

Abstract
One major factor that contributes to the virulence of Pseudomonas aeruginosa is its ability to reside and replicate unchallenged inside airway epithelial cells. The mechanism by which P. aeruginosa escapes destruction by intracellular host defense mechanisms, such as autophagy, is not known. Here, we show that the type III secretion system effector protein ExoS facilitates P. aeruginosa survival in airway epithelial cells by inhibiting autophagy in host cells. Autophagy inhibition is independent of mTOR activity, as the latter is also inhibited by ExoS, albeit by a different mechanism. Deficiency of the critical autophagy gene Atg7 in airway epithelial cells, both in vitro and in mouse models, greatly enhances the survival of ExoS‐deficient P. aeruginosa but does not affect the survival of ExoS‐containing bacteria. The inhibitory effect of ExoS on autophagy and mTOR depends on the activity of its ADP‐ribosyltransferase domain. Inhibition of mTOR is caused by ExoS‐mediated ADP ribosylation of RAS, whereas autophagy inhibition is due to the suppression of autophagic Vps34 kinase activity.
Keywords: ADP‐ribosyltransferase, autophagy, ExoS, mTOR, Pseudomonas aeruginosa
Subject Categories: Microbiology, Virology & Host Pathogen Interaction; Post-translational Modifications, Proteolysis & Proteomics; Signal Transduction
Virulence of Pseudomonas aeruginosa depends on its ability to reside and replicate unchallenged inside airway epithelial cells. The type III secretion system effector ExoS enables this bacterial survival by inhibiting autophagy and mTOR.
Introduction
The luminal surface of the respiratory tract is covered with airway epithelial cells, including ciliated, basal, and Clara cells, which share the common goal of maintaining an antimicrobial environment in the respiratory tract (Crystal et al, 2008). Airway epithelial cells employ various methods to prevent pathogen colonization (Diamond et al, 2000; Li et al, 2012). They act as a cellular barrier, generate antimicrobial agents, and recruit neutrophils and macrophages to the site of infection. In addition, studies have suggested that airway epithelial cells are able to promote the clearance of internalized bacteria (Cortes et al, 2002). A damaged airway epithelial barrier or a weakened immune system allows opportunistic pathogens to invade and persist in airway epithelial cells (Williams et al, 2010).
Pseudomonas aeruginosa bacterium is an opportunistic pathogen that typically targets the respiratory tract in patients who are immunocompromised and is frequently responsible for hospital‐acquired pneumonia (Yasmin et al, 2006). In these conditions, severe pneumonia and bronchitis can develop with a high rate of mortality (Scheetz et al, 2009). Chronic P. aeruginosa infection occurs in subjects with cystic fibrosis (Goldberg & Pier, 2000; Scheetz et al, 2009). Pseudomonas aeruginosa has evolved methods to successfully colonize and damage airway epithelium (Hauser, 2009). The type III secretion system (T3SS) is a virulence mechanism that allows P. aeruginosa to inject up to four cytotoxins ExoS, ExoT, ExoU, and ExoY into host cells (Vance et al, 2005). ExoS and ExoT are bifunctional enzymes that contain an N‐terminal small GTPase‐activating protein (GAP) domain and C‐terminal ADP‐ribosyltransferase (ADPRT) domain (Barbieri & Sun, 2004). The GAP domains in ExoS and ExoT inhibit Rho‐like GTPases, including Rho, Rac1, and CDC42 (Kazmierczak et al, 2004). The ADPRT domain of ExoT targets CrkI and CrkII, whereas that of ExoS negatively regulates several host cell substrates including Ras‐like GTPases (Ganesan et al, 1998; Ganesan et al, 1999b). Importantly, ExoS is critical for the survival of P. aeruginosa in epithelial cells, though the underlying mechanisms remain unidentified (Angus et al, 2010).
Autophagy is characterized by the formation of double‐membrane organelles called autophagosomes, which engulf cytoplasmic components and induce their degradation after fusion with lysosomes. Autophagy is a key process in innate immunity and pathogen clearance, and pathogens have evolved ways to evade autophagy (Levine & Deretic, 2007; Rao & Eissa, 2020). During lung infection by P. aeruginosa, autophagy plays an important role in mast cells and alveolar macrophages in the clearance of the bacteria (Yuan et al, 2012). However, the role of autophagy in innate immune functions of airway epithelium is not well studied. Further, the relationship between autophagy and P. aeruginosa in airway epithelial cells has not been elucidated. Specifically, it is not known if P. aeruginosa could modulate autophagy to facilitate its survival in airway epithelial cells. Previous studies showed that P. aeruginosa could survive and multiply in airway epithelial cells without any evidence of elimination by the lysosomes (Chi et al, 1991). In corneal epithelial cells, the mutant P. aeruginosa lacking the T3SS showed more colocalization with the lysosomal markers compared to the WT P. aeruginosa (Angus et al, 2010). In addition, an in vivo study indicated that T3SS mutant P. aeruginosa is more susceptible to elimination by host cells than its wide type counterpart (Vance et al, 2005). These studies suggest that the T3SS of P. aeruginosa might play a role in overcoming the host defense mechanisms to facilitate its survival in epithelial cells. In addition to the need to evade host defense, pathogens can benefit from inhibiting host protein synthesis. However, inhibition of the mTOR pathway, as a pathogen strategy to inhibit host protein synthesis, could endanger pathogen survival because mTOR inhibition would activate autophagy (Kim et al, 2011). Here, we show that P. aeruginosa simultaneously inhibits both the mTOR pathway and autophagy through independent mechanisms. Remarkably, the T3SS effector protein ExoS mediates both the inhibitory mechanisms through its ExoS ADPRT activity. These mechanisms are critical for the survival of P. aeruginosa in airway epithelial cells in vitro and in vivo.
Results
Pseudomonas aeruginosa T3SS protected Pseudomonas aeruginosa from autophagy elimination
Pathogens are commonly targeted to the autophagy‐lysosomal pathway by host cells as a strategy to contain their replication and limit the infection (Nakagawa et al, 2004). In order to persist inside host cells, pathogens have evolved strategies to evade or inhibit autophagy (Ogawa et al, 2005; Jia et al, 2009). Existing evidence suggests that P. aeruginosa T3SS is required for their intracellular survival (Angus et al, 2010) but the underlying mechanisms are not known. We hypothesized that P. aeruginosa T3SS promoted the survival of P. aeruginosa in airway epithelial cells by suppressing autophagy. To test this hypothesis, we generated a cell line of type II alveolar human epithelial cells line A549 that stably expressed shRNA against autophagy‐related gene 7 (Atg7) we termed A549‐(Atg7−) and a control cell line expressing non‐target shRNA, termed A549‐(Atg7+). We confirmed the reduction of autophagy in A549‐(Atg7−) cells by immunoblotting. Compared to the control cells, A549‐(Atg7−) cells showed an accumulation of autophagy substrate SQSTM1 (also known as p62), and a reduction of Atg7 and LC3 type II (Fig 1A). To investigate if autophagy in airway epithelial cells was required to control bacterial pathogens, we first tested the effect of autophagy deficiency on the intracellular survival of airway epithelium pathogen Klebsiella pneumoniae (Cortes et al, 2002). We infected both A549‐Atg7− and A549‐Atg7+ cells with K. pneumoniae for 1 and 4 h, followed by 1 h of gentamycin treatment to kill extracellular bacteria. We then evaluated the intracellular bacterial load using colony‐forming units (CFU) assay. After 4 h of infection, a significantly higher number of bacteria was recovered from the autophagy‐deficient A549‐Atg7− cells, compared to their control counterpart (Fig 1B). This result showed that autophagy was important for K. pneumoniae elimination in airway epithelial cells. We then conducted similar experiments utilizing wild‐type P. aeruginosa or ΔpscD P. aeruginosa, a mutant strain that contains the three cytotoxins but is deficient in the T3SS translocation apparatus (Stover et al, 2000; Miao et al, 2008; Sun et al, 2012; Fig 1C). There was no significant difference between the CFU counts of WT P. aeruginosa recovered from A549‐Atg7+ and A549‐Atg7− cells 4 h post‐infection (Fig 1D). In contrast, CFU counts of the mutant ΔpscD P. aeruginosa were markedly reduced in A549‐Atg7+ cells compared to autophagy‐deficient A549‐Atg7− cells (Fig 1E). In both bacterial infections, there was no substantial difference in recovered CFU after 1‐h infection period, indicating that autophagy deficiency, rather than an internalization defect, was responsible for the reduction in bacterial survival. Taken together, these data showed that WT, but not T3SS‐deficient mutant P. aeruginosa, could resist elimination by autophagy.
Figure 1. Pseudomonas aeruginosa T3SS protected P. aeruginosa from autophagy elimination.
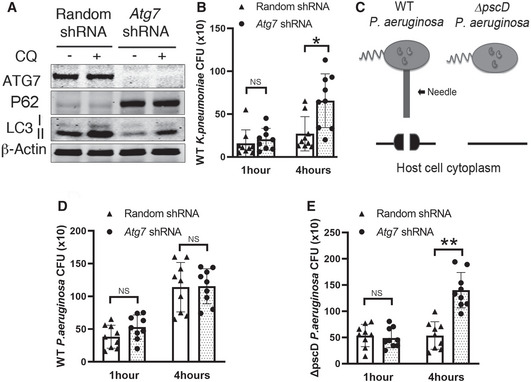
A549 cells stably expressing lentivirus vector containing random shRNA or Atg7 specific shRNA were generated.
-
ACells were incubated for 2 h in the presence or absence of 50 nM of lysosomal inhibitor chloroquine (CQ). Cell lysates were evaluated by immunoblotting.
-
BCells were infected, for 1 and 4 h, with K. pneumonia at MOI of 10 followed by 1 h of gentamycin treatment. Cell lysates were plated in LB agar for 18 h and CFU were determined.
-
CSchematic of WT P. aeruginosa and ΔpscD P. aeruginosa mutant.
-
D, ECells were infected, for 1–4 h, with WT (D) or ΔpscD P. aeruginosa (E) at MOI of 10. All infections were followed by 1 h of gentamycin treatment. Cell lysates were plated in LB agar for 18 h and CFU were determined.
Data information: In (B) and (D, E), the triangles and dots represent the individual test (three technical replicates per individual test) on control and Atg7 knock down cells. The bars represent the mean CFU of each group (three biological replicates each group), error bars represent standard deviation. Statistical significance between assays was calculated using two‐tailed Student’s t‐test with Welch’s correction. NS: not significant; *P ≤ 0.05; **P ≤ 0.01.
Source data are available online for this figure.
