

INTRODUCTION
Sepsis and severe sepsis continue to burden the health care system and are among the greatest medical concerns, with a mortality rate ranging from 30% to 50% in North America.1 Sepsis is the most common cause of death in intensive care units and has been shown to result in an annual health care cost of $14 to $16 billion.1,2 Between 1979 and 2000, sepsis accounted for an incidence rate of 500,000 cases in the United States. More recently, that figure has increased to onward of 750,000 cases annually.3 Sepsis can be defined as a systemic inflammatory response syndrome (SIRS) occurring in the presence of an infectious source.4 The most frequently observed gram-positive and gram-negative bacteria in septic patients include Staphylococcus aureus, Streptococcus pyogenes, Escherichia coli, Klebsiella species, and Pseudomonas aeruginosa.5–7 It has long been understood that gram-negative bacteria are the most common sources of bacteremia in sepsis; however, Martin and colleagues8 showed that the last 25 years has witnessed a shift in gram-positive dominance, with an average annual increase of 26% in cases within the study period.
In 1992, as part of an international effort to standardize the classification process, the American College of Chest Physicians and Critical Care Consensus Conference proposed diagnostic guidelines for SIRS, sepsis, severe sepsis, and septic shock.9 Generally speaking, the SIRS response is triggered by nontraumatic causes; sepsis can be considered the activation of SIRS inflicted by microbial infections (eg, bacterial, fungal, viral); severe sepsis has the inclusion of multiple organ dysfunction; and the manifestation of hypotension with a lack of responsiveness to fluid resuscitation is septic shock. The specific criteria for each condition are listed later in this article. In 2003, another consensus panel revisited these in an attempt to clarify categorical ambiguity. It was determined that some of the symptoms of SIRS, such as tachycardia, manifest in other septic and nonseptic conditions, and are poor at differentiating it from other conditions. Thus, the terms sepsis and severe sepsis can be used interchangeably when there are organ complications present as a result of infection.10 More recently, the Surviving Sepsis Campaign produced updated guidelines in an effort to improve the management of sepsis.11 Suggestions were noted for all categories of sepsis, with an emphasis placed on early quantitative resuscitation of the septic patient within the first 6 hours of positive blood cultures. In light of these multiple paradigm shifts in defining clinical sepsis, this review attempts to provide a summation of the innate immunologic alterations that manifest during sepsis, establish and compare mouse models of sepsis with the clinical course, and discuss the authors’ views on additional elements that should be considered in modeling and predicting clinical sepsis from a basic research setting.
Systemic Inflammatory Response Syndrome
A patient must demonstrate at least 2 of the following criteria:
Temperature less than 36°C or greater than 38.3°C
Heart rate greater than 90 beats/min
Respiratory rate greater than 20 breaths/min or PaCO2 (partial pressure of arterial carbon dioxide) less than 32 mm Hg
White blood cell counts less than 4000 cells/μL, greater than 12,000 cells/μL, or more than 10% bands
Sepsis
SIRS with the inclusion of an infection.
Severe Sepsis
Sepsis with the association of hypotension, hypoperfusion, or multiple organ dysfunction.
Septic Shock
Sepsis with hypotension (blood pressure <90 mm Hg) or a deviation from baseline of 40 mm Hg or greater despite fluid resuscitation.
IMMUNE PATHOPHYSIOLOGY
The traditional and most widely accepted perspective regarding the immunologic alterations during the septic response includes the hyperinflammatory response initiated from multiple factors. More recently, this paradigm has shifted with a competing theory regarding a compensatory response. Both of these theories along with the pathophysiology of sepsis are discussed in this section.
The host immune response initiates a surge of proinflammatory cytokines that collectively encompass the “cytokine storm.” Endotoxins found on the bacterial cell wall serve as danger-associated molecular patterns (DAMP) and pathogen-associated molecular patterns (PAMP) that are detected by pattern recognition receptors of the innate immune system.12 For example, the lipopolysaccharide (LPS) component of gram-negative Pseudomonas aeruginosa is recognized by toll-like receptor (TLR)4 on the cell surface of antigen-presenting cells (monocytes/macrophages), which in turn stimulates the production of proinflammatory mediators such as tumor necrosis factor (TNF), interleukin (IL)-1b, and IL-6. The severity of this response has been attributed to numerous factors including comorbidities, pathogen load, and virulence.13 This response elicits a cascade of systemic and tissue-specific events including homing of chemokines to the injury site, phagocytosis, and vascular damage.14–16 The response is not limited to the site of infection; elevated levels of IL-6 stimulate the liver’s production of C-reactive protein, which functions as an acute-phase reactant and is a biomarker of interest for predicting sepsis.10,17,18 This sequence of events contains numerous alterations on immune cell behavior and function, outlined in Table 1.
Table 1.
