

Abstract
Mutations in the nuclear trypsin‐like serine protease FAM111A cause Kenny–Caffey syndrome (KCS2) with hypoparathyroidism and skeletal dysplasia or perinatally lethal osteocraniostenosis (OCS). In addition, FAM111A was identified as a restriction factor for certain host range mutants of the SV40 polyomavirus and VACV orthopoxvirus. However, because FAM111A function is poorly characterized, its roles in restricting viral replication and the etiology of KCS2 and OCS remain undefined. We find that FAM111A KCS2 and OCS patient mutants are hyperactive and cytotoxic, inducing apoptosis‐like phenotypes such as disruption of nuclear structure and pore distribution, in a protease‐dependent manner. Moreover, wild‐type FAM111A activity causes similar nuclear phenotypes, including the loss of nuclear barrier function, when SV40 host range mutants attempt to replicate in restrictive cells. Interestingly, pan‐caspase inhibitors do not block these FAM111A‐induced phenotypes, implying it acts independently or upstream of caspases. In this regard, we identify nucleoporins and the associated GANP transcription/replication factor as FAM111A interactors and candidate targets. Overall, we reveal a potentially unifying mechanism through which deregulated FAM111A activity restricts viral replication and causes KCS2 and OCS.
Keywords: FAM111A, Kenny–Caffey syndrome, nuclear pore complex, Osteocraniostenosis, restriction of polyomavirus replication
Subject Categories: Microbiology, Virology & Host Pathogen Interaction; Molecular Biology of Disease; Post-translational Modifications, Proteolysis & Proteomics
The protease FAM111A was identified as an antiviral restriction factor, and as the gene mutated in two multisystem human syndromes, KCS2 and OCS. Patient mutations and polyomavirus replication in restrictive cells hyperactivate FAM111A, resulting in compromised nuclear barrier function, DNA replication, and cell viability.
Introduction
Family with sequence similarity 111 member A (FAM111A) was identified as an interactor of the SV40 large T antigen (LT) and a host range (HR) restriction factor that antagonizes productive infection by certain SV40 mutants (Fine et al, 2012). Specifically, FAM111A depletion rescues replication center formation and gene expression defects of LT mutants lacking C‐terminal sequences (Tarnita et al, 2019). Further analysis indicated that FAM111A executes its restrictive function during or following SV40 viral genome replication (Tarnita et al, 2019).
Expanding the role of FAM111A in protecting against viral challenge, it was identified along with the PCNA clamp‐loading complex RFC1‐5 as a restriction factor for orthopoxvirus HR mutants lacking the serine protease inhibitor SPI‐1 (Panda et al, 2017). Early studies on rabbitpox virus (RPXV) HR mutants lacking SPI‐1 suggested that restrictive human A459 cells exhibit apoptosis‐like features upon infection, which required RPXV DNA synthesis (Brooks et al, 1995). However, a subsequent study found that the related vaccinia virus (VACV) lacking SPI‐1 caused nuclear morphological changes such as chromatin condensation and membrane invagination, without the normal biochemical features of apoptosis (Shisler et al, 1999).
Thus, viruses with diverse life cycles, e.g., nuclear replication sites (polyomavirus) versus replication in the cytoplasm (orthopoxvirus), possess a mechanism to overcome inhibition by FAM111A. It is likely that FAM111A activity is triggered during viral replication, but how it combats viral challenge remains poorly understood (Luttge & Moyer, 2005; Panda et al, 2017; Tarnita et al, 2019). Consistent with its apparent replication‐dependent role in viral restriction, FAM111A was identified in high‐throughput screens as a protein enriched on nascent chromatin, i.e., replication forks (Alabert et al, 2014; Wessel et al, 2019). Exogenous FAM111A associated with replication sites via a PCNA interacting motif (Fig 1A; Alabert et al, 2014), whereas endogenous FAM111A was detected in nucleoli until S phase when, despite elevated expression, it appeared to become undetectable (Tarnita et al, 2019).
Figure 1. FAM111A localization in multiple cell lines.
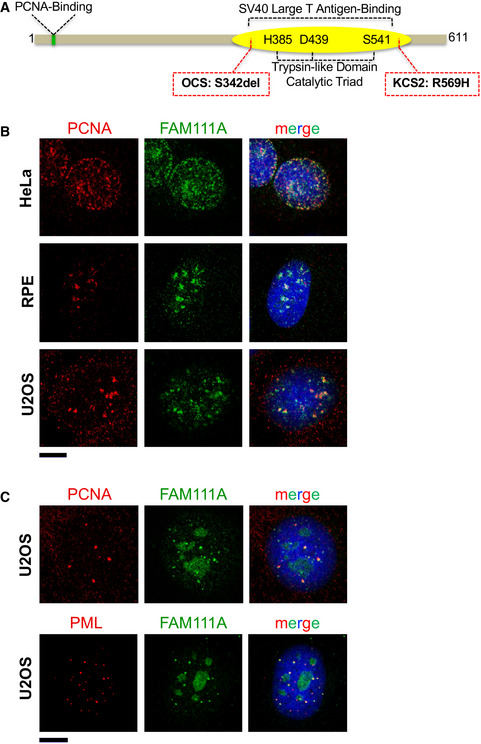
- Schematic representation of human FAM111A domains and mutations that compromise FAM111A functions. The FAM111A N‐terminus contains a functional PCNA interaction motif (PIP box; Alabert et al, 2014). The FAM111A C‐terminal trypsin‐like protease domain contains a typical H385‐D439‐S541 catalytic triad (Unger et al, 2013) and binds the C‐terminus of the SV40 Large T antigen (Fine et al, 2012; Tarnita et al, 2019). We used an S541A mutation to disrupt the catalytic triad and inactivate FAM111A’s trypsin‐like protease activity. Finally, the KCS2 and OCS patient mutations are indicated in red boxes. See text and (Unger et al, 2013) for phenotypic details.
- Confocal microscopy images of HeLa, RPE, and U2OS cell lines that were pre‐extracted with Triton X‐100 before fixed with formaldehyde and costained with DAPI and antibodies against PCNA and FAM111A. Scale bar: 10 μm.
- U2OS cells were stained with antibodies against PML and FAM111A and imaged by confocal microscopy. Scale bar: 10 μm.
Source data are available online for this figure.
The FAM111A C‐terminus contains a predicted trypsin‐like serine protease domain with an as yet untested catalytic triad (Fig 1A). However, the role of vaccinia virus SPI‐1 in antagonizing FAM111A is consistent with the presence of protease activity (Panda et al, 2017). Notably, dominant mutations in the FAM111A protease domain cause Kenny–Caffey syndrome type 2 (KCS2) and osteocraniostenosis (OCS), which present with hypoparathyroidism, short stature, and skeletal defects (Unger et al, 2013; Isojima et al, 2014; Nikkel et al, 2014). How the KCS2 and OCS mutations impact FAM111A activity remains unknown but due to their position in the protease domain, and apparent dominance over wild‐type, they were suggested to be gain‐of‐function (Unger et al, 2013).
Here, we show that the FAM111A KCS2 and OCS patient mutants are more cytotoxic than wild‐type FAM111A, due to deregulation of their trypsin‐like protease activity. Ectopic expression of the KCS2/OCS FAM111A mutants disrupts nuclear morphology, nuclear pore distribution, DNA replication, and cell‐substrate attachment in a protease activity‐dependent manner. Similarly, in response to the attempted replication of polyomavirus (SV40) HR mutants in restrictive cells, endogenous FAM111A impairs nuclear barrier function, morphology, and pore distribution. Surprisingly, these phenotypes are insensitive to pan‐caspase inhibitors, suggesting that FAM111A may trigger them more directly. In this regard, we identify several nuclear pore associated factors as candidate substrates of FAM111A. Overall, our data provide unifying insights into the pathological effects of KCS2/OCS FAM111A mutants, and the functions of FAM111A in viral restriction.
Results and Discussion
Endogenous FAM111A colocalizes with PCNA during replication
Earlier high‐throughput studies found that FAM111A associates with nascent chromatin, but the presence of endogenous FAM111A at replication forks had not been visualized (Alabert et al, 2014; Wessel et al, 2019). Using a validated commercial FAM111A antibody in multiple cell lines, we detected the previously reported nucleolar localization (Tarnita et al, 2019) and also observed a focal nuclear pattern in some cells that colocalized with PCNA (Fig 1B). Based on the characteristic patterns of nuclear PCNA during DNA replication, our data confirm that FAM111A is associated with sites of replication.