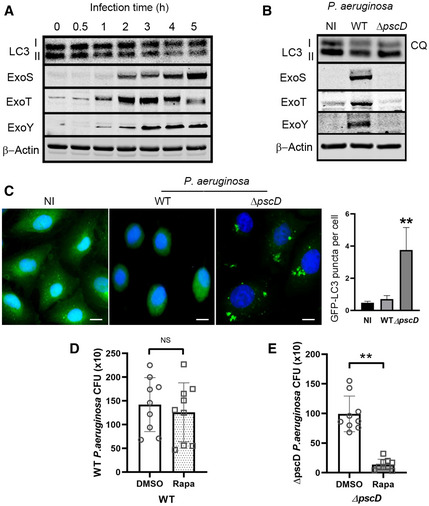
We then speculated that autophagy inhibition by WT P. aeruginosa allowed the bacteria to survive in A549 cells. To determine the effect of P. aeruginosa on autophagy, we infected A549 cells with WT P. aeruginosa for various time points and evaluated the generation of the autophagy marker LC3 type II (LC3II) by immunoblotting of cell lysates (Fig 2A). The T3SS of P. aeruginosa PAO1 strain secretes the effector proteins ExoS, ExoT, and ExoY. Using specific antibodies, we were able to detect the presence of the effector toxins in cell lysates of A549 cells as early as 2 h post‐infection (Fig 2A). More importantly, concomitant with the expression of T3SS effector proteins following infection, there was a corresponding reduction in autophagy marker LC3II (Fig 2A). Given autophagy marker protein LC3II are affected by LC3I to LC3II conversion, as well as lysosomal mediated degradation. We infected A549 cells with either P. aeruginosa WT or ΔpscD mutant under the treatment of lysosomal inhibitor chloroquine (CQ) (Klionsky et al, 2008). Levels of autophagy marker LC3II were decreased in cells infected with WT P. aeruginosa compared with cells infected with ΔpscD mutant or with non‐infected cells (Fig 2B). This finding suggested that LC3II reduction found in WT P. aeruginosa‐infected cells was caused by the decreased generation of LC3II, rather than by increased lysosomal degradation of LC3II. P. aeruginosa’ T3SS is known to induce cell death in the host cells (Jia et al, 2006). We thus determined cell death and find that although cell death was higher in cells infected with T3SS‐competent P. aeruginosa compared to those infected T3SS‐deficient P. aeruginosa, the overall cell death rate was less than 10% in both conditions (Fig EV1A and B). These data suggested that the significant reduction of LC3II caused by WT P. aeruginosa was not simply caused by increased cell death.
Figure 2. Pseudomonas aeruginosa inhibited autophagy by a T3SS‐dependent mechanism.
-
AA549 cells were infected with WT P. aeruginosa for various time points and cell lysates were evaluated by immunoblotting.
-
BA549 cells infected with WT or ΔpscD P. aeruginosa for 4 h or left uninfected (NI) in the presence of lysosomal inhibitor CQ (50 nM). The cell lysates were evaluated by immunoblotting.
-
CA549 cells stably expressing GFP‐LC3 were left non‐infected or infected, for 4 h, with WT or ΔpscD P. aeruginosa, and evaluated by fluorescence microscopy. Cell nuclei were counterstained with DAPI (blue) and autophagosome were visualized as green puncta. Graph represents quantitative analysis of GFP‐LC3‐positive autophagosome per cell, scale bars: 10 µm. Each bar represents mean value of three independent experiments, error bars represent standard deviation. Statistical significance between assays was determined using One‐way ANOVA followed by Dunn's Multiple‐comparison test. **P ≤ 0.01.
-
D, EA549 cells were treated for 16 h with rapamycin 0.8 μg/ml or DMSO, then infected with either WT (D) or ΔpscD P. aeruginosa and followed by 1 h of gentamycin treatment (E). The circles and squares represent the individual test (three technical replicates per individual test) with DMSO and rapamycin treatment. The bars represent the means of CFU from three independent experiments, error bars represent standard deviation. The significance of differences between different drug treatment was determined using two‐tailed Student’s t‐test with Welch’s correction. NS: not significant; *P ≤ 0.05; **P ≤ 0.01.
Source data are available online for this figure.
Figure EV1. Evaluation of cell death after P. aeruginosa infection.
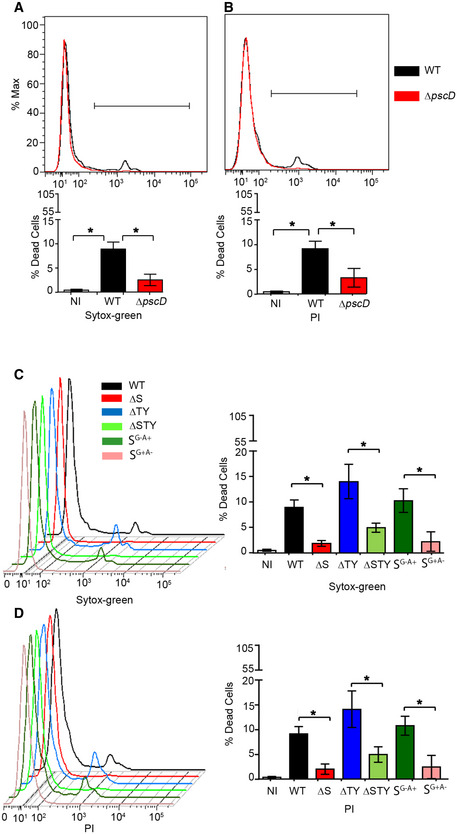
-
A–DA549 cells were either left uninfected (NI) or infected for 4 h with WT and different P. aeruginosa mutants. Cells were then stained with Sytox‐green (A, C) or propidium iodide (PI) (B, D) and the percentage of dead cells was analyzed by flow cytometry. Data are mean +/− SD from three independent experiments; one‐way ANOVA analysis followed Dunn's multiple‐comparison posttest t, *P ≤ 0.05.
To further evaluate the effect of P. aeruginosa’ T3SS on autophagy, we generated A549 cell line stably expressing the autophagy marker LC3 fused to green fluorescent protein (GFP‐LC3). In these cells, the formation of autophagosomes is accompanied by changes in the distribution of GFP‐LC3 from diffuse cytosolic staining to punctate fluorescence bodies (Levine & Deretic, 2007). We then infected these cells with WT or ΔpscD P. aeruginosa. There was increased GFP‐LC3 autophagosomes only in cells infected with the ΔpscD P. aeruginosa mutant strain, compared to non‐infected cells or to cells infected with WT P. aeruginosa (Fig 2C). These data confirmed that autophagy was attenuated by WT P. aeruginosa T3SS.
We then decided to investigate whether or not the induction of autophagy by rapamycin (an mTOR inhibitor) could overcome the inhibitory effect of P. aeruginosa T3SS. We treated A549 cells with DMSO or rapamycin before bacterial infection. We found no difference in the survival of WT P. aeruginosa in cells treated with rapamycin compared to cells treated with vehicle (Fig 2D). In contrast, in A549 cells infected with ΔpscD P. aeruginosa, there was a marked reduction in the viability of bacteria in A549 cells treated with rapamycin (Fig 2E). The immunoblotting assay showed rapamycin inhibited mTOR and induced autophagy, as evidenced by the reduction of mTOR downstream target phospho‐S6 and increased LC3II respectively (Fig EV2A). Interestingly, A549 cells treated with rapamycin and infected with WT P. aeruginosa still showed a reduced LC3II when compared to non‐infected cells (Fig EV2A). Similar results were found following exogenous overexpression of the ExoS‐encoding construct in A549 cells (Fig EV2B). These findings suggested that rapamycin could not restore autophagy inhibition caused by T3SS. Taken together, these results indicated that T3SS facilitates the intracellular survival of WT P. aeruginosa by inhibiting autophagy. They further implied that P. aeruginosa inhibited autophagy by a mechanism independent of mTOR signaling pathway.
Figure EV2. Induction of autophagy was attenuated by P. aeruginosa T3SS ExoS.
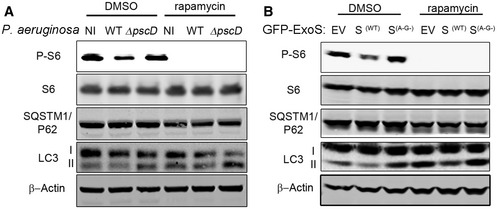
- A549 cells were treated for 16 h with DMSO or rapamycin. Cells were then left uninfected (NI) or infected for 4 h with WT or ΔpscD P. aeruginosa and cell lysates were evaluated by immunoblotting.
- A549 cells were transfected, for 24 h, with a plasmid containing an empty vector (EV), GFP‐ExoS or an inactive mutant (GFP‐ExoSG−A−). Cells were then treated for 16 h with DMSO or rapamycin and cell lysates were evaluated by immunoblotting.
Source data are available online for this figure.
The Pseudomonas aeruginosa ExoS was required and sufficient for autophagy inhibition
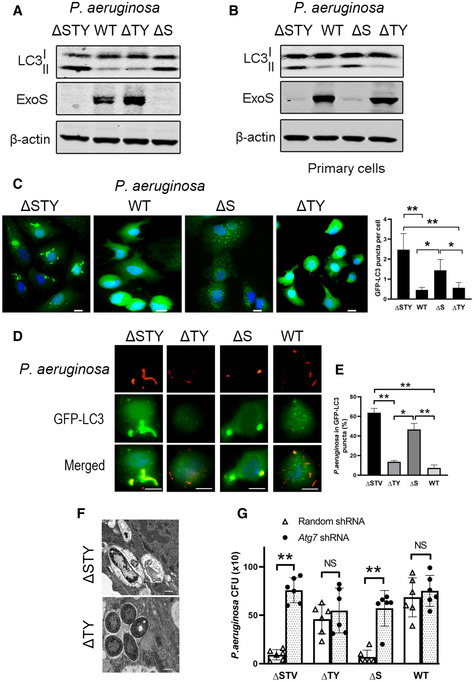
The P. aeruginosa T3SS is composed of several proteins including translocation proteins, chaperones and effectors proteins (Hauser, 2009). It has been shown that P. aeruginosa T3SS effector proteins contribute the most to P. aeruginosa pathogenesis (Williams et al, 2010). They are also found to negatively modulate different intracellular pathways (Hauser, 2009). Therefore, we hypothesized that P. aeruginosa T3SS effector proteins were responsible for autophagy inhibition. To determine which effectors proteins were implicated in such effect, we chose different P. aeruginosa mutants (deficient in one or more of the three T3SS effectors proteins ExoS, ExoT, and ExoY) to infect A549 cell line and primary human bronchial epithelial cells (NHBE). By evaluating formation of LC3II (Fig 3A and B) and GFP‐LC3 puncta (Fig 3C), we found cells infected with bacteria containing ExoS P. aeruginosa (WT, ΔTY) had reduced autophagy compared to cells infected with bacteria not containing ExoS P. aeruginosa (ΔS and ΔSTY) (Fig 3A–C). Importantly, ExoS was the only toxin, among those tested, that was required for the inhibition of autophagy. Bacteria containing only ExoT (ΔSY) or ExoY (ΔST) but lacking ExoS did not reduce LC3II formation (Fig EV3A). Limited cell death (< 15% of whole cell population) (Fig EV1C and D) was detected in these experiments suggesting that the marked reduction in LC3II and GFP‐LC3 puncta could not be simply the result of cell death caused by ExoS‐containing P. aeruginosa.
