

Abstract
HEPN (Higher Eukaryotes and Prokaryotes Nucleotide-binding) RNases are an emerging class of functionally diverse RNA processing and degradation enzymes. Members are defined by a small α-helical bundle encompassing a short consensus RNase motif. HEPN dimerization is a universal requirement for RNase activation as the conserved RNase motifs are precisely positioned at the dimer interface to form a composite catalytic center. While the core HEPN fold is conserved, the organization surrounding the HEPN dimer can support large structural deviations that contribute to their specialized functions. HEPN RNases are conserved throughout evolution and include bacterial HEPN RNases such as CRISPR-Cas and toxin-antitoxin associated nucleases, as well as eukaryotic HEPN RNases that adopt large multi-component machines. Here we summarize the canonical elements of the growing HEPN RNase family and identify molecular features that influence RNase function and regulation. We explore similarities and differences between members of the HEPN RNase family and describe the current mechanisms for HEPN RNase activation and inhibition.
Keywords: HEPN, Endoribonuclease, RNA processing, RNA degradation, NiKs, Toxin-Antitoxin, CRISPR-Cas
Introduction
Ribonucleases are central to RNA processing and degradation pathways, driving important cellular activities such as viral RNA decay for adaptive immunity, mRNA splicing for alternative gene expression, and precursor ribosomal RNA maturation for ribosome production. The HEPN (Higher Eukaryotes and Prokaryotes Nucleotide-binding) family is a class of RNA binding proteins with a subset of members possessing endoribonuclease activity (Grynberg, Erlandsen, and Godzik, 2003). The HEPN family was first established based on a canonical α-helical fold and is now referred to as the HEPN domain. The poor sequence conservation of HEPN domains has historically presented challenges with their identification; however, many well-characterized endoribonucleases have been reclassified as HEPN RNases, such as the eukaryotic UPR (unfolded protein response) factor Ire1, as well as novel HEPN endoribonucleases have been identified, including the bacterial Cas13 subfamily responsible for coordinating phage defense (Anantharaman et al., 2013; Shmakov et al., 2015). As the HEPN RNase family continues to expand into new areas of RNA biology, there is a growing need to understand the structure and regulation of these functionally diverse enzymes.
The HEPN RNase family features a broad class of endoribonucleases that can play critical roles in human health and disease. The recent explosion of newly classified HEPN RNases yielded over 38 unique members (Anantharaman et al., 2013). Several HEPN RNases have been linked to human disease, including neurological disorders and cancer, and hold therapeutic potential in the mitigation of viral illnesses and the advancement of transcriptome editing technologies. HEPN RNases are found in all kingdoms of life and process distinct RNA substrates, including tRNA, rRNA, mRNA, and viral RNA targets (Anantharaman et al., 2013). Despite their functional diversity, all HEPN RNases are metal-ion independent and catalyze RNA cleavage through a shared mechanism of 2′-O-transesterification resulting in 2′−3′ cyclic phosphate and 5′-hydroxyl RNA ends (Pillon and Stanley, 2018; East-Seletsky et al., 2016; Shigematsu, Kawamura, and Kirino, 2018). Over the last 5 years, seminal contributions in HEPN RNase biology have led to significant advances in the field, yet how HEPN RNases recognize their distinct RNA targets and catalyze RNA cleavage remains poorly defined. In this review we focus on recent advances pertaining to the structure and function of HEPN RNases, as well as summarize the current models for HEPN RNase regulation.
Molecular characteristics of HEPN RNase domains
The HEPN domain is a universal signature of all HEPN family members and harbors key molecular features necessary for endoribonuclease activity. HEPN domains are small α-helical domains that typically range from 100 to 120 residues (Anantharaman et al., 2013). The HEPN domain core adopts a compact architecture formed by four α-helices (α1, α2, α3, α4) of variable length that fold into a canonical up-down arrangement (Figure 1). The RNase class of HEPN members also encode the consensus RNase motif RϕxxxH (where ϕ is the polar residue N, H, or D and x is any amino acid ranging between 3–5 residues) (Anantharaman et al., 2013). The highly conserved RNase motif is surface exposed and located at the junction between α3 and the α3-α4 insertion element of the core HEPN domain (Figure 1). The arginine residue at the first position (R1) and histidine residue at the last position (H6) are exclusively conserved and indispensable for HEPN endoribonuclease activity (Pillon et al., 2019; Lee et al., 2008; Huang et al., 2014; Liu et al., 2017). Moreover, there is mounting evidence indicating that the polar residue at the second position (ϕ2) also plays an important role in RNA cleavage (Pillon et al., 2019; Lee et al., 2008; Pillon et al., 2020). Considering there are numerous HEPN members that lack the RϕxxxH motif, future studies are needed to determine whether they are catalytically inactive or divergent forms of HEPN RNases that rely on an RNA cleavage mechanism independent of the RϕxxxH motif (Anantharaman et al., 2013).
Figure 1.
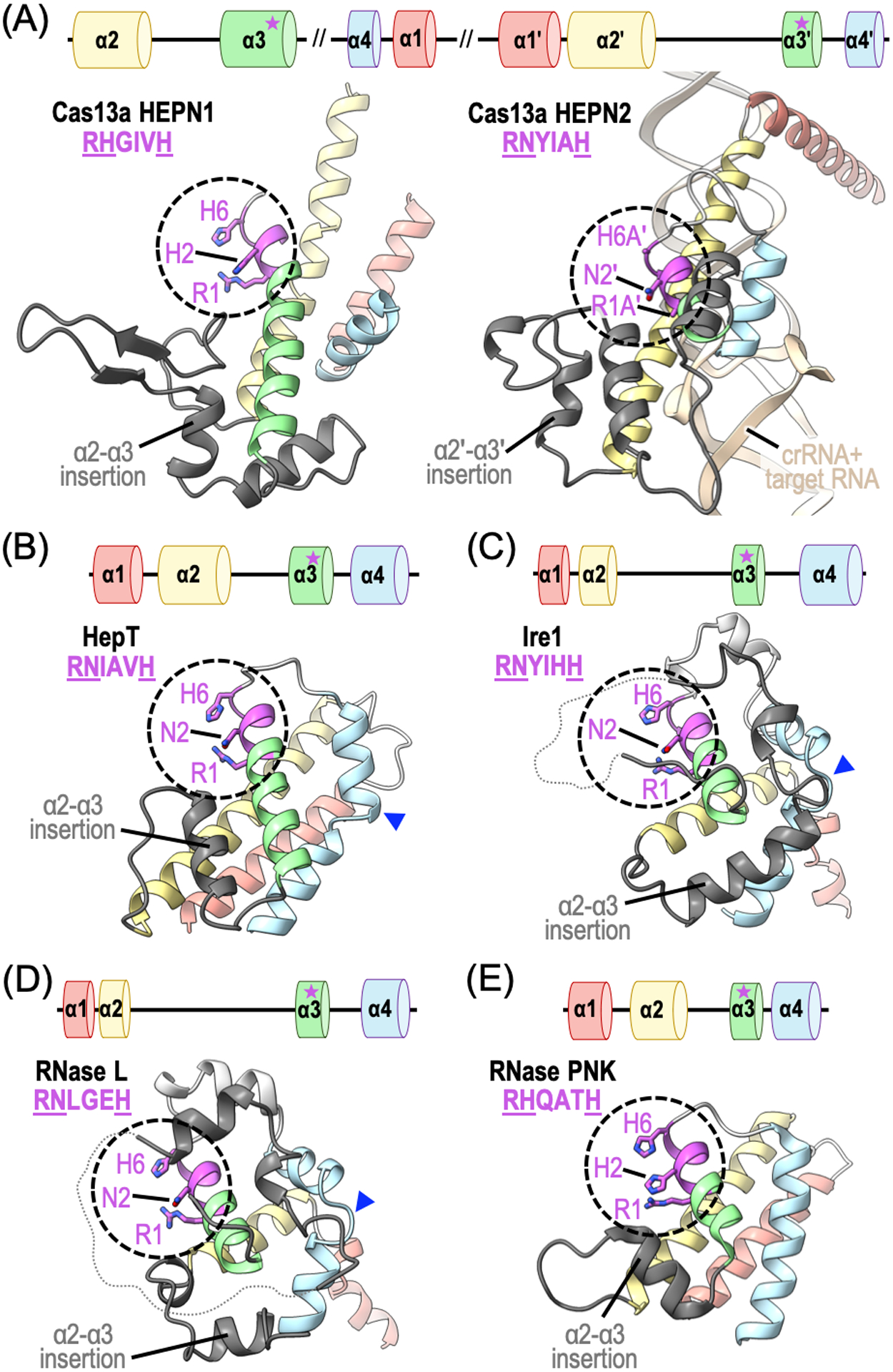
Structural organization of HEPN domains. Ribbon diagrams of HEPN domains from HEPN RNase family members: (A) Leptotrichia buccalis Cas13a (PDB ID: 5XWP), (B) Shewanella oneidensis HepT (PDB ID: 5YEP), (C) Saccharomyces cerevisiae Ire1 (PDB ID: 2RIO), (D) Sus scrofa RNase L (PDB ID: 4O1P), and (E) Chaetomium thermophilum RNase PNK (PDB ID: 6OF3). The canonical four α-helical bundle (α1, α2, α3, α4) is colored in red, yellow, green and blue, respectively. Insertions between α2 and α3 are shown in dark grey and kinks to α4 are highlighted using a blue arrowhead. The canonical HEPN RNase RϕxxxH motif is circled and conserved residues are colored in magenta.
HEPN domains can accommodate permutations that likely contribute to their functional versatility. The canonical HEPN domain is most often formed from the consecutive arrangement of its four core α-helices in an N- to C-terminal direction (α1 → α2 → α3 → α4). However, there are several HEPN RNases that reveal exceptions to this rule. For instance, HEPN α2-α4 precedes α1 in the primary sequences of Cas13a and Cas13d orthologs, yet they retain the canonical α-helical bundle fold (Figure 1A). HEPN α-helical arrays are also disrupted by large domain insertions, as seen with many Cas13 HEPN1 domains (Liu et al., 2017; Zhang et al., 2018). Smaller insertion elements (up to 60 residues) are also seen in many HEPN RNases and are often located between α2 and α3 of the domain core (Anantharaman et al., 2013). The α2-α3 insertion elements are structurally diverse and can take on the form of a single loop, α-helix, or β-hairpin, as well as appear as a complex mixture of secondary structure elements (Figure 1) (Anantharaman et al., 2013). The relative positioning of α1 can also deviate dramatically to allow the formation of an RNA binding site or accommodate structural features from neighboring domains (Figure 1A). Many HEPN RNases also harbor a discrete kink within α4 resulting in an alternative trajectory for downstream helical residues (Figure 1B–D) (Anantharaman et al., 2013). In addition to structural deviations, a small subset of HEPN RNases also encode a second consensus motif Exxx[K/R] (where x is any amino acid residue) upstream of the RNase RϕxxxH motif (Anantharaman et al., 2013). While this second motif is thought to play an important role in RNA cleavage, its precise function and limited retention among HEPN RNases remains unexplained. With the growing list of distinct HEPN folds, it is clear that some HEPN members emulate the canonical fold, while others display dramatic structural deviations. As the repertoire of high-resolution structures of HEPN members continues to expand, we will likely continue to discover new ways the HEPN domain can accommodate structural permutations for its repurposing in diverse RNA processing and degradation pathways.
A single HEPN domain is insufficient for RNA cleavage and must dimerize in trans or in cis to form a composite RNase active site. Both homodimerization and heterodimerization have been observed amongst members of the HEPN RNase family, however trans-homodimerization appears to be more common. Upon proper association of the HEPN domains, the variable HEPN α2-α3 insertion element forms a prominent cleft with the RNase RϕxxxH residues lining the base of the cleft (Figure 2). To form the catalytic center, the neighboring copies of α3 cluster in an antiparallel configuration at the dimerization interface to align the conserved RϕxxxH motifs. As a result, the HEPN arginine (R1) and polar (ϕ2) residues are precisely positioned across from the conserved histidine (H6) residue in the neighboring HEPN monomer (Figure 2). The molecular basis for RϕxxxH mediated RNA cleavage has yet to be ascribed, however recent work suggests H6 likely promotes 2′-OH nucleophilic attack, R1 stabilizes the transition state, and R1 along with ϕ2 supports proper coordination of the RNA within the active site (Anantharaman et al., 2013; Pillon et al., 2020). To understand the requirement for two juxtaposed copies of the RϕxxxH motif, biochemical studies have characterized chimeric variants of distinct HEPN RNases where a single copy of the invariant arginine (R1) or histidine (H6) residue were mutated. While some chimeric HEPN RNases, such as the Cas13 subfamily and RNase PNK, were sensitive to these changes, others, including Ire1 and RNase L, retained their RNA cleavage activity suggesting there is variability amongst HEPN RNase active sites (East-Seletsky et al., 2016; Pillon et al., 2020; Abudayyeh et al., 2016; Korennykh and Walter, 2012; Han et al., 2014).
Figure 2.
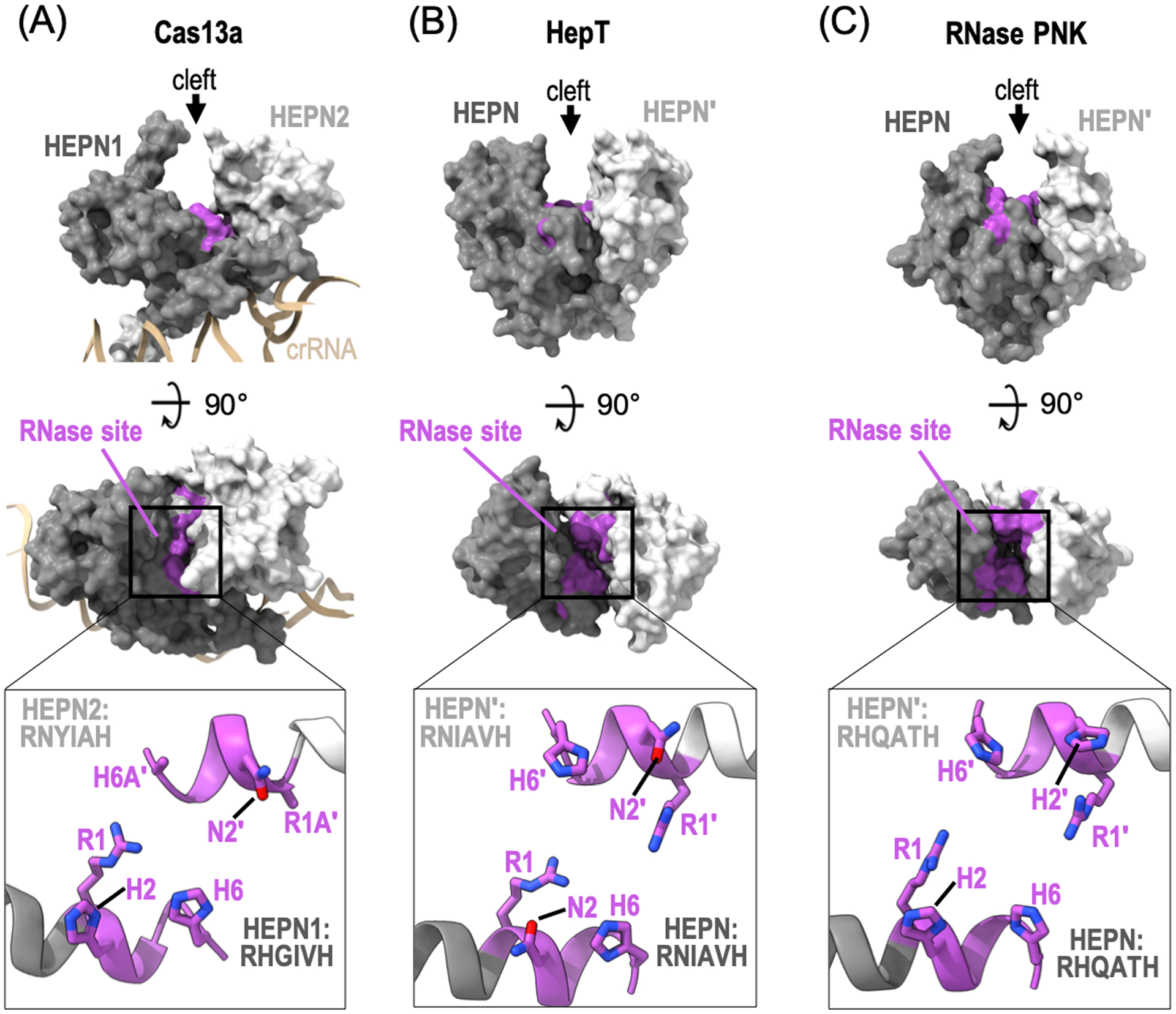
Composite HEPN RNase active sites. Orthogonal views of the surface representation of dimeric HEPN RNase family members: (A) L. buccalis Cas13a (PDB ID: 5XWP), (B) S. oneidensis HepT (PDB ID: 5YEP), and (C) C. thermophilum RNase PNK (PDB ID: 6OF3). Each HEPN monomer is colored in dark (HEPN) or light grey (HEPN′). The canonical HEPN RNase motifs are colored in magenta. Insets are zoom in views of HEPN α3 which encompass the conserved HEPN RϕxxxH motif.
