

SUMMARY
Human aging is frequently accompanied by the acquisition of somatic mutations in the hematopoietic system that induce clonal hematopoiesis, leading to the development of a mutant clone of hematopoietic progenitors and leukocytes. This somatic-mutation-driven clonal hematopoiesis has been associated with an increased incidence of cardiovascular disease and type 2 diabetes, but whether this epidemiological association reflects a direct, causal contribution of mutant hematopoietic and immune cells to age-related metabolic abnormalities remains unexplored. Here, we show that inactivating mutations in the epigenetic regulator TET2, which lead to clonal hematopoiesis, aggravate age- and obesity-related insulin resistance in mice. This metabolic dysfunction is paralleled by increased expression of the pro-inflammatory cytokine IL-1β in white adipose tissue, and it is suppressed by pharmacological inhibition of NLRP3 inflammasome-mediated IL-1β production. These findings support a causal contribution of somatic TET2 mutations to insulin resistance and type 2 diabetes.
Graphical Abstract
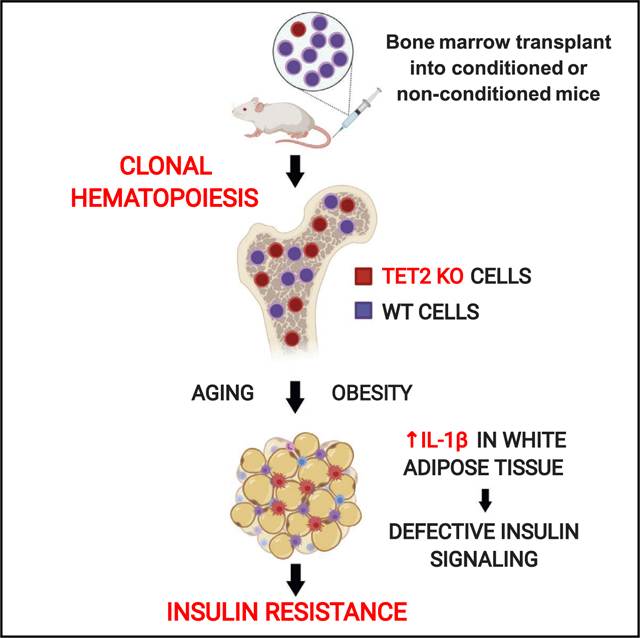
In Brief
Somatic-mutation-driven clonal hematopoiesis is emerging as a potent risk factor for a variety of age-related conditions. Fuster et al. show that clonal hematopoiesis driven by TET2 mutations aggravates insulin resistance in mice. These findings support a causal contribution of somatic TET2 mutations to metabolic syndrome and type 2 diabetes.
INTRODUCTION
Advanced age as a primary risk factor for cardiovascular and metabolic disease is now more apparent due to the progressive aging of the world population. Surprisingly, there is a limited understanding of how aging contributes to the pathophysiology of these conditions. In this context, recent human studies suggested an unexpected potential role of age-related somatic mutations in the hematopoietic system. Exome sequencing studies unveiled that hematopoietic stem cells accumulate somatic DNA mutations with aging, which sometimes confer a competitive advantage to the mutant cell, leading to its clonal expansion in the stem cell population and its leukocyte progeny (Buscarlet et al., 2017; Fuster and Walsh, 2018; Genovese et al., 2014; Jaiswal et al., 2014; Xie et al., 2014; Zink et al., 2017). This somatic-mutation-driven clonal hematopoiesis (CH) has been associated with a higher risk of cardiovascular disease (CVD) and type 2 diabetes (T2DM). Specifically, sequencing studies focused on the identification of single-nucleotide variants and small insertions/deletions in CH-driver genes revealed an association between the presence of these mutations in blood and a higher incidence of T2DM and atherosclerotic CVD (Jaiswal et al., 2014, 2017). Consistent with these findings, a previous study had found that clonally expanded chromosomal alterations in blood cells—some of them affecting CH-driver genes—are associated with a higher prevalence of T2DM and its vascular complications (Bonnefond et al., 2013). Collectively, these epidemiological associations suggested an unexpected connection between somatic-mutation-driven CH and the development of T2DM and CVD. However, whether these somatic mutations in blood cells contribute directly to the pathogenesis of these conditions remains a matter of debate and requires further investigation.
We recently provided experimental evidence supporting that somatic mutations in TET2 (ten-eleven translocation 2, one of the most frequently mutated genes in individuals exhibiting CH) accelerate atherosclerosis development (Fuster et al., 2017), supporting a causal role of these mutations in atherosclerotic CVD. The main objective of the experimental study presented herein was to investigate whether hematopoietic TET2 mutations and associated CH also impact glucose homeostasis and other T2DM-related metabolic conditions. Our findings demonstrate that CH driven by inactivation of this transcriptional regulator aggravates insulin resistance in mice in conditions of physiological aging and diet-induced obesity, which could partly explain the association between CH and T2DM observed in humans.
RESULTS
TET2-Loss-of-Function-Driven CH Exacerbates Age-Related Insulin Resistance in Mice
The prevalence of somatic TET2 mutations in hematopoietic cells is associated with age (Buscarlet et al., 2017; Fuster and Walsh, 2018; Genovese et al., 2014; Jaiswal et al., 2014; Xie et al., 2014; Zink et al., 2017). Therefore, we first evaluated the effects of TET2-mutation-driven CH on glucose homeostasis in conditions of physiological aging. CH is frequently modeled in mice by performing competitive bone marrow transplantation (BMT) experiments that involve recipient BM ablation through γ-radiation. While this approach is suitable for short-term experiments, it has limitations in the context of aging studies due to the confounding long-term side effects of genotoxic γ-radiation and the increasing evidence that post-BMT hematopoiesis and unperturbed hematopoiesis exhibit substantial differences (Busch and Rodewald, 2016; Lu et al., 2019). Therefore, we used an adoptive transfer strategy in non-conditioned, non-irradiated mice (Shimoto et al., 2017; Wang et al., 2020) to introduce a small proportion of Tet2−/− or Tet2+/+ hematopoietic cells in recipients and monitor the effects of TET2 mutant cells on glucose homeostasis and other metabolic parameters in the context of aging. Each recipient mouse was injected intravenously with a total of 1.5 × 107 unfractionated Tet2−/− or Tet2+/+ BM cells, split into three doses of 5 × 106 cells delivered on consecutive days (Figure 1A; see details in STAR Methods). To distinguish donor and recipient cells, C57BL/6 Pep Boy mice, which carry the CD45.1 isoform of the CD45 antigen, were used as recipients, whereas donor cells were obtained from mice carrying the CD45.2 isoform. Following transfer at 10 weeks of age, mice were monitored until the age of 84 weeks (approximately 1.6 years), with periodic evaluation of basic hematologic and metabolic parameters. Adoptive transfer led to stable engraftment of donor hematopoietic cells, as CD45.2+ white blood cells (WBCs) were detectable in blood at all timepoints following delivery (Figure 1B). Importantly, Tet2−/− CD45.2+ cells expanded progressively in this experimental setting (from ~6% of the WBC population 4 weeks after transfer to ~60% 73 weeks after transfer), in contrast to Tet2+/+ cells, which typically did not exceed 11% of peripheral WBCs. Tet2−/− cell expansion was detectable in Lin− Sca1+ cKit+ hematopoietic stem and progenitor cells (HSPCs) in the BM (Figure S1A) and in all peripheral WBC lineages (Figures 1C and S1B), although with varying kinetics depending on cell type. Expansion in the T-lymphoid lineage was limited, particularly in the CD8+ T cell population (~14%) (Figures 1C and S1B). This pattern of expansion may reflect the myeloid bias of hematopoiesis in aged mice and/or the different turnover rates and differentiation dynamics of the various BM-derived cell lineages, given that TET2-deficient blood cells were distributed in the myeloid and lymphoid compartment similar to wild-type (WT) cells (Figure S1C). Regardless, these mouse data are consistent with recent human studies reporting that most cancer-free individuals carrying somatic TET2 mutations harbor the mutation preferentially in myeloid and B-lymphoid populations, with a minor presence in the T-lymphoid lineage (Arends et al., 2018; Buscarlet et al., 2018). Similarly consistent with human data, the expansion of TET2-deficient cells in our experimental setting did not affect absolute numbers of WBCs or specific blood cell populations (Figure S1D), except for a trend of decreased neutrophil counts (p = 0.08, unpaired t test), which was also observed in TET2-mutation carriers in a previous human study (Buscarlet et al., 2017).
Figure 1. TET2-Loss-of-Function-Driven CH Aggravates Insulin Resistance and Hyperglycemia in Aged Mice.
