

Abstract
A dysfunctional immune response in coronavirus disease 2019 (COVID-19) patients is a recurrent theme impacting symptoms and mortality, yet a detailed understanding of pertinent immune cells is not complete. We applied single-cell RNA sequencing to 284 samples from 196 COVID-19 patients and controls and created a comprehensive immune landscape with 1.46 million cells. The large dataset enabled us to identify that different peripheral immune subtype changes are associated with distinct clinical features, including age, sex, severity, and disease stages of COVID-19. Severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) RNA was found in diverse epithelial and immune cell types, accompanied by dramatic transcriptomic changes within virus-positive cells. Systemic upregulation of S100A8/A9, mainly by megakaryocytes and monocytes in the peripheral blood, may contribute to the cytokine storms frequently observed in severe patients. Our data provide a rich resource for understanding the pathogenesis of and developing effective therapeutic strategies for COVID-19.
Keywords: COVID-19, SARS-CoV-2, single-cell RNA-seq, single-cell transcriptomics, host cell range, cell-cell interaction, B cell receptor sequencing, T cell receptor sequencing, cytokine storm, ligand-receptor interaction
Graphical Abstract
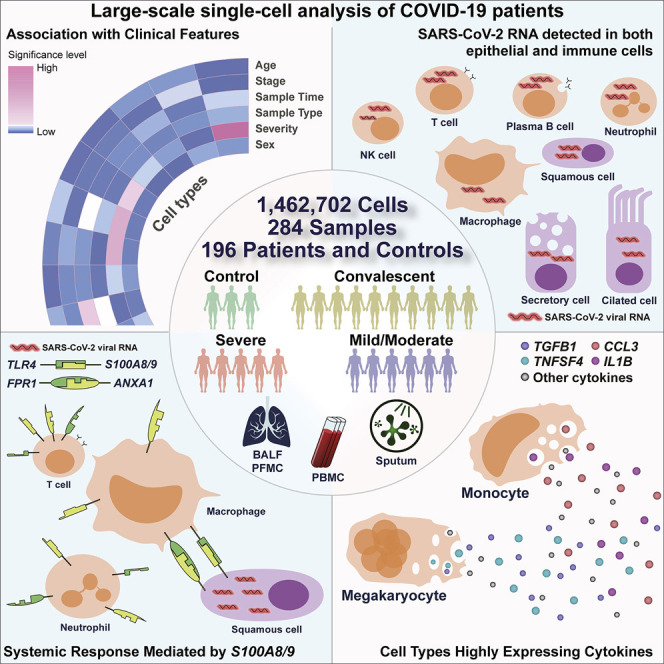
Analysis of the immune landscape in the lung and peripheral blood of COVID patients across different regions in China at the single-cell level documents the presence of viral RNAs in diverse cell types and highlights the potential contribution of megakaryocytes and monocyte subsets to cytokine storms.
Introduction
Coronavirus disease 2019 (COVID-19), caused by severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2), has caused more than 98 million infections and more than 2.1 million deaths according to the statistics of World Health Organization (WHO) as of January 24, 2021. Although many COVID-19 patients are asymptomatic or experience only mild or moderate symptoms, some patients progress to severe disease or even death. It is thus important to understand the disease mechanisms to control the pandemic. Multiple studies have suggested the alterations of immune responses as one of the key mechanisms for severe symptoms (Guo et al., 2020; Schulte-Schrepping et al., 2020; Silvin et al., 2020; Wen et al., 2020; Zhang et al., 2020a, 2020b). While recent studies have offered deeper insights (Blanco-Melo et al., 2020; Mathew et al., 2020; Su et al., 2020), a detailed immune landscape of COVID-19 patients in both lung and peripheral blood is still needed to dissect the potential changes associated with disease severity and illustrate the potential sources of the inflammatory storm in COVID-19.
Single-cell RNA sequencing (scRNA-seq) is powerful at dissecting the immune responses and has been applied to COVID-19 studies (Cao et al., 2020; Chua et al., 2020; Fan et al., 2020; Su et al., 2020; Wen et al., 2020; Xie et al., 2020; Zhang et al., 2020a, 2020b). While the current single-cell studies of COVID-19 have provided important cellular and molecular insights, such studies are often limited by the cohort size and thus the levels of robustness. Here, we obtained scRNA-seq data for a cohort of 196 individuals, including hospitalized COVID-19 patients with moderate or severe disease, and patients in the convalescent stage, as well as healthy controls. We reveal that SARS-CoV-2 RNA could be detected in a wide range of cell types, accompanied by distinct transcriptomic changes between SARS-CoV-2-RNA-positive and negative cells. We also observed critical changes to COVID-19 clinical features. Further, our data provide a resource to reveal the characteristics of cytokine storms in patients. Our data and findings may have important implications for understanding and controlling COVID-19.
Results
Integrated analysis of COVID-19 scRNA-seq data
To characterize the immune properties of COVID-19, we formed a Single Cell Consortium for COVID-19 in China (SC4), which consisted of researchers from 39 institutes or hospitals from different regions of China. SC4 generated a scRNA-seq dataset for 171 COVID-19 patients, including 22 patients with mild or moderate symptoms, 54 hospitalized patients with severe symptoms, and 95 recovered convalescent persons (57 with mild or moderate symptoms and 38 with severe symptoms), as well as 25 healthy controls according to the WHO classification (Figure 1 A; Table S1). 186 out of 284 samples were unpublished. Patients with mild or moderate symptoms were merged as one group but were further divided into progression (moderate) or convalescence (moderate) based on the time of sample collection (Figure 1A; Table S1). Similarly, we merged patients with severe symptoms or in the critical stage into one group (Figure 1A; Table S1) and focused on analyzing the molecular and cellular mechanisms underlying distinctions between mild/moderate and severe/critical symptoms. This cohort covered an age range from 6 to 92 years (Figure S1 A), in which aged patients were enriched in the severe groups, consistent with a previous report (Huang et al., 2020). Additionally, no significant difference was noted in the sex composition between the moderate and severe groups (Figure S1B).
Figure 1.

Multi-tissue and multi-stage single-cell atlas of COVID-19 patients and healthy controls
(A) Flowchart depicting the overall experimental design of this study. Cells circled with dash lines were enriched in samples from the disease progression stage.
(B) Overview of the cell clusters in the integrated single-cell transcriptomes of 1,462,702 cells derived from COVID-19 patients and healthy controls. Clusters were named based on the cluster-specific gene expression patterns, in which we used “high” or “low” labels to indicate the relative expression levels in the corresponding clusters. Genes without high or low labels were specifically expressed in the corresponding clusters.
(C) Tissue preference of each cluster measured by the ratio of observed to randomly expected cell numbers (RO/E) (Zhang et al., 2018).
(D) Patient group preference of each cluster measured by RO/E.
Figure S1.

Basic characteristics of the integrated dataset and selected markers of cell subsets in different major cell lineages, related to Figure 1
(A) The age distribution of the dataset (color-coded by disease conditions). (B) Distribution of sex. Chi-square test. (C-E) Distribution of unique molecular identifier (UMI) counts per cell (C), gene counts per cell (D), and percentage of mitochondrial transcripts per cell (E) detected for cells in various tissue types. PBMC, peripheral blood mononuclear cells; BALF, bronchoalveolar lavage fluid; PFMC/Sputum, pleural effusion/sputum. (F-J) Violin plots of selected marker genes (rows) for cell subsets (columns) within each cell lineage, including 6 B/plasma B cell clusters (F), 23 Myeloid cell clusters (G), 3 NK cell clusters (H), 4 Epithelial cell clusters (I) and 28 T cell clusters (J).
Among the 284 samples, 249 were from peripheral blood mononuclear cells (PBMCs) with or without further sorting for B or T cells, and 35 were from the respiratory system, including 12 bronchoalveolar lavage fluid (BALF) samples, 22 sputum samples, and 1 sample for pleural fluid mononuclear cells (PFMCs) (Table S1). Seven patients had matching BALF and PBMC samples collected. Most samples were subjected to scRNA-seq based on the 10x Genomics 5′ sequencing platform to generate both the gene expression and T cell receptor (TCR) or B cell receptor (BCR) data (Table S1). Gene expression data were obtained by the kallisto and bustools programs (Bray et al., 2016; Melsted et al., 2019), and TCR and BCR sequences were obtained by the CellRanger program.
We applied stringent quality-control criteria to ensure that the selected data were from single and live cells (Table S1). A total of 1,462,702 high-quality single cells were ultimately obtained, with an average of 4,835 unique molecular identifiers (UMIs), representing 1,587 genes (Figures S1C–S1E). 64 cell clusters were derived, covering diverse cell types in the respiratory system and peripheral blood (Figure 1B). Such an information-rich resource (available at http://covid19.cancer-pku.cn/ for quick browsing) enabled accurate annotation and analysis of these cell clusters at different resolutions (Figures S1F–S1J; Table S2).
Notable differences could be observed based on the t-distributed stochastic neighbor embedding (t-SNE) projection (Figure 1A). The tissue preference of each cluster was illustrated based on RO/E (Figure 1C), i.e., the ratio of observed to randomly expected cell numbers used for removing the technical variations on tissue preference estimation (Zhang et al., 2018). Notably, various clusters of proliferating CD8+ and CD4+ T and plasma B cells were more enriched in BALF than PBMCs (Figure 1C). Similarly, the preference of each cluster in different patient groups was also illustrated (Figure 1D), with proliferative and activated B and T cells and macrophages more enriched in severe COVID-19 patients in the disease progression stage.
Association of patient age, sex, COVID-19 severity, and stage with PBMC compositions
We first analyzed the compositional changes of the broad categories of immune cells in PBMC. Notably, the percentages of megakaryocytes and CD14+ monocytes in PBMCs were elevated, particularly in severe COVID-19 patients during the disease progression stage (Figure S2 A). While natural killer (NK) cells did not show significant changes among the different groups, B cells were significantly increased in severe COVID-19 patients, but T cells and DCs were decreased (Figure S2A), consistent with the lymphopenia phenomenon previously reported (Chen and John Wherry, 2020).
Figure S2.

Comparison of different immune cell types among patient groups, related to Figure 2
(A) Comparison on the major cell type level based on 159 unsorted PBMC samples with at least one thousand cells available in the scRNA-seq data. NK, natural killer cells; Mono, monocytes; DC, dendritic cells; Mega, megakaryocytes. (B) State transition between B_c05-MZB1-XBP1 and other B cell sub clusters quantified by the STARTRAC algorithm based on TCR clonotypes (Zhang et al., 2018). Clonotypes with more than 5 cells were shown in the right panel. (C) Percentage of B_c06-MKI67 in PBMC across disease conditions based on the same cohort with (A). (D) RNA velocity analysis shows the transition potential from B_c03-CD27-AIM2 to B_c05-MZB1-XBP1. Cell pairs transiting from B_c03-CD27-AIM2 to B_c05-MZB1-XBP1 or vice versa were quantified in the bar plot. (E) Percentage of B_c03-CD27-AIM2 in PBMC across disease conditions based on the same cohort with (A). (F) State transition quantified by STARTRAC (Zhang et al., 2018) between T_CD4_c13-MKI67-CCL5low proliferating cells and other CD4 cell sub-clusters (left) and clones containing T_CD4_c13-MKI67-CCL5low cells with more than 5 cells (right). (G) Percentage of T_CD4_c04−ANXA2 across disease conditions based on the same cohort with (A). (H) Sex differences of T_CD4_c04-ANXA2. Single-side Wilcoxon test. (I and J) Percentage of T_gdT_c14-TRDV2 and T_CD8_c09-SLC4A10 across disease conditions based on the same cohort with (A). Adjusted P-values smaller than 0.05 are indicated (two-sided unpaired Wilcoxon test).
The large cohort size enabled us to dissect the associations of age, sex, disease severity, and stage with the compositional changes of immune cells in PBMCs. We applied analysis of variance (ANOVA) to interrogate such associations based on 159 PBMC samples each with >1,000 single cells available. We also incorporated two technical factors in the ANOVA model for controlling technical variations, i.e, sample type (fresh or frozen PBMCs) and sample time (days after symptom onset). After multiple testing correction, significant associations were found (Figure 2 A). Notably, while most B cell clusters were associated with disease recovery status, XBP1 + plasma cells (B_c05-MZB1-XBP1) showed an association with COVID-19 severity (Figure 2A). XBP1, POU2AF1, PRDM1, and IRF4 were highly expressed in B_c05-MZB1-XBP1 (Table S2), confirming this cluster as plasma cells (Todd et al., 2009). The percentage of plasma cells in PBMCs could reach 15% in severe COVID-19 patients, but none of the other individuals could reach 3% (Figure 2B). This increase was observed irrespective of sample type (fresh or frozen; Figure 2B), indicating the robustness of this observation. Similarly, this increase was also irrespective of sampling time (Figures 2A and S3A). These plasma B cells in PBMCs highly expressed the genes encoding the constant regions of immunoglobulin A1 (IgA1), IgA2, IgG1, or IgG2 (Figure 2C), implying their function in the secretion of antigen-specific antibodies. This observation is consistent with the previous finding that serum of severe COVID-19 patients had high titers of SARS-CoV-2-specific antibodies (Ni et al., 2020).
Figure 2.

