

Key Points
Question
What is the molecular diagnostic yield of exome sequencing (prevalence of pathogenic and likely pathogenic variants) among patients with cerebral palsy?
Findings
In this cross-sectional study that included 2 independent cohorts of 1526 patients with cerebral palsy, the molecular diagnostic yield of exome sequencing was 32.7% in a cohort that predominantly consisted of pediatric patients and 10.5% in a cohort that predominantly consisted of adult patients.
Meaning
This study identified pathogenic and likely pathogenic variants among some patients with cerebral palsy, although further research is needed to understand the clinical implications of these findings.
Abstract
Importance
Cerebral palsy is a common neurodevelopmental disorder affecting movement and posture that often co-occurs with other neurodevelopmental disorders. Individual cases of cerebral palsy are often attributed to birth asphyxia; however, recent studies indicate that asphyxia accounts for less than 10% of cerebral palsy cases.
Objective
To determine the molecular diagnostic yield of exome sequencing (prevalence of pathogenic and likely pathogenic variants) in individuals with cerebral palsy.
Design, Setting, and Participants
A retrospective cohort study of patients with cerebral palsy that included a clinical laboratory referral cohort with data accrued between 2012 and 2018 and a health care–based cohort with data accrued between 2007 and 2017.
Exposures
Exome sequencing with copy number variant detection.
Main Outcomes and Measures
The primary outcome was the molecular diagnostic yield of exome sequencing.
Results
Among 1345 patients from the clinical laboratory referral cohort, the median age was 8.8 years (interquartile range, 4.4-14.7 years; range, 0.1-66 years) and 601 (45%) were female. Among 181 patients in the health care–based cohort, the median age was 41.9 years (interquartile range, 28.0-59.6 years; range, 4.8-89 years) and 96 (53%) were female. The molecular diagnostic yield of exome sequencing was 32.7% (95% CI, 30.2%-35.2%) in the clinical laboratory referral cohort and 10.5% (95% CI, 6.0%-15.0%) in the health care–based cohort. The molecular diagnostic yield ranged from 11.2% (95% CI, 6.4%-16.2%) for patients without intellectual disability, epilepsy, or autism spectrum disorder to 32.9% (95% CI, 25.7%-40.1%) for patients with all 3 comorbidities. Pathogenic and likely pathogenic variants were identified in 229 genes (29.5% of 1526 patients); 86 genes were mutated in 2 or more patients (20.1% of 1526 patients) and 10 genes with mutations were independently identified in both cohorts (2.9% of 1526 patients).
Conclusions and Relevance
Among 2 cohorts of patients with cerebral palsy who underwent exome sequencing, the prevalence of pathogenic and likely pathogenic variants was 32.7% in a cohort that predominantly consisted of pediatric patients and 10.5% in a cohort that predominantly consisted of adult patients. Further research is needed to understand the clinical implications of these findings.
This cross-sectional study describes the prevalence of pathogenic and likely pathogenic variants detected on exome sequencing of children and adults with cerebral palsy referred for genetic testing.
Introduction
Cerebral palsy is a major nonprogressive neurodevelopmental disorder affecting early motor development and characteristically impairs movement and posture. Cerebral palsy is frequently accompanied by other neurodevelopmental disorders, including intellectual disability, epilepsy, and autism spectrum disorder.1 The prevalence of cerebral palsy in the US was estimated as 2.6 to 2.9 per 1000 (data collected between 2011 and 2013).2 Due to the increasing life expectancy of individuals with cerebral palsy, the adult population with this disorder has been increasing, accompanied by changes in medical and social care needs.3
Although the etiology of cerebral palsy has been attributed to a variety of factors, the specific causal mechanism remains unknown in many individual cases. There has been a pervasive belief in the medical, scientific, and lay communities that birth asphyxia, secondary to adverse intrapartum events, is the leading cause of cerebral palsy. However, large population–based studies showed that birth asphyxia accounts for less than 10% of cerebral palsy cases.4
A small but growing body of evidence suggests that a proportion of cerebral palsy cases may be secondary to rare genomic variants of large effect size, including copy number variants (CNVs) and single nucleotide variants (SNVs),5 as is the case in other neurodevelopmental disorders, such as intellectual disability, autism spectrum disorder, and epilepsy.6,7,8 The rate of positive genomic findings in prior studies with small sample sizes (50 to 250 cases of cerebral palsy) ranged from 9.6% to 31.0% for CNVs detected using chromosomal microarray analysis9,10,11,12 and from 14% to 19% for SNVs identified using exome sequencing.13,14,15
The purpose of this cross-sectional study was to determine the molecular diagnostic yield of exome sequencing (prevalence of pathogenic and likely pathogenic variants) in individuals with cerebral palsy.
Methods
Setting and Study Participants
Two independent cerebral palsy cohorts (a clinical laboratory referral cohort and a health care–based cohort) were evaluated in this study (Table 1 and Figure 1).
Table 1. Demographics and Neurodevelopmental Comorbidities in 2 Cerebral Palsy Cohorts.
| Clinical laboratory referral cohort | Health care–based cohort | |
|---|---|---|
| No. of patients | 1345 | 181 |
| No. of triosa | 1009 | 0 |
| Age, median (IQR) [range], y | 8.8 (4.4-14.7) [0.1-66] | 41.9 (28.0-59.6) [4.8-89] |
| Sex, No. (%) | ||
| Female | 601 (45) | 96 (53) |
| Male | 744 (55) | 85 (47) |
| Neurodevelopmental comorbidities, No. (%)b | ||
| Intellectual disability or developmental delayc | 1243 (92) | 42 (23) |
| Epilepsyd | 666 (50) | 77 (43) |
| Autism spectrum disordere | 285 (21) | 11 (6) |
Abbreviation: IQR, interquartile range.