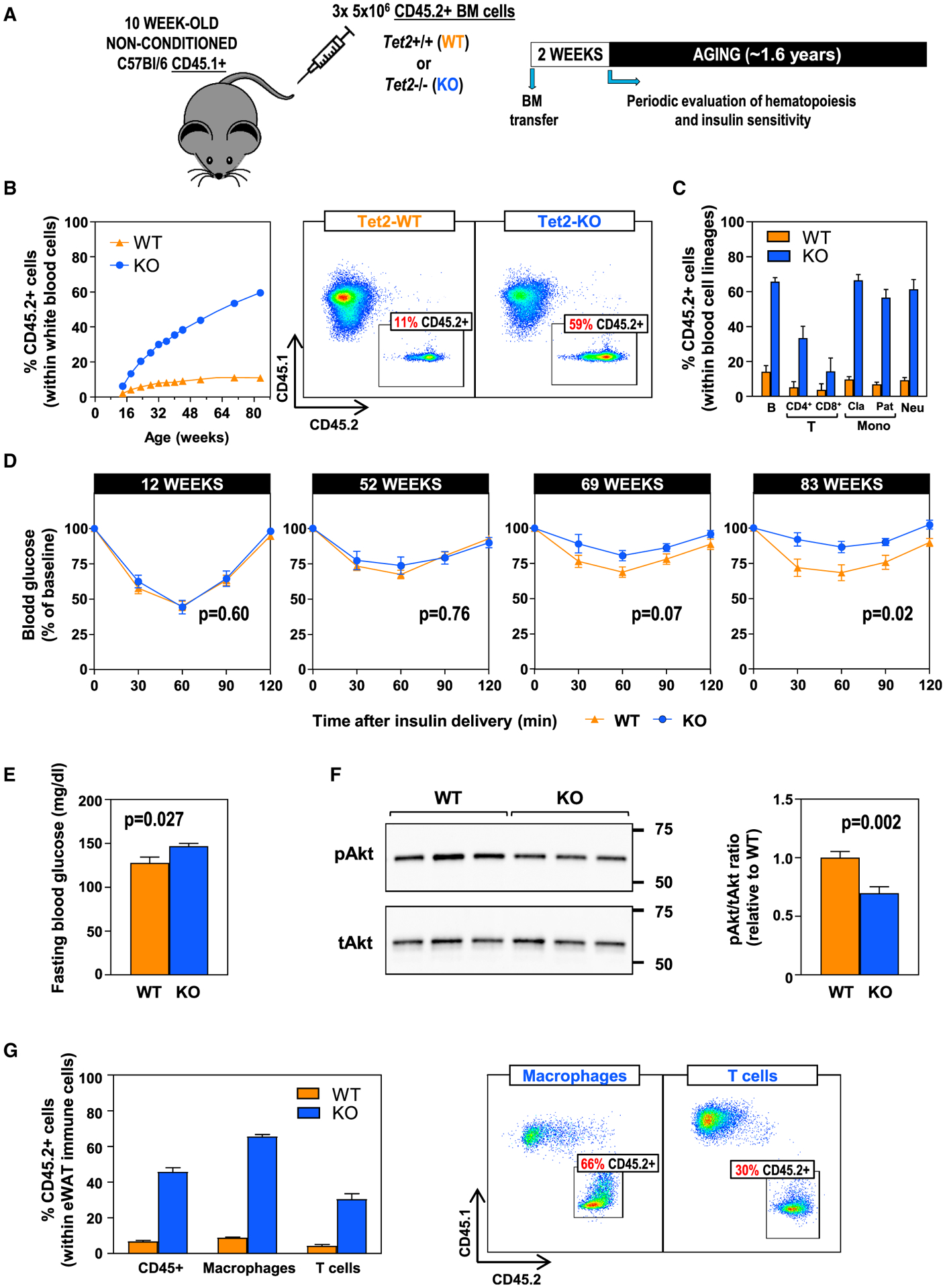
Tet2−/− (KO) or +/+ (WT) CD45.1−/CD45.2+ bone marrow (BM) cells were delivered via adoptive transfer to non-conditioned 10-week-old CD45.1+/CD45.2− recipient mice, which were then monitored until the age of 84 weeks (~1.6 years). n = 9 WT, 6 KO mice.
(A) Summary of the adoptive BM transfer approach in non-conditioned, non-irradiated mice and the timeline of these studies.
(B) Percentage of CD45.2+ cells in the WBC population in blood, evaluated by flow cytometry. Representative images of CD45.1/CD45.2 analysis in WBCs are shown.
(C) Percentage of CD45.2+ cells within main WBC lineages at 82 weeks of age, measured by flow cytometry. Cla, classical; Pat, patrolling monocytes.
(D) Insulin tolerance tests at different ages.
(E) Fasting blood glucose levels at 83 weeks of age.
(F) Western blot analysis of Akt phosphorylation in eWAT.
(G) Percentage of CD45.2+ cells within F4/80+ macrophage and CD3+ T cell populations in eWAT, evaluated by flow cytometry. Representative cytometry dot plots are shown.
Data are represented as mean ± SEM.
See also Figure S1.
Having validated the use of an adoptive transfer strategy to model TET2-inactivation-driven CH with aging, we next evaluated the impact of TET2-deficient cell expansion on age-related changes in body composition and metabolic function. There was a non-statistically significant trend toward increased mortality in mice that received TET2-deficient BM cells (Figure S1E). In mice that survived to the end of the experiment, the expansion of TET2-deficient hematopoietic cells had no effect on body weight or composition (Figures S1F–S1H). In contrast, a notable effect on the development of insulin resistance was observed. While the adoptive transfer of BM cells had no acute effect on insulin sensitivity in young mice, mice carrying TET2-deficient cells developed a progressive aggravation of systemic insulin resistance with aging (Figure 1D), which was accompanied by increased fasting blood glucose levels (Figure 1E). In addition, a significant reduction of steady-state Akt Ser473-phosphorylation was observed in epididymal white adipose tissue (eWAT) of these mice (Figure 1F), suggesting reduced insulin signaling in this tissue. TET2-deficient cells expanded into macrophage and T cell populations in eWAT to a degree consistent with their expansion in blood cells (Figure 1G). No differences in Akt phosphorylation were observed in the liver (Figure S1I). Collectively, these findings demonstrate that TET2-loss-of-function-driven CH aggravates insulin resistance in conditions of normal aging, without affecting body composition.
TET2-Loss-of-Function-Driven CH Exacerbates Obesity-Related Insulin Resistance in Mice
To better understand the effects of TET2-mutation-driven CH on glucose homeostasis, we validated our above-described findings in a model of obesity-induced insulin resistance. Given the shorter time frame for the development of insulin resistance in obesogenic diet-fed mice, we used a BMT strategy (Fuster et al., 2017) to mimic CH in these experiments (Figure 2A). CD45.1/CD45.2 heterozygous mice were irradiated with two doses of 400 rad, 3 h apart to ablate recipient BM cells, and were then transplanted with suspensions of unfractionated BM cells containing 90% CD45.1+ Tet2+/+ cells and 10% CD45.2+ Tet2−/− cells (10% knockout [KO] BMT) or 90% CD45.1+ Tet2+/+ cells and 10% CD45.2+ Tet2+/+ cells (10% WT BMT controls; see details in STAR Methods and Figure S2A). Starting 5 weeks post-irradiation, mice were fed high-fat/high-sucrose (HF/HS) obesogenic diet for 14 weeks. Over this time course, Tet2−/− BM cells expanded progressively into all blood cell lineages to an extent similar to that observed in our aging studies, with preferential expansion into the myeloid and B-lymphoid populations and a reduced expansion into the T-lymphoid lineage, both in blood (Figures 2B and 2C) and eWAT (Figure 2D). This relative expansion did not affect blood cell counts (Figure S2B), and no intrinsic myeloid or lymphoid bias was observed in TET2-deficient cells (Figure S2C). Tet2−/− cell expansion did not affect body weight (Figure 2E) or composition (Figures 2F and S2D). In contrast, it aggravated obesity-induced systemic insulin resistance, as assessed by insulin tolerance tests (Figure 2G), which was paralleled by increased fasting blood glucose levels (Figure 2H) and reduced insulin-induced Akt phosphorylation in eWAT (Figure 2I). No differences were observed in insulin-induced Akt phosphorylation in liver (Figure S2E), in spite of the similar percentage of TET2-deficient cells within macrophage populations in both WAT and liver (Figure S2F). Overall, these findings are in agreement with the results of the above-described aging studies and demonstrate that TET2-loss-of-function-driven CH also aggravates insulin resistance induced by HF/HS diet feeding and obesity.
Figure 2. TET2-Loss-of-Function-Driven CH Aggravates Insulin Resistance and Hyperglycemia in Obese Mice.
