

Abstract
Coronavirus disease 2019 (COVID-19) is a current global health threat caused by the novel coronavirus severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2). Emerging evidence indicates that SARS-CoV-2 elicits a dysregulated immune response and a delayed interferon (IFN) expression in patients, which contribute largely to the viral pathogenesis and development of COVID-19. However, underlying mechanisms remain to be elucidated. Here, we report the activation and repression of the innate immune response by SARS-CoV-2. We show that SARS-CoV-2 RNA activates the RIG-I-MAVS-dependent IFN signaling pathway. We further uncover that ORF9b immediately accumulates and antagonizes the antiviral type I IFN response during SARS-CoV-2 infection on primary human pulmonary alveolar epithelial cells. ORF9b targets the nuclear factor κB (NF-κB) essential modulator NEMO and interrupts its K63-linked polyubiquitination upon viral stimulation, thereby inhibiting the canonical IκB kinase alpha (IKKα)/β/γ-NF-κB signaling and subsequent IFN production. Our findings thus unveil the innate immunosuppression by ORF9b and provide insights into the host-virus interplay during the early stage of SARS-CoV-2 infection.
Keywords: COVID-19, SARS-CoV-2, innate immune response, RIG-I-MAVS signaling, interferon
Graphical Abstract
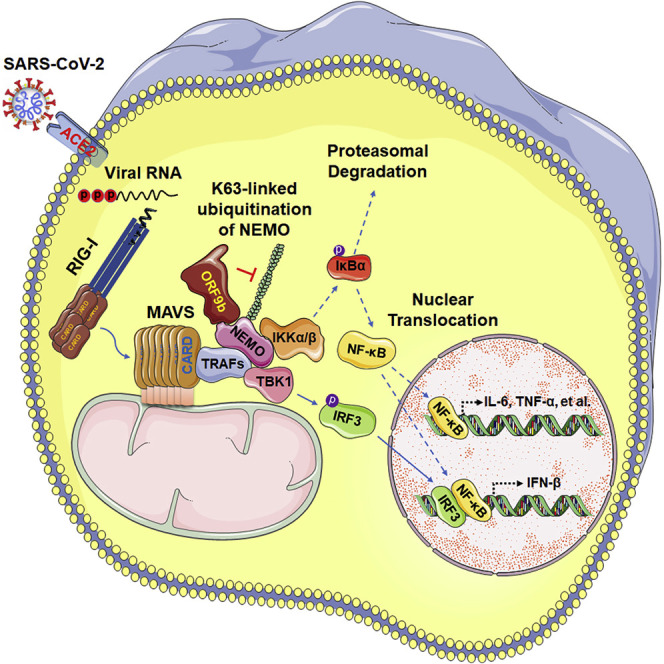
The reason why COVID-19 patients experience innate immunosuppression at the early stage of SARS-CoV-2 infection is unclear. Wu et al. report that ORF9b accumulates immediately during SARS-CoV-2 infection and antagonizes the RIG-I-MAVS antiviral type I interferon response by interrupting the K63-linked polyubiquitination of the interferon signaling modulator NEMO.
Introduction
Since the outbreak of severe acute respiratory syndrome (SARS) and Middle East respiratory syndrome (MERS), coronavirus (CoV)-associated diseases have emerged as a major threat to public health. Coronavirus disease 2019 (COVID-19), a novel coronavirus disease caused by SARS-CoV-2 (also referred to as 2019-nCoV), was first reported in Wuhan, China, in December 2019 and was later declared by the World Health Organization as a global pandemic (Xu et al., 2020; Zhu et al., 2020). Growing evidence supports the notion that SARS-CoV-2 had been circulating unnoticed around the world before the first official detection in China (Amendola et al., 2021). It is suggested that SARS-CoV-2 probably originates from bats, the same natural reservoirs of the highly pathogenic SARS-CoV and MERS-CoV (Wölfel et al., 2020; Zhou et al., 2020). In comparison with SARS and MERS, COVID-19 has a lower mortality rate but transmits much more efficiently, leading to a global case count of 90,054,813 and death count of 1,945,610 (as of January 13, 2021; https://covid19.who.int). To date, there are no effective antiviral drugs approved for COVID-19, which is largely attributed to the specific viral pathogenesis and host immunity.
The clinical features of COVID-19 patients and their immune response to SARS-CoV-2 are quite different from other respiratory viruses. Most COVID-19 cases are asymptomatic or exhibit mild to moderate symptoms during the early phase of SARS-CoV-2 infection. Pharyngeal virus shedding is very high during the first week of symptoms, with the peak of viral RNA concentrations reached at day 4 (Wölfel et al., 2020). However, for SARS, it takes 7–10 days to reach the peak (Peiris et al., 2003). Notably, no significant difference was detected in the viral load in upper respiratory specimens between asymptomatic patients and symptomatic patients, suggesting the high transmission potential of asymptomatic patients (Zou et al., 2020). It is thus believed that SARS-CoV-2 subverts patients’ innate immune response, the first line of host defense for rapidly eliminating viruses. Along with the development of COVID-19, ~20% patients rapidly progress to acute respiratory distress syndrome (ARDS) and experience a “cytokine storm,” which is characterized by the excessive levels of pro-inflammatory cytokines (e.g., tumor necrosis factor alpha [TNF-α], interleukin-6 [IL-6], IL-1β, IL-2, IL-8, IL-17, G-CSF, GM-CSF, IP10, MCP1, MIP-1α, and MIP-1β) in the plasma (Huang et al., 2020; Qin et al., 2020; Tan et al., 2020). Type I and III interferons (IFN-β and IFN-γ), however, stayed at the basal level in COVID-19 patients, as well as in SARS-CoV-2-infected cell and animal models, indicating a dysregulated innate immune defense (Blanco-Melo et al., 2020). The interrupted and imbalanced host response to SARS-CoV-2 drives the development of COVID-19, whereas the underlying mechanisms remain unknown.
Entry of SARS-CoV and SARS-CoV-2 into host cells relies on the same receptor, surface angiotensin-converting enzyme 2 (ACE2), which recognizes the viral spike protein primed by the serine protease TMPRSS2 (Hoffmann et al., 2020; Wan et al., 2020; Yan et al., 2020). After viral entry, RIG-I-like receptors (RLRs) (RIG-I and MDA5) and endosomal Toll-like receptors 3, 7, and 8 (TLR3, TLR7, and TLR8, respectively) sense the CoV genomic single-stranded RNA (ssRNA) or replication-intermediate double-stranded RNA (dsRNA) and subsequently transduce the signal to downstream adaptors (MAVS, MyD88, and TRIF) (Park and Iwasaki, 2020). Two signaling cascades involving diverse IκB kinases (IKKs), namely, TBK1/IKKε and IKKα/β/γ (IKKγ is also known as NEMO), are activated through the signal transduction, leading to the activation of transcription factors IRF3 and nuclear factor κB (NF-κB) that are required for type I IFN and pro-inflammatory cytokines expression (Fitzgerald et al., 2003; Rothwarf et al., 1998). To keep pace with host defenses in the arms race, CoVs use a variety of evasion strategies to interrupt diverse processes in innate immunity. SARS-CoV nonstructural protein 16 (nsp16) methylates the 5′ cap of viral mRNAs to mimic cellular mRNAs, thus facilitating the viral evasion of recognition by MDA5 (Bouvet et al., 2010; Menachery et al., 2014). SARS-CoV nsp3/papain-like protease (PLpro), ORF3b, and ORF6 interfere with the phosphorylation and the nuclear translocation of IRF3, which is the same target of MERS-CoV structural protein M and ORF4b/4b/5 (Devaraj et al., 2007; Kopecky-Bromberg et al., 2007; Yang et al., 2013). SARS-CoV ORF6 can further limit the IFN effector function by blocking the nuclear import of STAT1 and IFN-stimulated gene (ISG) expression (Frieman et al., 2007). Although SARS-CoV-2 is genetically related to SARS-CoV, it is imperative to determine whether SARS-CoV-2 stimulates the activation of shared signaling pathways. In particular, the low amino acid homology shared by accessory proteins from SARS-CoV and SARS-CoV-2 might translate into different pathophysiological outcomes.
The accessory protein ORF9b was reported to accumulate rapidly within the 24-h time of SARS-CoV-2 infection, during which the delayed IFN-β production was observed (Bojkova et al., 2020; Lei et al., 2020). Robust antibody response to SARS-CoV-2 ORF9b was identified in COVID-19 patients, suggesting a critical role of ORF9b in the host-virus interplay (Jiang et al., 2020a). ORF9b from SARS-CoV has already been defined as an IFN antagonist by targeting the mitochondrial protein MAVS, an adaptor essential for the RIG-I/MDA5 antiviral signaling and IFN-β production (Shi et al., 2014). Similarly, an interaction between SARS-CoV-2 ORF9b and another mitochondrial protein TOM70 was revealed, implying that SARS-CoV-2 ORF9b might interfere with the IFN signaling (Gordon et al., 2020a, 2020b; Jiang et al., 2020b). Here, we report the activation of a certain antiviral signaling by SARS-CoV-2 RNA and the innate immunosuppression by ORF9b during viral infection of human airway epithelial cells. We further dissect the molecular mechanism by which ORF9b inhibits the antiviral signal transduction and antagonizes the IFN response. Moreover, the target signaling molecule and functional motif of ORF9b have been uncovered. Our studies thus extend the understanding of SARS-CoV-2 immunopathology and shed light on the screening of COVID-19 drug targets.