Interestingly, we also detected FAM111A at distinct subnuclear foci when U2OS cells were not in S phase, as judged by the lack of nuclear PCNA staining and the predominant nucleolar localization of FAM111A (Fig 1C). Such foci were not detected in the other cell lines tested. A key difference is that U2OS cells maintain their telomeres via a recombination‐based replication process called alternative lengthening of telomeres (ALT), as they do not have active telomerase (Bryan et al, 1997; Cesare & Reddel, 2010). ALT telomeres undergo DNA synthesis in the G2/M phases of the cell cycle at ALT‐associated PML bodies (APBs) that contain PCNA among other DNA replication and repair factors (Dilley et al, 2016; Min et al, 2017; Pan et al, 2017; Zhang et al, 2019; Sobinoff & Pickett, 2020; Loe et al, 2020). Further analysis revealed that the atypical FAM111A foci in U2OS cells colocalize with both PCNA and PML (Fig 1C). Therefore, FAM111A localizes to sites of DNA synthesis during both bulk genome duplication and the unscheduled DNA replication/repair events at ALT telomeres.
Protease‐dependent cytotoxicity of FAM111A
Genetic analysis of Kenney–Caffey syndrome 2 (KCS2) and osteocraniostenosis (OCS) revealed causative mutations in FAM111A (R569H and S342del, respectively; Fig 1A; Unger et al, 2013; Isojima et al, 2014)). Based on genetic considerations, these mutations were hypothesized to generate hyperactive variants of FAM111A (Unger et al, 2013). To test this, we generated a panel of inducible cell lines to study the effects of ectopically expressed wild‐type and mutant FAM111A.
Ectopic expression of wild‐type FAM111A strongly inhibited cell growth in HEK293 cells, whereas it had only a mild effect on U2OS cells and no significant effect on RPE cells (Fig 2A). These different responses to the ectopic expression of wild‐type FAM111A may be due to the extremely low levels of endogenous FAM111A present in HEK293 versus U2OS and RPE cells (Fig EV1). We attribute the growth inhibition by FAM111A to its protease activity, since the absence of toxicity of the protease‐dead mutant FAM111AS541A rules out any negative effects of it competing with other proteins for PCNA binding (Fig 2A). Notably, the KCS2 (FAM111AR569H) and OCS (FAM111AS342del) mutants were more potent inhibitors of cell growth than wild‐type FAM111A, strongly impacting all three cell lines (Fig 2A). These results are consistent with the gain‐of‐function hypothesis for FAM111A in KCS2 and OCS (Unger et al, 2013).
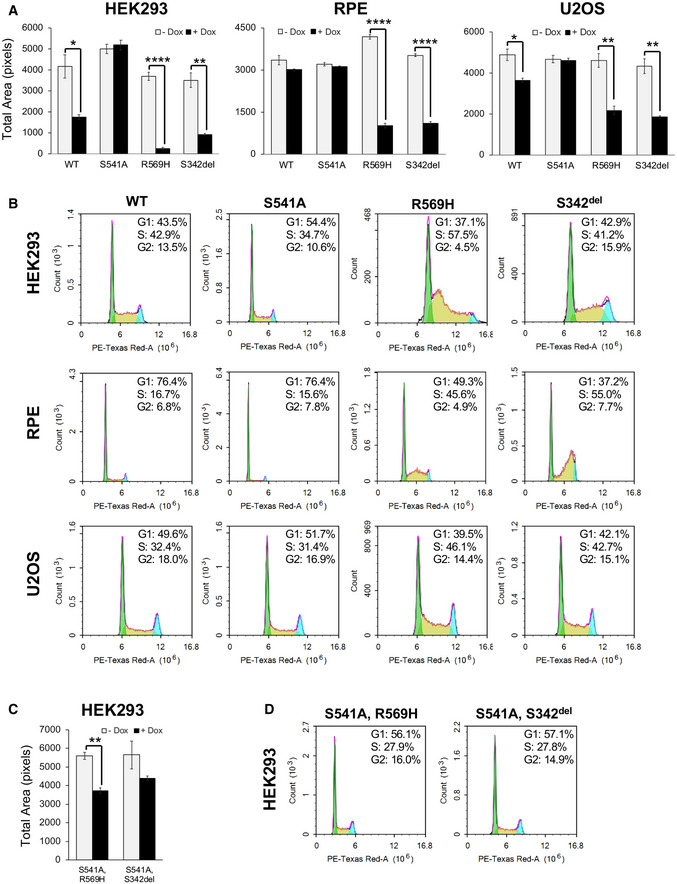
Figure 2. The effect of overexpressing wild‐type or mutant FAM111A on cell growth and cell cycle profiles in multiple cell lines.
-
A, CHEK293, RPE, or U2OS/MycBioID‐FAM111A cell lines were induced with doxycycline to express wild‐type or mutant FAM111A for 24 h (HEK293), 48 h (RPE), or 72 h (U2OS). The colonies were fixed then stained with crystal violet. Cell growth was quantitated as total area on ImageJ. Values are mean ± s.e.m. of independent experiments (n = 3). *P < 0.05, **P < 0.01, ****P < 0.0001 (two‐tailed unpaired t‐test).
-
B, DCell cycle profiles of HEK293, RPE, and U2OS FAM111A‐expressing cell lines that were induced with doxycycline for 48 h and analyzed by flow cytometry.
Figure EV1. Endogenous FAM111A levels in various cell lines.

Western blot analysis of FAM111A levels in the indicated cell lines. Total protein from an equal number of cells was loaded in each lane. The blots were probed with FAM111A and Tubulin antibodies.
Paralleling its cytotoxicity, the ectopic expression of FAM111A but not protease‐dead FAM111AS541A also perturbed cell cycle profiles (Fig 2B, FACS ‐Dox in Fig EV2). In particular, FAM111AR569H and FAM111AS342del expression caused cells to accumulate in S phase (Fig 2B). Given the localization of FAM111A to replication forks and its protease‐dependent toxicity, these results suggest that the KCS2 and OCS patient mutants disrupt replication, conceivably through the unscheduled removal of replication factors.
Figure EV2. FACS analysis of cell cycle in control conditions (‐ Dox).
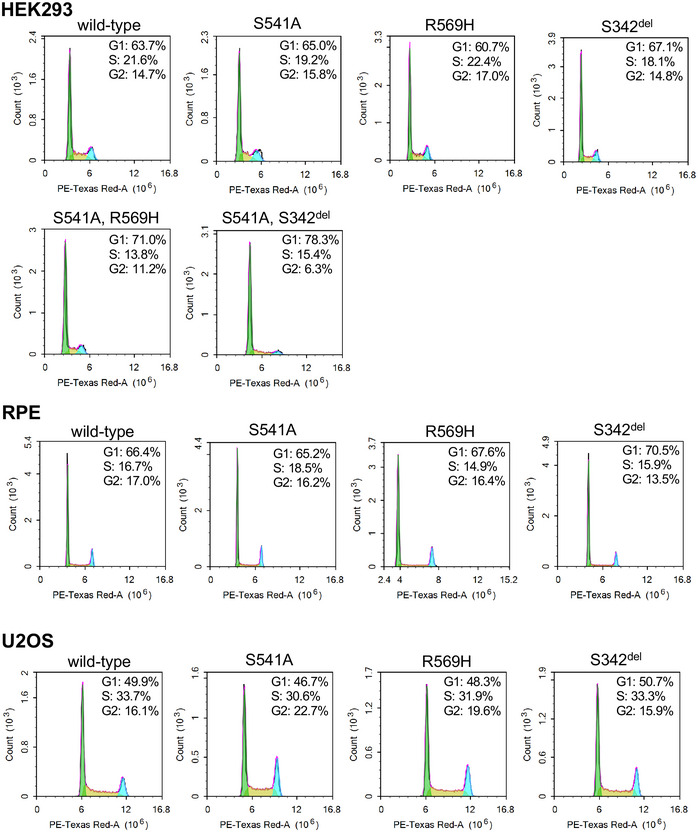
Flow cytometry analysis of the cell cycle profiles of HEK293, RPE, and U2OS FAM111A cell lines without doxycycline induction was performed as described in Fig 2B.
Ablating the protease activity of FAM111AR569H and FAM111AS342del with the S541A mutation mitigated both their toxicity (Fig 2C) and effects on cell cycle distribution (Fig 2D). Moreover, HEK293T cells that constitutively express the SV40 large T antigen were relatively more resistant to wild‐type, FAM111AR569H or FAM111AS342del overexpression than HEK293 cells (Fig EV3). This supports the suspected role of the SV40 Large T antigen in inactivating FAM111A protease activity (Tarnita et al, 2019).
Figure EV3. Cytotoxicity of FAM111A overexpression in HEK293T cells.
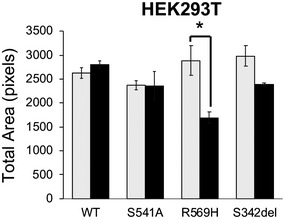
HEK293T/MycBioID‐FAM111A cell lines were induced with doxycycline to express for 24 h. The colonies were fixed then stained with crystal violet. Cell growth was quantitated as total area on ImageJ. Values are mean ± s.d. of independent experiments (n = 3). *P < 0.05 (two‐tailed unpaired t‐test).