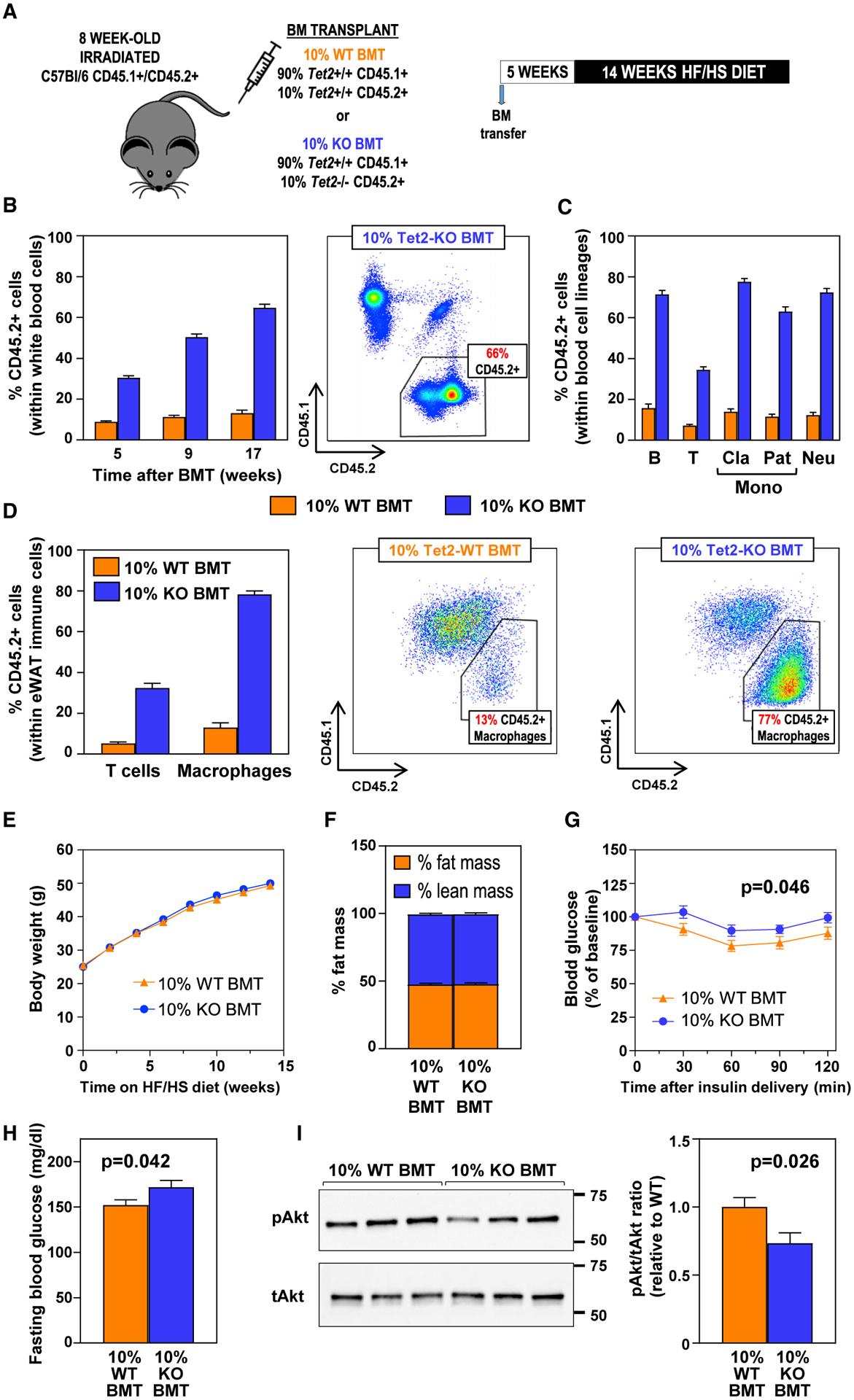
BMTs following whole-body irradiation were used to generate mice carrying 10% Tet2−/− (10% KO BMT) or +/+ (10% WT BMT) hematopoietic cells, which were then fed HF/HS diet for 14 weeks. n = 15 mice per BM genotype unless otherwise noted.
(A) Summary of the competitive BM transplantation approach and the timeline of these studies.
(B) Percentage of CD45.2+ cells in the WBC population in blood, evaluated by flow cytometry. A CD45.1/CD45.2 dot plot representative of 10% KO BMT mice is shown.
(C) Percentage of CD45.2+ cells within main WBC lineages after 12 weeks on HF/HS diet (17 weeks post-BMT), measured by flow cytometry.
(D) Percentage of CD45.2+ cells within F4/80+ macrophage and CD3+ T cell populations in eWAT after 14 weeks on HF/HS diet (19 weeks post-BMT), measured by flow cytometry (n = 7 for 10% WT BMT, n = 6 for 10% KO BMT mice). Representative dot plots of macrophage populations are shown.
(E) Body weight.
(F) Percentage of body fat and lean mass, assessed by magnetic resonance imaging (n = 11 mice per BM genotype).
(G) Insulin tolerance tests.
(H) Fasting blood glucose levels.
(I) Western blot analysis of insulin-induced Akt phosphorylation in eWAT (n = 6 mice per BM genotype).
Data are represented as mean ± SEM.
See also Figure S2.
NLRP3 Inflammasome-/IL-1β-Mediated Inflammation Is Essential for the Effects of TET2-Loss-of-Function-Driven CH on Glucose Homeostasis
Extensive evidence supports that adipose tissue inflammation is directly associated with insulin resistance, due in large part to the effects of macrophage-derived cytokines on adipocytes (Fuster et al., 2016; Saltiel and Olefsky, 2017). In this regard, we and others have previously shown that TET2 regulates macrophage expression of IL-1β (Cull et al., 2017; Fuster et al., 2017; Sano et al., 2018), a pro-inflammatory cytokine known to interfere with insulin signaling (Gao et al., 2014; Jager et al., 2007). In agreement with these previous results, ELISA analysis of eWAT samples revealed increased IL-1β protein levels in aged and obese mice carrying TET2-deficient cells (Figures 3A and 3B). Consistent changes in Il1b transcript levels were observed in eWAT of obese mice by quantitative PCR (qPCR), whereas no statistically significant change was detected in the transcript expression of a selected group of other pro-inflammatory cytokines and chemokines known to play a role in adipose tissue inflammation (Figure S2G). No differences in IL-1β or other cytokine expression were observed in liver or subcutaneous WAT (Figures S2H and S2I). Consistent with findings in eWAT, primary TET2-deficient macrophages in culture exhibited increased IL-1β secretion after lipopolysaccharide (LPS)/interferon (IFN)-γ priming and treatment with ceramide (Figure 3C), a lipid metabolite known to activate the NLRP3 inflammasome, a macromolecular complex that catalyzes IL-1β proteolytic processing and secretion in conditions of metabolic dysfunction (Vandanmagsar et al., 2011).
Figure 3. NLRP3-/IL-1β-Driven Inflammation Is Essential for the Effects of TET2-Loss-of-Function-Induced CH on Glucose Homeostasis.
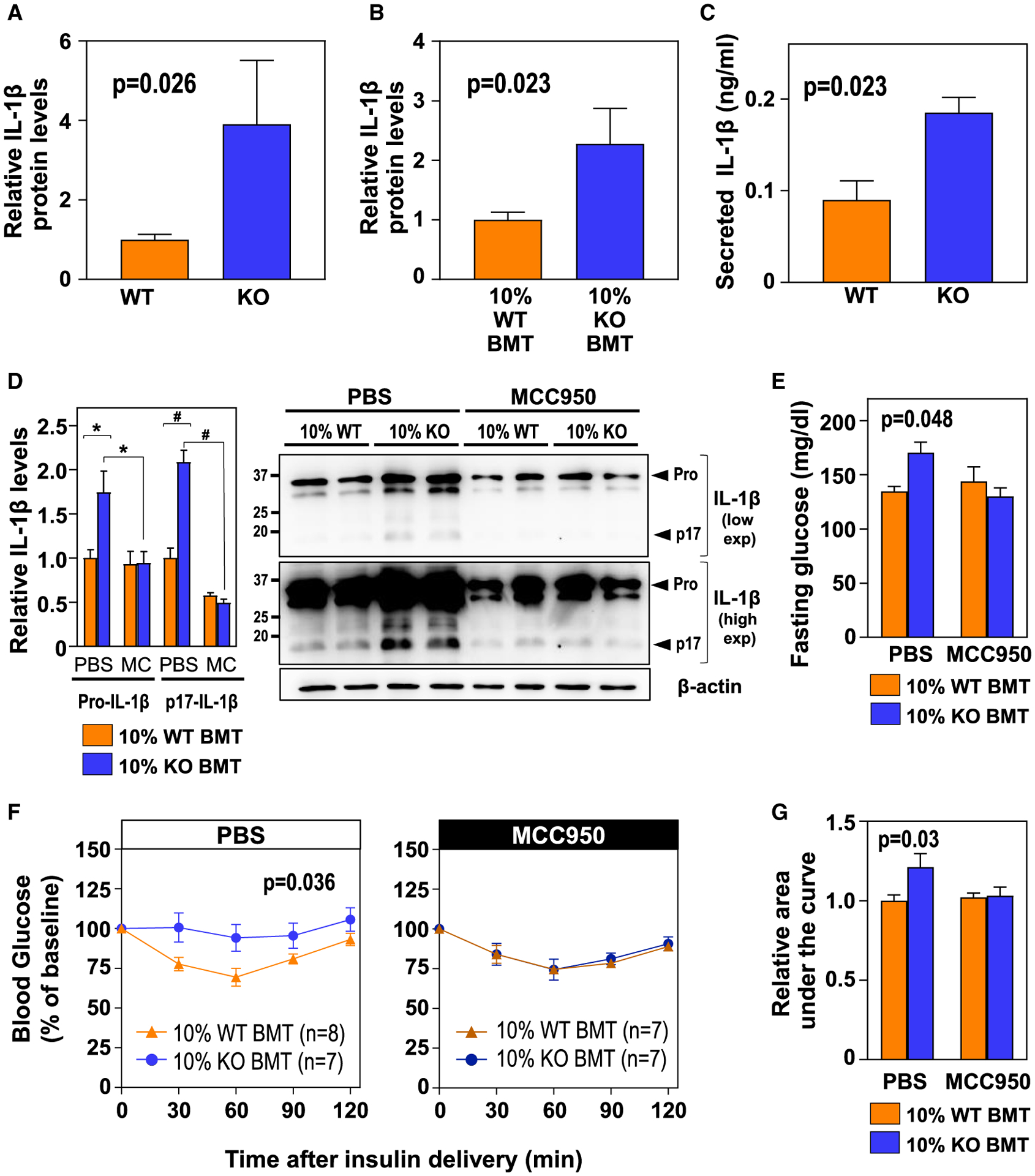
(A and B) ELISA analysis of IL-1β protein levels in eWAT in conditions of aging (A) (n = 9 WT, 6 KO BM) and HF/HS diet-induced obesity (B) (n = 11 mice per BM genotype).