Associations of patient age, sex, COVID-19 severity, and stage with cellular compositions in PBMCs
(A) Heatmap for q values of ANOVA. Sample type, fresh or frozen; sample time, days after symptom onset.
(B) Composition comparison for plasma B cells (B_c05-MZB1-XBP1) based on 159 unsorted PBMC samples with at least 1,000 cells available in the scRNA-seq data.
(C) Classes of heavy chains for B_c05-MZB1-XBP1.
(D–G) Composition comparison for DC_c4−LILRA4, Neu_c3−CST7, T_CD4_c13-MKI67-CCL5 low, and T_CD8_c10-MKI67-GZMK.
(H) Associations between age and T_CD8_c01−LEF1 (Spearman’s correlation).
(I) Sex differences of T_CD4_c08−GZMK−FOShigh. Adjusted p values < 0.05 are indicated (two-sided unpaired Wilcoxon tests).
Figure S3.

Effects of sampling time and sample processing methods (fresh or frozen) on immune cell composition and the BCR/TCR diversity, related to Figures 2 and 3
(A) Gross relationship between B_c05−MZB1−XBP1 frequency in PBMC and sampling days. ANOVA rejected the association between B_c05−MZB1−XBP1 frequency and sampling days after incorporating age, sex, COVID-19 severity and stage (Figure 2A). (B) Gross relationship between DC_c4−LILRA4 frequency in PBMC and sampling days. (C) Gross relationship between Neu_c3−CST7 frequency in PBMC and sampling days. (D-G) Comparison among patient groups for T_CD4_c02−AQP3, T_CD8_c01−LEF1, T_CD8_c02−GPR183, and T_CD4_c08−GZMK−FOShigh via separating fresh and frozen PBMC samples. (H-K) Gross relationship of sampling time with frequencies of T_CD4_c02−AQP3, T_CD4_c08−GZMK−FOShigh, T_CD8_c01−LEF1, and T_CD8_c02−GPR183.
The increase of plasma B cells in PBMCs appeared to be derived from active proliferation and transitions from memory B cells based on BCR analysis (Figure S2B). Plasmablast cells (B_c06_MKI67), characterized by high expression of MKI67 and thus indicating a proliferative state, were elevated in the peripheral blood of severe COVID-19 patients and shared the most clonotypes with plasma cells (Figures S2B and S2C). The memory B cell cluster (B_c03-CD27-AIM2), expressing relatively high levels of CD27, CD80, AIM2, GRIP2, and COCH, was the second major source of plasma B cells and shared a large proportion of clonotypes with plasma cells and plasmablasts (Figure S2B). Transition from B_c03-CD27-AIM2 to B_c05-MZB1-XBP1 was supported by BCRs (Figure S2B) and RNA velocity analysis (Figure S2D). Similarly, the plasmacytoid dendritic cell cluster DC_c4−LILRA4 was decreased in severe COVID-19 patients in both the progression and convalescence stages, irrespective of sample processing methods or sampling time (Figures 2A, 2D, and S3 B). Neu_c3−CST7, the largest neutrophil cluster in PBMCs, was associated with patient age, COVID-19 severity, and stage after correcting technical covariates (Figures 2A, 2E, and S3C).
For T cells, diverse proliferative T cell subsets, marked by high levels of MKI67, exhibited distinct associations with COVID-19 severity and stage (Figure 2A). Two proliferative CD4+ T cell clusters were identified, with T_CD4_c13-MKI67-CCL5low characterized by high SELL and low CCL5 and T_CD4_c14-MKI67-CCL5high characterized by low SELL and high CCL5, among others (Table S2). T_CD4_c13-MKI67-CCL5low cells were elevated in COVID-19 patients, particularly in those with severe disease during the disease progression stage (Figure 2F). Three proliferative CD8+ T cell clusters were identified, including T_CD8_c10-MKI67-GZMK, T_CD8_c11-MKI67-FOS, and T_CD8_c12-MKI67-TYROBP. They were increased in COVID-19 patients but showed different associations with COVID-19 severity (Figure 2A). T_CD8_c10-MKI67-GZMK, a proliferative effector memory CD8+ T cell group characterized by high STMN1, HMGB2, MKI67, and GZMK, was increased in severe COVID-19 patients, particularly in the convalescence stage (Figure 2G). The variations of proliferative CD8+ T cell clusters in different severity and stages may indicate the complexity of T cell responses induced by SARS-CoV-2 infection.
Unlike the B cell cluster B_c03-CD27-AIM2, which increased in peripheral blood (Figure S2E), T_CD4_c04−ANXA2, a major source of proliferative CD4+ T cells, was decreased in COVID-19 patients, particularly in those with severe disease during the disease progression stage (Figures S2F and S2G). This increase in proliferative CD4 T cells and decrease in their precursor cells in severe COVID-19 patients may partially explain the dichotomous and incomplete adaptive immunity previously observed (Gao et al., 2020). Interestingly, naive CD8+ T cells (T_CD8_c01−LEF1) exhibited the most significant association with patient age among T cells (Figures 2A and 2H), providing a plausible explanation for the epidemiological observation of age biases. Furthermore, sex-associated T cell subsets were also observed, including T_CD4_c04−ANXA2, T_CD4_c08−GZMK−FOShigh, and T_CD8_c02−GPR183 (Figures S2H, 2A, and 2I).
In contrast to proliferative T cells that were elevated in PBMCs, most T cell clusters decreased in COVID-19 patients and varied in terms of association with disease severity. The significantly decreased T cell clusters included γδT cells (T_c14_gdT-TRDV2) and mucosal-associated invariant T (MAIT) cells (T_CD8_c09-SLC4A10); a CD8+ T cell cluster highly expressing TYROBP, KLRF1, CD247, and IL2RB (T_CD8_c08-IL2RB); and three CD4+ T cell clusters showing effector memory characteristics (Figure 2A). Accordingly, decreases of γδT cells and MAIT cells in the peripheral blood of severe COVID-19 patients (Figures S2I and S2J) have been supported by flow-cytometry-based analyses (Jouan et al., 2020).
Association of patient age, sex, COVID-19 severity, and stage with the diversity of B and T cell repertoires
Our scRNA-seq data also contained TCR and BCR sequence data and thus provided a rich resource to investigate the TCR/BCR usage of COVID-19 patients. ANOVA revealed that the diversity of B and T cell subsets in PBMC had heterogeneous associations with the various clinical features (Figure 3 A; Table S3). A diverse set of B and T cell subsets showed associations with COVID-19 severity (Figure 3A). As exemplified by T_CD4_c02−AQP3 (Figure 3B), T_CD4_c08−GZMK−FOShigh (Figure 3C), T_CD8_c01−LEF1 (Figure 3D), and T_CD8_c02−GPR183 (Figure 3E), the TCR diversity of these T cell subsets tended to be smaller in severe COVID-19 patients than those with moderate disease, particularly in the disease progression stage. Among these clusters, certain T cell subsets also showed sex- and age-associated variations (Figure 3A), as exemplified by T_CD4_c08−GZMK−FOShigh (Figure 3F), T_CD8_c01−LEF1 (Figure 3G), T_CD8_c05−ZNF683 (Figure 3H), and T_CD8_c09−SLC4A10 (Figure 3I). In general, the diversity of these T cell subsets was higher in females than in males and in younger than older individuals. While the diversity of certain B and T cell subsets was affected by technical variations, including sample processing methods and sampling time, ANOVA dissected the effects of clinical features from such technical considerations and identified associations robust to technical interference (Figures 3A and S3D–S3K).
Figure 3.

Associations of patient age, sex, COVID-19 severity, and stage with the diversity of B and T cell repertoires in PBMCs
(A) Heatmap for q values of ANOVA. Sample type, fresh or frozen; sample time, days after symptom onset.
(B–E) Comparison for T_CD4_c02−AQP3, T_CD4_c08−GZMK−FOShigh, T_CD8_c01−LEF1, and T_CD8_c02−GPR183.
(F) Sex differences for T_CD4_c08−GZMK−FOShigh.
(G–I) Age associations of the TCR diversity of T_CD8_c01−LEF1, T_CD8_c05-ZNF683, and T_CD8_c09-SLC4A10 (Spearman’s correlation).
(J) V gene usage of published SARS-CoV-2 neutralized antibodies and their relationship with those differentially used IGHV genes in our dataset. Gini-index was used to quantify the skewness of the V gene usage of the published SARS-CoV-2 neutralized antibodies. IGHV genes differentially used by moderate or severe COVID-19 patients compared with healthy controls and their intersections are shown with different colors. Venn diagram is used to show their overlaps with those published SARS-CoV-2 antibodies. Adjusted p values < 0.05 are indicated (two-sided unpaired Wilcoxon test).
We further examined whether identical TCRs or BCRs could be identified across COVID-19 patients but found very limited sharing (four BCR clonotypes in two patients). Only one non-clonal BCR had an identical CDR3 amino acid sequence in its heavy chain with a comprehensive compendium containing 1,505 SARS-CoV-2-specific antibodies (Raybould et al., 2020). Such scarcity of common BCRs was in contrast with previous studies on patients with severe disease who had recovered from enterovirus A71 infection and influenza vaccination (Chen et al., 2017; Jiang et al., 2013), suggesting that SARS-CoV-2 infection might not impose dramatic selective pressure on the somatic evolution of BCRs.
Although no identical BCRs were found, we noticed that the BCR repertoire of COVID-19 patients exhibited biased VDJ usage compared with that of healthy controls. We trained a random forest classifier with the VDJ usage frequencies to discriminate COVID-19 patients with moderate or severe symptoms from healthy controls and found that the classification accuracy measured by the values of area under curve was as high as 0.85. Among the top 20 VDJ combinations important to discriminate severe COVID-19 patients from healthy controls selected by random forests, 14 had identical VDJ usage with experimentally verified neutralizing antibodies (Figure 3J). Of note, the VDJ usage of the currently known SARS-CoV-2-neutralizing antibodies was biased toward IGHV3 and IGHV1 (Figure 3J). In particular, more than 40 neutralizing antibodies used IGHV3−53. Such observations and the data may be important for identifying new neutralizing antibodies.
SARS-CoV-2 RNAs detected in multiple epithelial and immune cell types
From the six BALF and two sputum samples of severe COVID-19 patients in the disease progression stage, we detected viral RNAs of SARS-CoV-2 in 3,085 cells from ciliated, secretory, and squamous epithelial cells and a diverse set of immune cells, including neutrophils, macrophages, plasma B cells, T cells, and NK cells (Figure 4 A; Table S4). Fewer cells were obtained in BALF from moderate COVID-19 patients, and no SARS-CoV-2 RNA was detected. The identity of these SARS-CoV-2-RNA-positive cells was confirmed by the corresponding marker genes (Figure 4B). Interestingly, immune cells harbored even more viral RNA sequences than epithelial cells (Figure 4C). Because ACE2 and TMPRSS2 play critical roles in mediating SARS-CoV-2 entry (Hoffmann et al., 2020; Zhou et al., 2020), we examined their expression levels in these cells (Figure 4D). We found that ACE2 and TMPRSS2 were expressed in a subset of these epithelial cells. However, immune cells did not express ACE2 or TMPRSS2. We then examined host factors recently reported to be relevant to SARS-CoV-2 entry in our data (Cantuti-Castelvetri et al., 2020; Daly et al., 2020; Singh et al., 2020; Tang et al., 2020) and found that BSG and TFRC demonstrated correlations with the abundance of viral RNA in different cell types (Figures 4E and S4 A). Consistently, independent scRNA-seq studies of COVID-19 patients also identified SARS-CoV-2 RNAs in neutrophils and macrophages (Bost et al., 2020; Chua et al., 2020). In BALF or sputum samples from patients with moderate symptoms, we did not detect SARS-CoV-2 RNA based on scRNA-seq.
Figure 4.

Cell types with SARS-Cov-2 RNA detected
(A) 3,085 cells with SARS-CoV-2 RNA detected (UMI > 0) from BALF (6/12) and sputum (2/22) samples. No cells from PBMCs or PFMCs were detected as SARS-CoV-2 positive.
(B) Markers used to determine cell types. Goblet and basal cells were merged as secretory epithelial cells for convenience in the subsequent analyses.
(C) Viral load in each cell quantified by log(CPM).
(D) Expression levels of host factors reported to associate with SARS-CoV-2 infection in literature.
(E) Pearson’s correlations of host factor expression with viral load (zero-expression cells were excluded from regression analysis to reduce the effects of dropouts).
(F) Expression levels of ISGs in cells with viral RNA detected.
(G) Detection rates of SARS-CoV-2 genes in different cell types on both 10x Genomics 5′ and 3′ platforms. Given a viral gene gv, the detection rate is defined as the ratio of the number of gv+ cells to the total viral-RNA-positive cells of the specific cell type and then normalized by the gene length in the SARS-CoV-2 genome.
(H) IHC staining of CD3 and SARS-CoV-2 spike protein in pulmonary tissue. Scale bar, 100 μM.
Figure S4.