The term trios refers to a patient with cerebral palsy plus both parents.
Some patients have more than 1 comorbidity so the comorbidity numbers may sum to more than 100%. For the clinical laboratory referral cohort, comorbidity information was extracted from the indication for referral as provided on the test requisition form and submitted via clinic notes, which were translated into Human Phenotype Ontology (HPO) terms. For the health care–based cohort, the diagnoses were extracted from the patient’s electronic health record using International Classification of Diseases, Ninth Revision (ICD-9) and International Statistical Classification of Diseases and Related Health Problems, Tenth Revision (ICD-10) codes. Any patients matching the HPO terms or ICD codes were included.
The HPO codes were HP:0001249 for intellectual disability and HP:0012758 for neurodevelopmental delay. For intellectual disability or developmental delay, the ICD-9 codes were 315.5, 315.8, 317, 318.0, 318.1, 318.2, and 319 and the ICD-10 codes were F70, F71, F72, F73, F78, F79, and F88.
The HPO term was HP:0001250. The ICD-9 codes were all 345 codes and the ICD-10 codes were all G40 codes.
The HPO term was HP:0000729. The ICD-9 codes were 299.00, 299.01, 299.80, 299.81, and 299.90 and the ICD-10 codes were F84.0, F84.5, F84.8, and F84.9.
Figure 1. Study Design and Flowchart.

Each cohort predominantly consisted of pediatric or adult patients.
aThe term trios refers to a patient with cerebral palsy plus both parents.
bRefers to the DiscovEHR project, which is an ongoing collaboration between Geisinger and the Regeneron Genetics Center.
cStandards and guidelines for the interpretation of sequence variants from the American College of Medical Genetics and Genomics and the Association for Molecular Pathology (ACMG/AMP) recommend the use of a 5-tier terminology system (ie, pathogenic, likely pathogenic, uncertain significance, likely benign, and benign) to describe variants identified in genes associated with mendelian disorders and describes a process for classifying variants into these 5 categories using evidence-based criteria.
dThe term pathogenic is used for variants with the strongest evidence of pathogenicity, whereas likely pathogenic is used to mean greater than 90% certainty of a variant causing a disease.
Clinical Laboratory Referral Cohort
The clinical laboratory referral cohort predominantly consisted of pediatric patients who had been referred for diagnostic exome sequencing at a clinical genetics laboratory (GeneDx) between August 31, 2012, and March 29, 2018. Written informed consent for genetic testing was obtained from the guardians of all pediatric individuals undergoing testing. The Western institutional review board waived authorization for the use of de-identified aggregate data for the purposes of this study.
The clinical diagnoses of cerebral palsy and neurodevelopmental comorbidities were made by the referring clinicians and documented on the test requisition form and submitted via clinic notes, which were translated into Human Phenotype Ontology terms (HP:0100021 for cerebral palsy; HP:0012758 for neurodevelopmental delay; HP:0001249 for intellectual disability; HP:0001250 for epilepsy; and HP:0000729 for autistic behavior). The final date of follow-up was March 29, 2018, for the clinical laboratory referral cohort.
Health Care–Based Cohort
The health care–based cohort predominantly consisted of adult patients who were recruited from the DiscovEHR project, which is an ongoing collaborative study between Geisinger and the Regeneron Genetics Center,16 between February 8, 2007, and April 24, 2017. Written informed consent was obtained from the adult patients and from the parents or guardians of the pediatric patients. The Geisinger institutional review board approved the study.
The diagnoses of cerebral palsy and neurodevelopmental comorbidities were made by the treating physicians and extracted from the patient’s electronic health record using International Classification of Diseases codes (International Classification of Diseases, Ninth Revision [ICD-9] codes 333.71, 343.0, 343.1, 343.2, 343.3, 343.8, and 343.9 and International Statistical Classification of Diseases and Related Health Problems, Tenth Revision [ICD-10] codes G80.0, G80.1, G80.2, G80.3, G80.4, G80.8, and G80.9 for cerebral palsy; ICD-9 codes 315.5, 315.8, 317, 318.0, 318.1, 318.2, and 319 and ICD-10 codes F70, F71, F72, F73, F78, F79, and F88 for intellectual disability; ICD-9 code 345 (all codes) and ICD-10 code G40 (all codes) for epilepsy; and ICD-9 codes 299.00, 299.01, 299.80, 299.81, and 299.90 and ICD-10 codes F84.0, F84.5, F84.8, and F84.9 for autism spectrum disorder). The final date of follow-up was April 24, 2017, for the health care–based cohort.
Exome Sequencing and Variant Detection
Exome sequencing was performed using previously reported protocols (additional information appears in the eMethods in the Supplement).17,18 Routine clinical exome analysis was conducted for the clinical laboratory referral cohort. The health care–based cohort underwent research-based exome sequencing analysis with CNV detection. Across cohorts, variants were evaluated for pathogenicity using standards and guidelines for the interpretation of sequence variants from the American College of Medical Genetics and Genomics and the Association for Molecular Pathology.19
Statistical Analysis
The molecular diagnostic yield was summarized by cohort and reported as frequency and percentage of pathogenic and likely pathogenic variants. Multivariable logistic regression, controlling for cohort, was used to estimate the association between comorbid neurodevelopmental disorders and the likelihood of detection of pathogenic and likely pathogenic variants. The presence or absence of intellectual disability, epilepsy, or autism spectrum disorder was coded as a binary variable.
