

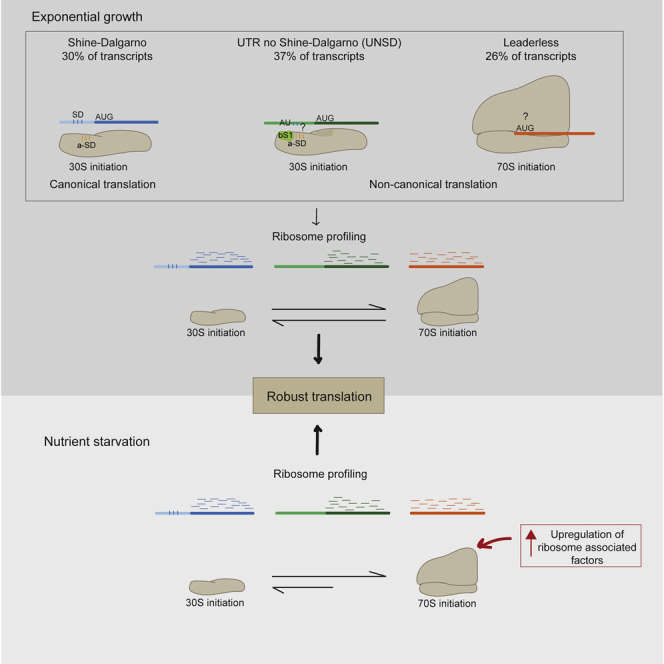
Summary
Mycobacterium tuberculosis, which causes tuberculosis, can undergo prolonged periods of non-replicating persistence in the host. The mechanisms underlying this are not fully understood, but translational regulation is thought to play a role. A large proportion of mRNA transcripts expressed in M. tuberculosis lack canonical bacterial translation initiation signals, but little is known about the implications of this for fine-tuning of translation. Here, we perform ribosome profiling to characterize the translational landscape of M. tuberculosis under conditions of exponential growth and nutrient starvation. Our data reveal robust, widespread translation of non-canonical transcripts and point toward different translation initiation mechanisms compared to canonical Shine-Dalgarno transcripts. During nutrient starvation, patterns of ribosome recruitment vary, suggesting that regulation of translation in this pathogen is more complex than originally thought. Our data represent a rich resource for others seeking to understand translational regulation in bacterial pathogens.
Keywords: Mycobacterium tuberculosis, ribosome profiling, translation, non-canonical translation, nutrient starvation
Graphical Abstract
Highlights
-
•
A resource for studying translation and its regulation in M. tuberculosis
-
•
Translation of non-canonical transcripts is robust and widespread
-
•
Positional data of initiating ribosomes reveal differences in initiation
-
•
Ribosome stabilization factors are upregulated in starvation
Sawyer et al. use ribosome profiling to characterize the translational landscape of Mycobacterium tuberculosis. They find widespread translation of non-canonical transcripts lacking canonical bacterial translation signals. Ribosome positioning reveals differences in initiation mechanisms, and factors that stabilize intact ribosomes may ensure continued robust translation during conditions of stress.
Introduction
Mycobacterium tuberculosis is an important human pathogen with a complex life cycle characterized by long periods of persistence, in which bacteria enter a non-replicative, antibiotic-tolerant state (Stewart et al., 2003; Gomez and McKinney, 2004). The mechanisms underlying bacterial adaptation to diverse environmental conditions encountered during infection—such as the acidification of phagosomes, the presence of reactive nitrogen and oxygen species, hypoxia, and nutrient starvation—are still poorly understood (Rengarajan et al., 2005). Recent findings have demonstrated differences in translational control between M. tuberculosis and the model organism Escherichia coli, and these mechanisms may play a role in the ability of M. tuberculosis to adapt to diverse environmental conditions (Trauner et al., 2012; Xue and Barna, 2012; Byrgazov et al., 2013; Bunker et al., 2015; reviewed in Sawyer et al., 2018).
The translation of messenger RNA (mRNA) into protein is a highly controlled process involving multiple layers of regulation at each step of initiation, elongation, termination, and ribosome recycling. In recent years, the heterogeneity of these regulatory mechanisms has begun to be appreciated and explored. In particular, there is increasing evidence to show that distinct populations of ribosomes preferentially translate mRNAs with certain features (Xue and Barna, 2012; Byrgazov et al., 2013; Shi et al., 2017). Not only do gene sequence features, such as the codon adaptation index, gene length, and functional category, influence translation levels (Dressaire et al., 2010; Picard et al., 2012), but untranslated regions (UTRs) of mRNAs can also influence the stability and translational efficiency of the transcript (Chen et al., 1991; Nguyen et al., 2020).
The development of ribosome profiling (Ribo-seq) techniques has proven to be powerful for studying translation (Ingolia et al., 2009). It involves isolation of ribosomes bound to mRNA, enzymatic digestion of mRNA unprotected by the ribosome, and sequencing the ribosome-protected fragments (RPFs or footprints) of the mRNA. This technique enables very precise identification of specific mRNA strands being actively translated at any given moment in time. Ribo-seq experiments performed in the non-pathogenic Mycobacterium smegmatis have uncovered important new details about translation, including the translation of hundreds of small, previously unannotated proteins at the 5′ ends of transcripts (Shell et al., 2015); the translation by alternative ribosomes that have distinct functionality compared with their canonical counterparts (Chen et al., 2020); as well as cysteine-responsive attenuation of translation (Canestrari et al., 2020).
The adaptive responses of mycobacteria to stress have been extensively analyzed by transcriptional profiling in well-defined experimental models (Stewart et al., 2002; Betts et al., 2002; Deb et al., 2009; Rohde et al., 2012; Galagan et al., 2013; Rustad et al., 2014; Namouchi et al., 2016; Aguilar-Ayala et al., 2017; Martini et al., 2019). However, due to the well-established poor correlation between levels of mRNA transcripts and proteins in the cell (Cortes et al., 2017; Haider and Pal, 2013) and differences in mRNA and protein half-lives (Taniguchi et al., 2010), it is unclear how the changes that occur in the transcriptome of bacteria that enter a persistent state play out in the proteome. Clearly, there are post-transcriptional control mechanisms that influence protein abundance, but these are currently poorly understood in mycobacteria. Previous genome-wide mapping of transcriptional start sites (TSSs) in M. tuberculosis has shown that this pathogen expresses a large number of transcripts that lack the sequence elements that usually play a role in ribosome recruitment and binding, such as the Shine-Dalgarno sequence that is normally present in the 5′ UTR of bacterial genes. In particular, around a quarter of the M. tuberculosis genes lack a 5′ UTR at all (referred to as leaderless transcripts), while approximately a further 25% have a 5′ UTR that does not contain a Shine-Dalgarno sequence (designated here as UTR-no-Shine-Dalgarno [UNSD]) (Cortes et al., 2013; Shell et al., 2015). It is known that under conditions of nutrient starvation, transcription of leaderless genes is upregulated and associated with genes expressed in nondividing cells (Cortes et al., 2013), but overall, understanding of the impact of the regulation of non-canonical genes on translation in this pathogen is lacking. Hence, further understanding of the quantitative correlations between transcription and translation, and the regulation of translation of non-canonical mRNAs, is needed in order to gain more insight into persistent infection with M. tuberculosis.
In this Resources article, we have applied parallel RNA sequencing (RNA-seq) and Ribo-seq in M. tuberculosis H37Rv (reference strain) to gain further insight into the translational landscape of this pathogen and to provide comprehensive measurements of translation under exponential growth and following nutrient starvation, which is a model for persistence. Our data provide a rich resource for studying translation and its regulation in M. tuberculosis.
Results
The translational landscape of M. tuberculosis during exponential growth
To better understand genome-wide translation in M. tuberculosis, we adapted a previously published protocol for Ribo-seq (Latif et al., 2015) to be suited for a Biosafety Level 3 (BSL3) laboratory environment. The main adaptations employed were the harvesting of cultures by centrifugation in sealed buckets and the sterilization of cell lysates by double filtration through a 0.2-μm membrane (ribosome footprint samples). Verification of sterility of samples was key to ensuring they could be removed from the BSL3 laboratory for downstream processing. Cultures of M. tuberculosis H37Rv were grown to an OD600 of ~0.6, and triplicate samples were processed for parallel Ribo-seq and RNA-seq (see STAR Methods). Chloramphenicol was used to halt elongation for the generation of Ribo-seq libraries (Becker et al., 2013). The libraries were multiplex sequenced on an Illumina NextSeq 500 by Cambridge Genomic Services. Samples were depleted of ribosomal RNA using the ribo-zero kit (see STAR Methods), and remaining ribosomal RNA reads were filtered out of the Ribo-seq dataset in silico. On average, we sequenced 36 million fragments, with 30% of the RPF reads aligning to non-ribosomal RNA genome locations (Table S1A). As our experimental procedure did not include a size selection step, we only considered RPF reads from 15–40 nucleotides for further analysis. The distribution of RPF reads was reproducible across replicates, presenting a broad distribution across the length range, but with a detectable peak at 26 nucleotides (Figure 1A). This footprint length of 26 nucleotides sits between the predominant footprint length identified in E. coli (24 nucleotides) (Hücker et al., 2017; Li et al., 2014; Mohammad et al., 2019) and in eukaryotes (28–30 nucleotides) (Brown et al., 2015; Ingolia et al., 2011), being closer to the predominant footprint length of 27 nucleotides recently described in archaea (Gelsinger et al., 2020). However, footprint lengths determined in different experiments and organisms are strongly impacted by experimental parameters (Mohammad et al., 2019), so although this comparison is interesting, it should not be overinterpreted.
Figure 1.

The translational landscape of M. tuberculosis during exponential growth
(A) Fraction of reads from 15 to 40 nucleotides in M. tuberculosis Ribo-seq libraries during exponential growth (three biological replicates).
(B) Pairwise correlation coefficients calculated between each pair of replicates for total RNA and RPFs based on number of counts per gene. Line and bars represent the mean with range across the three replicates.
(C) Fraction of RNA and RPF reads aligning to various genomic features, coding sequences (CDSs), 5′ UTRs, non-coding RNAs (ncRNA), and intergenic regions (IGRs). Values are represented as percentages. Error bars indicate standard deviation across the three replicates.
