

Abstract
Background and Aims:
In stomach, metaplasia can arise from differentiated chief cells that become mitotic via paligenosis, a stepwise program. In paligenosis, mitosis initiation requires reactivation of the cellular energy hub mTORC1 after initial mTORC1 suppression by DNA damage induced transcript 4 (DDIT4 aka REDD1). Here, we use DDIT4-deficient mice and human cells to study how metaplasia increases tumorigenesis risk.
Methods:
A tissue microarray of human gastric tissue specimens was analyzed by immunohistochemistry for DDIT4. C57BL/6 mice were administered combinations of intraperitoneal injections of high-dose tamoxifen (TAM) to induce Spasmolytic Polypeptide Expressing Metaplasia (SPEM) and rapamycin to block mTORC1 activity, and N-methyl-N-nitrosourea (MNU) in drinking water to induce spontaneous gastric tumors. Stomachs were analyzed for proliferation, DNA damage, and tumor formation. CRISPR/Cas9-generated DDIT4−/− and control human gastric cells were analyzed for growth in vitro and in xenografts with and without 5-fluorouracil (5-FU) treatment.
Results:
DDIT4 was expressed in normal gastric chief cells in mice and humans and decreased as chief cells became metaplastic. Paligenotic Ddit4−/− chief cells maintained constitutively high mTORC1, causing increased mitosis of metaplastic cells despite DNA damage. Lower DDIT4 expression correlated with longer survival of gastric cancer patients. 5-FU-treated DDIT4−/− human gastric epithelial cells had significantly increased cells entering mitosis despite DNA damage and increased proliferation in vitro and in xenografts. MNU-treated Ddit4−/− mice had increased spontaneous tumorigenesis after multiple rounds of paligenosis induced by TAM.
Conclusions:
During injury-induced metaplastic proliferation, failure of licensing mTORC1 reactivation correlates with increased proliferation of cells harboring DNA damage, as well as increased tumor formation and growth in mice and humans.
Keywords: Regeneration, IFRD1, gamma-H2Ax, cyclical hit model
Introduction
Tissue injury stimulates fully differentiated cells to proliferate and reenter the cell cycle to replace damaged cells and fuel regeneration1,2, 3. Such injury-induced reprogramming of mature cells into progenitors is executed by an evolutionarily conserved, stepwise cellular program called paligenosis (Figure S1). The stages of paligenosis are: 1) the cellular master energetics-regulator mTORC1 is quenched as lysosomal and autophagic activity ramps up to dismantle mature cell features; 2) expression of progenitor or embryonic genes is induced; 3) mTORC1 is reactivated to initiate S-phase of mitosis and repair the tissue4. Understanding how paligenosis works may be critical for understanding tumorigenesis, as precancerous lesions like metaplasia can arise from mature cells and are often characterized by expression of embryonic and progenitor genes5–7.
The idea that mature cells can retool their energetics to fuel regeneration and cause precancerous lesions is well over a century old. Pathologist George Adami thought injury invoked a program to retool mature cell energy usage from fueling physiological function to fueling proliferation8, 9. Specifically in the stomach, the link between dedifferentiation of mature cells into mucus-secreting, more primitive cells has been considered a key initiating event in gastric cancer since at least the time of Ménétrier10, 11. The lesion containing potentially precancerous mucous cells has been referred to as chronic atrophic gastritis (most commonly used by pathologists today), spasmolytic polypeptide expressing metaplasia (SPEM, based on the pattern of gene expression), and pseudopyloric metaplasia (based on histological similarity to antrum/pylorus)12, 13.
By dissecting the mechanisms that govern paligenosis, we can focus on the core set of genes that evolved specifically to regulate entry into metaplasia and govern how metaplasia normally resolves without becoming chronic or progressing to cancer. A key early event in the paligenosis that generates SPEM cells from digestive-enzyme-secreting, differentiated chief cells is activation of DDIT414. DDIT4 is an evolutionarily conserved, injury-induced scaffold that suppresses the cellular energy hub mTORC1 by blocking 14-3-3 proteins from interacting with the TSC1/2 complex, thereby inactivating Rheb, a small GTPase that activates mTORC115–18. Early after paligenotic injury, DDIT4 is induced to suppress mTORC114. DDIT4 slowly degrades as paligenosis progresses, but mTORC1 remains suppressed as the injury activates p53, an mTORC1 inhibitor. Healthy cells, however, concomitantly accumulate IFRD1, which suppresses p53, derepressing mTORC1 and permitting its reactivation to fuel S-phase entry.
Because p53 blocks only reactivation of mTORC1 after initial mTORC1 suppression by DDIT4, paligenotic cells in Ddit4−/− mice would be predicted to bypass p53 to re-enter the cell cycle. Long-lived, post-mitotic cells like chief cells may be more likely to have accumulated oncogenic mutations that are unmasked when the cells begin to proliferate13, 19. Thus, we reasoned that the aberrant paligenosis wherein DDIT4-deficient cells bypass a p53 checkpoint could be used as a tool to study the functional consequences of aberrant metaplasia.
Here, we focus on the failure of licensing in cells lacking DDIT4 during the paligenosis that causes SPEM. We show that DDIT4 is expressed specifically in normal mouse and human chief cells, and its expression decreases in patients with chronic metaplasia and dysplasia. Patients with low expression have poorer survival. In our acute, rapid, synchronous SPEM induction model using high-dose tamoxifen (TAM), we see early, brief induction of DDIT4 and later extinction. Loss of DDIT4, as expected, caused dysregulated, constitutively increased mTORC1 in paligenosis with a functional consequence of the failed checkpoint being increased cells proliferating despite DNA damage. In human gastric adenocarcinoma cells, loss of DDIT4 also increased mitosis despite DNA damage and despite chemotherapeutic agents in vitro and in xenografts into mice. Finally, we show that Ddit4−/− mice had significantly increased MNU-catalyzed spontaneous tumorigenesis after multiple rounds of TAM-induced, paligenosis-fueled SPEM.
Materials and Methods
Regulatory Approval
All human gastric pathological tissue specimens included in this paper were approved by the Institutional Review Board Ethics Committee of Washington University School of Medicine or China Medical University. All experiments using animals followed protocols approved by the Washington University School of Medicine or China Medical University Animal Studies Committees.
Animal studies and associated reagents
SPEM was induced by Tamoxifen (5 mg/20 g body weight; Toronto Research Chemicals) dissolved in 10% ethanol and 90% sunflower oil, injected intraperitoneally (IP) daily with mice euthanized at 12–72h as described previously20. Rapamycin (60 μg/20 g body weight; LC Laboratories) was injected IP in 0.25% Tween-20, 0.25% polyethylene glycol in PBS daily for 3 days pre-treatment and throughout the injury time course. 5-bromo-2’-deoxyuridine (BrdU; 120 mg/kg) and 5-fluoro-2’-deoxyuridine (12 mg/kg) in sterile water 90 min was injected IP before sacrifice for all BrdU labeling experiments.
Gastric cancer cell DDIT4 knockout
To generate a stable DDIT4 knockout MGC-803 cell line, a published protocol described previously21 was used with minor modifications. Three gRNA sets were designed and cloned into pSpCas9(BB)-2A-Puro plasmids (Addgene, PX459) to make double nicks on the DNA strand. These were applied to MGC-803 cells and selected for with puromycin (200 μg/ml) for two weeks. At least 20 single-cell derived clones were established and confirmed via PCR. The DDIT4 knockout MGC-803 cell lines (DDIT4−/−) and negative control (NC) cell lines (DDIT4+/+) were further confirmed via western blot against DDIT4.
Statistical analyses
All statistical analyses were performed using SPSS 19.0 software (IBM, only for Kaplan–Meier survival analysis) or GraphPad Prism 7 (La Jolla). Overall survival was determined using Kaplan–Meier curves, with an event defined as a cancer-related death. The log-rank test was used to assess differences between the survival curves in different groups. In histopathological univariate analysis, continuous data were analyzed by a two-tailed Student’s t-test, and categorical data were evaluated by a two-tailed χ2 test. For normally distributed mouse or cell data, statistical analysis of significance was by t-test in the case of 2-sample comparisons or ANOVA with post-hoc inter-sample comparison to evaluate significance when there were ≥3. For 2-sample data whose distribution was not demonstrated or expected to be normal, the Mann-Whitney Test was used. All data were expressed as mean±standard deviation or standard error of mean (SEM), depending on experiment design. P<0.05 was considered statistically significant.
Results
DDIT4 is expressed in healthy chief cells and early in paligenosis but lost in chronic metaplasia and dysplasia
We examined DDIT4 expression in a human tissue microarray representing 218 patients with normal and precancerous lesions, as well as a collection of >50 biopsy and resection specimens harboring regions of transition from normal to SPEM (Table S1). Most gastric adenocarcinomas are thought to arise via a defined sequence of lesions with normal stomach undergoing first chronic atrophy with SPEM, progressing to intestinal metaplasia and finally to dysplasia22, 23. DDIT4 expression progressively declined with each step (Figure 1A, B).
Figure 1. DDIT4 is expressed in human chief cells with expression decreasing in proliferative or dysplastic lesions.