The multivariable logistic regression model was used to estimate molecular diagnostic yield for each individual neurodevelopmental disorder as well as in combination. An initial model was fit that included the main effects for each neurodevelopmental comorbidity and all 2-way and 3-way interactions while controlling for cohort.
A joint test of the interaction effects was conducted using the likelihood ratio test and comparing the model with a nested model of only main effects. Model fit for the logistic regression model was assessed using the deviance statistic and the Hosmer-Lemeshow goodness-of-fit statistic. For the primary estimates, 95% CIs were reported.
Statistical significance was defined as P<.05 and all tests were 2-sided. Because of the potential for type I error due to multiple comparisons, the findings for the analyses of the secondary end points should be interpreted as exploratory. There were no missing or imputed data. SAS version 9.4 (SAS Institute Inc) was used for all the analyses.
Results
Demographics of the 2 Cohorts
The clinical laboratory referral cohort included 1345 patients (1009 parent-offspring trios) with a median age of 8.8 years (interquartile range, 4.4-14.7 years; range, 0.1-66 years) and 601 (45%) were female (Table 1). The health care–based cohort included 181 patients (parental samples were not available) with a median age of 41.9 years (interquartile range, 28.0-59.6 years; range, 4.8-89 years) and 96 (53%) were female.
Exome Sequencing Molecular Diagnostic Yield
Clinical Laboratory Referral Cohort
Exome sequencing yielded a positive diagnostic result in 440 of 1345 patients in the clinical laboratory referral cohort (32.7% [95% CI, 30.2%-35.2%]; Table 2 and eTable 1 in the Supplement), of which 94.3% (415/440) had SNVs, 4.3% (19/440) had CNVs, and 1.4% (6/440) had both SNVs and CNVs. Testing of a proband concurrently with parents (trio; n = 1009) significantly improved the molecular diagnostic yield from 23.3% (proband only) to 35.4% (Fisher exact test P = .003).
Table 2. Molecular Diagnostic Yield of Pathogenic and Likely Pathogenic Variants in 2 Cerebral Palsy Cohorts Using Exome Sequencing.
| Clinical laboratory referral cohort | Health care–based cohort | |
|---|---|---|
| Molecular diagnostic yield, No./total (%) [95% CI]a | 440/1345 (32.7) [30.2-35.2] | 19/181 (10.5) [6.0-15.0] |
| Single nucleotide variants | 415/440 (94.3) | 14/19 (73.7) |
| Copy number variants | 19/440 (4.3) | 5/19 (26.3) |
| Both single nucleotide and copy number variants | 6/440 (1.4) | 0/19 (0) |
| Inheritance pattern of pathogenic and likely pathogenic variants among trios in which the patient had positive diagnostic resultsb | ||
| De novo | 257/357 (72) | NAc |
| Autosomal recessive | 70/357 (19.6) | NAc |
| Autosomal dominant | 18/357 (5) | NAc |
| X-linked | 12/357 (3.4) | NAc |
Abbreviation: NA, not applicable.
Molecular diagnostic yield refers to the prevalence of pathogenic and likely pathogenic variants.
Pathogenicity was determined using standards and guidelines for the interpretation of sequence variants from the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. The term trios refers to a patient with cerebral palsy plus both parents.
Parental samples were not available.
Among trios with positive diagnostic results (n = 357), de novo variants were identified most frequently (72%; 257/357), whereas 5% (18/357) were inherited in an autosomal dominant manner, 19.6% (70/357) were inherited in an autosomal recessive manner, and 3.4% (12/357) were X-linked. Exome sequencing–derived CNV analysis identified 25 patients with pathogenic and likely pathogenic CNVs, ranging in size from 637 base pairs to 93.3 mega base pairs, of which 15 cases involved a single gene and 10 included multiple genes.
The identification of a molecular diagnosis allowed for revision of the 1% to 2% empirical recurrence risk of cerebral palsy for future children born to the same parents.20 In 72% (257/357) of trios with positive diagnostic results, the identification of a de novo pathogenic and likely pathogenic variant resulted in an adjusted recurrence risk of less than 1%, whereas the recurrence risk increased to 25% in cases with recessive variants (19.6%; 70/357) and to 50% in cases with dominant or X-linked pathogenic and likely pathogenic variants (8.4%; 30/357).
Health Care–Based Cohort
Pathogenic and likely pathogenic variants were identified in 19 of 181 patients in the health care–based cohort (10.5% [95% CI, 6.0%-15.0%]; Table 2 and eTable 2 in the Supplement), of which 73.7% (14/19) had SNVs and 26.3% (5/19) had CNVs. Of the 19 cases, 9 (47.4%) had pathogenic and likely pathogenic SNVs in genes implicated in disorders with dominant inheritance, 5.3% (1/19) with recessive inheritance, and 21% (4/19) with X-linked inheritance.
Exome sequencing–derived CNV analysis identified 5 individuals with pathogenic and likely pathogenic deletions, ranging in size from 6.4 kilobase pairs to 14.8 mega base pairs, including the recurrent deletions 16p13.11 and 17p11.2, a large deletion of chromosome 4, a heterozygous exonic deletion of TCF4 (NM_003199; Pitt-Hopkins syndrome), and a homozygous exonic deletion of TUSC3 (NM_006765).
Molecular Diagnostic Yield and Neurodevelopmental Comorbidities in Both Cohorts
Complete comorbidity data on the presence or absence of intellectual disability, epilepsy, or autism spectrum disorder was available for all 1526 patients from both cohorts. A test of the interactions among the 3 neurodevelopmental comorbidities was not significant (likelihood ratio test = 1.88; P = .76). Therefore, a main effect model was fitted.