Structural architecture of functionally distinct HEPN endoribonucleases
The HEPN domain is an ancient protein fold that has evolved to support a broad class of endoribonucleases that orchestrate diverse RNA processing and degradation pathways. For instance, prokaryotic HEPN endoribonucleases are CRISPR (clustered regularly interspaced short palindromic repeat)-associated nucleases, abortive infection system nucleases, and the toxic components to several bacterial toxin-antitoxin modules (Anantharaman et al., 2013). Moreover, eukaryotic HEPN endoribonucleases form large multi-subunit complexes and are members to the family of Nuclease-integrated Kinase super assemblies (NiKs) (Pillon and Stanley, 2018). NiKs-associated RNases support stress-activated pathways, such as the UPR and the antiviral response, as well as play an essential role in ribosome production. In the following section we will highlight select HEPN RNases to describe their distinct cellular roles and unique structural organization.
CRISPR-Cas associated HEPN nucleases.
There are two subsets of HEPN RNases that have recently been associated with CRISPR-Cas systems, including Cas13 and Csm6/Csx1. CRISPR-Cas systems provide microbial organisms with adaptive immunity against foreign genetic elements, such as RNA transcripts generated from phage genomic DNA, through a multi-step process of adaptation, expression, and interference (Knott and Doudna, 2018). Immunity is acquired through a process known as adaptation whereby foreign genetic elements are integrated into microbial genomic CRISPR-arrays, providing cells with a “molecular memory” of the foreign genetic element. The expression (or transcription) of microbial CRISPR arrays leads to the generation of precursor CRISPR-RNAs (crRNA) that are processed into mature crRNAs before they associate with CRISPR associated (Cas) nucleases to form “surveillance” or “interference” complexes. These active surveillance complexes scan for foreign nucleic acids by looking for complementarity to the crRNA. Once sufficient base-pairing has been achieved, the Cas nuclease is activated leading to cleavage (also referred to as interference) of the target and immunity against the foreign nucleic acid.
CRISPR-Cas systems can be divided into two classes based on the organization and architecture of the Cas effectors. Class 1 CRISPR-Cas systems are defined by the presence of multiple effector genes with differing functions, whereas the less common Class 2 systems have a single multifunctional effector (Makarova et al., 2020). There is extensive diversity amongst CRISPR-Cas systems and each class can be further divided into three types based on the effector proteins. Class 1 systems include Types I, III, and IV while Class 2 systems include Types II, V, and VI (Makarova et al., 2020; Koonin and Makarova, 2019). HEPN nucleases are found in both CRISPR classes. Cas13 nucleases, which are large multifunctional effectors are the defining members of Class 2 Type VI, whereas Csm6 and Csx1 are ancillary factors that belong to Class 1 Type III.
Cas13 subfamily −
Cas13 CRISPR effectors were identified only five years ago from a large-scale computational pipeline designed to identify previously undetected CRISPR-Cas systems (Shmakov et al., 2015). This approach predicted Cas13 (originally called C2c2 for class 2 candidate 2) candidates from a number of bacterial species. The only homology between these predicted effectors and other known proteins was the presence of two conserved HEPN RNase motifs (RϕxxxH), suggesting that Cas13 effectors possess RNase activity (Shmakov et al., 2015). This finding was soon confirmed by several independent studies that showed that the two HEPN domains of Cas13 support RNA-guided endoribonuclease activity (East-Seletsky et al., 2016; Abudayyeh et al., 2016). In addition to the two HEPN domains, Cas13 effectors also contain a second RNase active site that is responsible for processing the precursor crRNA (East-Seletsky et al., 2016). The discovery of this new class of HEPN-containing CRISPR effectors that target RNA instead of DNA has led to an explosion in the development of RNA-guided RNA-targeting applications (Patchsung et al., 2020; Abbott et al., 2020; Abudayyeh et al., 2019; Terns, 2018; Ackerman et al., 2020).
To date four different subtypes of Cas13 effectors have been identified named A, B, C, and D (Abudayyeh et al., 2016; O’Connell, 2019; Shmakov et al., 2017; Konermann et al., 2018; Yan et al., 2018; Smargon et al., 2017). Beyond the HEPN catalytic motifs, little sequence homology exists amongst the different Cas13 subtypes (Koonin and Makarova, 2019; O’Connell, 2019). With the exception of Cas13c, multiple structures of the different Cas13 subtypes have been determined in recent years (Table 1). Despite the lack of sequence identity, all Cas13 effectors adopt a bi-lobed architecture that is reminiscent of the Cas9 effectors. The bi-lobed organization includes a recognition lobe (REC) responsible for crRNA binding and processing, as well as a Nuclease lobe (NUC) comprised of two HEPN domains. Below we summarize the molecular details of the HEPN nuclease domains for Cas13 effector subtypes that have been characterized to date. For more specific details on pre-crRNA recognition and processing along with crRNA spacer organization, please see the following recent reviews on Cas13 effectors (O’Connell, 2019; Garcia-Doval and Jinek, 2017).
Table 1.
Molecular features of HEPN RNase structures.
| CRISPR associated HEPN RNases | |||||
| Cas13a (or C2C2) | Lachnospiracease bacterium | hetero-dimer | RNdsfH/RNyieH | 5WLH, 5W1L,5WIH | (Knott et al., 2017) |
| Leptotrichia shahii | hetero-dimer | RNrilH/RNyisH | 5WTK, 5WTJ | (Liu et al., 2017) | |
| Leptotrichia buccalis | hetero-dimer | RNgivH/RNyiaH | 5XWY, 5XWP | (Liu et al., 2017) | |
| Listeria seeligeri | hetero-dimer | RNeiiH/RNnisH | 6VRB, 6VRC | (Meeske et al., 2020) | |
| Cas13b | Bergeyella zoohelcum | hetero-dimer | RNfytH/RNkfaH | 6AAY | (Zhang et al., 2018) |
| Prevotella buccae | hetero-dimer | RNyysH/RNafsH | 6DTD | (Slaymaker et al., 2019) | |
| Cas13d | uncultured Ruminococcus sp. | hetero-dimer | RQcvfH/RNciaH | 6IV8, 6IV9 | (Zhang et al., 2019) |
| Eubacterium siraeum | hetero-dimer | RHwcvH/RNtycH | 6E9E, 6E9F | (Zhang et al., 2018) | |
| Csm6 | Thermus thermophilus | homo-dimer | RNsallvH | 5FSH | (Niewoehner and Jinek, 2016) |
| Thermococcus onnurineus | homo-dimer | RNylaH | 6OV0, 6O71, 6O70, 6O6Z, 6O6Y, 6OSX, 6O6V, 6O6T, 6O6S | (Jia et al., 2019) | |
| Enterococcus italicus | homo-dimer | RNkvaH | 6TUG | (Garcia-Doval et al., 2020) | |
| Csx1 | Sulfolobus solfataricus | homo-dimer | RIlyaH | 2I71 | Structural Genomics |
| Pyrococcus furiosus | homo-dimer | RNfiaH | 4EOG | (Kim, Kim, and Oh, 2013) | |
| Sulfolobus islandicus | homo-dimer | RNfiaH | 6R9R, 6R7B, 6QZT | (Molina et al., 2019) | |
| Toxin-antitoxin associated HEPN RNases | |||||
| HepT (or SO_3166) | Shewanella oneidensis | homo-dimer | RNiavH | 5YEP, 6M6V, 6W6U, 6M6W, 7BXO | (Yao et al., 2020; Jia et al., 2018) |
| LsoA | Escherichia coli | hetero-dimer with antitoxin | RHglfH | 5HY3 | (Wan et al., 2016) |
| RnlA | Escherichia coli | non-canonical homo-dimer | RHslfH | 4I8O | (Wei et al., 2013) |
| HI0074 | Haemophilus influenzae | homo-dimer | RNitsH | 1JOG | (Lehmann et al., 2003) |
| NiKs associated HEPN RNases | |||||
| Ire1 | Saccharomyces cerevisiae | homo-dimer (back-to-back) | RNkyhH | 2RIO, 3LJ0, 3LJ1, 3LJ2, 3SDM, 3SDJ, 3FBV, 2BE1* | (Lee et al., 2008; Korennykh et al., 2009; Credle et al., 2005; Wiseman et al., 2010; Korennykh et al., 2011; Korennykh et al., 2011) |
| Homo sapiens | homo-dimer (face-to-face) | RNkkhH | 2HZ6*, 6HV0, 6URC, 6HX1, 5HGI, 4U6R, 4YZC, 4YZ9, 4YZD, 3P23, 4Z7H, 4Z7G, 6SHC | (Ali et al., 2011; Feldman et al., 2016; Zhou et al., 2006; Colombano et al., 2019; Harnoss et al., 2019; Harrington et al., 2015; Concha et al., 2015; Joshi et al., 2015; Amin-Wetzel et al., 2019) | |
| Mus musculus | homo-dimer (face-to-face) | RNkkhH | 4PL3, 4PL4, 4PL5 | (Sanches et al., 2014) | |
| RNase L | Sus scrofa | homo-dimer | RNlgeH | 6M11, 6M12, 6M13, 4O1O, 4O1P | (Huang et al., 2014) |
| Homo sapiens | homo-dimer | RNlgeH | 4OAU, 4OAV | (Han et al., 2014) | |
| RNase PNK | Chaetomium thermophilum | homo-dimer | RHqatH | 6OF2, 6OF3, 6OF4 | (Pillon et al., 2019) |
Luminal domain structure of the corresponding cytosolic Ire1 protein kinase-HEPN RNase.
Cas13a (Type VI-A) was the first Cas13 effector to be identified and as such remains the most well characterized (East-Seletsky et al., 2016; Abudayyeh et al., 2016). Cas13a is the largest of all the Cas13 subtypes and is composed of six distinct domains including an N-terminal domain (NTD), two helical domains, two HEPN domains, and a linker domain (Figure 3A). The NTD and the first helical domain, which contains the crRNA processing active site, form the REC lobe. The NUC lobe is composed of the first HEPN domain, which is divided in half by the second helical domain, a linker domain, and the second HEPN domain. Both HEPN domains are significantly larger than the canonical HEPN domain, and contain several additional α-helices and a small β-hairpin insertion between α2 and α3 (Figure 1A and 3B). These extra components make important intramolecular contacts with other subdomains and are also critical for recognition of the crRNA stem-loop (Liu et al., 2017). The structures of Cas13a from Leptotrichia shaii (Lsh) and Lachno spiracease bacterium (Lba) revealed that the dual RNase active sites are completely independent of one another, with the HEPN active site positioned on an external concave surface (Figure 3B) (Liu et al., 2017; Knott et al., 2017).
Figure 3.
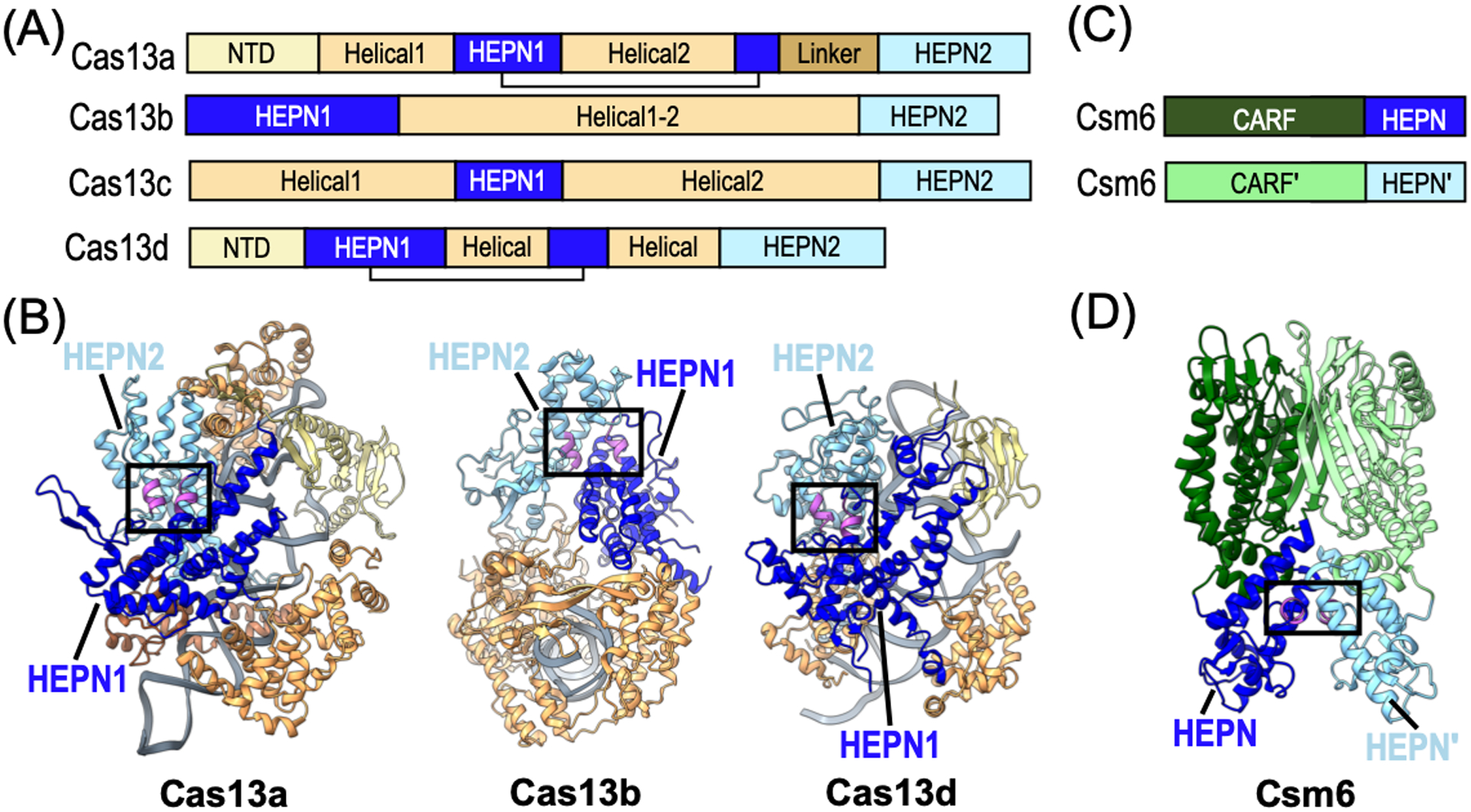
Structural architecture of CRISPR-Cas associated HEPN RNases. (A) Domain architecture of Cas13 subtypes A, B, C, and D. (B) Ribbon diagrams of L. buccalis Cas13a (PDB ID: 5XWP), Prevotella buccae Cas13b (PDB ID: 6DTD), and Eubacterium siraeum Cas13d (PDB ID: 6E9F) colored as seen in panel A. The HEPN RNase motifs are colored in magenta and the composite RNase active sites are boxed. The bound RNA is shown in grey. (C) Domain architecture of CRISPR-Cas associated Csm6 RNase. Abbreviation CARF defines CRISPR-associated Rossman-fold. (D) Ribbon diagram of Thermococcus onnurineus Csm6 (PDB ID: 6O6S) colored as seen in panel C.