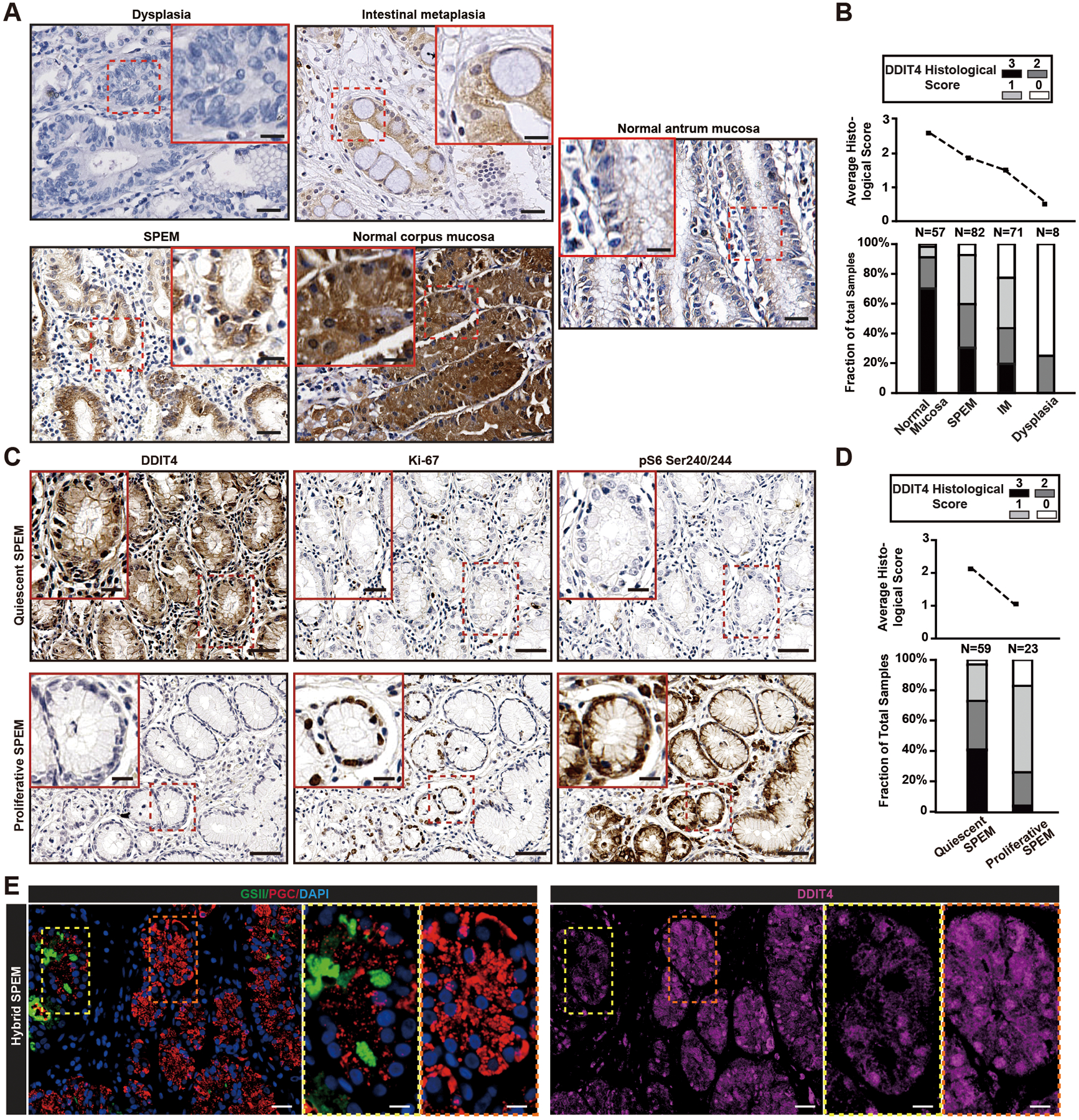
A. DDIT4 expression in human stomach mucosa tumor microarrays. Histological scores were: 0, no staining; 1, staining in ≤20% of cells; 2, moderate to strong staining in≥20% of cells; 3, strong staining in ≥50% of cells. Scale bar, 30 μm; boxed area insert, 15 μm.
B. Average histological score (upper plot) of DDIT4 expression scores in human stomach tissue microarrays of only regions with normal histology and precancerous lesions. Lower plot shows fractional distribution of each score within each lesion phenotype.
C. SPEM lesions categorized as Ki-67 positive (“proliferative”) and Ki-67 negative (“quiescent”) were examined in serial tissue sections for DDIT4 and phosphorylated-S6 (correlating with mTORC1 activity). Depicted are representative images of lesions with proliferative SPEM or quiescent SPEM in serial slides of a single core from the tissue microarray. Scale bar, 30 μm; boxed area, 15 μm.
D. Upper plot shows average DDIT4 score (0= no expression, 3=maximal) for the lesion type, and lower plot shows fraction of biopsy with a given DDIT score for each lesion.
E. Human gastric resection specimen in a region of normal chief cells, SPEM, and cells with hybrid, transitional phenotype. Green=GSII; Red=PGC; Blue=DAPI; Purple=DDIT4. Fluorescence channels are separated in inserts to show co-labeling patterns. Scale bar, 30 μm; boxed area insert, 15 μm.
Chronic metaplasia increases risk for tumorigenesis13, 24, 25. In humans, most SPEM lesions are mitotically quiescent at any one time4, 26, 27. Figure 1C and D show that quiescent SPEM resembled normal tissue in having higher DDIT4 expression, whereas proliferating SPEM had lower DDIT4. As expected, high DDIT4 in quiescent SPEM correlated with low mTORC1, whereas low DDIT4 in proliferating SPEM correlated with high mTORC14. Normal chief cells in human stomach had abundant DDIT4, but in regions where cells had transitional chief cell-SPEM cell morphology (see references4, 28) DDIT4 was decreased (Figure 1E).
Given that DDIT4 suppresses mTORC129, 30, and mTORC1 reactivation dictates which cells will proliferate during paligenosis and metaplasia14, we hypothesized we could use DDIT4 as a tool to dissect how metaplasia is regulated. We used high-dose tamoxifen (TAM), which causes SPEM and DNA damage across the whole gastric body of mice within 3 days, with resolution normally by 14 days20, 31, to clarify a role for DDIT4 in progression of metaplasia to dysplasia/neoplasia. DDIT4 expression in the body of the stomach was confined to chief cells. During Stage 1 of paligenosis, cells undergo dramatic loss of mTORC1 and massive autophagy to downscale differentiated cell architecture, whereas Stages 2 and 3 of paligenosis are characterized by cells expressing metaplastic/progenitor markers and then re-inducing mTORC1 to enter the cell cycle (Figure S1). During TAM-induced paligenosis, DDIT4 increased somewhat in Stage 1 of paligenosis, as mTORC1 was suppressed, and then was largely lost from the stomach by Stages 2 and 3 when mTORC1 reactivated (Figure 2A, Figure S1).
Figure 2. DDIT4 is expressed early in chief cell paligenosis is required for mTORC1 decrease, degradation of secretory cell architecture, and to moderate paligenotic proliferation.
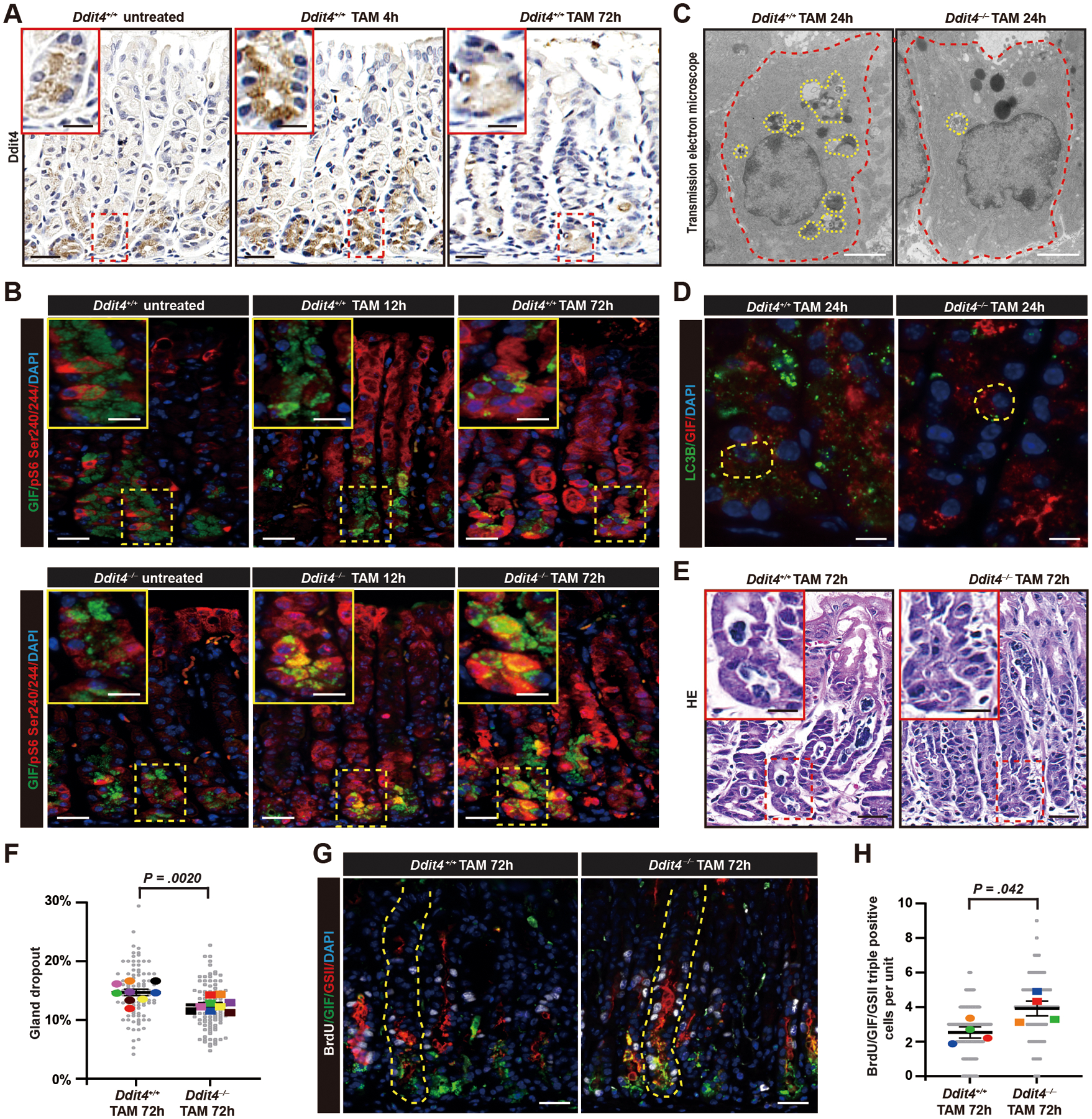
A. DDIT4 expression in stomach chief cells as they undergo paligenosis to SPEM after TAM. Scale bar, 30 μm; boxed area insert, 15 μm.