Caspase‐independent cytotoxicity of FAM111A hyperactivity
Considering the foregoing, we tested whether hyperactive FAM111A induces apoptosis. Cleavage of PARP is a well‐characterized marker of caspase‐dependent apoptosis (Soldani & Scovassi, 2002) and was weakly detected in cells expressing wild‐type FAM111A but not protease‐dead FAM111AS541A (Fig 3A). Notably, PARP cleavage was stronger in cells expressing FAM111AR569H or FAM111AS342del, despite their relatively low levels (Fig 3A). Further, the levels of ectopically expressed FAM111A were similar to those of endogenous FAM111A in HEK293 but much lower than those in U2OS cells, making their toxicity all the more striking (Fig 3A). These different ectopic/endogenous ratios of FAM111A expression may explain the higher sensitivity of HEK293 versus U2OS cells to ectopic FAM111A expression (Fig 2A).
Figure 3. Caspase inhibitor Z‐VAD‐FMK inhibits PARP cleavage, but not cell death, in cells overexpressing FAM111AR569H .
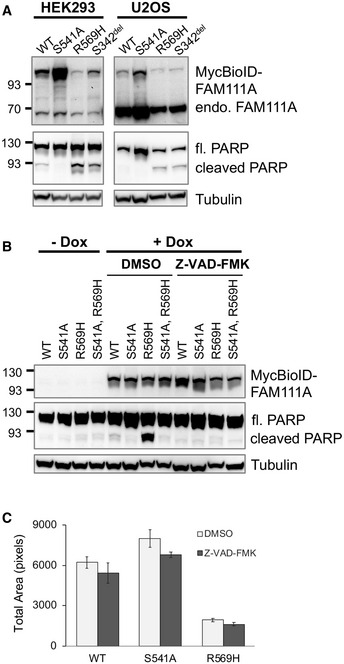
- HEK293 or U2OS/MycBioID‐FAM111A cell lines were induced with doxycycline for 24 h. Total protein from 105 cells was resolved by SDS–PAGE and probed in Western blots for both endogenous FAM111A (71 kDa) and MycBioID‐FAM111A (110 kDa) as well as full length and cleaved PARP (116 and 89 kDa, respectively). The Tubulin Western blot is a sample loading control.
- HEK293/MycBioID‐FAM111A cell lines with no treatment (‐ Dox) or treated with doxycycline, along with DMSO or 100 μM Z‐VAD‐FMK for 24 h (total inhibition of PARP cleavage was also observed when 50 μM Z‐VAD‐FMK was used). Total protein from ~105 cells was resolved by SDS–PAGE and probed in Western blots with Myc, PARP, or Tubulin antibodies.
- HEK293 cell lines were treated with doxycycline and Z‐VAD‐FMK for 24 h. The colonies were fixed and then stained with crystal violet. Cell growth was quantitated as total area on ImageJ. Values are mean ± s.e.m. of independent experiments (n = 3).
We next tested the contribution of apoptotic induction to the profound cytotoxic effects of FAM111AR569H. To this end, we used the well‐characterized pan‐caspase inhibitor Z‐VAD‐FMK to reduce apoptotic signaling in cells expressing FAM111A, FAM111AR569H, FAM111AS541A, or FAM111AS541A,R569H. As anticipated, PARP cleavage was enhanced by FAM111AR569H expression, but was largely absent in cells expressing the protease‐dead FAM111A constructs (Fig 3B, FAM111AS541A and FAM111AS541A,R569H). Therefore, the protease activity of FAM111A and FAM111AR569H can induce caspase‐dependent apoptosis. Notably, however, while Z‐VAD‐FMK treatment blocked PARP cleavage in both FAM111A and FAM111AR569H expressing cells (Fig 3B), it did not reduce the cytotoxicity of FAM111AR569H (Fig 3C).
Thus far, our data show that KCS2 and OCS FAM111A patient mutants are gain‐of‐function and that their elevated protease activity has caspase‐independent cytotoxic effects.
BioID‐based proteomics identifies candidate targets of FAM111A activity
To define FAM111A protease targets driving the above phenotypes, we used proximity labeling (BioID) of FAM111A and FAM111AS541A coupled to mass spectrometry‐based protein identification (Roux et al, 2012). We reasoned that protease‐dead FAM111S541A could generate enhanced or more durable biotin labeling of interactors than wild‐type, similar to a substrate trap approach.
Candidate FAM111A interactors were identified from both asynchronous and S phase cells (Dataset EV1). Consistent with FAM111A’s colocalization with PCNA, several replication‐associated proteins were identified including RFC1, RAD18, LIG1, and PCNA (Fig 4A). Despite being proximal to FAM111A, these factors were not consistently enriched in FAM111AS541A over wild‐type FAM111A purifications, suggesting that they may not be targets of its protease activity.
Figure 4. NPC components are potential targets of FAM111A activity.
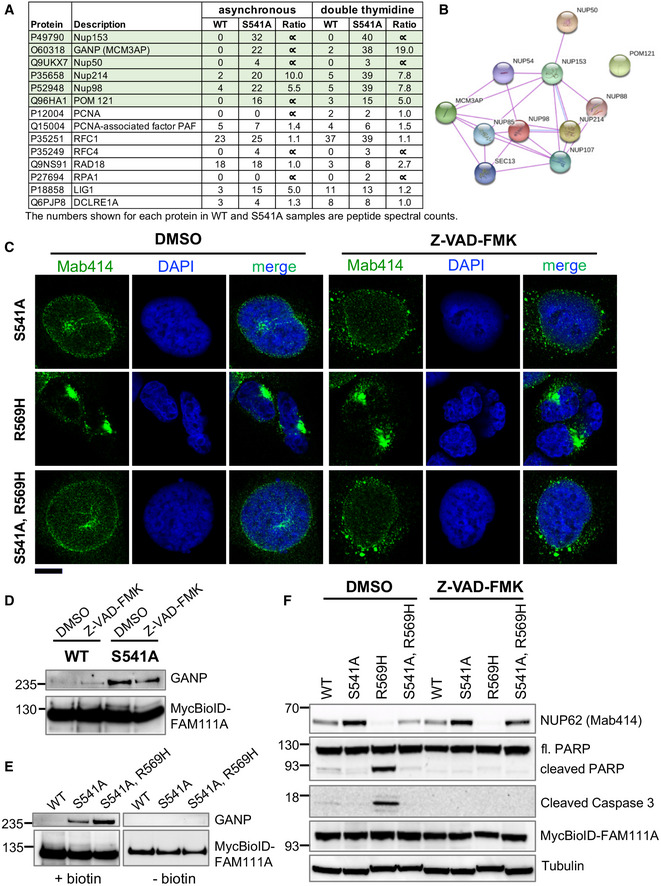
- A subset of FAM111A interacting proteins identified by BioID screen is listed along with the number of peptide spectral counts observed for each protein.
- A STRING diagram showing the interaction of NPC components identified in our FAM111A BioID screen.
- Cells were treated with doxycycline along with DMSO or Z‐VAD‐FMK for 24 h. They were fixed with formaldehyde and stained with NPC antibody Mab414 and DAPI. The images were captured by confocal microscopy. Scale bar: 10 μm.
- DMSO or Z‐VAD‐FMK‐treated HEK293/MycBioID‐FAM111A cell lines were induced with doxycycline for 24 h. Total protein from an equal number of cells was incubated with magnetic streptavidin beads, and pull downs were analyzed by Western blotting using GANP and Myc antibodies.
- HEK293/MycBioID‐FAM111A cell lines were induced with doxycycline for 24 h, while growing in the presence or absence of 50 µM supplemental biotin. Total protein from whole cell lysates was incubated with magnetic streptavidin beads. Pulldowns were analyzed by Western blotting using GANP and Myc antibodies.
- DMSO or Z‐VAD‐FMK‐treated HEK293/MycBioID‐FAM111A cell lines were induced with doxycycline for 24 h. Total protein from ~105 cells was resolved by SDS–PAGE and probed in Western blots using antibodies against NPC (Mab414), PARP, cleaved Caspase 3, Myc, or Tubulin.
Interestingly, however, among the most highly enriched proteins identified in FAM111AS541A over wild‐type FAM111A purifications were a number of nuclear pore complex (NPC) factors (Fig 4A). These included NUP153, POM121, NUP214, NUP50, NUP98, and the multifunctional mRNA export cofactor GANP (Fig 4A and B; Wickramasinghe et al, 2010b).