(D) Correlation between transcriptome and translatome assessed from plotting average RPKMs across the three replicates for mRNA and RPFs.
(E) Distribution of gene categories among the 3,569 genes for which reliable rates of translation were quantified. Shine-Dalgarno (SD, blue), leaderless (L, orange), 5′ UTR not including a Shine-Dalgarno (UNSD, green), and not assigned (gray). The suffix “op” refers to genes assigned to operons.
(F) Plot of ribosome occupancy (RO) values distribution of Shine-Dalgarno (SD, blue), leaderless (L, orange), UNSD (green), and not assigned (NA) genes during exponential growth. Bars denote median RO values for each group. Asterisk denotes significant differences between gene categories (Kruskal-Wallis test, p < 0.0018).
(G) Plots of mRNA versus RO with Spearman coefficients and linear regression included for each gene category.
(H) Top: RO values distribution among functional categories (see key below). Bars indicate median RO values. Bottom: proportion of Shine-Dalgarno (blue), leaderless (orange), UNSD (green), or not assigned (gray) genes representing each functional category. Key to functional categories: a, degradation; b, energy metabolism; c, central intermediary metabolism; d, amino acid biosynthesis; e, purines, pyrimidines, nucleosides, and nucleotides; f, biosynthesis of cofactors, prosthetic groups, and carriers; g, lipid biosynthesis; h, polyketide and non-ribosomal peptide synthesis; i, broad regulatory functions; j, synthesis and modification of macromolecules; k, ribosomal proteins; l, degradation of macromolecules; m, cell envelope; n, cell processes; o, other; p, conserved hypotheticals; q, unknowns; r, toxin-antitoxin systems.
See also Figures S1 and S2 and Tables S1 and S2.
To assess the quality and reproducibility of our data, we calculated the pairwise Spearman correlation for each pair of biological replicates for both the RNA-seq and Ribo-seq libraries, which showed a high degree of reproducibility between replicates (Spearman’s r > 0.97; Figure 1B; Table S1A). Next, we calculated the fraction of reads corresponding to various genomic regions for both the RNA and RPF reads (see STAR Methods). As RPF reads reflect translating ribosomes, they are expected to have a higher fraction of the reads mapped within or proximal to coding sequences (CDSs) and 5′ UTRs, whereas RNA reads will have a broader distribution, with a higher fraction of reads mapped to non-coding regions of the genome. As expected, whereas only 37% of the RNA reads accounted for CDSs, 73% of the RPF reads mapped within CDSs with a further 16% mapping within 5′ UTRs (Figure 1C). Mapping of RPF reads within 5′ UTRs is expected to mainly reflect ribosomal engagement near the annotated start codon for translation initiation. To verify this, we further looked at the 79 expressed 5′ UTRs shared across the top 100 most expressed 5′ UTRs among the three biological replicates, which accounted for 67.3% of the total fraction of reads mapped to 5′ UTRs. In 61 out of the 79 cases (77.2%), mapped RPF reads to these regions could be attributed to ribosomal recruitment for translation initiation (Figure S1).
The overall expression rate for each gene during exponential growth was calculated as read density across each gene in reads per kilobase per million (RPKM). Plotting of gene expression rates for RPF and RNA across replicates revealed a high correlation between measures of transcription and translation, consistent with the coupling of these processes in bacteria (Spearman’s r = 0.91; Figure 1D). We were able to reliably quantify translation rates for 3,569 genes, representing 88.5% of the annotated translatome (reliable translation is defined here as genes with 128 or more total read counts among the three replicates; see STAR Methods; Table S1C; Figures S2A and S2B). Based on the previous categorization of M. tuberculosis transcripts on the basis of TSS mapping (Cortes et al., 2013), we divided the translatome genes into those having a 5′ UTR region and those lacking a 5 ′UTR, designated as leaderless transcripts. The set of genes with a 5′ UTR were further categorized into “Shine-Dalgarno” if a Shine-Dalgarno sequence was previously predicted within the 5′ UTR region or “UNSD” when a Shine-Dalgarno sequence was not predicted within the UTR (classification of UTR categories was performed using the method described in Cortes et al. [2013]). Furthermore, the operon categorization for genes belonging to UNSD, Shine-Dalgarno, and leaderless operons was also used. The remaining genes for which a reliable translation rate was quantified during exponential growth but no previous categorization was available were classified as “not assigned” (1,200 out of 3,569 genes; Figure 1E). The classification “not assigned” is based on detection of a TSS for the gene. In cases where a TSS cannot be assigned, it becomes impossible to determine the length or content of any UTR since the mRNA upstream of the start codon is not defined (see Methods in Cortes et al. 2013). For all downstream analysis, genes assigned to operons, which represent 18.2% of the reliably quantified genes (Figure 1E), were excluded. We were able to quantify similar reliable rates of translation for leaderless mRNAs (497 out of 3,569; 14%), Shine-Dalgarno mRNAs (546 out of 3,569; 15%), and UNSD mRNAs (678 out of 3,569; 19%; Figure 1E), thereby corroborating that M. tuberculosis is able to efficiently initiate global translation of leaderless transcripts and UNSD transcripts without the requirement of a Shine-Dalgarno sequence during exponential growth (Shell et al., 2015; Nguyen et al., 2020).
We next assessed genome-wide rates of mRNA translation by calculating ribosome occupancies (ROs). These were calculated by dividing the RPKM of the translatome by the RPKM of the transcriptome (Table S1B). Ribo-seq does not distinguish actively translating ribosomes from those stalled or engaged in non-productive initiation; therefore, RO does not correspond precisely to active translation. However, with this caveat in mind, it is reasonable to compare ROs between gene classes. Comparison of RO values for the different transcript categories revealed significantly higher median ROs for Shine-Dalgarno genes (Kruskal-Wallis test, p < 0.0018; Figure 1F), consistent with previous observations of higher levels of expression for genes in this UTR category (Ma et al., 2002). Interestingly, UNSD transcripts showed significantly lower median ROs than did Shine-Dalgarno and leaderless genes (Kruskal-Wallis test, p < 0.0001; Figure 1F), suggesting that not having a 5′ UTR might be better than having one, but without a Shine-Dalgarno sequence. As RO values could be affected by mRNA stability, we used available data on mRNA half-lives in M. tuberculosis (Rustad et al., 2013) to look at the correlation between RO values and mRNA stability among the set of reliable translated genes. We found that Shine-Dalgarno genes had a significantly lower median half-life than did the other transcript categories (Figure S2C), but the correlation between RO and mRNA stability in the four gene categories was poor (Spearman’s r ranging from −0.2 to −0.04) (Figure S2D), suggesting that the overall effect of mRNA stability on the estimation of RO in our dataset is negligible.
Comparing gene RO values against mRNA gene expression rates should provide information on the regulation of gene expression at the level of translation and reveal potential differences in ROs among gene categories. Plotting of mRNA to RO ratios for the different gene categories provided no evidence for an overall difference in the translation of the different gene categories (Spearman’s r < 0.20) (Figure 1G). Overall, our data suggest that in contrast to E. coli, where the efficiency of leaderless translation is strongly dependent on the availability of initiation factors (Moll and Bläsi, 2002), M. tuberculosis can efficiently initiate and robustly translate non-canonical transcripts using the translational machinery available during in vitro growth conditions.
Next, we asked if there is an association of RO with different functional categories. For this, we analyzed the RO values of genes from various functional categories and found that overall, RO values are widely spread among functional categories (Figure 1H). One exception are genes involved in non-ribosomal peptide synthesis (h) that have significantly higher median RO values (Kruskal-Wallis test, p < 0.007; Table S2A). When we plotted the proportions of genes in each category that have a Shine-Dalgarno sequence (blue), are leaderless (orange), or are UNSD (green; Figure 1H), it is apparent that those functional categories with higher median RO values, like ribosomal protein synthesis (k) and non-ribosomal peptide synthesis (h), tend to have higher proportions of Shine-Dalgarno genes and are associated with higher RO than categories with more leaderless or UNSD genes, like toxin-antitoxin systems (r).
In summary, we have sequenced total RNA and ribosome footprints from exponentially growing cultures of M. tuberculosis H37Rv. Analysis of the sequencing data indicates that our RPF data likely reflect genuine translation events in the mycobacterial cell, with the majority of RPF reads mapping to CDSs and a small proportion also mapping to 5′ UTRs. Interestingly, we have been able to detect global genome-wide translation of non-canonical genes and found an association of RO values with both gene and functional categories during exponential growth.
Metagene analysis reveals differences in ribosome densities at the start codon between gene categories
As leaderless and UNSD transcripts lack the ribosomal recognition sequences for translation initiation, we hypothesized that maps of ribosome densities for genes aligned at their start codon will differ among the different transcript categories. In order to test this hypothesis, we used ribosome densities assigned to the 3′ end of reads to perform metagene analysis and generate maps of ribosome densities. For this analysis, we only considered genes longer than 300 nt and that had more than one RPF read per codon. The 3′ assignment of reads allowed us to identify a consensus distance of 14 nt from the 3′ end of mapped reads and the start codon in our dataset (see STAR Methods), which provides an estimate of the distance from the P-site codon to the 3′ boundary of the mycobacterial ribosome. Visualization of the metagene profiles obtained for leaderless, Shine-Dalgarno, and UNSD genes revealed striking differences in the ribosome densities around the start codons (Figure 2A). The metagene analysis of Shine-Dalgarno genes shows a predominant peak at the start codon, indicating a high ribosome density in that region, whereas the ribosome density at the 5′ end of leaderless genes increases gradually with distance from the start codon. In the case of UNSD genes, there is no predominant peak at the start codon, either. We propose that the higher density of ribosomes near the start codon of Shine-Dalgarno genes is due to a slower initiation associated with binding of the anti-Shine-Dalgarno and Shine-Dalgarno sequences during positioning of the start codon in the P-site, in contrast to the 70S ribosome binding to leaderless transcripts and initiation proceeding more rapidly to elongation.
Figure 2.

