

Abstract
Centrosomes, composed of two centrioles and pericentriolar material, organize mitotic spindles during cell division and template cilia during interphase. The first few divisions during mouse development occur without centrioles, which form around embryonic day (E) 3. However, disruption of centriole biogenesis in Sas‐4 null mice leads to embryonic arrest around E9. Centriole loss in Sas‐4 −/− embryos causes prolonged mitosis and p53‐dependent cell death. Studies in vitro discovered a similar USP28‐, 53BP1‐, and p53‐dependent mitotic surveillance pathway that leads to cell cycle arrest. In this study, we show that an analogous pathway is conserved in vivo where 53BP1 and USP28 are upstream of p53 in Sas‐4 −/− embryos. The data indicate that the pathway is established around E7 of development, four days after the centrioles appear. Our data suggest that the newly formed centrioles gradually mature to participate in mitosis and cilia formation around the beginning of gastrulation, coinciding with the activation of mitotic surveillance pathway upon centriole loss.
Keywords: centrosomes, SAS‐4, p53, USP28, 53BP1
Subject Categories: Cell Cycle, Development & Differentiation
This study shows that the de novo formed centrioles in the early developing mouse gradually mature to participate in mitosis and cilia formation around gastrulation, when the mitotic surveillance pathway is activated.

Introduction
Centrosomes are major microtubule organizing centers (MTOCs) of animal cells and are composed of two centrioles, one mature mother centriole with distal and subdistal appendages and one daughter centriole, surrounded by a proteinaceous pericentriolar material (PCM) (Conduit et al, 2015). During mitosis, centrosomes help assemble the mitotic spindle, and during interphase, the mother centriole forms the basal body template for cilia (Bornens, 2012). In proliferating cells, centrioles can form de novo without pre‐existing centrioles or use the scaffold of existing centrioles to duplicate once per cell cycle in late G1 and S phases (Loncarek & Khodjakov, 2009). The centriole formation pathway has been defined in cell culture and in different organisms and relies on a set of core proteins that include spindle assembly defective protein 4 (SAS‐4, also called CENPJ or CPAP; Kirkham et al, 2003; Leidel & Gonczy, 2003; Kleylein‐Sohn et al, 2007; Tang et al, 2009). The newly formed centrioles undergo maturation over two cell cycles to acquire appendages, such as ODF2 and CEP164, become MTOCs and template cilia (Kong et al, 2014). Cilia formation relies on docking of the mother centriole to the plasma membrane through distal appendage proteins, such as CEP164 (Graser et al, 2007; Siller et al, 2017), and on intra‐flagellar transport proteins, such as IFT88 (Haycraft et al, 2007).
During rodent development, and unlike the development of most organisms, the first cell divisions post‐fertilization occur without centrioles (Woolley & Fawcett, 1973; Gueth‐Hallonet et al, 1993; Manandhar et al, 1998; Courtois et al, 2012; Howe & FitzHarris, 2013). In the mouse embryo, centrioles first form by de novo biogenesis starting at the blastocyst stage around embryonic day (E) 3.5 (Courtois & Hiiragi, 2012). Before centriole formation, diffuse γ‐tubulin signals, a PCM component and microtubule nucleator, appear at the morula stage around E3, and γ‐tubulin signals become more focused as centrioles form; however, the newly formed centrioles do not seem to act as MTOCs in interphase cells (Howe & FitzHarris, 2013). In addition, the first cilia form almost two days post‐implantation around E6.5 in cells of the epiblast (Bangs et al, 2015).
Mouse embryonic stem cells (mESCs) are a well‐established in vitro model of embryo development that are derived from the pluripotent inner cell mass of blastocysts at E3.5 but molecularly resemble epiblast cells post‐implantation (Nichols & Smith, 2011). To maintain uniform pluripotency, mESCs are cultured with leukemia inhibitory factor (LIF) and two other differentiation inhibitors abbreviated as 2i (Williams et al, 1988; Ying et al, 2008). In pluripotency, the transcription factor NANOG is highly expressed in mESCs and regulates self‐renewal (Rosner et al, 1990). In this study, we used mESCs to complement our in vivo experiments by studying the growth dynamics of cells without centrioles.
We have previously shown that the genetic removal of SAS‐4 in the mouse resulted in the loss of centrioles and cilia (Bazzi & Anderson, 2014a). The Sas‐4 −/− embryos arrested development around E9.5 due to p53‐dependent cell death. The increase in p53 in Sas‐4 −/− embryos was not due the secondary loss of cilia, DNA damage or chromosome segregation errors. Also, these phenotypes are not specific to Sas‐4 −/− embryos because mutations in different genes, such as Cep152, that cause centriole loss show similar phenotypes (Bazzi & Anderson, 2014a, 2014b). Notably, the fraction of mitotic cells was higher in Sas‐4 −/− embryos at E7.5 and E8.5, indicating a longer mitotic duration of cells without centrioles, which was also confirmed by time‐lapse imaging of dividing cells. Because a short nocodazole treatment to prolong mitosis upregulated p53 in cultured wild‐type (WT) embryos, the data suggested that the less efficient mitosis without centrioles activated a novel p53‐dependent pathway (Bazzi & Anderson, 2014a). In cultured mammalian cell lines in vitro, a similar pathway that is activated by the loss of centrioles or prolonging mitosis leads to p53‐dependent cell cycle arrest and has been termed the mitotic surveillance pathway (Lambrus et al, 2015; Wong et al, 2015; Lambrus & Holland, 2017). Recently, p53‐binding protein 1 (53BP1) and ubiquitin specific peptidase 28 (USP28) have been shown to be essential for the conduction of this pathway in vitro (Fong et al, 2016; Lambrus et al, 2016; Meitinger et al, 2016). These studies showed that mutations in 53BP1 or USP28 rescued the growth arrest phenotype observed in cells without centrioles. However, whether a similar 53BP1‐ and USP28‐dependent pathway operates in vivo and can cause the p53‐dependent cell death phenotype in the mouse are still not known.
In this study, our data showed that the mitotic surveillance pathway is conserved in mice in vivo and that 53BP1 and USP28 are essential for its conduction upstream of p53. In order to explain the late onset of the phenotype upon the loss of centrioles, we also asked when during development this pathway is established. The data indicated that the newly formed centrioles around E3 are not fully mature and do not seem to be required for mitosis until around E7 of development, when the pathway is initiated. Our data suggest that once the cells start to depend on centrosomes as MTOCs in mitosis and ciliogenesis, then they sense the loss of centrioles and activate the p53‐dependent mitotic surveillance pathway.
Results and Discussion
Mutations in 53bp1 or Usp28 rescue the Sas‐4 mutant phenotype in vivo
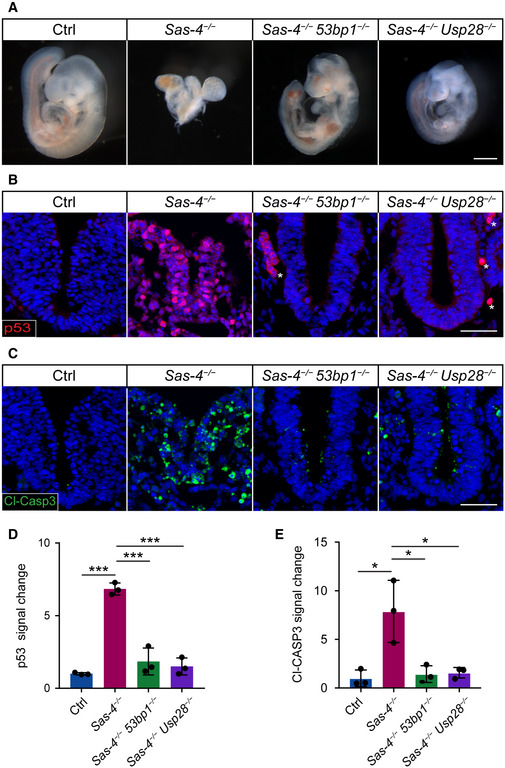
To test the conservation of the mitotic surveillance pathway and the involvement of 53BP1 and USP28 in its activation in vivo, 53bp1 +/− and Usp28 +/− null mouse alleles were generated using CRISPR/Cas9 gene‐editing (Fig EV1A and B; see Table 1 in Materials and Methods) and crossed to Sas‐4 +/− mice (Bazzi & Anderson, 2014a). Both Sas‐4 −/− 53bp1 −/− and Sas‐4 −/− Usp28 −/− embryos showed remarkable rescues of the morphology and size compared to the Sas‐4 −/− embryos at E9.5 (Fig 1A). Sas‐4 −/− 53bp1 −/− and Sas‐4 −/− Usp28 −/− embryos both underwent body turning, had visible somites and open heads, and were similar to Sas‐4 −/− p53 −/− mutants (Bazzi & Anderson, 2014a). At the molecular level, Sas‐4 −/− 53bp1 −/− and Sas‐4 −/− Usp28 −/− embryos showed highly reduced levels of p53 (Fig 1B and D) and cleaved‐Caspase 3 (Cl‐CASP3) (Fig 1C and E) and were more similar to controls when compared to Sas‐4 −/− embryos (Fig 1B–E). The data indicated that mutating 53bp1 or Usp28 suppressed both p53 stabilization and p53‐dependent cell death upon centriole loss in vivo and established the conservation of the mitotic surveillance pathway in the mouse.
