken to avoid this. This can be especially problematic in densely packed regions if labeling is not sparse enough, such as within the granule cell layer where cells are abutted next to one another. Many researchers in this case often draw a single “background” region where no such signals are expected. A useful alternative method for background subtraction that has been used recently is to subtract the intensity of the lower percentile of pixels from the whole dataset (Ayaz et al., 2019). For neurons that are more distributed, such as hilar cells of the dentate gyrus, or for astrocytes which are typically non-overlapping, we have found that individually drawn annular regions are most advantageous. The benefits of individual annular regions are that: (1) a non-signaling background region of the tissue may not be readily available; (2) illumination intensities across the field of view may not be uniform due to tissue scattering, uneven expression of reporters, or unique characteristics of the LED; and (3) contaminating out-of-focus signals from cells in other focal planes occurs locally can be therefore be subtracted appropriately.
-
3.Criteria for Inclusion/Exclusion.
-
a.Manual definition of ROIs depends on the analyzer’s ability to identify fluorescence fluctuations by eye. Some adjustment of brightness and contrast levels can assist in identification of fluorescent transients. Include any region (cellular or subcellular) that has a visible (increasing) signal in at least one of the datasets. Occasionally, unhealthy cells may maintain a high fluorescent state, only to suddenly lose intensity. This type of decreasing signal does not reflect a responsive cell and should be excluded.
-
b.If the tissue is drifting such that the cell does not remain within the ROI boundary, there are many drift correction algorithms built for ImageJ which may work.
-
c.If the drifting is too severe to correct, however, discard the datasets.
-
d.Similarly, if the fluorescent signal bleaches completely, or becomes suddenly unresponsive, discard the data sets. Typically, signal photobleaching/ phototoxicity only occurs when light intensities are too high due to attempted compensation for low expression of the calcium sensor, or if excessively repeated image acquisition of same tissue is attempted (>3 attempts).
-
a.
-
4.Measure intensities.
-
a.Define the measurement parameter in FIJI/ImageJ by going to the Analyze -> Set Measurements menu. Select only “Mean gray value.”
-
b.Get fluorescence intensity measurements by sequentially:
-
i.Opening the each of the dataset (Data_1, Data_2 etc…).
-
ii.Use the More -> “Multi Measure” buttons in the ROI Manager to analyze.
-
iii.Save the output file with an appropriate and unique filename.
-
i.
-
a.
-
5.Quantify in Igor Pro. Once measurements are made for each dataset, import them into an analysis environment, such as Igor Pro, MATLAB, or a custom program which allows development and running of customized analysis routines for calculating ΔF/F values and detecting events. See Figure 3.
-
a.Import Measurement data. Open .csv files (delimited text data), from each dataset in the sequence. ImageJ writes .csv files as delimited text, where columns correspond to each ROI that is defined in the ROI Manager, and rows correspond to the 1,050 image frames (5 z_planes and 210 time_points).
-
b.Background subtraction. Since ROIs are defined in FIJI/ImageJ as alternating Region_ROIs and Annular_backgrounds:
-
i.Extract the odd columns into a new array called Regions.
-
ii.Extract the even columns into a new array called Backgrounds.
-
iii.Subtract Background measurements from Region measurements.
-
iv.Prohibit negative fluorescence by setting a lower limit of 0 fluorescence.
-
i.
-
c.Best Plane Selection. If careful selection of the optimal z-plane is made during ROI creation, the appropriate z-values are written in the Overlay Elements text file.
-
i.Import the Overlay Elements file as a delimited text file.
-
ii.Extract only the odd rows from the Overlay Elements.
-
iii.Use the column entitled “Z” to get the numerical value corresponding to the z-plane to use for each ROI.
-
iv.Extract the correct z-plane from the background-subtracted measurements and save each ROI as a 210 point time series.
-
i.
-
d.Create F0. The penultimate step before calculating ΔF/F is the creation of the F0 trace, which helps account for bleaching of the signal over time, as well as other non-signal related changes in baseline.
-
i.Duplicate the 210 point ROI time series just created in step 5.c.iv.