Cas13b (Type VI-B) was identified from a bioinformatics analysis in 2017 designed to discover novel Class 2 effectors lacking the typical CRISPR associated proteins, Cas1 and Cas2 (Shmakov et al., 2017; Smargon et al., 2017). Cas13b effectors have two HEPN domains located at the N- and C-terminus of the protein, but aside from the HEPN domains their overall domain organization is distinct from other Cas13 effectors (Figure 3A). Two subclasses of Cas13b have been discovered thus far based on the presence of two different accessory proteins, Csx27 and Csx28, which effect the activity of Cas13b. Csx27 is predicted to form a membrane channel for single stranded DNA (Makarova et al., 2019) and has been shown to repress Cas13b activity (Smargon et al., 2017). In contrast, Csx28 enhances RNA cleavage activity of Cas13b through an unknown mechanism. Csx28 has a putative HEPN domain that has yet to be characterized, but it is tempting to speculate that this additional HEPN domain could also contribute to Cas13b-mediated RNA cleavage (Smargon et al., 2017). Structures of Cas13b from Berheyella zoohelcum and Prevotella buccae have shown that despite the location of the HEPN domains at the extreme N- and C-termini of the protein, the two HEPN domains come together to form a solvent exposed RNase active site (Figure 3B) (Zhang et al., 2018; Slaymaker et al., 2019).
Cas13d (Type VI-D) effectors were discovered in 2018 from an expanded bioinformatics search looking for proteins with two HEPN domains (Konermann et al., 2018; Yan et al., 2018). While they share less than 8% sequence similarity with Cas13a effectors, Cas13d effectors have a similar domain organization (Figure 3A–B). Cas13d is about 20% smaller than all other Cas13 effectors, which may be advantageous for in vivo RNA targeting applications. The other notable difference is the presence of members of a family of accessory proteins known as WYL-domain containing proteins, named after the presence of three conserved amino acids (Yan et al., 2018). WYL proteins are found in many other CRISPR-Cas and microbial defense systems. WYL has been shown to enhance the nuclease activity of Cas13d. While the precise mechanism is unclear, recent work revealed that WYL has a high affinity for single strand RNA, which may aid in recruitment of RNA substrates to Cas13d (Zhang et al., 2019). Structures of Cas13d from Ruminococcus and Eubacterium siraeum revealed a bi-lobed architecture that is reminiscent of the structural organization of Cas13a (Figure 3B) (Zhang et al., 2018; Zhang et al., 2019).
Csm6/Csx1 −
Csm6 and Csx1 form a distinct family of HEPN nucleases that function as ancillary factors for Type III CRISPR-Cas systems. Type-III CRISPR-Cas systems are defined by the presence of the Cas10 signature nuclease, and are further subdivided into four subtypes (A-D) based on the composition of additional proteins. Csm6 HEPN nucleases are found in Type III-A, while Csx1 HEPN nucleases are found in the other three subtypes (Makarova et al., 2020). One distinction of Type-III systems is that they degrade both DNA and RNA from foreign invaders through a unique signal transduction cascade (Niewoehner et al., 2017; Kazlauskiene et al., 2017). Type-III Csm CRISPR-Cas effectors have a large multi-subunit ribonucleoprotein complex that includes Cas10, crRNA, and a number of additional Csm factors. The nuclease domain of Cas10 is activated to degrade single strand DNA non-specifically upon target RNA binding, while additional Csm factors specifically cleave bound RNA. Beyond nuclease activity, an additional function of the Cas10 complex is the synthesis of a second messenger known as cyclic oligoadenylate (cOA). This cyclic messenger is synthesized by the PALM domain of Cas10 from individual ATP molecules following activation by target RNA binding (Niewoehner et al., 2017; Kazlauskiene et al., 2017). Following synthesis, cOA then binds to the HEPN nuclease Csm6/Csx1 and activates the HEPN domain to non-specifically cleave RNA (Niewoehner et al., 2017; Kazlauskiene et al., 2017).
Like most HEPN RNases, Csx1 and Csm6 rely on HEPN homodimerization for RNase activity. Structures of Csm6 and Csx1 have been determined from several organisms (Table 1). Crystal structures of Csm6 from Enteroccocus italicus and Thermococcus onnurineus revealed that it is composed of two domains including an N-terminal CARF (CRISPR Associated Rossman Fold) domain and a C-terminal HEPN domain (Figure 3C) (Garcia-Doval et al., 2020; Jia et al., 2019).. Csm6 assembles into a homodimer with both the CARF and HEPN domains participating in dimer formation (Figure 3D). Dimerization of the CARF domain forms the binding pocket for cOA whereas dimerization of the HEPN domains forms the RNase active site. In addition to the CARF and HEPN domains, Sulfolobus islandicus (Sis) Csx1 also contains a helix-turn-helix (HTH) domain between the CARF and HEPN domains. Similar to Csm6, both the CARF and HEPN domains homodimerize, however SisCsx1 further oligomerizes into a stable hexamer. The hexamer is formed by an extension (hexamerization helix) following α2 in the HEPN domain. Hexamerization of SisCsx1 is essential for RNase activity and is likely important for facilitating cooperativity between the three RNase active sites (Molina et al., 2019).
Toxin-antitoxin associated HEPN nucleases.
There are numerous HEPN RNases associated in toxin-antitoxin (TA) systems. TA systems are ubiquitous in prokaryotes and archaea and can support diverse cellular functions including cell survival during times of stress. All TA systems contain a toxin component along with its inhibitor, the antitoxin component. Based on the interaction mode of the toxin-antitoxin pair, TA systems can be classified into six discrete types. The Type II system is the largest and best characterized, comprising a small protein toxin and a small protein antitoxin. The toxin-antitoxin loci are co-expressed and its protein components form a stable toxin-antitoxin complex that sequesters and inhibits the toxin. Upon the detection of a stress signal, the antitoxin is degraded by proteases resulting in the release and activation of the toxin (Jurėnas and Van Melderen, 2020). While Type II toxins are diverse, they share a common role in suppressing protein synthesis by targeting rRNA, mRNA or tRNA substrates (Christensen and Gerdes, 2003; Jiang et al., 2002; Liu et al., 2008; Winther and Gerdes, 2011; Winther et al., 2013). By inhibiting protein translation, the toxin induces cell growth arrest and provides an opportunity for stress acclimation.
Toxins belonging to the Type II TA system are typically RNases regulated by their specialized antitoxin auxiliary factor. To safeguard cells from non-specific disruption of protein translation, Type II TA systems are encoded within bicistronic loci where the antitoxin often precedes the toxin to ensure the RNase toxin is immediately inhibited upon translation. Type II antitoxins typically encode two modules, a toxin binding domain for RNase inhibition and a DNA binding domain for TA operon autorepression. To inhibit the toxin RNase, the antitoxin often interacts with the RNase active site thereby mimicking substrate and sterically occluding the catalytic residues. While Type II TA pairs assemble into macromolecular complexes of varying size and complexity, the RNase toxin and its antitoxin often present in stoichiometric ratios. Likewise, Type VII TA systems can also feature RNases where the antitoxin neutralizes the nuclease toxin through an enzymatic reaction (Yao et al., 2020). In the section below, we will highlight specialized features of three HEPN containing TA systems: HepT-MntA, LsoA-LsoB, and RnlA-RnlB.
HepT −
The Type VII TA toxin HepT (HEPN toxin) of the two-gene module HEPN/MNT represents an unusual member of the HEPN RNase family. Unlike most HEPN RNases that are encoded within multi-domain proteins, HepT homologs are single domain proteins encoding a minimal HEPN core. The small footprint of HepT homologs likely reflects its role in TA systems, as toxins are often small in size (typically less than 20 kDa) (Otsuka and Yonesaki, 2012). For instance, HepT subgroup I homologs from Geobacillus harbors a remarkably small HEPN domain (10 kDa) along with the canonical HEPN RNase motif, whereas subgroup II homologs encode a slightly larger HEPN domain (16 kDa) and encompass both the RNase motif (RϕxxxH) and its less-prevalent secondary motif (Exxx[K/R]) (Anantharaman et al., 2013; Alkhalili, Wallenius, and Canbäck, 2019). Prokaryotic HepT homologs can also be found fused to a MNT (minimal nucleotidyltransferase) partner. In some cases the RNase motif has been lost in the HepT-MNT fusion pair, while others retain an intact RϕxxxH motif (Anantharaman et al., 2013). As limited work has focused on the characterization of prokaryotic HepT homologs, future studies are needed to confirm their RNase activity, as well as define the regulation of these atypical HEPN members.
Structural studies of the Shewanella oneidensis HepT toxin in complex with its MNT antitoxin (MntA) partner reveals an unexpected higher-order arrangement. S. oneidensis HepT interacts with MntA to form a hetero-octameric arrangement comprising 6 HepT toxins and 2 MntA antitoxins (Figure 4A) (Jia et al., 2018). This rare stoichiometry deviates from the general rule that TA macromolecular assemblies comprise one toxin for every 1–2 antitoxins. Correspondingly, the VapBC TA system also forms a hetero-octameric assembly, and yet it retains stoichiometric amounts of VapC toxin to VapB antitoxin (Bendtsen et al., 2017; Das et al., 2014; Dienemann et al., 2011). The HepT-MntA complex forms three discrete HepT homodimers interspaced by MntA antitoxins (Figure 4A) (Jia et al., 2018; Yao et al., 2015). The juxtaposed RNase motifs (RNxxxH) lie at the base of deep clefts decorated with positively charged residues that likely interact with the RNA backbone to aid proper substrate alignment. The shape and size of the HepT RNase cleft is also likely to be important for conferring its mRNA specificity as it could easily accommodate single strand RNA, but would occlude structured and double stranded RNAs (Jia et al., 2018).
Figure 4.
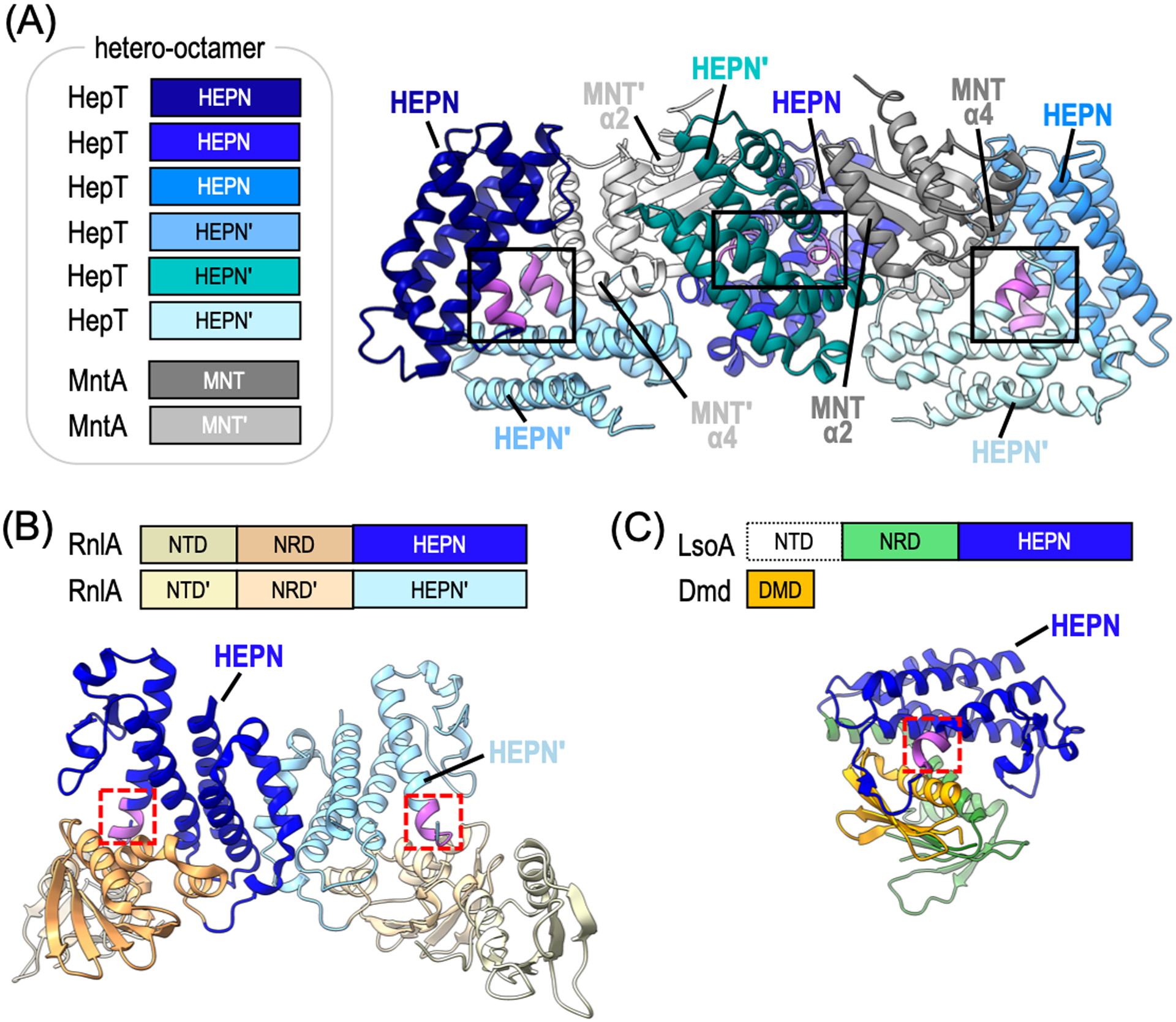
Architecture of toxin-antitoxin associated HEPN RNases. (A) Domain architecture and corresponding ribbon diagram of S. oneidensis HepT-MntA hetero-octamer (PDB ID: 5YEP). Toxin HepT protomers are colored in shades of blue and the antitoxin MntA protomers are shown in grey. The HEPN RNase motifs are highlighted in magenta and the composite HEPN RNase active sites are boxed. The MntA α-helices (α2 and α4) important for HEPN inactivation are labeled. (B) Domain architecture and corresponding ribbon diagram of the Escherichia coli RnlA homodimer (PDB ID: 4I8O). The RnlA homodimer does not form a composite HEPN RNase active site because the conserved motifs are on the opposite ends of the dimer interface (red dashed box). (C) Domain architecture and corresponding ribbon diagram of E. coli LsoA toxin in complex with T4 phage antitoxin Dmd (PDB ID: 5HY3). The LsoA NTD could not be resolved. The LsoA NRD and HEPN domains are shown in green and blue, respectively. Single HEPN RNase motif is colored in magenta and boxed in red. The Dmd viral antitoxin (orange) interacts with the HEPN motif, mimicking substrate binding.
LsoA/RnlA −
The Type II TA toxins LsoA and its functional homolog RnlA are HEPN RNases involved in phage defense (Otsuka and Yonesaki, 2012; Koga et al., 2011). LsoA has 20% sequence identity and 45% sequence similarity to RnlA (also known as RNase LS). Despite their low sequence identity, both LsoA and RnlA adopt the canonical HEPN domain fold, encode the conserved RNase RϕxxxH motif, and catalyze viral mRNA cleavage (Otsuka and Yonesaki, 2012). To prevent spurious RNase activity, LsoA and RnlA are inhibited by the bacterial antitoxins LsoB and RnlB, respectively. Viruses have evolved their own antitoxins to inhibit TA systems, such as the T4 phage antitoxin protein (Dmd) that can directly inactivate LsoA and RnlA RNase activity, thereby halting viral mRNA degradation and defeating the bacterial defense system (Otsuka and Yonesaki, 2012; Wan et al., 2016; Otsuka et al., 2007).
Structural studies of LsoA and RnlA toxins reveal that they contain inactive HEPN catalytic centers. Unlike HepT toxins, LsoA and RnlA toxins are larger (~ 40 kDa) multidomain proteins. LsoA and RnlA are characterized by their N-terminal domain (NTD), central N-repeated domain (NRD), and C-terminal HEPN domain (also known as the Dmd binding domain (DBD)) (Figure 4B–C) (Otsuka and Yonesaki, 2012; Wei et al., 2013). Structural studies of RnlA revealed an association between its HEPN domains, but through non-canonical HEPN α-helices. This novel HEPN dimerization mode prevents the formation of a composite RNase active site by positioning the RϕxxxH motifs at opposite ends of the macromolecule (Figure 4B) (Wan et al., 2016; Wei et al., 2013). As the field moves forward, it will be important to confirm this unusual structural architecture in cells and define the molecular basis for RNase activation. Similar to RnlA, structural characterization of LsoA bound to its viral Dmd antitoxin captures the HEPN RNase in an inactive state. The LsoA NTD was poorly resolved, however the central NRD and C-terminal HEPN domains were well defined. Interestingly, the structural fold of RnlA and LsoA are homologous, and yet the LsoA HEPN domain did not form a homodimer, but instead formed a complex with the viral Dmd antitoxin (Figure 4C). Dmd sits in a concave groove formed by the relative positioning of the LsoA NRD and HEPN domains where it associates with the RϕxxxH motif and sterically occludes formation of the composite HEPN RNase site (Wan et al., 2016).