Figure 1. The mitotic surveillance pathway is conserved in the mouse in vivo .
-
AGross morphology of control (Ctrl), Sas‐4 −/−, Sas‐4 −/− 53bp1 −/−, and Sas‐4 −/− Usp28 −/− embryos at E9.5. At least five embryos were considered per genotype and all the double mutant embryos exhibited the rescue criteria mentioned in the text. Scale bar = 500 μm.
-
B, CImmunostaining for p53 (B) and Cleaved‐Caspase3 (Cl‐CASP3, (C)) on transverse sections of Ctrl, Sas‐4 −/−, Sas‐4 −/− 53bp1 −/−, and Sas‐4 −/− Usp28 −/− embryos at E9.5. The area shown encompasses the ventral neural tube and surrounding mesenchyme. Asterisks in (B) denote non‐specific staining of blood cells. Scale bar = 50 μm.
-
D, EQuantifications of the p53 (D) and Cl‐CASP3 (E) fluorescence signals shown in (B) and (C), respectively. Three embryos per genotype were used for the quantifications. ***P < 0.001, *P < 0.05 (two‐tailed Student’s t‐test, one‐way ANOVA with Tukey’s multiple comparisons tests gave similar results). Bars represent mean ± s.d. (D) Ctrl: 1.00 ± 0.06 (n = 1,770 cells); Sas‐4 −/−: 6.84 ± 0.34 (n = 275), Sas‐4 −/− 53bp1 −/−: 1.85 ± 0.75 (n = 690), Sas‐4 −/− Usp28 −/−: 1.50 ± 0.48 (n = 975). (E) Ctrl: 1.00 ± 0.70 (n = 1,690); Sas‐4 −/−: 7.88 ± 2.62 (n = 175); Sas‐4 −/− 53bp1 −/−: 1.43 ± 0.71 (n = 1030); Sas‐4 −/− Usp28 −/−: 1.57 ± 0.44 (n = 950).
Table 1.
Information for CRISPR/Cas9 and genotyping of 53bp1 andUsp28 mouse alleles
| 53bp1 | Usp28 | |
|---|---|---|
| Exon | 4 | 2 |
| gRNA | TCTTCTCATTTGGGTACCAG | AATCAGCTGCGAGAAATCAC |
| Mutation | 5 bp deletion and 4 bp insertion (a net of 1 bp deletion) | 1 bp insertion |
| InDel | TCTTCTCATTTGTTCT‐CAG | AATCAGCTGCGAGAAATTCAC |
| Primer 1 | GTGTTTAAGGTCCTGTGGGG | TGATGCTCTGCTCCGAGAAA |
| Primer 2 | AGCTTTAATGTCCCTGCCCA | AAGCCCACTGTACATTCCCA |
The mitotic surveillance pathway is activated around E7
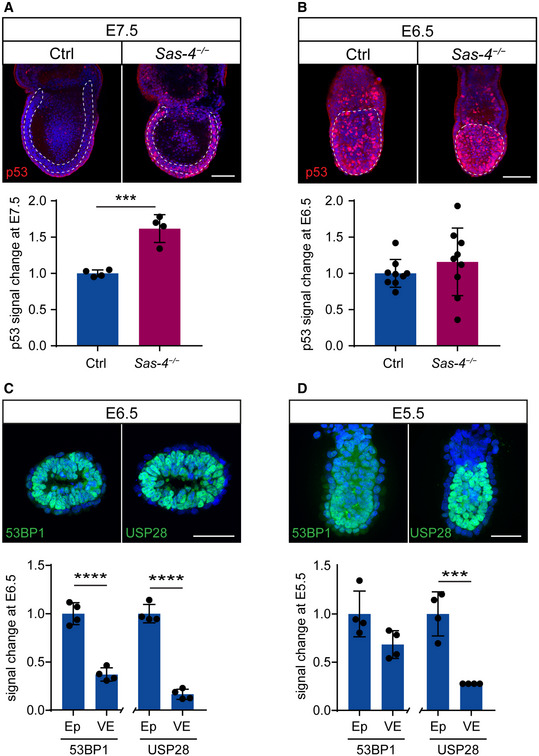
In order to determine when the mitotic surveillance pathway is activated in Sas‐4 −/− embryos, we used immunostaining and quantified nuclear p53 levels during development. At E7.5, Sas‐4 −/− embryos were smaller than control embryos (WT or Sas‐4 +/−) with around 1.5‐fold higher nuclear p53 in the epiblast (Fig 2A) (Bazzi & Anderson, 2014a). Earlier in development at E6.5, Sas‐4 −/− embryos were morphologically indistinguishable from control embryos, and nuclear p53 was not detectably different (Fig 2B). Ift88 null (cilia mutant) embryos were used as controls for Sas‐4 −/− centriole mutant embryos and were similar to WT embryos both morphologically and in terms of p53 nuclear levels at E7.5 (Fig EV2A) and E6.5 (Fig EV2B), confirming our earlier finding that p53 upregulation was due to centriole loss and not the secondary loss of cilia (Bazzi & Anderson, 2014a). The data suggested that the increased level of nuclear p53 in Sas‐4 −/− embryos starts around E7 of development and is independent of cilia loss per se.
Figure 2. p53 upregulation in Sas‐4 −/− embryos is evident by E7.5 and is not due to the onset of 53BP1 or USP28 expression.
-
A, BWhole‐mount immunostaining and quantification for p53 on Ctrl and Sas‐4 −/− embryos at E7.5 (A) and E6.5 (B). Representative sagittal planes are shown and the dotted lines demarcate the epiblast. Quantification of nuclear p53 fluorescence intensity in the epiblast is shown below, normalized to control embryos in the same batch at E7.5 (4 embryos) and at E6.5 (9 embryos). ***P < 0.001 (two‐tailed Student’s t‐test). Bars represent mean ± s.d. Scale bars = 100 μm. (A) Ctrl: 1.04 ± 0.00 (n = 495 cells); Sas‐4 −/−: 1.65 ± 0.19 (n = 850). (B) Ctrl: 1.00 ± 0.18 (n = 1,665); Sas‐4 −/−: 1.16 ± 0.44 (n = 1,330).
-
C, DImmunostaining for 53BP1 and USP28 on sagittal sections of WT embryos at E6.5 (C) or sagittal planes of whole‐mount immunostaining at E5.5 (D). Quantification of the fluorescent signals in the epiblast (Ep) versus visceral endoderm (VE) are shown below. Four embryos per genotype were used for the quantifications. ****P < 0.0001, ***P < 0.001 (two‐tailed Student’s t‐test). Bars represent mean ± s.d. Scale bar = 50 μm. (C) 53BP1 in Ep: 1.00 ± 0.10 (n = 225 cells); 53BP1 in VE: 0.37 ± 0.06 (n = 105); USP28 in Ep: 1.00 ± 0.08 (n = 235); USP28 in VE: 0.17 ± 0.05 (n = 90). (D) 53BP1 in Ep: 1.00 ± 0.24 (n = 225); 53BP1 in VE: 0.68 ± 0.14 (n = 115); USP28 in Ep: 1.00 ± 0.23 (n = 280); USP28 in VE: 0.28 ± 0.00 (n = 110).
USP28 and 53BP1 are expressed in the epiblast before E6
We next asked whether the upregulation of p53 in Sas‐4 −/− embryos around E7, and not before, coincided with the onset of expression of either 53BP1 or USP28, the upstream regulators of p53, in developing WT embryos. We performed immunostaining of 53BP1 and USP28 in WT embryos at E6.5 (Fig 2C) and E5.5 (Fig 2D) and found that both 53BP1 and USP28 were highly expressed in the epiblast of WT embryos at both stages. Of note, both proteins were clearly detectable in the embryonic epiblast but had much lower levels in the surrounding visceral endoderm (Fig 2C and D). The data indicated that the regulation of the onset of 53BP1 or USP28 expression does not seem to be responsible for p53 upregulation and activation of the mitotic surveillance pathway in Sas‐4 −/− embryos, suggesting that other mechanisms establish the pathway around E7.