Figure 3. The Pseudomonas aeruginosa ExoS was required and sufficient for autophagy inhibition.
-
A, B(A) A549 cells or (B) primary human bronchial epithelial cells were infected with one of several P. aeruginosa mutants. Cell lysates were evaluated by immunoblotting.
-
CA549 cells, stably expressing GFP‐LC3, were infected with WT or P. aeruginosa mutants and evaluated by fluorescence microscopy, scale bars: 10 µm. GFP‐LC3 puncta representing the autophagosomes was quantified and presented in the graph. Each bar represents mean value from three independent experiments and error bars represent standard deviation. Statistical significance between assays was determined using one‐way ANOVA followed by Dunn's multiple‐comparison test. *P ≤ 0.05; **P ≤ 0.01. P. aeruginosa mutant strains legend: ΔSTY (mutant deficient for the three cytotoxins), WT (contains all cytotoxin), ΔS (deficient for ExoS but still express ExoT and ExoY), ΔTY (express ExoS but deficient for ExoT and ExoY).
-
D, EA549 cells, stably expressing GFP‐LC3, were infected for 4 h with WT or with one of P. aeruginosa mutants (ΔSTY, ΔS and ΔTY) and evaluated by fluorescence microscopy (D). Pseudomonas aeruginosa were visualized using specific antibody (red) and the colocalized bacteria signals with GFP‐LC3 puncta (green) were merged and presented as yellow. Scale bars: 10 µm. (E) Quantitative analysis of colocalization of internalized P. aeruginosa with GFP‐LC3‐positive puncta. The colocalization of these two signals was quantified with 10 independent visual fields of more than 100 cells. Data represent mean ± SD of three independent experiments. Statistical significance between assays was determined using One‐way ANOVA followed by Dunn's multiple‐comparison test. *P ≤ 0.05; **P ≤ 0.01.
-
FRepresentative transmission electron microscopy images of A549 cells infected with P. aeruginosa containing ExoS (ΔTY) or deficient for the three T3SS effector proteins (ΔSTY) for 4 h. ΔSTY P. aeruginosa (white star) was engulfed by multiple membrane autophagosomes (black arrow). ΔTY P. aeruginosa (white star) were found in cytoplasm with single membrane surrounded, scale bars: 0.5 µm.
-
GA549 cells, stably expressing lentiviral vectors containing random shRNA or Atg7 shRNA, were infected with WT or P. aeruginosa mutants (ΔSTY, ΔTY, and ΔS) at MOI of 10. All infections were followed by 1 h of gentamycin treatment. Cell lysates were plated in LB agar for 18 h, and CFU were determined. The triangles and dots represent the individual test (three technical replicates per individual test) on control and Atg7 knock down cells from three biological replicates. The bars represent the means of CFU of each group (three biological replicates each group), error bars represent standard deviation. The significance of differences between different assay was determined using two‐tailed Student’s t‐test with Welch’s correction, NS: not significant; **P ≤ 0.01.
Source data are available online for this figure.
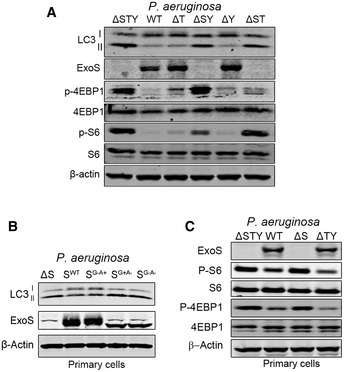
Figure EV3. ExoS ADP‐ribosyltransferase has the same effect on autophagy in primary epithelial cells; Single cytotoxin ExoT or ExoY does not affect mTOR or autophagy.
-
A–CA549 cells (A) and primary NHBE cells (B, C) were infected for 4 h, with WT or P. aeruginosa mutants (ΔSTY, ΔT, ΔSY, ΔY, ΔST SWT, SG−A−, SG−A+, and SG+A−), and cell lysate were evaluated by immunoblotting using indicated antibodies. P. aeruginosa mutant strains legend: SWT (P. aeruginosa contains only active ExoS), SG−A+ (P. aeruginosa contains only ExoS with loss‐of‐function mutations in the GTPase‐activating domain), SG+A− (P. aeruginosa contains ExoS with loss‐of‐function in the ADP ribosylation domain), SG−A− (P. aeruginosa contains a nonfunctional ExoS);ΔSTY (mutant deficient for the three cytotoxins), ΔT (deficient for ExoT but still express ExoS and ExoY cytotoxins), ΔSY (deficient for ExoS and ExoY only express ExoT), ΔY (deficient for ExoY but still express ExoS and ExoT cytotoxins), ΔST (deficient for ExoS and ExoT only express ExoY).
Source data are available online for this figure.
We also examined ExoS’s effect on autophagic flux in cells expressing a tandem fluorescent RFP‐GFP‐tagged LC3. As GFP fluorescence signal gets quenched in acidic environment inside lysosome lumen, autophagosomes are visualized as yellow puncta (the overlay of green and red fluorescence), while autolysosomes are visualized as red puncta only (Klionsky et al, 2008).We found cells infected with ExoS‐containing P. aeruginosa exhibited a marked reduction in both autophagosomes (yellow puncta) and autolysosomes (red puncta) (Fig EV4). It suggested ExoS affects autophagy by inhibiting autophagosome formation, instead of increasing autophagosome–lysosome fusion.
Figure EV4. ExoS reduced autophagosome and autolysosome formation.
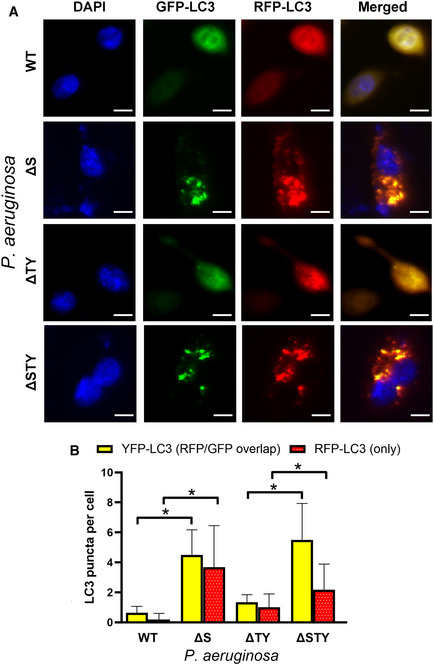
- A549 cells, transfected with RFP‐GFP‐LC3 vectors for 24 h, were infected with different P. aeruginosa mutant strains. Representative images form fluorescence microscopy assays were shown. Scale bars: 10 µm.
- Quantitative analysis of yellow puncta generated from overlapping GFP and RFP puncta (represent autophagosome) and RFP‐LC3 puncta (represent autolysosome). The puncta from more than 100 cells were counted and the ratios of these puncta per cell are shown. Data are mean +/− SD from three independent experiments. The significance of differences between treatments was determined using two‐tailed Student’s t‐test with Welch’s correction, *P ≤ 0.05.
Source data are available online for this figure.
ExoS protected Pseudomonas aeruginosa from elimination by host autophagy
Previous studies suggested ExoS is required for P. aeruginosa survival within epithelial cells (Angus et al, 2010). We reasoned that ExoS promoted intracellular P. aeruginosa viability by inhibiting autophagy. We infected A549 cells, stably expressing GFP‐LC3, with WT, ΔS, ΔTY, and ΔSTY P. aeruginosa and analyzed their colocalization with GFP‐LC3 autophagosomes. We found in infected cells, ExoS‐deficient P. aeruginosa (ΔS and ΔSTY) colocalized significantly more than ExoS‐containing P. aeruginosa (WT and ΔTY) with GFP‐LC3 autophagosomes (Fig 3D and E). Further, in cells infected with ExoS‐expressing P. aeruginosa (WT and ΔTY), the GFP‐LC3 remained mostly diffused indicating that they did not form autophagosomes (Fig 3D and E). This finding suggested that ExoS might protect P. aeruginosa from being engulfed into autophagosome. We also confirmed the result by electron microscopy assays. We found ΔSTY P. aeruginosa were engulfed by multiple membrane autophagosome, whereas ΔTY P. aeruginosa were found in cytoplasm (Fig 3F).
We then evaluated the ability of autophagy to eliminate intracellular P. aeruginosa by determining bacterial survival following infection of normal A549‐(Atg7+) or autophagy‐deficient cell line A549‐(Atg7−). ExoS‐expressing P. aeruginosa (WT or ΔTY) survived to a similar extent in both A549‐(Atg7+) and A549‐(Atg7−) cells. In contrast, ExoS‐deficient P. aeruginosa (ΔS or ΔSTY) viability was markedly reduced in A549‐(Atg7+) cells, compared to A549‐(Atg7−) (Fig 3G). These data suggested that reduction of autophagy in A549 cells enhanced the survival of ExoS‐deficient P. aeruginosa. Taken together, these results showed that ExoS is the main T3SS effector that protected P. aeruginosa from autophagy‐mediated elimination.
ExoS protected Pseudomonas aeruginosa from autophagy‐mediated elimination in vivo
The above in vitro study on epithelial cells indicated ExoS facilitates bacterial survival by inhibiting autophagy. We wonder if ExoS had a similar effect in vivo. We generated conditional knockout Atg7Δ mice with Atg7 deficient in ciliated airway epithelial cells by breeding FOXJ1‐Cre mice with Atg7 flox/flox mice. In transgenic Atg7Δ mice, Atg7 depletion in mouse tracheal airway epithelial cells was validated by immunoblotting and immunofluorescence microscopy (Fig 4A and B). Further, accumulation of P62 and diminished LC3II confirmed autophagy deficiency in these cells (Fig 4A). Examination of lung, spleen and liver from Atg7Δ mice revealed no gross abnormalities and Atg7Δ mice had similar life span to wild‐type Atg7 flox/flox mice. We infected both Atg7Δ and Atg7 flox/flox mice with a non‐lethal dose (3 × 107 CFU) of wild‐type or P. aeruginosa mutants. 24 h post‐infection, bacterial CFU in the lungs were quantitated. No significant difference in bacterial CFU was observed in mice infected with ExoS‐expressing P. aeruginosa (WT or ΔTY). In contrast, a higher bacterial count was observed in autophagy‐deficient Atg7Δ mice when infected with ExoS‐deficient (ΔS or ΔSTY) P. aeruginosa than in normal mice (Atg7 flox/flox) (Fig 4C). These results suggested that, in vivo, ExoS protects P. aeruginosa from elimination by autophagy. We also dissected bacterial dissemination in spleen and liver. Similar to the lungs, no difference in bacterial counts was found in liver and spleen from P. aeruginosa (ΔTY) infected mice. From P. aeruginosa (ΔSTY) infected mice, we also found higher bacterial counts in Atg7Δ mice compared to Atg7 flox/flox mice (Fig 4D). Moreover, higher P. aeruginosa (ΔTY) counts than P. aeruginosa (ΔSTY) was found in all these organs (Fig 4D) which is likely due to ExoS’s ability in facilitating P. aeruginosa dissemination by breaking pulmonary vascular barrier (Rangel et al, 2015).