Results
SARS-CoV-2 RNA activates the RIG-I-MAVS signaling pathway
It was supposed that SARS-CoV-2 and its counterpart SARS-CoV activate shared signaling pathways due to their high genomic similarity (Park and Iwasaki, 2020). We thus sought to determine whether the reported RIG-I/MDA5-MAVS signaling in response to SARS-CoV is also stimulated by SARS-CoV-2. To this end, RIG-I-, MDA5-, or MAVS-deficient HEK293T cell lines were generated using the CRISPR-Cas9 gene-editing system (Figure S1A). We stimulated wild-type (WT) and defective HEK293T cells with viral RNA isolated from Vero E6 cells infected with SARS-CoV-2 or vesicular stomatitis virus (VSV), a well-known stimulus of RIG-I-MAVS signaling. Compared with mock RNA, both SARS-CoV-2 and VSV RNA dramatically induced the expression of IFN-β-encoding gene IFNB1 in WT and MDA5 −/− cells in a dose-dependent manner (Figure 1 A). As a result of IFN-β production, expression of ISGs like RIG-I and MDA5 in WT cells was remarkably increased (Figure S1A). In contrast, no induction of IFNB1 was detected in either RIG-I −/− or MAVS −/− cells under viral RNA stimulation. Considering VSV is sensed by RIG-I rather than MDA5 (Kato et al., 2006), we thus conclude that RIG-I recognizes SARS-CoV-2 RNA and initiates the MAVS-dependent IFN signaling pathway.
Figure 1.

SARS-CoV-2 ORF9b suppresses viral-RNA-induced IFN production through RIG-I-MAVS signaling
(A) Induction of IFNB1 by SARS-CoV-2 RNA relies on RIG-I and MAVS. HEK293T wild-type (WT), DDX58−/− (RIG-I−/−), IFIH1−/− (MDA5−/−), and MAVS−/− cells were transfected for 12 h with indicated amounts of RNA from mock-infected Vero E6 cells; and viral RNA was isolated from either SARS-CoV-2- or VSV-infected cells. qPCR was conducted to determine the induction of IFNB1 mRNA. See also Figure S1A. Data are represented as means ± SDs calculated from three independent experiments.
(B–D) Induction of IFNB1 and accumulation of ORF9b during a 24-h period of SARS-CoV-2 infection. As shown in the experimental scheme, HPAEpiC and Caco-2 cells were either mock infected or infected with SARS-CoV-2 at a multiplicity of infection (MOI) of 1 for the indicated time and were then collected for subsequent measurement (B). SARS-CoV-2-induced IFNB1 mRNA levels were measured by qPCR, with the IFNB1 levels of VSV infection shown as a control (C). Cell lysates were subjected to fluorescence quantification immunoblotting for measuring the ORF9b protein levels in individual sample, which were converted into concentrations (ng/1 × 106 cells) (D). Data are represented as means ± SDs calculated from three biological replicates in the same experiment. See also Figures S1B and S1C.
(E–G) Inhibition of SARS-CoV-2 RNA-induced type I IFN response by ectopically expressed ORF9b under near-physiological levels. HPAEpiC were transfected with empty vector or increasing doses of expressing vector for FLAG-tagged ORF9b. At 24 h post-transfection, cells were transfected with 100 ng mock RNA or SARS-CoV-2 RNA as described in (A) for another 12 h. PCR was conducted to determine the expression of IFNB1 (E), ISG15 (F), and TNF (G). See also Figure S1D. Data are represented as means ± SDs calculated from three independent experiments (∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.001; t test).
(H) Dose-dependent inhibition of viral-RNA-induced IFNB1 activation by ORF9b in HEK293T cells. Similar to (A), except that HEK293T cells were transfected with increasing doses of empty vectors or FLAG-ORF9b expression vectors for 24 h before being stimulated with viral RNA. See also Figure S1E. Data are represented as means ± SDs calculated from three independent experiments (∗p < 0.05; t test).
(I) Inhibitory effects of ORF9b on SeV-, VSV-, or poly(I:C)-induced IFN-β promoter activation. HEK293T cells were co-transfected with luciferase reporter plasmids plus empty vector or FLAG-ORF9b-expressing plasmid for 24 h and were non-stimulated (mock) or stimulated with SeV, VSV, or poly(I:C) for another 12 h. IFN-β luciferase (IFN-β-Luc) reporter activity is normalized to that of Renilla luciferase and shown as fold induction. Data are represented as means ± SDs calculated from three independent experiments (∗p < 0.05, ∗∗p < 0.01; t test).
ORF9b immediately accumulates during SARS-CoV-2 infection and antagonizes the antiviral IFN response
ORF9b was reported to elicit a strong antibody response in COVID-19 patients (Jiang et al., 2020a), inspiring us to speculate that ORF9b might play key roles in the immunopathogenesis of SARS-CoV-2. We first established a SARS-CoV-2 infection model using permissive Caco-2 and primary human pulmonary alveolar epithelia cells (HPAEpiC). At different time points during a 24-h period of infection, the IFNB1 mRNA and ORF9b protein levels in cells, as well as the viral RNA copies in supernatants, were measured (Figure 1B). In both HPAEpiC and Caco-2 cells, a fast virus proliferation and an immediate accumulation of ORF9b were observed during the 24-h infection (Figures 1C, S1B, and S1C). However, the IFNB1 expression was barely stimulated by SARS-CoV-2 compared with that found under VSV stimulation (Figure 1D), appearing a previously reported IFN antagonistic activity upon SARS-CoV-2 infection (Blanco-Melo et al., 2020; Lei et al., 2020). The time of ORF9b accumulation overlapped that of IFN antagonism, suggesting that ORF9b may serve as an IFN antagonist.
We next examined whether ORF9b has an IFN antagonistic effect. HPAEpiC were introduced into ectopically expressed ORF9b and then stimulated with SARS-CoV-2 RNA. In the absence of ORF9b, SARS-CoV-2 RNA induced the expression of IFNB1 and downstream antiviral gene ISG15, as well as the inflammatory cytokine gene TNF (Figures 1E–1G). Under the near-physiological expression levels, ectopically expressed ORF9b exhibited an apparent IFN antagonistic activity by reducing the viral-RNA-induced IFN response (Figures 1E–1G and S1D). The IFN antagonistic activity of ORF9b was further tested in HEK293T cells stimulated by either SARS-CoV-2 RNA or VSV RNA (Figures 1H and S1E). In addition, ORF9b suppressed the activation of the IFN-β promoter in response to various stimuli of the RIG-I-MAVS signaling, such as Sendai virus (SeV), VSV, and poly(I:C) (Figure 1I). Given that the human airway epithelium BEAS-2B and Calu-3 cells are susceptible to both SARS-CoV-2 and VSV infection (Hoffmann et al., 2020), we established VSV-infected BEAS-2B and Calu-3 cell models to further study the IFN antagonistic activity of ORF9b. It was shown that the secretive expression of IFN-β in VSV-infected BEAS-2B, Calu-3, and HEK293T cells was inhibited by ORF9b (Figures 2 A and S2A). In line with the suppression of IFNB1 induction, expression of pro-inflammatory cytokine and chemokine genes IL-6, TNF, MCP-1 (CCL2), IP-10 (CXCL10), and ISG15 was hindered by ORF9b (Figure 2B). Due to the compromised IFN-β production and antiviral response, VSV amplified more efficiently in BEAS-2B, Calu-3, and HEK293T cells expressing ORF9b than the control group (Figures 2C and S2B). Consistently, VSV titers were elevated in the presence of ORF9b (Figures 2D and S2C). Taken together, our data demonstrate that SARS-CoV-2 ORF9b antagonizes the antiviral type I IFN response during viral infection.
Figure 2.

SARS-CoV-2 ORF9b antagonizes the antiviral IFN response in a variety of human cells
(A) ORF9b-mediated suppression of IFN-β production in human airway epithelial cells. At 24 h post-transfection of empty vector or FLAG-ORF9b-expressing plasmid, various human cells were uninfected (mock) or infected with VSV for 24 h. ELISA was conducted to measure the IFN-β production in BEAS-2B, Calu-3, and HEK293T cells. Data are represented as means ± SDs calculated from three independent experiments (∗p < 0.05, ∗∗p < 0.01; t test). See also Figure S2A.
(B) Inhibition of virally induced cytokine and chemokine expression by ORF9b. Experiments were conducted as described in (A). qPCR was conducted to determine the induction of IFNB1, IL-6, TNF, ISG15, IP-10, and MCP-1. Data are represented as means ± SDs calculated from three independent experiments (∗p < 0.05, ∗∗p < 0.01; t test).