Overview of systemic inflammatory response syndrome and sepsis effects on innate and adaptive immune cells
| Apoptosis | Cytokine Production | Reference | |
|---|---|---|---|
| Macrophage | ↑↓ Pro- (early/late) ↑ Anti- |
Cavaillon and Annane,21 2006 King et al,53 2014 Hotchkiss et al,54 2002 |
|
| Dendritic cell | ↓ | ↓ | Hotchkiss et al,54 2002 |
| Neutrophil | ↓ (delayed apoptosis) | ↑↓ Pro- (early/late) ↑ Anti- |
Drifte et al,55 2013 Taneja et al,56 2004 |
| Natural killer cell | ↑ | ↓ | Reviewed in Stearns-Kurosawa etal,57 2011 |
| T cell | ↑ | ↑ | Hotchkiss et al,58 2001 Venet et al,59 2004 |
| B cell | ↑ | ↑ Antibody production | Hotchkiss et al,58 2001 |
Another paradigm that has gained increasing acceptance is a delayed and potentially prolonged anti-inflammatory response that succeeds the initial hyperinflammation. The inability of SIRS to explain all of the pathologic alterations, and the lack of sepsis prevention after reducing hyperinflammation occurring in conjunction with anti-inflammatory mediator release,19 raised the possibility of a fundamental disconnect for a unilateral understanding of sepsis. This process was later described as the compensatory anti-inflammatory response syndrome (CARS).20 This biphasic view of the septic response has shed light on the increased susceptibility to secondary complications and immune dysfunction at variable times during the course of infection.14,21 Specifically, CARS has been described to include the following components20,22:
Reduced cytokine response in monocyte activation
Lymphocyte apoptosis and dysfunction
Reduction in monocyte human leukocyte antigen (HLA)-presenting receptors
Increased expression of anti-inflammatory IL-10
The cytokine components of the compensatory response include release of IL-10, IL-1 receptor antagonist (IL-1RA), and transforming growth factor-β (TGF-β),23–25 collectively representing a normal homeostatic response to limit inflammation.26 Other studies have used the CARS model to show that septic patients with low proportions of HLA-DR (a measure of immune challenge and hallmark of sepsis) produced low amounts of proinflammatory cytokines in response to bacterial challenge.27 IL-10 levels have been shown to correlate with multiple organ dysfunction and mortality.28 Lastly, when considering both models of the septic response, a high IL-10 to TNF ratio was a predictor of mortality in patients admitted with fever.29 Contrasting evidence for the capacity of anti-inflammatory soluble mediators to have therapeutic value has generally been limited to animal models (see later discussion). A prospective single-center clinical study conducted by Gomez and colleagues30 did not show any support for a 2-phase model underlying the pathophysiology of sepsis and, as expected, was only able to conclusively show that immune variables behaved in a “mixed and time-dependent manner.”30 Thus, neglecting all of the aforementioned studies supporting both sides of the spectrum, the compensatory response can be simplified to an adaptive reaction of the immune system to dampen inflammation (Table 2 summarizes the immune mediators). However, the distinguishable elevation in anti-inflammatory cytokines at early stages of trauma raises questions regarding the adaptive nature of this response, discussed further in subsequent sections.
Table 2.
Overview of compensatory anti-inflammatory response syndrome (CARS) effect on innate and adaptive immune cells
| Apoptosis | Cytokine Production | Reference | |
|---|---|---|---|
| Macrophage | ↑ Anti- | Opal and DePalo,60 2000 | |
| Dendritic cell | ↑ | ↑ Anti- | Ward et al,22 2008 |
| Neutrophil | ↓ | ↓ Pro- ↑ Anti- |
Reviewed in Hotchkiss and Karl,13 2003 |
| Natural killer cell | ↑ | ↓ | Reviewed in Hotchkiss and Karl,13 2003 |
| T cell | ↑ | ↓ | Reviewed in Adib-Conquy and Cavaillon,19 2009 |
| B cell | ↑ | ↓ | Reviewed in Adib-Conquy and Cavaillon,19 2009 |
WHAT ARE ANIMAL MODELS ACTUALLY MODELING?
Clinically, sepsis is physiologically defined by 2 hemodynamic phases: an early hyperdynamic phase and a subsequent late hypodynamic phase.31 Although the underlying mechanisms of these phases are beyond the scope of this review, it is pertinent that the hyperdynamic phase is characterized by low systemic vascular resistance (SVR) and increased cardiac output (CO), whereas the hypodynamic phase causes a decline in CO and SVR remains constantly low.32 As the hallmark of clinical sepsis, this parallel phenomenon is often used as the gold standard in animal models of sepsis to evaluate its clinical relevancy.33,34 However, in addition to the hemodynamic phases, extensive reports from the past 2 decades have suggested a complex and rivaling role of the immune-inflammatory responses of sepsis in defining the accuracy of these animal models in recapitulating human sepsis. Therefore, subsequent sections of this review are dedicated mainly to discussing the immunologic similarities and differences between mouse models and clinical sepsis.
IMMUNOLOGIC MOUSE MODELS OF SEPSIS
Fervent attempts to study and develop novel clinical therapeutics for sepsis have led to the implementation of mouse models. At present, there are 3 different approaches to induce sepsis in mice that are commonly practiced: exogenous administration of a toxin (such as LPS, other endotoxins, or zymosan); exogenous administration of live bacteria; and disturbance of the animals’ endogenous protective barrier (introducing colonic leakage to allow migration of bacteria). However, despite the definitive progression of our understanding about the physiologic and immune responses to sepsis with these models, the practicality of translating this knowledge to clinical applications is controversial. The source of this controversy lies within the inherent discrepancy of these models to recapitulate the course of human disease in the clinic. Specific types of models (eg, exogenous toxin administration) may depict only a particular type of sepsis or partial pathophysiology of septic patients. Thus, the main challenge faced by researchers is to select the appropriate model that most accurately reflects the human disease. Applying the knowledge generated from these models to dictate the mechanistic course for therapeutic development continues to be the overall goal. This section presents a comprehensive overview of the different mouse models of sepsis and their relevance to human sepsis.
Toxemia
The toxemia model is established by the injection of TLR agonists such as LPS (TLR4 agonist35) or CpG DNA (TLR9 agonist36) into mice via intravenous or intraperitoneal routes. The efficacy of this model in recapitulating clinical sepsis is summarized in Table 3. One of the major deviations from clinical sepsis of this model is the differences in the immune response, whereby a single bolus of endotoxin administration in mice results in a transient and rapid “spike” of systemic cytokine production, whereas clinical sepsis exhibits a prolonged elevation of systemic cytokine production and is typically lower in serum concentration by multiple orders of magnitude. Furthermore, neutralization of proinflammatory mediators ameliorated septic pathophysiology in endotoxicosis mouse models, whereas no success was observed in clinical trials.
Table 3.