Figure 4. ExoS protected Pseudomonas aeruginosa from autophagy elimination in vivo .
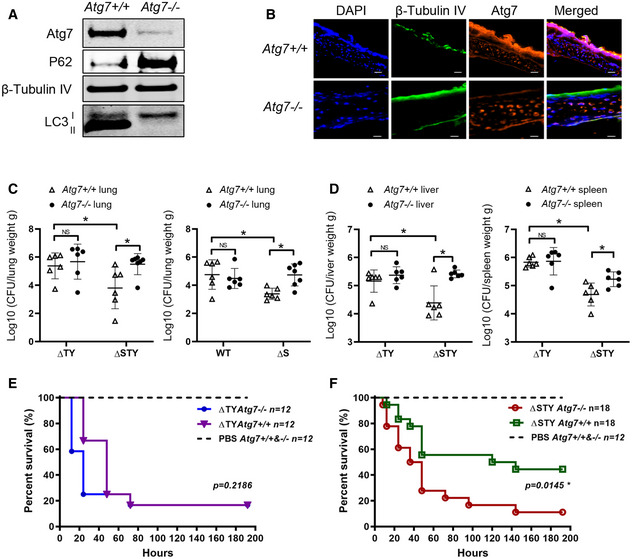
-
APrimary airway epithelial cells were isolated from tracheas of Atg7 +/+ and Atg7 −/− mouse and were evaluated by immunoblotting.
-
BSections of tracheas from Atg7 +/+ and Atg7 −/− mouse were evaluated by immunofluorescence microscopy following staining of nuclear DNA with DAPI (Blue), ciliated epithelial cell marker β‐tubulin IV (green) and Atg7 (red). Scale bars: 20 µm.
-
C, DAtg7 +/+ and Atg7 −/− mice were intranasally injected with 3 × 107 CFU of WT, ΔS, ΔTY or ΔSTY P. aeruginosa. After 24 h, lungs (C), spleens and livers (D) were harvested, homogenized and diluted for CFU counting. Each data point represents bacterial count (Log10 CFU/g) from individual mouse. The horizontal lines represent the mean value from each group (6 mice each group), error bars represent standard deviation. The significance of differences between different drug treatment was determined using one‐way ANOVA and Dunn's multiple‐comparison test., NS: not significant; *P ≤ 0.05
-
E, FAtg7 +/+ and Atg7 −/− mice were intranasally injected with PBS with or without 1 × 109 CFU of ΔTY (E) ΔSTY (F). Mice survival was monitored for 10 days post‐infection. Kaplan–Meier survival curves were made based on the observation of 12–18 mice/group from two independent experiments. The P‐values were calculated using the log‐rank test (ΔTYAtg7 +/+ vs. ΔTYAtg7 −/−, P = 0.2186; ΔSTYAtg7 +/+ vs. ΔSTYAtg7 −/−, P = 0.0145).
Source data are available online for this figure.
We evaluated survival of Atg7flox/flox and Atg7Δ mice in response to lethal dose (1 × 109 CFU) of P. aeruginosa infection. With P. aeruginosa (ΔTY) infection, there was no significant difference in survival rate between the two groups of mice (Fig 4E). In contrast, when infected with (ΔSTY) P. aeruginosa, survival rate was significantly reduced in Atg7Δ mice compared to wild‐type mice (Fig 4F). We believe the higher mortality resulted from higher bacterial burden inside of these autophagy‐deficient mice. Together, these experiments suggest that the P. aeruginosa effector protein ExoS protects the bacteria from elimination by autophagy in vivo. In addition, it also points out the importance of autophagy in airway epithelial cells for the clearance of P. aeruginosa.
ADP‐ribosyl transferase activity of ExoS was required for autophagy inhibition by Pseudomonas aeruginosa
ExoS is a bifunctional enzyme that contains both GTPase‐activating protein and ADP‐ribosyltransferase ADPRT domain (Barbieri & Sun, 2004). We wanted to determine which domain was responsible for autophagy inhibition. We infected cells with wild‐type P. aeruginosa or strains containing point mutations in the GAP or ADPRT domains of ExoS. We found that P. aeruginosa strains with intact ExoS ADPRT activity, that is, WT or P. aeruginosa with ExoS‐containing ADPRT activity but mutated for the GAP activity (SG−A+), were able to inhibit autophagy and reduce LC3II generation in both A549 and NHBE cells (Figs 5A and EV3B). In contrast, P. aeruginosa strains containing ExoS lacking ADPRT activity, that is, P. aeruginosa with ExoS mutated in ADRPT domain (SG+A−) or with ExoS lacking both GAP and ADPRT activity (SG−A−) were not able to inhibit autophagy (Figs 5A and EV3B). These data suggested that ADPRT activity of ExoS was required for P. aeruginosa inhibitory effect on autophagy.
Figure 5. ExoS ADP‐ribosyltransferase activity inhibited autophagy and mTOR signaling pathway.
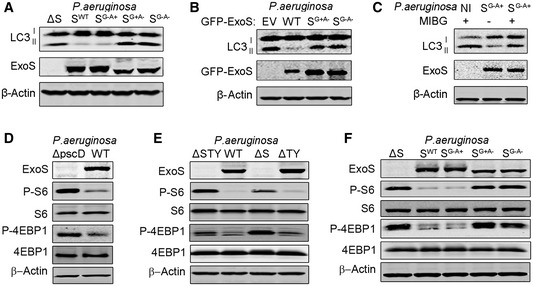
-
A, FA549 cells were infected for 4 h with ExoS‐deficient P. aeruginosa (ΔS), ExoS‐containing P. aeruginosa (SWT), P. aeruginosa containing ExoS with loss‐of‐function mutations in the GTPase‐activating domain (SG−A+), in the ADP ribosylation domain (SG+A−), or in both domains (SG−A−). Cell lysates were evaluated by immunoblotting.
-
BA549 cells were transfected, for 24 h, with a plasmid expressing empty vector (EV), WT GFP‐ExoS, a GFP‐ExoS mutant for ADPRT domain (SG+A−) or a GFP‐ExoS mutant deficient in both GAP and ADPRT domains (SG−A−). Cell lysates were evaluated by immunoblotting.
-
CA549 cells were treated for 24 h with 100 µM of the ADPRT activity inhibitor MIBG and then left uninfected (NI) or infected with P. aeruginosa containing ExoS with ADPRT activity only (SG−A+). Cell lysates were evaluated by immunoblotting.
-
D, EA549 cells were infected for 4 h with WT or P. aeruginosa mutants (ΔpscD, ΔSTY, ΔS, and ΔTY). Cell lysates were evaluated by immunoblotting.
Source data are available online for this figure.
We then wanted to determine if ADPRT activity of ExoS was sufficient to inhibit autophagy in the absence of any other bacterial products. We transfected A549 cells with plasmids expressing ExoS (WT), ExoS without ADPRT activity (SG+A−) or ExoS with no GAP or ADPRT activity (SG−A−). Transfection of cells with plasmids containing WT ExoS resulted in inhibition of autophagy, as evidenced by reduction of LC3II formation, whereas plasmids containing mutated ExoS lacking ADPRT activity had no such effect (Fig 5B). In additional experiments, A549 cells were pre‐treated with the ADPRT inhibitor m‐Iodobenzylguanidine hemisulfate salt (MIBG), which competes with the arginine residues of target proteins for the ADP‐ribose group, thereby inhibiting the ADPRT effect on target molecules (Loesberg et al, 1990). Pre‐treatment of cells with MIBG prevented the reduction of autophagy marker LC3II by P. aeruginosa containing ExoS with only ADPRT activity (SG−A+), (Fig 5C). These experiments suggested that only ADPRT activity of single cytotoxin ExoS was required for cellular autophagy inhibition.
ExoS ADP‐ribosyltransferase activity inhibited mTOR signaling pathway
Because mTOR is a major inhibitor of cellular autophagy (Kim et al, 2011), we initially thought that ExoS inhibitory effect on autophagy might be mediated by stimulation of mTOR activity.
We evaluated mTOR activity by monitoring the phosphorylation levels of its downstream targets S6 and 4EBP1. Unexpectedly, cells infected with ExoS‐expressing P. aeruginosa (WT or ΔTY) exhibited reduced mTOR activity reflected by decreased P‐S6 and P‐4EBP1, compared to cells infected with ExoS‐deficient P. aeruginosa (ΔpscD, ΔS or ΔSTY) (Fig 5D and E). Similar results were found in primary NHBE cells infected with various P. aeruginosa mutants (Fig EV3C). P. aeruginosa containing either ExoT (ΔSY) or ExoY (ΔST), as a single T3SS toxin, did not affect mTOR activity (Fig EV3A). We then wanted to determine if the ADPRT activity of ExoS was responsible for mTOR inhibition. We found that mTOR activity was reduced in cells infected with P. aeruginosa (SG−A+) but not in cells infected with P. aeruginosa (SG+A−) or P. aeruginosa (SG−A−) (Fig 5F). Taken together, these data suggested that ExoS ADPRT activity was required and sufficient to inhibit mTOR signaling pathway.