NiKs associated HEPN RNases.
To date, all confirmed eukaryotic HEPN RNases belong to the NiKs family of RNA processing and degradation machines (Anantharaman et al., 2013; Pillon and Stanley, 2018). A defining characteristic of NiKs members are their dual nuclease and kinase components. The nuclease component comprises the HEPN RNase, whereas the kinase component is variable and can encompass either a protein kinase, pseudo-protein kinase, or RNA kinase (Pillon and Stanley, 2018). To achieve RNase activity, the nuclease and kinase components assemble into higher-order structures (i.e. super assemblies) which are either constitutive in nature or induced upon ligand binding. These complex and intricate assemblies form the canonical HEPN RNase active site and also provide the structural infrastructure to allow for kinase-dependent regulation of the RNase catalytic center. NiKs members also harbor specialized features that are important for their diverse cellular functions. For instance, NiKs members include Ire1, involved in the UPR; RNase L, associated with the antiviral response; and RNase PNK, responsible for ribosome production. In the following section we will highlight the unique features of these bifunctional machineries and their roles in eukaryotic RNA processing and decay.
Ire1 −
The Ire1 endoribonuclease was characterized well before its classification as an HEPN RNase or NiKs family member (Cox, Shamu, and Walter, 1993; Sidrauski and Walter, 1997; Mori et al., 1993). Ire1 is conserved across eukaryotes and initiates a stress response upon the detection of unfolded and/or misfolded proteins in the endoplasmic reticulum (ER) (Korennykh and Walter, 2012; Coelho and Domingos, 2014; Maurel et al., 2014). Upon stress-dependent activation, Ire1 targets HAC1/XPB1 mRNA for alternative splicing and ER-bound mRNAs for decay to augment the protein folding capacity of the ER (Korennykh and Walter, 2012). Ire1 is a multi-domain protein harboring an N-terminal luminal domain followed by a transmembrane helix, a globular Ser/Thr protein kinase domain, and a C-terminal HEPN domain (also referred to as the kinase extension domain (KEN)) (Figure 5A). The transmembrane helix anchors Ire1 to the ER membrane positioning the luminal domain inside the ER in search of aberrant proteins whereas the protein kinase and HEPN RNase domains remain in the cytosol where they can access a variety of RNA targets (Korennykh and Walter, 2012; Maurel et al., 2014; Chalmers et al., 2019). Similar to other HEPN RNases, Ire1 relies on HEPN dimerization to form a functional composite RNase active site (Lee et al., 2008; Korennykh et al., 2009). Upon the detection of misfolded protein, the luminal domains associate and subsequently promote the formation of an extensive homodimerization interface that spans across the entire length of the protein, including its protein kinase and HEPN domains (Lee et al., 2008; Karagöz et al., 2017). Upon Ire1 dimerization, the juxtaposed protein kinase domains undergo trans-autophosphorylation followed by activation of the HEPN RNase catalytic center (Lee et al., 2008; Sidrauski and Walter, 1997; Korennykh et al., 2009; Aragon et al., 2009; Gardner et al., 2013; Gardner and Walter, 2011). Structures of dimeric Saccharomyces cerevisiae Ire1 reveal a parallel back-to-back dimer configuration where the HEPN RNase motifs are positioned at the dimer interface to form the composite RNase site (Figure 5B) (Lee et al., 2008; Credle et al., 2005). Interestingly, structures of mouse and human Ire1 have yet to produce the back-to-back arrangement, but instead show a face-to-face dimer where the HEPN RNase motifs are found at opposite ends of the HEPN homodimer (Table 1) (Ali et al., 2011; Sanches et al., 2014; Feldman et al., 2016). While the functional significance of the face-to-face arrangement remains unclear, it has been suggested to promote Ire1 trans-autophosphorylation (Adams et al., 2019). On the other hand, it is widely accepted that the back-to-back configuration represents the RNase active state and that all Ire1 orthologs are likely to adopt this arrangement during RNA cleavage.
Figure 5.
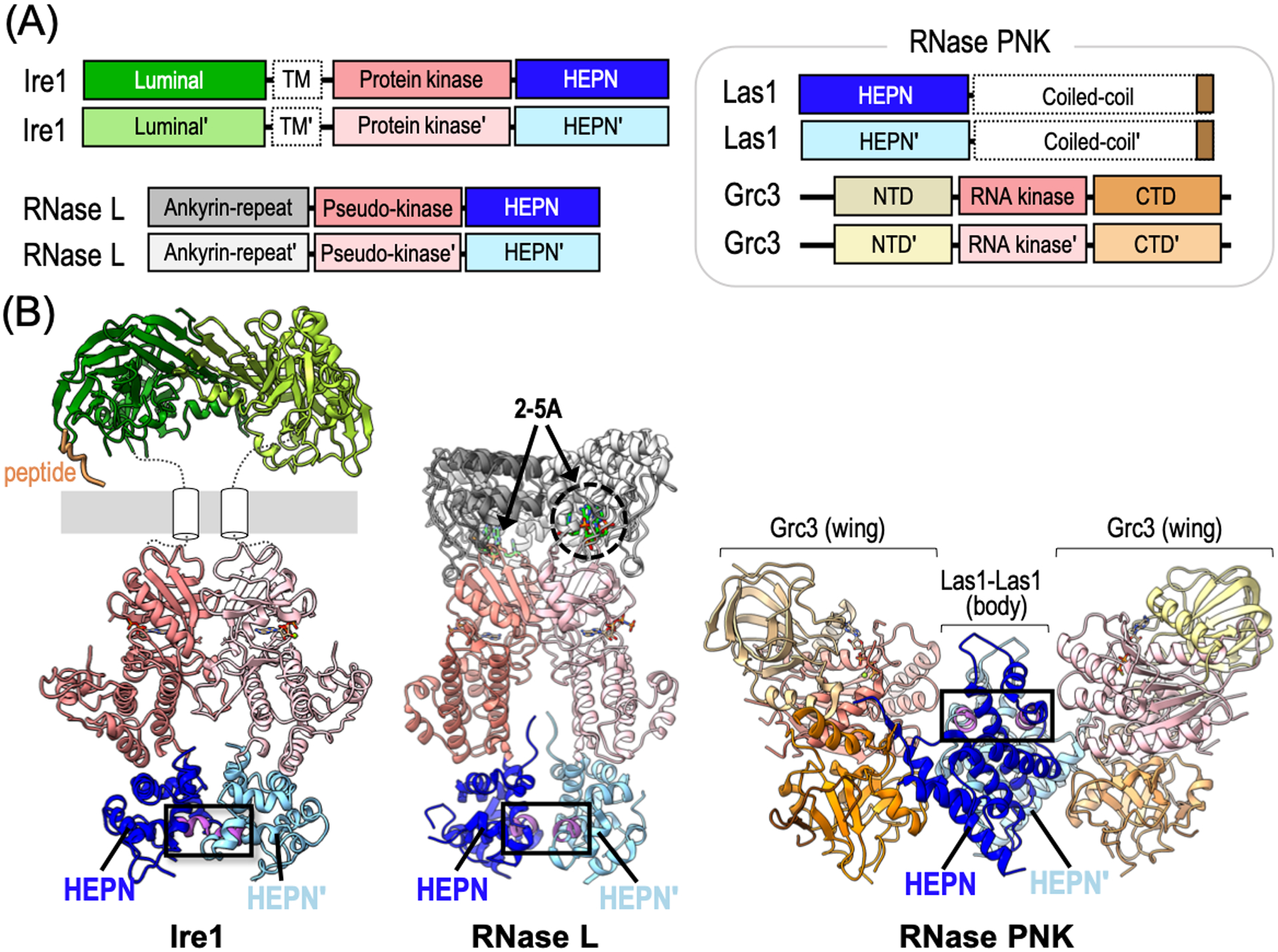
Structural architecture of NiKs-associated HEPN RNases. (A) Domain architecture of Ire1, RNase L and RNase PNK. Domains outlined with a dashed border have no available structural information. (B) Ribbon diagrams of S. cerevisiae Ire1 (PDB IDs: 2BE1, 2RIO), Sus scrofa RNase L (PDB ID: 4O1P), and C. thermophilum RNase PNK (PDB ID: 6OF3) colored as seen in panel A. The HEPN RNase motifs are colored in magenta and the composite RNase active sites are boxed. The poly-valine peptide (orange) mimics an unfolded protein in the Ire1 structure. White cylinders represent the Ire1 transmembrane domain and the grey bar depicts the ER membrane. RNase L ligand 2–5A is shown as greens sticks.
RNase L −
Since its discovery over two decades ago, extensive characterization of the RNase L endoribonuclease has revealed key insight into its intricate function and regulation (Wreschner et al., 1981; Floyd-Smith, Slattery, and Lengyel, 1981; Dong et al., 1994). RNase L is conserved in higher-eukaryotes where it localizes to the cytoplasm for antiviral signaling (Chakrabarti, Jha, and Silverman, 2011; Gusho, Baskar, and Banerjee, 2016; Khodarev, 2019). As seen with all NiKs members, RNase L is a multi-domain protein composed of an N-terminal ankyrin-repeat domain, central pseudo-protein kinase domain, and C-terminal HEPN endoribonuclease domain (Figure 5A) (Huang et al., 2014). The RNase L pseudo-kinase and HEPN domains have extensive sequence and structural homology to Ire1, although RNase L is thought to lack protein kinase activity due to nonconventional residues in its central domain (Huang et al., 2014; Han et al., 2014). The RNase L HEPN domain retains all the canonical HEPN RNase features, including its conserved RNase RϕxxxH motif. Upon detection of viral RNA, the interferon-induced oligoadenylate synthetase 3 (OAS3) produces the RNase L ligand, 2′,5′-oligoadenylate (2–5A) (Kristiansen et al., 2011; Li et al., 2016; Schwartz and Conn, 2019). The RNase L ankyrin-repeat domain binds 2–5A triggering RNase L homodimerization and priming of the HEPN RNase active site (Figure 5B) (Gusho, Baskar, and Banerjee, 2016; Dong and Silverman, 1995). In its dimeric state, RNase L targets a wide variety of viral and endogenous RNAs to promote translational reprogramming where proteins responsible for innate immunity are preferentially synthesized (Khodarev, 2019; Dong and Silverman, 1995; Andersen et al., 2009; Rath et al., 2015; Rath et al., 2019; Nogimori et al., 2019; Burke et al., 2019).
RNase PNK −
The HEPN nuclease of RNase PNK is critical for ribosome production. RNase PNK is conserved across eukaryotes and comprises the Las1 (Las1L in mammals) endoribonuclease and Grc3 RNA kinase (Figure 5A) (Pillon et al., 2017). Both its Las1 nuclease and Grc3 RNA kinase activities are essential for life and play integral roles in precursor rRNA processing (Gasse, Flemming, and Hurt, 2015; Heindl and Martinez, 2010; Pillon, Sobhany, and Stanley, 2018; Castle et al., 2010). During ribosome assembly, numerous RNA spacers are removed from the precursor rRNA transcript (for a comprehensive review please see (Henras et al., 2015)). RNase PNK targets the ITS2 (internal transcribed spacer 2) RNA spacer for degradation by initiating an rRNA processing cascade in the nucleolus. To mark the ITS2 for decay, the Las1 HEPN RNase first cleaves the ITS2 before the Grc3 RNA kinase phosphorylates the 5′ hydroxyl end of the ITS2 cut site (Gasse, Flemming, and Hurt, 2015; Fromm et al., 2017). The resulting monophosphate signals ITS2 RNA decay by a phosphate-dependent 5′-exoribonuclease and 3′-exoribonuclease (Fromm et al., 2017). Since the Grc3 RNA kinase must phosphorylate the ITS2 for precursor rRNA processing, RNase PNK represents the first NiKs family member where both the nuclease and kinase components directly modify the RNA target.
The RNase PNK structure adopts unique features that emphasize its distinction from other NiKs family members. Unlike Ire1 and RNase L, RNase PNK is composed of two independently translated enzymes. To promote proper assembly of the RNase PNK multienzyme complex, the Las1 RNase and Grc3 RNA kinase rely on each other for protein stability (Castle et al., 2013). Another notable difference is that RNase PNK is a constitutive complex, whereas Ire1 and RNase L rely on stress-induced signals for higher-order assembly (Pillon et al., 2017). Las1 is a multi-domain RNase encoding an N-terminal HEPN domain followed by a poorly characterized coiled-coil domain (Figure 5A). Cryo-EM structures of RNase PNK resolved the Las1 HEPN domain confirming its juxtaposed RNase RHxxxH motifs, however its C-terminal coiled-coil domain has remained refractory to traditional structural biology approaches (Pillon et al., 2019). The RNase PNK complex adopts a hetero-tetrameric butterfly-like assembly (Figure 5B). The ‘body’ is comprised of a Las1 homodimer whereas each ‘wing’ is formed by a protomer of the Grc3 RNA kinase. The relative positioning of Las1 and Grc3 forms two symmetric RNA binding clefts at the interface of the body (Las1) and wings (Grc3). While an RNA-bound structure of RNase PNK has yet to be determined, the cleft, lined by the Las1 nuclease and Grc3 RNA kinase active sites, is likely an important RNA binding site.
Regulation of HEPN RNases
To date, the mechanism for HEPN endoribonuclease regulation is poorly understood. High resolution structures have described in great detail the requirement for HEPN dimerization whereby the associating HEPN domains are precisely oriented to form a composite RNase site across the dimer interface (Pillon et al., 2019; Huang et al., 2014; Liu et al., 2017; Jia et al., 2018). And yet, it is clear that proper HEPN dimerization alone is insufficient for RNase activation. In the absence of endogenous RNA substrates, the juxtaposed RϕxxxH motifs are too far apart to support RNA cleavage (Lee et al., 2008; Zhang et al., 2018). This suggests there are additional HEPN active site rearrangements that are required for RNA catalysis. Therefore, HEPN RNase activation is likely a two-step process where HEPN dimerization primes the RNase active site, but that a secondary signal, likely the RNA substrate itself, induces additional conformational changes within the active site to license RNA cleavage. In addition to HEPN activation, cells also rely on precise and efficient mechanisms to inhibit HEPN RNases. Nuclease inactivation is vital for protecting the cell from catastrophic RNA destruction, however the modes of HEPN inhibition remain largely unclear. In the following section, we will summarize the current models for HEPN RNase activation and inhibition.
Prokaryotic HEPN RNase activation.
The success of CRISPR-Cas associated HEPN RNases Cas13 and Csm6 in transcriptome editing applications has led to advancements in our understanding of HEPN nuclease activation (Koonin and Makarova, 2019; O’Connell, 2019; Barrangou and Horvath, 2017). Comparison of LshCas13a structures in both the apo and crRNA-bound binary complex revealed that upon crRNA binding, LshCas13a undergoes a large conformational change leading to the creation of a closed crRNA channel, however crRNA binding alone does not cause large conformational changes within the HEPN RNase site (Liu et al., 2017). Structures of the ternary Leptotrichia buccalis (Lbu) Cas13a complex, including the crRNA and target RNA, revealed that upon target RNA binding both LbuCas13a and the crRNA undergo significant conformation changes (Liu et al., 2017). These conformational changes result in the movement of HEPN1 towards HEPN2 and the activation of non-specific cleavage of single strand RNA (Figure 6A). To date, only binary crRNA-bound structures of Cas13b have been solved, in which the two HEPN motifs are spaced too far apart to support RNA cleavage (Zhang et al., 2018). A combination of thermal denaturation and site-directed mutagenesis studies suggest that in contrast to Cas13a, which binds target RNA along a solvent exposed channel, Cas13b must undergo an opening of its central channel to allow for target RNA binding (Slaymaker et al., 2019). Ternary structures of Cas13d bound to both the crRNA and target RNA revealed that analogous to Cas13a, target RNA binding triggers the activation of the HEPN active site. Upon target RNA binding the catalytic HEPN motif from the HEPN2 domain moves approximately 4 Å closer to the catalytic HEPN motif from HEPN1 (Zhang et al., 2018). Therefore, HEPN RNase conformational rearrangements appears to be a common requirement for CRISPR-Cas associated HEPN activation.
Figure 6.
