

Abstract
Elongation factor 2 (EF-2), a five-domain, GTP-dependent ribosomal translocase of archaebacteria and eukaryotes, undergoes post-translational modification to form diphthamide on a specific histidine residue in domain IV prior to binding the ribosome. The first step of diphthamide biosynthesis in archaebacteria is catalyzed by Dph2, a homodimeric radical S-adenosylmethionine (SAM) enzyme having a non-canonical architecture. Here, we describe a 3.5 Å resolution crystal structure of the Methanobrevibacter smithii Dph2 homodimer bound to two molecules of MsEF-2, one of which is ordered and the other largely disordered. MsEF-2 is bound to both protomers of MsDph2, with domain IV bound to the active site of one protomer and domain III bound to a surface α-helix of an adjacent protomer. The histidine substrate of domain IV is inserted into the active site, which reveals for the first time the architecture of the Dph2 active site in complex with its target substrate. We also determined a high-resolution crystal structure of isolated MsDph2 bound to 5ʹ-methylthioadenosine that shows a conserved arginine residue pre-oriented by conserved phenylalanine and aspartate residues for binding the carboxylate group of SAM. Mutagenesis experiments suggest that the arginine plays an important role in the first step of diphthamide biosynthesis.
Graphical Abstract
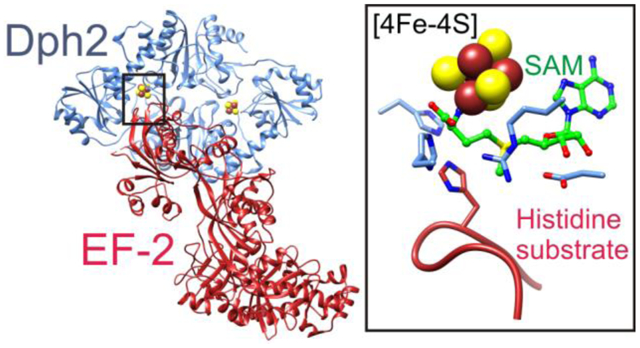
INTRODUCTION
Elongation factor 2 (EF-2) is a five-domain GTPase1–3 that catalyzes the translocation of tRNA and mRNA through the ribosome during translation in eukaryotes and archaebacteria.4–6 A conserved histidine residue in domain IV of EF-2 undergoes post-translational modification to diphthamide (2-(3-carboxyamido-3-(trimethylammonio)propyl)histidine),7 which has been suggested to reduce translational frameshift errors, promote selenoprotein translation, and promote internal ribosomal entry site (IRES) mediated translation.8–12 Diphthamide biosynthesis is thought to require seven proteins in eukaryotes (Dph1-Dph7) and requires three proteins in archaebacteria (Dph2, Dph5, and Dph6).13–15 In archaebacteria, the first reaction is catalyzed by Dph2, a non-canonical radical S-adenosylmethionine (SAM) enzyme that uses a [4Fe-4S] cluster to generate a 3-amino-3-carboxypropyl (ACP) radical16 instead of the 5′-deoxyadenosyl radical generated by canonical radical SAM enzymes.17, 18 The ACP radical then reacts with the EF-2 histidine substrate to form 2-(3-amino-3-carboxypropyl)histidine (ACPHis).16 Structurally, Dph2 is a homodimer with triangularly-shaped protomers, each having a centrally-located active site containing a [4Fe-4S] cluster ligated by three cysteine residues from three separate domains.16
Recently, the chemical mechanism of the reaction was shown to occur through the formation of a novel organometallic intermediate (intermediate I) consisting of the ACP moiety of SAM and the [4Fe-4S] cluster linked by a Fe-Cγ bond.19, 20 Intermediate I was also shown to react with the histidine substrate of EF-2 to produce a nitrogen histidine-based radical (intermediate II).19 In the same study, SAM was shown to display a binding mode distinct from that used by canonical radical SAM enzymes21–25 in which its Cγ atom is positioned in close proximity to the differentiated iron of the [4Fe-4S] cluster.
Although the chemical mechanism of the Dph2-catalyzed reaction and the SAM-bound structure of archaeal Dph2 have been determined, there are several details that we do not know about the reaction and structure. How does the enzyme bind to the substrate to ensure specific reaction on the desired histidine residue and how does the enzyme prevent the ACP radical from reacting with other residues? Here, we report the crystal structures of Methanobrevibacter smithii Dph2 (MsDph2) bound to MsEF-2 and MsDph2 bound to 5ʹ-methylthioadenosine (MTA). The structures reveal two protein-protein interfaces involved in the enzyme-substrate protein complex and the conformational changes that lead to insertion of the histidine substrate of EF-2 into the active site of Dph2. Along with previously reported structures of orthologous archaeal Dph2s bound to SAM,19 the structures of MsDph2 also suggest roles in substrate binding for several highly conserved active site residues. The structural analysis led us to perform a mutagenesis study of an arginine residue that interacts with SAM and whose side chain is oriented through interactions with other highly conserved active site residues.
MATERIALS AND METHODS
Preparation of MsEF-2.
A gene encoding MsEF-2 with a tobacco etch virus (TEV) protease-cleavable His6-tag and codon-optimized for expression in E. coli (GenScript Inc.; Table S1) was cloned into pET-28a(+) using NcoI and NotI restriction sites for overexpression of the product NH2-MGSDKIHHHHHHSSGENLYFQGSGM1…G730-COOH. Competent E. coli NiCo21(DE3) cells were transformed with plasmid pET-28a(+)-His6-MsEF-2 and spread onto agar plates containing Lysogeny Broth (LB) medium and kanamycin (40 mg/L). Large-scale cultures were grown in shaker flasks containing 1.35 L of LB medium by rotation at 200 rpm and 37 °C. When the optical density at 600 nm (OD600) reached 0.8–0.9, the flasks were moved to a cold room set at 4 °C for 2 h. Isopropyl β-D-1-thiogalactopyranoside (IPTG) was then added to the cultures to a final concentration of 0.5 mM, and the flasks were rotated at 200 rpm and 15 °C for 20 h. The cultures were moved to ice and centrifuged at 6,000 g and 4 °C for 15 min, and the cell pellets were flash frozen in liquid nitrogen. The cell pellets were thawed and resuspended in lysis buffer [20 mM Tris, 500 mM NaCl, 23 mM imidazole, 2.5 mM DTT, 0.3 mg/mL lysozyme, 28 units/mL benzonase (Sigma; ≥ 250 units/μL), pH 7.5] and then sonicated while kept on ice. The lysate was centrifuged at 50,000 × g and 4 °C for 30 min and the supernatant was subjected to immobilized nickel chelate chromatography using wash (20 mM Tris, 500 mM NaCl, 23 mM imidazole, 2.5 mM DTT, pH 7.5) and elution (20 mM Tris, 500 mM NaCl, 340 mM imidazole, 0.8 mM DTT, pH 7.5) buffers. The protein eluate was subjected to size exclusion chromatography using a HiLoad 26/600 Superdex200 column (GE Healthcare Life Sciences) equilibrated with His6-tag cleavage buffer (20 mM HEPES, 200 mM NaCl, 2 mM DTT, pH 7.5) and then incubated with TEV protease overnight at 18 °C. The reaction mixture was subjected to subtractive nickel chelate chromatography followed by size exclusion chromatography (using the buffers employed in the earlier chromatography steps) and then concentrated to 7 mL. The sample was moved into a Vinyl Anaerobic Chamber (Coy Laboratory Products, Inc.), passed over Bio-Rad Econo Pac 10DG desalting columns equilibrated with 5 mM HEPES, 28 mM NaCl, pH 7.0, and flash frozen in liquid nitrogen. The final concentration of MsEF-2 was estimated to be 0.14 mM based on the absorbance at 280 nm (A280) and an extinction coefficient of 59,000 M−1cm−1.