The tests of fit using the deviance statistic (6.68; P = .74) and the Hosmer-Lemeshow statistic (3.47; P = .48) were not significant. The estimated cross-cohort exome sequencing molecular diagnostic yield ranged from 11.2% (95% CI, 6.4%-16.2%) for patients with cerebral palsy but without intellectual disability, epilepsy, or autism spectrum disorder to 32.9% (95% CI, 25.7%-40.1%) for patients with cerebral palsy and all 3 comorbidities.
The association comparisons between neurodevelopmental comorbidities and the likelihood of having a pathogenic and likely pathogenic variant appear in Figure 2. For patients with cerebral palsy and intellectual disability vs patients with cerebral palsy but without intellectual disability, the adjusted odds ratio was 2.59 (95% CI, 1.61-4.17; P < .001) for the association between neurodevelopmental comorbidities and the likelihood of having a pathogenic and likely pathogenic variant; it was 1.01 (95% CI, 0.81-1.27; P = .91) for patients with cerebral palsy and epilepsy vs patients with cerebral palsy but without epilepsy; it was 1.04 (95% CI, 0.79-1.37; P = .78) for patients with cerebral palsy and autism spectrum disorder vs patients with cerebral palsy but without autism spectrum disorder; and it was 2.73 (95% CI, 1.58-4.72; P < .001) for patients with cerebral palsy and all 3 neurodevelopmental comorbidities vs patients with cerebral palsy but without the 3 neurodevelopmental comorbidities.
Figure 2. Association of Neurodevelopmental Comorbidities With Likelihood of Detection of Pathogenic and Likely Pathogenic Variants in Patients With Cerebral Palsy.
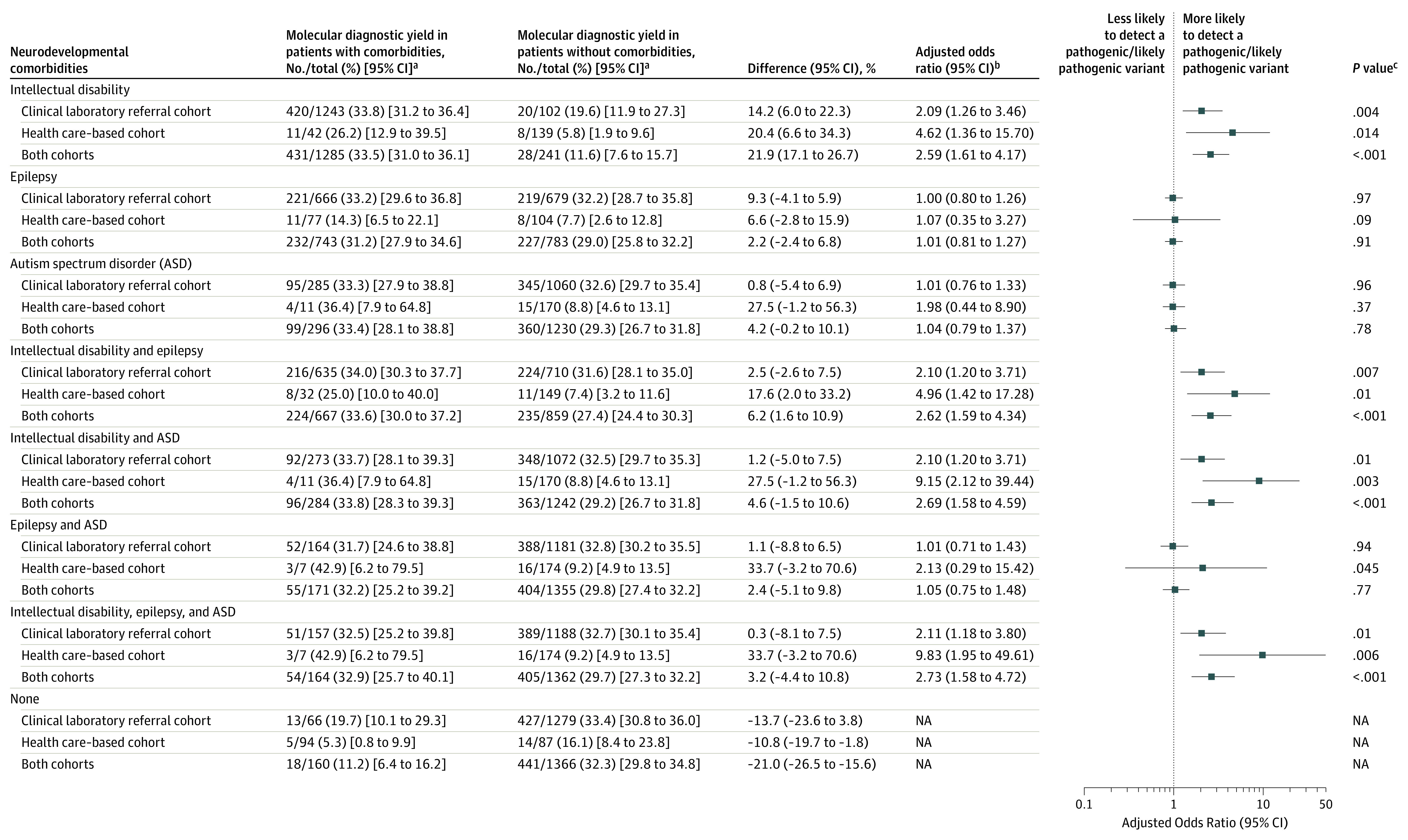
NA indicates not applicable.
aMolecular diagnostic yield refers to the prevalence of pathogenic and likely pathogenic variants.
bThe odds ratios were estimated from a logistic regression model that controlled for cohort and each individual neurodevelopmental comorbidity. The analysis assesses the likelihood of detection of pathogenic and likely pathogenic variants in patients with cerebral palsy and the neurodevelopmental comorbidity listed in the first column vs patients with cerebral palsy but without the comorbidity. When analyzing cohort-specific data, only the neurodevelopmental comorbidities were included in the model.
cCalculated using a χ2 test.