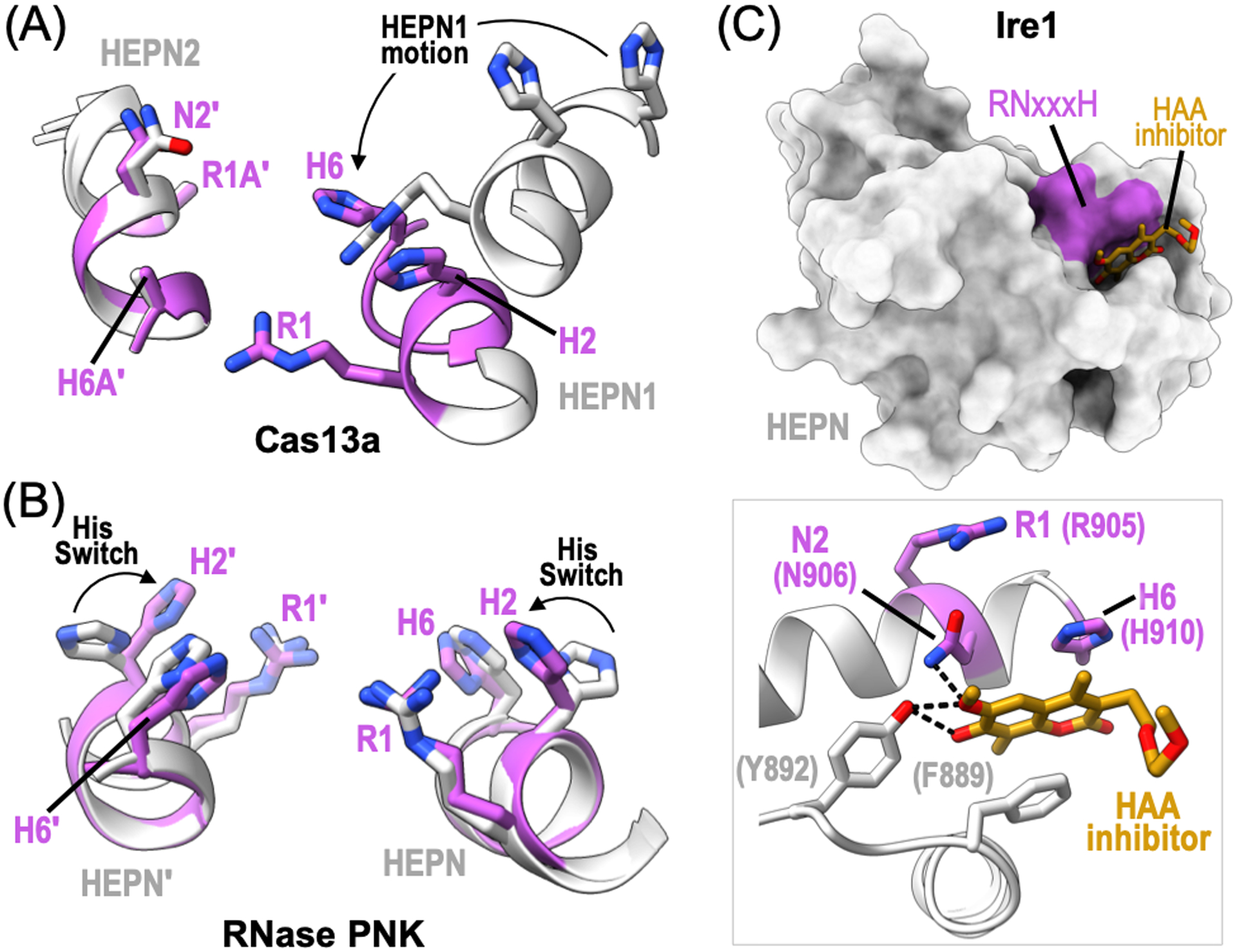
HEPN RNase active site regulation. (A) Ribbon diagram of the RNase active site from L. buccalis Cas13a bound to crRNA and target RNA (PDB ID: 5XWP, 5XWY). The inactive state is shown in light grey and the active state is colored magenta. HEPN1 undergoes conformational changes, such as rearrangement of its RHxxxH motif (black arrow), for RNase activation. (B) Ribbon diagram of C. thermophilum RNase PNK RNase active site (PDB ID: 6OF2, 6OF3). The polar residue (H2) from the RNase RHxxxH motif undergoes a conformational change to orient H2 towards the catalytic center. This conserved histidine is essential for rRNA processing and is referred to as the Histidine Switch (His Switch). (C) Surface rendering of the Mus musculus Ire1 HEPN domain bound to the HAA inhibitor, MKC9989 (orange) (PDB ID: 4PL3). Inset shows a zoom in view of the HAA inhibitor binding site. Ire1 HEPN residues, such as RNase N2 and H6 residues, along with surrounding aromatic residues (grey) directly coordinate the HAA inhibitor. Dotted black lines show hydrogen bonds.
Toxicity of the RnlA toxin is stimulated by the RNase HI nuclease suggesting a novel mechanism of HEPN activation (Naka et al., 2014). RNase HI associates with RnlA through its central NRD domain suggesting an allosteric mechanism for regulating its C-terminal HEPN domain. RNase HI is the only known factor to directly enhance toxin activity, although most RNase LS family members harboring an HEPN domain also encode an upstream RNase H domain (Anantharaman et al., 2013). While the precise mechanism for RnlA activation by RNase HI remains unclear, one possibility is that the RNase HI nuclease may pre-process RNA targets to aid with subsequent RNA cleavage by the RnlA HEPN RNase. Paradoxically, RNase HI also promotes RnlA inhibition by interacting with its antitoxin, RnlB (Naka et al., 2017). To define the molecular basis of RnlA regulation by RNase HI, comprehensive structural studies of RnlA in complex with its regulatory factors and RNA substrates are needed.
Eukaryotic HEPN RNase activation.
Distinct conformational states of RNase PNK shed light on a possible mechanism for Las1 RNase activation. Structures of RNase PNK in its apo and ATP-bound states reveals a conformational change that spans across the entire hetero-tetrameric complex. These global conformational changes are reminiscent of a butterfly flapping its wings and alter the width of the RNA binding cleft found at the junction between the Las1 nuclease and Grc3 RNA kinase. In addition to large scale changes, structural studies of RNase PNK also captured local changes at the HEPN RNase active site. In the presence of nucleotide, RNase PNK adopts two dominant conformations. In one state, the conserved polar residue (H2) of the Las1 HEPN RHxxxH motif is oriented away from the catalytic center where it is too far to coordinate RNA bound at the active site (Figure 6B). In the other state, H2 was captured pointing towards the center of the RNase active site (Pillon et al., 2019). Biochemistry and yeast genetics have confirmed the importance of this conserved histidine residue in ITS2 cleavage and ribosome production, suggesting conformational changes to H2 may allow RNase PNK to toggle between an inactive and active RNase state (Pillon et al., 2019; Pillon et al., 2020). While this working model will require follow-up studies, it appears that RNase activation of the RNase PNK complex is regulated by a conserved Histidine Switch.
Prokaryotic HEPN RNase inactivation.
Activation of the CRISPR-Cas associated HEPN nucleases Csm6/Csx1 provide immunity for microbial cells, however following the clearance of the foreign pathogen these RNases must be inactivated to avoid deleterious outcomes such as growth arrest (Rostøl and Marraffini, 2019). These specialized Type-III CRISPR-Cas effectors function as intricate ribonucleoprotein complexes where the associated Cas10 subunit synthesizes the Csm6/Csx1 HEPN activating signal, cOA. Recent work identified a ring nuclease that degrades cOA providing a molecular “off-switch” for Csm6/Csx1 nuclease activity (Athukoralage et al., 2018). This ring nuclease can either be translated independently from the CRISPR-Cas system or encoded within the Csm6 enzyme itself. Considering that this unique CRISPR cOA signaling pathway has been identified in a number of bacterial species, follow-up studies are needed to discover the precise mechanism for resetting CRISPR-Cas associated HEPN nucleases Csm6/Csx1. Recent work identified a new mechanism of viral inhibition of the CRISPR associated Cas13a nuclease from Listeria seeligeri. The listeriaphage (ϕLS46) encodes for a viral anti-CRISPR protein named AcrVIA1 that inhibits Cas13a and thus blocks CRISPR-Cas acquired immunity. A combination of cryo-EM and biochemistry revealed that AcrVIA1 binds to Cas13a and prevents binding of target RNA and HEPN nuclease activation (Meeske et al., 2020).
Antitoxins share a common mechanism of HEPN toxin inhibition by blocking its RNase motif. Structural characterization of the bacterial LsoA HEPN toxin bound to its viral Dmd antitoxin revealed an extreme case of HEPN steric occlusion. The interaction interface supporting the LsoA-Dmd heterodimer overlaps with the canonical interaction interface of HEPN dimers, preventing the formation of a LsoA homodimer and composite RNase active site (Wan et al., 2016). Moreover, the viral antitoxin encodes a well-defined α-helix which aligns to the LsoA HEPN RϕxxxH motif blocking its solvent accessibility. Similarly, access to the S. oneidensis HepT HEPN RNase site is obstructed by the MntA antitoxin through two distinct mechanisms. First, the rare hetero-octomeric HepT-MntA arrangement positions the MntA molecules at the entrance of the neighboring HepT RNase clefts. For example, the extended MntA α-helix (MNT-α4) directly inserts into the RNase clefts of the peripheral HepT dimers whereas a second MntA α-helix (MNT-α2) lies along the edge of the central HepT RNase cleft (Figure 4A) (Jia et al., 2018). Secondly, MntA catalyzes HepT adenylylation by modifying a highly conserved tyrosine residue found immediately downstream of the HepT HEPN motif (RϕxxxHxY) (Yao et al., 2020). While HepT polyadenylylation does not alter the conformation of the HEPN RNase site, it has been suggested to disrupt access of the HepT RNase cleft (Yao et al., 2020).
Eukaryotic HEPN RNase inactivation.
Ire1 activation is suppressed by the ER Hsp70 chaperone, BiP. To prevent spurious activation of the UPR, the luminal domain of inactive monomeric Ire1 associates with the BiP chaperone, thereby inhibiting Ire1 RNase activation (Oikawa et al., 2009; Pincus et al., 2010; Kopp et al., 2019). As such, BiP is a gatekeeper of Ire1 activation, where BiP dissociation is a prerequisite for initiating the Ire1 activation cascade. As an inhibitor of Ire1, the BiP chaperone promotes its inactive monomeric form by blocking Ire1 self-association (Gardner et al., 2013; Amin-Wetzel et al., 2017). It is only during ER stress that BiP dissociates from Ire1 allowing the luminal domain the opportunity to sense misfolded proteins and trigger Ire1-mediated RNA processing (Gardner et al., 2013; Gardner and Walter, 2011; Pincus et al., 2010). In addition to BiP and misfolded proteins, Ire1 also relies on additional signals to modulate RNase activity. For instance, the Ire1 transmembrane domain has been suggested to sense ER membrane distortions that would restrict Ire1 diffusion and promote Ire1 dimerization (Gardner et al., 2013; Liu, Schröder, and Kaufman, 2000; Promlek et al., 2011).
Pharmacological inhibition of Ire1 selectively targets small molecules to the HEPN RNase domain. A small group of hydroxyl-aryl-aldehyde (HAA) compounds (MKC9989, OICR464, OICR573) have been identified as potent inhibitors of Ire1 RNase activity (Sanches et al., 2014). Comparison of mouse Ire1 apo and HAA inhibitor-bound structures reveals a common binding mode within the HEPN domain and suggests a mechanism for Ire1 inactivation. The HAA inhibitors bind a shallow groove formed in part by the Ire1 HEPN RNxxxH motif (Figure 6C). The polar asparagine (N2) residue is within hydrogen bonding distance of the methoxy group of the HAA compounds, and the invariant histidine (R6) π-stacks with HAA aromatic rings (Figure 6C) (Sanches et al., 2014). Additional surrounding aromatic (F889, Y892) and basic residues contribute to HAA affinity and specificity for the Ire1 RNase site. While a ternary complex of Ire1 bound to both its HAA inhibitor and RNA target has yet to be determined, molecular modeling suggests HAA compounds do not occlude RNA binding, but induce subtle conformational changes to the conserved HEPN RNase N2 and H6 residues that dramatically reduce RNA cleavage efficiency (Sanches et al., 2014).
HEPN nucleases in human health and disease
Accurate RNA expression, processing, and decay are vital for fundamental cellular processes and human health. As a central player in RNA biology, the functionally diverse HEPN family is gaining attention with its disease-causing defects. Below we highlight links between dysregulated HEPN RNases and disease, as well as discuss their promise in RNA-based therapeutics.
Neurological dysfunction.
HEPN RNases Ire1 and Las1L have emerged as important determinants of neurological dysfunction. Ire1 is associated with several central nervous system (CNS) diseases, including Alzheimer’s, Parkinson’s, and Huntington’s diseases (for comprehensive review please see (Ni et al., 2018)). The precise mechanism for Ire1 dysregulation in CNS disease remains poorly defined, but has been linked to its role in XPB1 mRNA splicing. While XPB1 RNA processing has traditionally been viewed as having a protective role, recent evidence suggests its dysregulation may also have adverse effects in disease contexts, such as Alzheimer’s disease and Post-traumatic stress disorder (PTSD). For instance, Ire1 signaling in the brain shows a positive correlation between Ire1 RNase activation and Alzheimer’s disease progression (Duran-Aniotz et al., 2017). Correspondingly, conditional genetic knockouts of the Ire1 HEPN RNase domain alleviated Alzheimer’s behavioral phenotypes in mice (Duran-Aniotz et al., 2017). A similar trend is seen with rat models for PTSD where pharmacological inhibition of the Ire1 HEPN RNase attenuates the apoptotic phenotype driving pathogenesis (Li, Han, and Shi, 2015; Zhao, Han, and Shi, 2016). The Las1L HEPN RNase is also linked to neurological disorders, such as X-linked intellectual disability and congenital lethal motor neuron disease (Butterfield et al., 2014; Hu et al., 2016; Tran et al., 2020). While the molecular basis for Las1L dysregulation in neurological disorders remains undetermined, its disease-associated mutations cluster in the poorly characterized Las1L coiled-coil domain, underscoring the urgent need to understand the function and regulation of this elusive coiled-coil domain.
Innate immunity.
RNase L plays a central role in amplifying an antiviral response upon detection of foreign RNA. RNase L is activated in response to numerous viruses, including the encephalomyocarditis virus and West Nile virus (Flodström-Tullberg et al., 2005; Samuel et al., 2006; Scherbik et al., 2006). To escape detection, several viruses coordinate their own signaling cascade to inhibit 2–5A-dependent RNase L activation. Of particular note is the MERS (Middle East Respiratory Syndrome) coronavirus which encodes the NS4b viral protein responsible for 2–5A degradation and subsequent RNase L inhibition (Thornbrough et al., 2016). The TMEV (Theiler’s murine encepahalomyelitis virus) mouse virus also inhibits RNase L by encoding the L* viral protein responsible for interacting with the RNase L ankyrin-repeat domain to prevent 2–5A binding and RNase L dimerization (Sorgeloos et al., 2013; Drappier et al., 2018). Considering novel human viruses, such as the Saffold cardiovirus, also encode a homologous L* viral protein (Chiu et al., 2008; Zoll et al., 2009), follow-up studies are needed to determine the extent of RNase L inactivation mechanisms in human pathogens. This is especially critical in light of COVID-19 since therapeutic activation of the RNase L-directed innate immune response may provide an effective strategy for the clearance of SARS-CoV-2 (Severe Acute Respiratory Syndrome coronavirus 2) and other human RNA viruses (for more details please refer to (Park and Iwasaki, 2020)).
Cancer.
Ire1 and RNase L are associated with cancer risk and progression. RNase L has tumor suppressive activities in a variety of cancers, including prostate and lung cancers. For example, RNase L-mediated antiviral signaling is impaired in lung cancer. Recent studies exploring the effects of pharmacological induction of RNase L in lung cancer reports RNase L activation, irreversible DNA damage, cell cycle arrest, and apoptosis of cancer cells (Yin et al., 2019; Yin et al., 2019). Similar to its antitumor effects, RNase L is also a prostate cancer susceptibility gene. Hereditary prostate cancer patients often encode RNase L mutations that result in enhanced androgen receptor signaling and cell migration (Dayal et al., 2017). Unlike RNase L, the Ire1 HEPN RNase has cancer-promoting functions that have yet to be well-defined. Cancer cells are under chronic ER stress due to low nutrient and high cell proliferation conditions. Therefore, upregulated UPR signaling factors such as Ire1 can have prosurvival activities in cancer cells. To bypass canonical Ire1 activation, cervical cancer relies on the UPR modulator Yip1A to phosphorylate Ire1 for RNase activation (Taguchi et al., 2017). Constitutive Ire1 activation upregulates anti-apoptotic factors and increases nutrient availability by enhancing protein recycling through dysregulated autophagy pathways (Taguchi et al., 2017).