(C) ELISA analysis of secreted IL-1β protein levels in lipopolysaccharide (LPS)/IFNγ-primed peritoneal macrophages isolated from WT and KO mice (n = 3 biological replicates per genotype) and treated with ceramide for 20 h.
(D) Western blot analysis of pro-IL-1β and mature (p17) IL-1β levels in eWAT stromal vascular fractions (n = 4 mice per BM genotype and experimental condition; *p < 0.03; #p < 0.0001, two-way ANOVA with Tukey’s multiple comparisons test). Representative blots after high or low exposure are shown.
(E) Fasting blood glucose.
(F) Insulin tolerance tests.
(G) Area under the curve of insulin tolerance tests.
In (D)–(G), HF/HS-diet-fed 10% WT BMT and 10% KO BMT mice received a continuous infusion of MCC950 or vehicle via subcutaneous osmotic pumps (n = 7–8 mice, as indicated in the figure, unless otherwise noted). Data are represented as mean ± SEM.
See also Figure S3.
Based on these findings, we hypothesized that exacerbated expression and NLRP3-inflammasome-mediated secretion of IL-1β in TET2-deficient immune cells is a central driver of eWAT dysfunction and insulin resistance in conditions of TET2-loss-of-function-induced CH. To test this possibility, we investigated the effects of TET2-inactivation-mediated CH in HF/HS-fed mice treated chronically with MCC950 (Figure S3A), a specific pharmacological inhibitor of the NLRP3 inflammasome (Coll et al., 2015). MCC950 led to a ~50% reduction in mature (p17) IL-1β protein levels in eWAT stromal vascular fractions and completely suppressed the increased levels of IL-1β observed in mice carrying TET2-deficient hematopoietic cells (Figure 3D). While this inhibition of IL-1β production via MCC950 treatment had no effects on body weight (Figure S3B) or on the clonal expansion of TET2-deficient cells (Figure S3C), it suppressed the increased hyperglycemia (Figure 3E) and insulin resistance (Figures 3F and 3G) associated with the expansion of TET2-deficient cells. These in vivo data demonstrate that NLRP3 inflammasome-mediated production of IL-1β is essential for the glucose homeostasis derangement associated with TET2-loss-of-function-driven CH.
Our previous studies in the context of atherosclerosis development revealed that exacerbated IL-1β production by TET2-deficient macrophages promotes inflammation and vascular disease by increasing the expression of endothelial adhesion molecules, leading to greater recruitment of myeloid cells and increased macrophage burden in the vascular wall (Fuster et al., 2017). However, in the current aging and obesity studies, we did not find any effect of hematopoietic TET2 deficiency on the expression of endothelial adhesion molecules in eWAT (Figure S3D), and, accordingly, no statistically significant differences in macrophage content were detected in this tissue by flow cytometry or qPCR analysis of the expression of macrophage-related transcripts (Figures S3E–S3G). In contrast, eWAT of obese mice exhibiting TET2-inactivation-driven CH had reduced transcript levels of Irs1 (Figure S3H), a pivotal mediator of insulin signaling whose expression in mouse and human adipocytes has been previously shown to be inhibited by IL-1β (Gao et al., 2014; Jager et al., 2007). Based on this observation, we hypothesize that reduced insulin signaling in eWAT of mice carrying TET2-deficient hematopoietic cells is mainly due to the effects of exacerbated IL-1β production on Irs1 expression in adipose tissue. Supporting this possibility, obese mice transplanted with TET2-deficient cells, but not those transplanted with WT cells, exhibited a significant negative correlation between Il1b and Irs1 transcript levels in eWAT (Figure S3I). No correlation or difference in Irs1 expression was observed when IL-1β secretion was inhibited through treatment with MCC950 (Figures S3J and S3K). Similarly, no correlation was found between transcript levels of Irs1 and those of other cytokines known to interfere in insulin signaling, but not regulated by TET2, such as TNF, regardless of the genotype of transplanted cells (Irs1/Tnf Spearman’s correlation coefficients: r = −0.34, p = 0.3 in 10% WT BMT mice; r = −0.36, p = 0.3 in 10% KO BMT mice). Cultured 3T3-L1-derived adipocytes that were exposed to conditioned medium from ceramide-treated TET2-deficient macrophages showed reduced Irs1 expression and insulin-induced glucose uptake, compared to adipocytes treated with conditioned medium from WT controls, which was completely prevented by co-treatment with an IL-1β-neutralizing antibody (Figures S3L and S3M). Overall, these data strongly suggest that reduced adipocyte Irs1 expression secondary to IL-1β overproduction by TET2 mutant macrophages is a central driver of increased insulin resistance in conditions of TET2-deficiency-induced CH.
DISCUSSION
Our experimental studies show that CH driven by somatic TET2 mutations in the hematopoietic system aggravates age- and obesity-related insulin resistance. These findings represent experimental evidence supporting the possibility that the expansion of BM cells carrying a CH-related mutation impairs glucose homeostasis. Previous epidemiological studies suggested an association between CH and T2DM (Bonnefond et al., 2013; Jaiswal et al., 2014), but it remained unknown whether this association reflected a causal contribution of somatic mutations and CH to the pathology of T2DM. Given that insulin resistance plays a key pathogenic role in T2DM, our findings herein support that CH, at least when driven by TET2 mutations, is causally connected to the development of T2DM. Furthermore, abnormal glucose homeostasis and WAT inflammation are important contributors to CVD (Fuster et al., 2016); therefore, our findings support an indirect mechanism by which TET2 mutations and CH may contribute to CVD in addition to the direct effects on atherosclerotic plaques and the myocardium we previously reported (Fuster et al., 2017; Sano et al., 2018). The evaluation of this potential indirect effect of TET2 mutations will require additional studies in models of atherosclerosis accelerated by metabolic syndrome or T2DM.