(C and D) BEAS-2B cells were transfected with empty vector and FLAG-ORF9b-expressing plasmid for 24 h and then infected with VSV-GFP for another 24 h. Fluorescent images were taken to examine VSV proliferation (C). Plaque assay was conducted to quantitate VSV titers (D). Scale bar, 50 μm. Data are represented as means ± SDs calculated from three independent experiments (∗∗p < 0.01; t test). See also Figures S2B and S2C.
ORF9b interferes with the activation of NF-κB
Next, we investigated the mechanism by which ORF9b disrupts RIG-I-MAVS antiviral signaling. Induction of IFNB1 depends on the activation of two nuclear transcription factors, namely, NF-κB and IRF3. Thus, we examined the effects of ORF9b on the activation of IFN-β, NF-κB, and IRF3 that were induced by viral infection and the RIG-I-MAVS signaling component expression (Figure S3A). ORF9b inhibited the activation of IFN-β and NF-κB promoters following viral infection or expression of RIG-I(N) (the constitutively active form of RIG-I) and MAVS, whereas the IRF3 promoter activation was not affected (Figures 3 A–3C). This inhibitory effect, however, was not exhibited on IFN-β and NF-κB promoter activation induced by the expression of TBK1, IRF3(S396D), or IKKβ, the signaling components downstream of RIG-I and MAVS. It is thus supposed that SARS-CoV-2 ORF9b functions downstream of RIG-I/MAVS and specifically inhibits the NF-κB signaling pathway.
Figure 3.

The N-terminus of ORF9b mediates its interaction with NEMO upon viral infection
(A–C) Inhibitory effects of ORF9b on the activation of IFN-β/NF-κB/IRF3 promoters by SeV and the RIG-I-MAVS signaling components. Similar to Figure 1C, except that the luciferase reporter activities were induced by SeV infection for 12 h or by transfection of RIG-I(N)-, MAVS-, TBK1-, IRF3(S396D)-, and IKKβ-expressing vectors into HEK293T cells for 24 h. IFN-β-Luc (A), NF-κB-Luc (B), and IRF3-Luc (C) reporter activities are normalized to that of Renilla luciferase and shown as fold induction. Data are represented as means ± SDs calculated from three independent experiments (∗p < 0.05, ∗∗p < 0.01, N.S., non-significant; t test). See also Figure S3A.
(D) Co-immunoprecipitation (coIP) determining the interaction between ORF9b and the RIG-I-MAVS signaling components. HEK293T cells were transfected with plasmid encoding HA-ORF9b-GFP, together with various expressing vectors for the FLAG-tagged RIG-I-MAVS signaling components as indicated. At 24 h post-transfection, cells were infected with or without VSV for 12 h. Immunoprecipitation was conducted using anti-hemagglutinin (HA) beads. See also Figure S3B.
(E) Cellular colocalization of ORF9b and endogenous NEMO. NCI-H1299 cells were co-transfected with expressing vector for HA-ORF9b-GFP for 24 h and were mock-infected or infected with SeV for another 12 h. After immunofluorescent staining of cells with anti-NEMO and Alexa Fluor 594-conjugated secondary antibodies, fluorescent images were taken. Nucleus was labeled with DAPI. Scale bar, 10 μm. See also Figure S3C.
(F) CoIP mapping the motif of ORF9b that is essential for its interaction with NEMO. Plasmid encoding FLAG-NEMO was transfected into HEK293T cells together with various vectors expressing HA-tagged ORF9b and its mutants as indicated. At 24 h post-infection, cells were mock infected or infected with VSV for 12 h. Immunoprecipitation was conducted using anti-FLAG beads. See also Figures S3D and S3E.
(G) Immunoprecipitation of ORF9b with endogenous NEMO under SARS-CoV-2 RNA stimulation. HPAEpiC were transfected with expressing vectors for FLAG-tagged ORF9b or ORF9bΔN30 for 24 h and were then stimulated with SARS-CoV-2 RNA for the indicated time. Immunoprecipitation using anti-flag beads and subsequent immunoblotting was conducted to examine the endogenous NEMO protein levels in each individual sample.
ORF9b interacts with NEMO under viral stimulation through its N-terminus
To determine which signaling component (s) is/are targeted by ORF9b, we performed co-immunoprecipitation (coIP) by co-expressing HA-ORF9b-GFP and a variety of FLAG-tagged RIG-MAVS signaling components in HEK293T cells. As shown in Figure 3D, a detectable level of FLAG-NEMO was immunoblotted in the IP products of HA-ORF9b-GFP. It was notable that the interaction between ORF9b and NEMO was dramatically enhanced under the stimulation by VSV. On the contrary, no interaction was observed between GFP and NEMO in either mock-infected or VSV-infected cells (Figure S3B). The virally induced intracellular interaction between ORF9b and endogenous NEMO was further validated by colocalization assay, as the fluorescent signal of ORF9b-GFP almost overlapped with that of endogenous NEMO in the cytoplasm of human lung adenocarcinoma NCI-H1299 cells upon SeV infection (Figure 3E). Nevertheless, the intracellular colocalization of GFP and NEMO was not detected under viral stimulation (Figure S3C).
Next, we sought to map the motif of ORF9b that is essential for its interaction with NEMO. An unpublished crystallization study dissected the structure of ORF9b from SARS-CoV-2 (https://www.rcsb.org/; PDB: 6Z4U). SARS-CoV-2 ORF9b protein consists of two alpha helices (α1 and α2) and eight beta sheets (β1–β8), with ~72% amino acid (aa) identity to ORF9b from SARS-CoV (Figures S3D and S3E). Based on the sequence and structural analysis, deletions of the secondary elements in SARS-CoV-2 ORF9b were conducted, generating three ORF9b mutants. It was found that the N-terminal 1- to 30-aa deletion mutant ORF9bΔN30, which lacks α1, β1, and β2, cannot associate with NEMO under viral stimulation (Figure 3F). In contrast, deletions of other secondary elements in ORF9b had no impacts on the ORF9b-NEMO interaction. The virally induced and motif-dependent ORF9b-NEMO interaction was further validated in primary cells, as endogenous NEMO could be immunoprecipitated by overexpressed ORF9b rather than ORF9bΔN30 only if HPAEpiC were stimulated with SARS-CoV-2 viral RNA (Figure 3G). Our findings thus unveil that ORF9b targets NEMO through its N-terminus upon viral infection.
ORF9b interrupts the K63-linked polyubiquitination of NEMO and inhibits the IKKα/β/γ-NF-κB signaling
During the antiviral signal transduction, polyubiquitination of NEMO is a key event in the activation of NF-κB (Arimoto et al., 2010; Wu et al., 2006). Having demonstrated that ORF9b interacts with NEMO and interferes with the NF-κB activation, we next tested if ORF9b impairs the ubiquitination of NEMO. For this purpose, ubiquitin (Ub) and NEMO were ectopically expressed in virally stimulated HEK293T cells in the presence of ORF9b or ORF9bΔN30. We found that NEMO underwent massive ubiquitination under the VSV stimulation (Figure 4 A). Remarkably, the levels of Ub conjugation to NEMO were decreased by ORF9b but not ORF9bΔN30, which lost the capability of targeting NEMO. We further observed that ORF9b exhibited inhibitory effects on the ubiquitination of NEMO in a dose-dependent manner but had no impacts on the mRNA levels or the protein levels of endogenous NEMO (Figures 4B and S4A).
Figure 4.

ORF9b inhibits the canonical NF-κB signaling pathway by interrupting the K63-linked polyubiquitination of NEMO
(A) Inhibitory effects of ORF9b on the ubiquitination of NEMO under viral stimulation. Expressing vectors for HA-ubiquitin (HA-Ub), FLAG-NEMO, and V5-tagged ORF9b were transfected into HEK293T cells as indicated for 24 h. Cells were then infected with or without VSV for 12 h and subjected to immunoprecipitation using anti-FLAG beads.
(B) Dose-dependent inhibition of virally induced Ub conjugation to NEMO by ORF9b. Similar to (A), except that an increasing dose of the V5-ORF9b-expressing vector was transfected into HEK293T cells. See also Figure S4A.
(C) Effects of ORF9b on the conjugation of diverse polyubiquitin linkages to NEMO under viral stimulation. Plasmids encoding various HA-Ub (WT, KallR, K48 only, K63 only, K48R, and K63R as indicated), together with expressing vectors for FLAG-NEMO and V5-ORF9b, were co-transfected into HEK293T cells. Infection and immunoprecipitation were conducted as described in (A).
(D) Interruption of the IKKα/β/γ-NF-κB signaling by ORF9b. HEK293T cells were transfected for 24 h with empty vector or with V5-ORF9b- and V5-ORF9bΔN30-expressing vectors and were then infected with VSV for the indicated time (0, 4, 8, or 12 h). Cells were collected and subjected to immunoblotting analysis by using indicated antibodies. β-actin was immunoblotted as loading control.
(E) Inhibitory effects of ORF9b on the translocation of NF-κB/p65 into the nucleus. Transfection was performed as described in (D). HEK293T cells were then mock infected or infected with VSV for 12 h. Cytoplasmic (cytoplasm) and nuclear (nucleus) factions of cells were obtained using commercial reagents. Immunoblotting analysis was conducted using indicated antibodies.