The proper growth of Sas‐4 −/− mESCs is dependent on p53
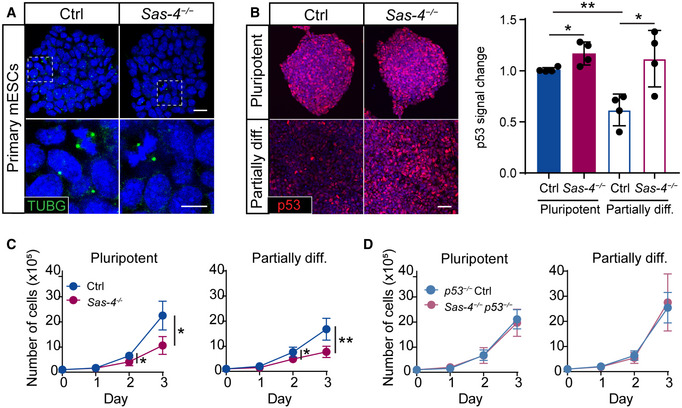
To study the dynamics of the mitotic surveillance pathway activation, we derived primary mESCs from WT and Sas‐4 −/− blastocysts at E3.5. Sas‐4 −/− primary mESCs were successfully derived and propagated in vitro and lacked detectable centrosomes in interphase cells, as judged by γ‐tubulin (TUBG) staining, compared to WT cells, which had centrosomes in every cell (Fig 3A). TUBG aggregates were seen only at the poles of mitotic cells in Sas‐4 −/− mESCs, consistent with our findings in Sas‐4 −/− embryos where these PCM aggregates lacked centrioles (Bazzi & Anderson, 2014a). Both WT and Sas‐4 −/− primary mESCs showed high levels of nuclear NANOG in media containing LIF and 2i, indicating their pluripotent potential (Fig EV3A). In pluripotent conditions (LIF and 2i), WT and Sas‐4 −/− primary mESCs had seemingly similar levels of p53, as judged by immunostaining (Fig 3B). Because Sas‐4 −/− embryos upregulated p53 starting after E6.5 (Fig 2A and B), we reasoned that Sas‐4 −/− mESC partial differentiation may trigger a similar response in vitro. Thus, we removed the pluripotency factors (LIF and 2i) for three days, and the pluripotency potential declined as shown by the decrease in NANOG nuclear signal in both WT and Sas‐4 −/− mESCs (Fig EV3A). The partially differentiated mESCs are not likely to represent a specific lineage because mESCs first move into a transitional state as they exit self‐renewal (Martello & Smith, 2014). Importantly, upon partial differentiation, nuclear p53 levels decreased in WT but not in Sas‐4 −/− mESCs (Fig 3B). Quantification of the normalized nuclear p53 levels revealed that they were slightly, but significantly, higher in Sas‐4 −/− mESCs compared to WT mESCs in pluripotent conditions, and this difference appeared more pronounced upon partial differentiation (Fig 3B). Also, the decrease in p53 in WT mESCs upon partial differentiation was significant (Fig 3B).
Figure 3. Sas‐4 −/− mESCs activate the mitotic surveillance pathway.
-
AImmunostaining for the centrosome marker γ‐tubulin (TUBG) on Ctrl and Sas‐4 −/− primary mESCs. The bottom panels are magnifications of the areas marked in the top panels. Scale bars = 20 μm (top) and 10 μm (bottom).
-
BImmunostaining and quantification for p53 on Ctrl and Sas‐4 −/− primary mESCs in pluripotent and partially differentiated (diff.) conditions. The quantification of p53 fluorescence intensities was normalized to Ctrl (Four independent experiments). **P < 0.01, *P < 0.05 (two‐tailed Student’s t‐test, the comparison of the genotypes in the “Pluripotent” condition was not significant using the one‐way ANOVA with Tukey’s multiple comparisons tests). Bars represent mean ± s.d. Scale bar = 50 μm. Pluripotent Ctrl: 1.00 ± 0.02 (n = 2,030 cells); pluripotent Sas‐4 −/−: 1.17 ± 0.10 (n = 2,170); partially diff. Ctrl: 0.62 ± 0.13 (n = 2,745); partially diff. Sas‐4 −/−: 1.12 ± 0.24 (n = 2,000).
-
C, DThree‐day growth curves of WT and Sas‐4 −/− (C) or p53 −/− and Sas‐4 −/− p53 −/− (D) primary mESCs in the indicated conditions starting with 105 cells on Day 0 (Four independent experiments). **P < 0.01, *P < 0.05 (two‐tailed Student’s t‐test). Bars represent mean ± s.d. For details of the measurements, see Materials and Methods (Tables 3 and 4).
Although Sas‐4 −/− mESCs could be derived and propagated in pluripotent condition cultures, we noticed that they grew slower than WT mESCs (Fig 3C). The growth defect became more obvious upon partial differentiation (Fig 3C). To check whether the slower growth in Sas‐4 −/− mESCs was dependent on p53 and the possible activation of the mitotic surveillance pathway, we generated Sas‐4 −/− p53 −/− and p53 −/− control mESCs using CRISPR/Cas9 (Fig EV3B, see Table 2 in Materials and Methods). The data showed that Sas‐4 −/− p53 −/− completely rescued the growth delay phenotype relative to p53 −/− and WT mESCs under pluripotent and partially differentiated conditions (Fig 3D). The pathways and molecular players operating upstream as well as downstream of p53 that lead to the growth defect in Sas‐4 −/− mESCs, and whether they involve cell cycle regulation or cell death, are currently not known.
Table 2.
Information for CRISPR/Cas9‐generated p53 alleles in mESCs
| p53 | ||||
|---|---|---|---|---|
| Exon | 4 | |||
| gRNA | AGGAGCTCCTGACACTCGGA | |||
| Cell line | p53 −/− | Sas‐4 −/− p53 −/− | ||
| Mutation | 16 bp deletion | 1 bp deletion | 2 bp deletion | 1 bp deletion |
| InDel |
TCC‐‐‐‐‐‐‐ ‐‐‐‐‐‐‐‐‐T |
T‐CGAGTGTC AGGAGCTCCT |
T‐‐GAGTGTC AGGAGCTCCT |
TCC‐AGTGTC AGGAGCTCCT |
| Primer 1 | TTGTTTTCCAGACTTCCTCCA | |||
| Primer 2 | CTGAAGAGGAACCCCCAAAT | |||
Mitotic surveillance pathway activation is associated with prolonged mitosis in vivo and in vitro
We have previously shown that prometaphase was prolonged in Sas‐4 −/− embryos at E7.5 and at E8.5 (Bazzi & Anderson, 2014a). To address whether the activation of the mitotic surveillance pathway around E7 coincided with the onset of prolonged mitosis in Sas‐4 −/− embryos, we performed immunostaining for the mitotic marker phospho‐histone H3 (pHH3) at E6.5. We calculated the mitotic index, the percentage of pHH3‐positive cells in the epiblast, as an indirect measure of mitotic duration and detected no difference between control and Sas‐4 −/− embryos at E6.5 (Fig EV3C). In contrast, our previous data showed that the mitotic index of Sas‐4 −/− embryos at E7.5 was significantly higher than that of control embryos (Fig EV3D) (Bazzi & Anderson, 2014a). The data indicated that the mitotic surveillance pathway activation through p53 upregulation temporally correlates with prolonged mitosis in vivo.
In line with the embryo data in vivo, the mitotic indices of WT and Sas‐4 −/− mESCs in vitro were similar in pluripotency. In contrast, upon partial differentiation, the mitotic index of Sas‐4 −/− mESCs was significantly higher than that of WT mESCs (Fig EV3E). These findings suggested that the potential enhanced activation of the mitotic surveillance pathway in mESCs also correlates with prolonged mitosis upon partial differentiation, and that the growth dynamics of Sas‐4 −/− mESCs largely resemble those of Sas‐4 −/− embryos. However, ~4% of WT mESCs were ciliated in pluripotency and the percentage doubled to ~8%, but was not significant, upon partial differentiation (Fig EV3F). Lineage‐directed differentiation methods of mESCs maybe required to uncover the full spectrum of defects in Sas‐4 −/− mESCs and assess whether they are dependent on p53 and/or cilia formation.