Pseudomonas aeruginosa ExoS ADP‐ribosyltransferase activity inhibited mTOR through the inhibition of Ras signaling pathway
Previous studies have shown that P. aeruginosa ExoS ADP‐ribosylates RAS and inhibits its activity by promoting the hydrolysis of Ras‐bound GTP to GDP (Barbieri & Sun, 2004). Because Ras signaling has been implicated in mTOR activation (Mihaylova & Shaw, 2011), we sought to determine if mTOR inhibition by ExoS was caused by Ras ADP ribosylation. We transfected A549 cells with a constitutively active form of Ras (G12V) (Ganesan et al, 1998) and found an increase in p‐ERK and p‐S6 (Fig 6A), suggesting that Ras (G12V) was functional in increasing the activity of Ras pathway in these cells. We then infected these cells with P. aeruginosa (ΔTY and ΔSTY). In A549 cells, transfected with empty vector only, infection of P. aeruginosa (ΔTY) resulted in inhibition of mTOR activity (reduced p‐S6) and autophagy (reduced LC3II) (Fig 6A). Interestingly, In A549 cells transfected with constitutively active Ras (G12V), the reduction of phospo‐S6 but not of LC3II was partially rescued (Fig 6A). These data suggested that ADP ribosylation of Ras contributed to the ExoS‐induced inhibition of mTOR activity but not to inhibition of autophagy. We then reasoned that Ras‐G12V only partially rescued mTOR activity because Ras‐G12V, though constitutively active, was still subjected to ADP ribosylation by ExoS on R41 residue, previously shown to be a target for ADP ribosylation (Ganesan et al, 1999a). Therefore, we mutated R41 to K in Ras‐G12V to prevent its ADP ribosylation (Fig EV5A). Similar to Ras‐G12V, exogenously expression of Ras‐G12V&R41K caused phosphorylation of ERK and mTOR activation (Fig EV5B). Importantly, in Ras‐G12V&R41K expressing cells, P. aeruginosa ΔTY infection could still cause significant autophagy inhibition but does not affect TOR activity as no change was found on p‐ERK and p‐S6 (Fig 6B). These data showed that ExoS’s inhibitory effect on mTOR activity was mediated by ExoS‐mediated ADP ribosylation of Ras. They further suggested that the ExoS inhibitory effect on autophagy is independent of the activity of either Ras or mTOR.
Figure 6. ExoS ADP‐ribosyltransferase activity inhibited mTOR through the inhibition of Ras signaling pathway.
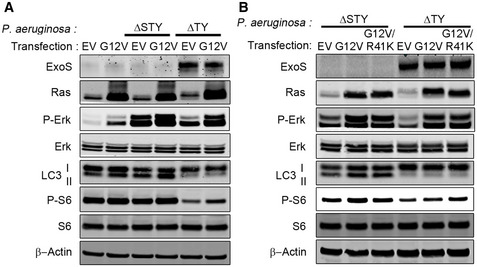
- A549 cells were transfected with empty vector (VE) and vector contains Ras‐G12V for 24 h, followed by a 4‐h infection with P. aeruginosa ΔSTY or ΔTY. Cell lysates were evaluated by immunoblotting.
- A549 cells were transfected, for 24 h, with empty vector (VE) and vectors contain Ras‐G12V or Ras‐G12V&R41K followed by a 4‐h infection with P. aeruginosa ΔSTY or ΔTY. Cell lysates were evaluated by immunoblotting.
Source data are available online for this figure.
Figure EV5. Evaluation of cellular activity of Ras mutants and Working model of ExoS facilitates intracellular survival of P. aeruginosa by inhibiting autophagy and mTOR.
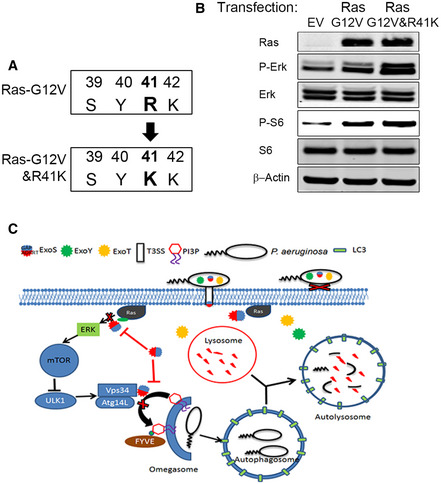
- Schematic of the generation of Ras‐G12V&R41K from Ras‐G12V.
- A549 cells were transfected, for 24 h, with plasmids containing empty vector (EV), Ras‐G12V or Ras‐G12V&R41K and cell lysates were evaluated by immunoblotting.
- Working model of P. aeruginosa ExoS role in inhibiting host cells autophagy and mTOR to facilitate intracellular bacterial survival. P. aeruginosa injects toxins including ExoS, ExoY, and ExoT through the T3SS. ExoS‐mediated ADP ribosylation of RAS and leads to inhibition of mTOR. In addition, ExoS’s ADP ribosylation activity suppresses autophagy initiation complex Atg14L‐Vps34 kinase activity preventing autophagosome formation. The inhibition of autophagy by ExoS ADP ribosylation protects bacteria from elimination by autolysosome digestion.
Source data are available online for this figure.
Pseudomonas aeruginosa ExoS ADP‐ribosyltransferase activity inhibited autophagic Vps34 lipid kinase activity
Our data above suggested that ExoS inhibited autophagy initiation rather than autophagic flux and that effect was mTOR‐independent. To elucidate the underlying mechanism for autophagy inhibition by ExoS, we evaluated the effect of ExoS on the early initiation events in autophagosome formation. Autophagy initiation depends on the formation of phosphatidylinositol 3‐phosphate (PI3P)‐enriched omegasomes, which act as platforms to recruit autophagic proteins (Itakura & Mizushima, 2010; Kim et al, 2015). Autophagic pool of PI3P specifically binds to proteins containing FYVE fingers domain such as double FYVE‐containing protein 1 (DFCP1); thus, fluorescent DFCP1 has been used as an indicator for PI3P’s localization (Itakura & Mizushima, 2010; Kim et al, 2015). We transfected mCherry‐DFCP1 into A549 cells, prior to infection with various P. aeruginosa mutants. mCHerry‐DFCP1 puncta were significantly reduced in cells infected with ExoS‐containing P. aeruginosa (WT and ΔTY), compared to cells infected with ExoS‐deficient P. aeruginosa (ΔS and ΔSTY) (Fig 7A and B). Furthermore, P. aeruginosa containing ADPRT‐competent ExoS (WT or SG−A+), but not P. aeruginosa containing ADPRT‐deficient ExoS (SG+A− or SG−A−), inhibited mCherry‐DFCP1 puncta’s formation (Fig 7A and B). These data suggested that ExoS affected autophagy PI3P production by the ADPRT activity. Class III phosphatidylinositol 3‐kinase Vps34 is the major kinase to produce PI3P (Itakura & Mizushima, 2010; Kim et al, 2015). Vps34 binding with Atg14L to form the Vps34‐Beclin1‐Atg14L complex, which is implicated in the initiation of autophagosome formation (Kim et al, 2013). We hypothesized that P. aeruginosa ExoS inhibited autophagy initiation by inhibiting the activity of Vps34 in the autophagy Atg14L‐Vps34 complex. To test this hypothesis, we transfected A549 cells with Flag‐tagged Atg14L prior to infecting the cells with various P. aeruginosa mutants. The Atg14L‐Vps34 complex was immunoprecipitated from cell lysates (Fig 8A). We then determined the kinase activity of Vps34 from the immunoprecipitated Atg14L‐Vps34 complex using in vitro lipid kinase assay. Atg14L‐Vps34 complex immunoprecipitated from cells infected with ExoS‐containing P. aeruginosa (WT and ΔTY) had reduced activity, measured by produced PI3P, compared to the immunoprecipitated complex from cells infected with ExoS‐deficient bacteria (ΔS and ΔSTY) (Fig 8B). These data suggested that ExoS inhibition of Atg14L‐associated Vps34 activity as an underlying mechanism for reduced autophagy in P. aeruginosa‐infected cells. In vitro kinase assay was also performed on Atg14L‐Vps34 complex derived from cells infected with P. aeruginosa containing various ExoS mutants (SG−A−, SG+A−, SG−A+). Importantly, only the complex derived from cells infected with P. aeruginosa with ADPRT‐active ExoS (SG−A+) exhibited reduced activity (Fig 8C and D). Taken together, these data suggested that P. aeruginosa inhibited host autophagy by a mechanism that involved inhibition of autophagic Vps34 activity by the ADRPT domain of ExoS.
Figure 7. ADP‐ribosyltransferase activity of ExoS was required for its effect on omegasome formation.
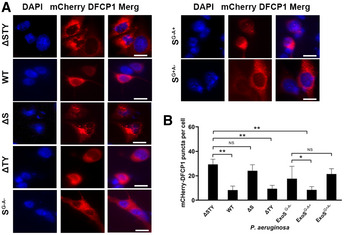
- A549 cells were transfected, for 48 h, with a mCherry‐DFCP1 plasmid and then infected, for 4 h, with P. aeruginosa (WT, ΔSTY, ΔS, ΔTY, SG−A−, SG−A+, or SG+A−) and the mCherry puncta number per cell was analyzed by fluorescence microscopy. Representative immunofluorescence images were showed, and scale bars represent 10 µm.
- Quantitative analysis of mCherry‐DFCP1 puncta by visually count more than 100 cells for each sample. Each bar represents mean value of from three independent experiments and error bars represent standard deviation. The significance of differences between assays was determined using ANOVA with Dunnett multiple‐comparison posttest, NS: not significant; *P ≤ 0.05; **P ≤ 0.01.
Source data are available online for this figure.
Figure 8. ExoS ADP‐ribosyltransferase activity inhibited Vps34 kinase activity.
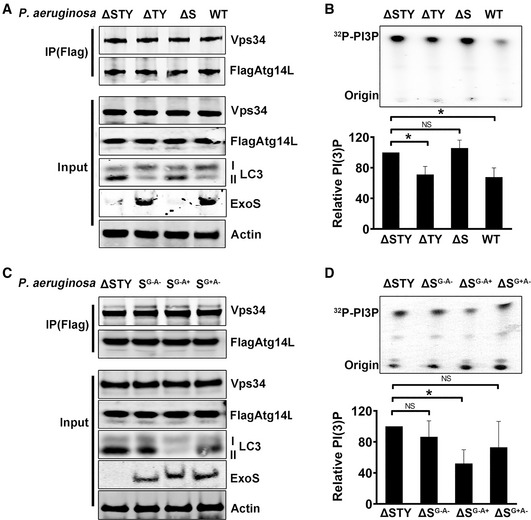
-
A, CA549 cells, transfected for 48 h with vector encoding Flag‐Atg14L, were infected with P. aeruginosa (WT, ΔSTY, ΔS, ΔTY, SG−A−, SG−A+, or SG+A−). Flag‐Atg14L‐Vps34 complex was immunoprecipitated (IP) by Flag‐antibody magnetic beads and evaluated by immunoblotting.
-
B, DThe IP complexes were subjected to in vitro kinase assay using phosphatidylinositol as substrate. The products radiolabeled PI3P were separated by TLC and detected by autoradiography. Data are mean ± SD of three independent experiments. The significance of differences between different assays was determined using one‐way ANOVA analysis followed Dunn's multiple‐comparison posttest, NS: not significant; *P ≤ 0.05.
Source data are available online for this figure.