Characteristics of SARS-CoV-2-RNA-positive epithelial and immune cells, related to Figure 4
(A) Associations of BSG with viral RNA load in neutrophils, plasma cells, T/NK cells, and ciliated epithelial cells (Person’s correlation). Grey points (no expression or dropouts) were excluded from the regression analysis to reduce the impacts of dropouts in scRNA-seq. (B) Violin plots showing the expression of ISGs in viral RNA-positive cells (from BALF) compared with viral RNA-negative cells from PBMC and BALF. Two-sided unpaired Wilcoxon test was used. (C) Pearson’s correlation between viral RNA load and the expression levels of ISGs. Grey points (no expression or dropouts) were excluded from the regression analysis to exclude the impacts of dropouts in scRNA-seq. (G) Detection rates of SARS-CoV-2 genes in different cell types on both 10x Genomics 5′ and 3′ platforms. (H) IHC staining of SARS-CoV-2 spike protein in lymphocytes in pulmonary tissue.
Since interferon-stimulated genes (ISGs) are typically associated with viral RNA sensing (Schoggins and Rice, 2011), we next examined the expression of ISGs in these cells (Figures 4F and S4B; Table S4). Compared with matched cell types in PBMCs, which were negative for SARS-CoV-2 RNA, ISG genes exhibited elevated expression in these viral-RNA-positive immune cells (Figure S4B; Table S5). Compared with viral-RNA-negative immune cells of the same types in the BALF, SARS-CoV-2-RNA-positive epithelial cells, including ciliated, secretory, and squamous cells, as well as virus-RNA-positive neutrophils, exhibited higher levels of ISG expression (Table S5). Positive correlations between the abundance of viral RNAs and ISG expression levels were observed for most cell types after removing potential dropouts in scRNA-seq (Figure S4C).
We then examined the detection rates of different SARS-CoV-2 genes in these cells (Figure 4G). In our cohort, SARS-CoV-2-RNA-positive immune cells were detected in different research centers based on either 10x Genomics 5′ or 3′ sequencing platforms. Since coronaviruses are characterized by subgenomic transcription and the genome of SARS-CoV-2 is a single and positive RNA strand, genes close to the 3′ end of the genome were expected to have higher detection rates than those close to the 5′ end if subgenomic transcription occurred. In fact, both 10x 5′ and 3′ sequencing data demonstrated a 3′-enriched detection pattern along the SARS-CoV-2 genome in viral-RNA-positive cells, reminiscent of subgenomic transcription, in contrast to those cells without detectable SARS-CoV-2 RNA (Figures 4G and S4D). Since both 5′ and 3′ sequencing platforms detected consistent patterns along the genome, the positive and negative strands of the SARS-CoV-2 genome (at least the subgenomes) likely both exist in these cell types, implicating active viral replication and transcription. We further validated the presence of a viral signal in lymphocytes with immunohistochemical (IHC) staining of the viral spike protein in lung tissue from a severe COVID-19 patient (Figures 4H and S4E).
Although type II alveolar (AT2) cells are vulnerable to SARS-CoV-2 infection (Hou et al., 2020), our study revealed few AT2 cells in the BALF and no detectable SARS-CoV-2 RNA in AT2 cells, consistent with the notion that lower respiratory tract cells are less likely to be infected with SARS-CoV-2 than those from the nasal and upper respiratory tract (Hou et al., 2020; Sungnak et al., 2020). The detection of SARS-CoV-2 RNA in epithelial and immune cells was independent of sample processing methods (all fresh samples) or sampling days (8–18 days after symptom onset) in our dataset.
Transcriptomic differences between SARS-CoV-2-RNA-positive and negative epithelial cells and the potential impact on cell-cell interactions
The presence of SARS-CoV-2 RNA in different epithelial cells seemed to be associated with additional transcriptomic changes. For squamous epithelial cells, SARS-CoV-2-RNA-positive cells exhibited elevated expression of a diverse set of genes, such as NT5E, CLCA4, and SULT2B1 (Figure 5 A). These genes were enriched in pathways such as “response to virus,” “response to type I interferon,” and “response to hypoxia” (Figure 5B). By contrast, the number of differential genes between SARS-CoV-2-RNA-positive and negative ciliated epithelial cells was much smaller, and few genes showed consistent changes in all the three epithelial cell types (Figure 5C).
Figure 5.

Impact of viral RNA presence on the expression and cell-cell interaction of epithelial subtypes
(A) Volcano plot showing differentially expressed genes between squamous cells with or without viral RNA detected. Adjusted p value < 0.05, two-sided unpaired Wilcoxon test. ANXA1 is denoted in dark blue.
(B) Enriched Gene Ontology (GO) terms in genes highly expressed in virus-positive squamous cells shown in (A).
(C) Venn plot showing the intersection of genes upregulated in different epithelial cells with viral detection.
(D) Cell-cell interaction networks of one severe COVID-19 patient (left) and one moderate COVID-19 patient (right) inferred by CSOmap-based on data from BALFs. Interactions with q values < 0.1 are shown. Significance: −log10(q values).
(E) Boxplots showing the self-distances among ciliated, secretory, and squamous cells with or without viral RNA detection in the pseudo-spaces predicted by CSOmap. Each dot represents an individual patient. Two-sided paired Wilcoxon test.
(F) Violin plot showing the self-distances of three types of epithelial cells with viral detection (exemplified by one patient). Two-sided unpaired Wilcoxon test.
(G) Boxplot showing the median self-distances of three type of epithelial cells with viral detection from all the patients with BALF samples. Each dot represents an individual patient. Two-sided unpaired Wilcoxon test.
(H) Pie charts showing the ligand-receptor contribution to the interaction between virus-positive squamous cells and virus-positive neutrophils (top) and virus-positive squamous cells and virus-positive macrophages (bottom).
(I) Boxplot showing the interactions between squamous cells (with and without viral detection) and macrophage (left) and neutrophils (left) with viral detection. Each dot represents an individual patient. Two-sided unpaired Wilcoxon test. Normalized connections: observed cell-cell interactions normalized by random expectation (nA × nB, where n is the cell number of type A or B).
See also Figures S5 and S6.
We next explored the potential impact of the above transcriptomic changes. Annexin A1 (ANXA1), upregulated in SARS-CoV-2 RNA-positive squamous epithelial cells (Figure S5 A), is known to regulate the functions of neutrophils in inflammation via its interactions with formyl peptide receptors (Sugimoto et al., 2016). This prompted us to investigate the potential cellular interaction differences between SARS-CoV-2-RNA-positive and negative cells. Based on CSOmap, a bioinformatics tool used to estimate cell-cell interactions in three-dimensional space via ligand-receptor-mediated cell self-organization and competition (Ren et al., 2020), we estimated the cellular interaction potentials in a computationally constructed pseudo-space. We found that SARS-CoV-2-RNA-positive ciliated, secretory, and squamous epithelial cells exhibited distinct interaction potentials between the severe and moderate groups (Figure 5D).
Figure S5.

Differences of various epithelial cells with viral RNA detected in the interaction potential with other cells, related to Figure 5
(A) Differential expression of ANXA1 in SARS-CoV-2 RNA-positive and negative squamous epithelial cells. (B) 2D visual view of the pseudo-space constructed by CSOmap with the location of ciliated cells highlighted. Each dot denotes a single cell and is colored by its cell type. (C and D) Self-distance of viral RNA-positive and negative ciliated and squamous cell groups in the pseudo-space shown in (B). Two-sided unpaired Wilcoxon test. (E) Comparison of interacting potentials of viral RNA-positive secretory epithelial cells with BALF Macro_c1-C1QC cells between moderate and severe patients. Spatial connections within the pseudo-space constructed by CSOmap were used for quantification, which were normalized by the cell numbers of both clusters. Error bar: s.e.m across different patients. (F) The ligand-receptor contribution between viral RNA-positive secretory epithelial cells and Macro_c6-VCAN cells. (G) Dot plot showing the expression level of MARCO in BALF samples. Pct, percentage of expressed cells.
SARS-CoV-2-RNA-positive ciliated epithelial cells exhibited lower interaction potentials with themselves and dispersed in the outer compartment of the pseudo-space topologically equivalent to the airway tract (Figures 5E, S5B, and S5C), similar to the pathological phenomenon of epithelial denudation of coronavirus infection in respiratory tract (Lee et al., 2003; Nicholls et al., 2003). By contrast, SARS-CoV-2 RNA-positive squamous epithelial cells showed enhanced interacting potentials with themselves compared with squamous cells without viral RNA detected (Figure S5D). Such changes were consistent across COVID-19 patients (Figure 5E). Comparison across ciliated, secretory, and squamous epithelial cells also highlighted the dispersing tendency of ciliated cells and the interacting potential among squamous cells themselves (Figures 5F and 5G).
Such distinctions existed not only in interactions among epithelial cells but also in interactions with immune cells. Consistent with the dispersing nature of ciliated cells in the outer compartment of the pseudo-space, no significant interactions were observed between viral-RNA-positive ciliated cells and immune cells. Viral-RNA-positive secretory epithelial cells showed interactions with neutrophils and macrophages in moderate COVID-19 patients via the SCGB3A1-MARCO axis (Figures S5E and S5F), but such interactions might be subdued in severe COVID-19 patients due to the downregulation of MARCO (Figure S5G). In severe patients, viral-RNA-positive squamous cells showed significant interactions with neutrophils and macrophages via the ANXA1-FPR1 and S100A9/A8-TLR4 axes (Figure 5H). Neutrophils and macrophages exhibiting high interacting potentials with viral-RNA-positive squamous epithelial cells were also prone to be SARS-CoV-2 RNA positive (Figure 5I). As ANXA1-FPR1 and S100A9/A8-TLR4 interactions have been reported to play critical roles in the recruitment of immune cells and inflammatory cascade in sepsis and tumors (Gavins et al., 2012; Laouedj et al., 2017; Osei-Owusu et al., 2019; Vogl et al., 2007), we hypothesize that they might also play important roles in the pathogenesis of COVID-19.
ANXA1, FPR1, S100A9, S100A8, and TLR4 also exhibited systemic changes in a wide range of immune cells between moderate and severe COVID-19 patients (Figures S6 A and S6B). Interestingly, for most immune cell clusters in BALF, the expression levels of ANXA1 and FPR1 were downregulated in severe COVID-19 patients compared with moderate COVID-19 patients (Figure S6B). But in PBMCs, except for MAIT cells (T_CD8_c09-SLC4A10) and γδT cells (T_gdT_c14-TRDV2), ANXA1 and FPR1 were significantly upregulated in multiple cell types in severe COVID-19 patients (Figure S6A). For severe COVID-19 patients in the disease progression stage, S100A9 and S100A8 were significantly upregulated in almost all cell clusters for both BALF and PBMCs (Figures S6A and S6B). In particular, the levels of S100A9 and S100A8 were significantly upregulated in T, B, NK, and dendritic cells compared to moderate patients (Figures S6A and S6B), indicating a systemic inflammatory response. TLR4 did not exhibit notable differences in PBMCs between severe and moderate COVID-19 patients (Figure S6A) but was significantly downregulated in certain BALF monocyte and macrophage subsets (Figure S6B).
Figure S6.

The expression of selected genes in PBMC and BALF samples, related to Figure 5
(A) Dot plots showing the expression of S100A9 in cell clusters found in PBMCs. Each dot is colored by the mean expression and sized by the scaled mean (Z scores). The blue box highlights the expressions in patients belonging to the progression (severe) group. (B) Dot plots showing the expression of ANXA1 (top), FPR1 (middle) and TLR4 (bottom) in clusters found in PBMCs. (B) Dot plots showing the expression of ANXA1 (first panel), FPR1 (second panel), S100A9 (third panel), S100A8 (fourth panel) and TLR4 (bottom panel) in clusters found in BALFs. Each dot is colored by the means of the expression and sized by the scaled means (Z scores).
Megakaryocytes and monocyte subsets as critical peripheral sources of cytokine storms
We next sought to investigate the potential sources of cytokine production. We first defined a cytokine score and inflammatory score for each cell based on the expression of the collected cytokine genes and reported inflammatory response genes (Liberzon et al., 2015) (Table S6), respectively, and used these two interrelated scores as indicators to evaluate the potential contribution to inflammatory cytokine storm for each cell. We found apparent elevated expression of cytokine and inflammatory genes in patients, especially at the severe progression stage (Figures 6 A and S7 A), indicating the existence of inflammatory cytokine storm. Seven cell subtypes, including three subtypes of monocytes (Mono_c1-CD14-CCL3, Mono_c2-CD14-HLA-DPB1, and Mono_c3-CD14-VCAN), three subtypes of T cells (T_CD4_c08-GZMK-FOShigh, T_CD8_c06-TNF, and T_CD8_c09-SLC4A10), and one subtype of megakaryocytes, were detected with significantly higher cytokine and inflammatory scores based on our scRNA-seq data for PBMC samples (Figure S7B; Table S7; p < 0.0001), indicating that these cells might be major sources of the inflammatory storm. Interestingly, megakaryocytes, which have been connected to the inflammatory response in COVID-19 patients (Manne et al., 2020; Thachil and Lisman, 2020), may affect the functions of platelets at the disease stage, consistent with a previous study (Manne et al., 2020). By contrast, eight cell subtypes exhibited higher cytokine scores even though their inflammatory scores showed no difference compared with other cell clusters (Figure S7B; Table S7; p < 0.0001).
Figure 6.