Gradual centriole maturation correlates with the establishment of the mitotic surveillance pathway in vivo
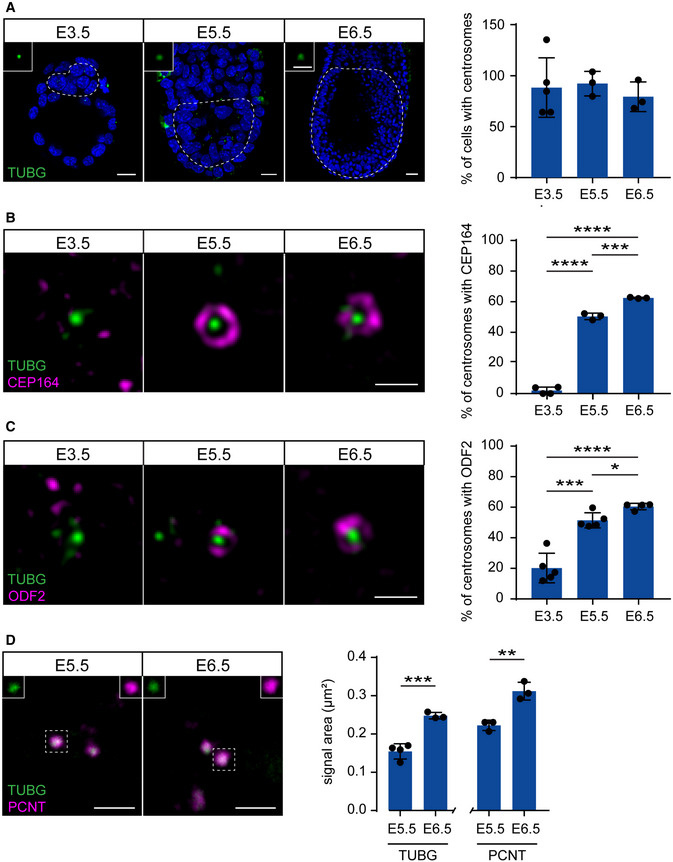
We next hypothesized that the centrioles that are first formed by de novo biogenesis around E3 were not fully mature yet and that their maturation correlates with the delayed response to centriole loss in Sas‐4 −/− embryos around E7. Therefore, we performed immunostaining, also combined with super‐resolution imaging, for TUBG together with either the distal appendage protein CEP164 or the subdistal appendage ODF2, both representing appendage markers of the more mature mother centrioles, on developing embryos between E3.5 and E6.5 (Fig 4A–C). Starting at E3.5, almost all the cells contained centrosomes marked by TUBG foci (Fig 4A). Intriguingly, neither CEP164 (Fig 4B) nor ODF2 (Fig 4C) localized to these centrosomes at E3.5, supporting our hypothesis that the centrioles were not mature. At E5.5, around 50% of the centrosomes in the epiblast colocalized with CEP164 or ODF2, and the percentage increased to around 60% at E6.5 (Fig 4B and C). In addition, the ability to recruit more PCM is another measure of centriole maturation; therefore, we quantified the areas of immunostaining signals for two PCM proteins, TUBG and PCNT, in developing WT embryos (Fig 4D). The data showed that the centrosomes of epiblast cells at E6.5 recruited significantly more TUBG and PCNT compared to those at E5.5 (Fig 4D). We concluded that the newly formed centrioles in mouse embryos gradually mature, acquire appendages, and recruit more PCM to participate in mitosis and cilia formation around gastrulation overlapping with the activation of the mitotic surveillance pathway in Sas‐4 −/− centriole mutant embryos around E7 (Fig 5). A causal relationship between centriole maturation and pathway activation is still elusive.
Figure 4. Centrioles gradually mature during mouse development.
-
ASagittal planes of whole‐mount immunostaining and quantification for TUBG on WT mouse embryos from E3.5 to E6.5. The insets are magnifications of selected centrosomes from the dotted areas denoting the inner cell mass (E3.5) or epiblast (E5.5 and E6.5). Bars represent mean ± s.d (two‐tailed Student’s t‐test or one‐way ANOVA with Tukey’s multiple comparisons tests). Scale bars: E3.5 and E5.5 = 15 μm; E6.5 = 30 μm and 3 μm (insets). E3.5: 88 ± 26% (n = 73 cells from 5 embryos); E5.5: 92 ± 10% (n = 270 from 3 embryos); E6.5: 79 ± 12% (n = 12,082 from 3 embryos).
-
B, CRepresentative gSTED super‐resolution images of immunostaining for TUBG and either CEP164 (B) or ODF2 (C). Whole‐mount staining was used for the CEP164 data as well as for the E3.5 blastocysts for ODF2, whereas staining on sections was used for ODF2 at E5.5 and E6.5. Quantification of the percentage of centrosomes (TUBG) with CEP164 or ODF2 are shown on the right. ****P < 0.0001, ***P < 0.001, *P < 0.05 (two‐tailed Student’s t‐test or one‐way ANOVA with Tukey’s multiple comparisons tests; the ODF2 data between E5.5 and E6.5 was not significant using the latter test). Bars represent mean ± s.d. Scale bar = 0.5 μm. CEP164 at E3.5: 2 ± 2%, (n = 92 centrosomes from 4 embryos); E5.5: 50 ± 2% (n = 249 from 3 embryos); E6.5: 62 ± 0% (n = 856 from 3 embryos). ODF2 at E3.5: 20 ± 9% (n = 108 from 5 embryos); E5.5: 51 ± 4% (n = 294 from 5 embryos); E6.5: 60 ± 2% (n = 1,183 from 4 embryos).
-
DImmunostaining and quantification of the area of TUBG and PCNT signals from E5.5 and E6.5 centrosomes in epiblast cells. The separate channels for the marked centriole are shown in the insets. ***P < 0.001, **P < 0.01 (two‐tailed Student’s t‐test). Bars represent mean ± s.d. Scale bar = 2 μm. Quantification of PCNT at E5.5: 0.22 ± 0.01 μm (n = 103 centrosomes from 3 embryos); E6.5: 0.31 ± 0.02 μm (n = 342 from 3 embryos). Quantification of TUBG at E5.5: 0.15 ± 0.02 μm (n = 108 from 4 embryos); E6.5: 0.25 ± 0.01 μm (n = 311 from 3 embryos).
Figure 5. A schematic model depicting the correlation between gradual centriole maturation and centrosome functions during mouse embryonic development.
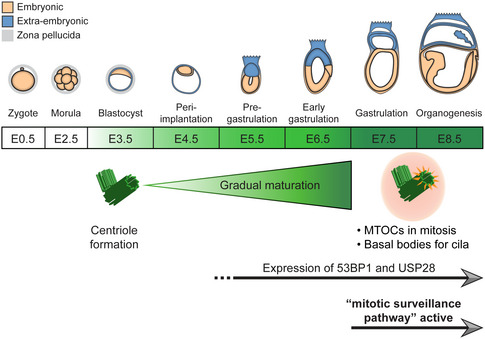
Centrioles first form by de novo biogenesis around E3 and gradually mature, acquire appendages (orange spokes) and recruit PCM (pink circle) to provide a basal body template for cilia and act as MTOCs for mitosis around E7, when the mitotic surveillance pathway is established. The expression of 53BP1 and USP28 precedes the establishment of the pathway and continues until at least E9.5. The embryo development timeline was adapted and modified from (Kojima et al, 2014).
Although mitosis is usually the shortest phase of the cell cycle and lasts only around half an hour, it is an essential phase where the segregation of DNA and other cellular components must be precisely accomplished. In addition to the well‐studied spindle assembly checkpoint (SAC), mammalian cells have developed a newly discovered pathway to monitor mitosis termed the mitotic surveillance pathway that is independent of the SAC (Lambrus & Holland, 2017). This pathway seems to be limited to mammalian systems because organisms such as Drosophila melanogaster lack 53PB1 and USP28 homologs (Lambrus & Holland, 2017), and zygotic Sas‐4 mutant flies survive until adulthood (Basto et al, 2006).