(F and G) At 24 h post-transfection of empty vector or V5-ORF9b- and V5-ORF9bΔN30-expressing plasmids, HEK293T cells were uninfected (mock) or infected with VSV for 12 h. qPCR was conducted to determine the expression of IFNB1 (F) and IL-6 (G). Data are represented as means ± SDs calculated from three independent experiments (∗∗p < 0.01, ∗∗∗p < 0.001, N.S., non-significant; t test). See also Figure S4B.
In general, the type of ubiquitination depends on the conjugation of specific polyubiquitin chains to the target protein, thus determining the fate of the substrates. To figure out which type of ubiquitylation in NEMO was compromised by ORF9b, we introduced a series of Ub mutants and linkage-specific polyubiquitin chains into the experiment, as indicated in Figure 4C. NEMO was showed to be conjugated with the K63-linked polyubiquitin but not the K48-linked polyubiquitin (Figure 4C), consistent with previous findings that NEMO cannot sense the K48-linked polyubiquitination (Arimoto et al., 2010). In the presence of ORF9b, the K63-linked polyubiquitination of NEMO was significantly reduced. The inhibitory effects of ORF9b were also exhibited on the conjugation of K48R-linked polyubiquitin chains to NEMO, as the K63 residues were still present in the K48R mutant. When the K63 residue of the WT Ub chain was mutated to R, however, ORF9b could not affect the remaining levels of Ub conjugated to NEMO. These results suggest that ORF9b specifically interrupts the K63-linked polyubiquitination of NEMO, which is the major type of ubiquitylation in NEMO during the antiviral signal transduction.
In the canonical IKKα/β/γ-NF-κB signaling pathway, activation of the IKK complex and phosphorylation of IKKβ depend on the K63-linked polyubiquitination of NEMO, leading to the phosphorylation and the Ub-proteasome degradation of IκBα and subsequent translocation of NF-κB/p65 into the nucleus (Bhoj and Chen, 2009). Along with the VSV infection from 0 h to 12 h, the phosphorylation of IKKβ and IκBα and the degradation of IκBα were gradually observed in HEK293T cells (Figure 4D). ORF9b but not ORF9bΔN30 substantially decreased the levels of phosphorylated IKKβ (p-IKKβ) and p-IκBα, thereby leading to the recovery of IκBα protein levels during the VSV infection. On the other side, the phosphorylation and dimerization of IRF3, as a result of the TBK1/IKKε-IRF3 signaling activation, were not affected by ORF9b. In line with the inhibitory effects on IκBα degradation, translocation of cytoplasmic NF-κB/p65 into the nucleus was compromised by ORF9b but not ORF9bΔN30, as we observed a retention of NF-κB/p65 in the cytoplasm of HEK293T cells expressing ORF9b (Figure 4E). Moreover, we found that the loss-of-function mutant ORF9bΔN30 cannot inhibit the VSV-stimulated expression of IFNB1, IL-6, and TNF, the transcriptional regulation target genes of NF-κB (Figures 4F, 4G, and S4B). Altogether, our collective data uncover that ORF9b disrupts the K63-linked polyubiquitination of NEMO and disturbs the activation of IKKα/β/γ-NF-κB signaling.
Discussion
The innate antiviral system uses pattern recognition receptors to detect viral molecular signatures. Sensing CoV genomic ssRNA or replicated dsRNA mainly occurs in the cytosolic or endosomal compartment by TLRs and RLRs. TLR3-TRIF and TLR7-MyD88 signaling are defined as major innate immune pathways against SARS-CoV and MERS-CoV (Cervantes-Barragan et al., 2007; Channappanavar et al., 2019; Sheahan et al., 2008; Totura et al., 2015); however, the role of RIG-I/MDA5 and their downstream adaptor MAVS in response to CoV infection has not been well established. HEK293T cells are equipped with RLRs but are deficient in TLR pathways, thus emerging as an ideal cell model for elucidating the RLR-MAVS signaling. By performing genetic manipulation on HEK293T cells, our present work reveals that RIG-I but not MDA5 is responsible for sensing SARS-CoV-2 RNA and inducing type I IFN expression in the presence of MAVS. The preferential recognition of the viral RNA ligand by RIG-I and MDA5 has mostly been attributed to specific features, such as the 5′-triphosphate (5′-ppp) end, the length, and the panhandle secondary structure (Kato and Fujita, 2016). Thus, it would be intriguing to dissect the signatures of SARS-CoV-2 viral RNA captured by RIG-I and study the binding properties in future work.
Using a SARS-CoV-2 infection cell model, we showed that ORF9b immediately accumulated during a 24-h infection time, within which the IFN antagonism was observed. In the absence of ORF9b, SARS-CoV-2 RNA was found to activate the RIG-I-MAVS signaling and induced the IFN production, which was then diminished by recovery of ORF9b. The IFN antagonistic activity of ORF9b was also validated in VSV-infected human airway epithelial cells BEAS-2B and Calu-3, which are the primary SARS-CoV-2 infection targets that are susceptible to viral entry (Hoffmann et al., 2020). It is thus speculated that the viral positive-sense ssRNA facilitates a fast translation of ORF9b, which in turn antagonizes the viral-RNA-induced RIG-I-MAVS IFN signaling. To further study the pathophysiological significance of ORF9b during SARS-CoV-2 infection, generation of a recombinant SARS-CoV-2 lacking the ORF9b-encoding gene is substantially in need. On the other side, SARS-CoV ORF9b promotes MAVS degradation and subverts its downstream TBK1-IRF3 and IKKα/β/γ-NF-κB signaling cascades (Shi et al., 2014), whereas SARS-CoV-2 uses ORF9b to target the IKK regulator subunit NEMO that is specifically essential for NF-κB activation but not IRF3 activation. The difference in physiological function most probably attributes to the low sequence identify (~72%) and structure homology shared by these two ORF9b proteins, implying that SARS-CoV-2 adopts an immune evasion strategy that is different from that of SARS-CoV in ong-term evolution. Further comparative studies among the accessory proteins from SARS-CoV-2 and its counterparts would help to interpret why this novel CoV triggers a unique host immune response.
Ectopically expressed ORF9b was shown to be located to mitochondria and interacts with the mitochondrial protein TOM70 in resting cells (Gordon et al., 2020a, 2020b; Jiang et al., 2020b). Here, we report that ORF9b can interact with NEMO upon virus infection or SARS-CoV-2 RNA stimulation, although NEMO is not a mitochondrial protein. An explanation interpreting the virally induced ORF9b-NEMO interaction is as follows: as a vital platform for antiviral signaling, mitochondria undergo morphological changes and alterations in dynamics and membrane potential during viral infection, which lead to the rearrangement of mitochondrial proteins and translocation of signaling proteins to mitochondria (Jacobs and Coyne, 2013). ORF9b is located to mitochondria in which the immune MAVS signaling complex is assembled (Gordon et al., 2020a; Jiang et al., 2020b; Liu et al., 2013). During the antiviral signal transduction, TRAF-mediated polyubiquitination and recruitment of NEMO to the mitochondrial MAVS signaling complex would spatially increase the contact of NEMO with ORF9b, thereby bridging NEMO to ORF9b. Therefore, the ORF9b-NEMO interaction on mitochondria could be targeted as a potential therapeutic strategy to disrupt the host immunosuppression and virus biology. In addition, our work has dissected the functional motif of ORF9b, which would shed light on the screening of inhibitors targeting ORF9b.
The infection of SARS-CoV-2 and progression of COVID-19 exhibit a “two-stage” pattern. During the early stage, mild or asymptomatic COVID-19 patients undergo immunosuppression, which facilitates significant viral shedding and transmission (Tian et al., 2020). Because ORF9b has emerged as a potent IFN antagonist during the early stage of SARS-CoV-2 infection, it is convincible that SARS-CoV-2 uses the early protein ORF9b to mask the antiviral defense and inflammatory response in early-stage patients. When COVID-19 progresses to the late stage, severe patients develop ARDS and exhibit a hyperinflammatory immune state with an extreme production of pro-inflammatory cytokines (Blanco-Melo et al., 2020; Huang et al., 2020; Qin et al., 2020; Tan et al., 2020). Despite the exuberant inflammatory response, however, the production of IFN is still blocked in late-stage patients. The delayed IFN expression and imbalanced host response are probably attributed to the compromised IRF3 signaling branch, which is targeted by other SARS-CoV-2 accessory proteins such as ORF6 (Lei et al., 2020; Yuen et al., 2020). It is thus suspected that SARS-CoV-2 adopts a variety of strategies for efficient immunosuppression and amplification during different phases of infection. Recombinant SARS-CoV-2 lacking IFN antagonists can serve as a vaccine candidate against COVID-19. To seek efficient therapeutic agents for the treatment of COVID-19, screening of inhibitors against these potential drug targets warrants further investigation.