Preparation of MsDph2.
A codon-optimized gene encoding MsDph2 (GenScript, Inc.; Table S1) was cloned into pET-28a(+) using NdeI and XhoI restriction sites for overexpression of the product NH2-MGSSHHHHHHSSGLVPRGSHM1…K334-COOH. Competent E. coli NiCo21(DE3) cells were transformed with plasmid pSuf,26, 27 made competent again and transformed with plasmid pET-28a(+)-His6-MsDph2, and spread onto agar plates containing LB medium (LB), kanamycin (40 mg/L), and chloramphenicol (34 mg/L). Large-scale cultures were grown in shaker flasks containing 1.8 L of selective minimal medium (1 X minimal medium salts, 40 mg/L kanamycin, 34 mg/L chloramphenicol, 6 g/L dextrose, 3 mM MgSO4, and 0.1 mM CaCl2) by rotation at 180 rpm and 37 °C. When the OD600 reached 0.5–0.55, the flasks were moved to a cold room set at 4 °C for 2.5 h. L-Cys, Fe(NH4)2(SO4)2, and IPTG were then added to the cultures to final concentrations of 0.29 mM, 0.089 mM, and 0.18 mM, respectively, and the cultures were rotated at 50 rpm and 15 °C for 20 h. The cultures were chilled in a 4 °C cold room, pelleted by centrifugation at 6,000 g and 4 °C for 15 min, and flash frozen in liquid nitrogen. The cell pellets were thawed in the anaerobic chamber, resuspended in lysis buffer (20 mM Tris-HCl, 500 mM NaCl, and 23 mM imidazole, pH 7.5) that was supplemented with 5 mM DTT, 0.4 mg/mL lysozyme, and 1.9 kU benzonase, and sonicated while kept on ice. The lysate was sealed in centrifuge bottles and removed from the anaerobic chamber for centrifugation at 60,000 g and 4 °C for 20 min. The spun lysate was placed back into the anaerobic chamber and the supernatant was subjected to immobilized nickel chelate affinity chromatography using lysis and elution (20 mM Tris-HCl, 500 mM NaCl, and 250 mM imidazole, pH 7.5) buffers. The protein eluate, identified by its color, was buffer exchanged into His6-tag cleavage buffer (20 mM HEPES and 200 mM NaCl, pH 7.5) using a Bio-Rad Econo-Pac 10DG desalting column and incubated for 7 h with 1 mg/mL bovine thrombin (Sigma; 52 NIH units/mg solid). The reaction mixture was subjected to subtractive immobilized nickel and benzamidine affinity chromatography, buffer exchanged into 5 mM HEPES and 300 mM NaCl, pH 7.8, and flash-frozen in liquid nitrogen. The final concentration of MsDph2 was estimated to be 0.388 mM based on the A280 and an extinction coefficient of 33,500 M−1cm−1.
Crystallization.
Protein crystals were grown using the hanging drop vapor diffusion method with drops formed by combining sample and reservoir solutions in a 1:1 ratio. The crystals were grown and then cryocooled in liquid nitrogen inside the anaerobic chamber, which was typically operated at room temperature (23 °C); however, for crystallization of the complex of MsEF-2 and MsDph2, the room and anaerobic chamber were maintained at 20 ± 1 °C.
For crystallization of the complex of MsDph2 and MsEF-2, the individually purified proteins were thawed and combined to give final concentrations of 0.18 mM and 0.14 mM, respectively. Prior to combining the samples, the NaCl concentration in the solution of MsEF-2 was increased to 300 mM. The protein solutions were also supplemented with L-Ala to a final concentration of 14 mM. The reservoir solution contained 100 mM Tris, pH 7.2, 200 mM MgSO4, and 21 % (w/v) PEG400; NaCl was added to the reservoir to a final concentration of 300 mM after forming the drop. The cryoprotectant solution contained 100 mM Tris, pH 7.2, 200 mM MgSO4, 300 mM NaCl, 26 % (w/v) PEG400, 7.2 % (v/v) glycerol, and 20 mM L-Ala.
For crystallization of MsDph2, protein samples were thawed and supplemented with MTA (solubilized in dimethylsulfoxide) and 2AB (solubilized in 300 mM NaCl and 5 mM HEPES, pH 7.8) at final concentrations of 6 mM and 14 mM, respectively. Typical reservoir solutions contained 100 mM bistrispropane, pH 6.5, and 16–17 % (w/v) PEG2000 monomethyl ether (MME). After drops were formed by combining the sample and reservoir solutions, 300 mM NaCl was added to the reservoirs prior to sealing. The crystals were cryoprotected by increasing the PEG2000 MME concentration to 40 % (w/v).
Data collection and structure determination.
X-ray diffraction experiments were performed at beam line 24-ID-E of the Advanced Photon Source (APS), which was equipped with an EIGER 16M detector. The experiments used X-rays having a wavelength of 0.979 Å and an oscillation range of 0.2°. The crystals of the MsEF-2-MsDph2 complex and MsDph2-MTA corresponding to the reported structures were placed 450 and 240 mm from the detector and were irradiated over angular ranges of 108 and 105°, respectively. Reflections were processed using HKL2000,28 XDS,29 AIMLESS,30 and SCALA.31
Initial phases were obtained by molecular replacement using PHASER32 within PHENIX.33 The structure of CmnDph2 bound to SAH (PDB entry 6BXO)19 was used as the search model for determining the initial phases for the structure of MsDph2-MTA. The structure of MsDph2-MTA and a homology model34 of MsEF-2 based on PhEF-2 (PDB entry 5H7J)35 were used as the search models for determining the initial phases for the structure of the MsEF-2-MsDph2 complex. The molecular replacement solution contained one molecule of MsEF-2 and two molecules of MsDph2. However, a large space in the crystal lattice remained that showed very weak Fo-Fc electron density, which was determined at a later stage of refinement to be a second molecule of MsEF-2.
The structure of MsDph2-MTA was determined by manual model building using COOT36 and automated structure refinement using PHENIX.33 Structure determination of the MsEF-2-MsDph2 complex required methods for low-resolution structure refinement. The molecular replacement solution for the MsEF-2-MsDph2 complex was subjected to rigid-body refinement of the individual domains followed by restrained refinement using Refmac537 with external distance restraints.38 Missing or poorly fitted segments were built or rebuilt using homology modeling,34 Autobuild,39 and/or flexible fitting (xMDFF).40, 41 Initial conformations for residues 425–434, 481–486, and 717–730 of MsEF-2 were obtained from a homology model based on ScEF-2 (PDB entry 1N0V).3 The conformations of a portion of the linker connecting domains II and III of MsEF-2 (residues 382–391) and the loop containing the histidine substrate of MsEF-2 (residues 591–601) could not be easily modeled based on fragments of other EF-2 structures and were selected (based on goodness of fit to the electron density and geometry considerations) from a set of conformations generated using simulated annealing in xMDFF. Electron density maps were sharpened to facilitate model building.36, 42 External distance restraints were applied during all stages of refinement using Refmac5. The external distance restraints based on the original homology model were maintained except those involving atoms whose positions changed through manual building. For these atoms, new restraints were added or replaced ones used in earlier refinement stages. PHENIX was used for performing group B-factor and TLS refinement.33, 43 The electron density showed no indication that MsEF-2 co-purified with a nucleotide; however, significant Fo-Fc electron density having the shape of SAM, SAM cleavage products, or structural analogs of these molecules was visible in the active site of MsDph2A. A ligand was not built into the final model because its identity could not be definitively discerned from the low resolution map, but this Fo-Fc electron density was useful for modeling the active site structure.