Discussion
The treatment of P. aeruginosa, one of the most common bacterial pathogens isolated from hospitalized patients, is often challenging due to the high number of multi‐drug resistant P. aeruginosa strains (Williams et al, 2010). New strategies are required to reduce the morbidity and mortality associated with antibiotic‐resistant P. aeruginosa infections. Interactions between P. aeruginosa and epithelial cells are critical in the development of most infections. These interactions depend largely on the spectrum of effectors secreted by the bacterial T3SS (Vance et al, 2005). The secretion systems in P. aeruginosa represent particularly appealing targets because they are essential for bacterial virulence, have conserved components, and depend on specific host factors for their toxicity.
The ability of P. aeruginosa to invade and replicate inside airway epithelial cells has been shown in several studies (Chi et al, 1991; Goldberg & Pier, 2000; Kazmierczak et al, 2004; Angus et al, 2010). Further, the requirement of P. aeruginosa T3SS for the survival of intracellular P. aeruginosa has also been suggested (Angus et al, 2010). However, the mechanisms by which P. aeruginosa T3SS promoted intracellular bacterial survival have not been fully elucidated. The formation of membranes blebs in epithelial cells by P. aeruginosa has been suggested as an intracellular survival strategy used by the bacteria (Angus et al, 2010). The fact that WT P. aeruginosa can survive in the cytoplasm of epithelial cells, whereas P. aeruginosa T3SS mutants cannot, suggests that inhibition of the host defense pathway by P. aeruginosa T3SS may account for P. aeruginosa intracellular survival.
Autophagy is a critical intracellular pathway for the elimination of bacteria (Klionsky & Emr, 2000; Rao & Eissa, 2020). K. pneumoniae and P. aeruginosa are both gram‐negative bacteria and are both typical opportunistic pathogens that cause pulmonary infection (Wang et al, 2019). Lung epithelial cells can actively degrade invading K. pneumoniae, and autophagy is critical for such innate host defense (Cortes et al, 2002; Ye et al, 2014). Our study revealed that P. aeruginosa, but not K. pneumoniae, were able to resist autophagy‐mediated elimination. This observation suggested that P. aeruginosa has adopted a protective strategy to counter cellular autophagy‐mediated elimination. That T3SS‐deficient P. aeruginosa could be eliminated by autophagy indicated that P. aeruginosa utilizes T3SS to resist autophagy‐mediated degradation.
Our study identified a new virulence mechanism of P. aeruginosa mediated by T3SS. Engulfment of ExoS‐deficient P. aeruginosa in autophagosomes suggests that the bacteria are eliminated through autolysosomal digestion. The inhibition of autophagy by P. aeruginosa ExoS explains why ExoS‐containing P. aeruginosa survive better within the epithelial cells compared to the ExoS‐deficient P. aeruginosa. Besides, we have identified ExoS from T3SS as the only toxin that inhibits autophagy. The most remarkable finding of this study is that a single domain of ExoS possessing the ADPRT activity is responsible for the negative regulation of both the mTOR and autophagy pathways in the host cells. The inhibition of mTOR is achieved by ADP ribosylation of Ras, and autophagy is inhibited by reducing the activity of autophagy‐associated kinase Vps34 through an ADP ribosylation mechanism. A working model of the roles of P. aeruginosa ExoS in inhibiting host cells autophagy and mTOR to facilitate intracellular bacterial survival is depicted in Fig EV5C.
Pseudomonas aeruginosa could inject up to four cytotoxins through the T3SS apparatus. The wild‐type strain of P. aeruginosa used in this study contains three cytotoxins ExoS, ExoT, and ExoY (Stover et al, 2000). Based on our results, only ExoS had an inhibitory effect on mTOR and autophagy. Although ExoT is highly homologous to ExoS and also contains the ADPRT domain, it did not have similar effects either on mTOR or autophagy. This differential effect is likely due to ExoS’s special molecular structure which leads to more promiscuous substrate specificity (Barbieri & Sun, 2004; Sun et al, 2004).
ExoS is a bifunctional protein that contains GAP and ADPRT activities, and it is one of the most well‐studied effector proteins (Ganesan et al, 1998; Miao et al, 2008). The ADPRT domain of ExoS has been implicated in several adverse effects in mammalian cells including cell death and cytoskeletal disruption (Hauser, 2009). It has also been linked to bacterial survival and dissemination in mouse models (Rangel et al, 2015). In our study, we have noticed the adverse effects caused by ExoS as described before, namely ExoS‐expressing P. aeruginosa caused increased cell death in vitro, enhanced bacterial dissemination, and significantly higher level of mortality in vivo compared with the ExoS‐deficient mutant (Fig 4). These findings are consistent with the notion that the cytotoxin ExoS contribute to P. aeruginosa virulence (Rangel et al, 2015). It also points out the importance of autophagy in airway epithelial cells for the clearance of P. aeruginosa. The in vivo results showing that ExoS‐deficient P. aeruginosa were eliminated more efficiently in normal mice than in autophagy‐deficient mice indicates that autophagy in airway epithelial cells is an important defense mechanism against pulmonary pathogens including P. aeruginosa.
The ADPRT domain of ExoS was also critical for the survival of intracellular P. aeruginosa as this domain inhibits vacuolar acidification. This, in turn, would eventually prevent the degradation of the internalized bacterial in the acidified phagolysosome (Angus et al, 2010; Heimer et al, 2013). Our finding that the ADPRT activity of ExoS inhibits autophagy revealed a new mechanism of how this toxin facilitates intracellular bacterial survival. Unlike Shigella virulence factor IcsB which facilitate intracellular Shigella survival by avoiding autophagy (Ogawa et al, 2005), ExoS from P. aeruginosa protects the bacteria by suppressing autophagy. The autophagy pathway involves a cascade of events comprising of multiple proteins and enzymes. In our study, there were no significant changes in protein levels of the key autophagy proteins (Atg7, Atg5, Atg4, Atg3, Beclin1, Atg14L, and LC3) following P. aeruginosa infection, suggesting that autophagy was regulated by ExoS at a posttranslational level. LC3II is known to be required for specific cargo recognition, and it is one of the first proteins recruited to the site of infection. The Legionella effector cytotoxin RavZ is able to inhibit autophagy by directly hydrolyzing the conjugated LC3II. Even though ExoS ADP ribosylation domain has a broad substrate specificity, we were not able to demonstrate that LC3I was ADP‐ribosylated by ExoS.
The mTOR pathway is a key regulator of autophagy and of cellular growth and metabolism. Some bacteria like Shigella and Salmonella inhibit mTOR and cause membrane damage. The host cells, in turn, trigger a protective innate immune response, including autophagy activation, to restrain the bacteria (Tattoli et al, 2012). In the case of P. aeruginosa, the inactivation of the Ras signaling pathway by ExoS leads to mTOR inhibition, which would otherwise lead to autophagy activation. However, we observed autophagy inhibition with the presence of ExoS which seemed counterintuitive. Our finding that ExoS directly influences the autophagic Atg14L‐Vps34 complex kinase activity explains how ExoS caused both mTOR inhibition and autophagy inhibition at the same time, though by different mechanisms. As mTOR’s effect on autophagy is also mediated by ATG14L‐Vps34 complex kinase activity (Russell et al, 2013), it is likely that the inhibitory effect of ExoS overcomes autophagy induction caused by the inhibition of mTOR.
The autophagic Vps34 complex activity is tightly regulated by ATG14L‐Vps34‐Beclin1 interaction (Kim et al, 2013; Russell et al, 2013; Qian et al, 2017). Through our study, we did not observe noticeable change in the interaction between Vps34, Beclin1, and ATG14L. Besides, we did not detect the ADP ribosylation of any of the components of the autophagic Vps34 complex. It suggests that autophagic Vps34 complex activity was not directly influenced by ExoS‐mediated ADP ribosylation. However, we could not rule out the possibility that ExoS ADPRT activity could indirectly affect Vps34 kinase molecular conformation, which could also result in the reduction of its kinase activity because conformational changes in Vps34 can affect its kinase activity without affecting its interactions with ATG14L or Beclin1 (Qian et al, 2017). A greater understanding of how exactly P. aeruginosa ExoS ADPRT activity inhibits autophagy could provide insights into novel therapeutic strategies to combat this difficult‐to‐treat pathogen. Additionally, a therapeutic compound targeting the ExoS ADPRT could be used not only to prevent ExoS‐mediated cytotoxic effects but also to benefit P. aeruginosa elimination via autophagy.
Materials and Methods
Antibodies and reagents
Antibodies used: rabbit anti‐LC3 (Novus); rabbit anti‐Atg7 (Santa Cruz, sc‐33211); mouse anti‐SQSTM1/P62 (Santa Cruz, sc‐28359); rabbit anti‐phospho‐S6 ribosomal protein (Cell Signaling, #2211); mouse anti‐S6 ribosomal protein (Cell Signaling, #2317); rabbit anti‐Beclin1(Cell signaling, #3495); rabbit anti‐phospho‐4EBP1 (Cell Signaling, #9451); rabbit anti‐4EBP1 (Cell Signaling, #9644); rabbit anti‐phospho‐ERK (Cell Signaling, # 4377); rabbit anti‐ERK (Cell Signaling, # 9102); rabbit anti‐Ras (Cell Signaling, #3965); mouse anti‐Actin (Ambion, #AM4302); mouse anti‐β‐Tubulin IV (Sigma, T7941); rabbit anti‐ExoS, ExoT, ExoY were gifts from Dr. Arne Rietsch (Case Western Reserve University, Ohio); rabbit anti‐Vps34 (Cell Signaling, #4263); rabbit anti‐Atg14L (Cell Signaling, #5504); rabbit anti‐Pseudomonas aeruginosa (Abcam, ab37057); Rapamycin (Sigma, R0395); Chloroquine, (Sigma, C6628); m‐Iodobenzylguanidine hemisulfate salt, (Sigma, I9890); Cetrimide Agar, (Sigma, 22470); Gentamycin, (Sigma, G1522‐10ML).
Plasmids
Ras‐G12V&R41K mutant was generated from a pCDNA3 plasmid containing the Ras G12V sequence (a gift from Dr. Seamus J. Martin). All mutant plasmids were verified by sequencing. pEGFP‐ExoS, pEGFP‐ExoSG+A−, pEGFP‐ExoSG−A− obtained from Shouguang Jin’s laboratory (University of Florida). Plasmids mCherry‐DFCP1 (item #86746) were purchased from Addgene. RFP‐GFP‐LC3 was constructed based on pEGFP‐LC3 (Plasmid #21073 Addgene) by in‐frame insertion of RFP gene between NheI and AgeI. pcDH GFP‐LC3‐B was obtained by subclone of GFP‐LC3‐B into a pcDH‐CMV‐mcs‐GF1‐puro vector (SBI System Biosciences, CD510B‐1). Plasmid express Flag‐Atg14L was provided by Dr. Xu Qian in Zhimin Lu’s laboratory from MD Anderson Cancer Center.