-
ii.Run a boxcar smoothing with boxsize = 20 points.
-
iii.For each timepoint, find the minimum smoothed value within a n-back window of 150 points. No negative fluorescence values are permitted.
-
i.
-
e.Calculate ΔF/F trace.
-
i.Use the background-subtracted values created in step 5.c.iv as the F values.
-
ii.Use the min_smoothed values created in step 5.d.iii as the F0 values.
-
iii.Execute a point by point computation of (F – F0)/F0 to get the ΔF/F trace.
-
i.
-
a.
-
6.Quantify frequencies of events. Event detection requires recognizing signal in the ΔF/F trace that rise above noise levels. Parameters for such detection may need to be carefully adjusted and will depend on the particularities of the equipment used. See Figures 3 and 4.
-
a.Trace concatenation. Extracting features from ΔF/F traces for a given ROI requires treating all conditions of that ROI equally. Begin by concatenating ΔF/Fs from each of Data_1 and Data_2 etc. for each ROI trace. (For example, if the experiment consisted of (1) baseline, (2) CNO, and (3) wash conditions; concatenate those three ΔF/Fs for each ROI).
-
b.Suppress unwanted detection of extra datapoints inside slow signals for the upcoming peak detection algorithms.
-
i.Smooth the concatenated ΔF/Fs by boxcar smoothing with box size = 200 points to get the general shape of the slow fluorescence responses.
-
ii.Take the difference of the ΔF/F data and the smoothed data to find the acute signal fluctuations. Smooth with a single-pass gaussian filter.
-
i.
-
c.Determine a cutoff threshold based on a 2-cycle analysis of the fluctuation data to determine signal and noise.
-
i.Sort fluctuation datapoints by intensity.
-
ii.Collect the 5th through 95th percentiles of the sorted data.
-
iii.Calculate the median and standard deviation for this middle 90%.
-
iv.Create an initial cutoff level at FCut = median + 0.25 × SD.
-
v.Recollect all fluctuation datapoints beneath Fcut level (non-signaling fraction).
-
vi.Take the 5th through 95th percentiles of only these points.
-
vii.Calculate a new median and standard deviation.
-
viii.Create a revised more stringent cutoff level at FCut = median + 1 SD.
-
i.
-
d.Run a peak detection algorithm with a minimum threshold set to the greater of Fcut or 0.2 ΔF/F units. The built-in peak detection in Igor Pro finds local maximums using the first and second derivatives, with a user defined box size. A box size of 5 datapoints provides reasonable detection of events at this time-scale.
-
i.Visual inspection of the detected events will help determine appropriateness of the algorithms. Adjustable parameters include:
- Smoothing size.
- Standard deviation adjustment factor (currently set to 1 SD).
- Box size of the peak detection algorithm.
- The min threshold parameter of 0.2 ΔF/F units.
-
ii.Once parameters are set, standardize them for the entire dataset to avoid any bias in analysis.
-
i.
-
e.Re-split the data into the separate conditions, and count the detected events. Divide by the time (approximately 10 min) to get the frequency of events in each ROI for baseline, CNO, and other optional conditions.
-
f.The mean frequency of events per minute at baseline and during stimulation can be calculated and plotted as bars. It is useful to plot individual datapoints connected with lines to show associations.
-
g.Statistical analysis comparing baseline to CNO conditions should use paired t tests, as the data come from 2 measurements of the same ROI.
-
a.
-
7.Quantifying size of events. In addition to frequency of events, amplitude measurements or area measurements are often reported.
-
a.Total area under the ΔF/F curve can be easily calculated by integrating the sum of the datapoints of the ΔF/F trace.
-
b.Restrict area measurement to be ≥ 0.
-
c.The mean area at baseline and during stimulation can be calculated and plotted as bars. It is useful to plot individual datapoints connected with lines to show associations.
-
d.Statistical analysis comparing baseline to CNO conditions should use paired t tests, as the data come from 2 measurements of the same ROI.
-
a.