B. Immunofluorescence for mature chief cell secretory granule protein GIF and a proxy for mTORC1 activity S6 phosphorylated one residues 240/244 in Ddit4+/+ and Ddit4−/− mice in stomach glands at various time points after TAM. Green=GIF; Red=pS6; Blue=DAPI. Scale bar, 30 μm; boxed area insert, 15 μm.
C. Transmission electron microscopy of single chief cells (outlined in red) from wild type and Ddit4−/− mice 24h post TAM (late Stage 1). Yellow outlines indicate autodegradative (autophagic/lysosomal) compartments engulfing organelles in reprogramming chief cells. Scale bar, 1 μm.
D. Immunofluorescence for GIF and autophagosome marker LC3B 24h after tamoxifen injury (late Stage 1). Yellow outlines single paligenotic chief cells. Green=LC3B; Red=GIF; Blue=DAPI. Scale bar, 30 μm.
E. H&E staining of Ddit4+/+ and Ddit4−/− mice 72h post TAM (Stage 3). Inserts highlight base of gland where chief cells have become cuboidocolumnar SPEM cells in controls but retain substantial cytoplasm in the absence of DDIT4. Scale bar, 30 μm; boxed area insert, 15 μm.
F. Quantification of gland dropout for data as in panel (E). Each colored datapoint represents mean of all counts from a single mouse (>11 microscopic fields counted per mouse); each gray datapoint = % dropout in a single field; black line indicates mean from all mice, with significance estimated by unpaired, two-tailed t test.
G. Immunofluorescence for GIF, GSII (progenitor and, when co-expressed with GIF, SPEM cell marker) and BrdU (proliferation marker) 72h post TAM. White=BrdU; Green=GIF; Red= GSII; Blue=DAPI. Scale bar, 30 μm; boxed area insert, 15 μm.
H. Quantification of BrdU+GIF+GSII+ cells (proliferating SPEM cells). Each colored datapoint represents mean of >40 gastric units counted from a single mouse; each gray datapoint = BrdU+ cells in a single unit; black line indicates mean from all mice, with significance estimated by unpaired, two-tailed t test.
DDIT4 is required for the mTORC1 suppression early in paligenosis that allows only healthy cells to later reactivate mTORC1
We examined the requirement for DDIT4 in our TAM model (Figure 2A). In homeostatic conditions, Ddit4–/– mice had a phenotype confined to chief cells with modest, but statistically significant, reduction in secretory granules (Figure S2A–E). Ddit4–/– mice showed much more dramatic defects during paligenosis. Paligenotic cells did not turn off mTORC1 in Stage 1 (Figure 2B; note that phospho-S6 is a proxy for mTORC1 activity4, 14). Lack of mTORC1 suppression correlated, as expected, with defective autophagy induction (Figure 2C, D). The defective autophagy led to preserved mature chief cell phenotype later on at Stage 3 (note retention of the mature chief cell granule marker GIF, Figure 2B, G). Stage 3 in Ddit4−/− mice also differed as proliferation was increased despite the persistent differentiated cell phenotype. Additionally, TAM injury normally causes about 15% of gland bases harboring chief cells to dropout in wildtype mice (Figure 2E, F), but Ddit4–/– mice had nearly no detectable gland loss. The loss of cells in control mice is due to p53-mediated apoptosis that occurs in Stage 2 to 3 transition as cells attempt to reactivate mTORC1 to drive proliferation14. Thus, the decreased cell loss and increased proliferation in Ddit4–/– mice were consistent with inappropriate entry into Stage 3 due to lack of initial suppression of mTORC1 in Stage 1 (Figure 2G, H).
We next determined the consequences of Ddit4–/– cells’ bypassing the licensing checkpoint for Stage 3 entry (Figure 3A). TAM causes DNA damage20, but cells with DNA damage executing the fully functioning paligenosis program would be expected to first repair the damage before entering S-phase, or they would undergo apoptosis32, 33. As anticipated, in Ddit4+/+ mice BrdU+ cells (ie those that had entered S-phase) were largely negative for the DNA damage-repair marker γH2AX. However, in Ddit4−/− mice, both the overall number of γH2AX+/BrdU+ cells and the fraction of γH2AX+ cells that were also BrdU+ were substantially and statistically increased (Figure 3B, C). The increase in both mitotic cells and in mitotic DNA-damage-harboring cells was mTORC1-dependent, as rapamycin blocked both phenotypes (Figure 3D, E). Overall, the results indicate that aberrant cell cycle entry in DDIT4-deficient cells is due to defective mTORC1 suppression and thus failed licensing in the Stage 2 to 3 transition.
Figure 3. Loss of DDIT4 increases entry of paligenotic cells into the cell cycle despite DNA damage, which is rescued by mTORC1 inhibition by rapamycin.
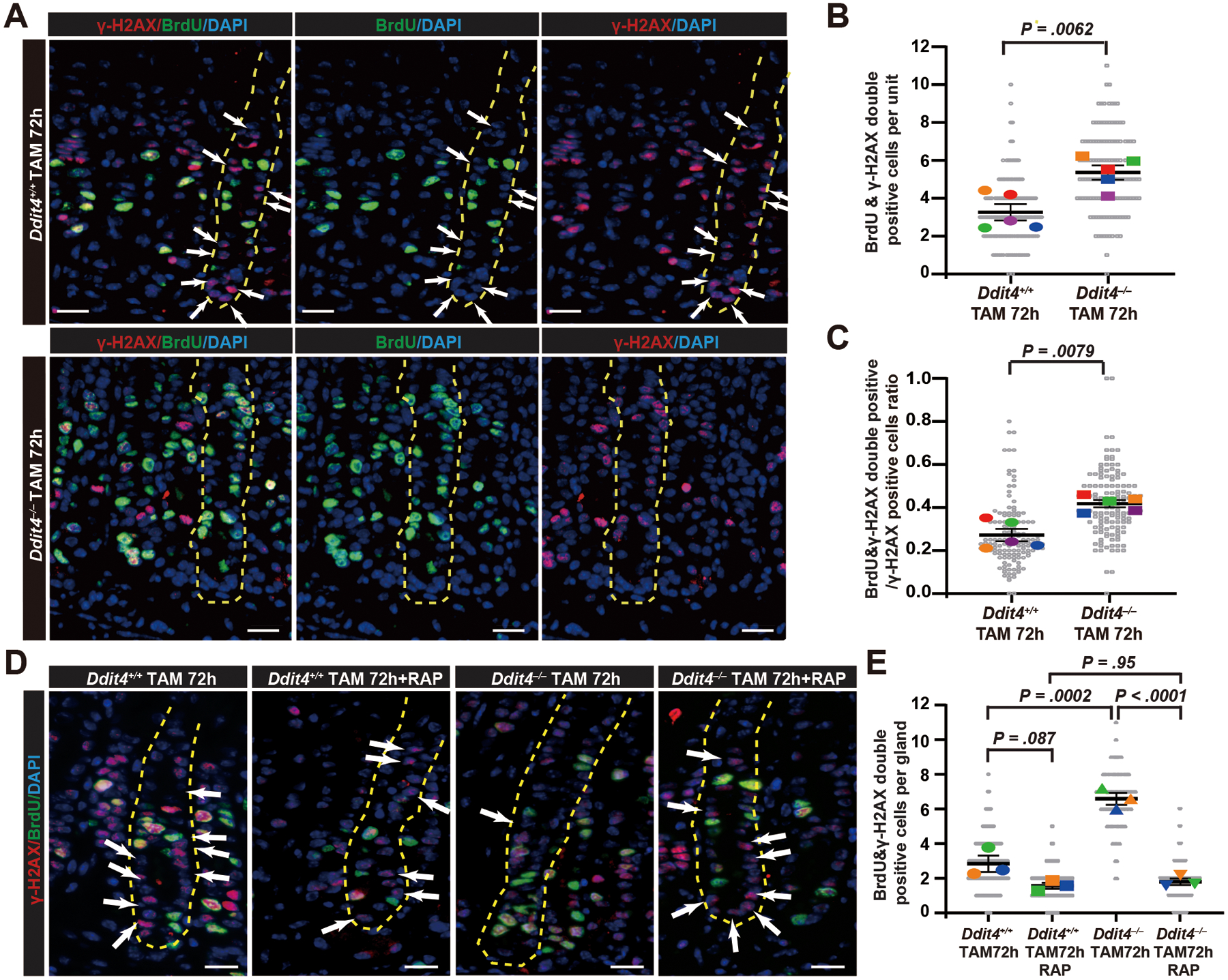
A. Immunofluorescence for BrdU and DNA damage marker γ-H2AX. Green, BrdU; Red, γ-H2AX; Blue, DAPI. BrdU+γ-H2AX− cells (ie cells with DNA damage that have not entered into cell cycle) in one full gastric unit are marked with white arrows. Yellow dashed line outlines a representative gastric gland. Scale bar, 30 μm.