Mono_c1-CD14-CCL3 and megakaryocytes in peripheral blood appear as a dominant source for the inflammatory cytokine storm
(A) t-SNE plots of PBMCs colored by major cell types (top left panel), inflammatory cell types (top right panel), cytokine score (middle panel), and inflammatory score (bottom panel).
(B) Heatmap and unsupervised clustering of cell proportion of seven hyper-inflammatory cell subtypes (row normalized).
(C) Boxplots of the cell proportion of Mono_c1-CD14-CCL3, Mega, and T_CD4_c08-GZMK-FOShigh clusters from healthy controls (n = 20), convalescent patients (moderate, n = 48), patients with progression (moderate, n = 18), convalescent patients (severe, n = 35), and patients with progression (severe, n = 38). Two-sided Wilcoxon test.
(D) Ordinary least-squares model of age to cell proportion of Mono_c1-CD14-CCL3, Mono_c2-CD14-HLA-DPB1, and Mono_c3-CD14-VCAN clusters from healthy controls (blue, n = 20), convalescent patients (purple, n = 83), and patients with progression (red, n = 56). p value was assessed with the F-statistic for ordinary least-squares model.
(E) Heatmap of cytokine expression among seven hyper-inflammatory cell subtypes (red font) and other clusters (gray font).
(F) Boxplots of cytokine expression based on scRNA-seq and plasma profiling for healthy controls (n = 20 for scRNA-seq, and n = 5 for plasma), convalescent patients (severe, n = 5, for both scRNA-seq and plasma), and patients with progression (severe, n = 14, for both scRNA-seq and plasma). Two-sided Wilcoxon test.
(G) Boxplots of the cytokine expression of Mono_c1-CD14-CCL3, Mega and T_CD8_c06-TNF clusters from healthy controls (n = 20), convalescent patients (moderate, n = 48), patients with progression (moderate, n = 18), convalescent patients (severe, n = 35), and patients with progression (severe, n = 38). Two-sided Wilcoxon test.
(H) Ordinary least-squares model of age to cytokine expression of Mono_c1-CD14-CCL3, Mega, and T_CD8_c06-TNF clusters from healthy controls (blue, n = 20) and patients with progression (n = 18 + 38). p value was assessed with the F-statistic for ordinary least-squares model.
In (C), (F), and (G), the box represents the second and third quartiles and median, whiskers each extend 1.5 times the interquartile range, and dots represent outliers. In (B) and (E), Mono_c1, Mono_c2, Mono_c3, T_CD4_c08, T_CD8_c09, T_CD8_c06, and Mega correspond to Mono_c1-CD14-CCL3, Mono_c2-CD14-HLA-DPB1, Mono_c3-CD14-VCAN, T_CD4_c08-GZMK-FOShigh, T_CD8_c09-SLC4A10, T_CD8_c06-TNF, and Mega, respectively. In (E), T_CD4_c11, T_CD8_c03, T_CD8_c04, T_CD8_c05, T_CD8_c07, T_gdT_c14, and T_CD8_c08, NK_c01 correspond to clusters of T_CD4_c11-GNLY, T_CD8_c03-GZMK, T_CD8_c04-COTL1, T_CD8_c05-ZNF683, T_CD8_c07-TYROBP, T_gdT_c14-TRDV2, T_CD8_c08-IL2RB, and NK_c01-FCGR3A, respectively. DC, dendritic cells; Mega, megakaryocytes; Mono, monocytes.
Figure S7.

Identification of hyper-inflammatory subtypes associated with cytokine storm in PBMCs, related to Figure 6
(A) t-SNE plots of PBMC cells colored by cytokine score (top panel) and inflammatory score (bottom panel). (B) The proportion of subtypes from healthy controls (n = 20), progression (severe, n = 38) and average of all samples (n = 159) (top panel); the inflammatory score (middle panel) and cytokine score (bottom panel) of subtypes from healthy controls (n = 20), convalescence (moderate, n = 48), progression (moderate, n = 18), convalescence (severe n = 35) and progression (severe, n = 38) patients. Significance was evaluated with Mann-Whitney rank test for each subtype versus all the other subtypes. ∗∗∗∗p < 0.0001. (C) Boxplots of the proportion of inflammatory cell-types and other cell-types from healthy controls (n = 20), convalescence (moderate, n = 48), progression (moderate n = 18), convalescence (severe, n = 35) and progression (severe, n = 38) patients. Two-sided Wilcoxon rank-sum test. (D) Pie chart showing the proportion of 4 classified groups (named ‘both’, ‘Mono’, ‘Mega’ and ‘neither’) based on the proportion of Mono_c1-CD14-CCL3 and Mega cell-types in patients at the progression (severe) stage. (E) Boxplots of the inflammatory and cytokine score within 4 classified groups (named ‘both’, ‘Mono’, ‘Mega’ and ‘neither’). (F) Bar graphs showing cytokine concentration at the plasma levels of CCL3, IFNG, IL1RN and TNF from healthy controls (n = 5), convalescent (n = 7), non-severe (n = 4), severe (n = 4), death case (n = 7) patients. Shown are P values by Student’s t test. (G) Boxplot of CXCL8 expression of Mono_c1-CD14-CCL3 subtype and IFNG expression of T_CD8_c06-TNF subtype from the scRNA-seq datasets with influenza (n = 5). (H) Ordinary least-squares model of age to IFNG signal from array data (n = 310) with influenza. P value was assessed with F-statistic for ordinary least-squares model. In panel (B), (C), (E) and (G), the box represents the second, third quartiles and median, whiskers each extend 1.5 times the interquartile range; dots represent outliers. In panel (F), all points are shown and bars represent mean with the 95% confidence intervals. DC, dendritic cells. Mega, megakaryocytes. Mono, monocytes.
We then investigated the proportion of each of the seven cell subtypes in patients and found that these hyper-inflammatory cell subtypes were enriched in patients with severe disease (Figure S7C). The proportion of these hyper-inflammatory cell subtypes in PBMCs showed different enrichment patterns in patient groups (Figure 6B). Mono_c1-CD14-CCL3 was highly enriched in a subpopulation of severe patients that were likely to be accompanied by an inflammatory storm (Figure 6C), and the proportion of these cells was also correlated with the age of the corresponding patients (Figure 6D). Hyper-inflammatory megakaryocytes were enriched in another batch of patients with severe disease who also could have been experiencing an excessive inflammatory response (Figures 6B and 6C). To determine whether the two cell types influence the immune state of severe COVID-19 patients, we further compared the inflammatory scores of patients with severe disease according to whether they showed enrichment with the two types of cells (Figure S7D). Patients with one or both cell types showed higher scores than those with neither of the cell types (Figure S7E).
By contrast, Mono_c2-CD14-HLA-DPB1 and Mono_c3-CD14-VCAN subtypes were widely distributed in every disease stage, and the proportion of hyper-inflammatory T cells (e.g., the T_CD4_c08-GZMK-FOShigh subtype) decreased in patients with severe disease at the progression stage (Figures 6B, 6C, and S7B), although both of these monocyte subtypes exhibited increased proportions in older convalescent patients (Figure 6D). Taken together, Mono_c1-CD14-CCL3 and megakaryocytes might be the major sources in PBMCs triggering the cytokine inflammatory storm, with cell ratios, inflammatory expression, or both elevated in severe COVID-19 patients. Certain T cell subtypes might also contribute to the inflammatory storm via enhanced expression of pro-inflammatory cytokines.
Next, we investigated the inflammatory signatures for each hyper-inflammatory cell subtype and found unique pro-inflammatory cytokine gene expression in each cell subtype (Figure 6E), such as TNF, CCL3, IL1B, CXCL8, IL6, TGFB1, LTB, and IFNG, suggesting diverse mechanisms potentially resulting in cytokine storms. The hyper-inflammatory Mono_c1-CD14-CCL3 largely expressed more cell-type-specific cytokines, suggesting central roles of the two cell types in driving the inflammatory storm. In particular, Mono_c1-CD14-CCL3 highly expressed CCL3, IL1RN, and TNF, which were detected at much higher levels in plasma in another cohort of patients at the severe stage, especially critically ill patients (Figures 6E and S7F). For 19 patients, we collected both scRNA-seq data and cytokine detection results using plasma (Table S7). Both data sources supported the finding that severe patients had higher level of multiple pro-inflammatory cytokines, such as IL1B, TNF, IL-6, and CCL3 (Figure 6F). This further confirmed the accuracy of our scRNA-seq analysis. Although the inflammatory megakaryocytes highly expressed cell-type identity marker genes such as PPBP (Zhang et al., 1997), the expression level of these genes was decreased in patients compared to healthy controls, indicating a loss of function of these cells after inflammatory activation (Figures 6E and 6G). Notably, the T_CD8_c06-TNF subtype specifically and highly expressed IFNG, a pro-inflammatory cytokine highly enriched in patients at the progression (severe) stage, was also confirmed by plasma cytokine detection (Figures 6E, 6G, and S7F). Moreover, pro-inflammatory cytokines CXCL8 and IFNG showed age-dependent expression in patients with disease progression, while no significance was observed in healthy controls (Figure 6H). This age-dependent expression of pro-inflammatory cytokines in COVID-19 patients could not be observed in influenza patients (Figures S7G and S7H), consistent with a recent study that reported similar patterns of immune cells between aging and COVID-19 patients (Zheng et al., 2020). PPBP showed no correlation with age in either patients or healthy controls, suggesting that the potential loss of function of megakaryocytes might not be age dependent (Figure 6H).
Interactions of hyper-inflammatory cell subtypes in lung and peripheral blood
A cytokine storm may cause immunopathological injury to the lung, and large amounts of infiltrating inflammatory immune cells have been demonstrated in the pulmonary tissue of COVID-19 patients (Cao, 2020). We compared the inflammatory and cytokine scores among all of the cell subtypes captured in BALF. No enrichment of cytokine genes was observed in epithelial cells, while subtypes of macrophages and monocytes had the highest cytokine and inflammatory scores in the progression (severe) samples (Figure 7 A). Similar to our analysis of PBMCs, we identified five hyper-inflammatory cell subtypes, including Macro_c2-CCL3L1, the three subtypes of monocytes, and neutrophils (Figure 7B), suggesting that these cell subtypes might be the major sources driving the inflammatory storm in lung tissue. None of the CD4+ or CD8+ T cells were detected with an elevated inflammatory score or cytokine score in BALF samples, which was different from those in PBMCs. Each hyper-inflammatory subtype highly expressed specific cytokines; for example, Macro_c2-CCL3L1 specifically expressed CCL8, CXCL10/11, and IL6. Mono_c1-CD14-CCL3, as one of the most notable pro-inflammatory cell types in both peripheral blood and BALF, uniquely expressed high levels of IL1B, CCL20, CXCL2, CXCL3, CCL3, CCL4, HBEGF, and TNF. Neutrophils also showed multiple uniquely expressed cytokines, including TNFSF13B, CXCL8, FTH1, and CXCL16 (Figure 7C).
Figure 7.

The interactions of hyper-inflammatory cell subtypes in lung and peripheral blood
(A) t-SNE plots of BALF cells colored by major cell types (top panel), cytokine score (middle panel), and inflammatory score (bottom panel).
(B) Boxplots of the inflammatory score (top panel) and cytokine score (bottom panel) of cell subtypes. Significance was evaluated with the Wilcoxon rank-sum test. ∗∗∗∗p < 0.0001.
(C) Heatmap and unsupervised clustering of cytokine expression of five hyper-inflammatory cell subtypes.
(D) Circos plot showing the prioritized interactions mediated by ligand-receptor pairs between inflammation-related cell types from BALF and PBMCs, respectively. The outer ring displays color-coded cell types, and the inner ring represents the involved ligand-receptor interacting pairs. The line width and arrow width are proportional to the log fold change between severe and moderate progression groups in ligand and receptor, respectively. Colors and types of lines are used to indicate different types of interactions as shown in the legend. The bar plot at bottom indicates the interaction score for each interaction, which serves to measure the interaction strength. DC, dendritic cells; Epi, epithelial cells; Macro, macrophage cells; Mono, monocytes; Neu, neutrophils.
We reasoned that the systematic inflammatory storm might also be associated with cellular cross-talk between lung and peripheral blood via secreting diverse cytokines. To examine this, we analyzed the ligand-receptor pairing patterns among hyper-inflammatory cell subtypes in severe and moderate samples within PBMCs and BALF, respectively (Figure S8 ). The interactions between PBMCs and BALF cells appeared to show significant alterations (Figure 7D). Our data revealed elevated ligand-receptor interactions of hyper-inflammatory cells in patients at the severe compared to moderate stage. Interestingly, cells in the peripheral blood of patients with severe disease showed much lower interactions with each other compared to those in BALF (Figure S8A), except for megakaryocytes, which expressed IL1B and could potentially stimulate Mono_c1-CD14-CCL3 cells. Mono_c1-CD14-CCL3 cells in BALF expressed CCR5, which could receive multiple cytokine stimuli secreted by other cell types in both the lung tissue and the peripheral blood. By contrast, the interactions of Macro_c2-CCL3L1 cells mainly relied on CCR2 and IL1R2. Collectively, these findings illustrate the molecular basis for the potential cell-cell interactions at the pulmonary interface in an inflamed state in COVID-19 patients.
Figure S8.