Cell culture
A549 cells were obtained from the American Type Culture Collection (ATCC) and cultured in Ham’s F‐12K media (Corning) supplemented with 10% fetal bovine serum (FBS), 50 IU/ml penicillin, and 50 µg/ml streptomycin. Normal human bronchial primary epithelial cells (NHBE) were obtained from Lonza (CC‐0225) and maintained in bronchial epithelial cell growth medium with Growth Supplements (Lonza CC‐3170). Differentiated epithelial cells were achieved as described previously by plated the cells onto semipermeable membrane inserts (Corning) and cultured at the air/liquid interface (Sha et al, 2009). All cells cultures were maintained at 37°C in humidified 5% CO2 cell culture incubator.
Generation of stable cell line
Atg7‐deficient A549 stable cell line A549‐(Atg7−), was generated using a shRNA against Atg7 (Sigma, Mission shRNA, TRCW0000007584). Atg7 shRNA was packed into a lentivirus system (SBI, System Bioscience, CD500) and used to transduce A549 cells. A549 cells stably expressing Atg7 shRNA were maintained in the presence of puromycin (1 µg/ml). A549 cells stably expressing a non‐targeting scrambled shRNA were used as control. A549 cell stably expressing GFP‐LC3 was generated using lentiviral vector pcDH GFP‐LC3‐B and maintained in the presence of puromycin (1 µg/ml). Other plasmids transfection was performed using Lipofectamine LTX (Life Technologies) according to its manufacturers' protocol.
Bacterial strains and infection
Klebsiella pneumonia (#43816™) obtained from ATCC. Pseudomonas aeruginosa wide type and mutants obtained from Dr Arne Rietsch’s laboratory (Cisz et al, 2008). Bacterial cultures were grown in Luria‐Bertani (LB) broth at 37°C with vigorous shaking. Overnight grown of bacterial were diluted 1:5 and grown for 2 more hours. Bacterial cultures were collected by centrifugation 2,000 × g for 5 min, washed in phosphate‐buffered saline (PBS), centrifuged and normalized to 2 × 109 colony‐forming unit (CFU)/ml. The general infection protocol for cell was as follow: (i) a monolayer of cells was washed three times with PBS, (ii) bacteria were suspended in F12K media supplemented with FBS but with no antibiotics at a multiplicity of infection of 10 bacteria per cell, (iii) cells and bacterial incubated in 5% CO2 at 37°C for 4 h unless otherwise indicated.
Colony‐forming unit assay
A549 cells (1 × 105 cells per well) were plated in 12‐well plate 1 day prior to infection. Cells were inoculated with bacteria at a multiplicity of infection (MOI) of 10 and incubated for 1 or 4 h at 37°C/5% CO2. Cells were then treated for 1 h with gentamycin 100 µg/ml to kill extracellular bacteria. Cells were then washed three times with PBS and lysed with 1 ml of 0.1% Trition X‐100 in PBS. Lysates (10 µl) were plated in LB agar in 10‐fold dilutions, and CFU were determined by direct counting after 18 h. In experiments involving rapamycin, cells were treated with rapamycin (0.8 μg/ml) 18 h prior to infection.
Immunoblot analysis
Cells were washed with PBS and then lysed in Triton X‐100 lysis buffer (40 mM Bis‐Tris propane, 150 mM NaCl, adjust pH to 7.7, 10% glycerol, 1% Triton X‐100) in presence of protease and phosphatase inhibitor cocktail (Thermo Scientific). Equal amount of protein was separated by NuPAGE Bis‐Tris‐Gel (Novex, life technologies) and transferred onto nitrocellulose membranes. The membranes were incubated with the appropriate antibodies, and the blots were analyzed with Odyssey imaging detection system (LI‐COR).
Fluorescence microscopy and flow cytometry
Cells were fixed with 4% paraformaldehyde for 10 min, permeabilized with 0.2% Triton X‐100 for 20 min and mounted using the Slow Fade Gold antifade reagent with DAPI (Invitrogen) and visualized with a Zeiss Axiovert 200M microscope. For trachea immunofluorescence staining, 6‐µm mouse trachea frozen sections were fixed in 4% paraformaldehyde for 10 min at room temperature, blocked with 10% normal goat serum (NGS), and then incubated with antibodies. Autophagy analysis was done based on published guideline (Klionsky et al, 2008) Fluorescence of GFP‐LC3 or mCherry‐DFCP1 puncta was counted with at least 10 independent visual fields at 400× magnification, and triplicate samples were counted in each experimental condition. To detect the cell viability, 1 × 106 cells were harvested and washed three times with PBS and then resuspend into flow cytometry buffer containing 5 µl of SYTOX™ Green (Thermo Fisher) or propidium iodide stock solution (Thermo Fisher). With an incubation of 5 min the stained cells were analyzed on LSR Fortessa flow cytometer (BD Biosciences). Data were acquired through FACS Diva (BD Biosciences) and analyzed with FlowJo software (TreeStar).
Transmission electron microscopy
Cells were fixed in 2% formaldehyde +2.5% glutaraldehyde in 0.1 M Millonig's phosphate buffer, pH 7.4 for 2 h at room temperature, postfixed in 1% OsO4 in 0.1 M Millonig's phosphate buffer for 1 h, dehydrated with increasing concentration of ethanol (70, 95, 100%), and infiltrated with Spurr's Low Viscosity Resin. The sections were cut at 60–70 nm on an RMC MT‐6000XL ultra‐microtome with a Diatome Ultra45 diamond knife. Cells were stained en bloc in saturated uranyl acetate dissolved in 50% ethanol during the dehydration phase of processing and were then visualized with a Hitachi H7500 transmission electron microscope equipped with a Gatan US1000 2 Mp camera and Digital Micrograph software, v1.82.366 to capture images.
Mice
FOXJ1‐Cre transgenic mice were purchased from Jackson Laboratory. Atg7flox/flox mice which containing Atg7 gene flanked by LoxP sites were obtained from Dr Masaaki Komatsu’s laboratory. Transgenic mice with Atg7 conditionally knockout in ciliated airway epithelial cell were generated by crossing FOXJ1‐Cre mice to Atg7 flox/flox mice. Mice were housed within a specific pathogen‐free vivarium and used at 4–12 weeks of age. The Baylor College of Medicine’s Institutional Animal Care and Use Committee approved all animal studies.
Isolation of tracheal epithelial cells
Tracheas were collected from mice, sliced longitudinally and kept overnight in DMEM/F12 media supplemented with pronase (1.5 mg/ml), DNase I (0.1 mg/ml), penicillin (50 IU) and streptomycin (50 µg/ml). Next day, protease activity was halted using 10% FBS and the mixture was inverted several times to dislodge cells from tracheas. Tracheas were removed using a cell strainer. Contaminating fibroblasts were removed by adherence to plastic. Epithelial cells in suspension were collected by centrifugation and cultured in bronchial epithelial cell growth media in collagen‐coated 6‐well plates.
In vivo P. aeruginosa infection
Mice were anesthetized with isoflurane, and 20 µl of PBS containing a non‐lethal dose of P. aeruginosa (3 × 107 CFU) was instilled to the airway by inhalation through the nasal opening. After 24 h, different organs were excised and homogenized in PBS solution and homogenized using an OMNI TIP Homogenizing kit (OMNI International, TH‐02) under aseptic condition. Lysates were plated in Cetrimide Agar plates in 10‐fold dilutions and CFU were determined by direct counting. For mice survival studies, a lethal dose of P. aeruginosa (1 × 109 CFU) were injected into mice intranasally and mice mortality were monitored for 10 days.
Immunoprecipitation and Vps34 lipid kinase assay
Flag‐tagged ATG14L in the Vps34 complex was immunoprecipitated using a Pierce crosslink Magnetic Co‐IP Kit (Thermo Scientific: 88805). Vps34 kinase activity was done, as previously described (Munson & Ganley, 2016). Briefly, phosphatidylinositol (Avanti, 840042) was dissolved in reaction buffer to produce stable 2 mg/ml phosphatidylinositol liposome by passing through Mini extruder set (Avanti, 610000). Immunoprecipitated ATG14L‐Vps34 complex was suspended in 60 µl of reaction buffer containing phosphatidylinositol. The reaction was started by adding 1 µM ATP and 1μCi 32P‐γ‐ATP (PerkinElmer, NEG002A) and incubated at ambient temperature for 30 min. The reaction was stopped by adding chloroform: methanol: 12 M HCl (100:200:3.5, v/v) and then subjected to phase‐split by the addition of 180 µl chloroform and subsequently 300 µl 0.1 M HCl. Extracted phospholipid products were separated by thin layer chromatography (TLC) using a coated silica gel and a solvent composed of Methanol/Chloroform/H2O/14.5 M NH4OH (47:60:11.2:2, v/v). The TLC gels were dried and exposed by autoradiography to visualize the production of isotope labeled PI3P.
Statistics
Statistical analyzed were performed with GraphPad Prism8. All data tested for significance are from at least three independent experiments. Differences between two groups were compared using Student's t‐test. Multiple comparison was analyzed using one‐way ANOVA analysis following different comparison test as specified in figure legend. P value of 0.05 was considered significant.
Author contributions
Conceptualization, LR, IDLR, and NTE; Performed experiments, LR, IDLR, YX; Methodology and Data analysis, LR, IDLR, YX, YS, AB; Reagents & Resource, MJH, BEG, and NTE, Writing—original draft, LR and IDLR; Writing—review & editing, LR, IDLR, MJH, BEG, and NTE.
Conflict of interest
The authors declare that they have no conflict of interest.
Supporting information
Expanded View Figures PDF
Source Data for Expanded View
Review Process File
Source Data for Figure 1
Source Data for Figure 2
Source Data for Figure 3
Source Data for Figure 4
Source Data for Figure 5
Source Data for Figure 6
Source Data for Figure 7
Source Data for Figure 8
Acknowledgements
We thank current and former members of the Eissa laboratory for useful discussions and technical assistance. We thank Dr. Arne Rietsch for wild‐type and mutants of Pseudomonas aeruginosa; Drs. Shouguang Jin and Joanne Engel for ExoS plasmids; Dr. Xu Qian and Zhimin Lu for Atg14L plasmid and Dr. Seamus J. Martin for the Ras vectors. This study was supported by funding from the National Heart, Lung and Blood Institute.
EMBO reports (2021) 22: e50613.
Data availability
All data of this study are present in the paper or provided as supporting materials. No data deposited in a public database.