B. Data as in (A) quantified, focusing on BrdU+γ-H2AX+ cells 72 hours post TAM Colored datapoint: mean of ≥ 28 glands quantified per each mouse; gray datapoint: count from each gland; dark black lines = mean ± SEM across all mice; significance estimated by unpaired, two-tailed t test.
C. Data as in (A) quantified, focusing on the ratio of BrdU+γ-H2AX+ cells to H2AX+ cells (ie fraction of cycling cells with DNA damage to total cells with DNA damage) per gastric gland. Datapoints and statistics as in (B)
D. Immunofluorescence for BrdU and γ-H2AX staining of Ddit4+/+ and Ddit4−/− mouse stomachs treated with rapamycin. Green, BrdU; Red, γ-H2AX; Blue, DAPI. BrdU+γ-H2AX− cells in one full gastric unit (outlined in yellow) are marked with white arrows; note outlined Ddit4−/− unit had no BrdU+γ-H2AX− cells, meaning all dividing cells had DNA damage. Scale bar, 30 μm.
E. Data as in (D), quantified for BrdU+γ-H2AX+ cells per gastric gland. Colored datapoint: mean of ≥ 42 glands quantified per each mouse; gray datapoint: count from each gland; dark black lines = mean ± SEM across all mice; significance estimated by one-way ANOVA with Tukey post hoc test.
Loss of DDIT4 causes cell-autonomous increased growth of human gastric cancer cells despite chemotherapy and DNA damage in vitro and in xenografts
To explore the cell-autonomous effects of DDIT4 on cell cycle entry and in the context of tumorigenesis, we returned to analysis of human patients and cells. We examined another annotated tissue microarray on which we had compiled 509 patients with gastric cancer. We saw statistically significant correlation of high DDIT4 expression with increased patient survival and more differentiated tumors, especially in patients who had received chemotherapy, consistent with DDIT4 slowing proliferation in response to DNA damage (Figure 4A–C; Table S3). To determine if loss of DDIT4 causes cell-autonomous effects on proliferation, we deleted DDIT4 from human MGC803 gastric cancer cells and treated them with the DNA damage-inducing, chemotherapeutic agent 5-FU (Figure S3A, B). 5-FU in control cells increased DDIT4 expression without initial effect on mTORC1 activity (Figure 4D). Contrastingly, DDIT4−/− cells had elevated mTORC1 at baseline that persisted even after 5-FU (Figure 4D); they continued to enter G2 and M-phases of the cell cycle and proliferate, whereas control cells were largely arrested (Figure 4E; Figure S3C, D).
Figure 4. In humans, loss of DDIT4 correlates with worse gastric cancer survival and causes cell-autonomous increased mTORC1 and proliferation despite chemotherapy.
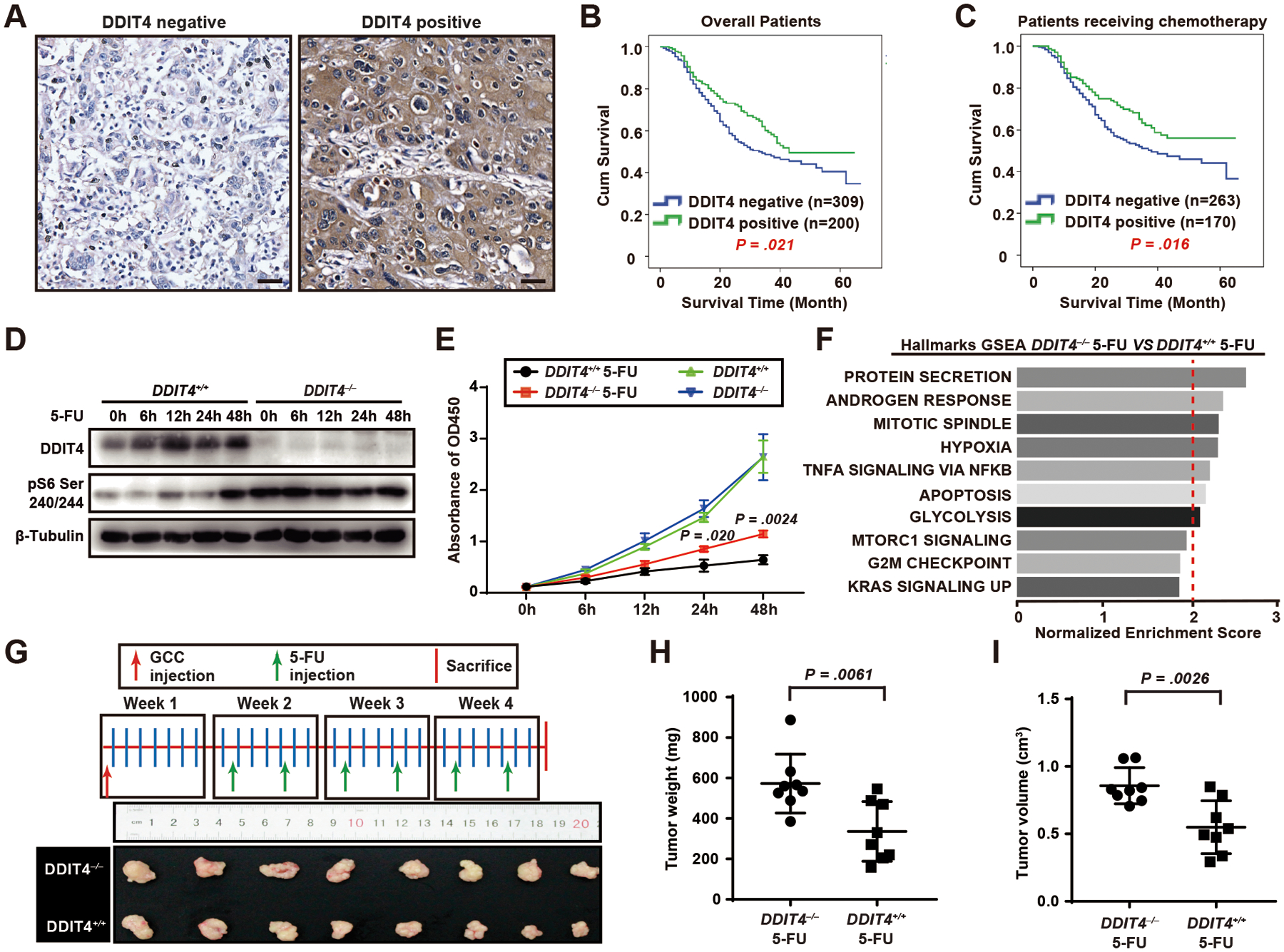
A. DDIT4 expression in representative cores scored as “negative” or “positive” from a human gastric cancer tissue microarray. Scale bar, 30 μm.
B. Overall survival determined via Kaplan-Meier curves separated by DDIT4 expression, with an event defined as a cancer-related death. The log-rank test was used to assess differences between the survival curves in different patient groups.
C. Overall survival determined using Kaplan-Meier curves in chemotherapy subgroups.
D. Western blot of DDIT4, pS6 Ser240/244 (proxy for mTORC1 activity) with β-tubulin loading control in CRIPSR-generated DDIT4 knockout (gRNA3-transfected cell line had the most effective knockout and was the one used for all experiments; see also Figure S3B) DDIT4−/−) and negative control (DDIT4+/+) MGC-803 gastric cancer cell lines at various time points after 5-FU treatment..
E. DDIT4−/− and DDIT4+/+ MGC-803 cells were treated with 5-FU, and proliferation measured via CCK-8 assay. Datapoint: mean ± SEM from 3 independent experiments with significance estimated by unpaired, two-tailed t test of the area under the curve (to avoid repeated measurement bias) of each individual experiment up until 24h and 48h.
F. GSEA of global mRNA expression profiling using all gene sets in Broad “Hallmark” compilation. All statistically significant gene sets (false discovery rate [FDR] and p value < 0.001) are shown with normalized enrichment score (NES)= 2.0 indicated by dashed red line.
G. DDIT4−/− and DDIT4+/+ cells were xenografted into the flanks of nude BALB/c mice and the mice treated with 5-FU according to the scheme at top. The growth of DDIT4−/− tumors (n=8) was less inhibited by 5-FU than those from DDIT4+/+ cells (n=8).
H. Tumor weight from (G) quantified. Each datapoint is a single tumor with mean of all tumors ±SD indicated and significance estimated by unpaired, two-tailed t test.
I. Tumor volume from (G) quantified. Tumor length (L) and width (W) were measured with calipers and volume by (L × W2) π/6. Statistics and plotting as for (H).
To identify pathways governed by DDIT4, we did a transcriptomic analysis of DDIT4+/+ and DDIT4−/− cells treated with 5-FU at 24h. An unbiased gene set enrichment analysis (GSEA) revealed increased proliferation-associated transcripts (mitotic spindle, G2M checkpoint, KRAS signaling), mTORC1 and biosynthetic/energetic transcripts (protein secretion, mTORC1 signaling, glycolysis), and transcripts associated with the known role of DDIT4 in regulating hypoxia and the role we are elucidating here that it may play in apoptosis of cells with DNA damage (Figure 4F). Unbiased analysis for Gene Ontology terms enriched in DDIT4−/− cells treated with 5-FU showed similar enrichment in proliferation-associated and bioenergetics-associated processes, molecular functions, and cellular compartments (Figure S3E). The increased proliferation of DDIT4−/− cells persisted even when cells were xenografted into the flanks of nude BALB/c mice that were then administered 5-FU (Figure 4G–I). As expected, both in vitro and in vivo, the effects of DDIT4 were mTORC1-dependent, as rapamycin normalized DDIT4−/− pS6 levels and cell proliferation rate (Figure 5A–F).