Metagene analysis of 3′ assigned genes aligned at the start codon
(A) Average ribosome density on genes aligned at the start codon during exponential growth (three biological replicates). Metagene maps for all genes are shown in black, Shine-Dalgarno genes in blue, UNSD genes in green, and leaderless genes in orange. For all maps, the number of genes included in the analysis is indicated.
(B) Distribution of RPF read lengths of initiating ribosomes for each gene category: Shine-Dalgarno, blue; UNSD, green; leaderless, orange.
(C) Influence of the degree of complementarity to the anti-Shine-Dalgarno on RO. Boxplots indicate median (horizontal line) and interquartile range (box), and whiskers and outliers are plotted following the Tukey method.
(D) Influence of the presence of the GGAGG core and degree of complementarity of the anti-Shine-Dalgarno sequence on RO. Boxplots indicate median (horizontal line) and interquartile range (box), and whiskers and outliers are plotted following the Tukey method.
See also Figures S3 and S4 and Table S2.
To further evaluate the contribution of different RPF read lengths to the ribosome densities observed in the three gene categories around the start codon, we performed metagene analysis on each set of read lengths and gene categories. We applied the appropriate offset between the initiation peak and the start codon (14 nt) and used the positions 0, 1, and 2 for the start codon to sum the ribosome densities for these three positions for each read length. The values were then normalized by the total ribosome density for each dataset, and the fraction of ribosome densities at the start codon was plotted for each RPF read length (Figure 2B; shown overlaid in Figure S3A). The resulting graphs show different predominant read lengths for ribosomes initiating translation of the three different gene categories. UNSD genes had the longest footprint lengths, around 34–35 nt; Shine-Dalgarno genes had a broader range of read lengths between 30 and 34 nt; and leaderless genes were associated with short read lengths, 17–18 nt. The short read lengths of leaderless genes occur because there is no RNA upstream of the start codon, and at initiation, the start codon is in the ribosomal P-site, which is ~14 nucleotides from the ribosome boundary. A similar analysis was performed for elongating ribosomes, and the results showed a mixture of all read lengths for each type of gene, with UNSD genes having the most even distribution of read lengths and Shine-Dalgarno and leaderless genes showing some bias toward shorter (<30 nt) read lengths (Figures S3B and S3C).
As Shine-Dalgarno genes are associated with higher median RO values and show a clear initiation peak in the metagene analysis, we next investigated the effect of the strength of the Shine-Dalgarno sequence on RO. Locations of Shine-Dalgarno sequences were found by searching for the canonical M. tuberculosis Shine-Dalgarno sequence (AGAAAGGAGG; complementary to the anti-Shine-Dalgarno sequence in the 16S ribosomal RNA of M. tuberculosis; Kempsell et al., 1992) in the 30 nucleotides upstream of the start codon for the set of reliably quantified Shine-Dalgarno and UNSD genes. The distance of the Shine-Dalgarno sequence from the start codon and the number of matches between the gene-specific and canonical Shine-Dalgarno sequence were extracted. As we have identified that UNSD genes have significantly reduced median RO values (Figure 1F), we also considered in our analysis instances with fewer than four matches, which should represent UNSD genes. These metrics were used to assess the relationship between RO and Shine-Dalgarno sequence strength. Indeed, Shine-Dalgarno sequences that are closer matches to the 16S rRNA anti-Shine-Dalgarno have higher ROs than those that are less well matched (linear regression RO versus number of matches; Figure 2C). The conservation of a core GGAGG motif within the Shine-Dalgarno sequence had no effect on RO (Figure 2D).
Translation efficiency (the ratio of RPF reads to RNA-seq reads) has been shown to be determined by both codon bias and mRNA folding energy (Tuller et al., 2010). To rule out the possibility that our RO results were unduly influenced by codon bias, we analyzed the codon composition of the 20 codons downstream of the start codon and compared leaderless, Shine-Dalgarno, and UNSD sequences. In general, there were few significant differences among the types of transcript (Table S2B), although it was interesting to note that it was sometimes the most abundant codons genome-wide that were significantly enriched among the types of transcript. For example, the codons CUG and CCG are the most used leucine and arginine codons in mycobacteria (Andersson and Sharp, 1996), and both were significantly over-represented (Fisher’s exact test p < 0.004; two-sided) in leaderless compared to Shine-Dalgarno and UNSD transcripts in our dataset (Table S2B).
Impact of nutrient starvation on mechanisms of translation
The concept of preferential translation of particular mRNA subsets by “specialized ribosomes” has been extensively explored in eukaryotic cells, and recent evidence suggests that it may also provide a central element in bacterial responses to stress (Berghoff et al., 2013; Picard et al., 2013; Vesper et al., 2011). We have previously characterized the transcriptional response of M. tuberculosis to nutrient starvation, showing that transcription of leaderless genes increases under conditions of nutrient starvation (Cortes et al., 2013). To further quantify the effect of translation of different classes of mRNA transcript on the stress translatome, we performed parallel RNA-seq and Ribo-seq on triplicate M. tuberculosis cultures that had been nutrient starved for 24 h (see STAR Methods; Tables S1D and S1E). We obtained a range of RPF read lengths from 15 to 40 nucleotides, but with a well-defined peak at 26 nucleotides (Figure 3A). We performed metagene analysis on the RPFs from nutrient-starved cultures of M. tuberculosis as described above and found that, as for exponentially growing cultures, the three classes of transcript showed distinctive patterns of ribosome densities around the start codon: Shine-Dalgarno and UNSD genes both have footprints upstream of the start codon, whereas leaderless genes show a small peak at the start codon followed by increased ribosome densities along the length of the gene (Figure 3B).
Figure 3.

Mechanisms of translation in starvation
(A) Fraction of reads from 15 to 40 nucleotides in M. tuberculosis Ribo-seq libraries during nutrient starvation (three biological replicates).
(B) Average ribosome density on genes aligned at the start codon during nutrient starvation. Metagene maps for all genes are shown in black, Shine-Dalgarno genes in blue, UNSD genes in green, and leaderless genes in orange. For all maps, the number of genes included in the analysis is indicated.
(C) Fraction of mapped reads around the start codon for Shine-Dalgarno, UNSD, and leaderless genes during exponential growth (blue) and nutrient starvation (purple).
(D) Distribution of RPF read lengths of initiating ribosomes for each gene category: Shine-Dalgarno, blue; UNSD, green; leaderless, orange.
See also Figures S3 and S4.
To better compare the differences in ribosome densities around the start codon for the different gene categories between exponential and nutrient starvation, we plotted the fraction of mapped reads corresponding to each nucleotide position from –20 to 20 nucleotides around the start codon (Figure 3C). This analysis revealed differences in the initiation patterns for the different gene categories upon starvation. For example, Shine-Dalgarno genes have an identifiable peak at the start codon in both datasets, but the initiation peak in the starvation dataset is less pronounced. Contrarily, the initiation peak for leaderless genes is more pronounced upon starvation, whereas the patterns for UNSD genes do not change considerably. The predominant read lengths for ribosomes initiating translation of the three different gene categories during nutrient starvation were similar to those described during exponential growth (Figure 3D; overlaid shown in Figure S3A).
Translational gene regulation during nutrient starvation
Having identified differences in the initiation profiles of Shine-Dalgarno transcripts versus non-canonical transcripts and how these vary upon stress, we next assessed the global translational response of M. tuberculosis to nutrient starvation.
RNA-seq analysis of nutrient-starved samples confirmed previous transcriptomic studies with mRNAs for 560 genes increasing >2-fold and 589 genes decreasing >2-fold (adjusted p < 0.01; Table S3), with downregulated genes including genes coding for ribosomal proteins and genes involved in energy metabolism (Betts et al., 2002; Dahl et al., 2003; Cortes et al., 2013). The global response at the level of the translatome was similar, with RPFs for 679 genes increasing >2-fold and 711 genes decreasing >2-fold, but the overlap between transcription and translation was moderate. In total, 414 genes were both significantly upregulated at the level of mRNA and RPFs, and 461 genes were significantly downregulated (Figure 4A), highlighting a potential scope for translational regulation. Of the mRNA transcripts that were more than 10-fold upregulated in our previous study (Cortes et al., 2013), 68% were also significantly upregulated at the level of the translatome in this dataset (Table S3).
Figure 4.

Differential expression in response to starvation
(A) Venn diagrams showing the up- and downregulation of gene expression at the level of RNA and RPFs in response to starvation (three biological replicates).
(B) Distribution of RO values across UTR categories in starvation. Horizontal bars represent median values. L, leaderless; SD, Shine-Dalgarno; UNSD, 5′ UTR no-Shine-Dalgarno; NA, not assigned (Kruskal-Wallis test, p < 0.001).
(C) Volcano plot and contingency plots showing the proportion of genes in each UTR category that were up- or downregulated in starvation. Orange, leaderless; blue, Shine-Dalgarno, green, UNSD; gray, not assigned. The bar above the plot indicates the representation of each functional category relative to its representation in the genome
(D) Scatterplot showing the changes in the transcriptome (RNA) and translatome (RPF) during starvation. Dark gray, co-regulated genes; yellow, genes regulated at the level of the translatome only; pink, genes regulated at the level of the transcriptome only; purple, genes changing in opposite directions.
(E) Changes in the expression of known and putative ribosome-associated factors in the translatome.
We were able to quantify reliable rates of translation for 3,032 genes, of which 477 (16%) were leaderless, 516 (17%) were Shine-Dalgarno genes, and 572 (19%) were UNSD, showing similar percentages of translation among gene categories to those reported during exponential growth. To check if different transcript categories are translated with differing ROs upon stress, we calculated RO values as previously described and compared their median RO values (Figure 4B). In the starved translatome, the median RO significantly decreased for all gene categories compared to exponential growth (Mann-Whitney test, p < 0.0001), but as for exponential growth, median RO values for Shine-Dalgarno genes remained significantly higher than for all other gene categories (Figure 4B) (Kruskal-Wallis test, p < 0.001). Next, to further evaluate if there was a global upregulation of any gene category in the starved translatome, we plotted the differentially regulated genes in the translatome in a volcano plot and overlaid the gene categories (Figure 4C). There was a statistically significant global upregulation of all types of transcript (leaderless, Shine-Dalgarno, and UNSD; Fisher’s exact test, p < 0.002) relative to those genes not assigned to a gene category, but the greatest effect was observed for leaderless genes (p < 0.0001), which represented 17% of the upregulated genes expressed during nutrient starvation, compared with just 7% of those downregulated (Figure 4C).