Domains III-V of the disordered molecule of MsEF-2 were built based on the polyalanine trace of the ordered molecule. The chain was initially fit into very weak Fo-Fc density using a twofold axis relating MsDph2A and MsDph2B and then subjected to rigid body refinement. Residues 716–730 were removed because a steric clash would result with the corresponding segment in the ordered molecule of MsEF-2. Distance restraints were identical to those used for the ordered molecule.
Illustrations of crystal structures and electron density maps were prepared using CHIMERA44 and PyMOL.45
Activity assays of SAM cleavage and ACP group transfer to PhEF-2 by wild type PhDph2 and PhDph2 R289A.
The PhDph2 R289A mutant was made by overlap extension polymerase chain reaction. PhDph2 R289A protein was expressed and purified using the procedures reported previously for production of the wild type protein.46 The reactions were set up under anaerobic conditions. The full SAM cleavage reaction contained 10 μM PhDph2 R289A, 1mM dithionite, 60 μM SAM, 150 mM NaCl, 1 mM DTT and 200 mM Tris-HCl at pH 7.4 in a final volume of 30 μL. The control samples (without PhDph2 R289A, dithionite, or SAM) were set up similarly by replacing the corresponding component with an equal volume of water respectively. The reactions were incubated at 65 °C for 10 min and quenched by adding 30 μL of 10% TFA in water. The protein was pelleted by centrifugation and the supernatant was applied to HPLC and resolved with a linear gradient of 0 to 40% of acetonitrile with 0.1% TFA over 15 min at a flow rate of 0.5 mL/min.
The ACP group transfer reactions contained 20 μM PhDph2 wild type or R289A, 7 μM of PhEF-2, 10 mM dithionite in 150 mM NaCl and 200 mM Tris-HCl at pH 7.4. The reaction vials were sealed with rubber stoppers before being taken out of the anaerobic chamber. 14C-SAM (final concentration of 18 μM) was injected using needles into each reaction vial to start the reaction. The reaction mixtures were vortexed briefly to mix and incubated at 65 °C for 30 min. The reactions were stopped by adding protein loading dye and subsequently heating at 95 °C for 5 min, followed by 4–20% SDS–polyacrylamide gradient gel electrophoresis. The dried gel was exposed to a PhosphorImaging screen and the radioactivity was detected using a Typhoon FLA 7000 (GE Healthcare Life Science).
RESULTS AND DISCUSSION
Crystal structure of the MsDph2-MsEF-2 complex.
We determined to 3.5 Å resolution a crystal structure belonging to space group C2221 and having an asymmetric unit containing two molecules of MsEF-2 and two molecules of MsDph2. To facilitate model building and analysis of conformational changes resulting from complex formation, we also determined to 1.8 Å resolution a crystal structure of an MsDph2 homodimer with MTA bound to each protomer. The crystals belong to space group P212121 with one homodimer per asymmetric unit (Tables 1 and 2).
Table 1.
Data collection statistics
| MsDph2 | MsDph2 | |
|---|---|---|
| Beamline | APS 24-ID-E | APS 24-ID-E |
| Wavelength (Å) | 0.97918 | 0.97918 |
| Space group | C2221 | P212121 |
| Unit cell dimensions (Å) | 100.9, 323.0, 172.4 | 63.1, 81.0, 131.0 |
| Resolution (Å) | 49.4–3.45 | 131.0–1.77 |
| Highest resolution shell (Å) | 3.45–3.64 | 1.77–1.86 |
| Total number of reflections | 152,333 | 254,208 |
| Number of unique reflections | 37,395 | 64,613 |
| Rmerge (%) | 7.5 (111.4)a | 3.3 (54.7) |
| < I/σ(I) > | 8.7 (1.1) | 19.3 (2.5) |
| Mn (I) half-set correlation CC(1/2) | 0.998 (0.568) | 0.999 (0.800) |
| Completeness (%) | 99.7 (99.7) | 97.6 (94.7) |
| Multiplicity | 4.1 (4.0) | 3.9 (3.9) |
Values in parentheses refer to the highest resolution shell.
Table 2.
Structure refinement statistics
| MsDph2 | MsDph2 | |
|---|---|---|
| Number of reflections | 37,270 | 64,546 |
| Number of reflections in working set | 35,406 | 61,389 |
| Rwork/Rfree (%) | 22.4/26.7 | 18.0/21.9 |
| Number of protein atoms | 12,169 | 5,183 |
| Number of ligand atoms | 16 | 57 |
| Number of water molecules | 0 | 565 |
| RMSD for bonds (Å) | 0.011 | 0.006 |
| RMSD for angles (°) | 1.568 | 0.840 |
| Ramachandran analysis |
In the structure of the MsDph2-MsEF-2 complex, one of the two molecules of MsEF-2 is mostly ordered with electron density visible for all five domains (Figure S1A). The electron density corresponding to the loop containing the histidine substrate is suitable for full chain tracing and its interpretability could be improved by application of map sharpening methods (Figure S1C and D).36, 42 For the disordered molecule, electron density is visible but very weak for domains III-V and mostly absent for domains I and II (Figure S1B). A polyalanine trace of these domains was generated by rotation of domains III-V of the ordered molecule about a twofold symmetry axis that relates the two MsDph2 protomers. However, the final 15 residues were omitted due to clashes with the equivalent segment of the ordered molecule of MsEF-2. Both MsDph2 molecules are well ordered and residues 1–329 and 5–329 and the [4Fe-4S] clusters of the two chains were modeled (Figure S2A and B). However, domains 1 and 2 of the molecule whose active site interacts with the disordered molecule of MsEF-2 (MsDph2B) have elevated average main chain B-factors compared to domain 3 and the adjacent MsDph2 protomer (MsDph2A) and contains multiple residues (including residues 48, 63, 101, 156, 159, 180, and 204) for which the electron density is absent. Unassigned Fo-Fc electron density in the active site of MsDph2A has a shape resembling SAM or SAM cleavage products (Figure S2C).
Overall structure.
The crystal structure of the complex shows that domain IV of MsEF-2 binds one protomer of the MsDph2 homodimer (MsDph2A) and that domain III binds the adjacent protomer (MsDph2B) (Figure 1A). The domain IV-MsDph2A interface has a buried surface area of 2,400 Å2 and consists mostly of three loop regions of domain IV (residues 481–485, 547–557, and 592–599) and the regions of the three domains of MsDph2A that form the active site opening (Figure 1B). The third loop, which contains the histidine substrate (His597), protrudes the most and inserts into the active site cavity near the [4Fe-4S] cluster. As a result, the [4Fe-4S] cluster and ACP radical binding sites are buried whereas the MTA binding site is solvent exposed (Figure S3). The domain III-MsDph2B interface has a buried surface area of 1,200 Å2 and is formed by two loop regions of domain III (residues 398–409 and 427–432) and the first α-helix and two flanking turns of the third domain of MsDph2B (Figure 1C). The differences in buried surface area and PISA analysis suggest a high affinity interaction of the MsDph2 homodimer with domain IV and a low affinity interaction with domain III. Analysis of the protein interfaces and surfaces using PISA predicts a ΔG for dissociation of 14.9 kcal/mol, providing theoretical support for stable complex formation in solution.47
Figure 1.