STAR★Methods
Key Resources Table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| RIG-I Rabbit mAb | Cell Signaling Technology | Cat# 3743 |
| MDA-5 Rabbit mAb | Cell Signaling Technology | Cat# 5321 |
| MAVS Rabbit mAb | Cell Signaling Technology | Cat# 24930 |
| SARS-CoV-2 ORF9b Rabbit pAb | ABclonal | Cat# A20260 |
| NF-κB p65 Rabbit mAb | Cell Signaling Technology | Cat# 8242 |
| IKKγ/NEMO Rabbit mAb | Abcam | Cat# ab178872 |
| IKKγ/NEMO Mouse mAb | Cell Signaling Technology | Cat# 2695 |
| PCNA Rabbit mAb | Cell Signaling Technology | Cat# 13110 |
| Phospho-IKKα/β (Ser176/180) Rabbit mAb | Cell Signaling Technology | Cat# 2697 |
| Phospho-IκBα (Ser32) Rabbit mAb | Cell Signaling Technology | Cat# 2859 |
| IκBα Rabbit mAb | Cell Signaling Technology | Cat# 4812 |
| IRF3 Rabbit mAb | Cell Signaling Technology | Cat# 4302 |
| Phospho-IRF3 (Ser396) Rabbit mAb | Cell Signaling Technology | Cat# 29047 |
| β-Actin Rabbit mAb | Cell Signaling Technology | Cat# 4970 |
| Flag-Tag Rabbit mAb | Cell Signaling Technology | Cat# 14793 |
| HA-Tag Rabbit mAb | Cell Signaling Technology | Cat# 3724 |
| V5-Tag Rabbit mAb | Cell Signaling Technology | Cat# 13202 |
| Anti-mouse IgG (H+L), Alexa Fluor® 594 Conjugate | Cell Signaling Technology | Cat# 8890 |
| Anti-Rabbit IgG (H+L), LICOR IRDye 800CW | Equl | Cat# 926-32211 |
| Anti-Rabbit IgG (H+L), HRP Conjugate | Promega | Cat# W4011 |
| Anti-Mouse IgG (H+L), HRP Conjugate | Promega | Cat# W4021 |
| Bacterial and virus strains | ||
| SARS-CoV-2 strain 2019-nCoV WIV04 | Wuhan Institute of Virology, Chinese Academy of Sciences | N/A |
| VSV-ΔM51-GFP | Wuhan Institute of Virology, Chinese Academy of Sciences | N/A |
| Sendai virus | Wuhan Institute of Virology, Chinese Academy of Sciences | N/A |
| Trans5α Chemically Competent Cell | TransGen Biotech | Cat# CD201-02 |
| Chemicals, peptides, and recombinant proteins | ||
| Dulbecco’s modified Eagle’s medium (DMEM) | Thermo Fisher Scientific | Cat# 11995073 |
| Minimum Essential Medium (MEM) | Thermo Fisher Scientific | Cat# 12571063 |
| DMEM: Nutrient Mixture F-12 (DMEM/F-12) | Thermo Fisher Scientific | Cat# 11320033 |
| RPMI-1640 medium | Thermo Fisher Scientific | Cat# 11875093 |
| Alveolar Epithelial Cell Medium (AEpiCM) | ScienCell Research Laboratories | Cat# 3201 |
| Certified Fetal Bovine Serum (FBS) | Biological Industries | Cat# 04-001-1A |
| Trypsin-EDTA (0.05%) | Thermo Fisher Scientific | Cat# 25300062 |
| Lipofectamine 3000 | Thermo Fisher Scientific | Cat# L3000015 |
| Penicillin-Streptomycin | Thermo Fisher Scientific | Cat# 15070063 |
| Phosphate-Buffered Saline (PBS) | Thermo Fisher Scientific | Cat# 10010023 |
| Dihydrochloride (DAPI) | Thermo Fisher Scientific | Cat# D1306 |
| Polyinosinic–polycytidylic acid sodium salt, poly(I:C) | Sigma-Aldrich | Cat# P1530 |
| HEPES | Sigma-Aldrich | Cat# H3375 |
| Triton X-100 | Sigma-Aldrich | Cat# X100 |
| Ampicillin | Sigma-Aldrich | Cat# A9518 |
| Protease Inhibitor Cocktail | Roche | Cat# 5892970001 |
| KpnI restriction enzyme | New England Biolabs | Cat# R3142S |
| HindIII restriction enzyme | New England Biolabs | Cat# R3104S |
| XhoI restriction enzyme | New England Biolabs | Cat# R0146S |
| XbaI restriction enzyme | New England Biolabs | Cat# R0145S |
| Agarose | Biowest | Cat# 3002 |
| Agarose (Low melting gel) | Solarbio | Cat# A8350 |
| ANTI-FLAG M2 Affinity Gel | Sigma | Cat# A2220 |
| Pierce Anti-HA Agarose beads | Thermo Fisher Scientific | Cat# 26181 |
| Critical commercial assays | ||
| Dual-Luciferase Reporter Assay System | Promega | Cat# E1910 |
| MiniBEST Viral RNA/DNA Extraction Kit | TAKARA | Cat# 9766 |
| PrimeScript™ RT reagent Kit with gDNA Eraser | TAKARA | Cat# RR047A |
| TB Green Premix Ex Taq II | TAKARA | Cat# RR820A |
| Human IFN-beta ELISA Kit | R&D Systems | Cat# 41410-1 |
| RNA simple total RNA kit | TIANGEN | Cat# DP419 |
| HyperScript 1st Strand cDNA Synthesis Kit | NovaBio | Cat# R201-2 |
| SYBR qPCR Mix | NovaBio | Cat# Q204-01 |
| High-Fidelity DNA Polymerase | NovaBio | Cat# G302 |
| Nuclear and Cytoplasmic Extraction Reagents | Thermo Fisher Scientific | Cat# 78833 |
| Deposited data | ||
| SARS-CoV-2 isolate Wuhan-Hu-1 genomic sequence | NCBI Reference Sequence | NC_045512 |
| SARS-CoV isolate Tor2 genomic sequence | NCBI Reference Sequence | NC_004718 |
| SARS-CoV-2 ORF9b protein sequence | NCBI Reference Sequence | P0DTD2.1 |
| SARS-CoV ORF9b protein sequence | NCBI Reference Sequence | YP_009825062 |
| SARS-CoV-2 ORF9b protein structure data | RSCB PDB database | PDB: 6Z4U |
| SARS-CoV ORF9b protein structure data | RSCB PDB database | PDB: 2CME |
| Experimental models: cell lines | ||
| Vero E6 cells | ATCC | Cat# CRL-1586 |
| HEK293T cells | ATCC | Cat# CRL-3216 |
| HPAEpiC | ScienCell Research Laboratories | Cat# 3200 |
| Caco-2 cells | ATCC | Cat# FS-0201 |
| BEAS-2B cells | ATCC | Cat# CRL-9609 |
| Calu-3 cells | ATCC | Cat# HTB-55 |
| NCI-H1299 cells | ATCC | Cat# CRL-5803 |
| HEK293T RIG-I−/− cells | This study | N/A |
| HEK293T MDA5−/− cells | This study | N/A |
| HEK293T MAVS−/− cells | This study | N/A |
| Oligonucleotides | ||
| For primer sequences see Table S1 | This study | N/A |
| Recombinant DNA | ||
| SARS-CoV-2 ORF9b cDNA | GENEWIZ | N/A |
| IFN-Beta_pGL3 | Gentili et al., 2015 | Addgene Depositor: Nicolas Manel, Cat# 102597 |
| pNF-κB-TA-luc | Beyotime Biotechnology | Cat# D2207 |
| pRL-TK | Beyotime Biotechnology | Cat# D2760 |
| pIRF3 RE-Luc | COBIOER | Cat# CBV20079 |
| pcDNA3.1 | Thermo Fisher Scientific | Cat# V79020 |
| pDsRed2-N1 | MIAOLINGBIO | Cat# P0139 |
| pRK5-HA-Ubiquitin-K0 | Lim et al., 2005 | Addgene Depositor: Ted Dawson Lab, Cat# 17603 |
| pCMV-HA-Ub-K48R | MIAOLINGBIO | Cat# P8355 |
| pCMV-HA-Ub-K63R | MIAOLINGBIO | Cat# P0855 |
| pcDNA-HA-Ub | Di Wang Lab (Guo et al., 2016) | N/A |
| pcDNA-HA-Ub K48-only | Di Wang Lab (Guo et al., 2016) | N/A |
| pcDNA-HA-Ub K63-only | Di Wang Lab (Guo et al., 2016) | N/A |
| pcDNA-Flag | This study | N/A |
| pcDNA-HA | This study | N/A |
| pcDNA-V5 | This study | N/A |
| pcDNA-Flag-ORF9b | This study | N/A |
| pcDNA-HA-ORF9b | This study | N/A |
| pcDNA-HA-ORF9b-GFP | This study | N/A |
| pcDNA-HA -GFP | This study | N/A |
| pcDNA-HA-ORF9bΔN30 | This study | N/A |
| pcDNA-HA-ORF9bΔ41-62 | This study | N/A |
| pcDNA-HA-ORF9bΔC30 | This study | N/A |
| pcDNA-V5-ORF9b | This study | N/A |
| pcDNA-V5-ORF9bΔN30 | This study | N/A |
| pcDNA-Flag-RIG-I(N) | This study | N/A |
| pcDNA-Flag-RIG-I | This study | N/A |
| pcDNA-Flag-MAVS | This study | N/A |
| pcDNA-Flag-NEMO | This study | N/A |
| pcDNA-Flag-TBK1 | This study | N/A |
| pcDNA-Flag-IRF3 | This study | N/A |
| pcDNA-Flag-IRF3S396D | This study | N/A |
| pcDNA-Flag-IKKβ | This study | N/A |
| Software and algorithms | ||
| GraphPad Prism 8 | GraphPad Prism | https://www.graphpad.com/ |
| ESPript | Robert and Gouet, 2014 | http://espript.ibcp.fr/ESPript/cgi-bin/ESPript.cgi |
| PyMOL | PyMOL | https://pymol.org/2/ |
| Adobe Photoshop CS5 Extended | Adobe | https://www.adobe.com/ |
Resource availability
Lead contact
Further information and requests for resources and reagents may be directed to and will be fulfilled by the lead contact, Nan Qi (qinan@zjut.edu.cn).