Note: Amplitude measurements can be difficult to interpret from single-photon datasets, as light scattering from different focal depths, or differences in expression of GCaMP indicator make absolute measurements of event amplitudes unreliable. Instead, use relative comparisons, making sure only to compare the effects of treatments “within ROIs.”
-
8.Calculate the fold change. It is useful to know what the fold change in frequency or area is for each ROI due to circuit activation (comparing baseline to CNO condition).
-
a.Fold Change in frequency is calculated as:
-
i.Frequency2/Frequency1.
-
ii.As frequencies can often be zero during Frequency1, infinite fold changes should be recalculated as 2 × Frequency2/minimum_Frequency1 (i.e., 1 event/imaging time period).
-
iii.Fold changes become zero if there are no events in Frequency2.
-
i.
-
b.Fold change in Area is calculated as Area2/Area1.
-
a.
-
9.Slice-wise comparisons. Recording many events within a slice, and then repeating the acquisition from multiple slices or animals is essentially a nested-study design. In order to account for bias that may exist within a particular slice due to peculiarities in local connectivity (such as one particularly active brain slice with an over-abundance of detected regions), we must determine whether changes in event frequency or event amplitude are independent of the sample origins. The correct statistical analysis process would be to use a Linear Mixed Effects model that takes the fixed and random effects of the study into account (for example, see Bojarskaite et al., 2020; Stobart et al., 2018). Such analysis is available using the fitlme function in MATLAB, or the lmer function in the lme4 package in R. We can also get a good idea of how similar or different individual slices are by doing statistical comparisons in a slice-wise manner. Using the fold changes calculated above, the percentage of regions displaying increasing or decreasing calcium responses can compared.
-
a.For all slices that are subjected to a given experimental condition, gather the Fold Changes, as calculated in step 8.
-
b.For one slice at a time, count the number of values with increasing (Fold > 1), unchanging (Fold = 1), and decreasing (Fold < 1) change.
-
c.Dividing by the total number or regions in that slice and multiplying by 100 will provide the corresponding %increasing, %unchanging, and %decreasing regions for that slice.
-
d.Compute the mean and standard error for all the %increases, %unchangings, and %decreases.
-
e.A stacked bar graph of these means should add up to 100%, and should include the error bars at the tops of each subsection of the bar. See (Asrican et al., 2020) for examples.
-
f.Statistics for comparing different conditional manipulations can use unpaired t tests, if particular parts (the increasing part, for example) are changing, or a Chi-squared test if the entire distribution should be compared.
-
a.
Note: Unlike frequency, area measurements do not have discrete counting of events, and therefore will not ever have fold changes exactly equal to 1. The %unchanging category should either be removed, or the criteria for %unchanging should have a defined “similarity” range acceptable enough to be considered unchanged.
Limitations
Somatic calcium events in neurons should currently be considered a proxy for neuronal action potentials and do not necessarily equal the spike rate. However, relative changes in calcium signals should follow the same trends as spiking, such that increases in calcium event frequency correlate with increases in spike rates.
Despite the well-known existence of dynamic calcium signals in astrocytes, there has been some controversy on the physiological role. Much of the confusion seems to be attributed to limiting analysis of only IP3 mediated calcium, or only consideration of somatic events (Khakh and McCarthy, 2015; Savtchouk and Volterra, 2018). By including the LCK tag on the GCaMP construct, we can circumvent some of this controversy and ask how signals in the fine processes are affected by neuronal circuitry.
Higher resolution techniques such as 2-photon or confocal microscopy could, of course, provide much finer details that the wide-field fluorescent technique lacks. For example, single-photon techniques that collect out-of-focus light from astrocytes above or below the plane of interest could obscure the ability to assign signals accurately. Despite this, it is still possible to extract meaningful signals from such datasets, provided that enough regions are included in analysis, enough time is allowed to collect longer data sets, and that the ROIs are drawn as carefully as possible.