Comparison of the toxemia model with clinical sepsis
| Similarities with Human Disease | Differences with Human Disease |
|---|---|
| Endotoxin induces shock state in both mice and humans | Humans more sensitive than mice to endotoxin The dose of lipopolysaccharide (LPS) resulting in 50% death (LD50) in mice is approximately 1–25 mg/kg body weight,61–63 whereas an LPS dose of 2–4 ng/kg body weight in humans can inflict severe sickness64,65 Model does not recapitulate the clinical hemodynamic phases Different cytokine responses |
In general, the culprit of the numerous discrepancies observed between the toxemia models of sepsis in comparison with clinical pathology is inherently attributed to the differential physiology between humans and mice. For example, hemopexin, an iron-binding acute-phase protein that is present in mouse serum but absent from human serum, may account for the difference in LPS sensitivity between mice and humans.37 These soluble factors have been suggested to suppress the production of proinflammatory cytokines by peripheral blood mononuclear cells when challenged by endotoxins such as LPS, or other exogenous danger signals recognized by pattern recognition receptors.38
Live Bacteria Infection
Another method to induce sepsis in mice is by inoculating viable bacteria into animals. Depending on the experimental design, single or mixed bacterial flora can be introduced into mice through either the intravenous or intraperitoneal route. In addition, topical administration of live bacteria at the injury site (eg. burns, trauma models) is also an option to mimic post-injury infections observed in clinical settings. However, when compared with clinical pathology, this method of sepsis induction still possesses several major pitfalls. In mice, low bacterial inoculant leads to clearance, whereas high bacterial load results in complement fixation, resulting in rapid bacteria lysis.39 Therefore, it is very difficult for this model to recapitulate the physiologic consequences of bacterial growth in the host and organ infiltration reported in the clinic, as the septic-like response observed in the mouse model is mainly inflicted by endotoxemia.
In addition to the aforementioned caveat, there have been reports describing differential routes of bacterial inoculation leading to unique cytokine responses. For example, bacterial inoculation into the blood compartment of animal models results in the strong generation of proinflammatory cytokines (eg, TNF-α, IL-6, IL-1), whereas peritoneal administration does not lead to such a robust cytokine response.40,41 Interestingly the anti-inflammatory cytokine IL-10 has a protective role in sepsis induced by peritoneal administration of bacteria, in contrast to its worsening effects in the lung infection models.42–44 Collectively, these differences suggest that there may be an organ-specific or site-specific immunity mounted against microbial infections, and these unique responses may account for the reported differences between mouse models of sepsis and clinical incidents, as outlined in Table 4.
Table 4.
Comparison of the live bacterial infection model with clinical sepsis
| Similarities with Human Disease | Differences with Human Disease |
|---|---|
| Induces a similar shock state that is observed in human bacterial infections This model is more accurate in recapitulating a clinical infectious scenario in contrast to the artificial LPS administration approach Dosage and the viable phases of bacteria inoculant can be manipulated to mimic clinical infection, sepsis, or SIRS in mice |
High inoculating bacterial dose in mice often results in rapid lysis of bacteria by complement39
Pathologic phenotypes observed in model is likely due to endotoxemia and not infection Route of bacterial administration (peritoneal cavity, blood, lungs) may have confounding roles in cytokine functions and the mediation/alleviation of sepsis pathology |
Host Barrier Disruption
The most credible animal model of sepsis involves the perturbation of endogenous physical barriers, consequently granting bacterial access to sterile compartments originally inaccessible (eg, peritoneal cavity). Two major host barrier-disruption mouse models are currently implemented in laboratory settings: cecal ligation and puncture (CLP)45 and colon ascendens stent peritonitis (CASP).41 Owing to the extensive amount of information and complexity of these models, a point-by-point depiction of the relevant details is listed here.
Cecal ligation and puncture: similarities with human disease
Considered the gold standard for sepsis research
Mimics human appendicitis or perforated diverticulitis
Model established by midline laparotomy followed by the exteriorization of cecum, ligation of the cecum distal to the ileocecal valve, and finally the puncturing of the ligated cecum45,46
Creates a “hole” in the cecum whereby the animal is infected by mouse stool gradually leaking into, and accumulating, in the intra-abdominal space of the animal, serving as an inflammatory source, and subsequently generates necrotic tissue (further propagates the inflammation)
Severity of the disease (assessed by mortality) can be manipulated by the size of the “hole”, which is determined by the needle gauge used for puncture, or the amount of punctures given to the animal46
This model can mimic both the early and late (irreversible) phases of sepsis, as surgical excision of necrotic tissue beyond certain time points is unable to improve survival
The CLP model recreates the hemodynamic, metabolic, and immunologic phases of human sepsis45
Cecal ligation and puncture: differences with human disease
Variable outcomes reported between laboratories, whereby some CLP models using 20-gauge needles for the puncture show higher survival rates than those using 22-gauge needles (would hypothesize the opposite)47
The strains of bacteria that cause the infection in CLP may not represent the flora that are commonly depicted as the culprits of infection, which leads to clinical sepsis (eg, P. aeruginosa)
Most reports of clinical sepsis and patients that exhibit septic shock are pediatric or elderly patients,1 whereas adult mice are the usual candidates when using mouse models; the age discrepancy of model versus clinic should not be overlooked
Cecal ligation and puncture: possible explanations for the observed differences
Despite the model being able to recapitulate the hemodynamic, metabolic, and immunologic phases of human sepsis, it is still very difficult to stage human sepsis with this model alone (especially the end stage of sepsis, such as severe sepsis and septic shock), because of the lack of the common and regular physiologic monitoring of these models that is available to humans in the clinic
The infectious bacterial flora in CLP may be different from those in natural infections, so the immunologic responses observed in these models in comparison with the clinic can obviously be different
The age discrepancy between mouse models of sepsis and clinical sepsis may suggest a differential composition and efficacy of their immune system in response to bacterial insult; therefore, results from adult mouse models may not match clinical reports, and should be interpreted with caution when extrapolating to pediatric/elderly patients