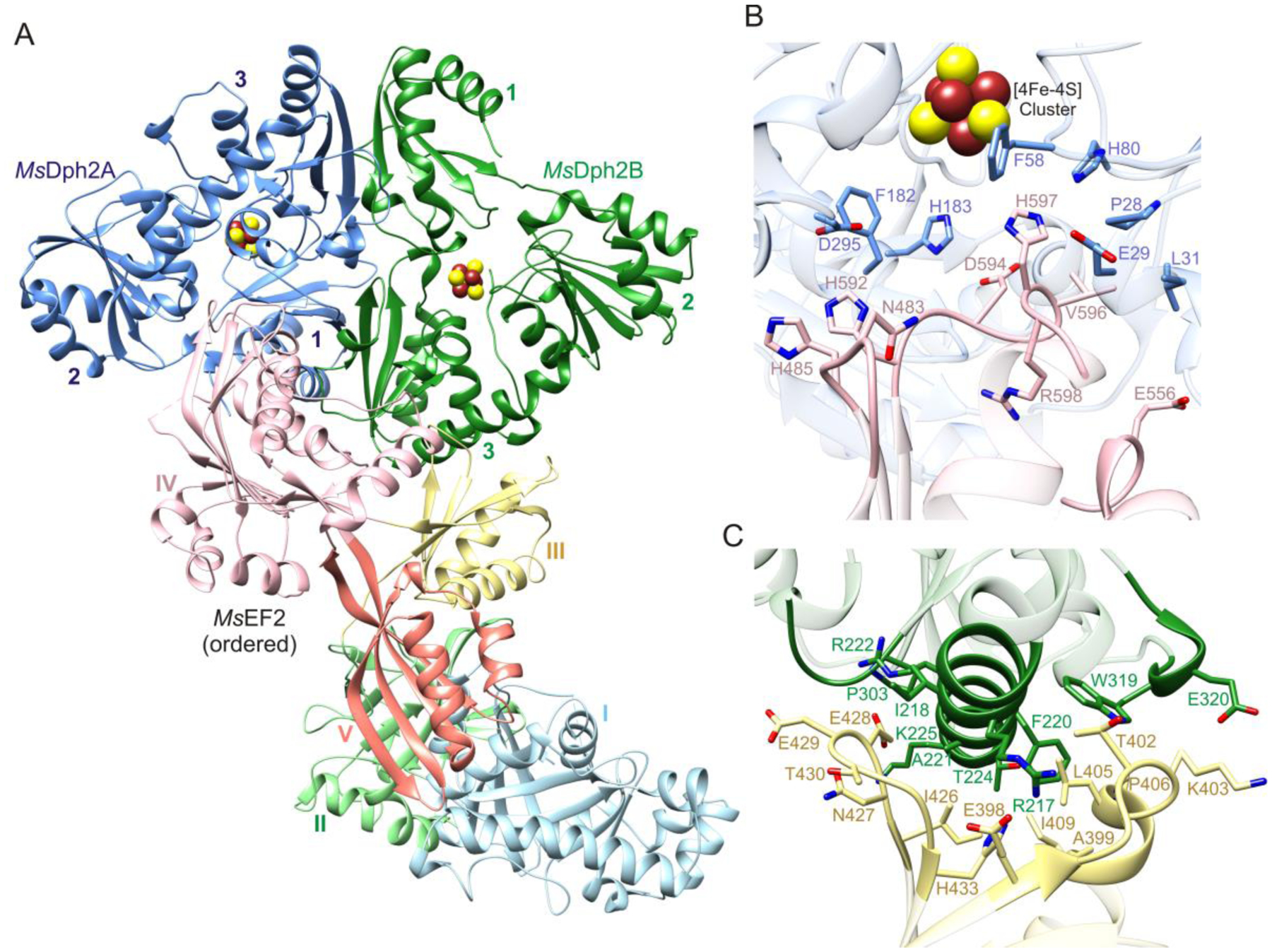
Structure and interfaces of the MsDph2-MsEF-2 complex. (A) Ribbon representation of the complex. The disordered molecule of MsEF-2 is omitted. Domains of MsEF-2 and MsDph2 are labeled with Roman and Arabic numerals, respectively. (B) Interface formed by domain IV of MsEF-2 and the active site of MsDph2A. (C) Interface formed by domain III of MsEF-2 and domain 3 of MsDph2B.
Conformational changes.
Comparisons of the structure of the crystallized complex with those of isolated Dph2 and EF-2 suggest that both proteins undergo significant conformational changes during complex formation (Figure S4). Comparisons with structures of isolated Pyrococcus horikoshii EF-2 and Saccharomyces cerevisiae EF-2 show structural changes in the loop of MsEF-2 inserted into MsDph2A and in an adjacent turn (residues 481–485) (Figure 2A). As a result, several interactions between these loops in the isolated structures, including the packing of side chains of Asn483, His485, His592, and His597 and hydrogen bonding interactions between the backbone of His597 and the side chains of Asn483 and Asn486 do not occur in the MsDph2-MsEF-2 complex. Comparisons with structures of isolated MsDph2-MTA, PhDph2, and Candidatus methanoperedens nitroreducens Dph2 (CmnDph2) show a rotation of domain 2 by approximately 7° (Figure 2B).48 The inserted loop of MsEF-2 binds between domains 1 and 2 of MsDph2A and forms backbone-backbone contacts with residues 29–30 and 179–182 (Figure 2C). The conformational changes in MsDph2 and MsEF-2 thus appear to be coupled, which may be important to ensure that SAM cleavage happens efficiently only when the substrate protein is bound with the histidine substrate properly oriented for reaction with the ACP radical.
Figure 2.