To further identify translationally regulated genes in nutrient starvation, we plotted the genes that were reliably expressed during starvation according to changes in mRNA and RPF levels (Figure 4D; significance criteria were an absolute log2 fold change > 1 and p-adjusted < 0.01; all genes with changes in RNA or RPF levels are shown, but only those meeting the thresholds for significant changes are colored) and looked at the representation of particular functional and UTR categories within each cluster of genes. There were only two genes that were dysregulated upon starvation: one gene was downregulated at the level of RNA but upregulated in the translatome (Rv2032), and one gene was upregulated at the level of RNA but downregulated in the translatome (Rv3182). We next performed an enrichment analysis of Gene Ontology (GO) terms for the set of genes that were significantly up- and downregulated at the level of the translatome only (see STAR Methods). Among the 264 genes that were significantly upregulated at the level of the translatome only, there was a significant overrepresentation of GO terms associated with monooxygenase and oxidoreductase activity, as well as with regulatory region DNA binding.
As potential mechanisms underlying differential translation of Shine-Dalgarno versus genes with other UTR types (including leaderless genes) could include quantitative differences in the abundance of components of the translational machinery, we looked for preferential translation of ribosomal proteins, ribosome-associated factors, as well as translation factors during starvation (Table S3). Analysis of the differential expression of ribosome-associated factors during starvation highlighted the significant upregulation of three genes: Rv1738, RafS, and Rv2632c (DESeq2; Love et al., 2014; Figure 4E). The function of the product of gene Rv1738 has not been biochemically determined, but structural studies suggest it may function as a ribosome hibernation-promoting factor (HPF) as it bears significant structural homology to the HPF proteins from E. coli and Vibrio cholerae, including docking of the Rv1738 protein dimer into the groove where HPFs bind the 70S ribosome (Bunker et al., 2015). The upregulation of Rv1738 in starvation is consistent with its potential role as an HPF and with a role in tolerance of early nutrient starvation. Rv2632c bears some homology to Rv1738 but lacks the ring of positively charged residues likely to be important for ribosomal RNA binding (Bunker et al., 2015). RafS (Rv3241c) was also significantly upregulated in starvation. RafS is homologous to MSMEG_3935, which contains an S30AE domain and a ribosome-binding domain and has been shown to bind mycobacterial ribosomes under conditions of hypoxic stress (Trauner et al., 2012). Biochemical characterization of RafS, including structural studies of this protein bound to mycobacterial ribosomes, indicates a mycobacterial-specific interaction that seems to prevent subunit dissociation and degradation during hibernation without the formation of 100S dimer (Mishra et al., 2018).
Discussion
In this manuscript, we present Ribo-seq data from M. tuberculosis H37Rv under conditions of exponential growth and nutrient starvation in vitro. Our data, which were collected in triplicate, show a high level of reproducibility and appear to represent genuine translation events. There is good agreement with the previously published transcriptomic response of M. tuberculosis to nutrient starvation: of the most highly upregulated mRNA transcripts in nutrient starvation (10-fold or more upregulation) previously reported (Cortes et al., 2013), 68% were also upregulated at the level of the translatome. Our data shed light on multiple aspects of translation in mycobacteria, from the role of the Shine-Dalgarno sequence in influencing RO to mechanisms of initiation.
Our data also provide biochemical evidence for a ~14-nt spacing between the P-site and the 3′ boundary of the mycobacterial ribosome, which is in agreement with estimates obtained in the attenuated M. tuberculosis mc27000 strain (Smith et al., 2019) and the distance found in E. coli (Woolstenhulme et al., 2015). This distance was obtained by 3′ assignment of RPF reads (Woolstenhulme et al., 2015), which has been shown to be the best method for determining the position of the ribosome on the mRNA. The need for 3′ assignment arises because unlike Ribo-seq libraries from Saccharomyces cerevisiase, which show strong three-nucleotide periodicity as the ribosome translocates codon by codon along the mRNA, bacterial Ribo-seq libraries show a broad distribution in RPF read lengths. However, when reads are aligned by the 3′ boundary, the start codon peak is reduced to 1–2 nt wide, and the position of the ribosome can be accurately inferred (Balakrishnan et al., 2014; Nakahigashi et al., 2014; Woolstenhulme et al., 2015). We therefore used 3′ assignment prior to metagene analysis of our data to obtain the position of the ribosome.
In our study, we used chloramphenicol to arrest translation. Chloramphenicol arrests elongation but has little effect on initiation, potentially leading to an accumulation of ribosome density at the 5′ end of CDSs (Mohammad et al., 2019). When we performed asymmetry analysis to assess the extent of this bias, we did indeed find an enrichment of the 5′ ends of genes (Figure S4A). However, when we repeated the DESeq2 analysis of differentially translated genes during nutrient starvation to account for reads across the first 50 codons, we found little difference in the results, suggesting that in this case, any bias toward the 5′ ends of genes has little influence on our analysis. Also, as chloramphenicol affects translation in a context-specific manner—preferentially arresting translation at alanine, serine, and threonine residues (Choi et al., 2020; Marks et al., 2016)—it could have potentially impacted our metagene analysis if these codons are represented differently across gene categories. After comparing the occurrence of these three codons across the first 20 amino acids for each gene category, we could not observe any significant bias, likely indicating the context-specific arrest of chloramphenicol has no influence on the interpretation of our metagene analysis (Figures S4B and S4C).
The role of translational reprogramming as a mechanism for shaping the proteome during stress to guarantee bacterial survival has been widely described in bacteria (reviewed in Starosta et al., 2014). In the context of M. tuberculosis, the robust translation of leaderless mRNA transcripts during exponential growth and their upregulation under stress conditions is likely to play a role in cell survival. Our data point toward mycobacterial ribosomes being somewhat promiscuous in terms of the types of mRNA transcript they are able to translate, with both Shine-Dalgarno and non-canonical transcripts being efficiently translated. Metagene analysis revealed differences in the initiation mechanisms of translation of these types of transcripts, with Shine-Dalgarno transcripts showing ribosomal pausing at the start codon—presumably representing assembly of the pre-initiation complex and Shine-Dalgarno to anti-Shine-Dalgarno binding to stabilize the complex. However, initiation of non-Shine-Dalgarno transcripts appears to lack this pause, consistent with a model in which pre-assembled 70S monosomes directly bind the mRNA and quickly become elongation competent. One limitation of our analysis is that by selecting only reads of 15 nucleotides or longer, we are able to capture only leaderless initiation complexes where the start codon is already in the P-site (14 nucleotides from the 3′ boundary of the ribosome), not any smaller intermediate complexes that may also be relevant for leaderless initiation. Thus, we may have selected against observing pauses near the start codon for leaderless transcripts. Finally, the changes observed in the initiation peaks for Shine-Dalgarno genes in starvation compared to exponential growth suggest that 30S initiation plays a less important role in the overall translational landscape of M. tuberculosis during starvation.
It is interesting that the putative ribosome stabilization factors RafS, Rv2632c, and Rv1738 were significantly upregulated during nutrient starvation. This observation is consistent with previous studies that have proposed roles for these proteins in 70S ribosome stabilization and tolerance of early nutrient starvation and hypoxic stress (Bunker et al., 2015; Trauner et al., 2012). The stabilization of 70S monosomes is relevant to the increase in leaderless translation under conditions of nutrient starvation because the assembled 70S ribosome has been proposed as the species that initiates leaderless translation. This is further supported by our metagene analysis, which indicates a smoother and more rapid initiation for leaderless translation compared to Shine-Dalgarno translation, which exhibits a characteristic pause at the start codon that we attribute to assembly of the pre-initiation complex involving binding of the Shine-Dalgarno-anti-Shine-Dalgarno sequences, 50S subunit joining, and GTP hydrolysis to render the ribosome elongation competent. Similar mechanisms for initiation seem to occur in both exponentially growing and nutrient-starved cultures; however, the proportion of leaderless-translated mRNA increased during nutrient starvation. This observation suggests that there are not distinct mechanisms of translation occurring during different growth conditions, but rather factors that stabilize intact ribosomes may play a role in ensuring continued robust translation during conditions of stress.
Both translation and decay affect expression and possibly also measures of RO, with naked mRNAs being more susceptible to decay than ribosome-protected molecules, potentially skewing RO values toward genes with less stable mRNAs appearing to have higher RO values than those with very stable mRNAs. As shown in Figure S2D, our analysis suggests that any such effect contributes only negligibly toward RO. The influence of the strength of the Shine-Dalgarno sequence on RO is a complex issue. Previous studies have reported little or no correlation between RO and the strength of the Shine-Dalgarno motif (Schrader et al., 2014; Li et al., 2014; Li, 2015; Del Campo et al., 2015) when looking at genome-wide effects. However, at the level of individual genes, mutation of the Shine-Dalgarno sequence to decrease binding of the mRNA to the ribosome has been shown to reduce protein expression (Woong Park et al., 2011), and mRNA transcripts with strong Shine-Dalgarno sequences and mutated start codons were shown to be protected from degradation in some cases (e.g., Bechhofer and Dubnau, 1987). In our study, closer complementarity to the anti-Shine-Dalgarno sequence (Kempsell et al., 1992) resulted in higher RO. How can these results be reconciled? A key study by Buskirk and co-workers dissected the influence of A-rich sequences in 5′ UTRs from the impact of Shine-Dalgarno strength (Saito et al., 2020). We found that UNSD genes have significantly reduced median levels of RO from those of Shine-Dalgarno genes and, in agreement with the results from Saito et al. (2020), could potentially reflect the importance that other components like the local RNA structure, RNA folding kinetics, and presence of A- or G-rich stretches around the start codon can play in translation initiation.