Colon ascendens stent peritonitis: similarities with human disease
A stent is inserted into the colon ascendens of the animal, which causes a continuous leakage of fecal matter into the peritoneal cavity41,46
The diameter of the inserted stent determines the pathologic severity of the sepsis: the bigger the diameter the greater the stool leakage, incidence of infection, and sepsis progression41
Lethality observed in CASP is usually a consequence of sepsis-induced multiorgan failure (lung, liver, and kidney), which is highly reminiscent of clinical reports of sepsis-induced death
CASP mimics the proinflammatory profile of clinical sepsis, as demonstrated by the rapid elevation of proinflammatory cytokines (eg, TNF-a, IL-1) 3 hours after stent insertion41,48
Colon ascendens stent peritonitis: differences with human disease
Does not recapitulate the hemodynamic phases of clinical sepsis46
The anti-inflammatory cytokine response occurs simultaneously with the proinflammatory (3 hours after stent insertion), which deviates from the 2-phase immune profile (proinflammatory [SIRS] followed by anti-inflammatory [CARS]) of clinical sepsis41,48,49
Like CLP, multiple bacterial flora originating from fecal matter of the animal will act in concert to induce sepsis, which may confound when comparing with the culprit bacterial species of clinical sepsis
Colon ascendens stent peritonitis: possible explanations for the observed differences
In comparison with other models, the CASP model is not as well characterized in its representation of the hemodynamic, metabolic, and immunologic phases of human sepsis
Cecal ligation and puncture versus colon ascendens stent peritonitis
- In CLP the disruption of the host barrier cannot be easily reversed (or reversed at all); in CASP the stent can be removed, thus reducing or eliminating the source of bacterial accumulation in the peritoneal cavity
- The option of reversing the disrupted host barrier in mouse models is to mimic surgical interventions in clinical sepsis
- In CLP the site of infection is the intra-abdominal space, as shown by the abscess formation postsurgical maneuver; in CASP, the peritoneal cavity is the susceptible niche where bacterial accumulation and infection takes place
- Therefore, CLP and CASP actually model 2 different kinds of disease that will both eventually lead to sepsis
As the pathogenesis between the models differs (peritoneal vs intra-abdominal), it is evident that the immunologic responses observed in both models can be different, and will be difficult to compare
CASP is more surgically challenging than CLP
DISCUSSION
As described earlier, the primary concern of using mouse models to simulate clinical sepsis is that a single model usually depicts only a particular type of sepsis, or delineates partially the clinical pathophysiology of septic patients. After surveying the common sepsis mouse models used in research laboratories, it is apparent that at least immunologically, most of these models can only mimic the acute septic response. This information may be trivially interesting, but lacks clinical applicability, considering that most clinical sepsis indicators are reported during the late or irreversible phases of sepsis. Nonetheless, this realization provides a rationale for explaining the poor efficacy thus far of pharmacologic interventions attempting to treat clinical sepsis, which mostly involve the neutralization of the acute proinflammatory response.50 It is inherently flawed to attempt to translate acute-phase sepsis regimens used in mouse models at the bench in the hopes of rescuing late-phase sepsis in the clinic.
The outstanding, yet fundamental question that still remains is: how do we accurately model clinical sepsis in animals to design appropriate and effective therapeutics? This question can potentially be approached in 2 ways: the development of additional mouse models to more accurately reflect the late phases of clinical sepsis; or finding a set of biomarkers that can detect clinical sepsis in the acute phase, which can be implemented to make comparisons with the abundance of acute-phase–centered sepsis data generated over the last 2 decades. In terms of developing or improving mouse models, progress by the scientific community has abated since the development of CLP and CASP, which were introduced and published in 198045 and 1998,41 respectively. Although these barrier-disruption models are acknowledged as the gold standard of laboratory sepsis models, they each possess multiple flaws in the simulation of clinical sepsis, particularly with the late phases. Lastly, a recent extensive study comparing the genomic regulation in response to trauma, burns, and infections between humans and mice showed very little correlation between the 2 species,51 suggesting, at least in the context of these insults, that a xenogeneic comparison of mouse with human may not be suitable.
Therefore, perhaps the best method to treat clinical sepsis should derive from a clinical research–based approach: the identification of a collection of biomarkers in preseptic patients that can predict their vulnerability to septic onset. Take, for example, a hypothetical course of hospitalization and complication of a thermally injured patient. Here, thermal injury serves as a clinical model whereby burned patients are now susceptible to opportunistic infections because of a damaged skin barrier. Subsequently, the immunologic profiles of these burned patients who eventually develop sepsis can be compared with their thermally injured, nonseptic counterparts (either infected or noninfected). The course of this patient study should be divided into at least 3 sections: preinfected (immediate post-thermal injury), infected, and septic, with blood work collected at all 3 stages. Preinfected, infected, and septic cytokine profiles of these patients can be generated via high-throughput enzyme-linked immunosorbent–based assays.
This approach offers several advantages over mouse modeling of sepsis: First, it circumvents the flaws of the mouse-human xenogeneic comparison (eg, genetic differences due to speciation, the confounding effects of clinical resuscitation maneuvers not given to mice). Second, the comparison of patient data can be stratified so that only patients of identical age range, gender, severity of burns, and sepsis diagnosis will be compared. Lastly, once a large enough sample size is achieved, statistical models can be tailored and implemented (eg, multiparameter analysis of variance) to evaluate the validity of this scientific design, while addressing the effects of the confounding factors such as comorbidities, ethnicities, and the different genetic background of this patient pool. Unpublished data (Chen et al. 2014) from the authors’ laboratory shows that there is a 5- to 7-fold increase of several anti-inflammatory cytokines in the plasma of preinfected burned patients who eventually develop sepsis (blood collected 96 hours after burn injury with no reported infection) in comparison with thermally injured patients who never develop sepsis. A robust plasma proinflammatory cytokine profile was detected in both groups, with no statistical difference.