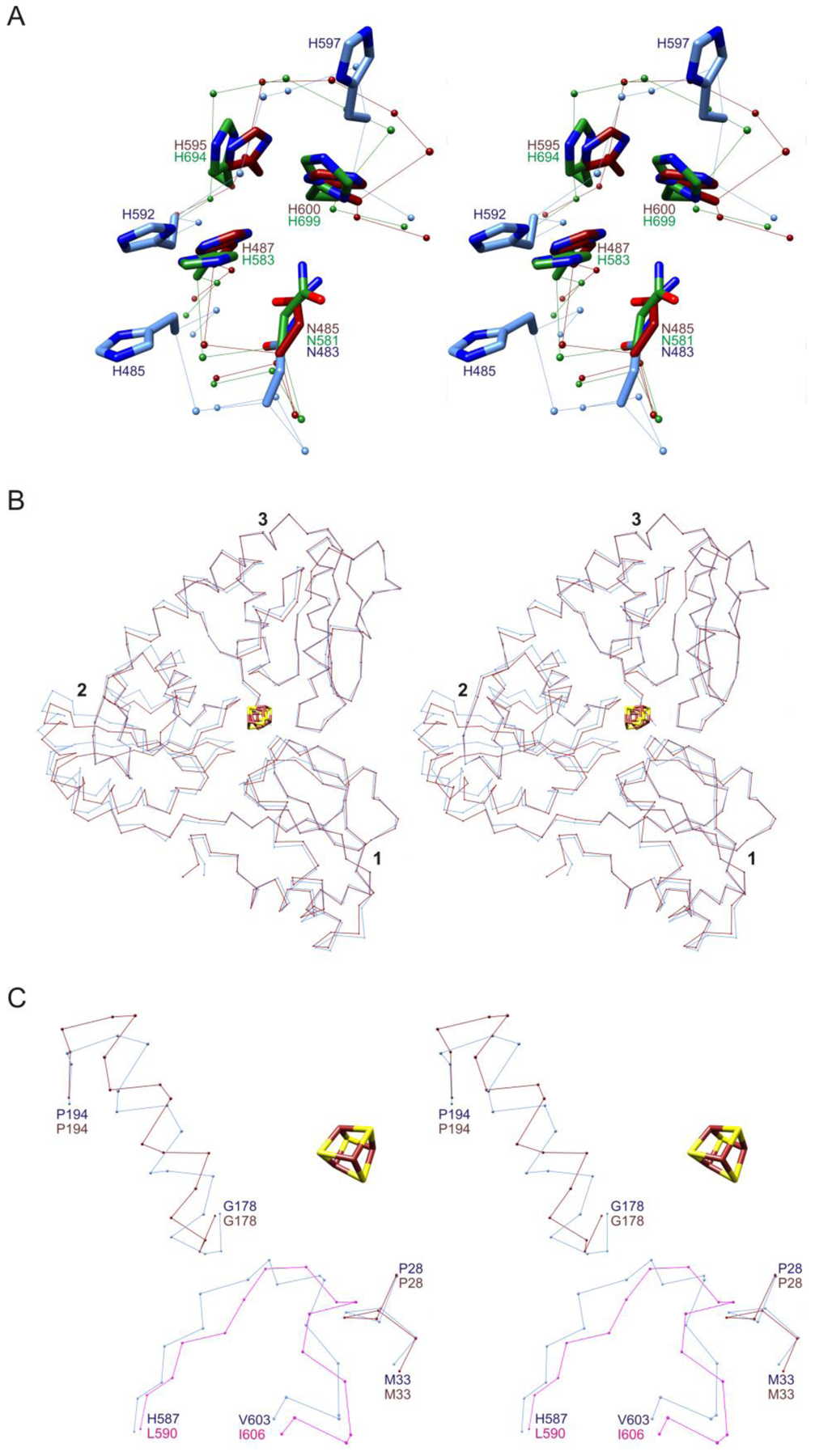
Stereoviews of conformational changes between MsDph2-MsEF-2 complex and isolated proteins. (A) Conformational differences in two turns of domain IV of MsEF-2 bound to MsDph2A (light blue) and corresponding turns of PhEF-2 (dark red; PDB entry 5H7J),35 and ScEF-2 (green; PDB entry 1N0V).3 The structures of domain IV of PhEF-2 and ScEF-2 were superimposed onto that of MsEF-2. (B) Conformational differences in MsDph2A (light blue) and isolated MsDph2-MTA (dark red). The structures were superimposed based on domains 1 and 3. (C) Conformational changes in inserted loop of MsEF-2 and nearby segments of MsDph2A. The corresponding segments of PhEF-2 (magenta) and isolated MsDph2-MTA (dark red) are superimposed onto the MsDph2-MsEF-2 complex (light blue). Protein chains are depicted as Cα atom traces with side chain models in Panel A shown in stick representation.
Active site and mutagenesis study.
Superimposition of the high resolution structure of CmnDph2 bound to MTA and 2-aminobutryrate (2AB)19 onto the low-resolution structure of the MsDph2-MsEF-2 complex shows that the histidine substrate is well-positioned for reaction with the ACP radical (Cγ-Cε1 atom separation distance = 1.5 Å; Figure 3A). This provides further support for the validity of the overall structure of the MsDph2-MsEF-2 complex.
Figure 3.
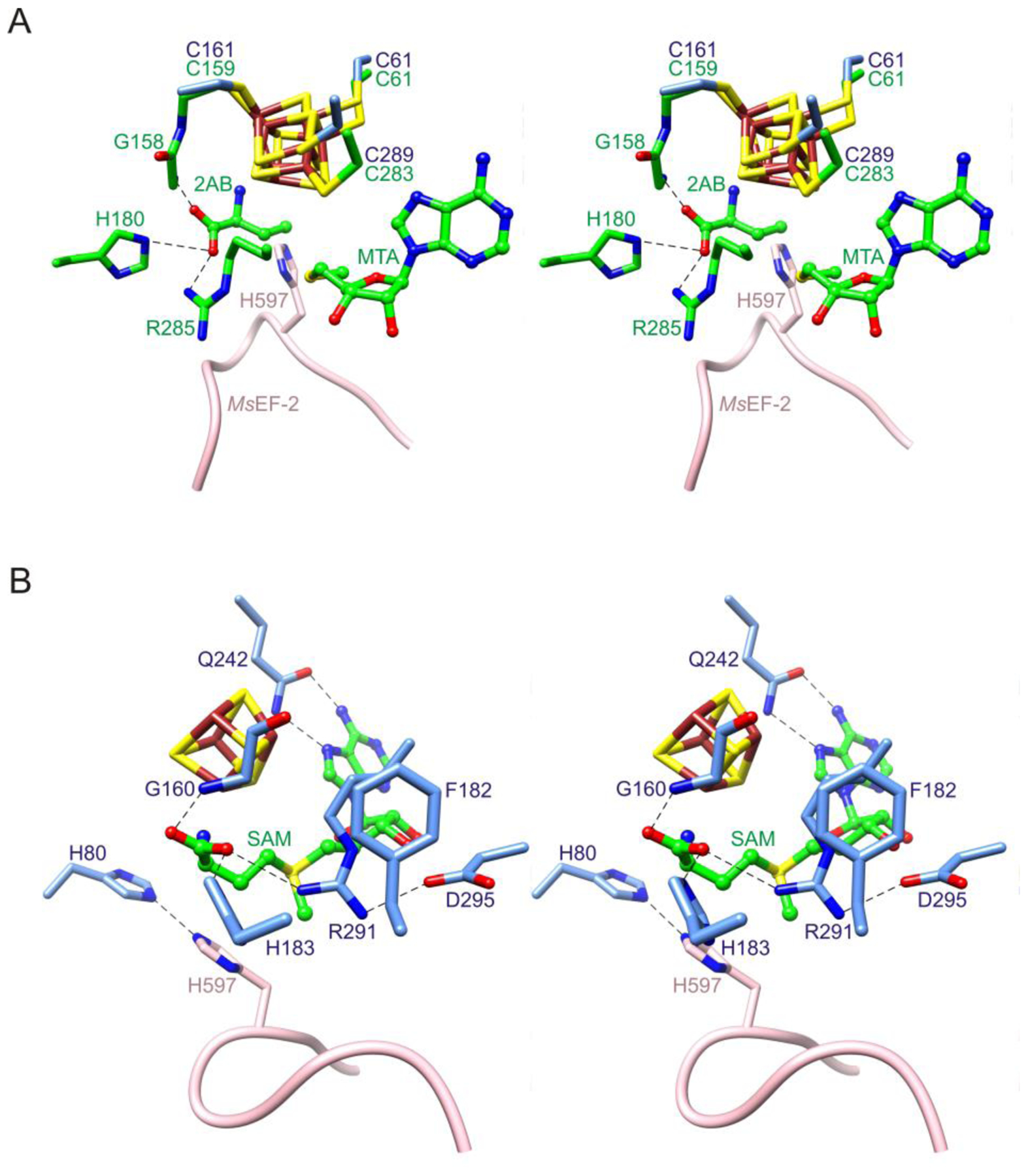
Stereoviews of Dph2 active site. (A) Insertion of the histidine substrate of MsEF-2 near the active site location of the ACP radical. The structure of the MsDph2-MsEF-2 complex was superimposed onto that of CmnDph2 bound to 2AB and MTA (PDB entry 6BXM; carbon atoms colored green).19 The resulting separation distance between the Cγ atom of 2AB and the Cε1 atom of His597 is 1.5 Å (this distance is only an approximation due to the use of orthologous structures and the uncertainty of the locations of these atoms, but highlights the close proximity of the substrates). (B) Composite model of the MsDph2 active site showing highly conserved residues. The model is based on the structures of isolated MsDph2-MTA, the histidine substrate loop from the MsDph2-MsEF-2 complex, and SAM from CmnDph2-SAM (PDB entry 6BXM)19 combined via structural superimpositions. Dashed lines indicate potential hydrogen bonds.