The seven genes with very high Watson-Crick complementarity to the anti-Shine-Dalgarno (Kempsell et al., 1992) had statistically significant higher RO than genes with fewer matches. These genes include a probable transcriptional regulatory protein (Rv2324), a phosphate starvation-inducible protein (Rv2368c), the 6-kDa early secretory antigenic target EsxA (Rv3875), a probable dehydrogenase (Rv2280), and three conserved hypothetical or unknown proteins (Rv0060, Rv1813c, and Rv0272c). As RO is a measure of RPF/mRNA and cannot account for protein degradation, the proteins with high ROs are not necessarily those most abundant in the cell. However, in terms of the mechanism of their translation, it is interesting to ask what the transcripts encoding these proteins have in common, and our data indicate that high complementarity of the Shine-Dalgarno sequence is one such feature.
To date, the development of novel therapeutics against M. tuberculosis has been slow, hindered in part by the difficulties of studying a slow-growing category 3 pathogen and by the challenges of genetic manipulation of this organism. The data obtained from our experiments provide a rich resource for studying translation and its regulation in M. tuberculosis that will be useful to other researchers.
STAR★methods
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Bacterial and virus strains | ||
| Mycobacterium tuberculosis: strain H37Rv STB | Laboratory stock | N/A |
| Chemicals, peptides, and recombinant proteins | ||
| Middlebrook 7H9 broth | BD Diagnostics | 271310 |
| Middlebrook ADC supplement | BD Diagnostics | 212352 |
| Tyloxapol | Sigma | T0307-10G |
| Chloramphenicol | Sigma | C0378-5G |
| Microccocal nuclease | NEB | M0247S |
| DNaseI | Thermo Fisher | 10849700 |
| Qiazol | QIAGEN | 79306 |
| HEPES | Sigma | H3375-100G |
| KCl | Sigma | P9333-500G |
| Triton X-100 | YorLab | X100-5ML |
| MgCl2 | Sigma | M8266-100G |
| CaCl2 | SLS | C1016-100G-D |
| Heparin | Fisher Scientific | 10239840 |
| Superase.in | Thermo Fisher | AM2694 |
| EGTA | Sigma | E3889-10G |
| Sucrose | Sigma | S9378-500G |
| RNase-free water | Ambion | AM9932 |
| Acid-phenol chloroform | Sigma | P3803-100ML |
| Isopropanol | Sigma | I9516-500ML |
| T4 PNK | NEB | M0201S |
| Sodium acetate 3M | Sigma | S7899-100ML |
| Chloroform | YorLab | 372978-100ML |
| Ethanol | Sigma | E7023-500ML-D |
| Critical commercial assays | ||
| miRNeasy mini kit | QIAGEN | 217004 |
| RiboZero rRNA removal kit for bacteria | Illumina | MRZMB126 |
| Fast RNApro blue kit | MPBio | 6025-050 |
| RNeasy mini kit | QIAGEN | 74104 |
| RNA fragmentation reagents | Ambion | AM8740 |
| NEBNext small RNA library prep set for Illumina | NEB | E7300S |
| Deposited data | ||
| Riboseq and RNaseq raw data | This paper | E-MTAB-8835 |
| Software and algorithms | ||
| Cutadapt | Martin, 2011 | https://cutadapt.readthedocs.io/en/stable/ |
| Bowtie | Langmead et al., 2009 | http://bowtie-bio.sourceforge.net/index.shtml |
| Trimmomatic | Bolger et al., 2014 | http://www.usadellab.org/cms/?page=trimmomatic |
| Bedtools | Quinlan and Hall, 2010 | https://bedtools.readthedocs.io/en/latest/ |
| DESeq2 | Love et al., 2014 | http://bioconductor.org/packages/release/bioc/html/DESeq2.html |
| BiNGO application software for Cytoscape | Maere et al., 2005 | http://apps.cytoscape.org/apps/bingo |
| GraphPad Prism version 8 for Mac | GraphPad Software | https://www.graphpad.com/ |
| Other | ||
| Lysing Matrix B | MPBio | 6911-050 |
| RNeasy MinElute Columns | QIAGEN | 74204 |
| Thinwall Ultra-Clear 14 ml ultracentrifuge tubes | Beckman | 344060 |
Resource availability
Lead contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Teresa Cortes (Teresa.cortes@lshtm.ac.uk).
Materials availability
This study did not generate new unique reagents.
Data and code availability
The accession number for the RNA-seq and Ribosome profiling data reported in this paper is ArrayExpress: E-MTAB-8335. https://www.ebi.ac.uk/arrayexpress
The code supporting the current study have not been deposited in a public repository but are available from the corresponding author on request.
Experimental model and subject details
Culture conditions
All materials and reagents were of analytical grade and obtained from Sigma-Aldrich (Merck, Darmstadt, Germany) unless otherwise specified. All buffers were prepared using water purified to a resistance of 18.2 MΩ cm-1 and filtered through a 0.2 μm membrane (Millipore, Billerica, MA, USA) before use.
M. tuberculosis H37Rv was grown in Middlebrook 7H9 medium (BD Diagnostics) supplemented with 10% Middlebrook albumin-dextrose-catalase (ADC) (BD Diagnostics), 0.2% glycerol and 0.05% Tween 80 at 37°C until OD600 was 0.4–0.6. For starvation experiments, exponentially growing bacteria were pelleted, washed twice in PBS and resuspended in PBS supplemented with 0.025% Tyloxapol, and maintained in culture for a further 24 h. Three independent cultures of exponentially growing and nutrient starved cells were prepared for the three biological replicates needed for parallel RNA and Ribosome profiling experiments. For ribosome profiling experiments, chloramphenicol was added to a final concentration of 100 μg/ml 2 min prior to harvesting cells. All bacteria were harvested by centrifugation at 4000 rpm and 4°C in a JS 4.0 swinging bucket rotor (Beckman Coulter, Brea California) and immediately processed as described below.
Method details
Ribosome profiling
Sample preparation for ribosome profiling was carried out according to a modified form of the method of Latif et al. (2015). All steps were performed on ice or using chilled buffers. M. tuberculosis cell pellets were resuspended in lysis buffer (20 mM HEPES, pH 7.4; 200 mM KCl, 1% Triton X-100, 12 mM MgCl2, 1 mM CaCl2, 1 mg/ml heparin, 100 μg/ml chloramphenicol and 5 U/ml DNase I) and transferred to ribolysing matrix tubes (Lysing Matrix B (cat no 6911-050) MPBio). Cell lysis was performed by ribolysing the sample three times for 30 s at power 6.5 (Fast Prep Hybaid FP120HY-230, MPBio). Ribolysing matrix and cell debris were removed by centrifugation at 12,000 rpm (JA-15 1.5 rotor, Beckman Coulter) for 10 min.
The ribosome profiling samples were then double filtered through a 0.2 mm membrane (Corning, New York) to ensure sterility before incubation for 45 min with 24 mL microccocal nuclease (NEB) and 15 mL DNaseI (Thermo Fisher, Waltham, Massachusetts) to digest mRNAs that are not bound by ribosomes. Nuclease digestion was stopped by addition of 10 mL Superase In (Thermo Fisher) and 5 mL 0.5 M EGTA and cell lysates loaded onto a 34% sucrose cushion. Ribosomes were harvested by centrifugation at 40,000 rpm for 15 h in an SW40Ti rotor (Beckman Coulter). To isolate the ribosome-bound mRNAs or ribosome footprints (RPF), the ribosomal pellet was then resuspended in Qiazol (QIAGEN, Hilden, Germany) and small RNA isolated using the miRNeasy mini kit (QIAGEN). Isolated RNA was precipitated and resuspended in 10 mM Tris, pH 8.0. Finally, RPF samples were depleted of ribosomal RNA using the RiboZero rRNA removal kit for bacteria (Illumina) and purified using RNeasy MinElute columns (QIAGEN). The quantity and quality of the RNA was assessed at each step using the bioanalyser and qubit.
RNA isolation
M. tuberculosis cell pellets were resuspended in RNApro solution (MPBio, Irvine, California) and ribolysed three times for 30 s at power 6.5 (FastPrep Hybaid FP120HY-230, MPBio). Ribolysing matrix and cell debris were removed by centrifugation at 12,000 rpm (JA-15 1.5 rotor, Beckman Coulter) 4°C for 10 min. The total RNA sample was then removed from the ribolysing tube and chloroform added. The sample was vortexed for 10 s and incubated at room temperature for 5 min before centrifugation at 12,000 rpm (JA-15 1.5 rotor, Beckman Coulter) 4°C for 5 min. The aqueous phase was then transferred to a new tube and 1.5 volumes of 100% ethanol added. Samples were incubated at −20°C overnight and spun to pellet the RNA. The pellet was washed with 75% ethanol, air-dried and resuspended in nuclease-free water. A further acid-phenol chloroform extraction was performed to improve purity of the RNA before resuspending the final pellet in nuclease-free water. Genomic DNA contamination was assessed by PCR for 16S genomic DNA and, if present, samples were treated with DNase I followed by a further acid-phenol chloroform extraction.
Next, samples were depleted of ribosomal RNA using the RiboZero rRNA removal kit for bacteria (Illumina) and purified using RNeasy MinElute columns (QIAGEN). Finally, total RNA samples were then subjected to fragmentation using RNA fragmentation reagents (Thermo Fisher) at 80°C for 20 min. The quantity and quality of the samples were assessed at each step using the bioanalyser and qubit.
Library construction and sequencing
RPF and total RNA samples were converted to cDNA libraries as previously described (Latif et al., 2015). Briefly, samples were treated with T4 PNK (NEB) at 37°C for 30 mins and purified using RNeasy MinElute columns (QIAGEN). The NEBnext small RNA library prep kit (NEB) was used according to manufacturer’s guidelines to construct the multiplexed cDNA libraries. Multiplexed sequencing was performed by Cambridge Genomic Services using a NextSeq 500 (Illumina, San Diego, California) with single end reads of 75 bp.