Both control and Sas‐4 −/− embryos show relatively high nuclear p53 around E6.5, which has been reported in WT embryos at E5.5 and E6.5 (Bowling et al, 2018). It has been suggested that p53 may be involved in cellular competition during this stage of development to eliminate less fit cells before the germline is selected (Zhang et al, 2017). Higher p53 levels in Sas‐4 −/− embryos around E7 coincide with the window of the initiation of gastrulation as well as the appearance of cilia on epiblast‐derived lineages (Bangs et al, 2015). Our data largely exclude the lack of cilia per se (Fig EV2) or the expression of the mitotic surveillance pathway components (Fig 2C and D) as determinants of pathway activation after a lag period and at a specific developmental window. In line with this, 53BP1 expression has been reported throughout mouse pre‐implantation development (Ziegler‐Birling et al, 2009). In addition, the expression of 53BP1 and USP28 was mostly restricted to the epiblast (Fig 2C and D), which may explain why the fast proliferating epiblast cells seem to be more affected by centriole loss compared to the visceral endoderm (Bazzi & Anderson, 2014a). Collectively, our data support a model whereby the newly formed centrioles around E3 gradually mature during development until around E7, when they are competent to become basal bodies and participate in cilia formation as well as act as efficient MTOCs during mitosis (Fig 5). As such, Sas‐4 −/− centriole mutant embryos may not activate the p53‐dependent mitotic surveillance pathway until centrioles are more mature and required for mitosis. This gradual transition is reminiscent of the earlier transition from meiotic‐ to mitotic‐like divisions during pre‐implantation and may be a general phenomenon in development including, for example, cilia formation, elongation and function (Courtois & Hiiragi, 2012; Bangs et al, 2015).
Figure EV2. Ift88 −/− cilia mutants do not upregulate p53.
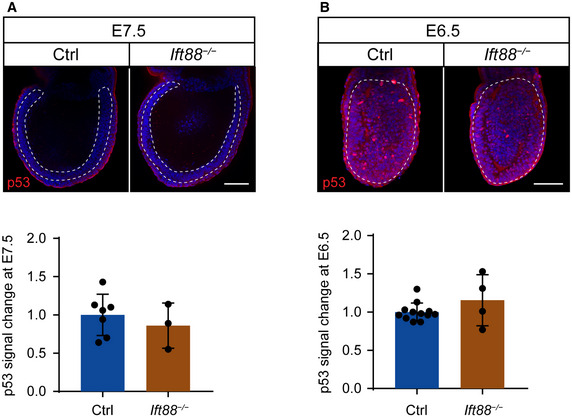
-
A, BImmunostaining and quantification for p53 on whole‐mount Ctrl and Ift88 −/− embryos at E7.5 (A) and E6.5 (B). Mid‐sagittal planes are shown with the dotted lines demarcating the epiblast. Scale bars = 100 μm. The quantification of p53 nuclear fluorescence intensity in the epiblast was normalized to Ctrl embryos in the same batch at E7.5 (A) and E6.5 (B). Bars represent mean ± s.d. (two‐tailed Student’s t‐test). (A) Ctrl: 1.00 ± 0.25 (n = 11,695 cells from 8 embryos); Ift88 −/− : 0.86 ± 0.24 (n = 40,815 from 3 embryos) (B) Ctrl: 1.00 ± 0.11 (n = 8,895 from 14 embryos); Ift88 −/− : 1.16 ± 0.29 (n = 3,470 from 4 embryos).
Materials and Methods
Animals and genotyping
The Sas‐4 −/− mice (Cenpjtm1d(EUCOMM)Wtsi/tm1d(EUCOMM)Wtsi) (Bazzi & Anderson, 2014a) and the Ift88 −/− null mouse allele generated from the Ift88fl/fl allele (Ift88tm1Bky) (Haycraft et al, 2007) were used in this study. The CRISPR/Cas9 endonuclease‐mediated knockouts of 53bp1 −/− and Usp28 −/− were generated by the CECAD in vivo Research Facility using microinjection or electroporation of the corresponding gRNA, Cas9 mRNA, and Cas9 protein into fertilized zygotes (Table 1) (Chu et al, 2016; Troder et al, 2018). The gRNA target sequence predictor tool developed by the Broad Institute was used to design gRNAs (Doench et al, 2016).
The animals were housed and bred under standard conditions in the CECAD animal facility, and the allele generation (84‐02.04.2014.A372) and experiments (84‐02.05.50.15.039) were approved by the Landesamt für Natur, Umwelt, und Verbraucherschutz Nordrhein‐Westfalen (LANUV‐NRW) in Germany. All the phenotypes were analyzed in the FVB/NRj background. Genotyping was carried out using standard and published PCR protocols. The PCR products for 53bp1‐ and Usp28‐mutant mice were digested with KpnI and ApoI restriction enzymes (New England BioLabs; Ipswich, MA, USA), respectively, to distinguish the WT and mutant alleles.
Wild type (WT) refers to unmodified +/+ alleles and Control (Ctrl) to combinations of +/+ and +/− (heterozygous) alleles for the respective genes.
Mouse embryonic stem cell culture
Primary mESCs were derived from E3.5 blastocysts as previously described (Bryja et al, 2006), cultured on feeder cells that were proliferation‐inactivated with mitomycin C (Sigma‐Aldrich; St. Louis, MO, USA) and 0.1% gelatin‐coated plates (Sigma‐Aldrich). They were maintained in media containing Knock‐Out DMEM (Thermo Fisher Scientific; Waltham, MA, USA), supplemented with 15% HyClone fetal bovine serum (FBS; VWR; Radnor, PA, USA), 2 mM l‐glutamine (Biochrom; Berlin, Germany), 1% penicillin/streptomycin (Biochrom), 0.1 mM MEM non‐essential amino acids (Thermo Fisher Scientific), 1 mM sodium pyruvate (Thermo Fisher Scientific), 0.1 mM β‐mercaptoethanol (Thermo Fisher Scientific), 1000 U/ml leukemia inhibitory factor (LIF; Merck; Darmstadt, Germany), and with 1 μM PD0325901 (Miltenyi Biotec; Bergisch Gladbach, Germany) and 3 μM CHIR99021 (Miltenyi Biotec), together abbreviated as 2i. Primary mESCs were gradually weaned off feeder cells and maintained in feeder‐free conditions. To induce partial differentiation, feeder‐free primary mESCs were split and cultured in media without LIF and 2i for three days.
Generating CRISPR/Cas9‐modified primary mESCs
The gRNA sequence for targeting p53 was cloned as double‐stranded DNA oligonucleotides into the BbsI restriction site of the pX330‐U6‐Chimeric_BB‐CBh‐hSpCas9 vector (Addgene; Watertown, MA, USA) modified with a Puro‐T2K‐GFP cassette containing puromycin‐resistance and eGFP expression by Dr. Leo Kurian’s research group (Center for Molecular Medicine Cologne).
p53 −/− and Sas‐4 −/− p53 −/− mESCs (Table 2) were generated by lipofection of the modified pX330 vector containing the gRNA target sequences using Lipofectamine 3000 (Thermo Fisher Scientific). One day after transfection, 2 μg/ml puromycin (Sigma‐Aldrich) was added to the medium for two days, and the cells were allowed to recover in regular medium up to one week after transfection. Single colonies were picked under a dissecting microscope and were expanded. p53 null cell lines were confirmed with sequencing (primers in Table 2), immunofluorescence, and Western blotting.
Growth assay
To determine the growth kinetics of mESCs over three days, WT, Sas‐4 −/−, p53 −/−, and Sas‐4 −/− p53 −/− mESCs were seeded at 105 cells per well of a 6‐well plate in media with or without LIF and 2i in triplicate. One set was counted every day for three days using a hemocytometer. Two pairs of each genotype from separate derivations were counted twice and constituted four biological replicates.
Table 3.
The measurements shown in Figure 3C
| Day | Pluripotent [×105 cells] | Partially diff. [×105 cells] | ||
|---|---|---|---|---|
| Ctrl | Sas‐4 −/− | Ctrl | Sas‐4 −/− | |
| 0 | 1.00 ± 0.00 | 1.00 ± 0.00 | 1.00 ± 0.00 | 1.00 ± 0.00 |
| 1 | 1.66 ± 0.21 | 1.59 ± 0.31 | 2.01 ± 0.45 | 1.53 ± 0.39 |
| 2 | 6.52 ± 1.01 | 4.13 ± 1.35 | 7.65 ± 1.73 | 4.90 ± 0.84 |
| 3 | 22.53 ± 4.95 | 10.64 ± 3.07 | 16.82 ± 3.76 | 7.80 ± 1.94 |
Table 4.