Materials availability
Cell lines and plasmids generated for this study are available from the lead contact with a completed Materials Transfer Agreement.
Data and Code Availability
This study did not generate any unique datasets or code.
Experimental model and subject details
Cell cultures and viral strains
All cell lines were incubated at 37°C in a 5% CO2 humidified atmosphere, and were tested for mycoplasma-free. Primary human pulmonary alveolar epithelia cells (HPAEpiC, from ScienCell Research Laboratories), which were isolated from human lung tissues and immediately cryopreserved at Passage 0 (P0), were maintained in the recommended AEpiCM medium. Vero E6 and HEK293T cells were cultured in DMEM. BEAS-2B cells were in incubated DMEM/F-12. Caco-2 and Calu-3 cells were grown in MEM. NCI-H1299 cells were cultured in RPMI-1640 medium. All media were supplemented with 10% FBS, and 100 U/ml of penicillin-streptomycin.
The SARS-CoV-2 strain 2019-nCoV WIV04 was isolated from the bronchoalveolar lavage fluid of a confirmed COVID-19 patient by inoculating onto Vero E6 cells (Zhou et al., 2020), and was propagated in Vero E6 cells in this study. Recombinant virus VSV-ΔM51-GFP from Wuhan Institute of Virology was amplified in Vero E6 cells. Sendai virus (Cantell strain) from Wuhan Institute of Virology was used at a concentration of 20 HA units/ml. The experiments involving the SARS-CoV-2 virus were conducted in the biosafety level 3 (BSL-3) laboratory of Wuhan Institute of Virology, Chinese Academy of Sciences.
Method details
Plasmids
Complementary DNA (cDNA) containing coding sequence of ORF9b was synthesized by GENEWIZ (Suzhou, China), and inserted into pcDNA3.1. Listed expressing vectors were constructed by fusing the PCR amplicons with various tags to the N-terminus (for Flag, HA and V5) or C-terminus (for GFP) of open reading frames (ORFs), and were then cloned into pcDNA3.1 between appropriate restriction enzyme sites. All constructs were confirmed by DNA sequencing. Primers are listed in Table S1.
Electrotransfection of viral RNA or plasmids into HPAEpiC and BEAS-2B were conducted using the Nucleofector™ 2b Device (Lonza Bioscience, Switzerland) according to manufacture’s instruction. For other cells, transfection was performed using Lipofectamine 3000.
Virus infection
HPAEpiC and Caco-2 cells were seeded in 12-well plates at a density of 4 × 105 cells/well, and were incubated with SARS-CoV-2 for 1 hour at a multiplicity of infection (MOI) of 1. After removing the infectious liquid, cells were washed with PBS and maintained in culture medium supplement with 2% FBS. At 1, 5, 9 and 24 h post-infection, cell supernatants and lysates were subjected to qPCR for measuring viral RNA copies and the IFN levels. Fluorescence quantification immunoblotting was conducted to examine the protein levels of ORF9b.
For quantifying the SARS-CoV-2 RNA copies, viral RNA was isolated from infected cell supernatants using the MiniBEST Viral RNA/DNA Extraction Kit (Takara), and were then converted into cDNA using the PrimeScript™ RT reagent Kit with gDNA Eraser (Takara). RNA copies were quantified from cDNA by a standard curve method qPCR targeting SARS-CoV-2 S gene (Wang et al., 2020), using the TB Green® Premix Ex Taq II kit (Takara).
Viral RNA for transfection into HEK293T cells and HPAEpiC were originated from Vero E6 cells infected with SARS-CoV-2 (MOI = 0.1) and VSV (MOI = 0.1). At 24 h post-infection, cells were collected for viral RNA extraction.
Plaque assay
Various cell lines were infected with VSV at a MOI = 0.1 for indicated time (related to Figure 2A). Culture media containing recovered viruses were collected, and used for incubating HEK293T cells in 6-well plates with serial dilutions for 1 h. Cells were overlaid with 1% soft agarose in DMEM and cultured for 48 h. Plaques were displayed by staining cells with 0.1% crystal violet in DMEM, and were then quantified.
Immunofluorescence microscopy
For examining VSV proliferation in various cell lines, experiments were performed as described (Shi et al., 2020). Briefly, cells were infected with VSV-ΔM51-GFP at a MOI = 0.1. At the indicated time (12 h or 24 h) after infection, fluorescence images were taken.
For cellular colocalization of ORF9b with endogenous NEMO, NCI-H1299 cells were transfected with plasmid expressing ORF9b-GFP for 24 h, and were infected with or without SeV for another 12h. After being washed with PBS and fixed by 4% Paraformaldehyde, cells were stained with anti-NEMO antibody (1:1000, Cat# 2695, Cell Signaling Technology) and Alexa Fluor® 594 Conjugated secondary antibody (1:2500, Cat# 8890, Cell Signaling Technology). Nucleus was labeled with DAPI at 10 μg/ml for 15 min. Fluorescence images were taken by OLYMPUS BX51 microscope with 1000X oil immersion lens.
Immunoprecipitations
Constructs were transfected into HEK293T cells or HPAEpiC. At 36 h after transfection, cells were harvested and lysed in lysis buffer [HEPES 20 mM pH = 7.5, KCl 10 mM, MgCl2 5 mM, EGTA 0.5 mM, 1% Triton X-100, and 1% protease inhibitor cocktail]. After a brief centrifugation, the lysates were immunoprecipitated with anti-HA agarose beads or anti-Flag agarose beads at 4°C for 4 h, and the precipitants were washed three times with lysis buffer at 4°C. The extract-bead mixture was then resuspended in loading buffer, followed by immunoblotting analysis.
Generation of the RIG-I−/−, MDA5−/−, MAVS−/− HEK293T cell lines
RIG, MDA5 and MAVS knockout HEK293T cell lines were custom-generated using CRISPR/Cas9 gene-editing system by NovaBio (Shanghai, China). Cell lines were validated by genomic sequencing and immunoblotting analysis.
Dual-luciferase reporter assays
HEK293T cells were seeded in 12-well plates and transfected with ORF9b expressing vector or empty vector, together with 100 ng luciferase reporter plasmid and 20 ng Renilla luciferase plasmid phRL-TK. At 24 h post-transfection, cells were transfected with plasmids encoding various RIG-I-MAVS signaling components for 24 h, or transfected with 10 μg/ml poly(I:C) for 8 h, or infected with SeV and VSV for 12 h. Following the manufacturer’s instructions for Dual-Luciferase reporter Assay System, cell lysates were obtained to measure the luciferase activity by Synergy H1 (BioTek, Vermont, US).
Quantitative PCR (qPCR)
RNA from various cells was extracted using the RNA simple total RNA kit (Tiangen) according to the manufacturer’s instructions. cDNA was synthesized from RNA by reverse transcribing using the HyperScript 1st Strand cDNA Synthesis Kit (NovaBio), and was then subjected to real-time qPCR using the SYBR Green PCR mix (NovaBio). Data was normalized by the level of internal control GAPDH expression in each individual sample. The relative expression of target gene was calculated using the 2-ΔΔCt method. For viral RNA copies quantification, a standard curve method qPCR targeting SARS-CoV-2 S gene was used (Wang et al., 2020). Primers are listed in Table S1.
Fluorescence quantification immunoblotting
An appropriate portion of lysates from SARS-CoV-2 infected cells were subjected to SDS-PAGE, and were transferred to PVDF membrane. Immunoblotting was conducted using the anti-ORF9b antibody (1:1000, Cat# A20260, ABclonal) and IRDye labeled secondary antibodies (1:5000, Cat# 926-32211, Equl). Fluorescence signal was detected by the Odyssey Infrared Imager (Gene company, Hong Kong, China), generating the fluorescence values. Serial dilutions (0.5 ng, 2.5 ng, 5 ng, 15 ng) of Flag-tagged ORF9b protein were used for accurate quantitative analysis. The concentration of individual sample (per 2 × 105 cells) was calculated by the linear equation.