Intercellular interaction alterations among cell types between severe and moderate progression sample groups, related to Figure 7
(A). Circos plot showing the prioritized interactions mediated by ligand-receptor pairs between inflammation-related cell subtypes for each tissue, namely, PBMC (left panel) and BALF (right panel). The outer ring displays color coded cell types and the inner ring represents the involved ligand-receptor interacting pairs. The line width and arrow width are proportional to the log fold change between severe progression and moderate progression patient groups in ligand and receptor, respectively. Colors and types of lines are used to indicate different types of interactions as shown in the legend. The barplot at bottom indicates the interaction score for each ligand-receptor interaction which serves to measure the interaction strength. (B) Summary illustration depicting the potential cytokine/receptor interactions of hyper-inflammatory cell subtypes involved in the cytokine storm. DC, dendritic cells. Epi, epithelial cells. Macro, macrophage cells. Mono, monocytes. Neu, neutrophils. Mega, megakaryocytes.
Discussion
Our SC4 alliance members generated scRNA-seq data for 284 clinical samples from 196 COVID-19 patients and healthy controls in China and constructed an information-rich data resource for dissecting the immune responses of COVID-19 patients at single-cell resolution. This dataset covered both lung and blood samples of COVID-19 patients with a wide age range, balanced sex ratio, moderate and severe symptoms, and both progression and convalescence stages. 64 well-annotated cell subsets were clustered, providing fine details of the cellular and molecular responses to SARS-CoV-2 infection.
The comprehensive nature of our dataset proved to be powerful at dissecting the associations of age, sex, disease severity, and stage with the diverse immune subsets in SARS-CoV-2 infection. In general, plasma B and proliferative T cells were associated with disease severity, while compositional differences of the precursor cells of these adaptive immune cell types were more prone to be influenced by sex and age. Of note, age and sex also seemed to impact the diversity of TCR/BCR repertoires for a wide range of T and B cells, which may have clinical implications.
We also demonstrated the presence of SARS-CoV-2 sequences in both epithelial and immune cells, along with any altered transcriptomic properties. The presence of viral sequences in multiple epithelial cell types in the human respiratory tract, including ciliated, secretory, and squamous cells, is likely explained by viral infection, although the consequences of viral presence appear to be distinct. The presence of SARS-CoV-2 RNA in various immune cell types, including neutrophils, macrophages, plasma B cells, T cells, and NK cells, was surprising to us initially, but the research community is beginning to appreciate this phenomenon. While it is still not clear how such immune cells would acquire viral sequences, our findings provide tractable angles to further explore these important questions. It appears to us that such viral presence in immune cells, through infection or otherwise, is not without functional consequences.
In our attempt to dissect the cellular origins of potential cytokine storms, we found that megakaryocytes and a few monocyte subsets might be key sources of a diverse set of cytokines highly elevated in COVID-19 patients with severe disease progression. Potential cross-talk between lung and peripheral blood could be abstracted from our dataset, as exemplified in Figure S8B, facilitating future studies.
In summary, the large scRNA-seq dataset covering diverse disease severity and stages has revealed multiple immune characteristics of COVID-19 that were not adequately appreciated previously. Such data provide a critical resource and important insights in dissecting the pathogenesis of COVID-19 and potentially help the development of effective therapeutics and vaccines against SARS-CoV-2.
Limitations of the study
Our data may have variations introduced by different sample processing methods (fresh or frozen) and the wide time range of sampling after symptom onset. While we have made efforts to explicitly include these factors in analyses by ANOVA, their potential impact needs to be considered. While our data show the presence of SARS-CoV-2 RNA and S proteins in epithelial and immune cells, follow-up studies are needed to explore whether these amount to direct infection and what the consequences therein are.
STAR★methods
Key Resources Table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Bio-Plex Pro Human Cytokine Screening Panel, 48-Plex | BIO-RAD | Cat #12007283 |
| Human Cytokine/Chemokine/Growth Factor Panel A Magnetic Bead Panel | LINCO Research, Inc. | Cat #HCYTA-60K-PX48 |
| Biological samples | ||
| PBMC, PFMC, BALF and sputum samples from 171 COVID-19 patients and 25 healthy donors | Single Cell Consortium for COVID-19 in China (SC4) | This paper (Table S1) |
| Critical commercial assays | ||
| Fixation/Permeabilization Solution Kit | BD Biosciences | Cat #554714 |
| SureSelectXT Target Enrichment System for Illumina Paired-End Multiplexed Sequencing Library Kit | Aglient | Cat #G9701 |
| TruePrep DNA Library Prep Kit V2 for Illumina | Vazyme Biotech | Cat #TD503 |
| Chromium Single Cell 3 0 Library and Bead kit | 10x Genomics | Cat #PN-120237 |
| Chromium Single Cell 30 Chip Kit v2 | 10x Genomics | Cat #PN-120236 |
| Chromium i7 Multiplex Kit | 10x Genomics | Cat #PN-120262 |
| Hiseq 3000/4000 SBS kit | Illunima | Cat #FC-410-1003 |
| Hiseq 3000/4000 PE cluster kit | Illunima | Cat #PE-410-1001 |
| Deposited data | ||
| Data files for single-cell RNA sequencing (processed data) | This paper | The NCBI GEO database (GSE158055) |
| Raw data | This paper | Genome Sequence Archive: HRA001149 |
| Software and algorithms | ||
| Harmony | (Korsunsky et al., 2019) | https://github.com/immunogenomics/harmony |
| Cellranger v3.0.2 | 10x Genomics | https://support.10xgenomics.com/single-cell-gene-expression/software/pipelines/latest/ahta-is-cell-ranger |
| kb v0.24.4 | Bray et al., 2016; Melsted et al., 2019 | https://github.com/pachterlab/kb_python |
| kallisto v0.46.1 | Bray et al., 2016 | https://github.com/pachterlab/kallisto |
| bustools v0.39.3 | Melsted et al., 2019 | https://github.com/BUStools/bustools |
| STARTRAC | Zhang et al., 2018 | https://github.com/Japrin/STARTRAC |
| Seurat 2.3.0/3.0 | (Butler et al., 2018) | http://satijalab.org/seurat |
| scanpy 1.4.6/1.5.1 | Wolf et al., 2018 | https://scanpy.readthedocs.io/en/latest/ |
| CSOmap | Ren et al., 2020 | https://github.com/zhongguojie1998/CSOmap |
| SCENIC 1.1.2-2 | Aibar et al., 2017 | https://github.com/aertslab/SCENIC |
| Scrublet | (Wolock et al., 2019) | https://github.com/AllonKleinLab/scrublet |
Resource availability
Lead contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Zemin Zhang (zemin@pku.edu.cn).
Materials availability
This study did not generate new unique reagents.
Data and code availability
The processed gene expression data in this paper have been deposited into the NCBI GEO database: GSE158055. Visualization of this dataset can be found at http://covid19.cancer-pku.cn. The raw data are available from Genome Sequence Archive for human with accession ID: HRA001149 (https://ngdc.cncb.ac.cn/gsa-human/browse/HRA001149). Additional Supplemental Items are also available at Mendeley Data: https://dx.doi.org/10.17632/dvp4y5ttd5.1.
Experimental model and subject details
Ethics statement
This study strictly follows the principles according to the Declaration of Helsinki, with written informed consents obtained from all participants before sample collection according to regular principles. Ethical approvals were gained from the Ethics Committees of 19 institutions, including State Key Laboratory of Ophthalmology of Sun Yat-sen University, Department of Infectious Diseases of Fifth Medical Center of Chinese PLA General Hospital, Eastern Hepatobiliary Surgery Hospital of Second Military Medical University, Guangzhou Regenerative Medicine and Health GuangDong Laboratory, Institute of Biophysics of Chinese Academy of Sciences, The First Affiliated Hospital of University of Science and Technology of China, Cancer Center of Renmin Hospital of Wuhan University, Department of Laboratory Medicine of Yuebei People’s Hospital of Shantou University Medical College, Shenzhen Third People’s Hospital, Center for Life Sciences of Harbin Institute of Technology, School of Life Science and Technology of Harbin Institute of Technology, Institute of Pathology and Southwest Cancer Center of Army Medical University, Southwest Hospital of Army Medical University of Center for Stem Cell Medicine and Department of Stem Cell & Regenerative Medicine, Chinese Academy of Medical Sciences and Peking Union Medical College, State Key Laboratory of Experimental Hematology and National Clinical Research Center for Blood Diseases, Institute of Hematology and Blood Diseases Hospital, Shanghai Institute of Immunology of Department of Microbiology and Immunology of Shanghai Jiao Tong University School of Medicine, Guangzhou Institutes of Biomedicine and Health of Chinese Academy of Sciences, Beijing Youan Hospital of Capital Medical University, State Key Laboratory of Oncology in South China, Collaborative Innovation Center for Cancer Medicine of Sun Yat-sen University Cancer Center.
Biological samples
A total of 171 patients with COVID-19 and 25 healthy individuals in this study were enrolled from 37 centers/ laboratories, with samples (n = 284) collected. Samples of COVID-19 were further categorized into groups of moderate convalescence (n = 89), moderate progression (n = 33), severe convalescence (n = 51) and severe progression (n = 83) according to disease severity (moderate or severe) and stages (progression and convalescence) based on the WHO guidelines (https://www.who.int/publications/i/item/clinical-management-of-covid-19). The sex ratio between female and male donor is 106:177. The age of the donors ranges from 6 to 92. Of all the 284 samples, 249 samples were collected from PBMC, among which 77 samples have sorted B/T cells or both. 13 samples were collected from lung tissues, including 12 BALF samples and 1 PFMC sample. We also collected 22 sputum samples from patients as well. Among all the samples, we have 7 paired lung BALF and PBMC samples. Single-cell transcriptome data for each sample was profiled using 10x Genomics scRNA-seq platform. Single-cell sequencing of TCRs (13 samples) and BCRs (53 samples) or both (11 samples) was also performed for part of the samples. Detailed clinical information and demographic characteristics of patient cohorts were shown in Table S1.
Method details
Sample collection
Blood samples that were not immediately processed for cell encapsulation were mixed with Whole Blood Cell Stabilizer (Cytodelics) and stored at −80°C freezer. The peripheral blood mononuclear cells (PBMCs) were isolated using standard density gradient centrifugation and then used for 10x single-cell RNA-seq. Bronchoalveolar lavage fluid (BALF) samples were collected from COVID-19 patients during intubation and processed with 2h according to WHO guidance. BALF was passed through 100-μm nylon cell strainer to obtain single cell suspensions with cooled RPMI 1640 complete medium. Cells in the BALF were freshly used for 10x single-cell RNA-seq. To reduce the possibility of blood cell contamination, we removed the red blood cell using Red Blood Cell Lysis Buffer in the BALF sample processing step. In the data quality step, we also checked the expression of blood cell marker such as HBB to confirm no contamination in the BALF data. Sputum samples were collected from COVID-19 patients using an oropharyngeal swab or hypertonic saline induction. To reduce squamous cell contamination, subjects were asked to rinse their mouth with water and clear their throat. Samples were incubated in Dulbecco’s Phosphate-Buffered Saline (DPBS) with agitation for 15 minutes and filtered through 40-micron strainers. Cells in the sputum were freshly used for 10x single-cell RNA-seq.
Single cell RNA library preparation and sequencing
Cell suspensions were barcoded through the 10x Chromium Single Cell platform using Chromium Single Cell 5′ Library, Chromium Single Cell 3′ Library, Gel Bead and Multiplex Kit, and Chip Kit (10x Genomics). The loaded cell numbers range from 300-500,000 aiming for 300-14,000 single cells per reaction. Single-cell RNA libraries were prepared using the Chromium Single Cell 3′ v2 Reagent (10x Genomics; PN-120237, PN-120236 and PN-120262), Chromium Single Cell 3′ v3 Reagent (10x Genomics; PN-1000075, PN-1000073 and PN-120262) the Chromium Single Cell 5′ v2 Reagent (10x Genomics, 120237), and Chromium Single Cell V(D)J Reagent kits (10x Genomics, PN-1000006, PN-1000014, PN-1000020, PN-1000005) was used to prepare single-cell RNA libraries according to the manufacturer’s instructions. Each sequencing library was generated with a unique sample index. The libraries were sequenced using either DIPSEQ, BGISEQ or Illumina platforms.
IHC staining for SARS-CoV-2+ lymphocytes