The collection of structures of Dph2 complexes reported previously and here identify several highly conserved active site residues involved in substrate binding. These include His80, Gly160, Phe182, His183, Gln242, Arg291 and Asp295 (Figure 3B and Figure S5). Gln242 is used for recognition of the adenine moiety of SAM and Gly160, His183, and Arg291 for recognition of the SAM carboxylate group. Phe182 and Asp295 make cation-π and salt bridge interactions with Arg291, respectively, which likely play a role in orienting the arginine side chain.49 The function of His80 is unclear; however, its side chain is positioned near that of the histidine substrate of EF-2 and the high resolution structure of MsDph2-MTA shows that its Nε2 atom is poised to act as a hydrogen bond acceptor with the Nδ1 atom anchored through hydrogen bonding with the side chain of His77. Thus, His80 could possibly play a role in orienting the imidazole ring of the histidine substrate through hydrogen bonding for reaction with the ACP radical.
Given the high degree of conservation of Arg291 and the interactions it makes with Phe182, Asp295, and SAM, we tested its significance by mutating the equivalent arginine of PhDph2 (Arg289) to alanine. Interestingly, the R289A mutant still had SAM cleavage activity (Figure 4A) but completely lost the ACP transfer activity in an EF-2 labeling experiment using 14C-SAM (Figure 4B). Given the orientational restraints imposed on the arginine side chain by Phe182 and Asp295 and the salt bridge formed between the arginine and the carboxylate group of SAM, the results suggest that Arg291 plays an important role in the transfer of the ACP radical to the histidine substrate of MsEF-2.19, 20, 50
Figure 4.
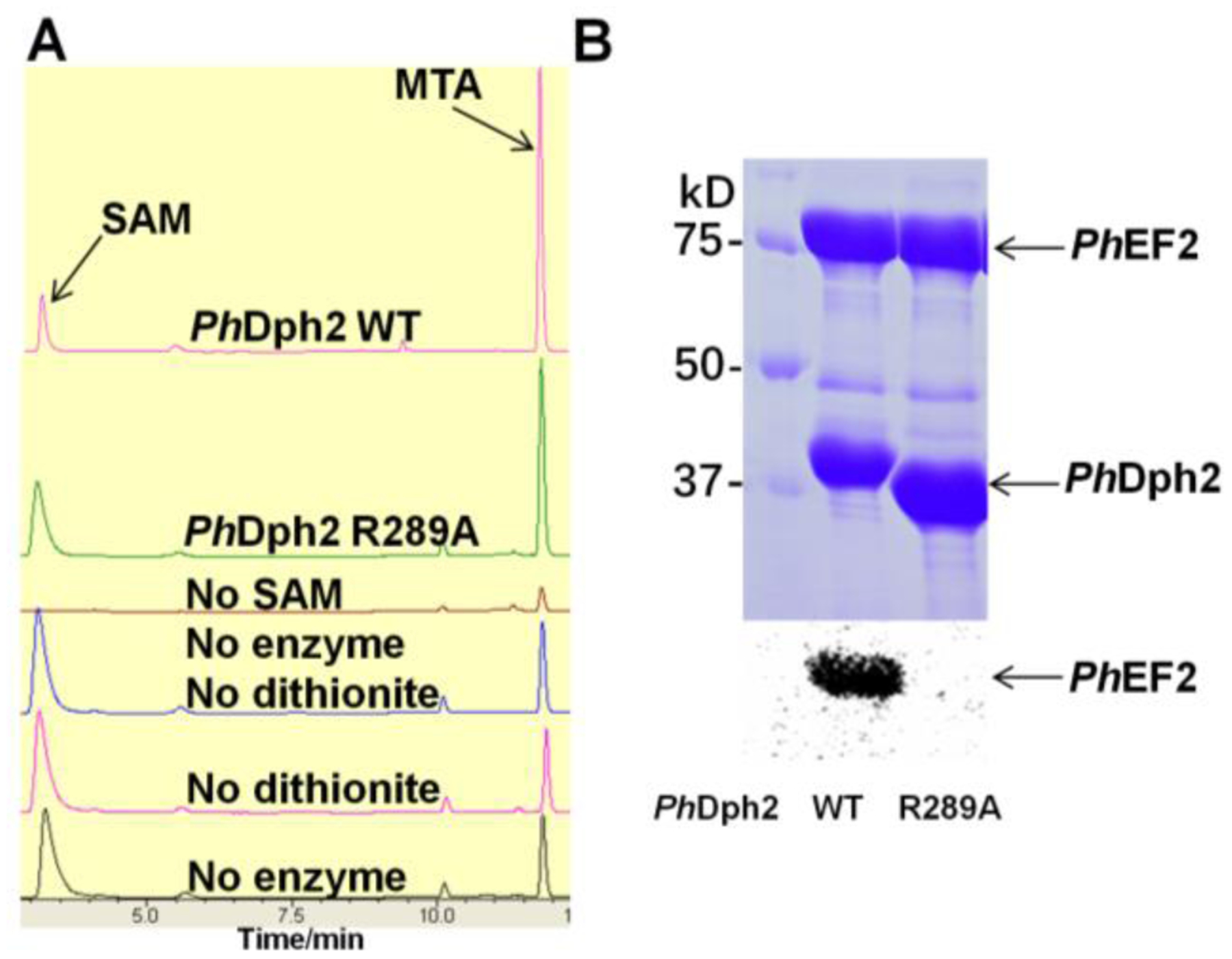
Mutagenesis study of PhDph2. (A) High-performance liquid chromatography showing PhDph2 R289A can cleave SAM and generate more MTA than controls. The MTA peaks in the control samples are from SAM decomposition. (B) In vitro reconstitution of PhDph2 wild type and R289A mutant activity using carboxy-14C-SAM shows R289A mutant lost the PhEF-2 labeling activity. The top panel shows the Coomassie-blue-stained gel; the bottom panel shows the autoradiography results. Both reactions contained PhEF-2, carboxy-14C-SAM and dithionite.
SUMMARY
The biosynthesis of diphthamide has been the subject of many research investigations in the last decade. These investigations have provided important insights into the mechanism of an unconventional radical SAM enzyme. It has been shown that the enzymes, the archaeal Dph2 homodimer or eukaryotic Dph1-Dph2 heterodimer, bind [4Fe-4S] clusters and use a radical mechanism for the first step of diphthamide biosynthesis. The reaction involves an organometallic intermediate I, which in the presence of the substrate is converted into a substrate-based organic radical intermediate II, in route to form the reaction product. This is a very interesting enzyme system and many questions remain to be addressed.
In this study, we report for the first time the structure of the archaeal Dph2 homodimer in complex with the substrate protein EF-2. The active site structure provides an important view on how the enzyme recognizes a specific histidine residue in the substrate that is consistent with the proposed reaction mechanism.19, 20, 31 As far as we know, diphthamide occurs only on EF-2. Given the strong and specific interactions observed between Dph2 and EF-2, it is not surprising that the diphthamide modification is so specific.
Compared to previously reported structures of Dph2 homodimers, binding of EF-2 induces substantial structural changes. Based on what is known about the Dph2 homodimer-catalyzed reaction, the enzyme needs to control its SAM cleavage activity depending on the availability of the substrate protein. It should thus minimize SAM cleavage when EF-2 is not present to prevent significant wasting of SAM molecules, but increase SAM cleavage to form reactive intermediate I in the presence of EF-2. The structural changes in Dph2 we observed upon EF-2 binding may thus be important for tuning the activity of Dph2 in response to EF-2 binding.