References
- Angus AA, Evans DJ, Barbieri JT, Fleiszig SM (2010) The ADP‐ribosylation domain of Pseudomonas aeruginosa ExoS is required for membrane bleb niche formation and bacterial survival within epithelial cells. Infect Immun 78: 4500–4510 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barbieri JT, Sun J (2004) Pseudomonas aeruginosa ExoS and ExoT. Rev Physiol Biochem Pharmacol 152: 79–92 [DOI] [PubMed] [Google Scholar]
- Chi E, Mehl T, Nunn D, Lory S (1991) Interaction of Pseudomonas aeruginosa with A549 pneumocyte cells. Infect Immun 59: 822–828 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cisz M, Lee PC, Rietsch A (2008) ExoS controls the cell contact‐mediated switch to effector secretion in Pseudomonas aeruginosa . J Bacteriol 190: 2726–2738 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cortes G, Alvarez D, Saus C, Alberti S (2002) Role of lung epithelial cells in defense against Klebsiella pneumoniae pneumonia. Infect Immun 70: 1075–1080 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crystal RG, Randell SH, Engelhardt JF, Voynow J, Sunday ME (2008) Airway epithelial cells: current concepts and challenges. Proc Am Thorac Soc 5: 772–777 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Diamond G, Legarda D, Ryan LK (2000) The innate immune response of the respiratory epithelium. Immunol Rev 173: 27–38 [DOI] [PubMed] [Google Scholar]
- Ganesan AK, Frank DW, Misra RP, Schmidt G, Barbieri JT (1998) Pseudomonas aeruginosa exoenzyme S ADP‐ribosylates Ras at multiple sites. J Biol Chem 273: 7332–7337 [DOI] [PubMed] [Google Scholar]
- Ganesan AK, Mende‐Mueller L, Selzer J, Barbieri JT (1999a) Pseudomonas aeruginosa exoenzyme S, a double ADP‐ribosyltransferase, resembles vertebrate mono‐ADP‐ribosyltransferases. J Biol Chem 274: 9503–9508 [DOI] [PubMed] [Google Scholar]
- Ganesan AK, Vincent TS, Olson JC, Barbieri JT (1999b) Pseudomonas aeruginosa exoenzyme S disrupts Ras‐mediated signal transduction by inhibiting guanine nucleotide exchange factor‐catalyzed nucleotide exchange. J Biol Chem 274: 21823–21829 [DOI] [PubMed] [Google Scholar]
- Goldberg JB, Pier GB (2000) The role of the CFTR in susceptibility to Pseudomonas aeruginosa infections in cystic fibrosis. Trends Microbiol 8: 514–520 [DOI] [PubMed] [Google Scholar]
- Hauser AR (2009) The type III secretion system of Pseudomonas aeruginosa: infection by injection. Nat Rev Microbiol 7: 654–665 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heimer SR, Evans DJ, Stern ME, Barbieri JT, Yahr T, Fleiszig SM (2013) Pseudomonas aeruginosa utilizes the type III secreted toxin ExoS to avoid acidified compartments within epithelial cells. PLoS One 8: e73111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Itakura E, Mizushima N (2010) Characterization of autophagosome formation site by a hierarchical analysis of mammalian Atg proteins. Autophagy 6: 764–776 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jia J, Wang Y, Zhou L, Jin S (2006) Expression of Pseudomonas aeruginosa toxin ExoS effectively induces apoptosis in host cells. Infect Immun 74: 6557–6570 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jia K, Thomas C, Akbar M, Sun Q, Adams‐Huet B, Gilpin C, Levine B (2009) Autophagy genes protect against Salmonella typhimurium infection and mediate insulin signaling‐regulated pathogen resistance. Proc Natl Acad Sci USA 106: 14564–14569 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kazmierczak BI, Mostov K, Engel JN (2004) Epithelial cell polarity alters Rho‐GTPase responses to Pseudomonas aeruginosa . Mol Biol Cell 15: 411–419 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim J, Kundu M, Viollet B, Guan KL (2011) AMPK and mTOR regulate autophagy through direct phosphorylation of Ulk1. Nat Cell Biol 13: 132–141 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim J, Kim YC, Fang C, Russell RC, Kim JH, Fan W, Liu R, Zhong Q, Guan KL (2013) Differential regulation of distinct Vps34 complexes by AMPK in nutrient stress and autophagy. Cell 152: 290–303 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim YM, Jung CH, Seo M, Kim EK, Park JM, Bae SS, Kim DH (2015) mTORC1 phosphorylates UVRAG to negatively regulate autophagosome and endosome maturation. Mol Cell 57: 207–218 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klionsky DJ, Abeliovich H, Agostinis P, Agrawal DK, Aliev G, Askew DS, Baba M, Baehrecke EH, Bahr BA, Ballabio A et al (2008) Guidelines for the use and interpretation of assays for monitoring autophagy in higher eukaryotes. Autophagy 4: 151–175 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klionsky DJ, Emr SD (2000) Autophagy as a regulated pathway of cellular degradation. Science 290: 1717–1721 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levine B, Deretic V (2007) Unveiling the roles of autophagy in innate and adaptive immunity. Nat Rev Immunol 7: 767–777 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Y, Wang Y, Liu X (2012) The role of airway epithelial cells in response to mycobacteria infection. Clin Dev Immunol 2012: 791392 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loesberg C, van Rooij H, Smets LA (1990) Meta‐iodobenzylguanidine (MIBG), a novel high‐affinity substrate for cholera toxin that interferes with cellular mono(ADP‐ribosylation). Biochem Biophys Acta 1037: 92–99 [DOI] [PubMed] [Google Scholar]
- Miao EA, Ernst RK, Dors M, Mao DP, Aderem A (2008) Pseudomonas aeruginosa activates caspase 1 through Ipaf. Proc Natl Acad Sci USA 105: 2562–2567 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mihaylova MM, Shaw RJ (2011) The AMPK signalling pathway coordinates cell growth, autophagy and metabolism. Nat Cell Biol 13: 1016–1023 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Munson MJ, Ganley IG (2016) Determination of VPS34/PIK3C3 Activity in vitro Utilising 32P‐gammaATP. Bio Protoc 6: e1904 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakagawa I, Amano A, Mizushima N, Yamamoto A, Yamaguchi H, Kamimoto T, Nara A, Funao J, Nakata M, Tsuda K et al (2004) Autophagy defends cells against invading group A Streptococcus . Science 306: 1037–1040 [DOI] [PubMed] [Google Scholar]
- Ogawa M, Yoshimori T, Suzuki T, Sagara H, Mizushima N, Sasakawa C (2005) Escape of intracellular Shigella from autophagy. Science 307: 727–731 [DOI] [PubMed] [Google Scholar]
- Qian X, Li X, Cai Q, Zhang C, Yu Q, Jiang Y, Lee JH, Hawke D, Wang Y, Xia Y et al (2017) Phosphoglycerate kinase 1 phosphorylates Beclin1 to induce autophagy. Mol Cell 65: 917–931.e6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rangel SM, Diaz MH, Knoten CA, Zhang A, Hauser AR (2015) The role of ExoS in dissemination of Pseudomonas aeruginosa during pneumonia. PLoS Pathog 11: e1004945 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rao L, Eissa NT (2020) Autophagy in pulmonary innate immunity. J Innate Immun 12: 21–30 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Russell RC, Tian Y, Yuan H, Park HW, Chang YY, Kim J, Kim H, Neufeld TP, Dillin A, Guan KL (2013) ULK1 induces autophagy by phosphorylating Beclin‐1 and activating VPS34 lipid kinase. Nat Cell Biol 15: 741–750 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scheetz MH, Hoffman M, Bolon MK, Schulert G, Estrellado W, Baraboutis IG, Sriram P, Dinh M, Owens LK, Hauser AR (2009) Morbidity associated with Pseudomonas aeruginosa bloodstream infections. Diagn Microbiol Infect Dis 64: 311–319 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sha Y, Pandit L, Zeng S, Eissa NT (2009) A critical role for CHIP in the aggresome pathway. Mol Cell Biol 29: 116–128 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stover CK, Pham XQ, Erwin AL, Mizoguchi SD, Warrener P, Hickey MJ, Brinkman FS, Hufnagle WO, Kowalik DJ, Lagrou M et al (2000) Complete genome sequence of Pseudomonas aeruginosa PAO1, an opportunistic pathogen. Nature 406: 959–964 [DOI] [PubMed] [Google Scholar]
- Sun J, Maresso A, Kim J‐J, Barbieri J (2004) How bacterial ADP‐ribosylating toxins recognize substrates. Nat Struct Mol Biol 11: 868–876 [DOI] [PubMed] [Google Scholar]
- Sun Y, Karmakar M, Taylor PR, Rietsch A, Pearlman E (2012) ExoS and ExoT ADP ribosyltransferase activities mediate Pseudomonas aeruginosa keratitis by promoting neutrophil apoptosis and bacterial survival. J Immunol 188: 1884–1895 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tattoli I, Sorbara MT, Vuckovic D, Ling A, Soares F, Carneiro LA, Yang C, Emili A, Philpott DJ, Girardin SE (2012) Amino acid starvation induced by invasive bacterial pathogens triggers an innate host defense program. Cell Host Microbe 11: 563–575 [DOI] [PubMed] [Google Scholar]
- Vance RE, Rietsch A, Mekalanos JJ (2005) Role of the type III secreted exoenzymes S, T, and Y in systemic spread of Pseudomonas aeruginosa PAO1 in vivo . Infect Immun 73: 1706–1713 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang K, Chen Y, Zhang P, Lin P, Xie N, Wu M (2019) Protective features of autophagy in pulmonary infection and inflammatory diseases. Cells 8: 123 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Williams BJ, Dehnbostel J, Blackwell TS (2010) Pseudomonas aeruginosa: host defence in lung diseases. Respirology 15: 1037–1056 [DOI] [PubMed] [Google Scholar]
- Yasmin L, Jansson AL, Panahandeh T, Palmer RH, Francis MS, Hallberg B (2006) Delineation of exoenzyme S residues that mediate the interaction with 14‐3‐3 and its biological activity. FEBS J 273: 638–646 [DOI] [PubMed] [Google Scholar]
- Ye Y, Li X, Wang W, Ouedraogo KC, Li Y, Gan C, Tan S, Zhou X, Wu M (2014) Atg7 deficiency impairs host defense against Klebsiella pneumoniae by impacting bacterial clearance, survival and inflammatory responses in mice. Am J Physiol Lung Cell Mol Physiol 307: L355–363 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yuan K, Huang C, Fox J, Laturnus D, Carlson E, Zhang B, Yin Q, Gao H, Wu M (2012) Autophagy plays an essential role in the clearance of Pseudomonas aeruginosa by alveolar macrophages. J Cell Sci 125: 507–515 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Expanded View Figures PDF
Source Data for Expanded View
Review Process File
Source Data for Figure 1
Source Data for Figure 2
Source Data for Figure 3
Source Data for Figure 4
Source Data for Figure 5
Source Data for Figure 6
Source Data for Figure 7
Source Data for Figure 8
Data Availability Statement
All data of this study are present in the paper or provided as supporting materials. No data deposited in a public database.