The measurements shown in Figure 3D
| Day | Pluripotent [×105 cells] | Partially diff. [×105 cells] | ||
|---|---|---|---|---|
| p53 −/−_Ctrl | Sas‐4 −/− p53 −/− | p53 −/−_Ctrl | Sas‐4 −/− p53 −/− | |
| 0 | 1.00 ± 0.00 | 1.00 ± 0.00 | 1.00 ± 0.00 | 1.00 ± 0.00 |
| 1 | 1.50 ± 0.54 | 2.00 ± 1.01 | 2.03 ± 0.54 | 1.88 ± 0.43 |
| 2 | 6.81 ± 1.82 | 6.68 ± 2.69 | 6.39 ± 1.82 | 5.46 ± 1.87 |
| 3 | 21.09 ± 3.31 | 19.73 ± 4.67 | 25.42 ± 3.31 | 27.52 ± 9.83 |
Embryo dissection, immunofluorescence and imaging
Pregnant female mice (E3.5 ‐ E9.5) were sacrificed by cervical dislocation for embryo dissections under a dissecting microscope (M165C or M80, Leica Microsystems; Wetzlar, Germany) as previously described (Bryja et al, 2006; Behringer et al, 2014). The embryos were fixed in 4% paraformaldehyde (PFA; Carl Roth; Karlsruhe, Germany) for 2 h at room temperature or overnight at 4°C. Embryos between E3.5–E6.5 were either fixed in 4% PFA for 30 min and in ice‐cold methanol for 15 min, or in pure methanol for 1 h at −20°C for cleaner centrosomal staining. After fixing, the embryos were washed with phosphate‐buffered saline (PBS; VWR), and then either used for whole‐mount immunostaining or cryoprotected in 30% sucrose overnight at 4°C. The embryos were transferred from 30% sucrose and embedded in optimum cutting temperature (OCT) compound (Sakura Finetek; Alphen an den Rijn, Netherlands) for cryosectioning.
Whole‐mount immunofluorescence staining of intact mouse embryos was performed as previously described (Xiao et al, 2018). The embryos were then mounted in 1% low‐melting agarose (Lonza; Basel, Switzerland) on a glass bottom dish (Thermo Fisher Scientific), covered in VectaShield mounting medium (Linaris; Dossenheim, Germany) and kept cold and protected from light until imaging. After imaging, the embryos were removed from the agarose, washed, and digested for genotyping. Embryos at E3.5 were either directly imaged in PBS in a glass bottom dish for confocal microscopy or mounted in Prolong Gold (Cell Signaling Technology; Danvers, MA, USA) on a slide for gSTED super‐resolution microscopy.
OCT‐embedded embryos were cryosectioned at 8 μm thickness and the slides were fixed with ice‐cold methanol for 10 min at −20°C, then washed two times with wash buffer containing 0.2% Triton‐X in PBS while shaking and blocked with wash buffer with 5% heat‐inactivated goat serum for 1 h at room temperature. The slides were incubated with primary antibodies diluted in blocking solution overnight at 4°C. After washing two times with wash buffer, the slides were incubated with secondary antibodies and DAPI in blocking solution for 1 h at room temperature. After washing, the slides were mounted with coverslips using Prolong Gold (Cell Signaling Technology).
For immunostaining of mESCs, 2 × 104 cells were seeded per chamber onto pre‐gelatinized Lab‐Tek II chamber slides (Thermo Fisher Scientific). After three days of culturing in corresponding media, the cells were washed with PBS, fixed with 4% PFA for 10 min at room temperature and washed three times with PBS. Next, the cells were fixed with ice‐cold methanol for 10 min at −20°C, permeabilized with 0.5% Triton‐X in PBS for 5 min at room temperature, and blocked with 5% heat‐inactivated goat serum (Thermo Fisher Scientific) for at least 15 min at room temperature. The cells were incubated with primary antibodies diluted in blocking solution overnight at 4°C. After washing three times with wash buffer, the cells were incubated with secondary antibodies and DAPI diluted 1:1,000 in blocking solution for 1 h at room temperature. After washing, the chamber was removed from the glass slide and coverslips were mounted using Prolong Gold anti‐fade reagent (Cell Signaling Technology). The images were obtained using a TCS gSTED SP8 confocal microscope equipped with gSTED super‐resolution (Leica Microsystems). For gSTED imaging, an HC PL APO 93X/1.3 GLYC motCORR objective was used.
Antibodies
Primary antibodies used in this study and their dilutions and sources are listed in Table 5. The secondary antibodies used for confocal microscopy were Alexafluor® 488, 568, or 647 conjugates (Life Technologies) and diluted at 1:1,000, in combination with DAPI (AppliChem) at 1:1,000. For gSTED imaging, the following secondary antibodies and conjugates were used: Abberior® STAR 580 anti‐rabbit IgG (Sigma‐Aldrich) and FluoTag®‐X2 anti‐mouse IgG1 coupled to Atto 647N (NanoTag Biotechnologies, Göttingen, Germany).
Table 5.
List of primary antibodies used in this study
| Antigen | Host | Dilution | Company | Catalog Number |
|---|---|---|---|---|
| 53BP1 | Rabbit | 1:1,000 | Novus Biologicals | NB100‐304 |
| CEP164 | Rabbit | 1:2,000 | Proteintech | 22227‐1‐AP |
| Cl‐CASP3 | Rabbit | 1:400 | Cell Signaling | 9661 |
| NANOG | Rat | 1:200 | Affymetrix | 14‐5761 |
| ODF2 | Rabbit | 1:500 | Proteintech | 12058‐1‐AP |
| p53 | Rabbit | 1:500 | Leica | P53‐CM5P‐L |
| PCNT | Rabbit | 1:3,000 | BioLegend | 923701 |
| pHH3 | rabbit | 1:400 | Merck | 06‐570 |
| TUBG | mouse IgG1 | 1:1,000 | Sigma‐Aldrich | T6557 |
| USP28 | rabbit | 1:1,000 | Sigma‐Aldrich | HPA006778 |
Image analyses
Images of whole‐mount embryos, cryostat sections, or mESCs were quantified using ImageJ (NIH, Maryland, USA). Representative mid‐sagittal or cross sections of the embryos, and a maximum projection image of mESCs were used for quantification (Table 5). The DAPI channel was used to set a threshold to obtain a region of interest. For example, p53 and DAPI fluorescence intensities were measured, and the p53 intensity was normalized to the DAPI intensity. The average p53 intensity of controls was set to 1.0, and a fold‐change p53 intensity was calculated. Centrosomes (defined as one TUBG focus or two close TUBG foci) and CEP164 or ODF2 foci were manually quantified using ImageJ (NIH). The number of nuclei was quantified using the image‐based tool for counting nuclei (ITCN) ImageJ plug‐in. For gSTED images, image deconvolution was performed using Huygens Essentials (Scientific Volume Imaging, Hilversum, Netherlands).
Western blotting
mESCs were scraped in radioimmunoprecipitation assay (RIPA) buffer containing 150 mM NaCl, 50 mM Tris pH 7.6, 1% Triton X‐100 (Sigma‐Aldrich), 0.25% sodium deoxycholate, and 0.1% sodium dodecyl sulfate (SDS; AppliChem; Darmstadt, Germany) with an ethylenediaminetetraacetic acid (EDTA)‐free protease inhibitor cocktail (Merck), phosphatase inhibitor cocktail sets II (Merck) and IV (Merck), and phenylmethylsulfonyl fluoride (PMSF; Sigma‐Aldrich). Protein concentration was measured with an RC DC protein assay kit (Bio‐Rad; Feldkirchen, Germany). 10 μg protein per sample was loaded. SDS‐polyacrylamide gel electrophoresis (PAGE) and immunoblotting were performed following standard procedures (Towbin et al, 1979; Kurien & Scofield, 2006). Following SDS–PAGE, the proteins were transferred to polyvinylidene fluoride (PVDF) membranes (Merck) that were activated in methanol (Carl Roth) for 1 min, blocked in 5% milk (Carl Roth) for 1 h, and incubated with an anti‐p53 antibody (1:5,000; Leica Biosystems; Buffalo Grove, IL, USA) or an anti‐GAPDH antibody (1:104; Merck) overnight at 4°C. The membranes were washed with Tris‐buffered saline containing Tween 20 (AppliChem; TBST) and incubated with secondary anti‐rabbit (GE Healthcare; Chicago, IL, USA) or anti‐mouse (GE Healthcare) antibodies linked with horseradish peroxidase (HRP) at 1:104 for 1 h at room temperature. Finally, the membranes were washed with TBST and incubated with enhanced chemiluminescence (ECL; GE Healthcare; Chicago, IL, USA) and signals were detected with on film (GE Healthcare) in a dark room.