Albeit preliminary, what do these data mean? The authors hypothesize that perhaps the pivotal determinant of sepsis progression may not be the proinflammatory response, but the degree of foreshadowing anti-inflammatory response before or during bacterial infection, which will ultimately affect the extent of bacterial clearance in these hosts. As shown in Fig. 1A, clinical patients who do not develop sepsis have a blood microenvironment that is characterized by a slightly proinflammatory state in the preinfected phase (the net result of medium-grade proinflammatory and low-grade anti-inflammatory response). This concept is intuitive because most burned patients (or trauma patients) have suffered some form of insult, which will lead to the production of proinflammatory cytokines in response to endogenous DAMP molecules as a result of inflammasome activation.52 These patients, usually with wound openings, are now more susceptible to microbial infections in comparison with healthy individuals. On bacterial invasion of the wound site, the microbes will elevate and perpetuate the proinflammatory response of the host, thus facilitating bacterial clearance. After bacterial clearance an anti-inflammatory response follows, resulting in the neutralization of the proinflammatory response, ultimately restoring the neutral immunologic state of the host. By contrast, an abundance of anti-inflammatory cytokines is already present in the plasma of patients who eventually develop sepsis (see Fig. 1B). In this scenario, the blood microenvironment is in an anti-inflammatory state. If bacteria invade the wound of this host, the host’s immunologic status is not conducive to bacterial clearance; thus the bacterial may be able to escape the immune surveillance of the host, and quickly establish logarithmic growth phase in the host, consequently leading to organ infiltration and, ultimately, sepsis.
Fig. 1.
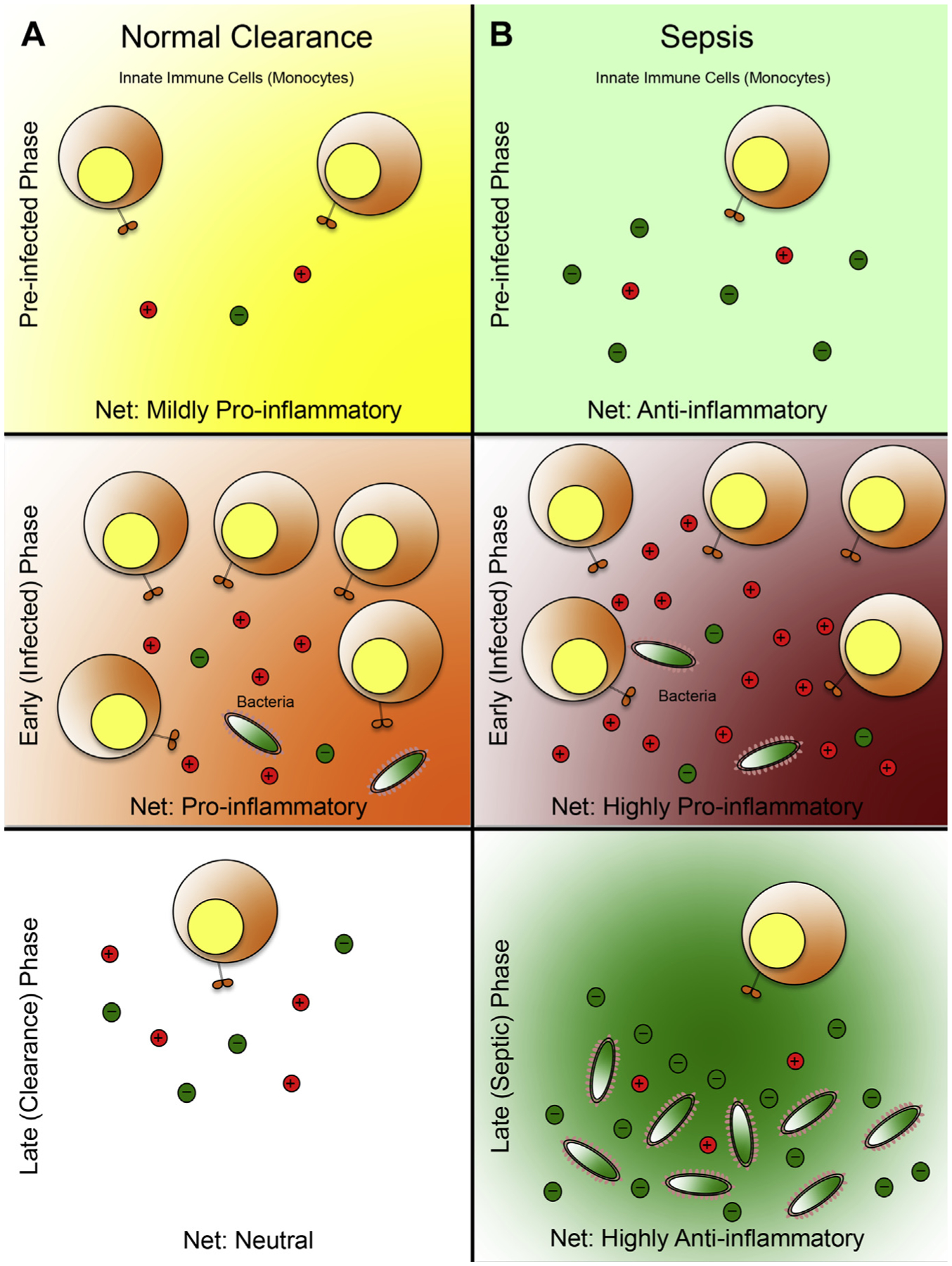
Systemic immunologic profile as a predictor of sepsis: a working model. (A) Microbial clearance of a patient who experiences opportunistic infection. In the preinfected phase, the patient blood microenvironment is in a mild proinflammatory state resulting from the mounted immune response against danger-associated molecular patterns (DAMP) molecules (generated in the process of the trauma or burn). This low-grade proinflammatory state primes the patient’s immune system to sense external insults, thus facilitating efficient detection of microbial infiltration. During the early, or infected phase, the invading bacteria are detected by the circulating leukocytes that were recruited to combat the original insult (trauma, burn). This detection results in a heightened proinflammatory response, which leads to additional proinflammatory cytokine production and recruitment of leukocytes, ultimately leading to bacterial eradication. In the late or clearance phase the invading microbes are eliminated, and the immune system is restored to a neutral state by a balance of proinflammatory and anti-inflammatory signals. (B) Sepsis development in an infected patient. In the preinfected phase, the net anti-inflammatory state of the blood microenvironment is defined by low-grade proinflammatory response mounted against the DAMP molecules, and an abundance of anti-inflammatory cytokines, produced possibly by the liver (the trigger for this response remains elusive). During the early/infected phase, a proinflammatory response is mounted against the invading microbes by the recruited leukocytes. However, because the blood microenvironment before the infected phase was anti-inflammatory, these recruited leukocytes must produce aberrantly higher levels of proinflammatory cytokines to achieve a net proinflammatory state to offset the effects of the preexisting anti-inflammatory soluble factors. As a result, these leukocytes will likely overexert their effector function and rapidly deplete their effector function potential, leading to immune exhaustion. This state is reminiscent of the systemic immune response syndrome (SIRS), or the classic cytokine storm that is observed in the acute phases of sepsis. Importantly this strong, yet relatively brief proinflammatory state does not lead to sterile immunity, which would lead to bacterial evasion from the host’s immune surveillance. In the late/sepsis phase, the immune system is exhausted and is therefore unable to produce additional proinflammatory mediators, despite having the residual bacteria from the infected phase. Furthermore, to counter the spike of proinflammatory signals in the infected phase, a massive surge in anti-inflammatory response is produced, resulting in a net anti-inflammatory microenvironment, similar to the compensatory anti-inflammatory response syndrome, which is conducive to bacterial growth, dissemination, and, ultimately, sepsis. Spheres with plus symbols denote proinflammatory mediators; spheres with minus symbols denote anti-inflammatory mediators. The immunologic state of each phase is determined by the ratio of proinflammatory mediators relative to anti-inflammatory mediators (mildly proinflammatory, 2:1; proinflammatory, 3:1; neutral, 1:1; anti-inflammatory, 1:3; highly proinflammatory, 5:1; highly anti-inflammatory, 1:5).