Figure 5. Rapamycin rescues the phenotype of human DDIT4−/− gastric cancer cells indicating effects of DDIT4 loss on proliferation are via mTORC1.
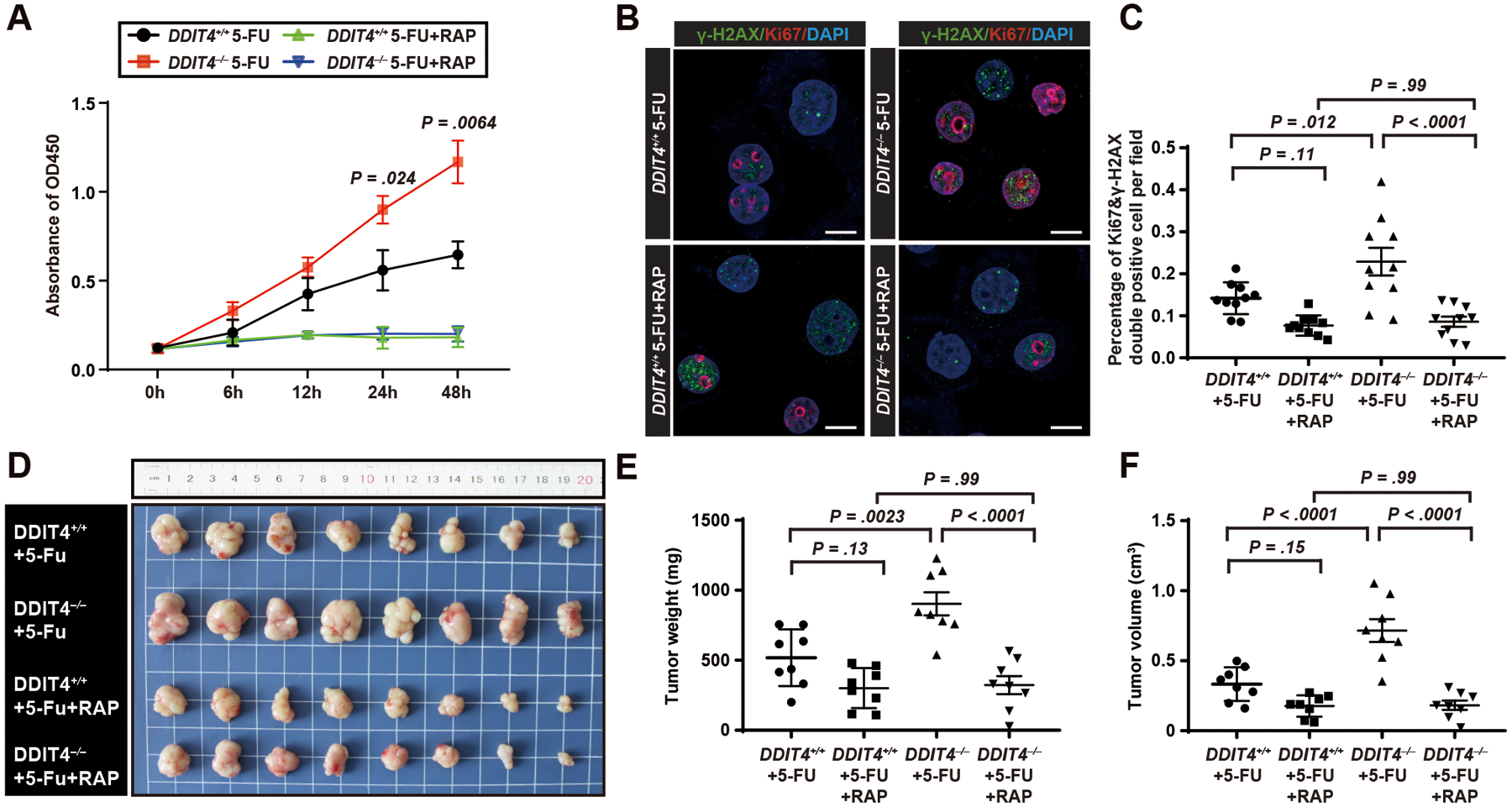
A. DDIT4−/− and DDIT4+/+ MGC-803 gastric cancer cell lines were treated with 5-FU or 5-FU with rapamycin, and proliferation measured by CCK-8 assay. Datapoint: mean ± SEM of 3 independent experiments with significance estimated by unpaired, two-tailed t test of the area under the curve (to avoid repeated measurement bias) of each individual experiment up until 24h and 48h.
B. Confocal immunofluorescence for Ki-67 (proliferation) and γ-H2AX (DNA damage) of MGC-803 cells treated with 5-FU or 5-FU with Rapamycin. Green, γ-H2AX; Red, Ki67; Blue, DAPI. Scale bar, 5μm.
C. Quantification of Ki67+γ-H2AX+ cells (ie proliferating cells with DNA damage) per field in (B). Data point: mean ± SD from 10 independent fields with significance estimated by one-way ANOVA with Tukey post hoc test.
D. DDIT4−/− and DDIT4+/+ cells were xenografted into the flanks of nude BALB/c mice which were then treated with 5-FU or 5-FU with Rapamycin (see Figure 4G for treatment scheme). n=8 for each group.
E. Tumor weight in (D) quantified. Each datapoint represents a single tumor with mean ± SD indicated and significance estimated by one-way ANOVA with Tukey post hoc test.
F. Quantification of (D) for tumor volume estimated from tumor length (L) and width (W) as: (L × W2) π/6. Statistics and plotting as for (E).
Loss of DDIT4 increases risk for metaplasia advancing to invasive gastric adenocarcinomas
Finally, we tested the functional consequences of unlicensed paligenosis for tumorigenesis. SPEM is a precursor lesion for gastric cancer in humans, but it does not efficiently progress to tumors in mice. The rate of spontaneous tumor formation can be increased by mutagens like N-methyl-N-nitrosourea (MNU)34, 35, which, we reasoned, would afford us a route to determine if lack of paligenosis licensing in the absence of DDIT4 increased risk for tumorigenesis (Figure 6A). In pilot experiments (not shown), we determined that treating mice both with multiple rounds of metaplasia-inducing TAM and MNU caused dysplastic, polypoid gastric lesions within 36 weeks, earlier than reported for MNU alone in C57/B6 mice. We next performed three independent experiments treating Ddit4−/− and control mice with cycles of TAM and MNU (experiment #1: n=10 mice per genotype; #2: n= 15 mice; #3: n= 15 mice, Figure 6A; Figure S5A, B). Overall, 24 of 30 surviving Ddit4−/− mice developed grossly-identifiable gastric tumors vs. 16 of 31 surviving controls (Figure 6B; Figure S5A, B). Wildtype tumors were mostly flat, raised lesions with focal cellular and glandular atypia; they were located in the corpus or overlapped corpus and antrum (Figure 6C). There were no truly invasive carcinomas in Ddit4+/+ mice (Figure S6A).
Figure 6. Loss of DDIT4 increases spontaneous tumorigenesis after multiple rounds of paligenosis in a mouse model.