Statistical analyses
Statistical analyses comparing two groups or more of data were performed using pair‐wise two‐tailed Student’s t‐tests or one‐way ANOVA with Tukey’s multiple comparisons tests which gave consistent and similar results. The statistics with a cutoff for significance of < 0.05 and the graphic representations with standard deviations (s.d.) were all performed using GraphPad Prism 7 (GraphPad Software, San Diego, CA, USA).
Author contributions
Conceptualization: CX, MG and HB; Methodology: CX, MG, CM‐G, MM, RF and HB; Software: CX, MG, CM‐G; Formal Analysis: CX, MG, CM‐G; Investigation: CX, MG, CM‐G, MM, RF and HB; Writing: CX and HB; Visualization: CX, MG, CM‐G; Supervision, Project administration and Funding Acquisition: HB.
Conflict of interest
The authors declare that they have no conflict of interest.
Figure EV1. 53bp1 −/− and Usp28 −/− are null alleles.
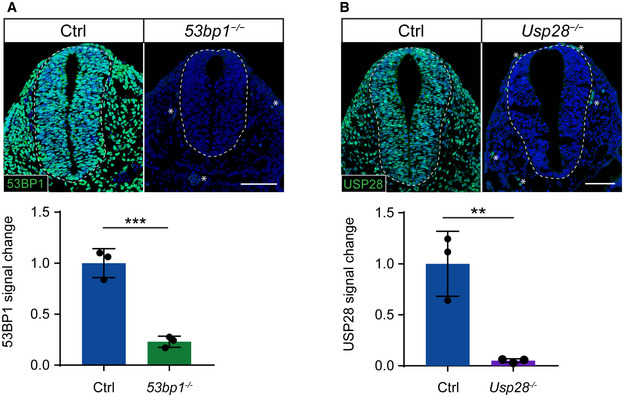
-
A, BImmunostaining and quantifications for 53BP1 (A) or USP28 (B) on sagittal sections of control (Ctrl) and Sas‐4 −/− 53bp1 −/− (A) or Usp28 −/− (B) embryos at E9.5. The signals for the corresponding proteins in the mutants compared to controls are not detectable and close to background staining of secondary antibody‐only negative controls, which were used as baseline for quantification. The cervical or brachial neural tube (the dotted quantified area) and underlying mesenchyme are shown. Asterisks indicate non‐specific staining in blood cells. Three embryos per genotype were used for the quantifications. ***P < 0.001, **P < 0.01 (two‐tailed Student’s t‐test). Bars represent mean ± s.d. Scale bars = 100 μm. (A) Ctrl: 1.00 ± 0.12; 53bp1 ‐/‐: 0.23 ± 0.04 (B) Ctrl: 1.00 ± 0.26; Usp28 ‐/‐: 0.05 ± 0.02.
Figure EV3. Pluripotency, p53 −/− null alleles and the mitotic indices in controls and Sas‐4 −/− mutants.
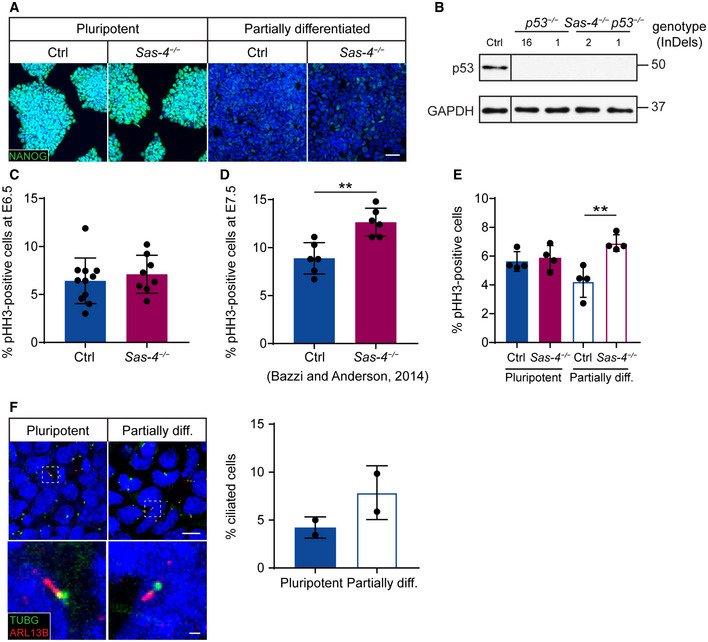
-
AImmunostaining for NANOG on Ctrl and Sas‐4 −/− primary mESCs in pluripotent and partially differentiated conditions. Scale bar = 50 μm.
-
BWestern blot analysis for p53 and GAPDH loading control on Ctrl, p53 −/−, and Sas‐4 −/− p53 −/− mESC lysates. The numbers below p53 −/− and Sas‐4 −/− p53 −/− indicate the number of base pairs deleted.
-
C–FThe mitotic index, or percentage of pHH3‐positive cells, of Ctrl and Sas‐4 −/− embryo epiblast at E6.5 (n = 8, (C)) and E7.5 (n = 6, (D)) or mESCs (Four independent experiments) in pluripotent or partially differentiated (diff.) conditions (E). The graph in (D) represents our previously published data (Bazzi & Anderson, 2014). (F) Immunostaining and quantification of the cilia marker ARL13B, together with the basal body marker TUBG, in Ctrl mESCs in pluripotent and partially diff. conditions (Two independent experiments). The selected areas in the top panels are magnified in the bottom panels. Scale bars = 10 µm (top) and 1 µm (bottom). **P < 0.01 (two‐tailed Student’s t‐test or one‐way ANOVA with Tukey’s multiple comparisons tests for (E)). Bars represent mean ± s.d. (C) Ctrl: 6 ± 2 (n = 9,545 cells); Sas‐4 −/−: 7 ± 2 (n = 5,875). (D) Ctrl: 9 ± 2 (n = 5,150), Sas‐4 −/−: 13 ± 1 (n = 4,990); (E) Ctrl pluripotent: 6 ± 1 (n = 2,445), Sas‐4 −/− pluripotent: 6 ± 1 (n = 2,330); Ctrl partially diff.: 4 ± 1 (n = 3,015); Sas‐4 −/− partially diff.: 7 ± 0 (n = 2,280). (F) Pluripotent mESCs: 4 ± 1 (n = 1,553) Partially diff. mESCs: 8 ± 2 (n = 1,538).
Supporting information
Expanded View Figures PDF
Review Process File
Acknowledgements
We thank the CECAD in vivo research facility for generating and maintaining our mouse lines and the CECAD imaging facility for microscopy support. The work was supported by the Deutsche Forschungsgemeinschaft (DFG, German Research Foundation) [BA 5810/1‐1 to H.B]. The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript. Open Access funding enabled and organized by ProjektDEAL.
EMBO reports (2021) 22: e51127.
Data availability
This study includes no data deposited in external repositories.