Formalin-fixed paraffin-embedded pulmonary tissue blocks were cut into 3 μm-thick serial sections. After block with streptavidin peroxidase, heat-induced antigen epitope retrieval in citrate buffer (pH: 6.0) was performed. Sections were incubated overnight at 4°C with primary antibodies against SARS-CoV-2 spike protein (Cat 40150-T62-COV2, Sino Biological). Staining was visualized by Dako REAL EnVision Detection System, Peroxidase/DAB+, Rabbit/Mouse (K5007, Dako) followed by counterstaining with hematoxylin. Images were captured using a digital camera (DP73, Olympus) under a light microscope (BX53, Olympus).
Cytokine analysis of plasma by using multiplex bead-based immunoassay
Human cytokines in the plasma were measured by Bio-plex Pro TM Human Cytokine Screening 48 plex Bio-PlexTM 200 System (# 12007283, Bio-Rad, US) and Human Cytokine/Chemokine Magnetic Bead Panel (#HCYTA-60K-PX48). The experiments were performed by following the manufacturers’ instructions. Whole blood from COVID-19 patients and healthy controls were drawn into collection tubes containing anticoagulant. Centrifugation the tubes at 1,000 x g for 15 min at 4°C and transfer the plasma to a clean polypropylene tube, followed by another centrifugation at 10,000 x g for 10 min at 4°C to completely remove platelets and precipitates. Dilute samples fourfold (1:4) by adding 1 volume of sample to 3 volumes of sample diluent. Fifty microliter of each sample were used to assay. Paired software was used for data acquisition and analysis.
Quantification and statistical analysis
Single-cell RNA-seq data processing
Single-cell sequencing data were aligned and quantified using kallisto/bustools (KB, v0.24.4) (Bray et al., 2016) against the GRCh38 human reference genome downloaded from 10x Genomics official website. Preliminary counts were then used for downstream analysis. Quality control was applied to cells based on three metrics step by step: the total UMI counts, number of detected genes and proportion of mitochondrial gene counts per cell. Specifically, cells with less than 1000 UMI counts and 500 detected genes were filtered, as well as cells with more than 10% mitochondrial gene counts. To remove potential doublets, for PBMC samples, cells with UMI counts above 25,000 and detected genes above 5,000 are filtered out. For other tissues, cells with UMI counts above 70,000 and detected genes above 7,500 are filtered out. Additionally, we applied Scrublet (Wolock et al., 2019) to identify potential doublets. The doublet score for each single cell and the threshold based on the bimodal distribution was calculated using default parameters. The expected doublet rate was set to be 0.08, and cells predicted to be doublets or with doubletScore larger than 0.25 were filtered. After quality control, a total of 1,598,708 cells were remained. The stepwise quality control metrics used for individual samples were listed in Table S1. The resulting distribution of UMI counts, gene counts as well as mitochondrial gene percentage were shown in Figures S1C–S1E. We normalized the UMI counts with the deconvolution strategy implemented in the R package scran. Specifically, cell-specific size factors were computed by computeSumFactors function and further used to scale the counts for each cell. Then the logarithmic normalized counts were used for the downstream analysis.
Batch effect correction and cell subsets annotations
To integrate cells into a shared space from different datasets for unsupervised clustering, we used the harmony algorithm (Korsunsky et al., 2019) to do batch effect correction. To detect the most variable genes used for harmony algorithm, we performed variable gene selection separately for each sample. A consensus list of 1,500 variable genes was then formed by selecting the genes with the greatest recovery rates across samples, with ties broken by random sampling. All ribosomal, mitochondrial and immunoglobulin genes were then removed from the list. Next, we calculate a PCA matrix with 20 components using such informative genes and then feed this PCA matrix into HarmonyMatrix() function implemented in R package Harmony. We set sample and dataset as two technical covariates for correction with theta set as 2.5 and 1.5, respectively. The resulting batch-corrected matrix was used to build nearest neighbor graph using Scanpy (Wolf et al., 2018). Such nearest neighbor graph was then used to find clusters by Louvain algorithm (Traag et al., 2019). The cluster-specific marker genes were identified using the rank_genes_groups function.
The first round of clustering (resolution = 0.3) identified six major cell types including T cells, NK cells, B cells, plasma B cells, myeloid cells and epithelial cells. To identify clusters within each major cell type, we performed a second round of clustering on T/NK, B/plasma B, myeloid and epithelial cells separately. The procedure of the second round of clustering is the same as first round, starting from low-rank harmony output (30 components) on the highly variable genes chosen as described above, with resolution ranging from 0.3 to 1.5. Each sub cluster was restrained to have at least 30 significantly highly expressed genes (FDR < 0.01, logFC > 0.25, t test) compared with other cells. Annotation of the resulting clusters to cell types was based on the known markers. Meanwhile, single cells expressing two sets of well-studied canonical markers of major cell types were labeled as doublets and excluded from the following analysis. Also, cells highly expressed HBA, HBB and HBD, which are the markers for erythrocytes, were also excluded. 136,006 cells were removed and a total of 1,462,702 cells were retained for downstream analysis. In total, we identified 6 major cell types including B cells (MS4A1, CD79A, CD79B), myeloid cells (CST3, LYZ), NK cells (GNLY, NKG7, TYROBP), epithelial cells (KRT18, KRT19), CD4 and CD8 T cells (CD3D, CD3E, CD3G, CD40LG, CD8A, CD8B). These major cell types were further classified into 64 clusters representing different cell types within major cell lineages (Figures 1B and S1F–S1J). A full list of canonical and signature marker genes for each cluster was deposited in Table S2.
Detection and processing scRNA-seq data with viral RNA
To identify single cells with viral RNA, we aligned raw scRNA-seq reads using kallisto/bustools (KB) against a customized reference genome, in which the SARS-CoV-2 genome (NC_045512, NCBI Refseq) was added as an additional chromosome to the human reference genome. Single cells with viral reads (UMI > 0) were retained. Cells with less than 200 genes expressed or more than 20% mitochondrial counts were excluded, as well as those labeled as doublet following aforementioned protocol (Figure 4A).
The remaining cells were then used for dimension reduction and unsupervised clustering using Python package Scanpy. In brief, the top 500 genes with the highest variance were selected and the dimensionality of the data was reduced by principal component analysis (PCA) (30 components) first and then with t-SNE, followed by Louvain clustering performed on the 30 principal components (resolution = 1). For t-SNE visualization, we directly fit the PCA matrix into the scanpy.api.tl.tsne function with perplexity of 30. To identify cell-type-specific gene markers, we selected genes that were differentially expressed across different cell types (FDR < 0.01, log fold change > 0.5) using the rank_genes_groups function. Clusters were annotated based on the expression of known marker genes (Figure 4B).
To confirm whether the virus detection rate is related to library preparation methods, we further aligned raw scRNA-seq reads against the same reference genome with additional annotation information of the 11 SARS-CoV-2 genes. We then calculated detection rates in 10x 3-prime and 10X 5-prime sequencing samples respectively (Figures 4F and S4G). We could directly calculate the detection rate () of each gene by the equation:
denotes the number of cells within cell type while denotes the number of cells with gene detected within . is the length of gene.
TCR and BCR analysis
TCR/BCR sequences were assembled and quantified following Cell Ranger (v.3.0.2) vdj protocol against GRCh38 reference genome. Assembled contigs labeled as low-confidence, non-productive or with UMIs < 2 were discarded. To identify TCR clonotype for each T cell, only cells with at least one TCR α chain (TRA) and one TCR β-chain (TRB) were remained. For a given T cell, if there are two or more α or β chains assembled, the highest expression level (UMI or reads) α or β chains was regarded as the dominated α or β chain in the cell. Each unique dominated α-β pair (CDR3 nucleotide sequences and rearranged VDJ genes included) was defined as a clonotype. T cells with exactly the same clonotype constituted a T cell clone.
BCR clonotypes were identified similar to TCR. Only cells with at least one heavy chain (IGH) and one light chain (IGL or IGK) were kept. For a given B cell, if there are two or more IGH or IGL/IGK assembled, the highest expression level (UMI or reads) IGH or IGL/IGK was defined as the dominated IGH or IGL/IGK in the cell. Each unique dominated IGH-IGL/IGK pair (CDR3 nucleotide sequences and rearranged VDJ genes) was defined as a clonotype. B cells with exactly the same clonotype constituted a B cell clone.
220,968 T cells with TCR information and 282,464 B cells with BCR information were used to perform the STARTRAC analysis as we previously described (Zhang et al., 2018). STARTRAC-expa was used to quantified the potential clonal expansion level. TCR/BCR diversity was calculated as Shannon’s entropy shown below:
The p(x) represents the frequency of a given TCR/BCR clone among all T/B cells with TCR/BCR identified.
Transcriptional factor analysis
Both activated transcriptional factors, differentially activated transcriptional factors and activated regulons in each cluster were clarified. The activated transcriptional factors in each cluster were chosen according to mean expression level calculated from scaled expression matrix, and the differentially activated transcriptional factors in each cluster were identified by wilcox.test using cells from other clusters. Activated regulons in each cluster were analyzed using SCENIC (Aibar et al., 2017) with raw count matrix as input.
Comparing immune cell proportion
For samples from PBMC and BALF tissue, we calculated immune cell proportions for each major cell type and underlying cell subsets. In order to avoid bias caused by samples dominated by a few cell types, we filtered samples containing FACS-sorted B/T cells and retained those samples with cells > 1000. For each sample, cell type proportion was calculated by number of cells in certain cell type divided by total number of cells. To identify changes in cell proportions between samples in different disease severity states, disease progression stages and sex, we performed Wilcoxon rank-sum test on the proportions of each major cell type and underlying cell subset across different phenotype groups (Figures 2B–2G, 2I, and 3B–3F). We performed correlation analysis to assess the association between cell type proportion and patient age (Figures 2H and 3G–3I). Only those cell types with statistically significant differences (FDR < 0.05) in proportions were shown.
ANOVA analysis
To further assess how different patients’ phenotypes and their potential interactions influence cell type proportions, we performed multivariate ANOVA on cell type proportions and on diversity of BCR/TCR based on different patient phenotypes (Figures 2A and 3A), including disease severity, disease progression stage, sex and age together with technical factors including sample type (fresh or frozen) and sampling time (days after symptom onset). Interactions between these variables were excluded. To convert age into a categorical variable, we binned patient age into four groups: young (< 18 years old), middle-age (18-50 years old), old-age (50-70 years old) and the elderly (70+ years old). Interactions between variables were regarded as significantly associated with cell type proportions when FDR < 0.05. Sampling time was binned into five groups: controls; < 10 days; 10-30 days; 30-60 days; > 60 days.
Differential expression and Gene Ontology enrichment analysis
To investigate the impact of the presence of the viral RNA on epithelial cells, we identify differential expressed genes by performing two-sided unpaired Wilcoxon tests on all the expressed genes (expressed in at least 10% cells in either group of cells). P values were adjusted following Benjamini & Hochberg protocol. Top 100 highly expressed genes of each group were shown in the volcano plots (Figure 5A). Based on these genes, enriched GO terms were then acquired for each group of cells using R package clusterProfiler (Yu et al., 2012) following the default parameters. Annotation Dbi R package “org.Hs.eg.db” was used to map gene identifiers. The results were visualized as bar plots (Figure 5B).
Cell-cell interaction analysis by CSOmap