Extraction of cytoplasmic and nuclear proteins
Following the manufacturer’s instructions for Nuclear and Cytoplasmic Extraction Reagents (Thermo Fisher Scientific), cells were harvested with trypsin-EDTA and centrifuged at 500 g for 5 mins. According to the number of cells, an appropriate amount of Cytoplasmic Extraction Reagent containing protease inhibitors was added to extract cytoplasmic protein. The insoluble fraction containing nuclei was suspended in Nuclear Extraction Reagent for extracting nuclear proteins.
ELISA
Concentrations of the IFN-β in cell culture supernatants were measured using the Human IFN-beta ELISA Kit (R&D Systems) according to the manufacturer’s instructions.
Quantification and statistical analysis
Data are represented as mean values with error bars indicating standard deviations (±SD) calculated from three independent experiments, or from three biological replicates in the same experiment. Statistical significance between two groups was determined by unpaired two-tailed Student’s t test using GraphPad Prism 8. Differences were significant when p < 0.05.
Acknowledgments
This work was supported by grants from the National Key Research and Development Program of China (2018YFA0900404); National Natural Science Foundation of China (31870864); National Science Foundation for Young Scientists of China (31901011); Fundamental Research Funds for the Provincial Universities of Zhejiang (RF-B2020003); and Research Program on COVID-19 of Zhejiang University of Technology. We thank Prof. Di Wang (Zhejiang University School of Medicine), Prof. Xi Zhou (Wuhan Institute of Virology), and Dr. Yanjing Li (National Facility for Protein Science in Shanghai) for generously providing experimental materials and insightful discussion.
Author contributions
N.Q., B.-C.Y., L.-K.Z., and Y.S. conceived and designed the study. J. Wu, Y.S., X.P., S.W., R.H., T.Z., H.T., W.D., L.W., and J. Wo. conducted experiments. L.-K.Z., X.P., J.M., and Y.Q. established the cell model of SARS-CoV-2 infection. J. Wu., Y.S., X.P., K.Y., B.-C.Y., and N.Q. analyzed the data. N.Q. and Y.S. prepared the manuscript. All authors discussed the results and commented on the manuscript.
Declaration of interests
The authors declare no competing interests.
Published: February 16, 2021
Footnotes
Supplemental Information can be found online at https://doi.org/10.1016/j.celrep.2021.108761.
Supplemental information
References
- Amendola A., Bianchi S., Gori M., Colzani D., Canuti M., Borghi E., Raviglione M.C., Zuccotti G.V., Tanzi E. Evidence of SARS-CoV-2 RNA in an Oropharyngeal Swab Specimen, Milan, Italy, Early December 2019. Emerg. Infect. Dis. 2021;27:648–650. doi: 10.3201/eid2702.204632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arimoto K., Funami K., Saeki Y., Tanaka K., Okawa K., Takeuchi O., Akira S., Murakami Y., Shimotohno K. Polyubiquitin conjugation to NEMO by triparite motif protein 23 (TRIM23) is critical in antiviral defense. Proc. Natl. Acad. Sci. USA. 2010;107:15856–15861. doi: 10.1073/pnas.1004621107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bhoj V.G., Chen Z.J. Ubiquitylation in innate and adaptive immunity. Nature. 2009;458:430–437. doi: 10.1038/nature07959. [DOI] [PubMed] [Google Scholar]
- Blanco-Melo D., Nilsson-Payant B.E., Liu W.C., Uhl S., Hoagland D., Møller R., Jordan T.X., Oishi K., Panis M., Sachs D. Imbalanced Host Response to SARS-CoV-2 Drives Development of COVID-19. Cell. 2020;181:1036–1045.e9. doi: 10.1016/j.cell.2020.04.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bojkova D., Klann K., Koch B., Widera M., Krause D., Ciesek S., Cinatl J., Münch C. Proteomics of SARS-CoV-2-infected host cells reveals therapy targets. Nature. 2020;583:469–472. doi: 10.1038/s41586-020-2332-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bouvet M., Debarnot C., Imbert I., Selisko B., Snijder E.J., Canard B., Decroly E. In vitro reconstitution of SARS-coronavirus mRNA cap methylation. PLoS Pathog. 2010;6:e1000863. doi: 10.1371/journal.ppat.1000863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cervantes-Barragan L., Züst R., Weber F., Spiegel M., Lang K.S., Akira S., Thiel V., Ludewig B. Control of coronavirus infection through plasmacytoid dendritic-cell-derived type I interferon. Blood. 2007;109:1131–1137. doi: 10.1182/blood-2006-05-023770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Channappanavar R., Fehr A.R., Zheng J., Wohlford-Lenane C., Abrahante J.E., Mack M., Sompallae R., McCray P.B., Jr., Meyerholz D.K., Perlman S. IFN-I response timing relative to virus replication determines MERS coronavirus infection outcomes. J. Clin. Invest. 2019;129:3625–3639. doi: 10.1172/JCI126363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Devaraj S.G., Wang N., Chen Z., Chen Z., Tseng M., Barretto N., Lin R., Peters C.J., Tseng C.T., Baker S.C., Li K. Regulation of IRF-3-dependent innate immunity by the papain-like protease domain of the severe acute respiratory syndrome coronavirus. J. Biol. Chem. 2007;282:32208–32221. doi: 10.1074/jbc.M704870200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fitzgerald K.A., McWhirter S.M., Faia K.L., Rowe D.C., Latz E., Golenbock D.T., Coyle A.J., Liao S.M., Maniatis T. IKKepsilon and TBK1 are essential components of the IRF3 signaling pathway. Nat. Immunol. 2003;4:491–496. doi: 10.1038/ni921. [DOI] [PubMed] [Google Scholar]
- Frieman M., Yount B., Heise M., Kopecky-Bromberg S.A., Palese P., Baric R.S. Severe acute respiratory syndrome coronavirus ORF6 antagonizes STAT1 function by sequestering nuclear import factors on the rough endoplasmic reticulum/Golgi membrane. J. Virol. 2007;81:9812–9824. doi: 10.1128/JVI.01012-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gentili M., Kowal J., Tkach M., Satoh T., Lahaye X., Conrad C., Boyron M., Lombard B., Durand S., Kroemer G. Transmission of innate immune signaling by packaging of cGAMP in viral particles. Science. 2015;349:1232–1236. doi: 10.1126/science.aab3628. [DOI] [PubMed] [Google Scholar]
- Gordon D.E., Hiatt J., Bouhaddou M., Rezelj V.V., Ulferts S., Braberg H., Jureka A.S., Obernier K., Guo J.Z., Batra J. Comparative host-coronavirus protein interaction networks reveal pan-viral disease mechanisms. Science. 2020;370:eabe9403. doi: 10.1126/science.abe9403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gordon D.E., Jang G.M., Bouhaddou M., Xu J., Obernier K., White K.M., O’Meara M.J., Rezelj V.V., Guo J.Z., Swaney D.L. A SARS-CoV-2 protein interaction map reveals targets for drug repurposing. Nature. 2020;583:459–468. doi: 10.1038/s41586-020-2286-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo C., Xie S., Chi Z., Zhang J., Liu Y., Zhang L., Zheng M., Zhang X., Xia D., Ke Y. Bile acids control inflammation and metabolic disorder through inhibition of NLRP3 inflammasome. Immunity. 2016;45:802–816. doi: 10.1016/j.immuni.2016.09.008. [DOI] [PubMed] [Google Scholar]
- Hoffmann M., Kleine-Weber H., Schroeder S., Krüger N., Herrler T., Erichsen S., Schiergens T.S., Herrler G., Wu N.H., Nitsche A. SARS-CoV-2 Cell Entry Depends on ACE2 and TMPRSS2 and Is Blocked by a Clinically Proven Protease Inhibitor. Cell. 2020;181:271–280.e8. doi: 10.1016/j.cell.2020.02.052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang C., Wang Y., Li X., Ren L., Zhao J., Hu Y., Zhang L., Fan G., Xu J., Gu X. Clinical features of patients infected with 2019 novel coronavirus in Wuhan, China. Lancet. 2020;395:497–506. doi: 10.1016/S0140-6736(20)30183-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jacobs J.L., Coyne C.B. Mechanisms of MAVS regulation at the mitochondrial membrane. J. Mol. Biol. 2013;425:5009–5019. doi: 10.1016/j.jmb.2013.10.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang H.W., Li Y., Zhang H.N., Wang W., Yang X., Qi H., Li H., Men D., Zhou J., Tao S.C. SARS-CoV-2 proteome microarray for global profiling of COVID-19 specific IgG and IgM responses. Nat. Commun. 2020;11:3581. doi: 10.1038/s41467-020-17488-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang H., Zhang H., Meng Q., Xie J., Li Y., Chen H., Zheng Y., Wang X., Qi H., Zhang J. SARS-CoV-2 Orf9b suppresses type I interferon responses by targeting TOM70. Cell. Mol. Immunol. 2020;17:998–1000. doi: 10.1038/s41423-020-0514-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kato H., Fujita T. Cytoplasmic Viral RNA Sensors: RIG-I-Like Receptors. Encyclopedia of Immunobiology. 2016;2:352–359. [Google Scholar]