Cross-Cohort Spectrum of Genomic Variation
Combining positive results from both cohorts resulted in the identification of 229 genes (29.5% of 1526 patients) harboring pathogenic and likely pathogenic variants (eTable 3 in the Supplement). There were 143 genes (62.4%) that were mutated in single cases and 86 genes (37.6%) that had pathogenic and likely pathogenic variants in 2 or more patients (20.1% of 1526 patients).
The most frequently mutated genes were CTNNB1 (NM_001098210; 18/450 [4.0%] of positive cases) followed by KIF1A (NM_004321; 8/450 [1.8%] of positive cases). The following mutated genes were found among 7 of 450 [1.6%] positive cases each: COL4A1 (NM_001303110), GNAO1 (NM_020988), KCNQ2 (NM_172109), MECP2 (NM_004992), and STXBP1 (NM_003165). There were 10 genes in which pathogenic and likely pathogenic variants were independently identified in both cohorts (2.9% of 1526 patients; Table 3).
Table 3. Genes With Pathogenic and Likely Pathogenic Variants Identified in Both Cohorts Among Unrelated Patients With Cerebral Palsy.
| Genea | RefSeq accession No. | No. of cases | OMIM phenotypeb | Inheritance | ||
|---|---|---|---|---|---|---|
| Clinical laboratory referral cohort | Health care–based cohort | Total | ||||
| MECP2 | NM_004992 | 5 | 2 | 7 | Rett syndrome | X-linked |
| Intellectual disability with spasticity | X-linked | |||||
| Intellectual disability, syndromic, Lubs type | X-linked | |||||
| Encephalopathy, neonatal severe | X-linked | |||||
| TCF4 | NM_003199 | 5 | 1 | 6 | Pitt-Hopkins syndrome | Autosomal dominant |
| TUBA1A | NM_006009 | 5 | 1 | 6 | Lissencephaly type 3 | Autosomal dominant |
| SLC2A1 | NM_006516 | 4 | 1 | 5 | GLUT1 deficiency syndrome type 1, infantile onset, severe | Autosomal dominant or autosomal recessive |
| GLUT1 deficiency syndrome type 2, childhood onset | Autosomal dominant | |||||
| Dystonia type 9 | Autosomal dominant | |||||
| Stomatin-deficient cryohydrocytosis with neurological defects | Autosomal dominant | |||||
| KMT2A | NM_005933 | 4 | 1 | 5 | Wiedemann-Steiner syndrome | Autosomal dominant |
| CAMTA1 | NM_015215 | 3 | 1 | 4 | Cerebellar ataxia, nonprogressive with intellectual disability | Autosomal dominant |
| ATL1 | NM_001127713 | 2 | 2 | 4 | Spastic paraplegia type 3A | Autosomal dominant |
| Neuropathy, hereditary-sensory–type intellectual disability | Autosomal dominant | |||||
| IQSEC2 | NM_001111125 | 2 | 1 | 3 | Intellectual disability type 1/78 | X-linked |
| ASXL3 | NM_030632 | 2 | 1 | 3 | Bainbridge-Ropers syndrome | Autosomal dominant |
| L1CAM | NM_024003 | 1 | 1 | 2 | CRASH syndrome | X-linked |
| Corpus callosum, partial agenesis | X-linked | |||||
| Hydrocephalus due to aqueductal stenosis | X-linked | |||||
The full lists of genes with pathogenic and likely pathogenic variants appear in eTables 1-3 in the Supplement.
The OMIM is an online catalog of human genes and genetic disorders. The phenotypes represent well-documented clinical manifestations of mendelian disorders (genotype-phenotype relationship). For some of the genes listed, the OMIM phenotype includes neuromotor disorders, whereas for other genes, the neuromotor manifestations have not been thoroughly characterized.
Discussion
The findings of this study indicated that, similar to other neurodevelopmental disorders, exome sequencing in patients with cerebral palsy identified pathogenic and likely pathogenic variants in hundreds of genes with a prevalence of 32.7% in the cohort that predominantly consisted of pediatric patients and a prevalence of 10.5% in the cohort that predominantly consisted of adult patients. Prior large-scale studies of individuals with neurodevelopmental disorders showed that detecting independent de novo pathogenic variants in the same gene among unrelated individuals provides strong evidence to reliably identify disease-related genes.21
Multiple genes were mutated in 2 or more unrelated patients with cerebral palsy in this study, including CTNNB1, KIF1A, GNAO1, and TUBA1A (NM_006009). Mutations in CTNNB1 have been previously reported in patients with cerebral palsy15 and an autosomal dominant neurodevelopmental disorder with intellectual disability, spasticity, microcephaly, and visual defects.22 Pathogenic variants in KIF1A have been identified in patients with autosomal dominant intellectual disability with spastic paraparesis, axonal neuropathy, cerebellar atrophy,23 and autosomal recessive spastic paraplegia.24
Mutations in GNAO1 have been reported in patients with cerebral palsy25 and an autosomal dominant neurodevelopmental disorder with epileptic encephalopathy and movement disorder.26 Mutations in TUBA1A have been reported in patients with cerebral palsy15 and autosomal dominant brain malformations, microcephaly, intellectual disability, and epilepsy.27 The identification of genes mutated in 2 or more patients and the replication of findings across different settings and cohort types provided strong evidence for their role in cerebral palsy and, in some cases, expanded the phenotype associated with these known neurodevelopmental disorder genes to include cerebral palsy phenotypes.