To illustrate the cell-cell interaction potential of cells with viral detection, we first created a set of datasets by joining 7 BALF samples with the virus+ dataset separately. Then, we used CSOmap (Ren et al., 2020) to construct a 3D pseudo space and calculate the significant interaction for each dataset. To investigate the interaction potentials of the cell types, we used two indexes, distances within cell type and normalized connection. Distance within each cell type is calculated based on the aforementioned 3D coordinates (Figures 5E–5G). The shorter the distance, the closer the cells are located in the 3D space, which indicates that they are more likely to interact with each other. To further investigate the interaction between different cell types, we made use of the CSOmap output connection matrix (Figure 5I). For a cluster pair, normalized connection was calculated by dividing its corresponding connection value by the product of their respective cell numbers. Normalized connections were then multiplied by 10,000. Meanwhile, to highlight the key ligand-receptor pairs function in the interaction, we also examine the contribution output by CSOmap (Figure 5H).
Inflammatory and cytokine score related subtypes analysis
Briefly, we first filtered out samples with fewer than 1000 cells available. For PBMC, only subtypes with more than 1000 cells were included in the subsequent analysis. For BALF data analysis, we removed major cell types with fewer than 500 cells. To define inflammatory and cytokine score, we downloaded a gene set termed ‘HALLMARK_INFLAMMATORY_RESPONSE’ from MSigDB (Liberzon et al., 2015) and collected cytokine genes based on these references (see Table S1). Cytokine and inflammatory score were evaluated with the AddModuleScore function built in the Seurat (Stuart et al., 2019). To select the most promising hyper-inflammatory cell types, we performed Mann-Whitney rank test (single-tail) for each subtype’s score versus all the other subtypes’ score. Seven subtypes (Mono_c1-CD14-CCL3, Mono_c2-CD14-HLA-DPB1, Mono_c3-CD14-VCAN, T_CD4_c08-GZMK-FOShigh, T_CD8_c06-TNF, T_CD8_c09-SLC4A10 and Mega) in PBMC were defined as hyper-inflammatory cell types with significantly statistical parameters (p < 0.0001) in both cytokine and inflammatory score. In addition, we defined 8 subtypes (T_CD8_c08-IL2RB, T_CD4_c11-GNLY, NK_c01-FCGR3A, T_CD8_c05-ZNF683, T_CD8_c04-COTL1, T_CD8_c07-TYROBP, T_CD8_c03-GZMK and T_gdT_c14-TRDV2) with significantly statistical parameters (p < 0.0001) only in cytokine score. For subtypes in BALF, we defined 5 subtypes (Macro_c2-CCL3L1, Mono_c1-CD14-CCL3, Mono_c2-CD14-HLA-DPB1, Mono_c3-CD14-VCAN, Neu) as hyper-inflammatory types with the same standard threshold as PBMC (p < 0.0001).
Cell ratio and cytokine marker analysis of hyper-inflammatory subtypes
To explore whether there are state-specific of COVID-19 patients enriched subtypes, we performed hierarchical clustering with setting standard scale (0-1) for 7 hyper-inflammatory subtypes (Figure 6B). Then, we used the Wilcoxon rank-sum test to calculate the significance of cell proportion of each subtype in states (moderate convalescent, moderate progression, severe convalescent, severe progression) compared with healthy control (Figures 6C and 6G). We also applied the ordinary least square method to calculate the correlation between age and cell proportion in different states of COVID-19 patients (Figures 6D and 6H). For the significance of cytokine expression level with state and age, we performed Wilcoxon rank-sum test and ordinary least square to assess the p values.
To determine whether Mono_c1-CD14-CCL3 and Mega cell-types influence the inflammatory state of the severe patients, we classified patients of the progression (severe) stage into 4 groups (named ‘both’, ‘Mono’, ‘Mega’ and ‘neither’) based on the proportion of Mono_c1-CD14-CCL3 and Mega cell-types in each sample. Specially, we normalized the ratio of each subtype among 7 hyper-inflammatory subtypes for each sample with setting standard scale (0-1). Then, we identified and named ‘both’ group which was enriched both Mono_c1-CD14-CCL3 and Mega cell-types subtypes (threshold > 0.5), ‘Mono’ group only enriched Mono_c1-CD14-CCL3 subtype cells, ’Mega’ only enriched Mega cell-types subtype cells, and ’neither’ group which was neither enriched Mono_c1-CD14-CCL3 nor Mega cell-types subtypes.
For the analysis of single-cell datasets with influenza from Lee et al.(Lee et al., 2020), we applied the function ‘ingest’ built in the Scanpy package for projection based on results for the COVID-19 data. In detail, raw data were processed and normalized with default parameters. As recommended, we used PCA space to map cells of influenza into each cluster of COVID-19.
Cell-cell communication analysis between PBMC and BALF by iTALK
To identify and visualize the possible cell-cell interactions in terms of cytokine storm between the highly inflammation-correlated cell types evaluated by the inflammation score within each tissue and the crosstalk between lung and circulating blood, we employed an R package iTALK introduced by (Wang et al., 2019). Cytokine/chemokine category (n = 320) in the ligand-receptor database was selectively used for our purpose. Wilcoxon rank sum test was used to identify the differentially expressed genes (DEGs) between the progression (severe) and progression (moderate) patient groups for each cell type. DEGs were then matched and paired against the ligand-receptor database to construct a putative cell-cell communication network. An interaction score defined as the product of the log fold change of ligand and receptor was used to rank these interactions (Figure 7D). In addition, the expression level of both ligand and receptor were also considered. We defined severe gained interaction if a ligand gene was upregulated in the progression (severe) group and its paired gene upregulated or remains no change. We defined severe lost interaction if a ligand (receptor) gene was downregulated in the progression (severe) group regardless of the expression level of its paired gene.
Acknowledgments
We thank the COVID-19 patients from Wuhan Jinyintan Hospital and their families for their dedication and Professor Dingyu Zhang and his colleagues at Wuhan Jinyintan Hospital for their contribution to the study. We thank the Computing Platform of the Center for Life Science for their contributions. We also thank the 10x Genomics China team for their strong support in the coordination and discussion of this study. We thank Analytical BioSciences for their help in building the visualization website of this dataset. We thank Zhuo Zhou of BIOPIC, Peking University for helpful discussion regarding the interferon responses of SARS-CoV-2 infection. We thank the USTC supercomputing center and the School of Life Science Bioinformatics Center for providing supercomputing resources for this project. We thank the CAS interdisciplinary innovation team for helpful discussion. We also thank W. Chen, C. Xiao, Y. Zheng, W. Su, Y. Zhang, C. Zhang, X. Wang, H. Ma, T. Jin, X. Wang, H. Wei, B. Fu, L. Liu, J. Weng, and X. Ma for their support of this project. This work was supported by the Beijing Advanced Innovation Centre for Genomics at Peking University, the Ministry of Science and Technology of the People's Republic of China (grants 2020YFC0848700, 2016YFA0100600, and CSTC2020jscx-fyzxX0037), the National Key R&D Program of China (grant 2017YFA0102900), the National Natural Science Foundation of China (grants 91940306, 81788101, 31970858, 32022016, 31771428, 91640113, 31700796, 81871479, 81772165, 81974303, 81988101, 31991171, 31530036, 91742203, 31825008, 31422014, 61872117, 61822108, and 62032007), the China Primary Health Care Foundation-Youan Medical Development Fund (BJYAYY-2020PY-01), the Beijing Municipal of Science and Technology Major Project (Z201100005320014, Z201100005420018, and Z201100005420022), the CAMS Initiative for Innovative Medicine (2017-I2M-1-015), the Fundamental Research Funds for the Central Universities (YD2070002019), the Research Project for Outstanding Scholar of Yuebei People’s Hospital, Shantou University Medical College (RS202001), and the Chang Jiang Scholars Program (J.-X.B.).
Author contributions
Conceptualization, Zemin Zhang, X.R., R.J., J.C., X. Wang, K.Q., Zheng Zhang, H.W., F.-S. Wang., Pingsen Zhao, X.B., X. Li, T.C., X. Liu, L.W., J.B., Z.H., Q.J., and P. Zhou; resources, Zemin Zhang, X.R., R.J., J.C., X. Wang, K.Q., Zheng Zhang, H.W., F.-S. Wang, Pingsen Zhao, X.B., X. Li, T.C., X. Liu, L.W., J.B., Z.H., Q.J., and P. Zhou; methodology, Zemin Zhang, X.R., R.J., J.C., X. Wang, K.Q., Zheng Zhang, H.W., F.-S. Wang, Pingsen Zhao, X.B., X. Li, T.C., X. Liu, L.W., J.B., Z.H., Q.J., P. Zhou, W.W., X.F., W.H., B.S., P.C., J. Li, Y. Liu, F.T., F.Z., Y.Y., Jiangping He, W.M., Jingjing He, and P.W.; investigation,Zemin Zhang, X.R., R.J., J.C., X. Wang, K.Q., Zheng Zhang, H.W., F.-S. Wang, Pingsen Zhao, X.B., X. Li, T.C., X. Liu, L.W., J.B., Z.H., Q.J., P. Zhou, W.W., X.F., W.H., B.S., P.C., J. Li, Y. Liu, F.T., F.Z., Y.Y., Jiangping He, W.M., Jingjing He, and P.W.; validation, P.Z., W.W., and W.Y.; writing – original draft, Zemin Zhang, X.R., R.J., J.C., X. Wang, K.Q., Zheng Zhang, H.W., F.-S. Wang, Pingsen Zhao, X.B., X. Li, T.C., X. Liu, L.W., J.B., Z.H., Q.J., P. Zhou, W.W., X.F., W.H., B.S., P.C., J. Li, Y. Liu, F.T., F.Z., Y.Y., Jianping He, W.M., Jingjing He, and P.W..
Declaration of interests
Zemin Zhang is a founder of Analytical Bioscience and an advisor for InnoCare. All financial interests are unrelated to this study. The remining authors declare no competing interests.
Published: February 3, 2021
Footnotes
Supplemental Information can be found online at https://doi.org/10.1016/j.cell.2021.01.053.
Supplemental information
References
- Aibar S., González-Blas C.B., Moerman T., Huynh-Thu V.A., Imrichova H., Hulselmans G., Rambow F., Marine J.-C., Geurts P., Aerts J., et al. SCENIC: single-cell regulatory network inference and clustering. Nat. Methods. 2017;14:1083–1086. doi: 10.1038/nmeth.4463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blanco-Melo D., Nilsson-Payant B.E., Liu W.C., Uhl S., Hoagland D., Møller R., Jordan T.X., Oishi K., Panis M., Sachs D., et al. Imbalanced Host Response to SARS-CoV-2 Drives Development of COVID-19. Cell. 2020;181:1036–1045.e9. doi: 10.1016/j.cell.2020.04.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bost P., Giladi A., Liu Y., Bendjelal Y., Xu G., David E., Blecher-Gonen R., Cohen M., Medaglia C., Li H., et al. Host-Viral Infection Maps Reveal Signatures of Severe COVID-19 Patients. Cell. 2020;181:1475–1488.e12. doi: 10.1016/j.cell.2020.05.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bray N.L., Pimentel H., Melsted P., Pachter L. Near-optimal probabilistic RNA-seq quantification. Nat. Biotechnol. 2016;34:525–527. doi: 10.1038/nbt.3519. [DOI] [PubMed] [Google Scholar]
- Butler A., Hoffman P., Smibert P., et al. Integrating single-cell transcriptomic data across different conditions, technologies, and species[J] Nature biotechnology. 2018;36:411–420. doi: 10.1038/nbt.4096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cantuti-Castelvetri L., Ojha R., Pedro L.D., Djannatian M., Franz J., Kuivanen S., van der Meer F., Kallio K., Kaya T., Anastasina M., et al. Neuropilin-1 facilitates SARS-CoV-2 cell entry and infectivity. Science. 2020;370:856–860. doi: 10.1126/science.abd2985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cao X. COVID-19: immunopathology and its implications for therapy. Nat. Rev. Immunol. 2020;20:269–270. doi: 10.1038/s41577-020-0308-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cao Y., Su B., Guo X., Sun W., Deng Y., Bao L., Zhu Q., Zhang X., Zheng Y., Geng C., et al. Potent Neutralizing Antibodies against SARS-CoV-2 Identified by High-Throughput Single-Cell Sequencing of Convalescent Patients’ B Cells. Cell. 2020;182:73–84.e16. doi: 10.1016/j.cell.2020.05.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen Z., John Wherry E. T cell responses in patients with COVID-19. Nat. Rev. Immunol. 2020;20:529–536. doi: 10.1038/s41577-020-0402-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen Z., Ren X., Yang J., Dong J., Xue Y., Sun L., Zhu Y., Jin Q. An elaborate landscape of the human antibody repertoire against enterovirus 71 infection is revealed by phage display screening and deep sequencing. MAbs. 2017;9:342–349. doi: 10.1080/19420862.2016.1267086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chua R.L., Lukassen S., Trump S., Hennig B.P., Wendisch D., Pott F., Debnath O., Thürmann L., Kurth F., Völker M.T., et al. COVID-19 severity correlates with airway epithelium-immune cell interactions identified by single-cell analysis. Nat. Biotechnol. 2020;38:970–979. doi: 10.1038/s41587-020-0602-4. [DOI] [PubMed] [Google Scholar]
- Daly J.L., Simonetti B., Klein K., Chen K.E., Williamson M.K., Antón-Plágaro C., Shoemark D.K., Simón-Gracia L., Bauer M., Hollandi R., et al. Neuropilin-1 is a host factor for SARS-CoV-2 infection. Science. 2020;370:861–865. doi: 10.1126/science.abd3072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fan X., Chi X., Ma W., Zhong S., Dong Y., Zhou W., Ding W., Fan H., Yin C., Zuo Z., et al. Single-cell RNA-seq and V(D)J profiling of immune cells in COVID-19 patients. medRxiv. 2020 2020.2005.2024.20101238. [Google Scholar]