References
- Bangs FK, Schrode N, Hadjantonakis AK, Anderson KV (2015) Lineage specificity of primary cilia in the mouse embryo. Nat Cell Biol 17: 113–122 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Basto R, Lau J, Vinogradova T, Gardiol A, Woods CG, Khodjakov A, Raff JW (2006) Flies without centrioles. Cell 125: 1375–1386 [DOI] [PubMed] [Google Scholar]
- Bazzi H, Anderson KV (2014a) Acentriolar mitosis activates a p53‐dependent apoptosis pathway in the mouse embryo. Proc Natl Acad Sci USA 111: E1491–E1500 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bazzi H, Anderson KV (2014b) Centrioles in the mouse: cilia and beyond. Cell Cycle 13: 2809 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Behringer R, Gertsenstein M, Vintersen Nagy K, Nagy A (2014) Manipulating the mouse embryo: a laboratory manual, 4th edn pp 198–200, 268–271. Cold Spring Harbor, NY: Cold Spring Harbor Laboratory Press; [Google Scholar]
- Bornens M (2012) The centrosome in cells and organisms. Science 335: 422–426 [DOI] [PubMed] [Google Scholar]
- Bowling S, Di Gregorio A, Sancho M, Pozzi S, Aarts M, Signore M, Schneider MD, Martinez‐Barbera JP, Gil J, Rodriguez TA(2018) P53 and mTOR signalling determine fitness selection through cell competition during early mouse embryonic development. Nat Commun 9: 1763 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bryja V, Bonilla S, Arenas E (2006) Derivation of mouse embryonic stem cells. Nat Protoc 1: 2082–2087 [DOI] [PubMed] [Google Scholar]
- Chu VT, Weber T, Graf R, Sommermann T, Petsch K, Sack U, Volchkov P, Rajewsky K, Kuhn R (2016) Efficient generation of Rosa26 knock‐in mice using CRISPR/Cas9 in C57BL/6 zygotes. BMC Biotechnol 16: 4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Conduit PT, Wainman A, Raff JW (2015) Centrosome function and assembly in animal cells. Nat Rev Mol Cell Biol 16: 611–624 [DOI] [PubMed] [Google Scholar]
- Courtois A, Hiiragi T (2012) Gradual meiosis‐to‐mitosis transition in the early mouse embryo. Results Probl Cell Differ 55: 107–114 [DOI] [PubMed] [Google Scholar]
- Courtois A, Schuh M, Ellenberg J, Hiiragi T (2012) The transition from meiotic to mitotic spindle assembly is gradual during early mammalian development. J Cell Biol 198: 357–370 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doench JG, Fusi N, Sullender M, Hegde M, Vaimberg EW, Donovan KF, Smith I, Tothova Z, Wilen C, Orchard R et al (2016) Optimized sgRNA design to maximize activity and minimize off‐target effects of CRISPR‐Cas9. Nat Biotechnol 34: 184–191 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fong CS, Mazo G, Das T, Goodman J, Kim M, O'Rourke BP, Izquierdo D, Tsou MF (2016) 53BP1 and USP28 mediate p53‐dependent cell cycle arrest in response to centrosome loss and prolonged mitosis. Elife 5: e16270 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Graser S, Stierhof YD, Lavoie SB, Gassner OS, Lamla S, Le Clech M, Nigg EA (2007) Cep164, a novel centriole appendage protein required for primary cilium formation. J Cell Biol 179: 321–330 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gueth‐Hallonet C, Antony C, Aghion J, Santa‐Maria A, Lajoie‐Mazenc I, Wright M, Maro B (1993) gamma‐Tubulin is present in acentriolar MTOCs during early mouse development. J Cell Sci 105(Pt 1): 157–166 [DOI] [PubMed] [Google Scholar]
- Haycraft CJ, Zhang Q, Song B, Jackson WS, Detloff PJ, Serra R, Yoder BK (2007) Intraflagellar transport is essential for endochondral bone formation. Development 134: 307–316 [DOI] [PubMed] [Google Scholar]
- Howe K, FitzHarris G (2013) A non‐canonical mode of microtubule organization operates throughout pre‐implantation development in mouse. Cell Cycle 12: 1616–1624 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kirkham M, Müller‐Reichert T, Oegema K, Grill S, Hyman AA (2003) SAS‐4 is a C. elegans centriolar protein that controls centrosome size. Cell 112: 575–587 [DOI] [PubMed] [Google Scholar]
- Kleylein‐Sohn J, Westendorf J, Le Clech M, Habedanck R, Stierhof YD, Nigg EA (2007) Plk4‐induced centriole biogenesis in human cells. Dev Cell 13: 190–202 [DOI] [PubMed] [Google Scholar]
- Kojima Y, Tam OH, Tam PP (2014) Timing of developmental events in the early mouse embryo. Semin Cell Dev Biol 34: 65–75 [DOI] [PubMed] [Google Scholar]
- Kong D, Farmer V, Shukla A, James J, Gruskin R, Kiriyama S, Loncarek J (2014) Centriole maturation requires regulated Plk1 activity during two consecutive cell cycles. J Cell Biol 206: 855–865 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kurien BT, Scofield RH (2006) Western blotting. Methods 38: 283–293 [DOI] [PubMed] [Google Scholar]
- Lambrus BG, Daggubati V, Uetake Y, Scott PM, Clutario KM, Sluder G, Holland AJ (2016) A USP28‐53BP1‐p53‐p21 signaling axis arrests growth after centrosome loss or prolonged mitosis. J Cell Biol 214: 143–153 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lambrus BG, Holland AJ (2017) A New Mode of Mitotic Surveillance. Trends Cell Biol 27: 314–321 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lambrus BG, Uetake Y, Clutario KM, Daggubati V, Snyder M, Sluder G, Holland AJ (2015) p53 protects against genome instability following centriole duplication failure. J Cell Biol 210: 63–77 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leidel S, Gonczy P (2003) SAS‐4 is essential for centrosome duplication in C. elegans and is recruited to daughter centrioles once per cell cycle. Dev Cell 4: 431–439 [DOI] [PubMed] [Google Scholar]
- Loncarek J, Khodjakov A (2009) Ab ovo or de novo? Mechanisms of centriole duplication. Mol Cells 27: 135–142 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manandhar G, Sutovsky P, Joshi HC, Stearns T, Schatten G (1998) Centrosome reduction during mouse spermiogenesis. Dev Biol 203: 424–434 [DOI] [PubMed] [Google Scholar]
- Martello G, Smith A (2014) The nature of embryonic stem cells. Annu Rev Cell Dev Biol 30: 647–675 [DOI] [PubMed] [Google Scholar]
- Meitinger F, Anzola JV, Kaulich M, Richardson A, Stender JD, Benner C, Glass CK, Dowdy SF, Desai A, Shiau AK et al (2016) 53BP1 and USP28 mediate p53 activation and G1 arrest after centrosome loss or extended mitotic duration. J Cell Biol 214: 155–166 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nichols J, Smith A (2011) The origin and identity of embryonic stem cells. Development 138: 3–8 [DOI] [PubMed] [Google Scholar]
- Rosner MH, Vigano MA, Ozato K, Timmons PM, Poirier F, Rigby PW, Staudt LM (1990) A POU‐domain transcription factor in early stem cells and germ cells of the mammalian embryo. Nature 345: 686–692 [DOI] [PubMed] [Google Scholar]
- Siller SS, Sharma H, Li S, Yang J, Zhang Y, Holtzman MJ, Winuthayanon W, Colognato H, Holdener BC, Li FQ et al (2017) Conditional knockout mice for the distal appendage protein CEP164 reveal its essential roles in airway multiciliated cell differentiation. PLoS Genet 13: e1007128 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang CJ, Fu RH, Wu KS, Hsu WB, Tang TK (2009) CPAP is a cell‐cycle regulated protein that controls centriole length. Nat Cell Biol 11: 825–831 [DOI] [PubMed] [Google Scholar]
- Towbin H, Staehelin T, Gordon J (1979) Electrophoretic transfer of proteins from polyacrylamide gels to nitrocellulose sheets: procedure and some applications. Proc Natl Acad Sci USA 76: 4350–4354 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Troder SE, Ebert LK, Butt L, Assenmacher S, Schermer B, Zevnik B (2018) An optimized electroporation approach for efficient CRISPR/Cas9 genome editing in murine zygotes. PLoS One 13: e0196891 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Williams RL, Hilton DJ, Pease S, Willson TA, Stewart CL, Gearing DP, Wagner EF, Metcalf D, Nicola NA, Gough NM (1988) Myeloid leukaemia inhibitory factor maintains the developmental potential of embryonic stem cells. Nature 336: 684–687 [DOI] [PubMed] [Google Scholar]
- Wong YL, Anzola JV, Davis RL, Yoon M, Motamedi A, Kroll A, Seo CP, Hsia JE, Kim SK, Mitchell JW et al (2015) Cell biology. Reversible centriole depletion with an inhibitor of Polo‐like kinase 4. Science 348: 1155–1160 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Woolley DM, Fawcett DW (1973) The degeneration and disappearance of the centrioles during the development of the rat spermatozoon. Anat Rec 177: 289–301 [DOI] [PubMed] [Google Scholar]
- Xiao C, Nitsche F, Bazzi H (2018) Visualizing the node and notochordal plate in gastrulating mouse embryos using scanning electron microscopy and whole mount immunofluorescence. J Vis Exp. 10.3791/58321 [DOI] [PubMed] [Google Scholar]
- Ying QL, Wray J, Nichols J, Batlle‐Morera L, Doble B, Woodgett J, Cohen P, Smith A (2008) The ground state of embryonic stem cell self‐renewal. Nature 453: 519–523 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang G, Xie Y, Zhou Y, Xiang C, Chen L, Zhang C, Hou X, Chen J, Zong H, Liu G (2017) p53 pathway is involved in cell competition during mouse embryogenesis. Proc Natl Acad Sci USA 114: 498–503 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ziegler‐Birling C, Helmrich A, Tora L, Torres‐Padilla ME (2009) Distribution of p53 binding protein 1 (53BP1) and phosphorylated H2A.X during mouse preimplantation development in the absence of DNA damage. Int J Dev Biol 53: 1003–1011 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Expanded View Figures PDF
Review Process File
Data Availability Statement
This study includes no data deposited in external repositories.