Genome mapping
For the RPF reads, adaptors were removed from the 3′end of sequencing reads using Cutadapt (Martin, 2011) and only trimmed sequences longer than 15nt were kept for further analysis. Bowtie (Langmead et al., 2009) with default parameters was then used to align the trimmed sequences against a reference database containing M. tuberculosis ribosomal RNAs (rRNAs) and stable RNAs sequences downloaded from the Mycobrowser website (Kapopoulou et al., 2011). All aligned reads to rRNA were discarded, and the pool of rRNA-depleted RPF reads were finally aligned to the M. tuberculosis H37Rv reference genome (AL123456) using Bowtie and allowing for up to two mismatches in a seed length of 28 nt. Reads not aligning to a unique location and reads aligned longer than 40nt were discarded
For the total RNA sequencing reads, Trimmomatic software (Bolger et al., 2014) was used to remove bases from the start and the end of the reads when its quality was below 20, and trimmed reads were mapped against the M. tuberculosis H37Rv reference genome as described above.
Read densities analyses
To gain more insight into ribosome position, a custom phyton script was used to assign ribosome occupancy to the 3′end of RPF reads as previously described (Balakrishnan et al., 2014; Nakahigashi et al., 2014; Woolstenhulme et al., 2015). This method yields higher resolution into ribosome positioning than the previous center-weighting method commonly applied to ribosome profiling studies in bacteria (Becker et al., 2013; Oh et al., 2011). After 3′ assignment, a custom phyton script was used to perform metagene analysis, which gives information on the distribution of ribosomes along an average message by averaging the read densities across all genes (Becker et al., 2013). Only genes greater than 300 nt and which are well-expressed (having more than 100 reads mapped and a minimum number of 1 read per codon along the gene region) were considered for analysis. This was used to generate plots of average ribosome occupancy for genes aligned at their start and stop codons.
In E. coli, 3′assignment defines a strong peak of 1-2 nt width located 15 nt downstream of the first nucleotide of the start codon (Woolstenhulme et al., 2015), which corresponds to the distance from the P-site codon to the 3′ boundary of the E. coli ribosome (Hartz et al., 1988; Yusupov et al., 2001). In order to determine the P-site offset in our dataset, we used the 3′ assignment to perform metagene analysis at the stat codon for each read length between 15-40 nt, and then measured the distance between the highest 3′ peak of the start codon and the start codon. This analysis revealed two populations of RPFs, with footprints between 28-40 nt showing clearly defined initiation peaks located approximately 14nt downstream of the start codon, and shorter footprints showing sometimes less pronounced and defined initiation peaks which were located closer to the start codon. After measuring the distances from the 3′end peaks to the start codon we could establish a default p-site offset of 14 nt for our dataset. Next we used this offset value of 14 nt to re-run the metagene analysis generating a plot of average ribosome occupancy for genes aligned at their start codon that clearly confirmed a pronounced peak at the first nucleotide of the start codon (Figure S4D).
Quantification and statistical analysis
Quantification of gene expression
For the RPFs and total RNA reads, Bedtools (Quinlan and Hall, 2010) was used to calculate the read coverage for each genomic feature. Mycobrowser annotation (Release R1, November 2017) was used as the annotation reference of the M. tuberculosis genome (Kapopoulou et al., 2011). The overall expression rate for each gene was then calculated as reads per kilobase per million (RPKM) values for both RPFs and total RNA reads as previously described (Becker et al., 2013; Mortazavi et al., 2008). Asymmetry scores for each gene were calculated as log2 values of the ratio of RPKM in the second half of the gene over RPKM in the first half (Mohammad et al., 2019). For the definition of 5′UTR regions, available data for transcriptional start sites in M. tuberculosis (Cortes et al., 2013) was integrated with the Mycobrowser annotation. Reads spanning across the 5′UTR-CDS boundary were counted as both UTR-mapping reads and CDS-mapping reads. For the definition of gene categories as leaderless, Shine-Dalgarno and UTR, the classification from Cortes et al. (2013) was also used. Reliably quantified genes were those genes with at least 128 total read counts between the three replicates, ensuring that measurements were reliable and not dictated by sampling error (Ingolia et al., 2009) (Figures S2A and S2B). Ribosome occupancies (ROs) for reliable quantified genes were calculated by dividing the expression rates (RPKMs) of translation by those of transcription.
Differential expression analysis
For the differential expression analysis of RPFs and total RNA reads under nutrient starvation, genome coverage of reads mapping to genes was used for statistical testing using the DESeq2 R package (Love et al., 2014). Differentially expressed genes were considered when fold changes between exponential growth and starvation were greater or equal to two and the corresponding adjusted p value was less than 0.01. The enrichment analysis in GO terms was performed using the BiNGO application software from Cytoscape (Maere et al., 2005).
Shine-Dalgarno specificity analysis
The Shine-Dalgarno sequence for each gene was found by searching for the canonical Shine-Dalgarno motif (AGAAAGGAGG) in the 30 nucleotides upstream of the start coding using blast (-task blastn -word_size 4). The number of matches and location of the Shine-Dalgarno sequence was extracted by analyzing the high-scoring segment pair output by blast.
Statistical analysis
Unless otherwise stated, GraphPad Prism version 8 for Mac (GraphPad Software, San Diego, California) was used to calculate all statistical tests as described in the text. For Figure 1B, data represent the Spearman’s correlation coefficient for the RPF (n = 3) and RNA libraries (n = 3). For Figures 1F and 4B, data represent the distribution of RO values across UTR categories with horizontal bars representing the median, and distribution of mRNA half-lives in Figure S2. Statistical analysis was performed using the Kruskal-Wallis test. Correlation plots of mRNA versus RO are presented in Figure 1G and of mRNA half-lives versus RO in Figure S2, with Spearman coefficients and linear regression included for each gene category in both Figures. Figure 1H represents distribution of RO values across different functional categories with horizontal bars indicating median values. Statistical analysis was performed using Dunn’s multiple comparisons for RO values across functional categories and results are available in Table S2A. For Figure 2C, the association between TE and Shine-Dalgarno strength was characterized by performing ordered linear regression using R (v3.6.3) by applying the linear regression (lm function) using the RO as the outcome variable and number of matches as a predictor. The number of matches were treated as an ordered categorical variable with 0-3 matches aggregated into a single category. For Figure 2D, linear regression between RO and the number of matches and presence of a core using the lm function in R was performed. For Figures 4C–4E, changes in expression were quantified using DESeq2, with significant changes occurring when absolute log2 fold-change > 1 and adjusted p value < 0.01. Bars in Figure 4C represent results of contingency analysis between the proportion of genes in each UTR category. In Figure S2, simple binomial partitioning of the total number of reads between two replicates was used to predict standard deviation values. Codon usage analysis was quantified by calculating frequency of codons across the first 20 amino acids after the start codon for the different gene categories. Differences in the representation of codons were analyzed by performing Fisher’s exact tests and associated p values are available in Table S2B. Figure S4C shows boxplots comparing the median levels of percentage representation of alanine, serine or threonine codons over the first 20 amino acids across the gene categories. Horizontal bars indicate median levels. Differences between median levels between categories were assessed using ANOVA.
Acknowledgments
The authors thank and acknowledge the support of Cambridge Genomic Services (Department of Pathology, University of Cambridge), who carried out the library quality control, pooling, and sequencing. We thank Brendan Wren, Kristine Arnvig, and Sam Willcocks for critical reading of the manuscript. This work was supported by funding from the European Research Council (ERC) under the European Union’s Horizon 2020 research and innovation programme (grant agreement 637730). T.G.C. is funded by the Medical Research Council UK (grants MR/M01360X/1, MR/N010469/1, MR/R025576/1, and MR/R020973/1) and BBSRC UK (grant BB/R013063/1).
Author contributions
Conceptualization, E.B.S. and T.C.; Methodology, E.B.S and T.C.; Software, J.E.P., T.G.C., and T.C.; Validation, E.B.S. and T.C.; Formal Analysis, E.B.S., J.E.P., and T.C.; Investigation, E.B.S.; Resources, T.G.C.; Writing—Original Draft, E.B.S. and T.C.; Writing—Review and Editing, E.B.S., J.E.P., T.G.C., and T.C.; Visualization, E.B.S., J.E.P., and T.C.; Supervision, T.G.C. and T.C.; Project Administration, T.C.; Funding Acquisition, T.G.C. and T.C.
Declaration of interests
The authors declare no competing interests.
Published: February 2, 2021
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.celrep.2021.108695.
Supplemental information
S1A. Mapping statistics for data from exponential cultures. Related to Figure 1B. S1B Reads per kilobase per million (RPKM) and count data from exponential cultures and S1C set of reliably quantified genes. Related to Figures 1D, 1F–1H, 2C, 2D, and S2. S1D Reads per kilobase per million (RPKM) and count data from nutrient-starved cultures and S1E set of reliably quantified genes. Related to Figure 4B.
S2A Results of Dunn’s multiple comparisons test for RO values across functional categories in exponential cultures. Related to Figure 1H and STAR methods. S2B Results of contingency analysis for the codon usage analysis in the first 20 amino acids. Related to Figures 2A and 2B and STAR methods.
DESeq2 data for RNA-seq (S3A) and Ribo-seq (S3B). Related to Figures 4A and 4C–4E.