Although there are several unanswered questions about this model (eg, what is the factor or factors that are triggering the initial production and accumulation of anti-inflammatory cytokines in the septic patient?), the primary goal of this schematic is to highlight the potential importance of the preinfected, or prelude, phase of sepsis pathology, which seems to have been be underappreciated in the past. Here the authors propose to adjust the conventional biphasic definition of sepsis (defined by early and late) into a multiphasic trilogy (preinfected, infected/early, sepsis onset/late) via the incorporation of this prelude phase. In conjunction with the immunologic cytokine profiles associated with each phase, it may help shed light on the development of novel animal models that can more accurately reflect clinical sepsis, which one day might serve as the foundation for the development of effective regimens for septic patients.
KEY POINTS.
Murine models do not accurately reflect human sepsis.
Detection of clinical sepsis usually occurs during the late phases of the disease progression, and is often associated with irreversible damage to patients.
Most of the existing mouse models of sepsis reflect only the immunologic aspect of the disease, and this xenogeneic comparison is suitable only when comparing the early phases of this clinical disorder.
Instead of the conventional biphasic approach to modeling sepsis, sepsis should be perceived as a triphasic phenomenon by incorporating a preinfected state as a part of the pathologic simulation.
Progression of sepsis may be foreshadowed by the immunologic state of the patient before infection, which is determined by the net effect of proinflammatory and anti-inflammatory modulators.
Acknowledgments
This work is supported by grants from the National Institutes of Health (R01 GM087285-01); Canadian Institutes of Health Research Funds (123336), CFI Leader’s Opportunity Fund (Project #25407); Physician’s Services Incorporated Foundation: Health Research Grant Program.
REFERENCES
- 1.Angus DC, Linde-Zwirble WT, Lidicker J, et al. Epidemiology of severe sepsis in the United States: analysis of incidence, outcome, and associated costs of care. Crit Care Med 2001;29(7):1303–10. [DOI] [PubMed] [Google Scholar]
- 2.HCUP Facts and Figures, 2006. Statistics on hospital-based care in the United States. Rockville (MD): Agency for Healthcare Research and Quality (US), 2008. [PubMed] [Google Scholar]
- 3.Funk DJ, Parrillo JE, Kumar A. Sepsis and septic shock: a history. Crit Care Clin 2009;25(1):83–101, viii. [DOI] [PubMed] [Google Scholar]
- 4.LaRosa SP, Opal SM. Sepsis strategies in development. Clin Chest Med 2008; 29(4):735–47, x–xi. [DOI] [PubMed] [Google Scholar]
- 5.Opal SM, Garber GE, LaRosa SP, et al. Systemic host responses in severe sepsis analyzed by causative microorganism and treatment effects of drotrecogin alfa (activated). Clin Infect Dis 2003;37(1):50–8. [DOI] [PubMed] [Google Scholar]
- 6.Gram-positive Ramachandran G. and gram-negative bacterial toxins in sepsis: a brief review. Virulence 2013;5(1):213–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Ranieri VM, Thompson BT, Barie PS, et al. Drotrecogin alfa (activated) in adults with septic shock. N Engl J Med 2012;366(22):2055–64. [DOI] [PubMed] [Google Scholar]
- 8.Martin GS, Mannino DM, Eaton S, et al. The epidemiology of sepsis in the United States from 1979 through 2000. N Engl J Med 2003;348(16): 1546–54. [DOI] [PubMed] [Google Scholar]
- 9.American College of Chest Physicians/Society of Critical Care Medicine Consensus Conference: definitions for sepsis and organ failure and guidelines for the use of innovative therapies in sepsis. Crit Care Med 1992;20(6):864–74. [PubMed] [Google Scholar]
- 10.Levy MM, Fink MP, Marshall JC, et al. 2001 SCCM/ESICM/ACCP/ATS/SIS International Sepsis Definitions Conference. Crit Care Med 2003;31(4):1250–6. [DOI] [PubMed] [Google Scholar]
- 11.Dellinger RP, Carlet JM, Masur H, et al. Surviving Sepsis Campaign guidelines for management of severe sepsis and septic shock. Crit Care Med 2004;32(3): 858–73. [DOI] [PubMed] [Google Scholar]
- 12.Calfee CS, Matthay MA. Clinical immunology: culprits with evolutionary ties. Nature 2010;464(7285):41–2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Hotchkiss RS, Karl IE. The pathophysiology and treatment of sepsis. N Engl J Med 2003;348(2):138–50. [DOI] [PubMed] [Google Scholar]