Mechanistically, our data suggest that aggravated insulin resistance in conditions of TET2-loss-of-function in hematopoietic cells is mainly mediated by exacerbated IL-1β expression in BM-derived macrophages in WAT, although we cannot rule out some contribution from other immune cells or other metabolic tissues. This NLRP3-/IL-1β-driven mechanism is consistent with our observation that aggravated insulin resistance in mice carrying TET2-deficient cells and fed a normal diet is only observed at an advanced age. IL-1β maturation and secretion requires the accumulation of “danger signals” that activate NLRP3 or other inflammasomes, such as ceramides and other lipids, which may accumulate rapidly in HF-diet-fed mice, but more gradually in aging mice fed a normal diet. Indeed, ceramide levels have been reported to be elevated in aged tissues and senescent cultured cells (Claycombe et al., 2002; Lightle et al., 2000; Ohanian et al., 2014; Wu et al., 2007; Youm et al., 2012). Further supporting our proposed mechanism, human sequencing studies strongly support the existence of heightened Il-1β production in TET2 mutation carriers (Abplanalp et al., 2020; Bick et al., 2019). These findings may have implications in the context of personalized medicine and the application of anti-inflammatory drugs to T2DM and vascular disease prevention. Several strategies targeting the NLRP3/IL-1β pathway have been developed and tested pre-clinically or clinically in the context of insulin resistance, T2DM, and its vascular complications, with a variety of results (Cavelti-Weder et al., 2012; Donath et al., 2019; Everett et al., 2018; Kataria et al., 2019; Peiró et al., 2017; Sloan-Lancaster et al., 2013). However, such anti-inflammatory drugs face numerous challenges in the setting of chronic disorders, considering the expected side effects of immunosuppression. Thus, it would be desirable to administer NLRP3/IL-1β inhibitors exclusively to those patients that are predicted to show the greatest response to this therapy. In this regard, our findings suggest that IL-1β-targeted drugs may be particularly effective for the prevention of T2DM and vascular complications in individuals carrying somatic TET2 mutations. Indeed, a recent exploratory analysis of the Canakinumab Anti-inflammatory Thrombosis Outcome Study (CANTOS) suggests that treatment with an IL-1β-neutralizing antibody leads to greater prevention of atherosclerotic CVD in TET2 mutation carriers than in the overall CANTOS cohort (Svensson et al., 2018). Our data herein suggest that individuals with hematopoietic TET2 mutations might also receive increased benefit in the context of T2DM prevention. Carefully designed clinical trials will be needed to examine this possibility.
STAR★METHODS
RESOURCE AVAILABILITY
Lead contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, José J. Fuster (jjfuster@cnic.es).
Materials availability
This study did not generate new unique reagents.
Data and code availability
This study did not generate any unique datasets or code.
EXPERIMENTAL MODELS AND SUBJECT DETAILS
Mice
Animal experiments followed protocols approved by Institutional Animal Care and Use Committees at Boston University and Centro Nacional de Investigaciones Cardiovasculares. All mice were maintained on a 12-h light/dark schedule in a specific pathogen-free animal facility and given food and water ad libitum. The following strains were obtained from the Jackson Laboratory: C57BL/6 Pep Boy Cd45.1+ mice and TET2-KO mice (Ko et al., 2011). All experiments were performed in male mice that were between the ages of 8 to 10 weeks at the time of BM transfer. Unless otherwise noted, mice were fed a standard chow diet (18% protein rodent diet). The number of mice included in each study is indicated in the figures or the associated legends.
Cultured cells
All cell culture experiments were done at 37°C and 5% CO2.Thioglycollate-elicited macrophages were obtained from the peritoneal cavity of littermate mice 4 days after intraperitoneal injection of 1 mL aged 4% Brewer’s thioglycollate broth. Macrophages were allowed to adhere for 6 h and cultured in RPMI medium supplemented with 100UI/mL penicillin/100 μg/mL streptomycin and 10% FBS. Mouse 3T3-L1 pre-adipocyte cells were cultured in DMEM supplemented with 10% fetal calf serum and 100UI/mL penicillin/100 μg/mL streptomycin. 3T3-L1 cell adipocytic differentiation was induced two days after reaching confluence by culturing the cells for three days in DMEM supplemented with 10% FBS, 100UI/mL penicillin/100 μg/mL streptomycin, 1 μg/ml insulin, 1 μM dexamethasone and 0.52 mM Isobutylmethylxanthine; followed by culturing the cells for two additional days in DMEM supplemented with 10% FBS, 100UI/mL penicillin/100 μg/mL streptomycin and 1 μg/ml insulin. Assays with differentiated 3T3-L1 cells were done at day 8 post-differentiation induction.
METHOD DETAILS
Adoptive (non-conditioned) bone marrow cell transfer
An adoptive bone marrow (BM) transfer (i.e., a non-conditioned BM transplantation) was used to model TET2 loss of function-driven clonal hematopoiesis in mice. 10 week-old unconditioned (i.e., non-irradiated) C57BL/6 Pep Boy CD45.1+ mice were injected intravenously with a total of 1.5×107 unfractionated CD45.2+ Tet2−/− or Tet2+/+ BM cells, split into 3 doses of 5×106 cells delivered in consecutive days. Donor cells were obtained from littermate mice. BM cells were isolated from femurs and tibias of 8–10 week-old donor mice after euthanasia.
Competitive bone marrow transplantations and induction of obesity
C57BL/6 CD45.1/CD45.2 heterozygous recipient mice in a C57BL/6 background were irradiated to achieve myeloablation and transplanted with suspensions of BM cells containing a mixture of 10% CD45.2+ homozygous Tet2−/− cells and 90% CD45.1+ homozygous Tet2+/+ cells (10% KO-BMT mice), or 10% CD45.2+ homozygous Tet2+/+ cells and 90% CD45.1+ homozygous Tet2+/+ cells (10% WT-BMT mice). CD45.2+ donor cells were obtained from littermate mice. BM cells were isolated from femurs and tibias of donor mice after euthanasia; donor CD45.2+ cells were obtained from Tet2+/+ or −/− littermates, whereas donor CD45.1+ cells were obtained from C57BL/6 Pep Boy mice. Recipient mice were irradiated in a pie cage (Braintree Scientific) to limit mobility and ensure equal dose of irradiation, and were exposed to two radiation doses of 400 rad 3 h apart using an X-RAD 320 Biological Irradiator. After the second irradiation, each recipient mouse was injected with 107 BM cells i.v. Sterilized caging, food, and water were provided during the first 14 days post-transplant and water was supplemented with antibiotics (sulfamethoxazole-trimethoprim). Mice that did not recover full pre-irradiation body weight 28 days after transplant were excluded from further analysis. The 2×400 rad dose was selected after titration of different doses in order to minimize potentially confounding gastrointestinal side effects of radiation, but still achieve efficient myeloablation (typically less than 0.5% recipient WBCs could be detected 4 weeks post-transplantation). Starting 5 weeks after BMT, mice were fed an obesogenic high fat and high sucrose diet (HF/HS, 5.51 kcal/g) for 14 weeks to induce obesity. The composition of the HF/HS diet was 35.8% fat (primarily lard), 36.8% carbohydrate (primarily sucrose), and 20.3% protein.
Non-invasive body composition analysis
Body composition (lean and fat mass percentage) was evaluated by magnetic resonance imaging (EchoMRI-700, Echo Medical System) at baseline and approximately every 6 months after the adoptive transfer for the aging experiment, and at baseline and endpoint for the obesity experiments.
Metabolic measurements
Insulin tolerance tests were performed on 5 h-fasted mice injected intraperitoneally with 1.2 U/Kg human insulin (Humulin R, Eli Lilly). Blood glucose levels were measured with an Accu-Chek glucometer (Roche Diagnostics Corp.) immediately before and 15, 30, 60, 90, and 120 min after glucose injection. Area under the curve values were calculated with the GraphPad Prism software (GraphPad Software Inc.). Fasting glucose levels were obtained after a 16-h fast with an Accu-Chek glucometer (Roche Diagnostics Corp.). Tissue weights were recorded at the time of euthanasia.
Adipose tissue stromal vascular fraction isolation
Stromal vascular fractions from eWAT were obtained as previously described (Fuster et al., 2015). Briefly, adipose tissue was minced in PBS and digested with 1 mg/ml collagenase Type 1 at 37°C for 30 min. The digested tissue was filtered through a cell strainer (100 μm mesh size) and centrifuged at 1000 rpm for 5 min to separate floating adipocytes from the stromal vascular fraction (pellet).
Flow cytometry analyses of bone marrow blood, liver, and stromal vascular fractions from adipose tissue
Bone marrow cells were flushed out of two femurs and two tibias per mouse in RPMI. Liver samples were digested in PBS containing 1 mg/ml collagenase Type 1 and 1 U/ml DNase at 37°C for 15 min. Peripheral blood was obtained from the facial vein and collected into EDTA-coated tubes. Stromal vascular fractions were isolated from adipose tissue as described above. Red blood cells were lysed in all samples by treatment with 1X Red Blood Cell Lysis Buffer for 2–5 min on ice. Samples were stained with combinations of biotinylated and/or fluorescently labeled antibodies in PBS with 1% FBS for 30 min on ice. Antibodies are listed in the Key Resources Table. Blood, liver and adipose tissue samples were stained in the presence of FcR Blocking reagent. Bone Marrow lineage-negative cells were defined as negative for CD11b, Gr-1, Ter119, B220, CD3e and CD127. Bone marrow HSPCs, were defined as: Lineage−, c-Kit+, Sca1+. Blood classical monocytes were identified as CD45+, CD115Hi, CD43Lo, Ly6cHi; patrolling monocytes, as CD45+, CD115Hi, CD43Hi, Ly6cLo; neutrophils, as CD45+, CD115Int, Ly6g+; T lymphocytes, as CD45+, CD115−, B220−, CD3+; B lymphocytes, as CD45+, CD115−, CD3−, B220+; and tissue macrophages, as CD45+, CD11B+, F4/80Hi. Gating strategies can be found in our previous publications (Fuster et al., 2017; Wang et al., 2020). Dead cells were excluded from analysis by DAPI staining. BD LSR II and BD FACSymphony Cytometers (BD Bioscience) were used for data acquisition. Data were analyzed with FlowJo Software.