References
- Aguilar-Ayala D.A., Tilleman L., Van Nieuwerburgh F., Deforce D., Palomino J.C., Vandamme P., Gonzalez-Y-Merchand J.A., Martin A. The transcriptome of Mycobacterium tuberculosis in a lipid-rich dormancy model through RNAseq analysis. Sci. Rep. 2017;7:17665. doi: 10.1038/s41598-017-17751-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andersson G.E., Sharp P.M. Codon usage in the Mycobacterium tuberculosis complex. Microbiology (Reading) 1996;142:915–925. doi: 10.1099/00221287-142-4-915. [DOI] [PubMed] [Google Scholar]
- Balakrishnan R., Oman K., Shoji S., Bundschuh R., Fredrick K. The conserved GTPase LepA contributes mainly to translation initiation in Escherichia coli. Nucleic Acids Res. 2014;42:13370–13383. doi: 10.1093/nar/gku1098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bechhofer D.H., Dubnau D. Induced mRNA stability in Bacillus subtilis. Proc. Natl. Acad. Sci. USA. 1987;84:498–502. doi: 10.1073/pnas.84.2.498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Becker A.H., Oh E., Weissman J.S., Kramer G., Bukau B. Selective ribosome profiling as a tool for studying the interaction of chaperones and targeting factors with nascent polypeptide chains and ribosomes. Nat. Protoc. 2013;8:2212–2239. doi: 10.1038/nprot.2013.133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berghoff B.A., Konzer A., Mank N.N., Looso M., Rische T., Förstner K.U., Krüger M., Klug G. Integrative “omics”-approach discovers dynamic and regulatory features of bacterial stress responses. PLoS Genet. 2013;9:e1003576. doi: 10.1371/journal.pgen.1003576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Betts J.C., Lukey P.T., Robb L.C., McAdam R.A., Duncan K. Evaluation of a nutrient starvation model of Mycobacterium tuberculosis persistence by gene and protein expression profiling. Mol. Microbiol. 2002;43:717–731. doi: 10.1046/j.1365-2958.2002.02779.x. [DOI] [PubMed] [Google Scholar]
- Bolger A.M., Lohse M., Usadel B. Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics. 2014;30:2114–2120. doi: 10.1093/bioinformatics/btu170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown A., Shao S., Murray J., Hegde R.S., Ramakrishnan V. Structural basis for stop codon recognition in eukaryotes. Nature. 2015;524:493–496. doi: 10.1038/nature14896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bunker R.D., Mandal K., Bashiri G., Chaston J.J., Pentelute B.L., Lott J.S., Kent S.B.H., Baker E.N. A functional role of Rv1738 in Mycobacterium tuberculosis persistence suggested by racemic protein crystallography. Proc. Natl. Acad. Sci. USA. 2015;112:4310–4315. doi: 10.1073/pnas.1422387112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Byrgazov K., Vesper O., Moll I. Ribosome heterogeneity: another level of complexity in bacterial translation regulation. Curr. Opin. Microbiol. 2013;16:133–139. doi: 10.1016/j.mib.2013.01.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Canestrari J.G., Lasek-Nesselquist E., Upadhyay A., Rofaeil M., Champion M.M., Wade J.T., Derbyshire K.M., Gray T.A. Polycysteine-encoding leaderless short ORFs function as cysteine-responsive attenuators of operonic gene expression in mycobacteria. Mol. Microbiol. 2020;114:93–108. doi: 10.1111/mmi.14498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen L.-H., Emory S.A., Bricker A.L., Bouvet P., Belasco J.G. Structure and function of a bacterial mRNA stabilizer: analysis of the 5′ untranslated region of ompA mRNA. J. Bacteriol. 1991;173:4578–4586. doi: 10.1128/jb.173.15.4578-4586.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen Y.-X., Xu Z.Y., Ge X., Sanyal S., Lu Z.J., Javid B. Selective translation by alternative bacterial ribosomes. Proc. Natl. Acad. Sci. USA. 2020;117:19487–19496. doi: 10.1073/pnas.2009607117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choi J., Marks J., Zhang J., Chen D.H., Wang J., Vázquez-Laslop N., Mankin A.S., Puglisi J.D. Dynamics of the context-specific translation arrest by chloramphenicol and linezolid. Nat. Chem. Biol. 2020;16:310–317. doi: 10.1038/s41589-019-0423-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cortes T., Schubert O.T., Rose G., Arnvig K.B., Comas I., Aebersold R., Young D.B. Genome-wide mapping of transcriptional start sites defines an extensive leaderless transcriptome in Mycobacterium tuberculosis. Cell Rep. 2013;5:1121–1131. doi: 10.1016/j.celrep.2013.10.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cortes T., Schubert O.T., Banaei-Esfahani A., Collins B.C., Aebersold R., Young D.B. Delayed effects of transcriptional responses in Mycobacterium tuberculosis exposed to nitric oxide suggest other mechanisms involved in survival. Sci. Rep. 2017;7:8208. doi: 10.1038/s41598-017-08306-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dahl J.L., Kraus C.N., Boshoff H.I.M., Doan B., Foley K., Avarbock D., Kaplan G., Mizrahi V., Rubin H., Barry C.E., 3rd The role of RelMtb-mediated adaptation to stationary phase in long-term persistence of Mycobacterium tuberculosis in mice. Proc. Natl. Acad. Sci. USA. 2003;100:10026–10031. doi: 10.1073/pnas.1631248100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deb C., Lee C.M., Dubey V.S., Daniel J., Abomoelak B., Sirakova T.D., Pawar S., Rogers L., Kolattukudy P.E. A novel in vitro multiple-stress dormancy model for Mycobacterium tuberculosis generates a lipid-loaded, drug-tolerant, dormant pathogen. PLoS ONE. 2009;4:e6077. doi: 10.1371/journal.pone.0006077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Del Campo C., Bartholomäus A., Fedyunin I., Ignatova Z. Secondary Structure across the Bacterial Transcriptome Reveals Versatile Roles in mRNA Regulation and Function. PLoS Genet. 2015;11:e1005613. doi: 10.1371/journal.pgen.1005613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dressaire C., Laurent B., Loubière P., Besse P., Cocaign-Bousquet M. Linear covariance models to examine the determinants of protein levels in Lactococcus lactis. Mol. Biosyst. 2010;6:1255–1264. doi: 10.1039/c001702g. [DOI] [PubMed] [Google Scholar]
- Galagan J.E., Minch K., Peterson M., Lyubetskaya A., Azizi E., Sweet L., Gomes A., Rustad T., Dolganov G., Glotova I. The Mycobacterium tuberculosis regulatory network and hypoxia. Nature. 2013;499:178–183. doi: 10.1038/nature12337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gelsinger D.R., Dallon E., Reddy R., Mohammad F., Buskirk A.R., DiRuggiero J. Ribosome profiling in archaea reveals leaderless translation, novel translational initiation sites, and ribosome pausing at single codon resolution. Nucleic Acids Res. 2020;48:5201–5216. doi: 10.1093/nar/gkaa304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gomez J.E., McKinney J.D. Tuberculosis. Churchill Livingstone; 2004. Mycobacterium tuberculosis persistence, latency, and drug tolerance; pp. 29–44. [DOI] [PubMed] [Google Scholar]
- Haider S., Pal R. Integrated analysis of transcriptomic and proteomic data. Curr. Genomics. 2013;14:91–110. doi: 10.2174/1389202911314020003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hartz D., McPheeters D.S., Traut R., Gold L. Extension inhibition analysis of translation initiation complexes. Methods Enzymol. 1988;164:419–425. doi: 10.1016/s0076-6879(88)64058-4. [DOI] [PubMed] [Google Scholar]
- Hücker S.M., Simon S., Scherer S., Neuhaus K. Transcriptional and translational regulation by RNA thermometers, riboswitches and the sRNA DsrA in Escherichia coli O157:H7 Sakai under combined cold and osmotic stress adaptation. FEMS Microbiol. Lett. 2017;364:262. doi: 10.1093/femsle/fnw262. [DOI] [PubMed] [Google Scholar]
- Ingolia N.T., Ghaemmaghami S., Newman J.R.S., Weissman J.S. Genome-wide analysis in vivo of translation with nucleotide resolution using ribosome profiling. Science. 2009;324:218–223. doi: 10.1126/science.1168978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ingolia N.T., Lareau L.F., Weissman J.S. Ribosome profiling of mouse embryonic stem cells reveals the complexity and dynamics of mammalian proteomes. Cell. 2011;147:789–802. doi: 10.1016/j.cell.2011.10.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kapopoulou A., Lew J.M., Cole S.T. The MycoBrowser portal: a comprehensive and manually annotated resource for mycobacterial genomes. Tuberculosis (Edinb.) 2011;91:8–13. doi: 10.1016/j.tube.2010.09.006. [DOI] [PubMed] [Google Scholar]
- Kempsell K.E., Ji Y.E., Estrada I.C., Colston M.J., Cox R.A. The nucleotide sequence of the promoter, 16S rRNA and spacer region of the ribosomal RNA operon of Mycobacterium tuberculosis and comparison with Mycobacterium leprae precursor rRNA. J. Gen. Microbiol. 1992;138:1717–1727. doi: 10.1099/00221287-138-8-1717. [DOI] [PubMed] [Google Scholar]
- Langmead B., Trapnell C., Pop M., Salzberg S.L. Ultrafast and memory-efficient alignment of short DNA sequences to the human genome. Genome Biol. 2009;10:R25. doi: 10.1186/gb-2009-10-3-r25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Latif H., Szubin R., Tan J., Brunk E., Lechner A., Zengler K., Palsson B.O. A streamlined ribosome profiling protocol for the characterization of microorganisms. Biotechniques. 2015;58:329–332. doi: 10.2144/000114302. [DOI] [PubMed] [Google Scholar]
- Li G.W. How do bacteria tune translation efficiency? Curr. Opin. Microbiol. 2015;24:66–71. doi: 10.1016/j.mib.2015.01.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li G.W., Burkhardt D., Gross C., Weissman J.S. Quantifying absolute protein synthesis rates reveals principles underlying allocation of cellular resources. Cell. 2014;157:624–635. doi: 10.1016/j.cell.2014.02.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Love M.I., Huber W., Anders S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014;15:550. doi: 10.1186/s13059-014-0550-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma J., Campbell A., Karlin S. Correlations between Shine-Dalgarno Sequences and Gene Features such as predicted expression levels and Operon Structures. J. Bacteriol. 2002;184:5733–5745. doi: 10.1128/JB.184.20.5733-5745.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maere S., Heymans K., Kuiper M. BiNGO: a Cytoscape plugin to assess overrepresentation of gene ontology categories in biological networks. Bioinformatics. 2005;21:3448–3449. doi: 10.1093/bioinformatics/bti551. [DOI] [PubMed] [Google Scholar]