- Gao L., Zhou J., Yang S., Chen X., Yang Y., Li R., Pan Z., Zhao J., Li Z., Huang Q., et al. The dichotomous and incomplete adaptive immunity in COVID-19. medRxiv. 2020 doi: 10.1038/s41392-021-00525-3. 2020.2009.2005.20187435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gavins F.N., Hughes E.L., Buss N.A., Holloway P.M., Getting S.J., Buckingham J.C. Leukocyte recruitment in the brain in sepsis: involvement of the annexin 1-FPR2/ALX anti-inflammatory system. FASEB J. 2012;26:4977–4989. doi: 10.1096/fj.12-205971. [DOI] [PubMed] [Google Scholar]
- Guo C., Li B., Ma H., Wang X., Cai P., Yu Q., Zhu L., Jin L., Jiang C., Fang J., et al. Single-cell analysis of two severe COVID-19 patients reveals a monocyte-associated and tocilizumab-responding cytokine storm. Nat. Commun. 2020;11:3924. doi: 10.1038/s41467-020-17834-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoffmann M., Kleine-Weber H., Schroeder S., Krüger N., Herrler T., Erichsen S., Schiergens T.S., Herrler G., Wu N.-H., Nitsche A., et al. SARS-CoV-2 Cell Entry Depends on ACE2 and TMPRSS2 and Is Blocked by a Clinically Proven Protease Inhibitor. Cell. 2020;181:271–280.e8. doi: 10.1016/j.cell.2020.02.052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hou Y.J., Okuda K., Edwards C.E., Martinez D.R., Asakura T., Dinnon K.H., 3rd, Kato T., Lee R.E., Yount B.L., Mascenik T.M., et al. SARS-CoV-2 Reverse Genetics Reveals a Variable Infection Gradient in the Respiratory Tract. Cell. 2020;182:429–446.e14. doi: 10.1016/j.cell.2020.05.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang C., Wang Y., Li X., Ren L., Zhao J., Hu Y., Zhang L., Fan G., Xu J., Gu X., et al. Clinical features of patients infected with 2019 novel coronavirus in Wuhan, China. Lancet. 2020;395:497–506. doi: 10.1016/S0140-6736(20)30183-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang N., He J., Weinstein J.A., Penland L., Sasaki S., He X.-S., Dekker C.L., Zheng N.-Y., Huang M., Sullivan M., et al. Lineage structure of the human antibody repertoire in response to influenza vaccination. Sci. Transl. Med. 2013;5:171ra19. doi: 10.1126/scitranslmed.3004794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jouan Y., Guillon A., Gonzalez L., Perez Y., Boisseau C., Ehrmann S., Ferreira M., Daix T., Jeannet R., François B., et al. Phenotypical and functional alteration of unconventional T cells in severe COVID-19 patients. J. Exp. Med. 2020;217:217. doi: 10.1084/jem.20200872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Korsunsky I., Millard N., Fan J., Slowikowski K., Zhang F., Wei K., Baglaenko Y., Brenner M., Loh P.R., Raychaudhuri S. Fast, sensitive and accurate integration of single-cell data with Harmony. Nat. Methods. 2019;16:1289–1296. doi: 10.1038/s41592-019-0619-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laouedj M., Tardif M.R., Gil L., Raquil M.A., Lachhab A., Pelletier M., Tessier P.A., Barabé F. S100A9 induces differentiation of acute myeloid leukemia cells through TLR4. Blood. 2017;129:1980–1990. doi: 10.1182/blood-2016-09-738005. [DOI] [PubMed] [Google Scholar]
- Lee N., Hui D., Wu A., Chan P., Cameron P., Joynt G.M., Ahuja A., Yung M.Y., Leung C.B., To K.F., et al. A major outbreak of severe acute respiratory syndrome in Hong Kong. N. Engl. J. Med. 2003;348:1986–1994. doi: 10.1056/NEJMoa030685. [DOI] [PubMed] [Google Scholar]
- Lee J.S., Park S., Jeong H.W., Ahn J.Y., Choi S.J., Lee H., Choi B., Nam S.K., Sa M., Kwon J.S., et al. Immunophenotyping of COVID-19 and influenza highlights the role of type I interferons in development of severe COVID-19. Sci. Immunol. 2020;5:5. doi: 10.1126/sciimmunol.abd1554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liberzon A., Birger C., Thorvaldsdóttir H., Ghandi M., Mesirov J.P., Tamayo P. The Molecular Signatures Database (MSigDB) hallmark gene set collection. Cell Syst. 2015;1:417–425. doi: 10.1016/j.cels.2015.12.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manne B.K., Denorme F., Middleton E.A., Portier I., Rowley J.W., Stubben C.J., Petrey A.C., Tolley N.D., Guo L., Cody M.J., et al. Platelet Gene Expression and Function in COVID-19 Patients. Blood. 2020;136:1317–1329. doi: 10.1182/blood.2020007214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mathew D., Giles J.R., Baxter A.E., Oldridge D.A., Greenplate A.R., Wu J.E., Alanio C., Kuri-Cervantes L., Pampena M.B., D’Andrea K., et al. UPenn COVID Processing Unit Deep immune profiling of COVID-19 patients reveals distinct immunotypes with therapeutic implications. Science. 2020;369:369. doi: 10.1126/science.abc8511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Melsted P., Ntranos V., Pachter L. The barcode, UMI, set format and BUStools. Bioinformatics. 2019;35:4472–4473. doi: 10.1093/bioinformatics/btz279. [DOI] [PubMed] [Google Scholar]
- Ni L., Ye F., Cheng M.L., Feng Y., Deng Y.Q., Zhao H., Wei P., Ge J., Gou M., Li X., et al. Detection of SARS-CoV-2-Specific Humoral and Cellular Immunity in COVID-19 Convalescent Individuals. Immunity. 2020;52:971–977.e3. doi: 10.1016/j.immuni.2020.04.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nicholls J.M., Poon L.L., Lee K.C., Ng W.F., Lai S.T., Leung C.Y., Chu C.M., Hui P.K., Mak K.L., Lim W., et al. Lung pathology of fatal severe acute respiratory syndrome. Lancet. 2003;361:1773–1778. doi: 10.1016/S0140-6736(03)13413-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Osei-Owusu P., Charlton T.M., Kim H.K., Missiakas D., Schneewind O. FPR1 is the plague receptor on host immune cells. Nature. 2019;574:57–62. doi: 10.1038/s41586-019-1570-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raybould M.I.J., Kovaltsuk A., Marks C., Deane C.M. CoV-AbDab: the coronavirus antibody database. Bioinformatics. 2020 doi: 10.1093/bioinformatics/btaa739. Published online August 17, 2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ren X., Zhong G., Zhang Q., Zhang L., Sun Y., Zhang Z. Reconstruction of cell spatial organization from single-cell RNA sequencing data based on ligand-receptor mediated self-assembly. Cell Res. 2020;30:763–778. doi: 10.1038/s41422-020-0353-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schoggins J.W., Rice C.M. Interferon-stimulated genes and their antiviral effector functions. Curr. Opin. Virol. 2011;1:519–525. doi: 10.1016/j.coviro.2011.10.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schulte-Schrepping J., Reusch N., Paclik D., Bassler K., Schlickeiser S., Zhang B., Kramer B., Krammer T., Brumhard S., Bonaguro L., et al. Severe COVID-19 Is Marked by a Dysregulated Myeloid Cell Compartment. Cell. 2020;182:1419–1440.e23. doi: 10.1016/j.cell.2020.08.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Silvin A., Chapuis N., Dunsmore G., Goubet A.G., Dubuisson A., Derosa L., Almire C., Henon C., Kosmider O., Droin N., et al. Elevated Calprotectin and Abnormal Myeloid Cell Subsets Discriminate Severe from Mild COVID-19. Cell. 2020;182:1401–1418.e18. doi: 10.1016/j.cell.2020.08.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singh M., Bansal V., Feschotte C. A Single-Cell RNA Expression Map of Human Coronavirus Entry Factors. Cell Rep. 2020;32:108175. doi: 10.1016/j.celrep.2020.108175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stuart T., Butler A., Hoffman P., Hafemeister C., Papalexi E., Mauck W.M., 3rd, Hao Y., Stoeckius M., Smibert P., Satija R. Comprehensive Integration of Single-Cell Data. Cell. 2019;177:1888–1902.e1821. doi: 10.1016/j.cell.2019.05.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Su Y., Chen D., Yuan D., Lausted C., Choi J., Dai C.L., Voillet V., Duvvuri V.R., Scherler K., Troisch P., et al. ISB-Swedish COVID19 Biobanking Unit Multi-Omics Resolves a Sharp Disease-State Shift between Mild and Moderate COVID-19. Cell. 2020;183:1479–1495.e20. doi: 10.1016/j.cell.2020.10.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sugimoto M.A., Vago J.P., Teixeira M.M., Sousa L.P. Annexin A1 and the Resolution of Inflammation: Modulation of Neutrophil Recruitment, Apoptosis, and Clearance. J. Immunol. Res. 2016;2016:8239258. doi: 10.1155/2016/8239258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sungnak W., Huang N., Bécavin C., Berg M., Queen R., Litvinukova M., Talavera-López C., Maatz H., Reichart D., Sampaziotis F., et al. HCA Lung Biological Network SARS-CoV-2 entry factors are highly expressed in nasal epithelial cells together with innate immune genes. Nat. Med. 2020;26:681–687. doi: 10.1038/s41591-020-0868-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang X., Yang M., Duan Z., Liao Z., Liu L., Cheng R., Fang M., Wang G., Liu H., Xu J., et al. Transferrin receptor is another receptor for SARS-CoV-2 entry. bioRxiv. 2020 2020.2010.2023.350348. [Google Scholar]
- Thachil J., Lisman T. Pulmonary Megakaryocytes in Coronavirus Disease 2019 (COVID-19): Roles in Thrombi and Fibrosis. Semin. Thromb. Hemost. 2020;46:831–834. doi: 10.1055/s-0040-1714274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Todd D.J., McHeyzer-Williams L.J., Kowal C., Lee A.H., Volpe B.T., Diamond B., McHeyzer-Williams M.G., Glimcher L.H. XBP1 governs late events in plasma cell differentiation and is not required for antigen-specific memory B cell development. J. Exp. Med. 2009;206:2151–2159. doi: 10.1084/jem.20090738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Traag V.A., Waltman L., van Eck N.J. From Louvain to Leiden: guaranteeing well-connected communities. Sci. Rep. 2019;9:5233. doi: 10.1038/s41598-019-41695-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vogl T., Tenbrock K., Ludwig S., Leukert N., Ehrhardt C., van Zoelen M.A.D., Nacken W., Foell D., van der Poll T., Sorg C., Roth J. Mrp8 and Mrp14 are endogenous activators of Toll-like receptor 4, promoting lethal, endotoxin-induced shock. Nat. Med. 2007;13:1042–1049. doi: 10.1038/nm1638. [DOI] [PubMed] [Google Scholar]
- Wang Y., Wang R., Zhang S., et al. iTALK: an R package to characterize and illustrate intercellular communication[J] BioRxiv. 2019:507871. [Google Scholar]
- Wen W., Su W., Tang H., Le W., Zhang X., Zheng Y., Liu X., Xie L., Li J., Ye J., et al. Immune cell profiling of COVID-19 patients in the recovery stage by single-cell sequencing. Cell Discov. 2020;6:31. doi: 10.1038/s41421-020-0168-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wolf F.A., Angerer P., Theis F.J. SCANPY: large-scale single-cell gene expression data analysis. Genome Biol. 2018;19:15. doi: 10.1186/s13059-017-1382-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wolock S.L., Lopez R., Klein A.M. Scrublet: Computational Identification of Cell Doublets in Single-Cell Transcriptomic Data. Cell Syst. 2019;8:281–291.e9. doi: 10.1016/j.cels.2018.11.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xie X., Liu M., Zhang Y., Wang B., Zhu C., Wang C., Li Q., Huo Y., Guo J., Xu C., et al. Single-cell Transcriptomic Landscape of Human Blood Cells. Natl. Sci. Rev. 2020 doi: 10.1093/nsr/nwaa180. Published online August 24, 2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu G., Wang L.G., Han Y., He Q.Y. clusterProfiler: an R package for comparing biological themes among gene clusters. OMICS. 2012;16:284–287. doi: 10.1089/omi.2011.0118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang C., Gadue P., Scott E., Atchison M., Poncz M. Activation of the megakaryocyte-specific gene platelet basic protein (PBP) by the Ets family factor PU.1. J. Biol. Chem. 1997;272:26236–26246. doi: 10.1074/jbc.272.42.26236. [DOI] [PubMed] [Google Scholar]
- Zhang L., Yu X., Zheng L., Zhang Y., Li Y., Fang Q., Gao R., Kang B., Zhang Q., Huang J.Y., et al. Lineage tracking reveals dynamic relationships of T cells in colorectal cancer. Nature. 2018;564:268–272. doi: 10.1038/s41586-018-0694-x. [DOI] [PubMed] [Google Scholar]
- Zhang F., Gan R., Zhen Z., Hu X., Li X., Zhou F., Liu Y., Chen C., Xie S., Zhang B., et al. Adaptive immune responses to SARS-CoV-2 infection in severe versus mild individuals. Signal Transduct. Target. Ther. 2020;5:156. doi: 10.1038/s41392-020-00263-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang J.Y., Wang X.M., Xing X., Xu Z., Zhang C., Song J.W., Fan X., Xia P., Fu J.L., Wang S.Y., et al. Single-cell landscape of immunological responses in patients with COVID-19. Nat. Immunol. 2020;21:1107–1118. doi: 10.1038/s41590-020-0762-x. [DOI] [PubMed] [Google Scholar]
- Zheng Y., Liu X., Le W., Xie L., Li H., Wen W., Wang S., Ma S., Huang Z., Ye J., et al. A human circulating immune cell landscape in aging and COVID-19. Protein Cell. 2020;11:740–770. doi: 10.1007/s13238-020-00762-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou P., Yang X.-L., Wang X.-G., Hu B., Zhang L., Zhang W., Si H.-R., Zhu Y., Li B., Huang C.-L., et al. A pneumonia outbreak associated with a new coronavirus of probable bat origin. Nature. 2020;579:270–273. doi: 10.1038/s41586-020-2012-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The processed gene expression data in this paper have been deposited into the NCBI GEO database: GSE158055. Visualization of this dataset can be found at http://covid19.cancer-pku.cn. The raw data are available from Genome Sequence Archive for human with accession ID: HRA001149 (https://ngdc.cncb.ac.cn/gsa-human/browse/HRA001149). Additional Supplemental Items are also available at Mendeley Data: https://dx.doi.org/10.17632/dvp4y5ttd5.1.