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Anti-Phospho-Akt Ser473 (D9E) | Cell Signaling Technology | Cat# 4060; RRID: AB_2315049 |
| Anti-Akt (pan) (C67E7) | Cell Signaling Technology | Cat# 8596; RRID: AB_10890703 |
| Anti-IL-1 β (for Western Blot) | Genetex | Cat# GTX74034; RRID: AB_378141 |
| Anti-β-Actin (13E5) | Cell Signaling Technology | Cat# 5125; RRID: AB_1903890 |
| Anti-IL-1 β (for neutralization) | Invitrogen | Cat# MM425B; RRID: AB_223529 |
| Mouse IgG1 kappa (Isotype Control for neutralization studies) | Invitrogen | Cat# 14-4714-82; RRID: AB_470111 |
| FcR Blocking Reagent, mouse | Miltenyi Biotec | Cat# 130-092-575 |
| eFluor 450 anti-CD45.2 | eBioscience | Cat# 48-0454-82; RRID: AB_11042125 |
| PE-Cy7 anti-CD45.1 | eBioscience | Cat# 25-0453-82; RRID: AB_469629 |
| PE anti-CD115 (CSF-1R) | BioLegend | Cat# 135505; RRID: AB_1937254 |
| PerCP/Cyanine5.5 anti-Ly-6G | BioLegend | Cat# 127615; RRID: AB_1877272 |
| BV711 Rat Anti-Mouse CD43 | BD Biosciences | Cat# 740668; RRID: AB_2740356 |
| APC Rat Anti-Ly-6C | BD Biosciences | Cat# 560595; RRID: AB_1727554 |
| APC/Cy7 anti-B220 | BioLegend | Cat# 103223; RRID: AB_313006 |
| PE-eFluor 610 anti-CD3e | eBioscience | Cat# 61-0031-82; RRID: AB_2574514 |
| Brilliant Violet 510 anti-CD8a | Biolegend | Cat# 100751; RRID: AB_2561389 |
| FITC anti-CD4 | eBioscience | Cat# 11-0042-82; RRID: AB_464896 |
| Biotin anti-CD127 (IL-7Rα) | Biolegend | 135005; RRID: AB_1953262 |
| Biotin Mouse Lineage Panel (biotinylated antibodies against CD3e, CD45R, Ly-6C/G, CD11b and TER-119) | BD Biosciences | Cat# 559971; RRID: AB_10053179 |
| BV421 Streptavidin | BD Biosciences | 563259 |
| PerCP/Cyanine5.5 anti-CD45.1 | BioLegend | Cat# 110727; RRID: AB_893348 |
| APC anti-CD45.2 | BioLegend | 109813; RRID: AB_389210 |
| PE anti-cKit | BD Biosciences | Cat# 553355; RRID: AB_394806 |
| PE-Cy7 anti-Sca-1 | BD Biosciences | Cat# 558162; RRID: AB_647253 |
| BV711 anti-CD115 | Biolegend | Cat# 135515; RRID: AB_2562679 |
| Chemicals, Peptides, and Recombinant Proteins | ||
| Humulin R 100 IU/ml (for in vivo studies) | Lilly | HI0210 |
| C16 Ceramide (d18:1/16:0) | Avanti | 860516P |
| LPS-EK (LPS from E. coli K12) | InVivoGen | tlrl-peklps |
| Recombinant Murine IFN-γ | Peprotech | 315–05 |
| 3-Isobutyl-1-methylxanthine (IBMX) | Sigma-Aldrich | I5879 |
| Dexamethasone | Sigma-Aldrich | D4902 |
| Human Insulin (for cell culture studies) | Sigma-Aldrich | I9278 |
| DMEM (Dulbecco’s Modified Eagle Medium) | GIBCO | 41965–039 |
| DMEM, low glucose, pyruvate | GIBCO | 31885–023 |
| RPMI Medium 1640 | GIBCO | 11875093 |
| Fetal Bovine Serum | Sigma-Aldrich | F7524 |
| Calf Bovine Serum, Iron Fortified | ATCC | 30–2030 |
| Penicillin-Streptomycin | GIBCO | 15140122 |
| Phosphate-Buffered Saline | GIBCO | 10010023 |
| Fluid Thioglycollate Medium | Difco BD | 225650 |
| ECL Select Western Blotting Detection Reagent | Sigma-Aldrich | GERPN2235 |
| Cell Lysis Buffer (10X) | Cell Signaling Technology | 9803S |
| phosSTOP (phosphatase inhibitor tablets) | Roche | 4906845001 |
| cOmplete (protease inhibitor cocktail tablets) | Roche | 11697498001 |
| SDS Solution 20% (w/v) | BioRad | 1610418 |
| Restore Western Blot Stripping Buffer | Thermo Scientific | 21059 |
| Restore PLUS Western Blot Stripping Buffer | Thermo Scientific | 46430 |
| Sulfatrim Pediatric (sulfamethoxazole-trimethoprim oral suspension) | STI Pharma, LLC | 54879-007-16 |
| eBioscience 1X RBC Lysis Buffer | Invitrogen | 00-4333-57 |
| UltraComp eBeads (compensation beads) | Invitrogen | 01-2222-42 |
| Collagenase Type 1 | Worthington Chemical Corporation | LS004196 |
| DNase I | Roche | 10104159001 |
| TRIzol | Invitrogen | 15596018 |
| DAPI | Thermo Scientific | 62248 |
| MCC950 | Provided by Dr. M.A. Cooper | N/A |
| Critical Commercial Assays | ||
| Pierce BCA Protein Assay Kit | Thermo Scientific | TJ268818 |
| Glucose Uptake-Glo Assay | Promega | J1341 |
| RNeasy Lipid Tissue Mini Kit | QIAGEN | 74804 |
| High-Capacity cDNA Reverse Transcription Kit | Applied Biosystems | 4368814 |
| SYBR Green PCR Master Mix | Applied Biosystems | 4309155 |
| Mouse IL-1 beta/IL-1F2 Quantikine ELISA | Bio-Techne R&D Systems | SMLB00C |
| Experimental Models: Cell Lines | ||
| 3T3-L1 cell line | ATCC | CL-173 |
| Experimental Models: Organisms/Strains | ||
| CD45.1 (B6.SJL-Ptprca Pepcb/BoyJ) mice | The Jackson Laboratory | Stock No: 002014 |
| TET2-KO (B6(Cg)-Tet2tm1.2Rao/J) mice | The Jackson Laboratory | Stock No: 023359 |
| Oligonucleotides | ||
| Primer 1 for TET2 KO mice genotyping (forward, WT allele): AGCTGATGGAAAATGCAAGC; | Integrated DNA Technologies | N/A |
| Primer 2 for TET2 KO mice genotyping (forward, KO allele): GCCACTTTAGAA GCCTATTGGA; | Integrated DNA Technologies | N/A |
| Primer 3 for TET2 KO mice genotyping (reverse, WT/KO alleles): TCTCAGAGCAAAGAGGACTGC. | Integrated DNA Technologies | N/A |
| Primers for Real-Time PCR: see Supplemental Table | This paper | N/A |
| Software and Algorithms | ||
| FlowJo software | FLOWJO | RRID: SCR_008520 |
| GraphPad Prism 7 software | GraphPad | RRID: SCR_002798 |
| QuantStudio software | Applied Biosystems | RRID: SCR_014246 |
| ImageQuant analysis software | GE HEalthcare | RRID: SCR_014246 |
| Other | ||
| BD LSRII Flow Cytometer | BD Biosciences | N/A |
| BD FACSymphony Flow Cytometer | BD Biosciences | N/A |
| ViiA 7 Real-Time PCR System | Applied Biosystems | N/A |
| ImageQuant LAS 4000 Biomolecular imager | GE Healthcare | N/A |
| Microtainer Tubes (EDTA-coated) | BD Biosciences | 365974 |
| Mini-PROTEAN TGX Stain-Free Precast Gels 10% | Bio-Rad | 456–8033 |
| Immobilon-P Transfer Membrane; PVDF | Millipore | IPVH85R |
| Mini-osmotic pumps, model 2006 | Alzet | 2006 |
| Mouse Diet, High Fat, 60% Fat Calories | Bio-Serv | F1850 |
| Teklad global 18% protein rodent diet | Envigo | 2018 |
Gene expression analysis by qPCR
Total RNA from tissues or cultured adipocytes was isolated using Trizol reagent and RNeasy kits (QIAGEN). RNA (1 μg) was reverse transcribed with High-Capacity cDNA Reverse Transcription Kit (Applied Biosystems). qPCR was performed with SYBR® Green PCR Master Mix (Applied Biosystems) in a ViiA7 Real-time PCR system. Results were analyzed with the ΔΔCt method. 36B4, β-actin or the combination of both were used as reference genes for normalization. Primer sequences are listed in the Supplemental Table.