Transcriptome editing.
Over the past few years the Cas13 effectors have emerged as an important molecular toolkit for detecting and manipulating RNA in vivo, opening the door for many clinical applications (East-Seletsky et al., 2016; O’Connell, 2019; Pickar-Oliver and Gersbach, 2019; Murugan et al., 2017; Zhang, 2019; Gootenberg et al., 2018; Gootenberg et al., 2017). For example, one key application of Cas13 technology is the detection and inhibition of single stranded RNA viruses, such as SARS-CoV-2. Recent experiments have shown that Cas13 can be programmed to cleave multiple types of single stranded RNA viruses and inhibit viral replication (Freije et al., 2019). Moreover, a Cas13 based RNA detection system named SHERLOCK (specific high-sensitivity enzymatic reporter unlocking), as well as variants to this approach have been shown to be powerful alternatives to quantitative PCR for detection of SARS-CoV-2 RNA (Patchsung et al., 2020; Joung et al., 2020; Arizti-Sanz et al., 2020; Fozouni et al., 2020).
Conclusion and perspectives
Ground-breaking work in the past few years has led to the identification and characterization of the emerging family of HEPN RNases, and yet many outstanding questions remain. Structural studies of HEPN members have been powerful in uncovering canonical and specialized elements of HEPN RNases (Table 1), however substrate-bound structures encompassing endogenous RNA targets have thus far proven elusive. As the HEPN field moves forward, significant effort is needed to reveal RNA-bound structures of HEPN RNases at discrete states along their RNA processing and degradation pathways. Furthermore, it will be important to apply non-traditional approaches to map proteome-wide HEPN interactomes to identify novel and dynamic auxiliary factors. The role of HEPN RNase paralogs and isoforms is another aspect of HEPN RNase biology that has yet to be explored in great detail. Characterization of human Ire1 paralogs Ire1α and Ire1β, encoded by ERN1 and ERN2, has recently uncovered a novel mode of HEPN RNase regulation. While both Ire1 paralogs harbor protein kinase and HEPN RNase domains, Ire1β was shown to bind Ire1α and negatively regulate its RNase activity through a poorly defined mechanism (Grey et al., 2020). Moreover, publicly available databases describe four predicted Las1L isoforms that have yet to be confirmed or biochemically characterized. As several HEPN RNases are linked to tissue-specific diseases (i.e. brain), it will be important to understand the specialized roles of HEPN RNases across tissue types. Finally, computational analysis of the HEPN family has formed a long list of putative HEPN RNases that have yet to be confirmed as genuine nucleases (Anantharaman et al., 2013). These candidates will require validation and comprehensive characterization before we can begin to grasp the far-reaching impact of the HEPN RNase family in shaping the RNA world.
Acknowledgements
We thank Dr. Andrea Kaminski and Dr. Andrew Sikkema for their critical review of this manuscript. This work was supported by the US National Institute of Environmental Health Sciences (ZIA ES103247 to R.E.S. and 1K99-ES030735 to M.C.P.).
Footnotes
Disclosure of interest
The authors declare no competing interests.
References
- 1.Grynberg M, Erlandsen H, Godzik A. (2003).HEPN: a common domain in bacterial drug resistance and human neurodegenerative proteins. Trends in biochemical sciences, 28, 224–6 [DOI] [PubMed] [Google Scholar]
- 2.Anantharaman V, Makarova KS, Burroughs AM, Koonin EV, Aravind L. (2013).Comprehensive analysis of the HEPN superfamily: identification of novel roles in intra-genomic conflicts, defense, pathogenesis and RNA processing. Biology direct, 8, 15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Shmakov S, Abudayyeh OO, Makarova KS, Wolf YI, Gootenberg JS, Semenova E, Minakhin L, Joung J, Konermann S, Severinov K, Zhang F, Koonin EV. (2015).Discovery and Functional Characterization of Diverse Class 2 CRISPR-Cas Systems. Molecular cell, 60, 385–97 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Pillon MC, Stanley RE. (2018).Nuclease integrated kinase super assemblies (NiKs) and their role in RNA processing. Current genetics, 64, 183–90 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.East-Seletsky A, O’Connell MR, Knight SC, Burstein D, Cate JH, Tjian R, Doudna JA. (2016).Two distinct RNase activities of CRISPR-C2c2 enable guide-RNA processing and RNA detection. Nature, 538, 270–73 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Shigematsu M, Kawamura T, Kirino Y. (2018).Generation of 2’,3’-Cyclic Phosphate-Containing RNAs as a Hidden Layer of the Transcriptome. Frontiers in genetics, 9, 562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Pillon MC, Hsu AL, Krahn JM, Williams JG, Goslen KH, Sobhany M, Borgnia MJ, Stanley RE. (2019).Cryo-EM reveals active site coordination within a multienzyme pre-rRNA processing complex. Nature structural & molecular biology, 26, 830–39 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Lee KP, Dey M, Neculai D, Cao C, Dever TE, Sicheri F. (2008).Structure of the dual enzyme Ire1 reveals the basis for catalysis and regulation in nonconventional RNA splicing. Cell, 132, 89–100 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Huang H, Zeqiraj E, Dong B, Jha BK, Duffy NM, Orlicky S, Thevakumaran N, Talukdar M, Pillon MC, Ceccarelli DF, Wan LC, Juang YC, Mao DY, Gaughan C, Brinton MA, Perelygin AA, Kourinov I, Guarne A, Silverman RH, Sicheri F. (2014).Dimeric structure of pseudokinase RNase L bound to 2–5A reveals a basis for interferon-induced antiviral activity. Molecular cell, 53, 221–34 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Liu L, Li X, Wang J, Wang M, Chen P, Yin M, Li J, Sheng G, Wang Y. (2017).Two Distant Catalytic Sites Are Responsible for C2c2 RNase Activities. Cell, 168, 121–34 e12 [DOI] [PubMed] [Google Scholar]
- 11.Pillon MC, Goslen KH, Gordon J, Wells ML, Williams JG, Stanley RE. (2020).It takes two (Las1 HEPN endoribonuclease domains) to cut RNA correctly. The Journal of biological chemistry, 295, 5857–70 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Liu L, Li X, Ma J, Li Z, You L, Wang J, Wang M, Zhang X, Wang Y. (2017).The Molecular Architecture for RNA-Guided RNA Cleavage by Cas13a. Cell [DOI] [PubMed] [Google Scholar]
- 13.Zhang C, Konermann S, Brideau NJ, Lotfy P, Wu X, Novick SJ, Strutzenberg T, Griffin PR, Hsu PD, Lyumkis D. (2018).Structural Basis for the RNA-Guided Ribonuclease Activity of CRISPR-Cas13d. Cell, 175, 212–23.e17 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Abudayyeh OO, Gootenberg JS, Konermann S, Joung J, Slaymaker IM, Cox DB, Shmakov S, Makarova KS, Semenova E, Minakhin L, Severinov K, Regev A, Lander ES, Koonin EV, Zhang F. (2016).C2c2 is a single-component programmable RNA-guided RNA-targeting CRISPR effector. Science (New York, NY), 353, aaf5573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Korennykh A, Walter P. (2012).Structural basis of the unfolded protein response. Annual review of cell and developmental biology, 28, 251–77 [DOI] [PubMed] [Google Scholar]
- 16.Han Y, Donovan J, Rath S, Whitney G, Chitrakar A, Korennykh A. (2014).Structure of human RNase L reveals the basis for regulated RNA decay in the IFN response. Science (New York, NY), 343, 1244–8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Knott GJ, Doudna JA. (2018).CRISPR-Cas guides the future of genetic engineering. Science (New York, NY), 361, 866–69 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Makarova KS, Wolf YI, Iranzo J, Shmakov SA, Alkhnbashi OS, Brouns SJJ, Charpentier E, Cheng D, Haft DH, Horvath P, Moineau S, Mojica FJM, Scott D, Shah SA, Siksnys V, Terns MP, Venclovas Č, White MF, Yakunin AF, Yan W, Zhang F, Garrett RA, Backofen R, van der Oost J, Barrangou R, Koonin EV. (2020).Evolutionary classification of CRISPR-Cas systems: a burst of class 2 and derived variants. Nature reviews Microbiology, 18, 67–83 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Koonin EV, Makarova KS. (2019).Origins and evolution of CRISPR-Cas systems. Philosophical transactions of the Royal Society of London Series B, Biological sciences, 374, 20180087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Patchsung M, Jantarug K, Pattama A, Aphicho K, Suraritdechachai S, Meesawat P, Sappakhaw K, Leelahakorn N, Ruenkam T, Wongsatit T, Athipanyasilp N, Eiamthong B, Lakkanasirorat B, Phoodokmai T, Niljianskul N, Pakotiprapha D, Chanarat S, Homchan A, Tinikul R, Kamutira P, Phiwkaow K, Soithongcharoen S, Kantiwiriyawanitch C, Pongsupasa V, Trisrivirat D, Jaroensuk J, Wongnate T, Maenpuen S, Chaiyen P, Kamnerdnakta S, Swangsri J, Chuthapisith S, Sirivatanauksorn Y, Chaimayo C, Sutthent R, Kantakamalakul W, Joung J, Ladha A, Jin X, Gootenberg JS, Abudayyeh OO, Zhang F, Horthongkham N, Uttamapinant C. (2020).Clinical validation of a Cas13-based assay for the detection of SARS-CoV-2 RNA. Nature biomedical engineering [DOI] [PubMed] [Google Scholar]
- 21.Abbott TR, Dhamdhere G, Liu Y, Lin X, Goudy L, Zeng L, Chemparathy A, Chmura S, Heaton NS, Debs R, Pande T, Endy D, La Russa MF, Lewis DB, Qi LS. (2020).Development of CRISPR as an Antiviral Strategy to Combat SARS-CoV-2 and Influenza. Cell, 181, 865–76.e12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Abudayyeh OO, Gootenberg JS, Franklin B, Koob J, Kellner MJ, Ladha A, Joung J, Kirchgatterer P, Cox DBT, Zhang F. (2019).A cytosine deaminase for programmable single-base RNA editing. Science (New York, NY), 365, 382–86 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Terns MP. (2018).CRISPR-Based Technologies: Impact of RNA-Targeting Systems. Molecular cell, 72, 404–12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Ackerman CM, Myhrvold C, Thakku SG, Freije CA, Metsky HC, Yang DK, Ye SH, Boehm CK, Kosoko-Thoroddsen TF, Kehe J, Nguyen TG, Carter A, Kulesa A, Barnes JR, Dugan VG, Hung DT, Blainey PC, Sabeti PC. (2020).Massively multiplexed nucleic acid detection with Cas13. Nature, 582, 277–82 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.O’Connell MR. (2019).Molecular Mechanisms of RNA Targeting by Cas13-containing Type VI CRISPR-Cas Systems. Journal of molecular biology, 431, 66–87 [DOI] [PubMed] [Google Scholar]
- 26.Shmakov S, Smargon A, Scott D, Cox D, Pyzocha N, Yan W, Abudayyeh OO, Gootenberg JS, Makarova KS, Wolf YI, Severinov K, Zhang F, Koonin EV. (2017).Diversity and evolution of class 2 CRISPR-Cas systems. Nature reviews Microbiology, 15, 169–82 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Konermann S, Lotfy P, Brideau NJ, Oki J, Shokhirev MN, Hsu PD. (2018).Transcriptome Engineering with RNA-Targeting Type VI-D CRISPR Effectors. Cell, 173, 665–76.e14 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Yan WX, Chong S, Zhang H, Makarova KS, Koonin EV, Cheng DR, Scott DA. (2018).Cas13d Is a Compact RNA-Targeting Type VI CRISPR Effector Positively Modulated by a WYL-Domain-Containing Accessory Protein. Molecular cell, 70, 327–39.e5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Smargon AA, Cox DBT, Pyzocha NK, Zheng K, Slaymaker IM, Gootenberg JS, Abudayyeh OA, Essletzbichler P, Shmakov S, Makarova KS, Koonin EV, Zhang F. (2017).Cas13b Is a Type VI-B CRISPR-Associated RNA-Guided RNase Differentially Regulated by Accessory Proteins Csx27 and Csx28. Molecular cell, 65, 618–30.e7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Garcia-Doval C, Jinek M. (2017).Molecular architectures and mechanisms of Class 2 CRISPR-associated nucleases. Current opinion in structural biology, 47, 157–66 [DOI] [PubMed] [Google Scholar]
- 31.Knott GJ, East-Seletsky A, Cofsky JC, Holton JM, Charles E, O’Connell MR, Doudna JA. (2017).Guide-bound structures of an RNA-targeting A-cleaving CRISPR-Cas13a enzyme. Nature structural & molecular biology, 24, 825–33 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Makarova KS, Gao L, Zhang F, Koonin EV. (2019).Unexpected connections between type VI-B CRISPR-Cas systems, bacterial natural competence, ubiquitin signaling network and DNA modification through a distinct family of membrane proteins. FEMS microbiology letters, 366 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Zhang B, Ye W, Ye Y, Zhou H, Saeed A, Chen J, Lin J, Perčulija V, Chen Q, Chen CJ, Chang MX, Choudhary MI, Ouyang S. (2018).Structural insights into Cas13b-guided CRISPR RNA maturation and recognition. Cell research, 28, 1198–201 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Slaymaker IM, Mesa P, Kellner MJ, Kannan S, Brignole E, Koob J, Feliciano PR, Stella S, Abudayyeh OO, Gootenberg JS, Strecker J, Montoya G, Zhang F. (2019).High-Resolution Structure of Cas13b and Biochemical Characterization of RNA Targeting and Cleavage. Cell reports, 26, 3741–51.e5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Zhang H, Dong C, Li L, Wasney GA, Min J. (2019).Structural insights into the modulatory role of the accessory protein WYL1 in the Type VI-D CRISPR-Cas system. Nucleic acids research, 47, 5420–28 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Zhang B, Ye Y, Ye W, Perčulija V, Jiang H, Chen Y, Li Y, Chen J, Lin J, Wang S, Chen Q, Han YS, Ouyang S. (2019).Two HEPN domains dictate CRISPR RNA maturation and target cleavage in Cas13d. Nature communications, 10, 2544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Niewoehner O, Garcia-Doval C, Rostol JT, Berk C, Schwede F, Bigler L, Hall J, Marraffini LA, Jinek M. (2017).Type III CRISPR-Cas systems produce cyclic oligoadenylate second messengers. Nature [DOI] [PubMed] [Google Scholar]
- 38.Kazlauskiene M, Kostiuk G, Venclovas C, Tamulaitis G, Siksnys V. (2017).A cyclic oligonucleotide signaling pathway in type III CRISPR-Cas systems. Science (New York, NY) [DOI] [PubMed] [Google Scholar]
- 39.Garcia-Doval C, Schwede F, Berk C, Rostøl JT, Niewoehner O, Tejero O, Hall J, Marraffini LA, Jinek M. (2020).Activation and self-inactivation mechanisms of the cyclic oligoadenylate-dependent CRISPR ribonuclease Csm6. Nature communications, 11, 1596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Jia N, Jones R, Yang G, Ouerfelli O, Patel DJ. (2019).CRISPR-Cas III-A Csm6 CARF Domain Is a Ring Nuclease Triggering Stepwise cA(4) Cleavage with ApA>p Formation Terminating RNase Activity. Molecular cell, 75, 944–56.e6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Molina R, Stella S, Feng M, Sofos N, Jauniskis V, Pozdnyakova I, López-Méndez B, She Q, Montoya G. (2019).Structure of Csx1-cOA(4) complex reveals the basis of RNA decay in Type III-B CRISPR-Cas. Nature communications, 10, 4302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Jurėnas D, Van Melderen L. (2020).The Variety in the Common Theme of Translation Inhibition by Type II Toxin-Antitoxin Systems. Frontiers in genetics, 11, 262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Christensen SK, Gerdes K. (2003).RelE toxins from bacteria and Archaea cleave mRNAs on translating ribosomes, which are rescued by tmRNA. Molecular microbiology, 48, 1389–400 [DOI] [PubMed] [Google Scholar]
- 44.Jiang Y, Pogliano J, Helinski DR, Konieczny I. (2002).ParE toxin encoded by the broad-host-range plasmid RK2 is an inhibitor of Escherichia coli gyrase. Molecular microbiology, 44, 971–9 [DOI] [PubMed] [Google Scholar]