- 14.Boomer JS, Green JM, Hotchkiss RS. The changing immune system in sepsis: is individualized immuno-modulatory therapy the answer? Virulence 2013;5(1): 45–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Casey LC. Immunologic response to infection and its role in septic shock. Crit Care Clin 2000;16(2):193–213. [DOI] [PubMed] [Google Scholar]
- 16.Giamarellos-Bourboulis EJ, Raftogiannis M. The immune response to severe bacterial infections: consequences for therapy. Expert Rev Anti Infect Ther 2012;10(3):369–80. [DOI] [PubMed] [Google Scholar]
- 17.Christ-Crain M, Morgenthaler NG, Struck J, et al. Mid-regional proadrenomedullin as a prognostic marker in sepsis: an observational study. Crit Care 2005;9(6):R816–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Gabay C, Kushner I. Acute-phase proteins and other systemic responses to inflammation. N Engl J Med 1999;340(6):448–54. [DOI] [PubMed] [Google Scholar]
- 19.Adib-Conquy M, Cavaillon JM. Compensatory anti-inflammatory response syndrome. Thromb Haemost 2009;101(1):36–47. [PubMed] [Google Scholar]
- 20.Bone RC, Grodzin CJ, Balk RA. Sepsis: a new hypothesis for pathogenesis of the disease process. Chest 1997;112(1):235–43. [DOI] [PubMed] [Google Scholar]
- 21.Cavaillon JM, Annane D. Compartmentalization of the inflammatory response in sepsis and SIRS. J Endotoxin Res 2006;12(3):151–70. [DOI] [PubMed] [Google Scholar]
- 22.Ward NS, Casserly B, Ayala A. The compensatory anti-inflammatory response syndrome (CARS) in critically ill patients. Clin Chest Med 2008;29(4):617–25, viii. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Fischer E, Van Zee KJ, Marano MA, et al. Interleukin-1 receptor antagonist circulates in experimental inflammation and in human disease. Blood 1992; 79(9):2196–200. [PubMed] [Google Scholar]
- 24.Marchant A, Deviere J, Byl B, et al. Interleukin-10 production during septicaemia. Lancet 1994;343(8899):707–8. [DOI] [PubMed] [Google Scholar]
- 25.Marie C, Cavaillon JM, Losser MR. Elevated levels of circulating transforming growth factor-beta 1 in patients with the sepsis syndrome. Ann Intern Med 1996;125(6):520–1. [DOI] [PubMed] [Google Scholar]
- 26.Munford RS, Pugin J. Normal responses to injury prevent systemic inflammation and can be immunosuppressive. Am J Respir Crit Care Med 2001;163(2): 316–21. [DOI] [PubMed] [Google Scholar]
- 27.Astiz M, Saha D, Lustbader D, et al. Monocyte response to bacterial toxins, expression of cell surface receptors, and release of anti-inflammatory cytokines during sepsis. J Lab Clin Med 1996;128(6):594–600. [DOI] [PubMed] [Google Scholar]
- 28.Doughty L, Carcillo JA, Kaplan S, et al. The compensatory anti-inflammatory cytokine interleukin 10 response in pediatric sepsis-induced multiple organ failure. Chest 1998;113(6):1625–31. [DOI] [PubMed] [Google Scholar]
- 29.van Dissel JT, van Langevelde P, Westendorp RG, et al. Anti-inflammatory cytokine profile and mortality in febrile patients. Lancet 1998;351(9107):950–3. [DOI] [PubMed] [Google Scholar]
- 30.Gomez HG, Gonzalez SM, Londono JM, et al. Immunological characterization of compensatory anti-inflammatory response syndrome in patients with severe sepsis: a longitudinal study. Crit Care Med 2014;42(4):771–80. [DOI] [PubMed] [Google Scholar]
- 31.Parker M, Parrillo J. Septic shock. Hemodynamics and pathogenesis. JAMA 1983;250(24):3324–7. [PubMed] [Google Scholar]
- 32.Abraham E, Shoemaker W, Bland R, et al. Sequential cardiorespiratory patterns in septic shock. Crit Care Med 1983;11(10):799–803. [DOI] [PubMed] [Google Scholar]
- 33.Fink M, Heard S. Laboratory models of sepsis and septic shock. J Surg Res 1990;49(2):186–96. [DOI] [PubMed] [Google Scholar]
- 34.Deitch EA. Animal models of sepsis and shock: a review and lessons learned. Shock 1998;9(1):1–11. [DOI] [PubMed] [Google Scholar]
- 35.Poltorak A, He X, Smirnova I, et al. Defective LPS signaling in C3H/HeJ and C57BL/10ScCr mice: mutations in Tlr4 gene. Science 1998;282(5396):2085–8. [DOI] [PubMed] [Google Scholar]
- 36.Hemmi H, Takeuchi O, Kawai T, et al. A Toll-like receptor recognizes bacterial DNA. Nature 2000;408(6813):740–5. [DOI] [PubMed] [Google Scholar]
- 37.Liang X, Lin T, Sun G, et al. Hemopexin down-regulates LPS-induced proinflammatory cytokines from macrophages. J Leukoc Biol 2009;86(2):229–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Warren H, Fitting C, Hoff E, et al. Resilience to bacterial infection: difference between species could be due to proteins in serum. J Infect Dis 2010;201(2):223–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Cross AS, Opal SM, Sadoff JC, et al. Choice of bacteria in animal models of sepsis. Infect Immun 1993;61(7):2741–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Evans GF, Snyder YM, Butler LD, et al. Differential expression of interleukin-1 and tumor necrosis factor in murine septic shock models. Circ Shock 1989; 29(4):279–90. [PubMed] [Google Scholar]