Some patients had variants in genes previously associated with both cerebral palsy and hereditary spastic paraplegia, including AP4E1 (NM_001252127), AP4M1 (NM_004722), and AP4S1 (NM_001128126; AP-4 deficiency syndrome),5,28,29 ATL1 (NM_001127713),15,30 and SPAST (NM_199436).15,31 Even though cerebral palsy and hereditary spastic paraplegia are considered distinct clinical entities due to the nonprogressive, permanent nature of cerebral palsy and the neurodegenerative progression of hereditary spastic paraplegia, shared genomic findings have been previously reported between these and other neurodevelopmental disorders.32,33
Similarly, other patients had mutations in genes or regions associated with disorders frequently considered different from cerebral palsy, including Pitt-Hopkins (TCF4), Rett (MECP2), and Smith-Magenis (17p11.2 deletion) syndromes. As with other neurodevelopmental disorders, such as autism spectrum disorder, intellectual disability, and epilepsy, the diagnosis of cerebral palsy is based on clinical manifestations not etiology. As noted by the International Cerebral Palsy Genomics Consortium in their clinical consensus statement, when a pathogenic and likely pathogenic variant is identified in a patient with a phenotype consistent with current consensus definitions of cerebral palsy,1,34 the cerebral palsy diagnosis should not be removed, nor should the case be reclassified as cerebral palsy mimic, masquerader, or cerebral palsy–like.35
Exome sequencing is currently recommended as a first-tier clinical diagnostic test for individuals with neurodevelopmental disorders.6 The mean molecular diagnostic yield of exome sequencing in other neurodevelopmental disorders averaged across multiple studies was 35% for intellectual disability or neurodevelopmental delay, 45% for epilepsy, and 15% for autism spectrum disorder,7,36,37 all of which are known cerebral palsy comorbidities. Cerebral palsy can be identified earlier than other neurodevelopmental disorders (such as intellectual disability and autism spectrum disorder).
Among individuals in a Danish national registry, the median age for cerebral palsy diagnosis was 11 months,38 and cerebral palsy or a high risk of cerebral palsy can often be accurately predicted before the corrected age of 6 months.39 Therefore, performing genetic testing in individuals with cerebral palsy may have the potential for a more timely identification of pathogenic and likely pathogenic variants, facilitating early initiation of prevention and treatment strategies. Moreover, pathogenic and likely pathogenic variants were also identified in individuals with cerebral palsy who did not have other diagnoses (intellectual disability or autism spectrum disorder) that would have led to clinical genetic testing based on current recommendations.
The identification of pathogenic and likely pathogenic variants in patients with cerebral palsy may result in genomics-informed changes in clinical care for some patients. For example, a patient from the clinical laboratory referral cohort had a homozygous pathogenic variant in the SPR (NM_003124) gene, which established the molecular diagnosis of sepiapterin reductase deficiency. The dopamine deficiency associated with this disorder can be corrected with levodopa in combination with carbidopa, which often corrects the motor abnormalities.40 In addition, families with 1 child with cerebral palsy are typically counseled that their risk of recurrence is 1% to 2%20; however, the findings in this study indicate the possibility of a higher recurrence risk for some families, which may lead to changes in family planning counseling.
Limitations
This study has several limitations. First, the 2 independent cohorts were ascertained using different approaches and the extent of available clinical information for each patient was variable, which resulted in a more heterogeneous combined cohort. The correlation between different types of cerebral palsy (spastic, dyskinetic, ataxic, and mixed) and exome sequencing molecular diagnostic yield was not explored because detailed clinical information was not available for most patients.
Second, the exome sequencing capture reagents and analytical pipelines varied between cohorts; however, all variants were evaluated for pathogenicity using standards and guidelines for the interpretation of sequence variants from the American College of Medical Genetics and Genomics and the Association for Molecular Pathology.
Third, the health care–based cohort did not have available parental samples to assess for variant inheritance because it was a cohort that predominantly consisted of adult patients, which resulted in a more conservative exome sequencing molecular diagnostic yield because identifying de novo variants can increase the strength of evidence to classify variants as pathogenic. This was demonstrated in the clinical laboratory referral cohort for which trio testing was associated with significant improvement in molecular diagnostic yield compared with proband only testing.
Fourth, this was an observational study and a causal relationship between detected gene variants and phenotypes in participants is not definitively established.
Conclusions
Among 2 cohorts of patients with cerebral palsy who underwent exome sequencing, the prevalence of pathogenic and likely pathogenic variants was 32.7% in a cohort that predominantly consisted of pediatric patients and 10.5% in a cohort that predominantly consisted of adult patients. Further research is needed to understand the clinical implications of these findings.