Structures of the isolated Dph2 homodimer also identified an important active site Arg residue (Arg291 in MsDph2 and Arg289 in PhDph2). Without this Arg residue, SAM cleavage could still occur (in the absence of EF-2), but the ACP group failed to transfer to EF-2. Thus, the Arg residue may be important for guiding the ACP radical to react with EF-2. In a previous study on the 5’-dA radical in lysine 2,3-aminomutase the authors state that “the free radical is never free”.51 It appears that the same statement also applies to the ACP radical in Dph2. However, not only does Dph2 form an organometallic intermediate to tame the ACP radical’s reactivity, it also uses important active site residues to guide its reaction with the substrate.
Supplementary Material
ACKNOWLEDGEMENTS
This work was supported by a National Institutes of Health grants GM088276 to H.L. and by the Howard Hughes Medical Institute (H.L.). The work is based upon research conducted at the Advanced Photon Source on the Northeastern Collaborative Access Team beamlines, which are supported by award P30 GM124165 from the NIH. Use of the Advanced Photon Source is supported by the U.S. Department of Energy, Office of Basic Energy Sciences, under Contract No. DE-AC02-06CH11357.
Footnotes
Supporting Information
The Supporting Information containing Table S1 and Figures S1-S5 is available free of charge on the ACS Publications website at DOI: 10.1021/acs.biochem.9b00718
Codon optimized Msdph2 and Msef-2 (Table S1), electron density of MsEF-2 (Figure S1), electron density of MsDph2 (Figure S2), MsEF-2 domain IV-MsDph2A interface (Figure S3), difference distance matrices indicating structural changes in MsDph2 and MsEF-2 upon complex formation (Figure S4), and amino acid sequence alignment of archaebacterial Dph2 orthologs (Figure S5).
Accession codes
The coordinates of the MsDph2-MsEF-2 complex and MsDph2-MTA have been deposited in the Protein Data Bank under accession codes 6Q2D and 6Q2E, respectively. The NCBI accession numbers of MsDph2 and MsEF-2 are WP_011954460.1 and WP_011954157.1, respectively.
Bibliography
- (1).Aevarsson A, Brazhnikov E, Garber M, Zheltonosova J, Chirgadze Y, al-Karadaghi S, Svensson LA, and Liljas A (1994) Three-dimensional structure of the ribosomal translocase: elongation factor G from Thermus thermophilus, EMBO J. 13, 3669–3677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (2).Czworkowski J, Wang J, Steitz TA, and Moore PB (1994) The crystal structure of elongation factor G complexed with GDP, at 2.7 A resolution, EMBO J. 13, 3661–3668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (3).Jorgensen R, Ortiz PA, Carr-Schmid A, Nissen P, Kinzy TG, and Andersen GR (2003) Two crystal structures demonstrate large conformational changes in the eukaryotic ribosomal translocase, Nat. Struct. Biol 10, 379–385. [DOI] [PubMed] [Google Scholar]
- (4).Kessel M, and Klink F (1980) Archaebacterial elongation factor is ADP-ribosylated by diphtheria toxin, Nature 287, 250–251. [DOI] [PubMed] [Google Scholar]
- (5).Kessel M, and Klink F (1981) Two elongation factors from the extremely halophilic archaebacterium Halobacterium cutirubrum. Assay systems and purification at high salt concentrations, Eur. J. Biochem 114, 481–486. [DOI] [PubMed] [Google Scholar]
- (6).Tanaka M, Iwasaki K, and Kaziro Y (1977) Translocation reaction promoted by polypeptide chain elongation factor-2 from pig liver, J. Biochem 82, 1035–1043. [DOI] [PubMed] [Google Scholar]
- (7).Van Ness BG, Howard JB, and Bodley JW (1980) ADP-ribosylation of elongation factor 2 by diphtheria toxin. NMR spectra and proposed structures of ribosyl-diphthamide and its hydrolysis products, J. Biol. Chem 255, 10710–10716. [PubMed] [Google Scholar]
- (8).Abeyrathne PD, Koh CS, Grant T, Grigorieff N, and Korostelev AA (2016) Ensemble cryo-EM uncovers inchworm-like translocation of a viral IRES through the ribosome, eLife 5, e14874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (9).Arguelles S, Camandola S, Cutler RG, Ayala A, and Mattson MP (2014) Elongation factor 2 diphthamide is critical for translation of two IRES-dependent protein targets, XIAP and FGF2, under oxidative stress conditions, Free Radic. Biol. Med 67, 131–138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (10).Hawer H, Utkur K, Arend M, Mayer K, Adrian L, Brinkmann U, and Schaffrath R (2018) Importance of diphthamide modified EF2 for translational accuracy and competitive cell growth in yeast, PLoS One 13, e0205870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (11).Mayer K, Mundigl O, Kettenberger H, Birzele F, Stahl S, Pastan I, and Brinkmann U (2019) Diphthamide affects selenoprotein expression: Diphthamide deficiency reduces selenocysteine incorporation, decreases selenite sensitivity and pre-disposes to oxidative stress, Redox Biol. 20, 146–156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (12).Ortiz PA, Ulloque R, Kihara GK, Zheng H, and Kinzy TG (2006) Translation elongation factor 2 anticodon mimicry domain mutants affect fidelity and diphtheria toxin resistance, J. Biol. Chem 281, 32639–32648. [DOI] [PubMed] [Google Scholar]
- (13).Lin Z, Su X, Chen W, Ci B, Zhang S, and Lin H (2014) Dph7 catalyzes a previously unknown demethylation step in diphthamide biosynthesis, J. Am. Chem. Soc 136, 6179–6182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (14).Liu S, Milne GT, Kuremsky JG, Fink GR, and Leppla SH (2004) Identification of the proteins required for biosynthesis of diphthamide, the target of bacterial ADP-ribosylating toxins on translation elongation factor 2, Mol. Cell. Biol 24, 9487–9497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (15).Uthman S, Bar C, Scheidt V, Liu S, ten Have S, Giorgini F, Stark MJ, and Schaffrath R (2013) The amidation step of diphthamide biosynthesis in yeast requires DPH6, a gene identified through mining the DPH1-DPH5 interaction network, PLoS Genet. 9, e1003334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (16).Zhang Y, Zhu X, Torelli AT, Lee M, Dzikovski B, Koralewski RM, Wang E, Freed J, Krebs C, Ealick SE, and Lin H (2010) Diphthamide biosynthesis requires an organic radical generated by an iron-sulphur enzyme, Nature 465, 891–896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (17).Frey PA, Hegeman AD, and Ruzicka FJ (2008) The Radical SAM Superfamily, Crit. Rev. Biochem. Mol. Biol 43, 63–88. [DOI] [PubMed] [Google Scholar]
- (18).Moss M, and Frey PA (1987) The role of S-adenosylmethionine in the lysine 2,3-aminomutase reaction, J. Biol. Chem 262, 14859–14862. [PubMed] [Google Scholar]