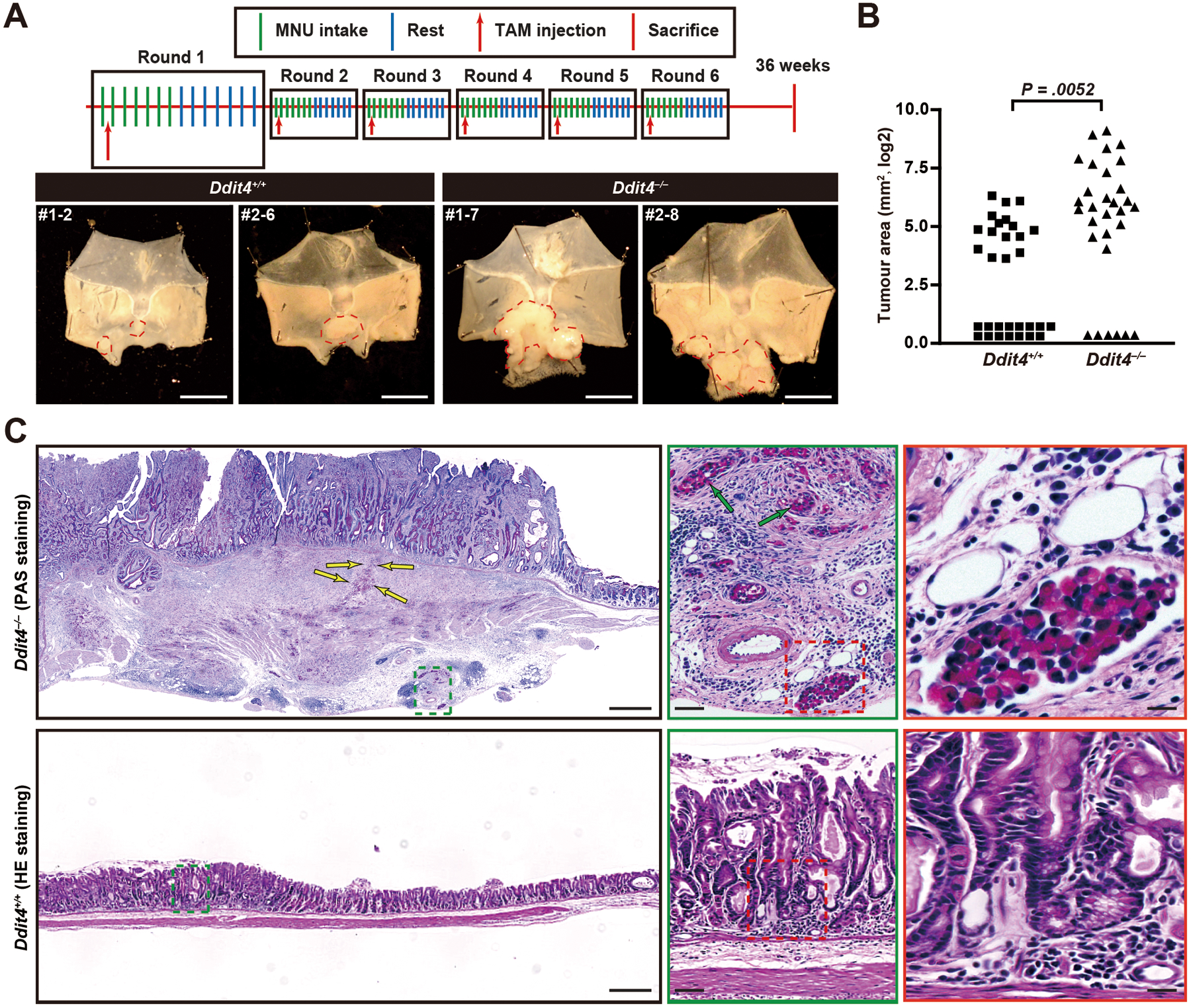
A. Scheme for TAM and MNU administration (top) and example tumors (bottom) in stomachs from 40 mice per genotype (30 Ddit4−/−, 31 Ddit4+/+ mice survived until endpoint of observation) in three independent cohorts. First number is cohort #, second is individual mouse number from that cohort. Tumor area outlined by red dashed line.
B. Total area per mouse occupied by tumors as measured under dissecting microscope from stomachs opened as for (B) quantified from all mice surviving until the endpoint of observation. Each datapoint: area of single mouse tumor area, log2 scale; significance estimated by Mann-Whitney test.
C. Representative H&E and PAS (for mucins) of one tumor from each mouse genotype. Tumors in wildtype mice were all raised, hyperplastic lesions with focal cellular and glandular atypia but no invasive features (red and green boxes show increased magnification). In the Ddit4−/− tumor depicted (1 of 6 invasive tumors, #1–7), a massive, poorly differentiated tumor with both irregular glandular and signet ring (PAS-staining positive) morphologies is seen invading through muscle to serosa. Invasive signet ring cells are marked by yellow arrows. Scale bar, 500 μm; green arrows = perineural infiltration, scale bar 50 μm; red box shows lymph-vascular space invasion, 25 μm.
Ddit4−/− lesions had similar location and morphology, but the size distribution was significantly shifted towards larger lesions (Figure 6A, B; Figure S5A, B). Moreover, 6 mice had invasive lesions, including poorly differentiated adenocarcinoma with “signet-ring” mucinous cells traveling alone or in nests and extending through the serosal surface, into duodenum and pancreas and penetrating lymph-vascular space (Figure S6A, B). The others were broadly invasive through muscle layers as irregular glands and as nests of cells (eg, see Figure S6A, B). Tumors in Ddit4+/+ mice were distributed similarly to those of wildtype mice and similar to previous reports36. The large and/or invasive tumors in the Ddit4−/− mice were found in a zone of transition between the corpus and antrum (eg note parietal cells present in glands at the tumor margin, Figure S6A, B) consistent with loss of DDIT4 specifically in chief cells (which do not occur in mouse antrum) increasing the risk of large, invasive tumors after multiple rounds of TAM-induced paligenosis.
Discussion
Here, we show that DDIT4 suppresses mTORC1 to license mature cells to enter the cell cycle during the gastric metaplastic response. Absence of DDIT4 leads to inappropriately elevated mTORC1 and aberrant, increased cell cycle re-entry of chief cells harboring DNA mutations. The phenotypes caused by loss of DDIT4 culminate in increased tumorigenesis. Our experiments in human tissue and in human gastric cancer cell lines in vitro and in xenografts corroborate our mouse results. DDIT4 is expressed specifically in differentiated chief cells, and DDIT4 phenotypes are limited to differentiated chief cells before and after injury. Moreover, our experiments with gastric cell lines indicate DDIT4 has cell-autonomous effects on mTORC1 and proliferation. Thus, we highlight a role for mature cells as cells of origin for metaplasia and a mechanism by which metaplasia arising from those cells carries risk for progression to cancer.
DDIT4 seems to have an evolutionarily conserved role in licensing during injury-induced proliferation. In Drosophila, for example, DDIT4 orthologues suppress TOR to prevent stress-induced recruitment of proliferative cells37. DDIT4 also plays a role in governing whether keratinocytes are activated in wound healing and in whether quiescent stem cells are recruited to become actively proliferating hematopoietic stem cells38, 39.
Dynamic mTORC1 regulation governs injury-induced recruitment of mature cells into the cell cycle (ie paligenosis) with cells necessarily turning mTORC1 off early and then reactivating it later (see model in Figure 7, Supplementary Figure 1). This mTORC1 pattern features in paligenosis-like regeneration programs such as: neuronal axon regrowth14, 40, liver and kidney repair4, satellite stem cell reactivation in muscle41. A critical role for mTORC1 in regeneration has also been shown across a broad range of species from hydra and sea anemone to humans42. We have shown that the mTORC1 fluctuation occurs because in differentiated cells, elevated mTORC1 maintains protein synthesis, which is needed in chief cells, for example, to secrete digestive enzymes. After paligenosis-inducing injury, DDIT4 is activated to suppress mTORC1 and shut off protein synthesis as well as to induce autophagy to recycle the elaborate rER and secretory granules into building blocks for subsequent cell division. The mTORC1 reactivation that later drives that cell division doesn’t happen until both DDIT4 is later degraded, and the p53 activated by injury is suppressed by IFRD1. We have shown recently that cells lacking IFRD1 fail mTORC1 reactivation and tend to undergo apoptosis instead of completing paligenosis14. Our results showing DDIT4 expression correlating with less proliferation in precancerous lesions and better survival of patients with gastric cancer confirm and extend on those of a previous report43. Similarly, in other tissues it has been shown that DDIT4 safeguards against cells with DNA damage entering mitosis by mediating DNA repair and inducing apoptosis44, 45. Additionally, dysregulated mTORC1 activity (eg as caused by loss of DDIT4) can cause genome instability in turn causing increased DNA damage46. Overall, thus, it seems constitutive mTORC1 may help tumor cells divide in the face of acute chemotherapeutic stress.
Figure 7. A model for how DDIT4 causes failure of mTORC1 reactivation checkpoint and increased tumorigenesis.
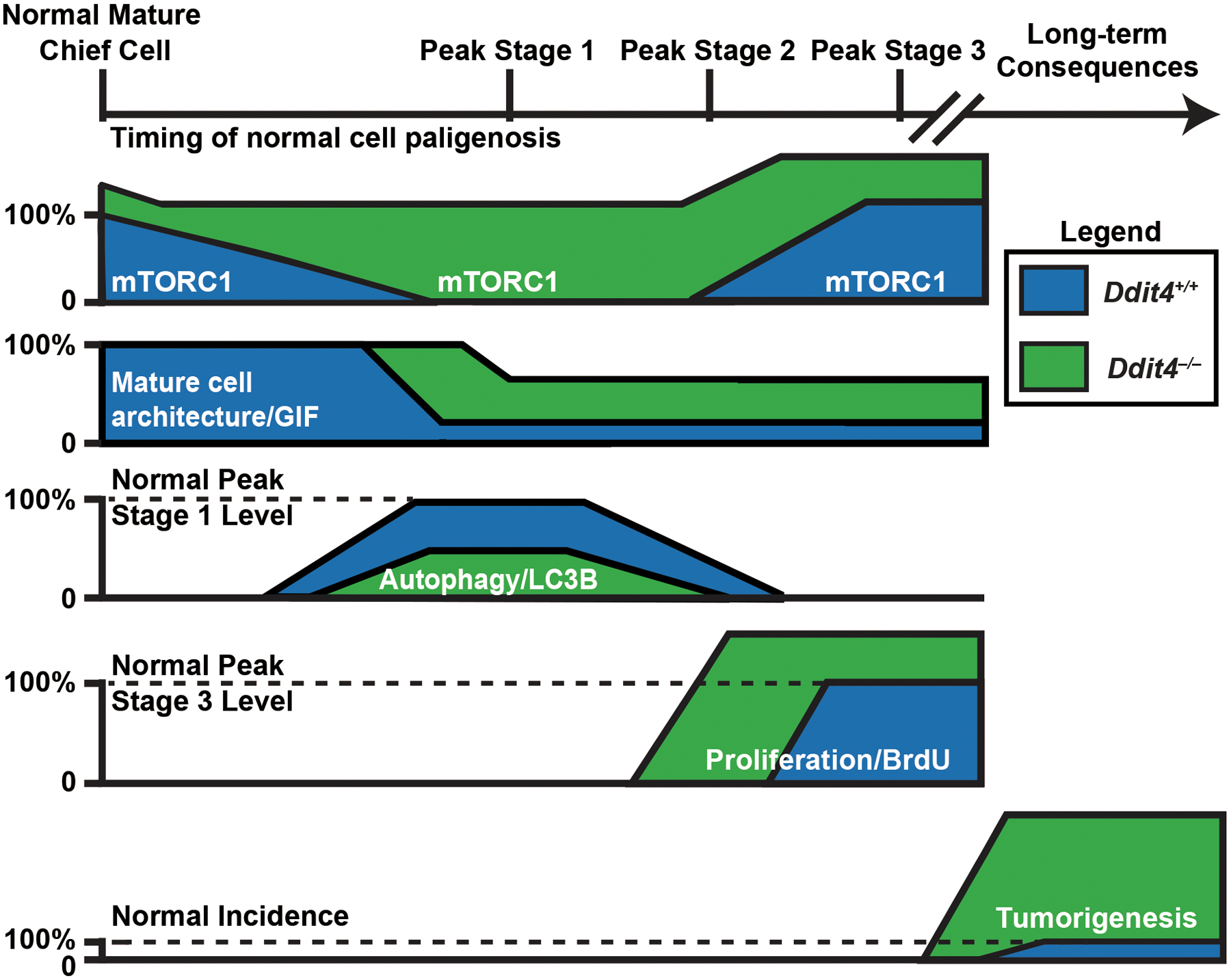
Schematic summarizing the molecular-genetic dynamics corresponding to the cellular changes over the timecourse of paligenosis with relative expression of each marker or phenotype in Ddit4+/+ (blue) vs Ddit4−/− (green) mice.