DiscovEHR collaboration contributors
eMethods. Exome sequencing and variant calling
eTable 1. Clinical laboratory referral cohort pathogenic/likely pathogenic variants
eTable 2. Healthcare-based cohort pathogenic/likely pathogenic variants
eTable 3. Cross-cohort spectrum of genomic variation
eReferences
References
- 1.Rosenbaum P, Paneth N, Leviton A, et al. A report: the definition and classification of cerebral palsy April 2006. Dev Med Child Neurol Suppl. 2007;109:8-14. [PubMed] [Google Scholar]
- 2.Maenner MJ, Blumberg SJ, Kogan MD, Christensen D, Yeargin-Allsopp M, Schieve LA. Prevalence of cerebral palsy and intellectual disability among children identified in two US national surveys, 2011-2013. Ann Epidemiol. 2016;26(3):222-226. doi: 10.1016/j.annepidem.2016.01.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Bromham N, Dworzynski K, Eunson P, Fairhurst C; Guideline Committee . Cerebral palsy in adults: summary of NICE guidance. BMJ. 2019;364:l806. doi: 10.1136/bmj.l806 [DOI] [PubMed] [Google Scholar]
- 4.Ellenberg JH, Nelson KB. The association of cerebral palsy with birth asphyxia: a definitional quagmire. Dev Med Child Neurol. 2013;55(3):210-216. doi: 10.1111/dmcn.12016 [DOI] [PubMed] [Google Scholar]
- 5.Moreno-De-Luca A, Ledbetter DH, Martin CL. Genetic [corrected] insights into the causes and classification of [corrected] cerebral palsies. Lancet Neurol. 2012;11(3):283-292. doi: 10.1016/S1474-4422(11)70287-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Srivastava S, Love-Nichols JA, Dies KA, et al. ; NDD Exome Scoping Review Work Group . Meta-analysis and multidisciplinary consensus statement: exome sequencing is a first-tier clinical diagnostic test for individuals with neurodevelopmental disorders. Genet Med. 2019;21(11):2413-2421. doi: 10.1038/s41436-019-0554-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Sánchez Fernández I, Loddenkemper T, Gaínza-Lein M, Sheidley BR, Poduri A. Diagnostic yield of genetic tests in epilepsy: a meta-analysis and cost-effectiveness study. Neurology. 2019;92(5):e418-e428. doi: 10.1212/WNL.0000000000006850 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Moreno-De-Luca A, Myers SM, Challman TD, Moreno-De-Luca D, Evans DW, Ledbetter DH. Developmental brain dysfunction: revival and expansion of old concepts based on new genetic evidence. Lancet Neurol. 2013;12(4):406-414. doi: 10.1016/S1474-4422(13)70011-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.McMichael G, Girirajan S, Moreno-De-Luca A, et al. Rare copy number variation in cerebral palsy. Eur J Hum Genet. 2014;22(1):40-45. doi: 10.1038/ejhg.2013.93 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Segel R, Ben-Pazi H, Zeligson S, et al. Copy number variations in cryptogenic cerebral palsy. Neurology. 2015;84(16):1660-1668. doi: 10.1212/WNL.0000000000001494 [DOI] [PubMed] [Google Scholar]
- 11.Oskoui M, Gazzellone MJ, Thiruvahindrapuram B, et al. Clinically relevant copy number variations detected in cerebral palsy. Nat Commun. 2015;6:7949. doi: 10.1038/ncomms8949 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Zarrei M, Fehlings DL, Mawjee K, et al. De novo and rare inherited copy-number variations in the hemiplegic form of cerebral palsy. Genet Med. 2018;20(2):172-180. doi: 10.1038/gim.2017.83 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.McMichael G, Bainbridge MN, Haan E, et al. Whole-exome sequencing points to considerable genetic heterogeneity of cerebral palsy. Mol Psychiatry. 2015;20(2):176-182. doi: 10.1038/mp.2014.189 [DOI] [PubMed] [Google Scholar]
- 14.Corbett MA, van Eyk CL, Webber DL, et al. Pathogenic copy number variants that affect gene expression contribute to genomic burden in cerebral palsy. NPJ Genom Med. 2018;3:33. doi: 10.1038/s41525-018-0073-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Jin SC, Lewis SA, Bakhtiari S, et al. Mutations disrupting neuritogenesis genes confer risk for cerebral palsy. Nat Genet. 2020;52(10):1046-1056. doi: 10.1038/s41588-020-0695-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Carey DJ, Fetterolf SN, Davis FD, et al. The Geisinger MyCode community health initiative: an electronic health record-linked biobank for precision medicine research. Genet Med. 2016;18(9):906-913. doi: 10.1038/gim.2015.187 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Retterer K, Juusola J, Cho MT, et al. Clinical application of whole-exome sequencing across clinical indications. Genet Med. 2016;18(7):696-704. doi: 10.1038/gim.2015.148 [DOI] [PubMed] [Google Scholar]
- 18.Dewey FE, Murray MF, Overton JD, et al. Distribution and clinical impact of functional variants in 50,726 whole-exome sequences from the DiscovEHR study. Science. 2016;354(6319):aaf6814. doi: 10.1126/science.aaf6814 [DOI] [PubMed] [Google Scholar]
- 19.Richards S, Aziz N, Bale S, et al. ; ACMG Laboratory Quality Assurance Committee . Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med. 2015;17(5):405-424. doi: 10.1038/gim.2015.30 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Korzeniewski SJ, Slaughter J, Lenski M, Haak P, Paneth N. The complex aetiology of cerebral palsy. Nat Rev Neurol. 2018;14(9):528-543. doi: 10.1038/s41582-018-0043-6 [DOI] [PubMed] [Google Scholar]