In vivo insulin signaling studies
Following a 5-hour fasting, mice were injected with 2 U/Kg human insulin. 5 min after injection, mice were euthanatized and epididymal white adipose tissue and liver samples were collected, snap-frozen in liquid nitrogen, and kept at −80°C until processing.
SDS-PAGE and Western Blot
Protein extracts were obtained using ice-cold lysis buffer supplemented with protease and phosphatase inhibitors. Tissue homogenization was achieved using a QIAGEN TissueLyser II tissue disruptor. Equal amounts of protein lysates were resolved by SDS-PAGE and analyzed by Western Blot (PVDF membranes) and immunostaining. Antibodies and other reagents are listed in the Key Resources Table.
Pro-inflammatory activation of cultured macrophages
Peritoneal macrophages were obtained from Tet2−/− and +/+ mice and cultured as described above. Pro-inflammatory priming was achieved by treating macrophages with 10 ng/ml LPS and 2 ng/ml IFNγ for 4h. NLRP3 inflammasome activation and IL-1β maturation in LPS/IFNγ-primed macrophages were induced by a subsequent 20-h treatment with 0.1 mM C16 ceramide.
IL-1β protein measurements by ELISA analysis
Whole eWAT samples were obtained from ad libitum-fed mice and protein extracts were prepared as described above. Cell culture supernatants were obtained from peritoneal macrophages. IL-1β protein levels in tissue extracts or cell culture supernatants were measured using ELISA assays. In the case of eWAT samples, IL-1β levels were normalized to whole tissue protein content and expressed relative to WT controls in each experiment.
Glucose uptake analysis in cultured adipocytes
3T3-L1 cells were differentiated into adipocytes as described above and treated for 16h with conditioned medium obtained from LPS/IFNγ-primed, ceramide-treated peritoneal macrophages isolated from Tet2−/− or +/+ mice. Conditioned media were supplement with 20 ng/ml mouse anti-mouse IL1β neutralizing IgG1 or mouse IgG1 isotype control 1 h before addition to 3T3-L1-derived adipocyte cultures. To determine insulin-induced glucose uptake, conditioned media was removed, and cells were washed in PBS and treated for one h with 10μM insulin in low-glucose DMEM. After medium removal and two washes in PBS, cells were treated with 1 mM 2-deoxyglucose for 10 min at room temperature. 2-deoxyglucose uptake was measured using a bioluminescence methods and a commercially available assay (Glucose Uptake-Glo Assay, Promega), following manufacturer’s instructions.
In vivo pharmacological NLRP3 inflammasome inhibition
MCC950, a small molecule inhibitor of the NLRP3 inflammasome, was synthesized and used as previously described (Coll et al., 2015; Fuster et al., 2017). In brief, MCC950 was diluted in PBS and delivered in vivo chronically at a dose of 10 mg/Kg/day via subcutaneous mini-osmotic pumps (Alzet 2006), starting 4 weeks after BMT. Pumps were replaced at weeks 10 and 16 post-BMT to allow a continuous infusion for the entire duration of the study. Control mice were infused with PBS.
QUANTIFICATION AND STATISTICAL ANALYSIS
Data are shown as mean ± SEM. Information on sample size can be found in figures and their associated legends. Statistical significance of differences in experiments with two groups and one variable was assessed by unpaired Student’s t tests (with Welch correction for unequal variance when appropriate) or Mann-Whitney U Tests. Differences in experiments with more than one independent variable were evaluated by two-way analysis of variance (ANOVA) with post hoc Sidak’s or Tukey’s multiple comparison tests. Results of ITT experiments were evaluated by two-way repeated-measures ANOVA. Statistical significance was defined as p < 0.05. Statistical tests were performed using GraphPad Prism software.
Supplementary Material
Highlights.
TET2-deficiency-driven clonal hematopoiesis aggravates insulin resistance in aged mice
TET2-deficiency-driven clonal hematopoiesis aggravates insulin resistance in obese mice
TET2-deficiency-driven clonal hematopoiesis increases IL-1β levels in adipose tissue
NLRP3 inhibition prevents the effects of TET2-deficient cells on insulin sensitivity
ACKNOWLEDGMENTS
This work was supported by a grant from the European Foundation for the Study of Diabetes and Lilly European Diabetes Research Programme (to J.J.F.); grants RYC-2016-20026 and RTI2018-093554-A-I00 from the Spanish Ministerio de Ciencia e Innovación (to J.J.F.); and National Institutes of Health grants HL152174 (to S.S.) and HL139819, HL138014, HL141256, and HL142650 (to K.W.). The project leading to these results also received funding from “la Caixa” Foundation (ID 100010434), under agreement HR17-00267. J.J.F. is a member of the Leducq Foundation Transatlantic Network on Clonal Hematopoiesis and Atherosclerosis and is also supported by a 2019 Leonardo Grant for Researchers and Cultural Creators from the BBVA Foundation, Madrid, Spain. The Centro Nacional de Investigaciones Cardiovasculares (CNIC) is supported by the Instituto de Salud Carlos III (ISCIII), the Ministerio de Ciencia e Innovación, and the Pro CNIC Foundation and is a Severo Ochoa Center of Excellence (SEV-2015-0505). The graphical abstract was created with BioRender (Biorender.com).
Footnotes
SUPPLEMENTAL INFORMATION
Supplemental Information can be found online at https://doi.org/10.1016/j.celrep.2020.108326.
DECLARATION OF INTERESTS
K.W. and J.J.F. are co-inventors on a patent related to the treatment of cardiometabolic diseases associated with somatic TET2 mutations. M.A.C. is co-founder and chief executive officer of Inflazome, a company developing drugs that target inflammasomes. These authors have no additional conflicts of interest. Other authors declare no competing interests.
REFERENCES
- Abplanalp WT, Mas-Peiro S, Cremer S, John D, Dimmeler S, and Zeiher AM (2020). Association of Clonal Hematopoiesis of Indeterminate Potential With Inflammatory Gene Expression in Patients With Severe Degenerative Aortic Valve Stenosis or Chronic Postischemic Heart Failure. JAMA Cardiol. Published online July 8, 2020. In press. 10.1001/jamacardio.2020.2468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arends CM, Galan-Sousa J, Hoyer K, Chan W, Jäger M, Yoshida K, Seemann R, Noerenberg D, Waldhueter N, Fleischer-Notter H, et al. (2018). Hematopoietic lineage distribution and evolutionary dynamics of clonal hematopoiesis. Leukemia 32, 1908–1919. [DOI] [PubMed] [Google Scholar]
- Bick AG, Weinstock JS, Nandakumar SK, Fulco CP, Leventhal MJ, Bao EL, Nasser J, Zekavat SM, Szeto MD, Laurie C, et al. (2019). Inherited Causes of Clonal Hematopoiesis of Indeterminate Potential in TOPMed Whole Genomes. bioRxiv. 10.1101/782748. [DOI] [Google Scholar]
- Bonnefond A, Skrobek B, Lobbens S, Eury E, Thuillier D, Cauchi S, Lantieri O, Balkau B, Riboli E, Marre M, et al. (2013). Association between large detectable clonal mosaicism and type 2 diabetes with vascular complications. Nat. Genet 45, 1040–1043. [DOI] [PubMed] [Google Scholar]
- Buscarlet M, Provost S, Zada YF, Barhdadi A, Bourgoin V, Lépine G, Mollica L, Szuber N, Dubé MP, and Busque L (2017). DNMT3A and TET2 dominate clonal hematopoiesis and demonstrate benign phenotypes and different genetic predispositions. Blood 130, 753–762. [DOI] [PubMed] [Google Scholar]
- Buscarlet M, Provost S, Zada YF, Bourgoin V, Mollica L, Dubé MP, and Busque L (2018). Lineage restriction analyses in CHIP indicate myeloid bias for TET2 and multipotent stem cell origin for DNMT3A. Blood 132, 277–280. [DOI] [PubMed] [Google Scholar]
- Busch K, and Rodewald HR (2016). Unperturbed vs. post-transplantation hematopoiesis: both in vivo but different. Curr. Opin. Hematol 23, 295–303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cavelti-Weder C, Babians-Brunner A, Keller C, Stahel MA, Kurz-Levin M, Zayed H, Solinger AM, Mandrup-Poulsen T, Dinarello CA, and Donath MY (2012). Effects of gevokizumab on glycemia and inflammatory markers in type 2 diabetes. Diabetes Care 35, 1654–1662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Claycombe KJ, Wu D, Nikolova-Karakashian M, Palmer H, Beharka A, Paulson KE, and Meydani SN (2002). Ceramide mediates age-associated increase in macrophage cyclooxygenase-2 expression. J. Biol. Chem 277, 30784–30791. [DOI] [PubMed] [Google Scholar]