- (19).Dong M, Kathiresan V, Fenwick MK, Torelli AT, Zhang Y, Caranto JD, Dzikovski B, Sharma A, Lancaster KM, Freed JH, Ealick SE, Hoffman BM, and Lin H (2018) Organometallic and radical intermediates reveal mechanism of diphthamide biosynthesis, Science 359, 1247–1250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (20).Dong M, Zhang Y, and Lin H (2018) Noncanonical Radical SAM Enzyme Chemistry Learned from Diphthamide Biosynthesis, Biochemistry 57, 3454–3459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (21).Berkovitch F, Nicolet Y, Wan JT, Jarrett JT, and Drennan CL (2004) Crystal structure of biotin synthase, an S-adenosylmethionine-dependent radical enzyme, Science 303, 76–79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (22).Layer G, Moser J, Heinz DW, Jahn D, and Schubert WD (2003) Crystal structure of coproporphyrinogen III oxidase reveals cofactor geometry of Radical SAM enzymes, EMBO J. 22, 6214–6224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (23).Lepore BW, Ruzicka FJ, Frey PA, and Ringe D (2005) The x-ray crystal structure of lysine-2,3-aminomutase from Clostridium subterminale, Proc. Natl. Acad. Sci. U. S. A 102, 13819–13824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (24).Vey JL, and Drennan CL (2011) Structural insights into radical generation by the radical SAM superfamily, Chem. Rev 111, 2487–2506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (25).Walsby CJ, Ortillo D, Broderick WE, Broderick JB, and Hoffman BM (2002) An anchoring role for FeS clusters: chelation of the amino acid moiety of S-adenosylmethionine to the unique iron site of the [4Fe-4S] cluster of pyruvate formate-lyase activating enzyme, J. Am. Chem. Soc 124, 11270–11271. [DOI] [PubMed] [Google Scholar]
- (26).Hanzelmann P, Hernandez HL, Menzel C, Garcia-Serres R, Huynh BH, Johnson MK, Mendel RR, and Schindelin H (2004) Characterization of MOCS1A, an oxygen-sensitive iron-sulfur protein involved in human molybdenum cofactor biosynthesis, J. Biol. Chem 279, 34721–34732. [DOI] [PubMed] [Google Scholar]
- (27).Mehta AP, Hanes JW, Abdelwahed SH, Hilmey DG, Hanzelmann P, and Begley TP (2013) Catalysis of a new ribose carbon-insertion reaction by the molybdenum cofactor biosynthetic enzyme MoaA, Biochemistry 52, 1134–1136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (28).Otwinoski Z, and Minor W (1997) Processing of X-ray diffraction data collected in oscillation mode, Methods Enzymol. 276, 307–326. [DOI] [PubMed] [Google Scholar]
- (29).Kabsch W (2010) XDS, Acta Cryst. D66, 125–132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (30).Evans PR, and Murshudov GN (2013) How good are my data and what is the resolution?, Acta Cryst. D69, 1204–1214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (31).Evans P (2006) Scaling and assessment of data quality, Acta Cryst. D62, 72–82. [DOI] [PubMed] [Google Scholar]
- (32).McCoy AJ, Grosse-Kunstleve RW, Adams PD, Winn MD, Storoni LC, and Read RJ (2007) Phaser crystallographic software, J. Appl. Cryst 40, 658–674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (33).Adams PD, Afonine PV, Bunkoczi G, Chen VB, Echols N, Headd JJ, Hung LW, Jain S, Kapral GJ, Grosse Kunstleve RW, McCoy AJ, Moriarty NW, Oeffner RD, Read RJ, Richardson DC, Richardson JS, Terwilliger TC, and Zwart PH (2011) The Phenix software for automated determination of macromolecular structures, Methods 55, 94–106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (34).Waterhouse A, Bertoni M, Bienert S, Studer G, Tauriello G, Gumienny R, Heer FT, de Beer TAP, Rempfer C, Bordoli L, Lepore R, and Schwede T (2018) SWISS-MODEL: homology modelling of protein structures and complexes, Nucleic Acids Res. 46, W296–W303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (35).Tanzawa T, Kato K, Girodat D, Ose T, Kumakura Y, Wieden HJ, Uchiumi T, Tanaka I, and Yao M (2018) The C-terminal helix of ribosomal P stalk recognizes a hydrophobic groove of elongation factor 2 in a novel fashion, Nucleic Acids Res. 46, 3232–3244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (36).Emsley P, Lohkamp B, Scott WG, and Cowtan K (2010) Features and development of Coot, Acta Cryst. D66, 486–501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (37).Murshudov GN, Skubak P, Lebedev AA, Pannu NS, Steiner RA, Nicholls RA, Winn MD, Long F, and Vagin AA (2011) REFMAC5 for the refinement of macromolecular crystal structures, Acta Cryst. D67, 355–367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (38).Nicholls RA, Long F, and Murshudov GN (2012) Low-resolution refinement tools in REFMAC5, Acta Cryst. D68, 404–417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (39).Terwilliger TC, Grosse-Kunstleve RW, Afonine PV, Moriarty NW, Zwart PH, Hung LW, Read RJ, and Adams PD (2008) Iterative model building, structure refinement and density modification with the PHENIX AutoBuild wizard, Acta Cryst. D64, 61–69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (40).McGreevy R, Singharoy A, Li Q, Zhang J, Xu D, Perozo E, and Schulten K (2014) xMDFF: molecular dynamics flexible fitting of low-resolution X-ray structures, Acta Cryst. D70, 2344–2355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (41).Trabuco LG, Villa E, Mitra K, Frank J, and Schulten K (2008) Flexible fitting of atomic structures into electron microscopy maps using molecular dynamics, Structure 16, 673–683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (42).Terwilliger TC, Sobolev OV, Afonine PV, and Adams PD (2018) Automated map sharpening by maximization of detail and connectivity, Acta Cryst. D74, 545–559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (43).Painter J, and Merritt EA (2006) Optimal description of a protein structure in terms of multiple groups undergoing TLS motion, Acta Cryst. D62, 439–450. [DOI] [PubMed] [Google Scholar]
- (44).Pettersen EF, Goddard TD, Huang CC, Couch GS, Greenblatt DM, Meng EC, and Ferrin TE (2004) UCSF Chimera--a visualization system for exploratory research and analysis, J. Comput. Chem 25, 1605–1612. [DOI] [PubMed] [Google Scholar]
- (45).DeLano WL (2002) The PyMOL Molecular Graphics System, DeLano Scientific, San Carlos, CA. [Google Scholar]
- (46).Dong M, Zhang Y, and Lin H (2018) Methods for Studying the Radical SAM Enzymes in Diphthamide Biosynthesis, Methods Enzymol. 606, 421–438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (47).Krissinel E, and Henrick K (2007) Inference of macromolecular assemblies from crystalline state, J. Mol. Biol 372, 774–797. [DOI] [PubMed] [Google Scholar]
- (48).Hayward S, and Lee RA (2002) Improvements in the analysis of domain motions in proteins from conformational change: DynDom version 1.50, J. Mol. Graph. & Model 21, 181–183. [DOI] [PubMed] [Google Scholar]
- (49).Flocco MM, and Mowbray SL (1994) Planar stacking interactions of arginine and aromatic side-chains in proteins, J. Mol. Biol 235, 709–717. [DOI] [PubMed] [Google Scholar]
- (50).Dong M, Horitani M, Dzikovski B, Freed JH, Ealick SE, Hoffman BM, and Lin H (2017) Substrate-Dependent Cleavage Site Selection by Unconventional Radical S-Adenosylmethionine Enzymes in Diphthamide Biosynthesis, J. Am. Chem. Soc 139, 5680–5683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (51).Horitani M, Shisler K, Broderick WE, Hutcheson RU, Duschene KS, Marts AR, Hoffman BM, and Broderick JB (2016) Radical SAM catalysis via an organometallic intermediate with an Fe-[5’-C]-deoxyadenosyl bond, Science 352, 822–825. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.