- Kato H., Takeuchi O., Sato S., Yoneyama M., Yamamoto M., Matsui K., Uematsu S., Jung A., Kawai T., Ishii K.J. Differential roles of MDA5 and RIG-I helicases in the recognition of RNA viruses. Nature. 2006;441:101–105. doi: 10.1038/nature04734. [DOI] [PubMed] [Google Scholar]
- Kopecky-Bromberg S.A., Martínez-Sobrido L., Frieman M., Baric R.A., Palese P. Severe acute respiratory syndrome coronavirus open reading frame (ORF) 3b, ORF 6, and nucleocapsid proteins function as interferon antagonists. J. Virol. 2007;81:548–557. doi: 10.1128/JVI.01782-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lei X., Dong X., Ma R., Wang W., Xiao X., Tian Z., Wang C., Wang Y., Li L., Ren L. Activation and evasion of type I interferon responses by SARS-CoV-2. Nat. Commun. 2020;11:3810. doi: 10.1038/s41467-020-17665-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lim K.L., Chew K.C., Tan J.M., Wang C., Chung K.K., Zhang Y., Tanaka Y., Smith W., Engelender S., Ross C.A. Parkin mediates nonclassical, proteasomal-independent ubiquitination of synphilin-1: implications for Lewy body formation. J. Neurosci. 2005;25:2002–2009. doi: 10.1523/JNEUROSCI.4474-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu S., Chen J., Cai X., Wu J., Chen X., Wu Y.T., Sun L., Chen Z.J. MAVS recruits multiple ubiquitin E3 ligases to activate antiviral signaling cascades. eLife. 2013;2:e00785. doi: 10.7554/eLife.00785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Menachery V.D., Yount B.L., Jr., Josset L., Gralinski L.E., Scobey T., Agnihothram S., Katze M.G., Baric R.S. Attenuation and restoration of severe acute respiratory syndrome coronavirus mutant lacking 2′-o-methyltransferase activity. J. Virol. 2014;88:4251–4264. doi: 10.1128/JVI.03571-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park A., Iwasaki A. Type I and Type III Interferons - Induction, Signaling, Evasion, and Application to Combat COVID-19. Cell Host Microbe. 2020;27:870–878. doi: 10.1016/j.chom.2020.05.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peiris J.S.M., Chu C.M., Cheng V.C.C., Chan K.S., Hung I.F.N., Poon L.L.M., Law K.I., Tang B.S.F., Hon T.Y.W., Chan C.S. Clinical progression and viral load in a community outbreak of coronavirus-associated SARS pneumonia: a prospective study. Lancet. 2003;361:1767–1772. doi: 10.1016/S0140-6736(03)13412-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qin C., Zhou L., Hu Z., Zhang S., Yang S., Tao Y., Xie C., Ma K., Shang K., Wang W., Tian D.S. Dysregulation of immune response in patients with COVID-19 in Wuhan, China. Clin. Infect. Dis. 2020;71:762–768. doi: 10.1093/cid/ciaa248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robert X., Gouet P. Deciphering key features in protein structures with the new ENDscript server. Nucleic Acids Res. 2014;42:W320–W324. doi: 10.1093/nar/gku316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rothwarf D.M., Zandi E., Natoli G., Karin M. IKK-gamma is an essential regulatory subunit of the IkappaB kinase complex. Nature. 1998;395:297–300. doi: 10.1038/26261. [DOI] [PubMed] [Google Scholar]
- Sheahan T., Morrison T.E., Funkhouser W., Uematsu S., Akira S., Baric R.S., Heise M.T. MyD88 is required for protection from lethal infection with a mouse-adapted SARS-CoV. PLoS Pathog. 2008;4:e1000240. doi: 10.1371/journal.ppat.1000240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi C.S., Qi H.Y., Boularan C., Huang N.N., Abu-Asab M., Shelhamer J.H., Kehrl J.H. SARS-coronavirus open reading frame-9b suppresses innate immunity by targeting mitochondria and the MAVS/TRAF3/TRAF6 signalosome. J. Immunol. 2014;193:3080–3089. doi: 10.4049/jimmunol.1303196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi Y., Wu J., Zhong T., Zhu W., She G., Tang H., Du W., Ye B.C., Qi N. Upstream ORFs Prevent MAVS Spontaneous Aggregation and Regulate Innate Immune Homeostasis. iScience. 2020;23:101059. doi: 10.1016/j.isci.2020.101059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tan M., Liu Y., Zhou R., Deng X., Li F., Liang K., Shi Y. Immunopathological characteristics of coronavirus disease 2019 cases in Guangzhou, China. Immunology. 2020;160:261–268. doi: 10.1111/imm.13223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tian W., Zhang N., Jin R., Feng Y., Wang S., Gao S., Gao R., Wu G., Tian D., Tan W. Immune suppression in the early stage of COVID-19 disease. Nat. Commun. 2020;11:5859. doi: 10.1038/s41467-020-19706-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Totura A.L., Whitmore A., Agnihothram S., Schäfer A., Katze M.G., Heise M.T., Baric R.S. Toll-Like Receptor 3 Signaling via TRIF Contributes to a Protective Innate Immune Response to Severe Acute Respiratory Syndrome Coronavirus Infection. mBio. 2015;6:e00638-15. doi: 10.1128/mBio.00638-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wan Y., Shang J., Graham R., Baric R.S., Li F. Receptor Recognition by the Novel Coronavirus from Wuhan: an Analysis Based on Decade-Long Structural Studies of SARS Coronavirus. J. Virol. 2020;94:e00127-20. doi: 10.1128/JVI.00127-20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang M., Cao R., Zhang L., Yang X., Liu J., Xu M., Shi Z., Hu Z., Zhong W., Xiao G. Remdesivir and chloroquine effectively inhibit the recently emerged novel coronavirus (2019-nCoV) in vitro. Cell Res. 2020;30:269–271. doi: 10.1038/s41422-020-0282-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wölfel R., Corman V.M., Guggemos W., Seilmaier M., Zange S., Müller M.A., Niemeyer D., Jones T.C., Vollmar P., Rothe C. Virological assessment of hospitalized patients with COVID-2019. Nature. 2020;581:465–469. doi: 10.1038/s41586-020-2196-x. [DOI] [PubMed] [Google Scholar]
- Wu C.J., Conze D.B., Li T., Srinivasula S.M., Ashwell J.D. Sensing of Lys 63-linked polyubiquitination by NEMO is a key event in NF-kappaB activation [corrected] Nat. Cell Biol. 2006;8:398–406. doi: 10.1038/ncb1384. [DOI] [PubMed] [Google Scholar]
- Xu Z., Shi L., Wang Y., Zhang J., Huang L., Zhang C., Liu S., Zhao P., Liu H., Zhu L. Pathological findings of COVID-19 associated with acute respiratory distress syndrome. Lancet Respir. Med. 2020;8:420–422. doi: 10.1016/S2213-2600(20)30076-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yan R., Zhang Y., Li Y., Xia L., Guo Y., Zhou Q. Structural basis for the recognition of SARS-CoV-2 by full-length human ACE2. Science. 2020;367:1444–1448. doi: 10.1126/science.abb2762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang Y., Zhang L., Geng H., Deng Y., Huang B., Guo Y., Zhao Z., Tan W. The structural and accessory proteins M, ORF 4a, ORF 4b, and ORF 5 of Middle East respiratory syndrome coronavirus (MERS-CoV) are potent interferon antagonists. Protein Cell. 2013;4:951–961. doi: 10.1007/s13238-013-3096-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yuen C.K., Lam J.Y., Wong W.M., Mak L.F., Wang X., Chu H., Cai J.P., Jin D.Y., To K.K., Chan J.F. SARS-CoV-2 nsp13, nsp14, nsp15 and orf6 function as potent interferon antagonists. Emerg. Microbes Infect. 2020;9:1418–1428. doi: 10.1080/22221751.2020.1780953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou P., Yang X.L., Wang X.G., Hu B., Zhang L., Zhang W., Si H.R., Zhu Y., Li B., Huang C.L. A pneumonia outbreak associated with a new coronavirus of probable bat origin. Nature. 2020;579:270–273. doi: 10.1038/s41586-020-2012-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu N., Zhang D., Wang W., Li X., Yang B., Song J., Zhao X., Huang B., Shi W., Lu R. A Novel Coronavirus from Patients with Pneumonia in China, 2019. N. Engl. J. Med. 2020;382:727–733. doi: 10.1056/NEJMoa2001017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zou L., Ruan F., Huang M., Liang L., Huang H., Hong Z., Yu J., Kang M., Song Y., Xia J. SARS-CoV-2 Viral Load in Upper Respiratory Specimens of Infected Patients. N. Engl. J. Med. 2020;382:1177–1179. doi: 10.1056/NEJMc2001737. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
This study did not generate any unique datasets or code.