- Coll RC, Robertson AA, Chae JJ, Higgins SC, Muñoz-Planillo R, Inserra MC, Vetter I, Dungan LS, Monks BG, Stutz A, et al. (2015). A small-molecule inhibitor of the NLRP3 inflammasome for the treatment of inflammatory diseases. Nat. Med 21, 248–255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cull AH, Snetsinger B, Buckstein R, Wells RA, and Rauh MJ (2017). Tet2 restrains inflammatory gene expression in macrophages. Exp. Hematol 55, 56–70.e13. [DOI] [PubMed] [Google Scholar]
- Donath MY, Meier DT, and Böni-Schnetzler M (2019). Inflammation in the Pathophysiology and Therapy of Cardiometabolic Disease. Endocr. Rev 40, 1080–1091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Everett BM, Donath MY, Pradhan AD, Thuren T, Pais P, Nicolau JC, Glynn RJ, Libby P, and Ridker PM (2018). Anti-Inflammatory Therapy With Canakinumab for the Prevention and Management of Diabetes. J. Am. Coll. Cardiol 71, 2392–2401. [DOI] [PubMed] [Google Scholar]
- Fuster JJ, and Walsh K (2018). Somatic Mutations and Clonal Hematopoiesis: Unexpected Potential New Drivers of Age-Related Cardiovascular Disease. Circ. Res 122, 523–532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fuster JJ, Zuriaga MA, Ngo DT, Farb MG, Aprahamian T, Yamaguchi TP, Gokce N, and Walsh K (2015). Noncanonical Wnt signaling promotes obesity-induced adipose tissue inflammation and metabolic dysfunction independent of adipose tissue expansion. Diabetes 64, 1235–1248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fuster JJ, Ouchi N, Gokce N, and Walsh K (2016). Obesity-Induced Changes in Adipose Tissue Microenvironment and Their Impact on Cardiovascular Disease. Circ. Res 118, 1786–1807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fuster JJ, MacLauchlan S, Zuriaga MA, Polackal MN, Ostriker AC, Chakraborty R, Wu CL, Sano S, Muralidharan S, Rius C, et al. (2017). Clonal hematopoiesis associated with TET2 deficiency accelerates atherosclerosis development in mice. Science 355, 842–847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao D, Madi M, Ding C, Fok M, Steele T, Ford C, Hunter L, and Bing C (2014). Interleukin-1b mediates macrophage-induced impairment of insulin signaling in human primary adipocytes. Am. J. Physiol. Endocrinol. Metab 307, E289–E304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Genovese G, Kähler AK, Handsaker RE, Lindberg J, Rose SA, Bakhoum SF, Chambert K, Mick E, Neale BM, Fromer M, et al. (2014). Clonal hematopoiesis and blood-cancer risk inferred from blood DNA sequence. N. Engl. J. Med 371, 2477–2487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jager J, Grémeaux T, Cormont M, Le Marchand-Brustel Y, and Tanti JF (2007). Interleukin-1beta-induced insulin resistance in adipocytes through down-regulation of insulin receptor substrate-1 expression. Endocrinology 148, 241–251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jaiswal S, Fontanillas P, Flannick J, Manning A, Grauman PV, Mar BG, Lindsley RC, Mermel CH, Burtt N, Chavez A, et al. (2014). Age-related clonal hematopoiesis associated with adverse outcomes. N. Engl. J. Med 371, 2488–2498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jaiswal S, Natarajan P, Silver AJ, Gibson CJ, Bick AG, Shvartz E, McConkey M, Gupta N, Gabriel S, Ardissino D, et al. (2017). Clonal Hematopoiesis and Risk of Atherosclerotic Cardiovascular Disease. N. Engl. J. Med 377, 111–121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kataria Y, Ellervik C, and Mandrup-Poulsen T (2019). Treatment of type 2 diabetes by targeting interleukin-1: a meta-analysis of 2921 patients. Semin. Immunopathol 41, 413–425. [DOI] [PubMed] [Google Scholar]
- Ko M, Bandukwala HS, An J, Lamperti ED, Thompson EC, Hastie R, Tsangaratou A, Rajewsky K, Koralov SB, and Rao A (2011). Ten-Eleven-Translocation 2 (TET2) negatively regulates homeostasis and differentiation of hematopoietic stem cells in mice. Proc. Natl. Acad. Sci. USA 108, 14566–14571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lightle SA, Oakley JI, and Nikolova-Karakashian MN (2000). Activation of sphingolipid turnover and chronic generation of ceramide and sphingosine in liver during aging. Mech. Ageing Dev 120, 111–125. [DOI] [PubMed] [Google Scholar]
- Lu R, Czechowicz A, Seita J, Jiang D, and Weissman IL (2019). Clonal-level lineage commitment pathways of hematopoietic stem cells in vivo. Proc. Natl. Acad. Sci. USA 116, 1447–1456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ohanian J, Liao A, Forman SP, and Ohanian V (2014). Age-related remodeling of small arteries is accompanied by increased sphingomyelinase activity and accumulation of long-chain ceramides. Physiol. Rep 2, e12015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peiró C, Lorenzo Ó, Carraro R, and Sánchez-Ferrer CF (2017). IL-1β Inhibition in Cardiovascular Complications Associated to Diabetes Mellitus. Front. Pharmacol 8, 363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saltiel AR, and Olefsky JM (2017). Inflammatory mechanisms linking obesity and metabolic disease. J. Clin. Invest 127, 1–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sano S, Oshima K, Wang Y, MacLauchlan S, Katanasaka Y, Sano M, Zuriaga MA, Yoshiyama M, Goukassian D, Cooper MA, et al. (2018). Tet2-Mediated Clonal Hematopoiesis Accelerates Heart Failure Through a Mechanism Involving the IL-1β/NLRP3 Inflammasome. J. Am. Coll. Cardiol 71, 875–886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shimoto M, Sugiyama T, and Nagasawa T (2017). Numerous niches for hematopoietic stem cells remain empty during homeostasis. Blood 129, 2124–2131. [DOI] [PubMed] [Google Scholar]
- Sloan-Lancaster J, Abu-Raddad E, Polzer J, Miller JW, Scherer JC, De Gaetano A, Berg JK, and Landschulz WH (2013). Double-blind, randomized study evaluating the glycemic and anti-inflammatory effects of subcutaneous LY2189102, a neutralizing IL-1β antibody, in patients with type 2 diabetes. Diabetes Care 36, 2239–2246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Svensson EC, Madar A, Campbell CD, He Y, Sultan M, Healey ML, D’Aco K, Fernandez A, Wache-Mainier C, Ridker PM, et al. (2018). Abstract 15111: TET2-Driven Clonal Hematopoiesis Predicts Enhanced Response to Canakinumab in the CANTOS Trial: An Exploratory Analysis. Circulation 138, A15111. [Google Scholar]
- Vandanmagsar B, Youm YH, Ravussin A, Galgani JE, Stadler K, Mynatt RL, Ravussin E, Stephens JM, and Dixit VD (2011). The NLRP3 inflammasome instigates obesity-induced inflammation and insulin resistance. Nat. Med 17, 179–188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y, Sano S, Yura Y, Ke Z, Sano M, Oshima K, Ogawa H, Horitani K, Min KD, Miura-Yura E, et al. (2020). Tet2-mediated clonal hematopoiesis in nonconditioned mice accelerates age-associated cardiac dysfunction. JCI Insight 5, e135204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu D, Ren Z, Pae M, Guo W, Cui X, Merrill AH, and Meydani SN (2007). Aging up-regulates expression of inflammatory mediators in mouse adipose tissue. J. Immunol 179, 4829–4839. [DOI] [PubMed] [Google Scholar]
- Xie M, Lu C, Wang J, McLellan MD, Johnson KJ, Wendl MC, McMichael JF, Schmidt HK, Yellapantula V, Miller CA, et al. (2014). Age-related mutations associated with clonal hematopoietic expansion and malignancies. Nat. Med 20, 1472–1478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Youm YH, Kanneganti TD, Vandanmagsar B, Zhu X, Ravussin A, Adijiang A, Owen JS, Thomas MJ, Francis J, Parks JS, and Dixit VD (2012). The Nlrp3 inflammasome promotes age-related thymic demise and immunosenescence. Cell Rep. 1, 56–68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zink F, Stacey SN, Norddahl GL, Frigge ML, Magnusson OT, Jonsdottir I, Thorgeirsson TE, Sigurdsson A, Gudjonsson SA, Gudmundsson J, et al. (2017). Clonal hematopoiesis, with and without candidate driver mutations, is common in the elderly. Blood 130, 742–752. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
This study did not generate any unique datasets or code.