Given our identification of NPC proteins as potential FAM111A targets, we tested the impact of FAM111A hyperactivity on NPC distribution using antibody Mab414, which recognizes several NPC components. Ectopic expression of FAM111AR569H caused the nuclei of most cells to “shrivel” and lose their characteristic smooth peripheries, resembling the appearance of early apoptotic nuclei (Fig 4C and Fig EV4A, Mab414). Also, nuclear pores underwent a dramatic redistribution, lost their typical granular appearance, and “clumped” in regions of nuclear invagination or distortion (Fig 4C and Fig EV4A). These phenotypes were not seen upon FAM111AS541A overexpression, were dependent on the protease activity of FAM111AR569H (see FAM111AS541A,R569H), and were consistent across different cell lines (Fig 4C and Fig EV4B). Notably, caspase inhibition again failed to block these FAM111AR569H‐induced phenotypes (Fig 4C, similar results were obtained with Q‐VD‐OPH). Therefore, the protease activity of FAM111AR569H appears to drive caspase‐independent apoptosis‐like phenotypes.
Figure EV4. Immunofluorescence of NPCs in cells expressing FAM111A mutants.
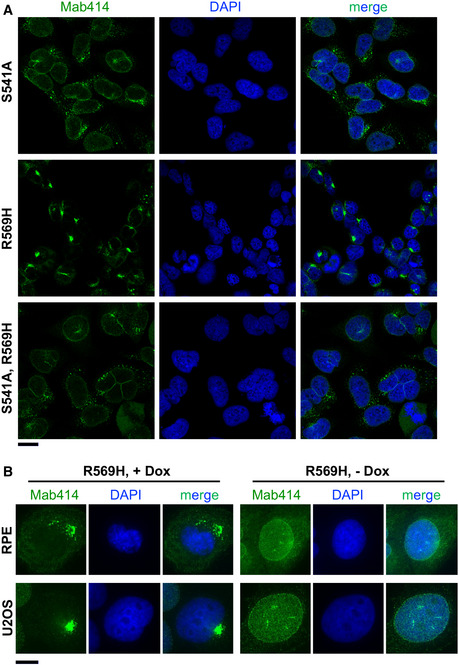
- Cells captured through a 63×/1.4 objective of an LSM880 confocal microscope (a selection of individual cells is shown in Fig 4C). Scale bar: 20 µm.
- Representative immunofluorescence images of RPE or U2OS/MycBioID‐FAM111A‐R569H stained with Mab414 and DAPI. Images were captured on a Zeiss Axio Imager through a 60x objective. Scale bar: 10 µm.
In this regard, there have been reports of a caspase‐independent change in nuclear structure and barrier function in early apoptosis similar to what we report, which is instead dependent on an unidentified serine protease (Ferrando‐May et al, 2001; Egger et al, 2003; O'Connell & Stenson‐Cox, 2007; Strasser et al, 2012; Kopeina et al, 2018). In the future, it will be interesting to determine if FAM111A is the serine protease in question.
Validating the NPC as a potential target of FAM111A protease activity
GANP is a multifunctional protein whose N‐terminus shares homology with the FG repeat‐containing nucleoporins (Wickramasinghe et al, 2010a). It functions as a scaffold for the TREX‐2 mRNA export complex at the NPC (Fischer et al, 2002; Wickramasinghe et al, 2010b; Bhatia et al, 2014), and its C‐terminal domain can interact with and acetylate a key replication factor, MCM3 (Kuwahara et al, 2000; Takei et al, 2001; Takei et al, 2002). To validate our FAM111A BioID results, we used a GANP antibody to test its copurification with FAM111A. Western blots revealed the expected enrichment of GANP in FAM111AS541A purifications over those of wild‐type FAM111A (Fig 4D). As FAM111A overexpression can induce apoptosis‐like phenotypes, we tested whether Z‐VAD‐FMK affects the interaction between wild‐type FAM111A and GANP, but found the results were unchanged (Fig 4D). We extended the analysis to include the FAM111AS541A,R569H double mutant. Again, GANP was only detected in BioID samples from FAM111A protease‐dead mutant expressing cells, including FAM111AS541A,R569H, indicating that this FAM111A patient mutation does not interfere with GANP interaction (Fig 4E). Thus, protease‐dead FAM111A spends longer in proximity to GANP, implying GANP may be proteolyzed by wild‐type FAM111A.
Given the dramatic NPC mislocalization caused by FAM111A hyperactivity (Fig 4C), we used the same antibody, Mab414, in Western analysis to detect possible changes in NPC factor abundance. Strikingly, NUP62, the NPC factor most robustly detected by Mab414 in whole cell extracts (Baquero‐Perez & Whitehouse, 2015), was strongly depleted upon FAM111AR569H expression (Fig 4F). This effect was lessened by ablating the protease activity of FAM111AR569H with the additional S541A mutation (Fig 4F, FAM111AS541A,R569H). Moreover, NUP62 abundance was lower in cells expressing wild‐type FAM111A as compared to those expressing the FAM111S541A protease‐dead mutant (Fig 4F). Importantly, while Z‐VAD‐FMK blocked PARP and caspase 3 cleavage, it failed to restore NUP62 levels in cells expressing FAM111AR569H (Fig 4F).
NUP62 (p62) is a phenylalanine–glycine (FG) repeat central channel nucleoporin that plays an essential role in gating and transport through the NPC (Rout et al, 2003; Lim et al, 2007; Tu et al, 2013; Chug et al, 2015). Although NUP62 was not identified in our FAM111A BioID screen, it localizes within the NPC near NUP153 and other NPC proteins identified. Such “missed” identifications occur with proximity labeling methods that require spatially available biotinylation sites and also with mass spectrometry‐based methods that more readily detect large and abundant proteins. Nevertheless, NUP62 is clearly depleted in a FAM111A‐dependent but caspase‐independent manner, making it the best candidate target of FAM111A protease activity described to date. NUP62 depletion by FAM111A also fits well with FAM111A’s induction of nuclear permeability during viral replication, as shown below. Whether NUP62 depletion contributes to KCS2/OCS development requires further study. However, it is interesting that clinically overlapping KCS1 is caused by mutations in a tubulin cofactor, TBCE (Parvari et al, 2002). Therefore, future studies on the etiology of KCS2/OCS should not only focus on the DNA replication defects caused by FAM111A hyperactivity.
FAM111A induces changes in nuclear permeability during viral replication
FAM111A is an SV40 HR restriction factor, which interacts with and is likely inactivated by the C‐terminus of wild‐type SV40 large T antigen (LT; (Fine et al, 2012)). That is, FAM111A antagonizes the formation of replication centers of SV40 HR mutants that contain LT C‐terminal mutations e.g. SV40HR684 (Tarnita et al, 2019). FAM111A also restricts an orthopoxvirus HR mutant that lacks the serpin family serine protease inhibitor SPI‐1, suggesting it has broad antiviral activity (Panda et al, 2017). Despite these advances, how FAM111A restricts viral challenges remains poorly defined.
Our results show that FAM111A activity can impair cellular viability and induce apoptosis‐like phenotypes. However, the data so far are most relevant to the situation in KCS2 or OCS patients, where FAM111A is hyperactive (Unger et al, 2013). We therefore tested the impact of endogenous wild‐type FAM111A on cells when challenged by SV40. To this end, we transfected U2OS cells with wild‐type SV40 or two HR mutants SV40HR684 and SV40dl1066 (Poulin & DeCaprio, 2006; Tarnita et al, 2019).
Indirect immunofluorescence (IF) analysis of wild‐type SV40 LT revealed a consistently nuclear localization in U2OS cells over the 72‐h time course, as reported previously (Figs 5A and B, and Fig EV5A; (Tarnita et al, 2019)). In contrast, the SV40HR684 and SV40dl1066 HR mutants developed a progressively more pan‐cellular LT distribution, after an initial nuclear localization at 24 h (Figs 5A and B, and Fig EV5A). In addition, the nuclei of cells transfected with SV40HR684 appeared to partially shrivel and nucleoporins underwent a dramatic redistribution (Fig EV5B), as seen with FAM111AR569H overexpression.
Figure 5. FAM111A affects nuclear barrier function during viral replication.
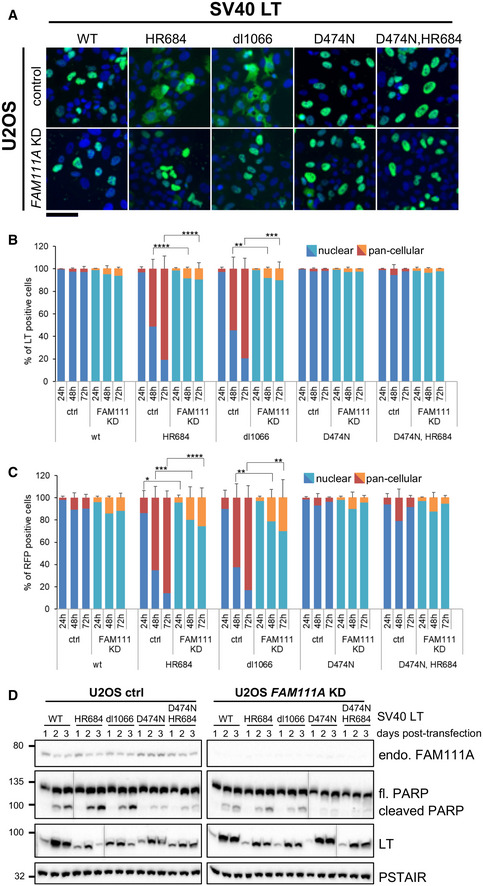
- Representative immunofluorescence images of SV40 LT localization 72 h after transfection into U2OS cells infected with control or FAM111A shRNA. Images were taken with a Zeiss Axio Imager. LT is shown in green, and DAPI blue. Scale bar: 100 μm.