Finally, there may be certain types of astrocyte calcium signals, such as growing or propagating signals, that are not able to be detected by region-based analysis routines. In this case, more sophisticated intensity-based algorithms, such as the ones developed by (Bojarskaite et al., 2020; or Wang et al., 2019) initially for analyzing multiphoton datasets in astrocytes, might be beneficial. It remains to be seen if such techniques can be adapted for use on wide-field, camera-based, datasets.
Troubleshooting
Problem 1
Calcium signals are weak or unstable and are unable to be reliably detected during analysis.
Potential solution
Signal to noise ratio can be the biggest difficulty to overcome in analysis of calcium events (steps 5–7 in Quantification and Statistical Analysis). During analysis, defining the best ROI to draw and optimizing the parameters in step 2 in “Quantification and Statistical Analysis” will help, but if the data set is sub-optimal, it will be more difficult. Some analysis parameters in step 5.d.ii-iii and in step 6.d.i in “Quantification and Statistical Analysis” can be adjusted for particular datasets.
Rundown of calcium fluorescence can be caused by too high an intensity of illumination for these longer data sets, or due to poor slice quality. Improving health of the slices, limiting unnecessary illumination and light intensity, and good transgene expression (see below) are all critical for getting quality data sets.
Physical instability of the tissue can also cause extraneous signal fluctuations, which are often due to expansion or contraction of the brain slice in solution. Allow some time, (∼5–10 min) after placing a slice into the perfusate within the recording chamber before initiating imaging sessions to allow for equilibration. A slice anchor is also useful to provide stability and prevent drift.
Problem 2
Poor or insufficient viral expression in brain slices (step 6.c in Step-by-step method details).
Potential solution
Ensure injection coordinates are correct. A test injection of a dye can quickly inform on targeting accuracy. Confirm genotype of mouse if using a Cre animal. Fresh virus may be needed, or additional days post injection to express a transgene may be necessary if expression is low. Preliminary titration of the viral titer is recommended. Occasionally, a different viral serotype may be beneficial. Alternatively, there are transgenic mice that are available commercially that express floxed GCaMP indicators (ex: from Jackson Laboratory).
Problem 3
Calcium datasets are prohibitively large. Data storage for the experiments described here may be a significant limitation.
Potential solution
The procedures described here use full frame images during acquisition. It may be necessary to bin pixels in order to provide a smaller dataset. However, this does sacrifice some imaging resolution. Additional hard drives may be needed to store and transfer files off of the acquisition computer. Sufficient RAM for loading data during analysis is required. Subsampling of the data is one possible post hoc solution if resources are limited. FIJI also provides a “virtual” mode of importing data, which streams the data from disk rather than loading entire datasets at once.
Resource availability
Lead contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the lead contact, Juan Song, juansong@email.unc.edu.
Materials availability
No new mouse lines or viral strains were generated in this study. The particular animals and reagents used here are reported in the Key resources table.
Data and code availability
Calcium data sets from this study have not been deposited publicly, but samples may be requested from the lead contact or the technical contact. Further information on specific analysis code written in Igor Pro may be made from the technical contact, Brent Asrican, basrican@email.unc.edu.
Acknowledgments
We thank the authors of Asrican et al. (2020) for application of this protocol. This work was supported by grants awarded to J.S. from American Heart Association, United States; Whitehall Foundation, United States; NIH (MH111773-01, MH122692-01 AG058160, NS104530), United States; and NARSAD Young Investigator Award (26017) to B.A from the Brain & Behavior Research Foundation, United States.
Author contributions
J.S. conceived and oversaw the original project published in Asrican et al. (2020). B.A. developed the protocols described here and authored this manuscript.
Declaration of interests
The authors declare no competing interests.
Contributor Information
Brent Asrican, Email: basrican@email.unc.edu.
Juan Song, Email: juansong@email.unc.edu.