Paradoxically, however, studies across multiple organs have shown disparate results for DDIT4 in tumorigenesis with some studies showing higher DDIT4 decreases cancer survival44, 47–50. The discrepancies may arise because mTORC1’s role in tumorigenesis and in tumor cells is complex, and constitutive loss of DDIT4, one of the most important mechanisms for stress-induced mTORC1 suppression, may handicap cells’ ability to regulate their metabolic activity to adapt to different conditions dynamically (eg chemotherapy). Even in the current study, we see a statistically significant but not dramatic difference in patient survival, based on DDIT4 expression, suggesting constitutively increased, dysregulated mTORC1 may not be uniformly advantageous for the long-term survival and expansion of tumor cells.
The increased tumorigenesis in the stomachs of MNU-treated Ddit4−/− mice is intriguing, because, to our knowledge, this is the first documentation that a specific aberration in the cellular program leading to metaplasia increases incidence of progression of metaplasia to tumors. There have been prior studies that have shown genes involved in progression of metaplasia to dysplasia and neoplasia; for example, Goldenring and colleagues have studied how TROP2 increases specifically in human lesions progressing between metaplasia to dysplasia51, which could eventually be developed as a tool or biomarker to follow metaplastic progression. But our studies here suggest a requirement for DDIT4 specifically in decreasing risk for subsequent tumorigenesis. And we note that in the Ddit4−/− mice, the 6 invasive tumors that were induced were heterogeneous in phenotype from a signet-ring-type diffuse carcinoma to various adenocarcinomas, invading through the musculature and exhibiting more glandular features. Thus, in this relatively small sample size, the MNU Ddit4−/− tumors resembled the invasiveness and morphological diversity that occur in human gastric cancer, which is not usually the case in mouse models of gastric cancer25.
Our results are consistent with the “Cyclical Hit Model” of tumorigenesis13, 19 in which mature cells undergoing paligenosis are uniquely situated to accumulate somatic mutations that put an organ at risk for tumorigenesis. Active stem cells, may not typically accumulate mutations because they proliferate rapidly, frequently generating differentiated progeny that are eventually shed from the tissue52, thereby ridding the tissue of mutations. On the other hand, most mature cells that undergo paligenosis to repair damage subsequently return, eventually, to being long-lived, functional cells that can accumulate mutations53. Any tumor-promoting mutations might eventually be unmasked in subsequent paligenosis events months or years later.
There is a limitation to the MNU plus TAM experiment in that we cannot confirm that chief cells were the cells of origin for the tumors, though we favor that as an interpretation. DDIT4 expression was restricted to chief cells (homeostatic and paligenotic), and the phenotype of DDIT4 loss in paligenosis was thus confined to chief cells. Moreover, in gastric cancer cells, loss of DDIT4 conferred increased tumor growth despite DNA damage, indicating DDIT4 works cell-autonomously. Additionally, the cells harboring DNA damage after TAM and loss of DDIT4 were the paligenotic chief cells. In the MNU experiments, the multiple rounds of paligenosis would put those cells at greatly increased risk for MNU-induced mutation (Figure 7). The location of the hyperplastic, non-invasive, raised lesions that appeared as tumors were relatively scattered throughout the stomach, but the invasive tumors were located in the transition between antrum and corpus, as many human tumors are13.
The loss of DDIT4 in later stages of paligenosis is not unexpected, as DDIT4 is controlled largely by protein turnover54, 55. The early inducing event that activates DDIT4 isn’t known, but ongoing work suggests the culprit may be reactive oxygen species (ROS), which are early and important upstream stressors in gastric paligenosis56 and are known to activate DDIT4.
Paligenosis likely evolved so multicellular organisms could recruit differentiated cells – the vast majority of cells in an adult – as regenerative progenitors. However, allowing long-lived cells to cyclically proliferate and then redifferentiate is dangerous, because such cells can accumulate mutations over the lifetime of the organism. Paligenosis thus must also involve strict licensing to ensure that damaged cells are not allowed to propagate. DDIT4 regulation of mTORC1 appears to be a central regulator of paligenosis that may help explain part of the mechanism of how cells progress from metaplasia to cancer.
Supplementary Material
Acknowledgments
We thank Quark Pharmaceuticals Inc for allowing the opportunity to use the Ddit4tm1.1 (KOMP)Vlcg/J (Ddit4−/−) mice. We thank Dr. Elena Feinstein (Quark Pharmaceuticals, Inc) and Prof. Rubin Tuder (University of Colorado Denver) for providing the Ddit4tm1.1 (KOMP)Vlcg/J (Ddit4−/−) mice. We are deeply grateful to Spencer G Willet (Washington University in St Louis) for critically revising the manuscript and figures.
Grant support:
The work is supported by AITAC of the Washington University Digestive Disease Center (DDRCC: P30 DK052574) and the NIH Shared Instrumentation Grant S10 RR0227552 (for Nanozoomer slide scanning). MAT by T32-DK077653 (NIH, NIDDK) and R25-GM103757 (NIGMS, NIH); ZNW by National Key R&D Program of China (MOST-2017YFC0908300), National Natural Science Foundation of China (81961128026, U1908207), Major Scientific and Technological Special Project of Liaoning Province of China (2019020176-JH1/103), Overseas training project of Liaoning general higher education (2019GJWYB022); JCM by R01DK094989, R01DK105129, R01DK110406, R01CA239645, by the Alvin J. Siteman Cancer Center-Barnes Jewish Hospital Foundation Cancer Frontier Fund, NIH National Cancer Institute P30 CA091842 and R01 CA246208, and the Barnard Trust.
Abbreviations:
- DDIT4
DNA Damage Inducible Transcript 4
- TAM
high-dose tamoxifen
- SPEM
Spasmolytic polypeptide-expressing metaplasia
- MNU
mutagen N-methyl-N-nitrosourea
- 5-FU
5-Fluorouracil
Footnotes
Disclosures:
The authors declare that they have no conflicts of interest.