- 41.Zantl N, Uebe A, Neumann B, et al. Essential role of gamma interferon in survival of colon ascendens stent peritonitis, a novel murine model of abdominal sepsis. Infect Immun 1998;66(5):2300–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Greenberger MJ, Strieter RM, Kunkel SL, et al. Neutralization of IL-10 increases survival in a murine model of Klebsiella pneumonia. J Immunol 1995;155(2):722–9. [PubMed] [Google Scholar]
- 43.van der Poll T, Marchant A, Buurman WA, et al. Endogenous IL-10 protects mice from death during septic peritonitis. J Immunol 1995;155(11):5397–401. [PubMed] [Google Scholar]
- 44.van der Poll T, Marchant A, Keogh CV, et al. Interleukin-10 impairs host defense in murine pneumococcal pneumonia. J Infect Dis 1996;174(5):994–1000. [DOI] [PubMed] [Google Scholar]
- 45.Wichterman KA, Baue AE, Chaudry IH. Sepsis and septic shock—a review of laboratory models and a proposal. J Surg Res 1980;29(2):189–201. [DOI] [PubMed] [Google Scholar]
- 46.Buras JA, Holzmann B, Sitkovsky M. Animal models of sepsis: setting the stage. Nat Rev Drug Discov 2005;4(10):854–65. [DOI] [PubMed] [Google Scholar]
- 47.Baker CC, Chaudry IH, Gaines HO, et al. Evaluation of factors affecting mortality rate after sepsis in a murine cecal ligation and puncture model. Surgery 1983; 94(2):331–5. [PubMed] [Google Scholar]
- 48.Maier S, Traeger T, Entleutner M, et al. Cecal ligation and puncture versus colon ascendens stent peritonitis: two distinct animal models for polymicrobial sepsis. Shock 2004;21(6):505–11. [DOI] [PubMed] [Google Scholar]
- 49.Emmanuilidis K, Weighardt H, Maier S, et al. Critical role of Kupffer cell-derived IL-10 for host defense in septic peritonitis. J Immunol 2001;167(7):3919–27. [DOI] [PubMed] [Google Scholar]
- 50.Fink MP. Animal models of sepsis. Virulence 2013;5(1):143–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Seok J, Warren HS, Cuenca AG, et al. Genomic responses in mouse models poorly mimic human inflammatory diseases. Proc Natl Acad Sci U S A 2013; 110(9):3507–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Stanojcic M, Chen P, Harrison R, et al. Leukocyte infiltration and activation of the nlrp3 inflammasome in white adipose tissue following thermal injury. Crit Care Med 2014;42(6):1357–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.King EG, Bauza GJ, Mella JR, et al. Pathophysiologic mechanisms in septic shock. Lab Invest 2014;94(1):4–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Hotchkiss RS, Tinsley KW, Swanson PE, et al. Depletion of dendritic cells, but not macrophages, in patients with sepsis. J Immunol 2002;168(5):2493–500. [DOI] [PubMed] [Google Scholar]
- 55.Drifte G, Dunn-Siegrist I, Tissieres P, et al. Innate immune functions of immature neutrophils in patients with sepsis and severe systemic inflammatory response syndrome. Crit Care Med 2013;41(3):820–32. [DOI] [PubMed] [Google Scholar]
- 56.Taneja R, Parodo J, Jia SH, et al. Delayed neutrophil apoptosis in sepsis is associated with maintenance of mitochondrial transmembrane potential and reduced caspase-9 activity. Crit Care Med 2004;32(7):1460–9. [DOI] [PubMed] [Google Scholar]
- 57.Stearns-Kurosawa DJ, Osuchowski MF, Valentine C, et al. The pathogenesis of sepsis. Annu Rev Pathol 2011;6:19–48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Hotchkiss RS, Tinsley KW, Swanson PE, et al. Sepsis-induced apoptosis causes progressive profound depletion of B and CD4+ T lymphocytes in humans. J Immunol 2001;166(11):6952–63. [DOI] [PubMed] [Google Scholar]
- 59.Venet F, Pachot A, Debard AL, et al. Increased percentage of CD4+CD25+ regulatory T cells during septic shock is due to the decrease of CD4+CD25− lymphocytes. Crit Care Med 2004;32(11):2329–31. [DOI] [PubMed] [Google Scholar]
- 60.Opal SM, DePalo VA. Anti-inflammatory cytokines. Chest 2000;117(4):1162–72. [DOI] [PubMed] [Google Scholar]
- 61.Glode LM, Mergenhagen SE, Rosenstreich DL. Significant contribution of spleen cells in mediating the lethal effects of endotoxin in vivo. Infect Immun 1976;14(3):626–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.McCuskey RS, McCuskey PA, Urbaschek R, et al. Species differences in Kupffer cells and endotoxin sensitivity. Infect Immun 1984;45(1):278–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Reynolds K, Novosad B, Hoffhines A, et al. Pretreatment with troglitazone decreases lethality during endotoxemia in mice. J Endotoxin Res 2002;8(4): 307–14. [DOI] [PubMed] [Google Scholar]
- 64.Barber AE, Coyle SM, Fischer E, et al. Influence of hypercortisolemia on soluble tumor necrosis factor receptor II and interleukin-1 receptor antagonist responses to endotoxin in human beings. Surgery 1995;118(2):406–10 [discussion: 410–1]. [DOI] [PubMed] [Google Scholar]
- 65.Suffredini AF, Reda D, Banks SM, et al. Effects of recombinant dimeric TNF receptor on human inflammatory responses following intravenous endotoxin administration. J Immunol 1995;155(10):5038–45. [PubMed] [Google Scholar]