- Marks J., Kannan K., Roncase E.J., Klepacki D., Kefi A., Orelle C., Vázquez-Laslop N., Mankin A.S. Context-specific inhibition of translation by ribosomal antibiotics targeting the peptidyl transferase center. Proc. Natl. Acad. Sci. USA. 2016;113:12150–12155. doi: 10.1073/pnas.1613055113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin M. Cutadapt removes adapter sequences from high-throughput sequencing reads. EMBnet. J. 2011;17:10. [Google Scholar]
- Martini M.C., Zhou Y., Sun H., Shell S.S. Defining the transcriptional and post-transcriptional landscapes of Mycobacterium smegmatis in aerobic growth and hypoxia. Front. Microbiol. 2019;10:591. doi: 10.3389/fmicb.2019.00591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mishra S., Ahmed T., Tyagi A., Shi J., Bhushan S. Structures of Mycobacterium smegmatis 70S ribosomes in complex with HPF, tmRNA, and P-tRNA. Sci. Rep. 2018;8:13587. doi: 10.1038/s41598-018-31850-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mohammad F., Green R., Buskirk A.R. A systematically-revised ribosome profiling method for bacteria reveals pauses at single-codon resolution. eLife. 2019;8:e42591. doi: 10.7554/eLife.42591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moll I., Bläsi U. Differential inhibition of 30S and 70S translation initiation complexes on leaderless mRNA by kasugamycin. Biochem. Biophys. Res. Commun. 2002;297:1021–1026. doi: 10.1016/s0006-291x(02)02333-1. [DOI] [PubMed] [Google Scholar]
- Mortazavi A., Williams B.A., McCue K., Schaeffer L., Wold B. Mapping and quantifying mammalian transcriptomes by RNA-Seq. Nat. Methods. 2008;5:621–628. doi: 10.1038/nmeth.1226. [DOI] [PubMed] [Google Scholar]
- Nakahigashi K., Takai Y., Shiwa Y., Wada M., Honma M., Yoshikawa H., Tomita M., Kanai A., Mori H. Effect of codon adaptation on codon-level and gene-level translation efficiency in vivo. BMC Genomics. 2014;15:1115. doi: 10.1186/1471-2164-15-1115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Namouchi A., Gómez-Muñoz M., Frye S.A., Moen L.V., Rognes T., Tønjum T., Balasingham S.V. The Mycobacterium tuberculosis transcriptional landscape under genotoxic stress. BMC Genomics. 2016;17:791. doi: 10.1186/s12864-016-3132-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nguyen T.G., Vargas-Blanco D.A., Roberts L.A., Shell S.S. The Impact of Leadered and Leaderless Gene Structures on Translation Efficiency, Transcript Stability, and Predicted Transcription Rates in Mycobacterium smegmatis. J. Bacteriol. 2020;202 doi: 10.1128/JB.00746-19. e00746-19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oh E., Becker A.H., Sandikci A., Huber D., Chaba R., Gloge F., Nichols R.J., Typas A., Gross C.A., Kramer G. Selective ribosome profiling reveals the cotranslational chaperone action of trigger factor in vivo. Cell. 2011;147:1295–1308. doi: 10.1016/j.cell.2011.10.044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Picard F., Milhem H., Loubière P., Laurent B., Cocaign-Bousquet M., Girbal L. Bacterial translational regulations: high diversity between all mRNAs and major role in gene expression. BMC Genomics. 2012;13:528. doi: 10.1186/1471-2164-13-528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Picard F., Loubière P., Girbal L., Cocaign-Bousquet M. The significance of translation regulation in the stress response. BMC Genomics. 2013;14:588. doi: 10.1186/1471-2164-14-588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quinlan A.R., Hall I.M. BEDTools: a flexible suite of utilities for comparing genomic features. Bioinformatics. 2010;26:841–842. doi: 10.1093/bioinformatics/btq033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rengarajan J., Bloom B.R., Rubin E.J. Genome-wide requirements for Mycobacterium tuberculosis adaptation and survival in macrophages. Proc. Natl. Acad. Sci. USA. 2005;102:8327–8332. doi: 10.1073/pnas.0503272102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rohde K.H., Veiga D.F.T., Caldwell S., Balázsi G., Russell D.G. Linking the transcriptional profiles and the physiological states of Mycobacterium tuberculosis during an extended intracellular infection. PLoS Pathog. 2012;8:e1002769. doi: 10.1371/journal.ppat.1002769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rustad T.R., Minch K.J., Brabant W., Winkler J.K., Reiss D.J., Baliga N.S., Sherman D.R. Global analysis of mRNA stability in Mycobacterium tuberculosis. Nucleic Acids Res. 2013;41:509–517. doi: 10.1093/nar/gks1019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rustad T.R., Minch K.J., Ma S., Winkler J.K., Hobbs S., Hickey M., Brabant W., Turkarslan S., Price N.D., Baliga N.S., Sherman D.R. Mapping and manipulating the Mycobacterium tuberculosis transcriptome using a transcription factor overexpression-derived regulatory network. Genome Biol. 2014;15:502. doi: 10.1186/s13059-014-0502-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saito K., Green R., Buskirk A.R. Translational initiation in E. coli occurs at the correct sites genome-wide in the absence of mRNA-rRNA base-pairing. eLife. 2020;9:e55002. doi: 10.7554/eLife.55002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sawyer E.B., Grabowska A.D., Cortes T. Translational regulation in mycobacteria and its implications for pathogenicity. Nucleic Acids Res. 2018;46:6950–6961. doi: 10.1093/nar/gky574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schrader J.M., Zhou B., Li G.-W., Lasker K., Childers W.S., Williams B., Long T., Crosson S., McAdams H.H., Weissman J.S., Shapiro L. The coding and noncoding architecture of the Caulobacter crescentus genome. PLoS Genet. 2014;10:e1004463. doi: 10.1371/journal.pgen.1004463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shell S.S., Wang J., Lapierre P., Mir M., Chase M.R., Pyle M.M., Gawande R., Ahmad R., Sarracino D.A., Ioerger T.R. Leaderless Transcripts and Small Proteins Are Common Features of the Mycobacterial Translational Landscape. PLoS Genet. 2015;11:e1005641. doi: 10.1371/journal.pgen.1005641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi Z., Fujii K., Kovary K.M., Genuth N.R., Röst H.L., Teruel M.N., Barna M. Heterogeneous Ribosomes Preferentially Translate Distinct Subpools of mRNAs Genome-wide. Mol. Cell. 2017;67:71–83.e7. doi: 10.1016/j.molcel.2017.05.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith C., Canestrari J.G., Wang J., Derbyshire K.M., Gray T.A., Wade J.T. Pervasive Translation in Mycobacterium tuberculosis. BioRxiv. 2019:665208. doi: 10.7554/eLife.73980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Starosta A.L., Lassak J., Jung K., Wilson D.N. The bacterial translation stress response. FEMS Microbiol. Rev. 2014;38:1172–1201. doi: 10.1111/1574-6976.12083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stewart G.R., Wernisch L., Stabler R., Mangan J.A., Hinds J., Laing K.G., Young D.B., Butcher P.D. Dissection of the heat-shock response in Mycobacterium tuberculosis using mutants and microarrays. Microbiology (Reading) 2002;148:3129–3138. doi: 10.1099/00221287-148-10-3129. [DOI] [PubMed] [Google Scholar]
- Stewart G.R., Robertson B.D., Young D.B. Tuberculosis: a problem with persistence. Nat. Rev. Microbiol. 2003;1:97–105. doi: 10.1038/nrmicro749. [DOI] [PubMed] [Google Scholar]
- Taniguchi Y., Choi P.J., Li G.W., Chen H., Babu M., Hearn J., Emili A., Xie X.S. Quantifying E. coli proteome and transcriptome with single-molecule sensitivity in single cells. Science. 2010;329:533–538. doi: 10.1126/science.1188308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trauner A., Lougheed K.E.A., Bennett M.H., Hingley-Wilson S.M., Williams H.D. The dormancy regulator DosR controls ribosome stability in hypoxic mycobacteria. J. Biol. Chem. 2012;287:24053–24063. doi: 10.1074/jbc.M112.364851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tuller T., Waldman Y.Y., Kupiec M., Ruppin E. Translation efficiency is determined by both codon bias and folding energy. Proc. Natl. Acad. Sci. USA. 2010;107:3645–3650. doi: 10.1073/pnas.0909910107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vesper O., Amitai S., Belitsky M., Byrgazov K., Kaberdina A.C., Engelberg-Kulka H., Moll I. Selective translation of leaderless mRNAs by specialized ribosomes generated by MazF in Escherichia coli. Cell. 2011;147:147–157. doi: 10.1016/j.cell.2011.07.047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Woolstenhulme C.J., Guydosh N.R., Green R., Buskirk A.R. High-precision analysis of translational pausing by ribosome profiling in bacteria lacking EFP. Cell Rep. 2015;11:13–21. doi: 10.1016/j.celrep.2015.03.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Woong Park S.W., Klotzsche M., Wilson D.J., Boshoff H.I., Eoh H., Manjunatha U., Blumenthal A., Rhee K., Barry C.E., 3rd, Aldrich C.C. Evaluating the sensitivity of Mycobacterium tuberculosis to biotin deprivation using regulated gene expression. PLoS Pathog. 2011;7:e1002264. doi: 10.1371/journal.ppat.1002264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xue S., Barna M. Specialized ribosomes: a new frontier in gene regulation and organismal biology. Nat. Rev. Mol. Cell Biol. 2012;13:355–369. doi: 10.1038/nrm3359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yusupov M.M., Yusupova G.Z., Baucom A., Lieberman K., Earnest T.N., Cate J.H.D., Noller H.F. Crystal structure of the ribosome at 5.5 A resolution. Science. 2001;292:883–896. doi: 10.1126/science.1060089. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
S1A. Mapping statistics for data from exponential cultures. Related to Figure 1B. S1B Reads per kilobase per million (RPKM) and count data from exponential cultures and S1C set of reliably quantified genes. Related to Figures 1D, 1F–1H, 2C, 2D, and S2. S1D Reads per kilobase per million (RPKM) and count data from nutrient-starved cultures and S1E set of reliably quantified genes. Related to Figure 4B.
S2A Results of Dunn’s multiple comparisons test for RO values across functional categories in exponential cultures. Related to Figure 1H and STAR methods. S2B Results of contingency analysis for the codon usage analysis in the first 20 amino acids. Related to Figures 2A and 2B and STAR methods.
DESeq2 data for RNA-seq (S3A) and Ribo-seq (S3B). Related to Figures 4A and 4C–4E.
Data Availability Statement
The accession number for the RNA-seq and Ribosome profiling data reported in this paper is ArrayExpress: E-MTAB-8335. https://www.ebi.ac.uk/arrayexpress
The code supporting the current study have not been deposited in a public repository but are available from the corresponding author on request.