- 45.Liu M, Zhang Y, Inouye M, Woychik NA. (2008).Bacterial addiction module toxin Doc inhibits translation elongation through its association with the 30S ribosomal subunit. Proceedings of the National Academy of Sciences of the United States of America, 105, 5885–90 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Winther KS, Gerdes K. (2011).Enteric virulence associated protein VapC inhibits translation by cleavage of initiator tRNA. Proceedings of the National Academy of Sciences of the United States of America, 108, 7403–7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Winther KS, Brodersen DE, Brown AK, Gerdes K. (2013).VapC20 of Mycobacterium tuberculosis cleaves the sarcin-ricin loop of 23S rRNA. Nature communications, 4, 2796. [DOI] [PubMed] [Google Scholar]
- 48.Yao J, Zhen X, Tang K, Liu T, Xu X, Chen Z, Guo Y, Liu X, Wood TK, Ouyang S, Wang X. (2020).Novel polyadenylylation-dependent neutralization mechanism of the HEPN/MNT toxin/antitoxin system. Nucleic acids research [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Otsuka Y, Yonesaki T. (2012).Dmd of bacteriophage T4 functions as an antitoxin against Escherichia coli LsoA and RnlA toxins. Molecular microbiology, 83, 669–81 [DOI] [PubMed] [Google Scholar]
- 50.Alkhalili RN, Wallenius J, Canbäck B. (2019).Towards Exploring Toxin-Antitoxin Systems in Geobacillus: A Screen for Type II Toxin-Antitoxin System Families in a Thermophilic Genus. International Journal of Molecular Sciences, 20 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Jia X, Yao J, Gao Z, Liu G, Dong YH, Wang X, Zhang H. (2018).Structure-function analyses reveal the molecular architecture and neutralization mechanism of a bacterial HEPN-MNT toxin-antitoxin system. The Journal of biological chemistry, 293, 6812–23 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Bendtsen KL, Xu K, Luckmann M, Winther KS, Shah SA, Pedersen CNS, Brodersen DE. (2017).Toxin inhibition in C. crescentus VapBC1 is mediated by a flexible pseudo-palindromic protein motif and modulated by DNA binding. Nucleic acids research, 45, 2875–86 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Das U, Pogenberg V, Subhramanyam UK, Wilmanns M, Gourinath S, Srinivasan A. (2014).Crystal structure of the VapBC-15 complex from Mycobacterium tuberculosis reveals a two-metal ion dependent PIN-domain ribonuclease and a variable mode of toxin-antitoxin assembly. Journal of structural biology, 188, 249–58 [DOI] [PubMed] [Google Scholar]
- 54.Dienemann C, Bøggild A, Winther KS, Gerdes K, Brodersen DE. (2011).Crystal structure of the VapBC toxin-antitoxin complex from Shigella flexneri reveals a hetero-octameric DNA-binding assembly. Journal of molecular biology, 414, 713–22 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Yao J, Guo Y, Zeng Z, Liu X, Shi F, Wang X. (2015).Identification and characterization of a HEPN-MNT family type II toxin-antitoxin in Shewanella oneidensis. Microbial biotechnology, 8, 961–73 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Koga M, Otsuka Y, Lemire S, Yonesaki T. (2011).Escherichia coli rnlA and rnlB compose a novel toxin-antitoxin system. Genetics, 187, 123–30 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Wan H, Otsuka Y, Gao ZQ, Wei Y, Chen Z, Masuda M, Yonesaki T, Zhang H, Dong YH. (2016).Structural insights into the inhibition mechanism of bacterial toxin LsoA by bacteriophage antitoxin Dmd. Molecular microbiology, 101, 757–69 [DOI] [PubMed] [Google Scholar]
- 58.Otsuka Y, Koga M, Iwamoto A, Yonesaki T. (2007).A role of RnlA in the RNase LS activity from Escherichia coli. Genes & genetic systems, 82, 291–9 [DOI] [PubMed] [Google Scholar]
- 59.Wei Y, Gao ZQ, Otsuka Y, Naka K, Yonesaki T, Zhang H, Dong YH. (2013).Structure-function studies of Escherichia coli RnlA reveal a novel toxin structure involved in bacteriophage resistance. Molecular microbiology, 90, 956–65 [DOI] [PubMed] [Google Scholar]
- 60.Cox JS, Shamu CE, Walter P. (1993).Transcriptional induction of genes encoding endoplasmic reticulum resident proteins requires a transmembrane protein kinase. Cell, 73, 1197–206 [DOI] [PubMed] [Google Scholar]
- 61.Sidrauski C, Walter P. (1997).The transmembrane kinase Ire1p is a site-specific endonuclease that initiates mRNA splicing in the unfolded protein response. Cell, 90, 1031–9 [DOI] [PubMed] [Google Scholar]
- 62.Mori K, Ma W, Gething MJ, Sambrook J. (1993).A transmembrane protein with a cdc2+/CDC28-related kinase activity is required for signaling from the ER to the nucleus. Cell, 74, 743–56 [DOI] [PubMed] [Google Scholar]
- 63.Coelho DS, Domingos PM. (2014).Physiological roles of regulated Ire1 dependent decay. Frontiers in genetics, 5, 76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Maurel M, Chevet E, Tavernier J, Gerlo S. (2014).Getting RIDD of RNA: IRE1 in cell fate regulation. Trends in biochemical sciences, 39, 245–54 [DOI] [PubMed] [Google Scholar]
- 65.Chalmers F, Mogre S, Son J, Blazanin N, Glick AB. (2019).The multiple roles of the unfolded protein response regulator IRE1α in cancer. Molecular carcinogenesis, 58, 1623–30 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Korennykh AV, Egea PF, Korostelev AA, Finer-Moore J, Zhang C, Shokat KM, Stroud RM, Walter P. (2009).The unfolded protein response signals through high-order assembly of Ire1. Nature, 457, 687–93 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Karagöz GE, Acosta-Alvear D, Nguyen HT, Lee CP, Chu F, Walter P. (2017).An unfolded protein-induced conformational switch activates mammalian IRE1. eLife, 6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Aragon T, van Anken E, Pincus D, Serafimova IM, Korennykh AV, Rubio CA, Walter P. (2009).Messenger RNA targeting to endoplasmic reticulum stress signalling sites. Nature, 457, 736–40 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Gardner BM, Pincus D, Gotthardt K, Gallagher CM, Walter P. (2013).Endoplasmic reticulum stress sensing in the unfolded protein response. Cold Spring Harb Perspect Biol, 5, a013169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Gardner BM, Walter P. (2011).Unfolded proteins are Ire1-activating ligands that directly induce the unfolded protein response. Science (New York, NY), 333, 1891–4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Credle JJ, Finer-Moore JS, Papa FR, Stroud RM, Walter P. (2005).On the mechanism of sensing unfolded protein in the endoplasmic reticulum. Proceedings of the National Academy of Sciences of the United States of America, 102, 18773–84 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Ali MM, Bagratuni T, Davenport EL, Nowak PR, Silva-Santisteban MC, Hardcastle A, McAndrews C, Rowlands MG, Morgan GJ, Aherne W, Collins I, Davies FE, Pearl LH. (2011).Structure of the Ire1 autophosphorylation complex and implications for the unfolded protein response. The EMBO journal, 30, 894–905 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Sanches M, Duffy NM, Talukdar M, Thevakumaran N, Chiovitti D, Canny MD, Lee K, Kurinov I, Uehling D, Al-awar R, Poda G, Prakesch M, Wilson B, Tam V, Schweitzer C, Toro A, Lucas JL, Vuga D, Lehmann L, Durocher D, Zeng Q, Patterson JB, Sicheri F. (2014).Structure and mechanism of action of the hydroxy-aryl-aldehyde class of IRE1 endoribonuclease inhibitors. Nature communications, 5, 4202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Feldman HC, Tong M, Wang L, Meza-Acevedo R, Gobillot TA, Lebedev I, Gliedt MJ, Hari SB, Mitra AK, Backes BJ, Papa FR, Seeliger MA, Maly DJ. (2016).Structural and Functional Analysis of the Allosteric Inhibition of IRE1α with ATP-Competitive Ligands. ACS chemical biology, 11, 2195–205 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Adams CJ, Kopp MC, Larburu N, Nowak PR, Ali MMU. (2019).Structure and Molecular Mechanism of ER Stress Signaling by the Unfolded Protein Response Signal Activator IRE1. Frontiers in molecular biosciences, 6, 11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Wreschner DH, McCauley JW, Skehel JJ, Kerr IM. (1981).Interferon action--sequence specificity of the ppp(A2’p)nA-dependent ribonuclease. Nature, 289, 414–7 [DOI] [PubMed] [Google Scholar]
- 77.Floyd-Smith G, Slattery E, Lengyel P. (1981).Interferon action: RNA cleavage pattern of a (2’−5’)oligoadenylate--dependent endonuclease. Science (New York, NY), 212, 1030–2 [DOI] [PubMed] [Google Scholar]
- 78.Dong B, Xu L, Zhou A, Hassel BA, Lee X, Torrence PF, Silverman RH. (1994).Intrinsic molecular activities of the interferon-induced 2–5A-dependent RNase. The Journal of biological chemistry, 269, 14153–8 [PubMed] [Google Scholar]
- 79.Chakrabarti A, Jha BK, Silverman RH. (2011).New insights into the role of RNase L in innate immunity. Journal of interferon & cytokine research : the official journal of the International Society for Interferon and Cytokine Research, 31, 49–57 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Gusho E, Baskar D, Banerjee S. (2016).New advances in our understanding of the “unique” RNase L in host pathogen interaction and immune signaling. Cytokine [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Khodarev NN. (2019).Intracellular RNA Sensing in Mammalian Cells: Role in Stress Response and Cancer Therapies. International review of cell and molecular biology, 344, 31–89 [DOI] [PubMed] [Google Scholar]
- 82.Kristiansen H, Gad HH, Eskildsen-Larsen S, Despres P, Hartmann R. (2011).The oligoadenylate synthetase family: an ancient protein family with multiple antiviral activities. Journal of interferon & cytokine research : the official journal of the International Society for Interferon and Cytokine Research, 31, 41–7 [DOI] [PubMed] [Google Scholar]
- 83.Li Y, Banerjee S, Wang Y, Goldstein SA, Dong B, Gaughan C, Silverman RH, Weiss SR. (2016).Activation of RNase L is dependent on OAS3 expression during infection with diverse human viruses. Proceedings of the National Academy of Sciences of the United States of America, 113, 2241–6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Schwartz SL, Conn GL. (2019).RNA regulation of the antiviral protein 2’−5’-oligoadenylate synthetase. Wiley interdisciplinary reviews RNA, 10, e1534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Dong B, Silverman RH. (1995).2–5A-dependent RNase molecules dimerize during activation by 2–5A. The Journal of biological chemistry, 270, 4133–7 [DOI] [PubMed] [Google Scholar]
- 86.Andersen JB, Mazan-Mamczarz K, Zhan M, Gorospe M, Hassel BA. (2009).Ribosomal protein mRNAs are primary targets of regulation in RNase-L-induced senescence. RNA biology, 6, 305–15 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Rath S, Donovan J, Whitney G, Chitrakar A, Wang W, Korennykh A. (2015).Human RNase L tunes gene expression by selectively destabilizing the microRNA-regulated transcriptome. Proceedings of the National Academy of Sciences of the United States of America, 112, 15916–21 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Rath S, Prangley E, Donovan J, Demarest K, Wingreen NS, Meir Y, Korennykh A. (2019).Concerted 2–5A-Mediated mRNA Decay and Transcription Reprogram Protein Synthesis in the dsRNA Response. Molecular cell, 75, 1218–28.e6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Nogimori T, Nishiura K, Kawashima S, Nagai T, Oishi Y, Hosoda N, Imataka H, Kitamura Y, Kitade Y, Hoshino SI. (2019).Dom34 mediates targeting of exogenous RNA in the antiviral OAS/RNase L pathway. Nucleic acids research, 47, 432–49 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Burke JM, Moon SL, Matheny T, Parker R. (2019).RNase L Reprograms Translation by Widespread mRNA Turnover Escaped by Antiviral mRNAs. Molecular cell, 75, 1203–17.e5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Pillon MC, Sobhany M, Borgnia MJ, Williams JG, Stanley RE. (2017).Grc3 programs the essential endoribonuclease Las1 for specific RNA cleavage. Proceedings of the National Academy of Sciences of the United States of America, 114, E5530–e38 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Gasse L, Flemming D, Hurt E. (2015).Coordinated Ribosomal ITS2 RNA Processing by the Las1 Complex Integrating Endonuclease, Polynucleotide Kinase, and Exonuclease Activities. Molecular cell, 60, 808–15 [DOI] [PubMed] [Google Scholar]
- 93.Heindl K, Martinez J. (2010).Nol9 is a novel polynucleotide 5’-kinase involved in ribosomal RNA processing. The EMBO journal, 29, 4161–71 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Pillon MC, Sobhany M, Stanley RE. (2018).Characterization of the molecular crosstalk within the essential Grc3/Las1 pre-rRNA processing complex. RNA (New York, NY), 24, 721–38 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Castle CD, Cassimere EK, Lee J, Denicourt C. (2010).Las1L is a nucleolar protein required for cell proliferation and ribosome biogenesis. Molecular and cellular biology, 30, 4404–14 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Henras AK, Plisson-Chastang C, O’Donohue MF, Chakraborty A, Gleizes PE. (2015).An overview of pre-ribosomal RNA processing in eukaryotes. Wiley interdisciplinary reviews RNA, 6, 225–42 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Fromm L, Falk S, Flemming D, Schuller JM, Thoms M, Conti E, Hurt E. (2017).Reconstitution of the complete pathway of ITS2 processing at the pre-ribosome. Nature communications, 8, 1787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Castle CD, Sardana R, Dandekar V, Borgianini V, Johnson AW, Denicourt C. (2013).Las1 interacts with Grc3 polynucleotide kinase and is required for ribosome synthesis in Saccharomyces cerevisiae. Nucleic acids research, 41, 1135–50 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Barrangou R, Horvath P. (2017).A decade of discovery: CRISPR functions and applications. Nature microbiology, 2, 17092. [DOI] [PubMed] [Google Scholar]
- 100.Naka K, Koga M, Yonesaki T, Otsuka Y. (2014).RNase HI stimulates the activity of RnlA toxin in Escherichia coli. Molecular microbiology, 91, 596–605 [DOI] [PubMed] [Google Scholar]
- 101.Naka K, Qi D, Yonesaki T, Otsuka Y. (2017).RnlB Antitoxin of the Escherichia coli RnlA-RnlB Toxin-Antitoxin Module Requires RNase HI for Inhibition of RnlA Toxin Activity. Toxins, 9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Rostøl JT, Marraffini LA. (2019).Non-specific degradation of transcripts promotes plasmid clearance during type III-A CRISPR-Cas immunity. Nature microbiology, 4, 656–62 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Athukoralage JS, Rouillon C, Graham S, Grüschow S, White MF. (2018).Ring nucleases deactivate type III CRISPR ribonucleases by degrading cyclic oligoadenylate. Nature, 562, 277–80 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Meeske AJ, Jia N, Cassel AK, Kozlova A, Liao J, Wiedmann M, Patel DJ, Marraffini LA. (2020).A phage-encoded anti-CRISPR enables complete evasion of type VI-A CRISPR-Cas immunity. Science (New York, NY), 369, 54–59 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Oikawa D, Kimata Y, Kohno K, Iwawaki T. (2009).Activation of mammalian IRE1alpha upon ER stress depends on dissociation of BiP rather than on direct interaction with unfolded proteins. Experimental cell research, 315, 2496–504 [DOI] [PubMed] [Google Scholar]