- Quantification of LT‐positive cells described in (A) and fixed at indicated times post‐transfection. Five hundred or more cells were counted for each condition.
- Quantification of RFP marker distribution upon cotransfection with SV40 LT variants into U2OS cells infected with control or FAM111A shRNA. Cells were fixed at the indicated times. Five hundred or more cells were counted for each condition.
- Western blot analysis of control or FAM111A‐depleted U2OS cells transfected with SV40 LT variants. Cells were collected and lysed at indicated timepoints after SV40 transfection, immunoblotted with FAM111A, LT, or PARP antibodies. Cdc2‐PSTAIR was used as a loading control.
Figure EV5. Representative immunofluorescence images of SV40 LT and NPC.
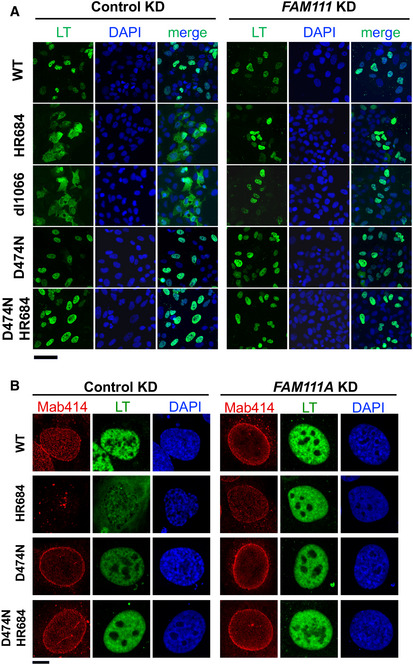
- SV40 LT (green) and DAPI (blue) staining in control or FAM111A shRNA infected U2OS cells 72 h after transfection with SV40 plasmids. Images were taken by Axio Imager 20×/0.6 objective. Scale bar: 100 µm.
- SV40 LT and NPC (Mab414) localization 72 h after transfection into U2OS cells infected with control or FAM111A shRNA. Images were captured through a 63×/1.4 objective of LSM880 confocal microscope. Scale bar: 10 µm.
To test whether SV40HR684 and SV40dl1066 transfected cells actually lose normal nuclear barrier function, we co‐expressed an independent 2XRFP‐NLS nuclear reporter (Hatch et al, 2013). The loss of reporter compartmentalization mirrored that of the SV40 HR LT mutants (Fig 5C). Therefore, rather than a specific defect in the nuclear retention of LT HR mutants, nuclear barrier function is generally compromised. Importantly, depletion of FAM111A rescued the above phenotypes, including increased nuclear permeability (Fig 5A–C). Therefore, the wild‐type, but not SV40HR684 and SV40dl1066, LT antagonizes FAM111A activity, which otherwise causes nuclear dysfunction.
We next analyzed LT levels by Western blot, as they reflect the ability of SV40 to transcribe and replicate its genome. As previously reported, wild‐type SV40 LT expression peaked at 48 h and sustained through 72 h, whereas SV40HR684 and SV40dl1066 LT expression was weaker overall and tapered off by 72 h (Fig 5D; (Tarnita et al, 2019)). Interestingly, PARP cleavage was higher in SV40HR684 and SV40dl1066 than wild‐type SV40 transfected cells, being strongest at 72 h when loss of nuclear barrier function peaks for the HR mutants (Fig 5D). Importantly, FAM111A depletion largely suppressed the differences in LT expression profiles of wild‐type and HR mutant SV40 and reduced PARP cleavage in SV40HR684 and SV40dl1066 cells (Fig 5D).
FAM111A appears to execute its restrictive function during viral DNA replication (Panda et al, 2017; Tarnita et al, 2019), which fits with its localization to cellular DNA replication sites and the negative effects of FAM111AR569H on processive replication. To determine whether SV40 replication triggers the FAM111A‐dependent loss of nuclear barrier function, we used an LT mutation (D474N) that abolishes its helicase activity and SV40 genome replication (Huang et al, 2010).
We generated the single SV40D474N LT mutant and the double SV40D474N,HR684 LT mutant for analysis by IF and Western blot. SV40D474N LT was almost exclusively nuclear at all time points, and Western analysis revealed a wild‐type expression profile that did not taper at 72 h (Fig 5A–D). Importantly, the SV40D474N,HR684 double mutant behaved like the SV40D474N single mutant in terms of nuclear localization and expression profile (Fig 5A–D). In addition, unlike the SV40HR684 LT, FAM111A depletion had no impact on the expression profile of SV40D474N or SV40D474N,HR684 LT (Fig 5A–D). Thus, it is not simply SV40HR684 LT expression that causes nuclear permeability changes, rather; it is SV40 replication in the presence of FAM111A activity.
Given that hyperactive FAM111AR569H disrupts cellular DNA replication, we envisioned that FAM111A could directly inhibit viral replication by degrading key replication factors. Crucially however, although others and we have identified replication factors as FAM111A interactors, to date none appear to be targets of its protease activity (Hoffmann et al, 2020). Instead, FAM111A causes nuclear barrier dysfunction, among other phenotypes, when SV40 HR mutants attempt to replicate in restrictive cells. These effects, which are strikingly similar to those caused by the KCS2 and OCS FAM111A patient mutants, could indirectly inhibit both viral and cellular DNA replication.
We do not believe that the induction of apoptosis is a primary restrictive function of FAM111A. Instead, FAM111A triggers early nuclear barrier dysfunction in the absence of late apoptotic phenotypes. As mentioned above, an unidentified serine protease was implicated in the caspase‐independent loss of nuclear barrier function in response to different types of cell stress (Ferrando‐May et al, 2001; Egger et al, 2003; O'Connell & Stenson‐Cox, 2007; Strasser et al, 2012; Kopeina et al, 2018). In addition, early studies revealed that RPXV and VACV HR mutants that lack the serine protease inhibitor SPI‐1 cause alterations in the nuclear morphology of restrictive cells (Brooks et al, 1995; Shisler et al, 1999). Together with the fact that FAM111A is a restriction factor for VACV SPI‐1∆ (Panda et al, 2017), our data suggest that the disruption of cell function by nuclear barrier loss could be a common function for FAM111A in restricting viral replication.
Together, our results demonstrate that FAM111A exhibits protease‐dependent cytotoxicity, which is amplified by the KCS2 and OCS patient mutants, consistent with recent studies (Kojima et al, 2020; Hoffmann et al, 2020). Thus, FAM111A hyperactivity can explain the dominant effects of FAM111AR569H and FAM111AS342del over wild‐type in the heterozygous disease state (Unger et al, 2013). Moreover, we show that hyperactive FAM111A compromises nuclear barrier function, S phase progression, and cell viability, possibly in part through the proteolysis of the NPC factors GANP and NUP62. Such effects could negatively impact parathyroid development (among other systems) causing KCS2 and OCS phenotypes (Unger et al, 2013). These same effects could also prevent SV40 HR mutant replication in restrictive cells.
Consistent with its S phase localization, FAM111A is in close proximity to DNA replication factors such as RFC1 and PCNA, but these do not appear to be targets of FAM111A protease activity (see also Hoffmann et al, 2020). Instead, FAM111A likely cooperates with PCNA and RFC1‐5 at both cellular and viral genome replication sites. Indeed, FAM111A appears to remove cellular replication fork‐blocking DNA‐protein complexes, e.g., TOP1cc, and collaborates with RFC1‐5 to restrict the replication of HR mutant poxviruses (Panda et al, 2017; Kojima et al, 2020). Therefore, FAM111A may be “activated” at replication forks to promote replication fork progression, and depending on the context, degrade NPC or other factors to control cell function.
At a glance, FAM111A’s localization to replication forks and its targeting of the NPC seem incompatible. Interestingly, however, nucleoporins and GANP make chromatin contacts to spatially organize chromatin, control its outputs such as transcription, and maintain genome stability (Ibarra & Hetzer, 2015). Moreover, when FAM111A is at the replication fork, several nucleoporins and GANP are actually depleted from nascent chromatin (Alabert et al, 2014; Wessel et al, 2019). Therefore, we speculate that FAM111A could help displace nuclear pore‐chromatin interactions to facilitate replication fork progression, as seen at actively transcribed genes (Brickner & Brickner, 2011).