- 21.Gonzalez-Mantilla AJ, Moreno-De-Luca A, Ledbetter DH, Martin CL. A cross-disorder method to identify novel candidate genes for developmental brain disorders. JAMA Psychiatry. 2016;73(3):275-283. doi: 10.1001/jamapsychiatry.2015.2692 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kharbanda M, Pilz DT, Tomkins S, et al. ; DDD Study . Clinical features associated with CTNNB1 de novo loss of function mutations in ten individuals. Eur J Med Genet. 2017;60(2):130-135. doi: 10.1016/j.ejmg.2016.11.008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Lee JR, Srour M, Kim D, et al. De novo mutations in the motor domain of KIF1A cause cognitive impairment, spastic paraparesis, axonal neuropathy, and cerebellar atrophy. Hum Mutat. 2015;36(1):69-78. doi: 10.1002/humu.22709 [DOI] [PubMed] [Google Scholar]
- 24.Klebe S, Lossos A, Azzedine H, et al. KIF1A missense mutations in SPG30, an autosomal recessive spastic paraplegia: distinct phenotypes according to the nature of the mutations. Eur J Hum Genet. 2012;20(6):645-649. doi: 10.1038/ejhg.2011.261 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Malaquias MJ, Fineza I, Loureiro L, Cardoso L, Alonso I, Magalhães M. GNAO1 mutation presenting as dyskinetic cerebral palsy. Neurol Sci. 2019;40(10):2213-2216. doi: 10.1007/s10072-019-03964-7 [DOI] [PubMed] [Google Scholar]
- 26.Schirinzi T, Garone G, Travaglini L, et al. Phenomenology and clinical course of movement disorder in GNAO1 variants: results from an analytical review. Parkinsonism Relat Disord. 2019;61:19-25. doi: 10.1016/j.parkreldis.2018.11.019 [DOI] [PubMed] [Google Scholar]
- 27.Hebebrand M, Hüffmeier U, Trollmann R, et al. The mutational and phenotypic spectrum of TUBA1A-associated tubulinopathy. Orphanet J Rare Dis. 2019;14(1):38. doi: 10.1186/s13023-019-1020-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Moreno-De-Luca A, Helmers SL, Mao H, et al. Adaptor protein complex-4 (AP-4) deficiency causes a novel autosomal recessive cerebral palsy syndrome with microcephaly and intellectual disability. J Med Genet. 2011;48(2):141-144. doi: 10.1136/jmg.2010.082263 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Ebrahimi-Fakhari D, Teinert J, Behne R, et al. Defining the clinical, molecular and imaging spectrum of adaptor protein complex 4-associated hereditary spastic paraplegia. Brain. 2020;143(10):2929-2944. doi: 10.1093/brain/awz307 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Rainier S, Sher C, Reish O, Thomas D, Fink JK. De novo occurrence of novel SPG3A/atlastin mutation presenting as cerebral palsy. Arch Neurol. 2006;63(3):445-447. doi: 10.1001/archneur.63.3.445 [DOI] [PubMed] [Google Scholar]
- 31.Takezawa Y, Kikuchi A, Haginoya K, et al. Genomic analysis identifies masqueraders of full-term cerebral palsy. Ann Clin Transl Neurol. 2018;5(5):538-551. doi: 10.1002/acn3.551 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Fink JK Hereditary spastic paraplegia: clinico-pathologic features and emerging molecular mechanisms. Acta Neuropathol. 2013;126(3):307-328. doi: 10.1007/s00401-013-1115-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Zouvelou V, Yubero D, Apostolakopoulou L, et al. The genetic etiology in cerebral palsy mimics: the results from a Greek tertiary care center. Eur J Paediatr Neurol. 2019;23(3):427-437. doi: 10.1016/j.ejpn.2019.02.001 [DOI] [PubMed] [Google Scholar]
- 34.Smithers-Sheedy H, Badawi N, Blair E, et al. What constitutes cerebral palsy in the twenty-first century? Dev Med Child Neurol. 2014;56(4):323-328. doi: 10.1111/dmcn.12262 [DOI] [PubMed] [Google Scholar]
- 35.MacLennan AH, Lewis S, Moreno-De-Luca A, et al. Genetic or other causation should not change the clinical diagnosis of cerebral palsy. J Child Neurol. 2019;34(8):472-476. doi: 10.1177/0883073819840449 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Finucane BM, Myers SM, Martin CL, Ledbetter DH. Long overdue: including adults with brain disorders in precision health initiatives. Curr Opin Genet Dev. 2020;65:47-52. doi: 10.1016/j.gde.2020.05.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Srivastava S, Cohen JS, Vernon H, et al. Clinical whole exome sequencing in child neurology practice. Ann Neurol. 2014;76(4):473-483. doi: 10.1002/ana.24251 [DOI] [PubMed] [Google Scholar]
- 38.Granild-Jensen JB, Rackauskaite G, Flachs EM, Uldall P. Predictors for early diagnosis of cerebral palsy from national registry data. Dev Med Child Neurol. 2015;57(10):931-935. doi: 10.1111/dmcn.12760 [DOI] [PubMed] [Google Scholar]
- 39.Novak I, Morgan C, Adde L, et al. Early, accurate diagnosis and early intervention in cerebral palsy: advances in diagnosis and treatment. JAMA Pediatr. 2017;171(9):897-907. doi: 10.1001/jamapediatrics.2017.1689 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Friedman J Sepiapterin reductase deficiency In: GeneReviews. University of Washington, Seattle; 2015. [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
DiscovEHR collaboration contributors
eMethods. Exome sequencing and variant calling
eTable 1. Clinical laboratory referral cohort pathogenic/likely pathogenic variants
eTable 2. Healthcare-based cohort pathogenic/likely pathogenic variants
eTable 3. Cross-cohort spectrum of genomic variation
eReferences