References
- Armbruster B.N., Li X., Pausch M.H., Herlitze S., Roth B.L. Evolving the lock to fit the key to create a family of G protein-coupled receptors potently activated by an inert ligand. Proc. Natl. Acad. Sci. U S A. 2007;104:5163–5168. doi: 10.1073/pnas.0700293104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Asrican B., Wooten J., Li Y.D., Quintanilla L., Zhang F., Wander C., Bao H., Yeh C.Y., Luo Y.J., Olsen R. Neuropeptides modulate local astrocytes to regulate adult hippocampal neural stem cells. Neuron. 2020;108:349–366. doi: 10.1016/j.neuron.2020.07.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ayaz A., Stauble A., Hamada M., Wulf M.A., Saleem A.B., Helmchen F. Layer-specific integration of locomotion and sensory information in mouse barrel cortex. Nat. Commun. 2019;10:2585. doi: 10.1038/s41467-019-10564-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bindocci E., Savtchouk I., Liaudet N., Becker D., Carriero G., Volterra A. Three-dimensional Ca(2+) imaging advances understanding of astrocyte biology. Science. 2017;356:eaai8185. doi: 10.1126/science.aai8185. [DOI] [PubMed] [Google Scholar]
- Bojarskaite L., Bjornstad D.M., Pettersen K.H., Cunen C., Hermansen G.H., Abjorsbraten K.S., Chambers A.R., Sprengel R., Vervaeke K., Tang W. Astrocytic Ca(2+) signaling is reduced during sleep and is involved in the regulation of slow wave sleep. Nat. Commun. 2020;11:3240. doi: 10.1038/s41467-020-17062-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khakh B.S., McCarthy K.D. Astrocyte calcium signaling: from observations to functions and the challenges therein. Cold Spring Harb. Perspect. Biol. 2015;7:a020404. doi: 10.1101/cshperspect.a020404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Linkert M., Rueden C.T., Allan C., Burel J.M., Moore W., Patterson A., Loranger B., Moore J., Neves C., Macdonald D. Metadata matters: access to image data in the real world. J. Cell Biol. 2010;189:777–782. doi: 10.1083/jcb.201004104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Savtchouk I., Volterra A. Gliotransmission: beyond black-and-white. J. Neurosci. 2018;38:14–25. doi: 10.1523/JNEUROSCI.0017-17.2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schindelin J., Arganda-Carreras I., Frise E., Kaynig V., Longair M., Pietzsch T., Preibisch S., Rueden C., Saalfeld S., Schmid B. Fiji: an open-source platform for biological-image analysis. Nat. Methods. 2012;9:676–682. doi: 10.1038/nmeth.2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shigetomi E., Kracun S., Khakh B.S. Monitoring astrocyte calcium microdomains with improved membrane targeted GCaMP reporters. Neuron Glia Biol. 2010;6:183–191. doi: 10.1017/S1740925X10000219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stobart J.L., Ferrari K.D., Barrett M.J.P., Gluck C., Stobart M.J., Zuend M., Weber B. Cortical circuit activity evokes rapid astrocyte calcium signals on a similar timescale to neurons. Neuron. 2018;98:726–735.e4. doi: 10.1016/j.neuron.2018.03.050. [DOI] [PubMed] [Google Scholar]
- Ting J.T., Daigle T.L., Chen Q., Feng G. Acute brain slice methods for adult and aging animals: application of targeted patch clamp analysis and optogenetics. Methods Mol. Biol. 2014;1183:221–242. doi: 10.1007/978-1-4939-1096-0_14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ting J.T., Lee B.R., Chong P., Soler-Llavina G., Cobbs C., Koch C., Zeng H., Lein E. Preparation of acute brain slices using an optimized N-methyl-D-glucamine protective recovery method. J. Vis. Exp. 2018;132:53825. doi: 10.3791/53825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y., DelRosso N.V., Vaidyanathan T.V., Cahill M.K., Reitman M.E., Pittolo S., Mi X., Yu G., Poskanzer K.E. Accurate quantification of astrocyte and neurotransmitter fluorescence dynamics for single-cell and population-level physiology. Nat. Neurosci. 2019;22:1936–1944. doi: 10.1038/s41593-019-0492-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
Calcium data sets from this study have not been deposited publicly, but samples may be requested from the lead contact or the technical contact. Further information on specific analysis code written in Igor Pro may be made from the technical contact, Brent Asrican, basrican@email.unc.edu.