Overall, we propose a model in which the loading of PCNA by RFC1‐5 acts as a DNA sensor that recruits FAM111A to endogenous replication sites or viral genomes that utilize/attract PCNA (Sowd & Fanning, 2012; Postigo et al, 2017). During unscheduled viral DNA replication or in KCS2/OCS patients, FAM111A is hyperactivated and cytotoxic due to the absence of normal protease regulation. Wild‐type SV40 LT and VACV SPI‐1 are able to inhibit FAM111A, preventing both local protease activity (at viral replication centers) and nuclear barrier dysfunction, thereby enabling viral replication. In the future, it will be interesting to define endogenous regulatory mechanisms for FAM111A, and if its protease activity can be therapeutically targeted.
Materials and Methods
Cell culture
Human embryonic kidney cell lines HEK293 and HEK293T, human osteosarcoma cell line U2OS, human colon cancer line HCT116, and human cervical adenocarcinoma cell line HeLa were obtained from American Type Culture Collection (ATCC). Human epithelial cells immortalized with hTERT cell line RPE was obtained from Denchi lab (Loe et al, 2020). These cell lines were cultured in Dulbecco’s modified Eagle’s medium (DMEM) supplemented with 10% fetal bovine serum (FBS).
Unless otherwise indicated, the following drug concentrations were used: doxycycline (1 μg/ml, Sigma‐Aldrich), biotin (50 μM, Sigma‐Aldrich, B4501), and Z‐VAD‐FMK (50 or 100 μM, APExBIO, A1902).
Plasmids
Full‐length human FAM111A cDNA was PCR‐amplified and inserted into destination vectors using In‐Fusion seamless cloning (Takara Bio). The pTRIPz‐MycBioID vector allows for doxycycline‐inducible expression of FAM111A with N‐terminal fusion of a Myc tag and a promiscuous biotin ligase (MycBioID; Roux et al, 2012). Point mutations in FAM111A and SV40 large T antigen (LT) were introduced using the QuikChange Site‐Directed Mutagen esis kit (Agilent Technologies) according to the manufacturer’s protocol. All cloning and mutagenesis were verified by sequencing. Plasmids and stable cell lines used in this study are listed in supplemental tables (Tables EV1 and EV2).
RNAi
Stable RNAi was achieved by viral shRNA generated in pLKO.1‐blast (Bryant et al, 2010). Knockdown was verified by standard Western blot, normalizing to Tubulin or Cdc2 expression. pLKO.1 lentiviruses were constructed according to the Addgene pLKO.1 protocol (www.addgene.org), with target sequences found in Mission shRNA database (Sigma‐Aldrich). RNAi target sequences used in this research include:
Control‐sh: 5’‐CCTAAGGTTAAGTCGCCCTCG‐3’.
FAM111A‐sh1: 5’‐GTCAATGTGTAAGGGTGACAT‐3’.
Transfection, virus production, and transduction
For lentivirus production, pTRIPz or pLKO.1 plasmid was cotransfected with lentiviral packaging vectors (pMD2.G and psPAX2) into HEK293T using TransIT‐LT1 (Mirus) transfection reagent following the manufacturer instruction. All supernatants were passed through 0.45‐μm filters to remove cell debris. The supernatants were either immediately applied to the target cells or frozen in liquid nitrogen for further use. For lentivirus transduction (pLKO.1, pTRIPz), subconfluent U2OS, RPE, HEK293, or 293T cultures were infected with virus‐containing supernatants for 12–16 h at 37°C. Viral supernatants were then replaced with fresh growth medium and cultured for a further 72 h with appropriate antibiotic selection. Puromycin (0.5 μg/ml for U2OS; 2 μg/ml for HEK293, 293T) and blasticidin (12 μg/ml) were used. To generate stable cell lines (Table EV2), the transduced cells were cultured in the presence of antibiotic selection until all control cells were killed. SV40 plasmid transfections were conducted using TransIT LT1 (Mirus) transfection reagent following the manufacturer’s instructions.
Immunofluorescence and microscopy
The cells grown on coverslips were fixed in 4% formaldehyde in PBS for 10 min and then permeabilized with PBS containing 0.2% Triton X‐100 for 5 min at room temperature. The cells stained for PCNA were pre‐extracted in Triton X‐100 buffer containing 0.5% Triton X‐100, 20 mM HEPES (pH 7.9), 50 mM NaCl, 3 mM MgCl2, and 300 mM Sucrose for 2 min on ice and then fixed in 3% formaldehyde and 2% sucrose in PBS for 10 min at room temperature. The cells were incubated with primary antibodies diluted in blocking solution of 5% Normal Goat Serum (BioLegend, 927502) or 0.2% (w/v) cold water fish gelatin (Sigma, G‐7765) and 0.5% (w/v) BSA (Sigma, A‐2153) in PBS for 1 h at room temperature or at 4°C overnight. Following staining with secondary antibodies and 0.1 μg/ml 4’,6‐diamidino‐2‐phenylindole dihydrochloride (DAPI, Sigma, D‐9542) diluted in 5% NGS‐PBS, cells were mounted onto glass slides using ProLong Gold Antifade (Invitrogen, #P36934). Primary antibodies used for immunofluorescence included: FAM111A (Abcam, ab184572; 1:100), SV40LT (Abcam, ab16879; 1:250), PCNA (Santa Cruz, sc‐56; 1:200), PML (Santa Cruz, sc‐966; 1:200), and anti‐Nuclear Pore Complex proteins (Mab414, Abcam, ab24609; 1:200). Secondary antibodies used were Alexa Fluor Secondary Antibodies (Life Technologies).
Confocal images were acquired with a Zeiss LSM 880 inverted spectral imaging confocal microscope (Carl Zeiss Microscopy) equipped with 63x/1.4 oil immersion high NA objective lens, using standard settings in Zen microscope software. Data were exported and processed using ImageJ software (NIH). Digital images were also taken using Zeiss Axio Imager M1 with 20x and 60x oil immersion objectives.
Clonogenic survival assays
Cells were counted and seeded in six‐well plates (seven hundred fifty cells for U2OS, five hundred cells for other cell lines), allowed to adhere overnight. The next day, 1 μg/ml of doxycycline was added to the media. Twenty‐four (HEK293, 293T), forty‐eight (RPE), or seventy‐two (U2OS) hours later, doxycycline was removed, and fresh medium was added. Cells were cultured for another four to five days until visible colonies have formed. Subsequently, the cells were fixed with 10% neutral‐buffered formalin and stained with 0.5% crystal violet. The plates were scanned on LI‐COR (LI‐COR Biosciences). Cell growth was quantitated as total area using ImageJ software (NIH).
Flow cytometry
For analysis of DNA content, cells were fixed with ice‐cold 70% EtOH, stained with 10 μg/ml propidium iodide, and treated with 100 μg/ml RNase A in PBS containing 0.1% Triton X‐100. Flow cytometry analysis was performed on a NovoCyte 3000 Flow Cytometer (ACEA Biosciences) using NovoExpress software.
BioID labeling and affinity purification of biotinylated proteins
Cells were incubated for 24 h in complete media supplemented with doxycycline and biotin. After washing with PBS, cells (2–5 × 106 cells for small‐scale analysis; 2 × 107 cells for large scale analysis) were collected and frozen. Cell pellets were lysed and processed as described (Roux et al, 2012). Briefly, each 106 cells were suspended in 100 μl of strong lysis buffer containing 50 mM Tris, pH 7.5, 500 mM NaCl, 0.4% SDS, 5 mM EDTA, 1 mM DTT, 1× Halt Protease Inhibitor Cocktail (Thermo Fisher, 78430), and 1 mM PMSF. The lysate was sonicated in a Misonix waterbath Sonicator. Triton X‐100 was added to 2% final concentration, and sonication was repeated. Equal volume of cold 50 mM Tris, pH 7.4 was added, followed by additional sonication. After centrifugation at 16,000 g for 10 min at 4°C, supernatants were incubated with Dynabeads MyOne Streptavidin C1 (Thermo Fisher, 65002) at 4°C for 2 h (20 μl of beads were used for small‐scale analysis; 100 μl for large scale preparation for mass spectrometry). Following incubation, the beads were washed at 25°C sequentially in 1 ml of 2% SDS (wash buffer 1) twice, once each in 0.1% deoxycholate, 1% Triton X‐100, 500 mM NaCl, 1 mM EDTA, and 50 mM HEPES, pH 7.5 (wash buffer 2), in 250 mM LiCl, 0.5% NP‐40, 0.5% deoxycholate, 1 mM EDTA, and 10 mM Tris, pH 8.1 (wash buffer 3), and finally twice in 50 mM Tris, pH 7.4, 50 mM NaCl (wash buffer 4). For Western blot analysis, proteins were eluted from the magnetic beads with 35 μl of 2× NuPAGE LDS Sample Loading Buffer (Thermo Fisher, NP0007) with 100 mM DTT at 100°C for 5 min. For mass spectrometry analysis, the beads were washed and stored in 100 ml of 8 M urea, 100 mM Tris, pH 8.5, until further processing.