REFERENCES:
- 1.Nam KT, Lee HJ, Sousa JF, et al. Mature chief cells are cryptic progenitors for metaplasia in the stomach. Gastroenterology 2010;139:2028–2037 e9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Karki A, Humphrey SE, Steele RE, et al. Silencing Mist1 Gene Expression Is Essential for Recovery from Acute Pancreatitis. PLoS One 2015;10:e0145724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Schaub JR, Huppert KA, Kurial SNT, et al. De novo formation of the biliary system by TGFbeta-mediated hepatocyte transdifferentiation. Nature 2018;557:247–251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Willet SG, Lewis MA, Miao ZF, et al. Regenerative proliferation of differentiated cells by mTORC1-dependent paligenosis. EMBO J 2018;37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Keeley TM, Samuelson LC. Cytodifferentiation of the postnatal mouse stomach in normal and Huntingtin-interacting protein 1-related-deficient mice. Am J Physiol Gastrointest Liver Physiol 2010;299:G1241–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Storz P Acinar cell plasticity and development of pancreatic ductal adenocarcinoma. Nat Rev Gastroenterol Hepatol 2017;14:296–304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Que J, Garman KS, Souza RF, et al. Pathogenesis and Cells of Origin of Barrett’s Esophagus. Gastroenterology 2019;157:349–364 e1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Adami JG. “Festschrift” in Honor of Abraham Jacobi, M.D., L.L.D.: To Commemorate the Seventieth Anniversary of His Birth Knickerbocker Press; 1900:422–432. [Google Scholar]
- 9.Adami JG. The principles of pathology: Philadelphia. Lea & Febiger, 1908- 1909. (January 1, 1908) [Google Scholar]
- 10.Ménétrier P ÉTIOLOGIE GÉNÉRALE DES TUMEURS In: Bouchard C, ed. TRAITE DE PATHOLOGIE GÉNÉRALE. Volume 3 Paris: G. Masson, 1900:762–796. [Google Scholar]
- 11.Lewis WG, Fred WS. Chronic atrophic gastritis and cancer of the stomach. Arch Surg 1943;46:823–843. [Google Scholar]
- 12.Goldenring JR. Pyloric metaplasia, pseudopyloric metaplasia, ulcer-associated cell lineage and spasmolytic polypeptide-expressing metaplasia: reparative lineages in the gastrointestinal mucosa. J Pathol 2018;245:132–137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Saenz JB, Mills JC. Acid and the basis for cellular plasticity and reprogramming in gastric repair and cancer. Nat Rev Gastroenterol Hepatol 2018;15:257–273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Miao ZF, Lewis MA, Cho CJ, et al. A Dedicated Evolutionarily Conserved Molecular Network Licenses Differentiated Cells to Return to the Cell Cycle. Dev Cell 2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Qiao S, Dennis M, Song X, et al. A REDD1/TXNIP pro-oxidant complex regulates ATG4B activity to control stress-induced autophagy and sustain exercise capacity. Nat Commun 2015;6:7014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Cam H, Easton JB, High A, et al. mTORC1 signaling under hypoxic conditions is controlled by ATM-dependent phosphorylation of HIF-1alpha. Mol Cell 2010;40:509–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.DeYoung MP, Horak P, Sofer A, et al. Hypoxia regulates TSC1/2-mTOR signaling and tumor suppression through REDD1-mediated 14-3-3 shuttling. Genes Dev 2008;22:239–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Vega-Rubin-de-Celis S, Abdallah Z, Kinch L, et al. Structural analysis and functional implications of the negative mTORC1 regulator REDD1. Biochemistry 2010;49:2491–501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Burclaff J, Mills JC. Plasticity of differentiated cells in wound repair and tumorigenesis, part II: skin and intestine. Dis Model Mech 2018;11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Huh WJ, Khurana SS, Geahlen JH, et al. Tamoxifen induces rapid, reversible atrophy, and metaplasia in mouse stomach. Gastroenterology 2012;142:21–24 e7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Ran FA, Hsu PD, Wright J, et al. Genome engineering using the CRISPR-Cas9 system. Nat Protoc 2013;8:2281–2308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Correa P, Piazuelo MB. The gastric precancerous cascade. J Dig Dis 2012;13:2–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Saenz JB, Vargas N, Mills JC. Tropism for Spasmolytic Polypeptide-Expressing Metaplasia Allows Helicobacter pylori to Expand Its Intragastric Niche. Gastroenterology 2019;156:160–174 e7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Shimizu T, Choi E, Petersen CP, et al. Characterization of progressive metaplasia in the gastric corpus mucosa of Mongolian gerbils infected with Helicobacter pylori. J Pathol 2016;239:399–410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Petersen CP, Mills JC, Goldenring JR. Murine Models of Gastric Corpus Preneoplasia. Cell Mol Gastroenterol Hepatol 2017;3:11–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Jeong S, Choi E, Petersen CP, et al. Distinct metaplastic and inflammatory phenotypes in autoimmune and adenocarcinoma-associated chronic atrophic gastritis. United European Gastroenterol J 2017;5:37–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Radyk MD, Burclaff J, Willet SG, et al. Metaplastic Cells in the Stomach Arise, Independently of Stem Cells, via Dedifferentiation or Transdifferentiation of Chief Cells. Gastroenterology 2018;154:839–843 e2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Lennerz JK, Kim SH, Oates EL, et al. The transcription factor MIST1 is a novel human gastric chief cell marker whose expression is lost in metaplasia, dysplasia, and carcinoma. Am J Pathol 2010;177:1514–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Foltyn M, Luger AL, Lorenz NI, et al. The physiological mTOR complex 1 inhibitor DDIT4 mediates therapy resistance in glioblastoma. Br J Cancer 2019;120:481–487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Brugarolas J, Lei K, Hurley RL, et al. Regulation of mTOR function in response to hypoxia by REDD1 and the TSC1/TSC2 tumor suppressor complex. Genes Dev 2004;18:2893–904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Huh WJ, Mysorekar IU, Mills JC. Inducible activation of Cre recombinase in adult mice causes gastric epithelial atrophy, metaplasia, and regenerative changes in the absence of “floxed” alleles. Am J Physiol Gastrointest Liver Physiol 2010;299:G368–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Chang HHY, Pannunzio NR, Adachi N, et al. Non-homologous DNA end joining and alternative pathways to double-strand break repair. Nat Rev Mol Cell Biol 2017;18:495–506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Hustedt N, Durocher D. The control of DNA repair by the cell cycle. Nat Cell Biol 2016;19:1–9. [DOI] [PubMed] [Google Scholar]
- 34.Yamamoto M, Furihata C, Ogiu T, et al. Independent variation in susceptibilities of six different mouse strains to induction of pepsinogen-altered pyloric glands and gastric tumor intestinalization by N-methyl-N-nitrosourea. Cancer Lett 2002;179:121–32. [DOI] [PubMed] [Google Scholar]
- 35.Nakamura Y, Sakagami T, Yamamoto N, et al. Helicobacter pylori does not promote N-methyl-N-nitrosourea-induced gastric carcinogenesis in SPF C57BL/6 mice. Jpn J Cancer Res 2002;93:111–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Tsukamoto T, Mizoshita T, Tatematsu M. Animal models of stomach carcinogenesis. Toxicol Pathol 2007;35:636–48. [DOI] [PubMed] [Google Scholar]
- 37.Reiling JH, Hafen E. The hypoxia-induced paralogs Scylla and Charybdis inhibit growth by down-regulating S6K activity upstream of TSC in Drosophila. Genes Dev 2004;18:2879–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Zhao H, Rieger S, Abe K, et al. DNA Damage-Inducible Transcript 4 Is an Innate Surveillant of Hair Follicular Stress in Vitamin D Receptor Knockout Mice and a Regulator of Wound Re-Epithelialization. Int J Mol Sci 2016;17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Basu S. A complex interplay between PGC-1 co-activators and mTORC1 regulates hematopoietic recovery following 5-fluorouracil treatment. Stem Cell Res 2014;12:178–93. [DOI] [PubMed] [Google Scholar]
- 40.Park KK, Liu K, Hu Y, et al. Promoting axon regeneration in the adult CNS by modulation of the PTEN/mTOR pathway. Science 2008;322:963–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Rodgers JT, King KY, Brett JO, et al. mTORC1 controls the adaptive transition of quiescent stem cells from G0 to G(Alert). Nature 2014;510:393–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Lund-Ricard Y, Cormier P, Morales J, et al. mTOR Signaling at the Crossroad between Metazoan Regeneration and Human Diseases. Int J Mol Sci 2020;21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Pinto JA, Rolfo C, Raez LE, et al. In silico evaluation of DNA Damage Inducible Transcript 4 gene (DDIT4) as prognostic biomarker in several malignancies. Sci Rep 2017;7:1526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Janic A, Valente LJ, Wakefield MJ, et al. DNA repair processes are critical mediators of p53-dependent tumor suppression. Nat Med 2018;24:947–953. [DOI] [PubMed] [Google Scholar]
- 45.Cam M, Bid HK, Xiao L, et al. p53/TAp63 and AKT regulate mammalian target of rapamycin complex 1 (mTORC1) signaling through two independent parallel pathways in the presence of DNA damage. J Biol Chem 2014;289:4083–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Xie X, Hu H, Tong X, Li L, et al. The mTOR-S6K pathway links growth signalling to DNA damage response by targeting RNF168. Nat Cell Biol 2018;20:320–331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Horak P, Crawford AR, Vadysirisack DD, et al. Negative feedback control of HIF-1 through REDD1-regulated ROS suppresses tumorigenesis. Proc Natl Acad Sci U S A 2010;107:4675–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Hwang-Verslues WW, Chang PH, Wei PC, et al. miR-495 is upregulated by E12/E47 in breast cancer stem cells, and promotes oncogenesis and hypoxia resistance via downregulation of E-cadherin and REDD1. Oncogene 2011;30:2463–74. [DOI] [PubMed] [Google Scholar]
- 49.Du F, Sun L, Chu Y, et al. DDIT4 promotes gastric cancer proliferation and tumorigenesis through the p53 and MAPK pathways. Cancer Commun (Lond) 2018;38:45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Tirado-Hurtado I, Fajardo W, Pinto JA. DNA Damage Inducible Transcript 4 Gene: The Switch of the Metabolism as Potential Target in Cancer. Front Oncol 2018;8:106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Riera KM, Jang B, Min J, et al. Trop2 is upregulated in the transition to dysplasia in the metaplastic gastric mucosa. J Pathol 2020;251:336–347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Mills JC, Sansom OJ. Reserve stem cells: Differentiated cells reprogram to fuel repair, metaplasia, and neoplasia in the adult gastrointestinal tract. Sci Signal 2015;8:re8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Burclaff J, Willet SG, Saenz JB, et al. Proliferation and Differentiation of Gastric Mucous Neck and Chief Cells During Homeostasis and Injury-induced Metaplasia. Gastroenterology 2020;158:598–609 e5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Pinno J, Bongartz H, Klepsch O, et al. Interleukin-6 influences stress-signalling by reducing the expression of the mTOR-inhibitor REDD1 in a STAT3-dependent manner. Cell Signal 2016;28:907–16. [DOI] [PubMed] [Google Scholar]
- 55.Thompson JW, Nagel J, Hoving S, et al. Quantitative Lys--Gly-Gly (diGly) proteomics coupled with inducible RNAi reveals ubiquitin-mediated proteolysis of DNA damage-inducible transcript 4 (DDIT4) by the E3 ligase HUWE1. J Biol Chem 2014;289:28942–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Meyer AR, Engevik AC, Willet SG, et al. Cystine/Glutamate Antiporter (xCT) Is Required for Chief Cell Plasticity After Gastric Injury. Cell Mol Gastroenterol Hepatol 2019;8:379–405. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.