- 106.Pincus D, Chevalier MW, Aragón T, van Anken E, Vidal SE, El-Samad H, Walter P. (2010).BiP binding to the ER-stress sensor Ire1 tunes the homeostatic behavior of the unfolded protein response. PLoS biology, 8, e1000415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Kopp MC, Larburu N, Durairaj V, Adams CJ, Ali MMU. (2019).UPR proteins IRE1 and PERK switch BiP from chaperone to ER stress sensor. Nature structural & molecular biology, 26, 1053–62 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Amin-Wetzel N, Saunders RA, Kamphuis MJ, Rato C, Preissler S, Harding HP, Ron D. (2017).A J-Protein Co-chaperone Recruits BiP to Monomerize IRE1 and Repress the Unfolded Protein Response. Cell, 171, 1625–37.e13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Liu CY, Schröder M, Kaufman RJ. (2000).Ligand-independent dimerization activates the stress response kinases IRE1 and PERK in the lumen of the endoplasmic reticulum. The Journal of biological chemistry, 275, 24881–5 [DOI] [PubMed] [Google Scholar]
- 110.Promlek T, Ishiwata-Kimata Y, Shido M, Sakuramoto M, Kohno K, Kimata Y. (2011).Membrane aberrancy and unfolded proteins activate the endoplasmic reticulum stress sensor Ire1 in different ways. Molecular biology of the cell, 22, 3520–32 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Ni H, Rui Q, Li D, Gao R, Chen G. (2018).The Role of IRE1 Signaling in the Central Nervous System Diseases. Current neuropharmacology, 16, 1340–47 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Duran-Aniotz C, Cornejo VH, Espinoza S, Ardiles Á O, Medinas DB, Salazar C, Foley A, Gajardo I, Thielen P, Iwawaki T, Scheper W, Soto C, Palacios AG, Hoozemans JJM, Hetz C. (2017).IRE1 signaling exacerbates Alzheimer’s disease pathogenesis. Acta neuropathologica, 134, 489–506 [DOI] [PubMed] [Google Scholar]
- 113.Li X, Han F, Shi Y. (2015).IRE1α-XBP1 Pathway Is Activated Upon Induction of Single-Prolonged Stress in Rat Neurons of the Medial Prefrontal Cortex. Journal of molecular neuroscience : MN, 57, 63–72 [DOI] [PubMed] [Google Scholar]
- 114.Zhao W, Han F, Shi Y. (2016).IRE1α pathway of endoplasmic reticulum stress induces neuronal apoptosis in the locus coeruleus of rats under single prolonged stress. Progress in neuro-psychopharmacology & biological psychiatry, 69, 11–8 [DOI] [PubMed] [Google Scholar]
- 115.Butterfield RJ, Stevenson TJ, Xing L, Newcomb TM, Nelson B, Zeng W, Li X, Lu HM, Lu H, Farwell Gonzalez KD, Wei JP, Chao EC, Prior TW, Snyder PJ, Bonkowsky JL, Swoboda KJ. (2014).Congenital lethal motor neuron disease with a novel defect in ribosome biogenesis. Neurology, 82, 1322–30 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Hu H, Haas SA, Chelly J, Van Esch H, Raynaud M, de Brouwer AP, Weinert S, Froyen G, Frints SG, Laumonnier F, Zemojtel T, Love MI, Richard H, Emde AK, Bienek M, Jensen C, Hambrock M, Fischer U, Langnick C, Feldkamp M, Wissink-Lindhout W, Lebrun N, Castelnau L, Rucci J, Montjean R, Dorseuil O, Billuart P, Stuhlmann T, Shaw M, Corbett MA, Gardner A, Willis-Owen S, Tan C, Friend KL, Belet S, van Roozendaal KE, Jimenez-Pocquet M, Moizard MP, Ronce N, Sun R, O’Keeffe S, Chenna R, van Bommel A, Goke J, Hackett A, Field M, Christie L, Boyle J, Haan E, Nelson J, Turner G, Baynam G, Gillessen-Kaesbach G, Muller U, Steinberger D, Budny B, Badura-Stronka M, Latos-Bielenska A, Ousager LB, Wieacker P, Rodriguez Criado G, Bondeson ML, Anneren G, Dufke A, Cohen M, Van Maldergem L, Vincent-Delorme C, Echenne B, Simon-Bouy B, Kleefstra T, Willemsen M, Fryns JP, Devriendt K, Ullmann R, Vingron M, Wrogemann K, Wienker TF, Tzschach A, van Bokhoven H, Gecz J, Jentsch TJ, Chen W, Ropers HH, Kalscheuer VM. (2016).X-exome sequencing of 405 unresolved families identifies seven novel intellectual disability genes. Mol Psychiatry, 21, 133–48 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Tran KT, Le VS, Bui HTP, Do DH, Ly HTT, Nguyen HT, Dao LTM, Nguyen TH, Vu DM, Ha LT, Le HTT, Mukhopadhyay A, Nguyen LT. (2020).Genetic landscape of autism spectrum disorder in Vietnamese children. Scientific reports, 10, 5034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Flodström-Tullberg M, Hultcrantz M, Stotland A, Maday A, Tsai D, Fine C, Williams B, Silverman R, Sarvetnick N. (2005).RNase L and double-stranded RNA-dependent protein kinase exert complementary roles in islet cell defense during coxsackievirus infection. Journal of immunology (Baltimore, Md : 1950), 174, 1171–7 [DOI] [PubMed] [Google Scholar]
- 119.Samuel MA, Whitby K, Keller BC, Marri A, Barchet W, Williams BR, Silverman RH, Gale M Jr., Diamond MS. (2006).PKR and RNase L contribute to protection against lethal West Nile Virus infection by controlling early viral spread in the periphery and replication in neurons. Journal of virology, 80, 7009–19 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Scherbik SV, Paranjape JM, Stockman BM, Silverman RH, Brinton MA. (2006).RNase L plays a role in the antiviral response to West Nile virus. Journal of virology, 80, 2987–99 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Thornbrough JM, Jha BK, Yount B, Goldstein SA, Li Y, Elliott R, Sims AC, Baric RS, Silverman RH, Weiss SR. (2016).Middle East Respiratory Syndrome Coronavirus NS4b Protein Inhibits Host RNase L Activation. mBio, 7, e00258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Sorgeloos F, Jha BK, Silverman RH, Michiels T. (2013).Evasion of antiviral innate immunity by Theiler’s virus L* protein through direct inhibition of RNase L. PLoS pathogens, 9, e1003474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Drappier M, Jha BK, Stone S, Elliott R, Zhang R, Vertommen D, Weiss SR, Silverman RH, Michiels T. (2018).A novel mechanism of RNase L inhibition: Theiler’s virus L* protein prevents 2–5A from binding to RNase L. PLoS pathogens, 14, e1006989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Chiu CY, Greninger AL, Kanada K, Kwok T, Fischer KF, Runckel C, Louie JK, Glaser CA, Yagi S, Schnurr DP, Haggerty TD, Parsonnet J, Ganem D, DeRisi JL. (2008).Identification of cardioviruses related to Theiler’s murine encephalomyelitis virus in human infections. Proceedings of the National Academy of Sciences of the United States of America, 105, 14124–9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Zoll J, Erkens Hulshof S, Lanke K, Verduyn Lunel F, Melchers WJ, Schoondermark-van de Ven E, Roivainen M, Galama JM, van Kuppeveld FJ. (2009).Saffold virus, a human Theiler’s-like cardiovirus, is ubiquitous and causes infection early in life. PLoS pathogens, 5, e1000416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Park A, Iwasaki A. (2020).Type I and Type III Interferons - Induction, Signaling, Evasion, and Application to Combat COVID-19. Cell host & microbe, 27, 870–78 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Yin H, Jiang Z, Wang S, Zhang P. (2019).IFN-γ restores the impaired function of RNase L and induces mitochondria-mediated apoptosis in lung cancer. Cell death & disease, 10, 642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Yin H, Jiang Z, Wang S, Zhang P. (2019).Actinomycin D-Activated RNase L Promotes H2A.X/H2B-Mediated DNA Damage and Apoptosis in Lung Cancer Cells. Frontiers in oncology, 9, 1086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Dayal S, Zhou J, Manivannan P, Siddiqui MA, Ahmad OF, Clark M, Awadia S, Garcia-Mata R, Shemshedini L, Malathi K. (2017).RNase L Suppresses Androgen Receptor Signaling, Cell Migration and Matrix Metalloproteinase Activity in Prostate Cancer Cells. International Journal of Molecular Sciences, 18 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Taguchi Y, Horiuchi Y, Kano F, Murata M. (2017).Novel prosurvival function of Yip1A in human cervical cancer cells: constitutive activation of the IRE1 and PERK pathways of the unfolded protein response. Cell death & disease, 8, e2718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Pickar-Oliver A, Gersbach CA. (2019).The next generation of CRISPR-Cas technologies and applications. Nature reviews Molecular cell biology, 20, 490–507 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Murugan K, Babu K, Sundaresan R, Rajan R, Sashital DG. (2017).The Revolution Continues: Newly Discovered Systems Expand the CRISPR-Cas Toolkit. Molecular cell, 68, 15–25 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Zhang F (2019).Exploration of Microbial Diversity to Discover Novel Molecular Technologies. The Keio journal of medicine, 68, 26. [DOI] [PubMed] [Google Scholar]
- 134.Gootenberg JS, Abudayyeh OO, Kellner MJ, Joung J, Collins JJ, Zhang F. (2018).Multiplexed and portable nucleic acid detection platform with Cas13, Cas12a, and Csm6. Science (New York, NY), 360, 439–44 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Gootenberg JS, Abudayyeh OO, Lee JW, Essletzbichler P, Dy AJ, Joung J, Verdine V, Donghia N, Daringer NM, Freije CA, Myhrvold C, Bhattacharyya RP, Livny J, Regev A, Koonin EV, Hung DT, Sabeti PC, Collins JJ, Zhang F. (2017).Nucleic acid detection with CRISPR-Cas13a/C2c2. Science (New York, NY), 356, 438–42 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Freije CA, Myhrvold C, Boehm CK, Lin AE, Welch NL, Carter A, Metsky HC, Luo CY, Abudayyeh OO, Gootenberg JS, Yozwiak NL, Zhang F, Sabeti PC. (2019).Programmable Inhibition and Detection of RNA Viruses Using Cas13. Molecular cell, 76, 826–37.e11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Joung J, Ladha A, Saito M, Segel M, Bruneau R, Huang MW, Kim NG, Yu X, Li J, Walker BD, Greninger AL, Jerome KR, Gootenberg JS, Abudayyeh OO, Zhang F. (2020).Point-of-care testing for COVID-19 using SHERLOCK diagnostics. medRxiv : the preprint server for health sciences [Google Scholar]
- 138.Arizti-Sanz J, Freije CA, Stanton AC, Boehm CK, Petros BA, Siddiqui S, Shaw BM, Adams G, Kosoko-Thoroddsen TF, Kemball ME, Gross R, Wronka L, Caviness K, Hensley LE, Bergman NH, MacInnis BL, Lemieux JE, Sabeti PC, Myhrvold C. (2020).Integrated sample inactivation, amplification, and Cas13-based detection of SARS-CoV-2. bioRxiv : the preprint server for biology [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Fozouni P, Son S, Díaz de León Derby M, Knott GJ, Gray CN, D’Ambrosio MV, Zhao C, Switz NA, Kumar GR, Stephens SI, Boehm D, Tsou C-L, Shu J, Bhuiya A, Armstrong M, Harris A, Osterloh JM, Meyer-Franke A, Langelier C, Pollard KS, Crawford ED, Puschnik AS, Phelps M, Kistler A, DeRisi JL, Doudna JA, Fletcher DA, Ott M. (2020).Direct detection of SARS-CoV-2 using CRISPR-Cas13a and a mobile phone. medRxiv : the preprint server for health sciences [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Grey MJ, Cloots E, Simpson MS, LeDuc N, Serebrenik YV, De Luca H, De Sutter D, Luong P, Thiagarajah JR, Paton AW, Paton JC, Seeliger MA, Eyckerman S, Janssens S, Lencer WI. (2020).IRE1β negatively regulates IRE1α signaling in response to endoplasmic reticulum stress. The Journal of cell biology, 219 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Niewoehner O, Jinek M. (2016).Structural basis for the endoribonuclease activity of the type III-A CRISPR-associated protein Csm6. RNA (New York, NY), 22, 318–29 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Kim YK, Kim YG, Oh BH. (2013).Crystal structure and nucleic acid-binding activity of the CRISPR-associated protein Csx1 of Pyrococcus furiosus. Proteins, 81, 261–70 [DOI] [PubMed] [Google Scholar]
- 143.Lehmann C, Lim K, Chalamasetty VR, Krajewski W, Melamud E, Galkin A, Howard A, Kelman Z, Reddy PT, Murzin AG, Herzberg O. (2003).The HI0073/HI0074 protein pair from Haemophilus influenzae is a member of a new nucleotidyltransferase family: structure, sequence analyses, and solution studies. Proteins, 50, 249–60 [DOI] [PubMed] [Google Scholar]
- 144.Wiseman RL, Zhang Y, Lee KP, Harding HP, Haynes CM, Price J, Sicheri F, Ron D. (2010).Flavonol activation defines an unanticipated ligand-binding site in the kinase-RNase domain of IRE1. Molecular cell, 38, 291–304 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Korennykh AV, Egea PF, Korostelev AA, Finer-Moore J, Stroud RM, Zhang C, Shokat KM, Walter P. (2011).Cofactor-mediated conformational control in the bifunctional kinase/RNase Ire1. BMC biology, 9, 48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Korennykh AV, Korostelev AA, Egea PF, Finer-Moore J, Stroud RM, Zhang C, Shokat KM, Walter P. (2011).Structural and functional basis for RNA cleavage by Ire1. BMC biology, 9, 47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Zhou J, Liu CY, Back SH, Clark RL, Peisach D, Xu Z, Kaufman RJ. (2006).The crystal structure of human IRE1 luminal domain reveals a conserved dimerization interface required for activation of the unfolded protein response. Proceedings of the National Academy of Sciences of the United States of America, 103, 14343–8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Colombano G, Caldwell JJ, Matthews TP, Bhatia C, Joshi A, McHardy T, Mok NY, Newbatt Y, Pickard L, Strover J, Hedayat S, Walton MI, Myers SM, Jones AM, Saville H, McAndrew C, Burke R, Eccles SA, Davies FE, Bayliss R, Collins I. (2019).Binding to an Unusual Inactive Kinase Conformation by Highly Selective Inhibitors of Inositol-Requiring Enzyme 1α Kinase-Endoribonuclease. Journal of medicinal chemistry, 62, 2447–65 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Harnoss JM, Le Thomas A, Shemorry A, Marsters SA, Lawrence DA, Lu M, Chen YA, Qing J, Totpal K, Kan D, Segal E, Merchant M, Reichelt M, Ackerly Wallweber H, Wang W, Clark K, Kaufman S, Beresini MH, Laing ST, Sandoval W, Lorenzo M, Wu J, Ly J, De Bruyn T, Heidersbach A, Haley B, Gogineni A, Weimer RM, Lee D, Braun MG, Rudolph J, VanWyngarden MJ, Sherbenou DW, Gomez-Bougie P, Amiot M, Acosta-Alvear D, Walter P, Ashkenazi A. (2019).Disruption of IRE1α through its kinase domain attenuates multiple myeloma. Proceedings of the National Academy of Sciences of the United States of America, 116, 16420–29 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Harrington PE, Biswas K, Malwitz D, Tasker AS, Mohr C, Andrews KL, Dellamaggiore K, Kendall R, Beckmann H, Jaeckel P, Materna-Reichelt S, Allen JR, Lipford JR. (2015).Unfolded Protein Response in Cancer: IRE1α Inhibition by Selective Kinase Ligands Does Not Impair Tumor Cell Viability. ACS medicinal chemistry letters, 6, 68–72 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Concha NO, Smallwood A, Bonnette W, Totoritis R, Zhang G, Federowicz K, Yang J, Qi H, Chen S, Campobasso N, Choudhry AE, Shuster LE, Evans KA, Ralph J, Sweitzer S, Heerding DA, Buser CA, Su DS, DeYoung MP. (2015).Long-Range Inhibitor-Induced Conformational Regulation of Human IRE1α Endoribonuclease Activity. Molecular pharmacology, 88, 1011–23 [DOI] [PubMed] [Google Scholar]
- 152.Joshi A, Newbatt Y, McAndrew PC, Stubbs M, Burke R, Richards MW, Bhatia C, Caldwell JJ, McHardy T, Collins I, Bayliss R. (2015).Molecular mechanisms of human IRE1 activation through dimerization and ligand binding. Oncotarget, 6, 13019–35 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Amin-Wetzel N, Neidhardt L, Yan Y, Mayer MP, Ron D. (2019).Unstructured regions in IRE1α specify BiP-mediated destabilisation of the luminal domain dimer and repression of the UPR. eLife, 8 [DOI] [PMC free article] [PubMed] [Google Scholar]