Western blotting
Cells were lysed in strong lysis buffer described in the section above. The whole cell lysate was combined with NuPAGE LDS Sample Loading Buffer and 100 mM DTT, separated by SDS–PAGE and transferred to nitrocellulose. Immunoblotting was performed as described (Nie et al, 2017). Antibodies used for immunoblotting included: cleaved Caspase‐3 (Asp175) (Cell Signaling, #9661; 1:1,000), FAM111A (Abcam, ab184572; 1:1,000), Myc 9E10 (Scripps Antibody Core Facility; 1:3,000), PARP (Cell Signaling, 9542; 1:1,000), anti‐Nuclear Pore Complex proteins (Mab414, Abcam, ab24609; 1:1,000), GANP (Bethyl, A303‐128A; 1:1,000), SV40LT (Abcam, ab16879; 1:3,000), tubulin alpha (Sigma, T9026; 1:10,000), and Cdc2‐PSTAIR (Abcam, ab10345; 1:15,000). Following incubation with HRP‐conjugated secondary antibodies, proteins were detected with SuperSignal West Dura Extended Duration Substrate (Thermo Fisher, 34076) on the ChemiDox XRS + System (BioRad).
Protein identification by mass spectrometry
Protein samples were reduced and alkylated by sequential incubation with 5 mM Tris (2‐carboxyethyl) phosphine for 20 minutes at room temperature and 10 mM iodoacetamide reagent in the dark at room temperature for 20 additional min. Proteins were digested sequentially at 37°C with lys‐C for 4 h followed by trypsin for 12 h. After quenching the digest by the addition of formic acid to 5% (v/v), peptides were desalted using Pierce C18 tips (Thermo Fisher Scientific), dried by vacuum centrifugation, and resuspended in 5% formic acid. Peptides were fractionated online using reversed phase chromatography on in‐house packed C18 columns. The 140‐min gradient of increasing acetonitrile was delivered using a Dionex Ultimate 3000 UHPLC system (Thermo Fisher Scientific). Peptides were electrosprayed into the mass spectrometer by the application of a distal 2.2 kV spray voltage. MS/MS data were acquired using an Orbitrap Fusion Lumos mass spectrometer operating in Data‐Dependent Acquisition (DDA) mode consisting of a full MS1 scan to identify peptide precursors that were subsequently targeted by MS2 scans (Resolution = 15,000) using high energy collision dissociation for the remainder of the 3 s cycle time. Data analysis was performed using the Integrated Proteomics bioinformatic pipeline 2 (Integrated Proteomics Applications, San Diego, CA). Database searching was performed using the ProLuCID algorithm against the EMBL Human reference proteome (UP000005640 9606). Peptides identifications were filtered using a 1% FDR as estimated using a decoy database. Proteins were considered present in a sample if they had 2 or more unique peptides mapping to them. Relative comparisons between samples to identify candidate FAM111A interacting proteins were done using raw peptide spectral counts.
Author contributions
MNB, MN, MO, EL‐D, YJ‐A, and JAW devised and executed experiments in support of the study. MN and MO performed most of the presented experiments. YJ‐A and JAW carried out mass spectrometry‐based protein identification analysis of our FAM111A BioID samples. All authors contributed to manuscript writing and figure preparation.
Conflict of interest
The authors declare that they have no conflict of interest.
Supporting information
Expanded View Figures PDF
Table EV1
Table EV2
Dataset EV1
Review Process File
Source Data for Figure 5
Acknowledgements
We thank Dr. James DeCaprio for the SV40 genome plasmids and invaluable discussions. We are grateful to Taylor Loe and the Scripps Research community for their support and helpful discussions. This study was supported by NIH grants GM089778 awarded to JAW, and R35GM136273, R01GM068608, and R01GM122987 awarded to MNB.
EMBO reports (2021) 22: e50803.
Data availability
The datasets produced in this study are available in the PRIDE database (Perez‐Riverol et al, 2019) as detailed below: Protein interaction AP‐MS data: PRIDE PXD020327 (http://www.ebi.ac.uk/pride/archive/projects/PXD020327).
References
- Alabert C, Bukowski‐Wills J‐C, Lee S‐B, Kustatscher G, Nakamura K, de Lima Alves F, Menard P, Mejlvang J, Rappsilber J, Groth A (2014) Nascent chromatin capture proteomics determines chromatin dynamics during DNA replication and identifies unknown fork components. Nat Cell Biol 16: 281–293 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baquero‐Perez B, Whitehouse A (2015) Hsp70 isoforms are essential for the formation of Kaposi's sarcoma‐associated herpesvirus replication and transcription compartments. PLoS Pathog 11: e1005274 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bhatia V, Barroso SI, Garcia‐Rubio ML, Tumini E, Herrera‐Moyano E, Aguilera A (2014) BRCA2 prevents R‐loop accumulation and associates with TREX‐2 mRNA export factor PCID2. Nature 511: 362–365 [DOI] [PubMed] [Google Scholar]
- Brickner DG, Brickner JH (2011) Gene positioning is regulated by phosphorylation of the nuclear pore complex by Cdk1. Cell Cycle 10: 392–395 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brooks MA, Ali AN, Turner PC, Moyer RW (1995) A rabbitpox virus serpin gene controls host range by inhibiting apoptosis in restrictive cells. J Virol 69: 7688–7698 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bryan TM, Englezou A, Dallapozza L, Dunham MA, Reddel RR (1997) Evidence for an alternative mechanism for maintaining telomere length in human tumors and tumor‐derived cell lines. Nat Med 3: 1271–1274 [DOI] [PubMed] [Google Scholar]
- Bryant DM, Datta A, Rodriguez‐Fraticelli AE, Peranen J, Martin‐Belmonte F, Mostov KE (2010) A molecular network for de novo generation of the apical surface and lumen. Nat Cell Biol 12: 1035–1045 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cesare AJ, Reddel RR (2010) Alternative lengthening of telomeres: models, mechanisms and implications. Nat Rev Genet 11: 319–330 [DOI] [PubMed] [Google Scholar]
- Chug H, Trakhanov S, Hulsmann BB, Pleiner T, Gorlich D (2015) Crystal structure of the metazoan Nup62*Nup58*Nup54 nucleoporin complex. Science 350: 106–110 [DOI] [PubMed] [Google Scholar]
- Dilley RL, Verma P, Cho NW, Winters HD, Wondisford AR, Greenberg RA (2016) Break‐induced telomere synthesis underlies alternative telomere maintenance. Nature 539: 54–58 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Egger L, Schneider J, Rheme C, Tapernoux M, Hacki J, Borner C (2003) Serine proteases mediate apoptosis‐like cell death and phagocytosis under caspase‐inhibiting conditions. Cell Death Differ 10: 1188–1203 [DOI] [PubMed] [Google Scholar]
- Ferrando‐May E, Cordes V, Biller‐Ckovric I, Mirkovic J, Gorlich D, Nicotera P (2001) Caspases mediate nucleoporin cleavage, but not early redistribution of nuclear transport factors and modulation of nuclear permeability in apoptosis. Cell Death Differ 8: 495–505 [DOI] [PubMed] [Google Scholar]
- Fine DA, Rozenblatt‐Rosen O, Padi M, Korkhin A, James RL, Adelmant G, Yoon R, Guo L, Berrios C, Zhang Y et al (2012) Identification of FAM111A as an SV40 host range restriction and adenovirus helper factor. PLoS Pathog 8: e1002949 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fischer T, Strasser K, Racz A, Rodriguez‐Navarro S, Oppizzi M, Ihrig P, Lechner J, Hurt E (2002) The mRNA export machinery requires the novel Sac3p‐Thp1p complex to dock at the nucleoplasmic entrance of the nuclear pores. EMBO J 21: 5843–5852 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hatch EM, Fischer AH, Deerinck TJ, Hetzer MW (2013) Catastrophic nuclear envelope collapse in cancer cell micronuclei. Cell 154: 47–60 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoffmann S, Pentakota S, Mund A, Haahr P, Coscia F, Gallo M, Mann M, Taylor NM, Mailand N (2020) FAM111 protease activity undermines cellular fitness and is amplified by gain‐of‐function mutations in human disease. EMBO Rep 21, e50662 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang H, Zhao K, Arnett DR, Fanning E (2010) A specific docking site for DNA polymerase {alpha}‐primase on the SV40 helicase is required for viral primosome activity, but helicase activity is dispensable. J Biol Chem 285: 33475–33484 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ibarra A, Hetzer MW (2015) Nuclear pore proteins and the control of genome functions. Genes Dev 29: 337–349 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Isojima T, Doi K, Mitsui J, Oda Y, Tokuhiro E, Yasoda A, Yorifuji T, Horikawa R, Yoshimura J, Ishiura H et al (2014) A recurrent de novo FAM111A mutation causes Kenny‐Caffey syndrome type 2. J Bone Miner Res 29: 992–998 [DOI] [PubMed] [Google Scholar]
- Kojima Y, Machida Y, Palani S, Caulfield TR, Radisky ES, Kaufmann SH, Machida YJ (2020) FAM111A protects replication forks from protein obstacles via its trypsin‐like domain. Nat Commun 11: 1318 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kopeina GS, Prokhorova EA, Lavrik IN, Zhivotovsky B (2018) Alterations in the nucleocytoplasmic transport in apoptosis: caspases lead the way. Cell Prolif 51: e12467 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuwahara K, Yoshida M, Kondo E, Sakata A, Watanabe Y, Abe E, Kouno Y, Tomiyasu S, Fujimura S, Tokuhisa T et al (2000) A novel nuclear phosphoprotein, GANP, is up‐regulated in centrocytes of the germinal center and associated with MCM3, a protein essential for DNA replication. Blood 95: 2321–2328 [PubMed] [Google Scholar]
- Lim RY, Fahrenkrog B, Koser J, Schwarz‐Herion K, Deng J, Aebi U (2007) Nanomechanical basis of selective gating by the nuclear pore complex. Science 318: 640–643 [DOI] [PubMed] [Google Scholar]
- Loe TK, Li JSZ, Zhang Y, Azeroglu B, Boddy MN, Denchi EL (2020) Telomere length heterogeneity in ALT cells is maintained by PML‐dependent localization of the BTR complex to telomeres. Genes Dev 34: 650–662 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luttge BG, Moyer RW (2005) Suppressors of a host range mutation in the rabbitpox virus serpin SPI‐1 map to proteins essential for viral DNA replication. J Virol 79: 9168–9179 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Min J, Wright WE, Shay JW (2017) Alternative lengthening of telomeres can be maintained by preferential elongation of lagging strands. Nucleic Acids Res 45: 2615–2628 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nie M, Moser BA, Nakamura TM, Boddy MN (2017) SUMO‐targeted ubiquitin ligase activity can either suppress or promote genome instability, depending on the nature of the DNA lesion. PLoS Genet 13: e1006776 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nikkel SM, Ahmed A, Smith A, Marcadier J, Bulman DE, Boycott KM (2014) Mother‐to‐daughter transmission of Kenny‐Caffey syndrome associated with the recurrent, dominant FAM111A mutation p.Arg569His. Clin Genet 86: 394–395 [DOI] [PubMed] [Google Scholar]
- O'Connell AR, Stenson‐Cox C (2007) A more serine way to die: defining the characteristics of serine protease‐mediated cell death cascades. Biochim Biophys Acta 1773: 1491–1499 [DOI] [PubMed] [Google Scholar]
- Pan X, Drosopoulos WC, Sethi L, Madireddy A, Schildkraut CL, Zhang D (2017) FANCM, BRCA1, and BLM cooperatively resolve the replication stress at the ALT telomeres. Proc Natl Acad Sci USA 114: E5940–E5949 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Panda D, Fernandez DJ, Lal M, Buehler E, Moss B (2017) Triad of human cellular proteins, IRF2, FAM111A, and RFC3, restrict replication of orthopoxvirus SPI‐1 host‐range mutants. Proc Natl Acad Sci USA 114: 3720–3725 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parvari R, Hershkovitz E, Grossman N, Gorodischer R, Loeys B, Zecic A, Mortier G, Gregory S, Sharony R, Kambouris M et al (2002) Mutation of TBCE causes hypoparathyroidism‐retardation‐dysmorphism and autosomal recessive Kenny‐Caffey syndrome. Nat Genet 32: 448–452 [DOI] [PubMed] [Google Scholar]
- Perez‐Riverol Y, Csordas A, Bai J, Bernal‐Llinares M, Hewapathirana S, Kundu DJ, Inuganti A, Griss J, Mayer G, Eisenacher M et al (2019) The PRIDE database and related tools and resources in 2019: improving support for quantification data. Nucleic Acids Res 47: D442–D450 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Postigo A, Ramsden AE, Howell M, Way M (2017) Cytoplasmic ATR activation promotes vaccinia virus genome replication. Cell Rep 19: 1022–1032 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Poulin DL, Decaprio JA (2006) The carboxyl‐terminal domain of large T antigen rescues SV40 host range activity in trans independent of acetylation. Virology 349: 212–221 [DOI] [PubMed] [Google Scholar]
- Rout MP, Aitchison JD, Magnasco MO, Chait BT (2003) Virtual gating and nuclear transport: the hole picture. Trends Cell Biol 13: 622–628 [DOI] [PubMed] [Google Scholar]
- Roux KJ, Kim DI, Raida M, Burke B (2012) A promiscuous biotin ligase fusion protein identifies proximal and interacting proteins in mammalian cells. J Cell Biol 196: 801–810 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shisler JL, Isaacs SN, Moss B (1999) Vaccinia virus serpin‐1 deletion mutant exhibits a host range defect characterized by low levels of intermediate and late mRNAs. Virology 262: 298–311 [DOI] [PubMed] [Google Scholar]
- Sobinoff AP, Pickett HA (2020) Mechanisms that drive telomere maintenance and recombination in human cancers. Curr Opin Genet Dev 60: 25–30 [DOI] [PubMed] [Google Scholar]
- Soldani C, Scovassi AI (2002) Poly(ADP‐ribose) polymerase‐1 cleavage during apoptosis: an update. Apoptosis 7: 321–328 [DOI] [PubMed] [Google Scholar]
- Sowd GA, Fanning E (2012) A wolf in sheep's clothing: SV40 co‐opts host genome maintenance proteins to replicate viral DNA. PLoS Pathog 8: e1002994 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Strasser C, Grote P, Schauble K, Ganz M, Ferrando‐May E (2012) Regulation of nuclear envelope permeability in cell death and survival. Nucleus 3: 540–551 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takei Y, Assenberg M, Tsujimoto G, Laskey R (2002) The MCM3 acetylase MCM3AP inhibits initiation, but not elongation, of DNA replication via interaction with MCM3. J Biol Chem 277: 43121–43125 [DOI] [PubMed] [Google Scholar]
- Takei Y, Swietlik M, Tanoue A, Tsujimoto G, Kouzarides T, Laskey R (2001) MCM3AP, a novel acetyltransferase that acetylates replication protein MCM3. EMBO Rep 2: 119–123 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tarnita RM, Wilkie AR, Decaprio JA (2019) Contribution of DNA replication to the FAM111A‐mediated simian virus 40 host range phenotype. J Virol 93: e01330‐18 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tu LC, Fu G, Zilman A, Musser SM (2013) Large cargo transport by nuclear pores: implications for the spatial organization of FG‐nucleoporins. EMBO J 32: 3220–3230 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Unger S, Gorna MW, le Bechec A, Do Vale‐Pereira S, Bedeschi MF, Geiberger S, Grigelioniene G, Horemuzova E, Lalatta F, Lausch E et al (2013) FAM111A mutations result in hypoparathyroidism and impaired skeletal development. Am J Hum Genet 92: 990–995 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wessel SR, Mohni KN, Luzwick JW, Dungrawala H, Cortez D (2019) Functional analysis of the replication fork proteome identifies BET proteins as PCNA regulators. Cell Rep 28: 3497–3509 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wickramasinghe VO, McMurtrie PI, Mills AD, Takei Y, Penrhyn‐Lowe S, Amagase Y, Main S, Marr J, Stewart M, Laskey RA (2010a) mRNA export from mammalian cell nuclei is dependent on GANP. Curr Biol 20: 25–31 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wickramasinghe VO, Stewart M, Laskey RA (2010b) GANP enhances the efficiency of mRNA nuclear export in mammalian cells. Nucleus 1: 393–396 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang JM, Yadav T, Ouyang J, Lan L, Zou L (2019) Alternative lengthening of telomeres through two distinct break‐induced replication pathways. Cell Rep 26: 955–968 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Expanded View Figures PDF
Table EV1
Table EV2
Dataset EV1
Review Process File
Source Data for Figure 5
Data Availability Statement
The datasets produced in this study are available in the PRIDE database (Perez‐Riverol et al, 2019) as detailed below: Protein interaction AP‐MS data: PRIDE PXD020327 (http://www.ebi.ac.uk/pride/archive/projects/PXD020327).